

Abstract
Malignant melanoma is highly lethal due to its aggressive invasive properties and metastatic dissemination. The transcription factor E2F1 is crucial for melanoma progression through poorly understood mechanisms. Here, we show that the miR-224/miR-452 cluster is significantly increased in advanced melanoma and invasive/metastatic cell lines that express high levels of E2F1. miR-224/miR-452 expression is directly activated by E2F1 through transactivation of the GABRE gene. Ectopic expression of miR-224/miR-452 in less aggressive cells induces EMT and cytoskeletal rearrangements and enhances migration/invasion. Conversely, miR-224/miR-452 depletion in metastatic cells induces the reversal of EMT, inhibition of motility, loss of the invasive phenotype and an absence of lung metastases in mice. We identify the metastasis suppressor TXNIP as new target of miR-224/miR-452 that induces feedback inhibition of E2F1 and show that miR-224/452-mediated downregulation of TXNIP is essential for E2F1-induced EMT and invasion. The E2F1-miR-224/452-TXNIP axis constitutes a molecular signature that predicts patient survival and may help to set novel therapies.
Subject Categories Cancer; Cell Adhesion, Polarity & Cytoskeleton; RNA Biology
Keywords: E2F1 transcription factor, epithelial-mesenchymal transition, melanoma metastasis, miRNA cluster, thioredoxin-interacting protein
Introduction
Malignant melanoma as a highly aggressive tumor is characterized by strong metastasis and a pronounced chemoresistance 1,2. Reasons for the poor therapeutic suggestibility in advanced stages of this cancer type are defects in apoptotic signaling pathways 2. Our group has shown first that the E2F1 transcription factor (TF) is key to driving metastasis of melanoma cells 3. E2F1 acts as tumor suppressor or oncogene 4,5. Cellular stress signals, such as uncontrolled cell proliferation and DNA damage, initiate activation of E2F1 as a mediator of apoptosis. However, E2F1 loses its tumor suppressor function in highly aggressive, apoptosis-resistant tumor types like malignant melanoma and contributes to tumor progression 3,6,7.
The exact mechanism of E2F1-induced metastasis is not completely understood. A promising approach to elucidate the molecular issues involved in the acquisition of an enhanced aggressive phenotype essential for E2F1-related tumor progression and metastasis of melanoma is the investigation of dysregulated microRNAs (miRs). MiRs represent a large class of non-protein-coding RNAs, which act as negative gene regulators 8. They bind their target mRNA sequence either with perfect or imperfect complementary, resulting in RNA degradation or inhibition of translation 8. Detailed descriptions about miRNA biogenesis and mode of action are given in numerous reviews. According to bioinformatic analysis, each miRNA is able to control hundreds of target mRNAs, allowing this class to manipulate almost every cellular scenario 9. MiRs are key players in carcinogenesis, as they participate in events that are impaired in cancer, for example proliferation and apoptosis 10. Since different reports hint toward an interaction of oncogenic miRNAs and E2F1 11, an involvement of miRNAs in E2F1-induced cancer metastasis is conceivable. To date, numerous miRs with therapeutic potential have been identified in a variety of tumor types 12. An interesting example is miR-224, which is well known for its tumorigenic function in hepatocellular carcinoma (HCC) 13. This miRNA is significantly upregulated during HCC development 14,15 and contributes to tumor-relevant processes like migration, invasion, proliferation and apoptosis inhibition 13,16. In line with these reports, miR-224 is also increased in other tumor types, like clear cell renal cell carcinoma (ccRCC) 17,18, colorectal cancer (CRC) 19–21 and glioma 22, having an impact on tumor progression. Nevertheless, miR-224 overexpression is also described as an indication for a more favorable prognosis in medullary thyroid carcinoma (MTC) patients, since tumors with high content of miR-224 did not show any node metastases but biochemical healing in post-treatment 23. Similar findings apply to medulloblastoma, particularly as enhanced miR-224 expression improves responsiveness to radiation and decreases proliferation as well as the transforming capability of medulloblastoma cells 24. Furthermore, underexpression of miR-224 was identified as a biomarker of oral squamous cell carcinoma (OSCC) 25. However, apart from the last-mentioned publications, there are considerably more, suggesting a tumorigenic function of miR-224. Interestingly, miR-224 is part of a miRNA cluster 24. Together with miR-452, it is located intronic in the GABRE gene, which encodes the epsilon subunit of the gamma-aminobutyric acid (GABA) A receptor 24. In most cases, closely arranged miRs are under the control of common regulatory elements and therefore possess similar biological functions 26. MiR-452 is dysregulated during diabetic wound healing 27 and decreased upon cigarette smoking in alveolar macrophages 28. In the course of myoblast differentiation, a reduction of miR-452 was recorded 29. Furthermore, miR-452 is inhibited by the tumorigenic SOX2 gene in glioblastoma cells 30,31 and assumed to have a tumor- suppressive potential in medulloblastomas 24. Intriguingly, miR-452 and miR-452* are described as prognostic markers in urothelial carcinoma, since strong expression of these miRs correlates with the incidence of lymph node metastases and an unfavorable prognosis for patients 32. MiR-452 was further validated as a urinary marker for bladder cancer determination 33. Moreover, this miRNA is highly expressed in esophageal cancer tissue 34, in prostate cancer stem/progenitor cells 35, as well as in neural crest cells 36. In the latter, miR-452 was shown to have an influence on an epithelial-mesenchymal signaling pathway in the first pharyngeal arch 36.
Regarding putative oncogenic activities of miR-224 in various malignancies and first hints about overexpression of miR-452 in some kinds of human cancer, we investigated the role of the whole cluster in melanoma progression. Here, we show that expression of miR-224/452 in invasive/metastatic melanoma is controlled by E2F1 causing a decrease of the metastasis suppressor TXNIP that blocks E2F1 in a regulatory loop. Our results demonstrate a novel transcription factor (E2F1)-miRNA axis that is activated during melanoma progression and promotes reversible phenotypic changes toward epithelial-mesenchymal transition (EMT) and invasion.
Results
E2F1 induces miR-224/452 expression during melanoma development
First, we analyzed the expression levels of potentially oncogenic miRs in primary and metastatic patient samples and found a high content of miR-224 and miR-452 in the latter (Fig1A, upper panel). These results were confirmed in established clinically relevant melanoma cell systems including SK-Mel-28, SK-Mel-29, SK-Mel-103 and SK-Mel-147 (Fig1A, lower panel). As described by Alla et al 3, the behavior of non-invasive SK-Mel-28/SK-Mel-29 and invasive/metastatic SK-Mel-103/SK-Mel-147 cells that correspond to primary tumors and metastases with low or high E2F1 expression, respectively, is clearly E2F1 dependent and thus represents an optimal model system to study the interaction between the transcription factor and the identified miRs.
Figure 1.
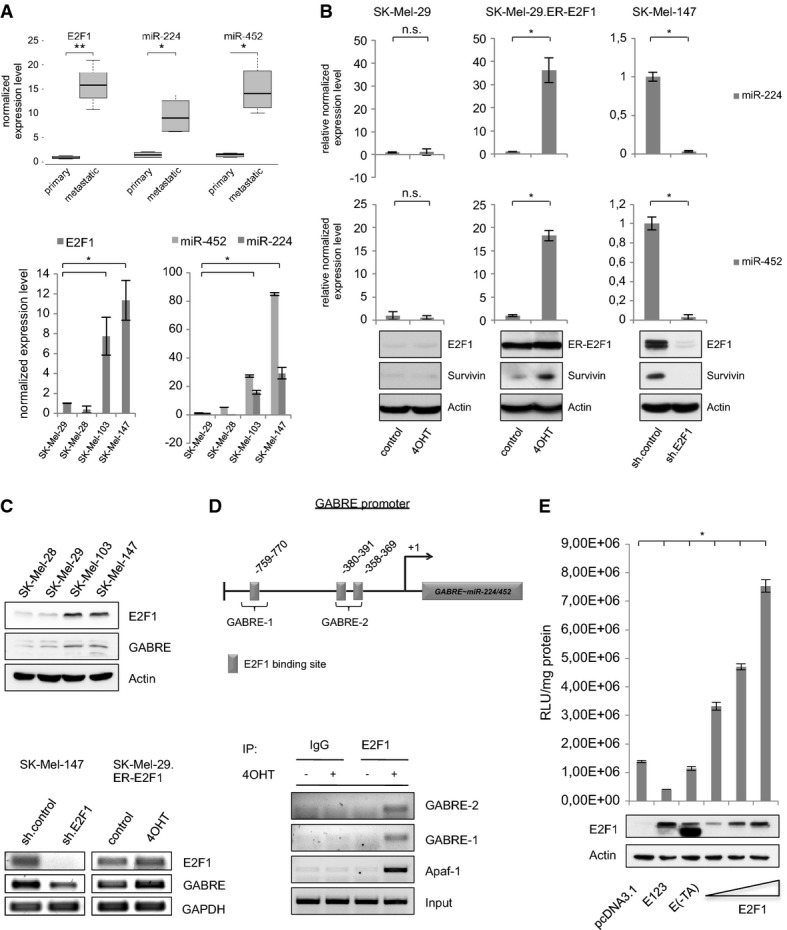
E2F1 regulates miR-224 and miR-452 by transactivating their host gene GABRE
A Quantification of E2F1, miR-224 and miR-452 expression determined by real-time PCR and TaqMan® MicroRNA single assays in primary tumor samples and non-invasive melanoma cell lines (SK-Mel-28, SK-Mel-29) versus metastases and highly metastatic SK-Mel-103 and SK-Mel-147 cells.
B E2F1 activates miR-224/452. Normalized miRNA expression is shown in stable SK-Mel-29.ER-E2F1 cells treated with 4OHT or solvent (control; middle panel) compared to parental SK-Mel-29, and in SK-Mel-147 cells expressing control or E2F1-specific shRNA. E2F1, ER-E2F1 and survivin expression was verified by Western blot. Actin served as a loading control.
C MiR-224/452 cluster is co-expressed with its host gene GABRE. GABRE and E2F1 expression in metastatic (SK-Mel-103 and -147) versus non-metastatic (SK-Mel-28 and -29) cells was verified by Western blot. GABRE transcript levels were analyzed after E2F1 knockdown in SK-Mel-147 and activation in SK-Mel-29.ER-E2F1.
D Scheme of the GABRE promoter showing relevant E2F-binding sites (GABRE-1, GABRE-2) upstream of the transcriptional start site (+1). Binding of E2F1 was verified by ChIP in SK-Mel-29.ER-E2F1 cells (treated with 4OHT (+), or solvent (−)) using E2F1 or IgG antibody. APAF-1 promoter served as a positive control.
E E2F1 directly activates GABRE. Relative luciferase activities (RLU) measured 36 h after co-transfection of GABRE promoter construct and increasing amounts (0.5, 1, 2 μg) of E2F1 expression plasmid or E2F mutants E(-TA) and E123 (2 μg). Protein expression was verified by Western blot. Actin served as a loading control.
Data information: Bar graphs are represented as means ± SD, n = 3, *P ≤ 0.05, two-sided Student's t-test. Box-whisker plots: Boxes indicate the 25 and 75% quartile surrounding the median (n = 4), *P ≤ 0.05, **P ≤ 0.01, two-sided Student's t-test. The lines represent the minimum and maximum transcript levels.
Since miR-224/452 expression correlates with E2F1 levels in primary versus metastatic patient samples as well as in invasive versus non-invasive melanoma cells, regulation of miR-224/452 by E2F1 is conceivable (Fig1A). In order to verify the effect of E2F1 on miR expression, SK-Mel-29.ER-E2F1 cells stably expressing E2F1 fused to the ligand-binding domain of the murine estrogen receptor (ER) were treated with tamoxifen (4-OHT). In parallel, we examined miR-224/452 expression in invasive/metastatic SK-Mel-147 cells after E2F1 knockdown using lentiviral pLKO.1-shE2F1 that ensures efficient depletion of E2F1 in melanoma cells. E2F1 activation and knockdown was confirmed by detection of the E2F1 target survivin. As shown in Fig1B, the miR-224/452 cluster is strongly upregulated upon E2F1 activation in SK-Mel-29.ER-E2F1 cells compared to parental SK-Mel-29, whereas knockdown of the transcription factor in SK-Mel-147 cells led to decreased expression of both miRs.
MiRNAs are located inside or outside of genes. Intergenic miRs are produced from their own transcriptional units. In contrast, miRNAs in introns of protein-coding genes are usually regulated by the gene promoter and processed with the precursor mRNA. This often results in co-expression of miR and host gene 37. Since miR-224 and miR-452 are located in the gamma-aminobutyric acid (GABA) A receptor epsilon gene (GABRE), we examined whether this gene is regulated through E2F1 (Fig1C). GABRE protein expression clearly correlates with the level of E2F1 in SK-Mel-103 and SK-Mel-147 cells (Fig1C, top panel) showing elevated miR-224/452 levels (Fig1A). Furthermore, decreased expression of the host gene after E2F1 ablation in invasive/metastatic SK-Mel-147 and upregulation of GABRE upon E2F1 activation in non-metastatic SK-Mel-29.ER-E2F1 indicates its transcriptional co-regulation with miR-224/452 by E2F1 (Fig1C, lower panel). In silico analysis of putative transcription factor binding sites in the GABRE promoter revealed three E2F elements in the region 812 bp upstream of the transcriptional start site (GABRE 1-759-770; GABRE 2-358-391 containing 2 E2F-motifs), which were confirmed using chromatin immunoprecipitation (ChIP) (Fig1D). To verify transcriptional activation by E2F1, the GABRE promoter region was cloned into the pGL3-basic reporter plasmid. Luciferase assay revealed a clear induction under E2F1 co-transfection in a concentration-dependent manner, whereas E2F1-mutants E(-TA) and E123 did not stimulate the promoter (Fig1E). Thus, in advanced tumors with high levels of E2F1, the transcription factor leads to the induction of miR-224/452 by transactivating their host gene GABRE.
MiR-224/452 cluster is an essential mediator of EMT and melanoma invasion
According to the cellular context, miR-224 and miR-452 show oncogenic as well as tumor suppressive properties. Therefore, functional analysis of the entire cluster is required to clearly assign potential cancer-promoting effects to one specific miR or the cooperation of both. To this end, we constructed lentiviral vectors for efficient and stable expression of individual candidates or the entire cluster in SK-Mel-29 (Fig2) and SK-Mel-28 cells (Supplementary Fig S2). Functional assays were performed in comparison with parental control cell lines expressing a randomly generated scrambled sequence (Scr). MiR expression levels were validated by TaqMan®MicroRNA single assays (Fig2A). Stable expression of miR-224/452 in non-invasive SK-Mel-29 resulted in increased migration and invasion (Fig2B and C; Supplementary Fig S1A and B). The same effect was observed in miR-expressing SK-Mel-28 cells (Supplementary Fig S2). In addition, we analyzed the impact of miR-224/452 on epithelial-mesenchymal transition, a highly conserved genetic program that enables epithelial tumor cells to migrate from the existing cell layer into surrounding tissues. This process includes loss of intercellular tight junctions and cell polarity, and the acquisition of mesenchymal properties 38–40. As reported previously, knockdown of endogenous E2F1 in metastatic melanoma cells restored expression of the epithelial marker E-cadherin 3. In accordance, non-invasive SK-Mel-29 cells that stably express miR-224/452 show loss of E-cadherin. These cells attain a mesenchymal state associated with increased levels of Slug, ZEB1 and vimentin that are substantially upregulated through miR-224 and/or miR-452 overexpression, whereas ZEB2, shown to be a differentiation factor in melanoma 41, is downregulated (Fig2D). Furthermore, the miR-224/452 cluster induces changes in the actin cytoskeleton toward a more aggressive phenotype as evident from phalloidin/TRITC staining (Fig2E).
Figure 2.

MiR-224/452 cluster induces EMT, migration and invasion of less aggressive melanoma cells
A Stable expression of miR-224 and miR-452 in SK-Mel-29 cells was verified by TaqMan® MicroRNA single assays.
B, C MiR-224 and miR-452 induce migration and invasion. Stable miR-expressing SK-Mel-29 cells were subjected to scratch (B) and Boyden chamber assay (C).
D MiR-224/452 induces EMT. Expression levels of epithelial (E-cadherin) and mesenchymal markers (vimentin, ZEB1 and Slug) as well as ZEB2 in stable SK-Mel-29.miR-224, SK-Mel-29.miR-452 and SK-Mel-29.miR-224/452 were verified by Western blot compared to miR-Scr cells. Actin was used for equal loading.
E MiR-224/452 expression leads to cytoskeleton rearrangements toward an aggressive phenotype. Stable miR-224/452-expressing SK-Mel-29 were stained for E-cadherin, vimentin (Cy3) and F-actin (Phalloidin/TRITC). Nuclei were counter-stained with DAPI, and fluorescence was visualized by confocal laser scanning microscopy (CLSM). Scale bar, 20 μm.
Data information: Bar graphs are represented as means ± SD, n = 3 (A, B) or n = 5 (C), *P ≤ 0.05, two-sided Student's t-test.
To directly assess the activity of endogenous miR-224/452 on the migratory/invasive capacity in the presence of high E2F1 levels, SK-Mel-147 cells were stably transduced with lentiviral vectors encoding shRNAs against miR-224 (miRZIP-224) and miR-452 (miRZIP-452) to specifically ablate miRNA expression. Efficient knockdown of miR-224/452 expression is shown in Fig3A. Compared to cells expressing control miRZIP-Scr, the ability of miR-224- and miR-452-depleted SK-Mel-147 cells to invade Matrigel (Fig3B; Supplementary Fig S3A) and migrate across the scratched area (Fig3C) was markedly reduced. A significantly impaired motility under miRZIP-224/452 expression was also observed in aggressive SK-Mel-103 cells (Supplementary Fig S4). Similar to miR-224/452 overexpression studies in SK-Mel-29, the strongest effect was achieved by the ablation of both miRs. In addition, knockdown of miR-224/452 in SK-Mel-147 caused an increase of epithelial E-cadherin and ZEB2, whereas mesenchymal vimentin, ZEB1 and Slug decreased compared to miRZIP-Scr control cells (Fig3D). Upregulation of E-cadherin and downregulation of vimentin as key epithelial and mesenchymal markers was also confirmed in miR-224/452-depleted SK-Mel-147 cells by immunofluorescence staining (Fig3E). These miR-224/452 knockdown cells showed clear changes in the actin cytoskeleton toward a less aggressive phenotype (Fig3E, lower panel). Whereas parental control cells exhibit actin-bearing membrane ruffles and thin F-actin filaments oriented parallel to the major cell axis as a typical feature of migrating cells, such characteristic structures are not visible after depletion of miR-224/452. Moreover, miR-depleted cells displayed actin staining predominantly at cell–cell contacts suggesting stronger cell–cell adhesions and impaired motility 42. Finally, in compliance with these morphological changes and the impaired migration/invasion, aggressive melanoma cells completely failed to form metastases in vivo when miR-224/452 expression is blocked (Fig3F). Notably, while invasive growth is largely abrogated after E2F1 depletion in SK-Mel-147.miR-Scr cells, those cells in which miR-224, miR-452 or both are re-expressed retain their aggressive behavior also in the absence of E2F1 (Fig3G; Supplementary Fig S3B), pointing out their autonomous oncogenic capabilities. The expression levels of miR-224/452 are shown in Supplementary Fig S3C.
Figure 3.

Depletion of miR-224/452 abolishes migration/invasion and lung metastasis
A Stable knockdown of miR-224/452 by specific miRZip anti-microRNA constructs was verified by TaqMan® MicroRNA single assays.
B, C Detection of the invasive and migratory potential of miR-224/452-depleted SK-Mel-147 cells by (B) Boyden chamber and (C) wound closure assay 24 h after seeding/scratching. Fold changes were calculated relative to SK-Mel-147.ZIP-Scr (set as 1). Representative images of three independent scratch assays for each ZIP-miR are shown.
D, E Ablation of miR-224/452 in SK-Mel-147 reverses EMT. Expression of the indicated proteins was measured by Western blot (D). Changes in E-cadherin (Cy3) and vimentin (Cy3) expression as well as cytoskeleton rearrangement (Phalloidin/TRITC) as visualized by confocal laser scanning microscopy (CLSM) (E).
F Knockdown of miR-224/452 leads to complete lack of pulmonary metastases. Representative hematoxylin and eosin sections of lungs (a–d). Visible metastatic tumor infiltrates with atypic large cell shape und nuclei with high mitotic activity in the miRZIP-Scr group (a, b). Normal lung structure without tumor infiltrates in mice (n = 5 per group) injected with miR-224/452 ablated SK-Mel-147 (c, d). Magnification (×20) of the marked area is shown (b, d).
G MiR-224/452 expression is sufficient to induce tumor invasion. The effect of miR-224/452 overexpression in shE2F1-treated SK-Mel-147 was investigated by Boyden chamber assay. Fold changes of invasion were calculated relative to SK-Mel-147.ZIP-Scr (set as 1). E2F1 expression was detected by immunoblotting. Actin served as a loading control.
Data information: Bar graphs are represented as means ± SD, n = 3 (A) or n = 5 (B, G), *P ≤ 0.05, two-sided Student's t-test.
MiR-224/452 regulates metastasis suppressor TXNIP
To further investigate the mechanism by which miR-224/452 promotes tumor progression, we analyzed putative targets of this cluster (Fig4A). In order to identify shared target genes of miR-224 and miR-452, we extracted from the starBase database (v1.0) 43 Argonaute-target interaction sites that match computationally predicted target sites of one of the two miRNAs. The data in starBase are derived from high-throughput CLIP-Seq experiments. Those targets in which binding sites for both miRNAs exist were considered for further analysis. Furthermore, we extracted predicted mutual targets of both miRNAs from the miRror Suite 44, a Web service that integrates predictions from twelve complementary algorithms and computes targets that are regulated by a set of miRNAs in a coordinated fashion. From these targets, we considered only those with approved AGO binding sites based on starBase. In total, we received a set of 20 target genes (Supplementary Table S1) most likely being targeted by both miR-224 and miR-452. From this set, we selected those genes with relevance in cancer based on their associated Gene Ontology terms 45. After GO filtering, a set of targets were subjected for experimental validation (Fig4A). The most notable one was thioredoxin-interacting protein TXNIP (also named as VDUP-1 or TBP-2), which is often downregulated in cancer and known as a proapoptotic factor and metastasis suppressor 46,47. Validation experiments revealed a clear inhibition of TXNIP upon miR-224 and miR-452 overexpression in SK-Mel-29, and its upregulation under knockdown of these miRs in metastatic SK-Mel-147 both on RNA (Fig4B, upper panel) and protein level (Fig4B, lower panel). Consistent with our data demonstrating that miR-224 and miR-452 are regulated by E2F1, knockdown of the transcription factor resulted in an increase of TXNIP, whereas target level declined in response to E2F1 activation through 4OHT addition (Fig4C). Furthermore, luciferase activity of the pMiR-Report construct containing the 3′ UTR of TXNIP was vigorously reduced after miR-224 and/or miR-452 co-transfection in SK-Mel-29 cells (Fig4D, left panel). In addition, we transfected pMiR-Report-TXNIP-3′ UTR together with miRZIP plasmids against miR-224/452 in SK-Mel-147 cells and saw a comparably strong, in this case stimulating effect on TXNIP 3′ UTR reporter activity after miR ablation (Fig4D, right panel). MiR-224/452 expression was confirmed by TaqMan Single Assays shown in Supplementary Fig S5. To further demonstrate a direct interaction of miR-224/452 with the TXNIP 3′ UTR, site-directed mutagenesis of the miR-224/452 binding sites was performed. These mutations completely abolished the repressive effects of miR-224 and miR-452 on TXNIP 3′ UTR (Fig4E) compared to the positive controls with intact miR-452 (left) and miR-224 (right) binding sites. In sum, these results indicate that TXNIP is a major target of the E2F1-miR-224/452 module.
Figure 4.

Identification of TXNIP as miR-224/452 target
A Scheme of target prediction for miR-224/452.
B MiR-224/452 regulates TXNIP. Overexpression of miR-224/452 in SK-Mel-29 resulted in less TXNIP expression, while inhibition of the endogenous miRNAs in SK-Mel-147 induced TXNIP. Transcript and protein levels were determined using actin as a control.
C Knockdown of E2F1 in SK-Mel-147 increases and E2F1 activation by addition of 4-OHT in SK-Mel-29.ER-E2F1 reduces TXNIP expression on RNA and protein level. GAPDH and actin served as loading controls.
D Luciferase reporter assay revealed a direct regulation of TXNIP by miR-224 and miR-452. Co-transfection of pMiR-Report-3′ UTR(TXNIP) and miR-224/452 plasmids in SK-Mel-29 results in less luciferase activity (in comparison with miR-Scr). Promoter activity is upregulated in miRZip-transfected SK-Mel-147.
E Mutation of miR-224/452 binding sites in the TXNIP-3′ UTR completely abolishes their repressive effects. Luciferase activity was measured after co-expression of pMiR-Report-3′ UTR(TXNIP-224-mut) or -(TXNIP-452-mut) with miR-224/452 in comparison with miR-Scr in SK-Mel-29.
Data information: Bar graphs are represented as means ± SD, n = 3, *P ≤ 0.05, two-sided Student's t-test.
Loss of TXNIP promotes EMT and invasion by the E2F1-miR-224/452 axis
Based on the results that TXNIP is a target of miR-224/452 and their function in melanoma progression, we speculated whether this oncogenic behavior is dependent on TXNIP inhibition. For this purpose, we expressed TXNIP (without 3′ UTR) in metastatic melanoma cells with low endogenous levels of this protein and analyzed changes in invasion and migration. Indeed, similar to the ablation of miR-224/452, overexpression of TXNIP in SK-Mel-147 led to an efficient reduction of migrating/invading cells (Fig5A and B; Supplementary Fig S6A and B). Furthermore, Western blot analysis indicated a gain of E-cadherin expression and loss of mesenchymal markers at levels comparable to miR-224/452-depleted cells (Fig5C). The effect of TXNIP on melanoma cell invasion became even more apparent in miR-224/miR-452-positive SK-Mel-29 cells, which showed enhanced migratory properties (Fig2). Ectopic overexpression of TXNIP in these cells suppressed miR-224/452-mediated invasiveness to the level detected for SK-Mel-29.miR-Scr control cells (Fig5D; Supplementary Fig S6C).
Figure 5.
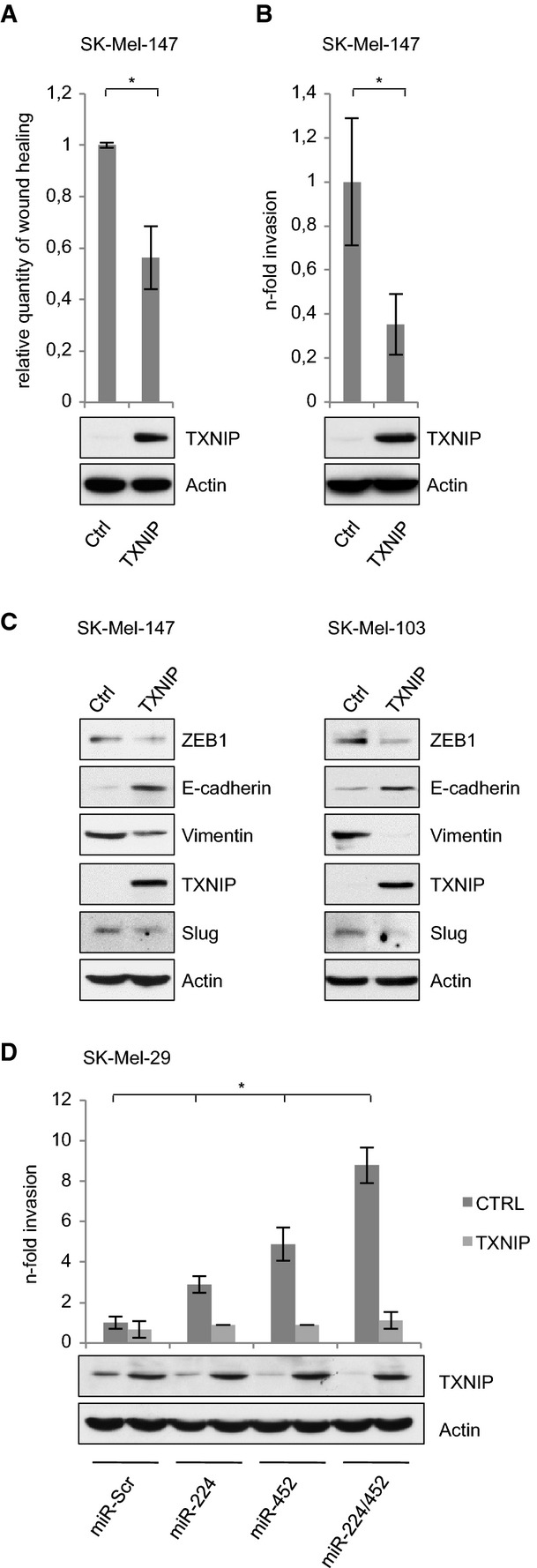
TXNIP repression is required for the oncogenic switch by miR-224/452
A, B Overexpression of TXNIP in aggressive melanoma cells with low endogenous levels reduces migration and invasion.
C Effect of ectopic TXNIP on EMT markers in SK-Mel-147 and SK-Mel-103 cells as indicated by Western blot.
D MiR-224/452-induced invasion is inhibited by TXNIP. TXNIP expression was determined by immunoblot using actin as a control. Fold changes were calculated relative to SK-Mel-29.miR-Scr (set as 1).
Data information: Bar graphs are represented as means ± SD, n = 3 (A) or n = 5 (B, D), *P ≤ 0.05, two-sided Student's t-test.
Importantly, increased invasion of less aggressive melanoma cells by overexpression of E2F1 was significantly reduced upon knockdown of miR-224 and miR-452. As demonstrated in Fig6A and Supplementary Fig S7A, this is due to the recovery of TXNIP expression upon miR-224/452 inhibition. In support of the relevance of TXNIP downregulation during E2F1-miR-224/452-induced malignant progression, E2F1-induced invasion of SK-Mel-29 cells is also markedly reduced when TXNIP (without 3′ UTR) is re-expressed (Fig6B, left; Supplementary Fig S7B). Moreover, depletion of endogenously high E2F1 in SK-Mel-147 cells which is associated with the upregulation of TXNIP protein expression (Fig6B, right) severely reduced their invasive capacity, whereas knockdown of TXNIP in these cells is needed to increase invasivity again (Fig6B, right; Supplementary Fig S7B). Similar effects were observed for the E2F1-mediated EMT phenotype where the E2F1-induced decrease of E-cadherin and increase of mesenchymal markers was to some extend reversed through re-expression of TXNIP (Fig6C, left). Furthermore, gain of epithelial and loss of mesenchymal markers in SK-Mel-147 cells ablated for E2F1 was to a significant part recovered by shTXNIP treatment (Fig6C, right). Hence, loss of TXNIP in melanoma cells due to aberrant E2F1-miR-224/452 expression is crucial for the transcription factor to induce cancer progression.
Figure 6.
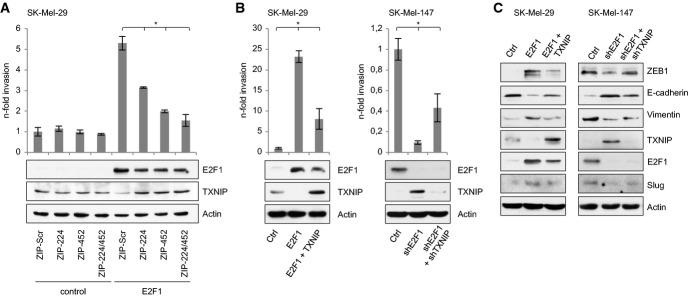
Loss of TXNIP due to aberrant E2F1-miR-224/452 expression is crucial for transcription factor-mediated cancer progression
A–C E2F1-induced EMT and invasion requires downregulation of TXNIP by miR-224/452. Invasion was measured by Boyden chamber, and fold changes were calculated relative to the controls (set as 1). Expression of indicated proteins is shown by Western blot.
Data information: Bar graphs are represented as means ± SD, n = 3 (A) or n = 5 (B), *P ≤ 0.05, two-sided Student's t-test.
Based on these findings, we compared the expression levels of E2F1 and TXNIP in vivo at the stages of melanoma progression where an EMT-like switch takes place, namely at the invasive front of primary melanomas, when they colonize the underlying dermis using expression data from a melanoma study (Xu et al 2008) available at Oncomine™ database (Compendia Bioscience, Ann Arbor, MI) 48 and own human samples of different Breslow depth. As shown in Fig7A, E2F1 levels positively correlate with Breslow depth of primary tumors (> 4 mm, high E2F1 expression versus < 1 mm, low E2F1 expression, P < 0.0053). Conversely, TXNIP expression and Breslow depth of invasion show an inverse correlation with low TXNIP levels at > 4 mm versus high levels at < 1 mm depth of invasion (P < 0.0039). This was confirmed by RT–PCR (Fig7B) and immunohistochemical analysis (Fig7C) in a statistically significant number of own primary patient tumors with low versus high Breslow Index. These data, clearly indicating a statistically significant association between high E2F1 versus low TXNIP expression and the highest invasion depth of primary melanoma predictive for progression and severity of the disease, again underscore the relevance of the uncovered E2F1-miR-224/452-TXNIP axis and support that the components of this module can serve as novel prognostic markers for melanoma patients at an early stage of metastasis initiation.
Figure 7.

TXNIP and E2F1 contribute to malignant progression in a regulatory loop
A–C Inverse correlation of E2F1 and TXNIP in primary melanoma of different Breslow depth obtained by Oncomine data analysis (A), real-time PCR (B) and immunohistochemistry (C).
D TXNIP inhibits E2F1 via the p16-CDK4-RB pathway. Levels of phospho-RB, CDK4, p16, E2F1 and TXNIP are indicated. The E2F1 target survivin was used as a positive control and actin for loading.
E Schematic illustration of the E2F1-miR-224/452-TXNIP axis in early- and late-stage primary melanoma. High TXNIP levels in early primary tumors restrict E2F1 activity through p16/CDK4-induced RB hypophosphorylation. During malignant progression, the miR-224/452 cluster is induced by increasing E2F1 levels, which leads to TXNIP suppression at the tumor invasion front associated with an EMT-like phenotype, enhanced migration/invasion and metastatic dissemination.
Data information: Boxes indicate the 25 and 75% quartile surrounding the median, *P ≤ 0.05 (n = 4), Mann–Whitney U-test; **P ≤ 0.01 (n = 9), two-sided Student's t-test. The lines represent the minimum and maximum transcript levels, and circles represent outliers.
E2F1 and TXNIP contribute to cancer progression in a regulatory feedback loop
Previously, TXNIP has been implicated in growth suppression in association with an increase of p16 expression and reduction of retinoblastoma (RB) phosphorylation 49. Since hypophosphorylated RB leads to the inhibition of E2F1 function, we suspected a mutual regulation of TXNIP and E2F1 to avoid uncontrolled E2F1 activity by building a negative feedback loop. Immunoblotting demonstrated a reduced expression of E2F1 in SK-Mel-103/-147 cells after TXNIP overexpression (Fig7D). To identify the possible mechanism for the TXNIP-induced decrease of E2F1, we tested its influence on different regulators of E2F1 activity. In accordance with the observations made by Nishinaka et al, p16 increased and phospho-RB decreased through TXNIP also in melanoma cells. The CDK inhibitor p16 inhibits different cyclin-dependent kinases, which are able to phosphorylate RB. We found that CDK4 was strongly downregulated under these circumstances (Fig7D). This argues that TXNIP indeed provides a negative feedback loop that prevents excessive E2F1 activity. In line with elevated levels of CDK4, hyperphosphorylation of RB and consecutive stimulation of E2F1 in both aggressive melanoma cell lines, miR-224/452 potentiates the accumulation of E2F1 by inhibiting TXNIP, which contributes to skin cancer progression (Fig7E). This finding hints toward a putative therapeutic approach to avoid EMT of tumor cells either by knockdown of miR-224/452 or addition of TXNIP.
Discussion
E2F1 is a critical factor for metastasis in melanoma. Since the exact mechanisms are incompletely understood, we investigated the role of miRs in E2F1-induced tumor progression. There are several reports on miRs regulating this transcription factor, but except some hints, it was less known about E2F1-induced miRs. Here, we uncovered the importance of E2F1-induced miRs for its oncogenic activity in melanoma progression. We could show that the miR-224/miR-452 cluster is upregulated in invasive/metastatic melanoma and controls as part of a gene regulatory module the stage of melanoma progression where an EMT-like switch takes place. The results demonstrate that expression of miR-224/miR-452 is necessary and sufficient to elicit an EMT phenotype and that this process is induced by the E2F1 TF via direct activation of the GABRE host gene and its intronic miRs. Increased miR-224/452 promotes oncogenic transition into a metastatic state by repressing the metastasis suppressor TXNIP. Interestingly, this factor in turn controls E2F1 activity in a negative regulatory loop.
As yet, there are only few studies on the whole cluster describing a correlation in miR-224/452 and GABRE expression, such as two recent publications on concomitant regulation of miR-224/452 and GABRE by epigenetic mechanisms, resulting in either high expression in HCC patients or low expression in prostate cancer 50,51. These findings already reflect the ability of both miRs to act as tumor suppressor or oncogene in a cell-context-dependent manner. In the present study, functional analyses on miR-224/452 clearly revealed tumorigenic actions of both effectors which are characterized by the stimulation of migratory and invasive properties of less aggressive melanoma cells and a decrease of motility and invasion when miR-224/452 was depleted in aggressive cell lines as evident from the complete lack of lung metastases in mice. As re-introduction of these miRs after E2F1 ablation in invasive/metastatic melanoma cells could recover their oncogenic effects, they are able to act independently of the transcription factor.
The epithelial-mesenchymal transition is an important prerequisite for metastatic cancer. As a process of epithelial plasticity, it includes dissolution of epithelial cell–cell adhesions, actin cytoskeleton reorganization, as well as an increase in cell–matrix contacts, leading to enhanced migration and invasion 42. It is well known that miRs are able to influence EMT 52. One of the first and well-described examples is the regulation of ZEB proteins by miR-205 as well as the miR-200 family. Inhibition of the E-cadherin repressors ZEB1 and ZEB2 by these miRs results in the stabilization of an epithelial phenotype of cancer cells 53–55. Concerning a putative role of miR-224/452 on EMT, there are only limited and controversial reports by Zhang et al 13 who found an increase of E-cadherin in miR-224-depleted Huh-7 cells, whereas miR-224 has been shown to correlate with high E-cadherin expression in normal breast epithelium 56. In addition, miR-452 is known to be highly expressed in neural crest cells having an influence on an epithelial-mesenchymal signaling pathway in the first pharyngeal arch 36. In our study, ectopic miR-224/452 mediates the EMT-like phenotype in non-invasive melanoma cells through increased expression of the transcription factors Slug and ZEB1, which are known repressors of epithelial E-cadherin 57. In line with E-cadherin decrease, the intermediate filament protein vimentin as mesenchymal marker that promotes cell migration and invasion is upregulated 58. The reverse effect on these EMT markers was observed when both miRs were knocked down in metastatic tumor cells. Consistent with previous studies indicating that remodeling of actin filaments is essential for EMT as it promotes cell migration and metastatic spread from primary tumors 42, we have shown that E2F1-miR-224/452-induced EMT involves cytoskeletal changes toward an aggressive cancer phenotype.
We uncovered TXNIP as an important target of both miRs. TXNIP was initially identified as thioredoxin-binding protein that inhibits thioredoxin (TRX), thereby contributing to redox homeostasis 59. Since TRX also promotes tumor progression by angiogenesis induction 60 and apoptosis inhibition 61,62, TXNIP, as its inhibitor, has great potential as a tumor suppressor gene. Several studies on reduced expression of TXNIP in different forms of aggressive cancers such as acute myeloid leukemia (AML), hepatocellular carcinoma (HCC), and breast and bladder cancer emphasize its relevance for tumor prevention, since less TXNIP expression clearly contributes to cancer malignancy 63–67. Interestingly, TXNIP is also described as a metastasis suppressor, which leads to less lung metastases in mice after overexpression in aggressive melanoma cells 47. In accordance with these findings, knockdown of miR-224/452 results in enhanced TXNIP expression, reduced migration and invasion, and the absence of metastases formation in vivo. Goldberg et al 47 detected low TXNIP expression in metastatic melanoma cells as a reason of Chr6 deletion. TXNIP itself is not located on Chr6; however, relevant regulators of this protein originate from Chr6, like CRSP3. In this regard, our data provide another way of TXNIP inhibition via E2F1-mediated miR-224/452 expression that is clearly important in melanoma cells without chromosome 6 deletion and supports the impact of silencing TXNIP to facilitate malignant progression. This is the case for SK-Mel-28/-29/-103 and -147 used in this study, which have apparently no Chr6 deletion as TXNIP is still expressed in SK-Mel-29 cells and can be recovered by inhibition of the E2F1-miR-224/452 module in SK-Mel-147 cells. In addition, we detected CRSP3 as a gene, which is located on Chr6 in these melanoma cell lines (data not shown). However, miR-224/452 induction by E2F1 is also essential in cells having such a modification, since TXNIP is still expressed and might be active at a certain threshold 47. Thus, the E2F1-miR-224/452-TXNIP axis represents a key regulatory mechanism to force tumor progression.
According to the role of miR-224/452 in an E2F1-induced EMT-like program, downregulation of its target TXNIP in non-invasive SK-Mel-29 cells upon E2F1 induction showed identical effects with an increase of mesenchymal and decrease of epithelial markers as described above. However, re-introduction of TXNIP rescued these effects to an incomplete but considerable extend, resulting in mesenchymal-epithelial transition and inhibition of invasion. In contrast, knockdown of E2F1 in highly aggressive melanoma cells leads to the induction of TXNIP and a more epithelial phenotype (gain of E-cadherin and loss of Slug, ZEB1 and vimentin). This process is partially reversible after ablation of the metastasis suppressor and shows that the removal of TXNIP during E2F1-induced melanoma progression is absolutely critical for the transcription factor to promote an EMT-like phenotype. The new E2F1 activity is also reflected by an inverse correlation between high E2F1 and low TXNIP expression in primary melanoma samples from patients with more than 4 mm Breslow depth of invasion compared to non-invasive tumor states. A previous study by Masaki et al 68 reported that loss of TXNIP enhances TGF-β-induced EMT, and this was associated with loss of E-cadherin and a gain of Slug and vimentin. In contrast, impaired TGF-β-mediated EMT has been detected in TXNIP-ablated cells in the context of diabetic nephropathy 69. These contradictory observations may result from the different cellular systems, which means breast and lung cancer cell lines versus proximal tubular cells derived from normal kidney. Thus, E2F1-induced oncogenic EMT bears some similarity with the signaling of the well-known EMT inducer TGF-β in cancer cells. As an inhibitor of the antioxidant protein TRX, overexpression of TXNIP in vitro increases the production of reactive oxygen species (ROS) and induces oxidative stress 70. Although ROS have controversial roles in tumorigenesis by inducing DNA mutations, genomic instability and aberrant pro-tumorigenic signaling on the one hand, as well as being toxic to cancer cells through the induction of cell death 71, oxidative stress apparently plays no role in mediating miR-224/452-induced tumor cell EMT and invasion. TXNIP has also critical functions in regulating glucose homeostasis, linking loss of TXNIP to the Warburg cancer phenotype. However, there was no evidence that supported a connection between TXNIP and metabolism through increased redox stress, suggesting that TXNIP does not act primarily as an inhibitor of thioredoxin 72. Given that miR-224/452-mediated TXNIP repression, which should reduce oxidative stress, drives EMT, the obvious implication here is that metastasis suppression by TXNIP similar to its metabolic function does not occur via TRX/ROS regulation. This view is also underscored by the finding that TXNIP depletion in the context of TGF-β-induced EMT did not result in altered TRX levels 68.
Strikingly, we observed that high levels of TXNIP cause a decrease of E2F1 expression. In order to elucidate the mechanism behind it, we investigated different upstream regulators of E2F1. In accordance with Nishinaka and colleagues who showed that TXNIP induces p16 resulting in reduced phospho-RB levels in HTLV-I positive T cells 49, the same effect on p16 and RB together with decreasing expression of cyclin-dependent kinase 4 was seen in melanoma cells. Hence, TXNIP is able to inhibit E2F1 via the p16/CDK4/RB axis. This interesting finding gives strong support that E2F1 and TXNIP are integral part of a regulatory loop in which increasingly high levels of E2F1 during cancer progression promote an EMT-like switch by miR-224/452-mediated repression of TXNIP, whereas high TXNIP expression in early primary tumors initially restricts malignant progression by inhibiting E2F1 (Fig7E).
In sum, our results demonstrate that E2F1 is a new EMT-promoting transcription factor. The miR-224/452 cluster represents a key mediator of E2F1-induced tumor progression, since it inhibits the metastasis suppressor TXNIP and contributes to EMT, invasion/migration and metastatic spread of melanoma cells. The uncovered mutual regulation between E2F1 and TXNIP points toward new prognostic and therapeutic options by using components of the E2F1-miR-224/452-TXNIP axis as potential key targets to restrict cancer progression.
Materials and Methods
Cell culture and lentiviral transduction
Melanoma cell lines SK-Mel-28, SK-Mel-29, SK-Mel-103 and SK-Mel-147 were maintained in Dulbecco's modified Eagle medium (high glucose, 4.5 g/l) containing sodium pyruvate as previously described 3. For E2F1 induction in SK-Mel-29.ER-E2F1 cells, 1 μM of 4-hydroxytamoxifen (4-OHT) was used 3. Stable cell lines expressing either miRNAs or anti-microRNAs were obtained by lentiviral transduction. Virus particle production required co-transfection of the expression plasmids, psPAX2 (‘packaging’) and pMD2.G vector (‘envelope’) in HEK 293T cells 73.
TaqMan®MicroRNA single assays and PCR
In general, large and small RNA was extracted using the NucleoSpin® miRNA kit (MACHEREY-NAGEL). MicroRNA expression levels were measured using TaqMan®MicroRNA single assays and the 7900HT Fast Real-Time PCR System (Applied Biosystems). For expression analysis, the comparative CT method was used with RNU6B as endogenous control 73. For semiquantitative PCR, 1 μg of RNA was reverse transcribed using First Strand cDNA Synthesis Kit (Thermo Scientific). cDNA was added to Thermo Scientific PCR Master Mix (2×) and amplified with specific primers in a MyCycler Thermal Cycler (Bio-Rad). Actin was used as a loading control. For qRT-PCR, cDNA was added to iQTM SYBR Green Supermix and analyzed using iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad). Relative gene expression was calculated by the comparative CT method using actin for normalization. For primer sequences, see Supplementary Table S2.
Western blotting and immunofluorescence
Protein analysis was performed as described previously 74. Briefly, cell lysis was carried out using RIPA buffer, containing PhosSTOP Phosphatase Inhibitor Cocktail (Roche), and protein concentration was determined by Bradford assay (Bio-Rad). The same quantity of different protein samples was separated by SDS–PAGE and transferred to nitrocellulose membranes (Amersham Biosciences) 74. Selected proteins were detected by the use of specific antibodies for E2F1 (KH-95, BD Biosciences), E-cadherin (Cell Signaling, BD), vimentin (V9, Santa Cruz), ZEB1 (H-102, Santa Cruz), Slug (H-140, Santa Cruz), GABRE (H-110, Santa Cruz), CDK4 (C-22, Santa Cruz), RB-P (Cell Signaling), survivin (Abcam), E2F1 (BD), TXNIP (K0205-3, MBL), p16 (M-156, Santa Cruz), ZEB2 (Sigma) and actin (Sigma) and their corresponding HRP-conjugated secondary antibodies. ECL Plus Western Blotting Detection Reagents (GE Healthcare) allowed detection of HRP activity with X-ray films.
For immunofluorescence, cells were grown on coverslips, fixed with 4% paraformaldehyde, permeabilized with Triton X-100 and blocked with bovine serum albumin (BSA). Incubation with primary antibodies was performed overnight. For visualization with the inverted confocal laser scanning microscope (Zeiss, ELYRA PS.1), fluorescence-labeled secondary antibodies (Cy3, Cy5; Molecular Probes) were used 74. Cytoskeleton staining was determined by Phalloidin/TRITC staining.
Chromatin immunoprecipitation (ChIP)
ChIP assay was carried out essentially as described 75 using SK-Mel-29.ER-E2F1 cells.
Cloning of expression plasmids and 3′ UTRs constructs and luciferase reporter assay
MiRNAs hsa-miR-224, hsa-miR-452, GABRE promoter and TXNIP 3′ UTR were amplified by PCR using genomic DNA as template and cloned into the pWPXL-, pGL3-basic- and pMiR-Report-vector. TXNIP was amplified from cDNA and cloned into pWPXL-expression vector. For primer sequences, see Supplementary Table S2. For luciferase assays, cells were transfected using Turbofect (Thermo Scientific). MiR knockdown was achieved with commercially available miRZip™ vectors (SBI System Biosciences). Luciferase activity was measured 36 h after transfection using the Luciferase Reporter Assay System (Promega) and normalized to total protein concentration in cell extract 73.
Animal studies and histological analysis
2 × 106 tumor cells stably expressing miRZIP-Scr or miRZIP-224/452 were injected into the tail vein of athymic NMRI nude mice at the age of 6 to 8 weeks (Charles River). Mice were monitored for lung metastases over several weeks. Lung tissue was surgically excised, fixed in 4% paraformaldehyde, paraffin-embedded and processed for histological analysis with hematoxylin and eosin staining. All animal experiments were performed according to the Institutional Animal Care and Use Committee.
Boyden chamber and scratch assays
For Boyden chamber assay, cells were seeded on a 8-μm PET membrane (BD BioCoat™ BD Matrigel™ Invasion Chamber, 6-well) covered with BD Matrigel™ Basement Membrane Matrix (BD Bioscience). A concentration gradient between media in cell culture inserts and surrounding well caused cell invasion through the pores of the membrane. Finally, cells of the upper membrane surface were detached, whereas the ones of the lower surface were stained with DAPI and documented by fluorescence microscopy. Wound healing assay (scratch assay) was performed with Ibidi's culture inserts that allow creating a defined gap of 500 μm between two areas with the same amount of confluent cells. Cell migration was determined by the extent of the gap closure, which was documented by microscopy recordings at certain times.
Human melanoma tissues and immunohistochemistry
For molecular analysis, primary melanoma and melanoma metastases were investigated by real-time RT–PCR and immunohistochemistry. This study was approved by the Ethics Committees of the Universities of Rostock and Kiel, Germany, and informed consent was obtained from all subjects.
Formaldehyde-fixed paraffin-embedded (FFPE) primary melanomas with different Breslow Index were immunohistologically analyzed. 4-μm-thick slices were cut and for heat-induced epitope retrieval pretreated with Tris/EDTA buffer pH 9.0. Tissue slides were blocked with BSA and incubated overnight using the following antibodies and dilutions: TXNIP (1:50, K0205-3, MBL) and E2F1 (1:50, KH-95, Santa Cruz). Finally, slides were washed and developed using LSAB+ System-HRP (Dako) and microscopically analyzed.
Identification of miR-224/452 candidate targets using bioinformatic tools
We extracted from the starBase database (v1.0) 43 Argonaute-target interaction sites that match computationally predicted target sites of miR-224 or miR-452. Those targets in which binding sites for both miRNAs exist where considered for further analysis. Furthermore, we retrieved predicted targets that are shared by both miRNAs from the miRror Suite 44. From these targets, we considered only those with approved AGO binding sites based on starBase. In total, we received a set of 20 target genes. From this set, we selected those genes with relevance in cancer based on their associated Gene Ontology terms 45 (Fig4A; Supplementary Table S1).
Statistical analysis
P-values were calculated using the Student's t-test or Wilcoxon–Mann–Whitney (Mann–Whitney) U-test as indicated. All bar graphs are plotted as mean ± SD. Box-whisker plots indicate the 25 and 75% quartile surrounding the median and the minimum and maximum as well as outliers. Statistical significance: n.s., no significance, *P ≤ 0.05; **P ≤ 0.01. Statistical significance was calculated using the R software package (http://www.r-project.org).
Acknowledgments
We thank Anja Stoll for excellent technical assistance and Ilona Klamfuß for help with animal experiments. Work related to this paper was supported by grants from Erich und Gertrud Roggenbuck-Stiftung, Federal Ministry of Education and Research (BMBF) as part of the project eBio:SysMet, and FORUN program of Rostock University Medical Faculty. SK received a fellowship of the Landesgraduiertenförderung des Landes Mecklenburg-Vorpommern.
Author contributions
SK and BMP conceived the idea and designed the experiments; SK, KF and BK executed the experiments; SK and SM analyzed the data; US performed target prediction and OW provided the general framework for bioinformatic study; HM analyzed tissue sections; SK, US and BMP wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
for this article is available online: http://embor.embopress.org
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Table S1
Supplementary Table S2
Review Process File
References
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- Soengas MS, Lowe SW. Apoptosis and melanoma chemoresistance. Oncogene. 2003;22:3138–3151. doi: 10.1038/sj.onc.1206454. [DOI] [PubMed] [Google Scholar]
- Alla V, Engelmann D, Niemetz A, Pahnke J, Schmidt A, Kunz M, Emmrich S, Steder M, Koczan D, Putzer BM. E2F1 in melanoma progression and metastasis. J Natl Cancer Inst. 2010;102:127–133. doi: 10.1093/jnci/djp458. [DOI] [PubMed] [Google Scholar]
- Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009;9:785–797. doi: 10.1038/nrc2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelmann D, Putzer BM. Translating DNA damage into cancer cell death-A roadmap for E2F1 apoptotic signalling and opportunities for new drug combinations to overcome chemoresistance. Drug Resist Updat. 2010;13:119–131. doi: 10.1016/j.drup.2010.06.001. [DOI] [PubMed] [Google Scholar]
- Lee JS, Leem SH, Lee SY, Kim SC, Park ES, Kim SB, Kim SK, Kim YJ, Kim WJ, Chu IS. Expression signature of E2F1 and its associated genes predict superficial to invasive progression of bladder tumors. J Clin Oncol. 2010;28:2660–2667. doi: 10.1200/JCO.2009.25.0977. [DOI] [PubMed] [Google Scholar]
- Tuve S, Wagner SN, Schittek B, Putzer BM. Alterations of DeltaTA-p 73 splice transcripts during melanoma development and progression. Int J Cancer. 2004;108:162–166. doi: 10.1002/ijc.11552. [DOI] [PubMed] [Google Scholar]
- Knoll S, Emmrich S, Putzer BM. The E2F1-miRNA cancer progression network. Adv Exp Med Biol. 2013;774:135–147. doi: 10.1007/978-94-007-5590-1_8. [DOI] [PubMed] [Google Scholar]
- Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- Hauptman N, Glavac D. MicroRNAs and long non-coding RNAs: prospects in diagnostics and therapy of cancer. Radiol Oncol. 2013;47:311–318. doi: 10.2478/raon-2013-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmrich S, Putzer BM. Checks and balances: E2F-microRNA crosstalk in cancer control. Cell Cycle. 2010;9:2555–2567. doi: 10.4161/cc.9.13.12061. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Purcell AL, Levin AA. Developing microRNA therapeutics. Circ Res. 2012;110:496–507. doi: 10.1161/CIRCRESAHA.111.247916. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Takahashi S, Tasaka A, Yoshima T, Ochi H, Chayama K. Involvement of microRNA-224 in cell proliferation, migration, invasion, and anti-apoptosis in hepatocellular carcinoma. J Gastroenterol Hepatol. 2013;28:565–575. doi: 10.1111/j.1440-1746.2012.07271.x. [DOI] [PubMed] [Google Scholar]
- Chen PJ, Yeh SH, Liu WH, Lin CC, Huang HC, Chen CL, Chen DS, Chen PJ. Androgen pathway stimulates microRNA-216a transcription to suppress the tumor suppressor in lung cancer-1 gene in early hepatocarcinogenesis. Hepatology. 2012;56:632–643. doi: 10.1002/hep.25695. [DOI] [PubMed] [Google Scholar]
- Gao P, Wong CC, Tung EK, Lee JM, Wong CM, Ng IO. Deregulation of microRNA expression occurs early and accumulates in early stages of HBV-associated multistep hepatocarcinogenesis. J Hepatol. 2011;54:1177–1184. doi: 10.1016/j.jhep.2010.09.023. [DOI] [PubMed] [Google Scholar]
- Scisciani C, Vossio S, Guerrieri F, Schinzari V, De Iaco R, D'Onorio de Meo P, Cervello M, Montalto G, Pollicino T, Raimondo G, et al. Transcriptional regulation of miR-224 upregulated in human HCCs by NFkappaB inflammatory pathways. J Hepatol. 2012;56:855–861. doi: 10.1016/j.jhep.2011.11.017. [DOI] [PubMed] [Google Scholar]
- Boguslawska J, Wojcicka A, Piekielko-Witkowska A, Master A, Nauman A. MiR-224 targets the 3′UTR of type 1 5′-iodothyronine deiodinase possibly contributing to tissue hypothyroidism in renal cancer. PLoS ONE. 2011;6:e24541. doi: 10.1371/journal.pone.0024541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Brannon AR, Reddy AR, Alexe G, Seiler MW, Arreola A, Oza JH, Yao M, Juan D, Liou LS, et al. Identifying mRNA targets of microRNA dysregulated in cancer: with application to clear cell Renal Cell Carcinoma. BMC Syst Biol. 2010;4:51. doi: 10.1186/1752-0509-4-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt GM, Dossey L, Cullen LM, Lai A, Druker R, Eisbacher M, Zhang C, Tran N, Fan H, Retzlaff K, et al. Characterization of global microRNA expression reveals oncogenic potential of miR-145 in metastatic colorectal cancer. BMC Cancer. 2009;9:374. doi: 10.1186/1471-2407-9-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Tang W, Du P, Wang G, Chen W, Li J, Zhu Y, Gao J, Cui L. Identifying microRNA-mRNA regulatory network in colorectal cancer by a combination of expression profile and bioinformatics analysis. BMC Syst Biol. 2012;6:68. doi: 10.1186/1752-0509-6-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Zhang XY, Zhang BF, Yang CQ, Chen XM, Gao HJ. Initial study of microRNA expression profiles of colonic cancer without lymph node metastasis. J Dig Dis. 2010;11:50–54. doi: 10.1111/j.1751-2980.2009.00413.x. [DOI] [PubMed] [Google Scholar]
- Lu S, Wang S, Geng S, Ma S, Liang Z, Jiao B. Upregulation of microRNA-224 confers a poor prognosis in glioma patients. Clin Transl Oncol. 2013;15:569–574. doi: 10.1007/s12094-012-0972-2. [DOI] [PubMed] [Google Scholar]
- Mian C, Pennelli G, Fassan M, Balistreri M, Barollo S, Cavedon E, Galuppini F, Pizzi M, Vianello F, Pelizzo MR, et al. MicroRNA profiles in familial and sporadic medullary thyroid carcinoma: preliminary relationships with RET status and outcome. Thyroid. 2012;22:890–896. doi: 10.1089/thy.2012.0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhale A, Kunder R, Goel A, Sarin R, Moiyadi A, Shenoy A, Mamidipally C, Noronha S, Kannan S, Shirsat NV. Distinctive microRNA signature of medulloblastomas associated with the WNT signaling pathway. J Cancer Res Ther. 2010;6:521–529. doi: 10.4103/0973-1482.77072. [DOI] [PubMed] [Google Scholar]
- Scapoli L, Palmieri A, Lo Muzio L, Pezzetti F, Rubini C, Girardi A, Farinella F, Mazzotta M, Carinci F. MicroRNA expression profiling of oral carcinoma identifies new markers of tumor progression. Int J Immunopathol Pharmacol. 2010;23:1229–1234. doi: 10.1177/039463201002300427. [DOI] [PubMed] [Google Scholar]
- Kim YK, Yu J, Han TS, Park SY, Namkoong B, Kim DH, Hur K, Yoo MW, Lee HJ, Yang HK, et al. Functional links between clustered microRNAs: suppression of cell-cycle inhibitors by microRNA clusters in gastric cancer. Nucleic Acids Res. 2009;37:1672–1681. doi: 10.1093/nar/gkp002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhyastha R, Madhyastha H, Nakajima Y, Omura S, Maruyama M. MicroRNA signature in diabetic wound healing: promotive role of miR-21 in fibroblast migration. Int Wound J. 2012;9:355–361. doi: 10.1111/j.1742-481X.2011.00890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff JW, Powers LS, Dickson AM, Kim J, Reisetter AC, Hassan IH, Kremens K, Gross TJ, Wilson ME, Monick MM. Cigarette smoking decreases global microRNA expression in human alveolar macrophages. PLoS ONE. 2012;7:e44066. doi: 10.1371/journal.pone.0044066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dmitriev P, Barat A, Polesskaya A, O'Connell MJ, Robert T, Dessen P, Walsh TA, Lazar V, Turki A, Carnac G, et al. Simultaneous miRNA and mRNA transcriptome profiling of human myoblasts reveals a novel set of myogenic differentiation-associated miRNAs and their target genes. BMC Genomics. 2013;14:265. doi: 10.1186/1471-2164-14-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Yoon JG, Li L, Yu W, Shao J, Hua D, Zheng S, Hood L, Goodlett DR, Foltz G, et al. The SOX2 response program in glioblastoma multiforme: an integrated ChIP-seq, expression microarray, and microRNA analysis. BMC Genomics. 2011;12:11. doi: 10.1186/1471-2164-12-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Schooneveld E, Wouters MC, Van der Auwera I, Peeters DJ, Wildiers H, Van Dam PA, Vergote I, Vermeulen PB, Dirix LY, Van Laere SJ. Expression profiling of cancerous and normal breast tissues identifies microRNAs that are differentially expressed in serum from patients with (metastatic) breast cancer and healthy volunteers. Breast Cancer Res. 2012;14:R34. doi: 10.1186/bcr3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veerla S, Lindgren D, Kvist A, Frigyesi A, Staaf J, Persson H, Liedberg F, Chebil G, Gudjonsson S, Borg A, et al. MiRNA expression in urothelial carcinomas: important roles of miR-10a, miR-222, miR-125b, miR-7 and miR-452 for tumor stage and metastasis, and frequent homozygous losses of miR-31. Int J Cancer. 2009;124:2236–2242. doi: 10.1002/ijc.24183. [DOI] [PubMed] [Google Scholar]
- Puerta-Gil P, Garcia-Baquero R, Jia AY, Ocana S, Alvarez-Mugica M, Alvarez-Ossorio JL, Cordon-Cardo C, Cava F, Sanchez-Carbayo M. miR-143, miR-222, and miR-452 are useful as tumor stratification and noninvasive diagnostic biomarkers for bladder cancer. Am J Pathol. 2012;180:1808–1815. doi: 10.1016/j.ajpath.2012.01.034. [DOI] [PubMed] [Google Scholar]
- Liu SG, Qin XG, Zhao BS, Qi B, Yao WJ, Wang TY, Li HC, Wu XN. Differential expression of miRNAs in esophageal cancer tissue. Oncol Lett. 2013;5:1639–1642. doi: 10.3892/ol.2013.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Kelnar K, Vlassov AV, Brown D, Wang J, Tang DG. Distinct microRNA expression profiles in prostate cancer stem/progenitor cells and tumor-suppressive functions of let-7. Cancer Res. 2012;72:3393–3404. doi: 10.1158/0008-5472.CAN-11-3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehy NT, Cordes KR, White MP, Ivey KN, Srivastava D. The neural crest-enriched microRNA miR-452 regulates epithelial-mesenchymal signaling in the first pharyngeal arch. Development. 2010;137:4307–4316. doi: 10.1242/dev.052647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, Naar AM. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science. 2010;328:1566–1569. doi: 10.1126/science.1189123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock MD, Sayan AE, Packham GK, Mirnezami AH. MicroRNAs: critical regulators of epithelial to mesenchymal (EMT) and mesenchymal to epithelial transition (MET) in cancer progression. Biol Cell. 2012;104:3–12. doi: 10.1111/boc.201100115. [DOI] [PubMed] [Google Scholar]
- Sreekumar R, Sayan BS, Mirnezami AH, Sayan AE. MicroRNA control of invasion and metastasis pathways. Front Genet. 2011;2:58. doi: 10.3389/fgene.2011.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013;19:1438–1449. doi: 10.1038/nm.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denecker G, Vandamme N, Akay O, Koludrovic D, Taminau J, Lemeire K, Gheldof A, De Craene B, Van Gele M, Brochez L, et al. Identification of a ZEB2-MITF-ZEB1 transcriptional network that controls melanogenesis and melanoma progression. Cell Death Differ. 2014;21:1250–1261. doi: 10.1038/cdd.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes J, Srivastava J, Madson N, Wittmann T, Barber DL. Dynamic actin remodeling during epithelial-mesenchymal transition depends on increased moesin expression. Mol Biol Cell. 2011;22:4750–4764. doi: 10.1091/mbc.E11-02-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JH, Li JH, Shao P, Zhou H, Chen YQ, Qu LH. starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res. 2011;39:D202–D209. doi: 10.1093/nar/gkq1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman Y, Naamati G, Linial M. MiRror: a combinatorial analysis web tool for ensembles of microRNAs and their targets. Bioinformatics. 2010;26:1920–1921. doi: 10.1093/bioinformatics/btq298. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Chng WJ. Roles of thioredoxin binding protein (TXNIP) in oxidative stress, apoptosis and cancer. Mitochondrion. 2013;13:163–169. doi: 10.1016/j.mito.2012.06.004. [DOI] [PubMed] [Google Scholar]
- Goldberg SF, Miele ME, Hatta N, Takata M, Paquette-Straub C, Freedman LP, Welch DR. Melanoma metastasis suppression by chromosome 6: evidence for a pathway regulated by CRSP3 and TXNIP. Cancer Res. 2003;63:432–440. [PubMed] [Google Scholar]
- Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishinaka Y, Nishiyama A, Masutani H, Oka S, Ahsan KM, Nakayama Y, Ishii Y, Nakamura H, Maeda M, Yodoi J. Loss of thioredoxin-binding protein-2/vitamin D3 up-regulated protein 1 in human T-cell leukemia virus type I-dependent T-cell transformation: implications for adult T-cell leukemia leukemogenesis. Cancer Res. 2004;64:1287–1292. doi: 10.1158/0008-5472.can-03-0908. [DOI] [PubMed] [Google Scholar]
- Wang Y, Toh HC, Chow P, Chung AY, Meyers DJ, Cole PA, Ooi LL, Lee CG. MicroRNA-224 is up-regulated in hepatocellular carcinoma through epigenetic mechanisms. FASEB J. 2012;26:3032–3041. doi: 10.1096/fj.11-201855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen H, Haldrup C, Strand S, Mundbjerg K, Mortensen MM, Thorsen K, Ostenfeld MS, Wild PJ, Arsov C, Goering W, et al. Hypermethylation of the GABRE∼miR-452∼miR-224 promoter in prostate cancer predicts biochemical recurrence after radical prostatectomy. Clin Cancer Res. 2014;20:2169–2181. doi: 10.1158/1078-0432.CCR-13-2642. [DOI] [PubMed] [Google Scholar]
- Diaz-Lopez A, Moreno-Bueno G, Cano A. Role of microRNA in epithelial to mesenchymal transition and metastasis and clinical perspectives. Cancer Manag Res. 2014;6:205–216. doi: 10.2147/CMAR.S38156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910–14914. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giricz O, Reynolds PA, Ramnauth A, Liu C, Wang T, Stead L, Childs G, Rohan T, Shapiro N, Fineberg S, et al. Hsa-miR-375 is differentially expressed during breast lobular neoplasia and promotes loss of mammary acinar polarity. J Pathol. 2012;226:108–119. doi: 10.1002/path.2978. [DOI] [PubMed] [Google Scholar]
- Bolos V, Peinado H, Perez-Moreno MA, Fraga MF, Esteller M, Cano A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci. 2003;116:499–511. doi: 10.1242/jcs.00224. [DOI] [PubMed] [Google Scholar]
- Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. 2011;68:3033–3046. doi: 10.1007/s00018-011-0735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama A, Matsui M, Iwata S, Hirota K, Masutani H, Nakamura H, Takagi Y, Sono H, Gon Y, Yodoi J. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J Biol Chem. 1999;274:21645–21650. doi: 10.1074/jbc.274.31.21645. [DOI] [PubMed] [Google Scholar]
- Welsh SJ, Bellamy WT, Briehl MM, Powis G. The redox protein thioredoxin-1 (Trx-1) increases hypoxia-inducible factor 1alpha protein expression: Trx-1 overexpression results in increased vascular endothelial growth factor production and enhanced tumor angiogenesis. Cancer Res. 2002;62:5089–5095. [PubMed] [Google Scholar]
- Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Min W. Thioredoxin promotes ASK1 ubiquitination and degradation to inhibit ASK1-mediated apoptosis in a redox activity-independent manner. Circ Res. 2002;90:1259–1266. doi: 10.1161/01.res.0000022160.64355.62. [DOI] [PubMed] [Google Scholar]
- Zhou J, Bi C, Cheong LL, Mahara S, Liu SC, Tay KG, Koh TL, Yu Q, Chng WJ. The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood. 2011;118:2830–2839. doi: 10.1182/blood-2010-07-294827. [DOI] [PubMed] [Google Scholar]
- Kwon HJ, Won YS, Suh HW, Jeon JH, Shao Y, Yoon SR, Chung JW, Kim TD, Kim HM, Nam KH, et al. Vitamin D3 upregulated protein 1 suppresses TNF-alpha-induced NF-kappaB activation in hepatocarcinogenesis. J Immunol. 2010;185:3980–3989. doi: 10.4049/jimmunol.1000990. [DOI] [PubMed] [Google Scholar]
- Sheth SS, Bodnar JS, Ghazalpour A, Thipphavong CK, Tsutsumi S, Tward AD, Demant P, Kodama T, Aburatani H, Lusis AJ. Hepatocellular carcinoma in Txnip-deficient mice. Oncogene. 2006;25:3528–3536. doi: 10.1038/sj.onc.1209394. [DOI] [PubMed] [Google Scholar]
- Butler LM, Zhou X, Xu WS, Scher HI, Rifkind RA, Marks PA, Richon VM. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc Natl Acad Sci USA. 2002;99:11700–11705. doi: 10.1073/pnas.182372299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa K, Nishiyama H, Matsui Y, Kobayashi T, Saito R, Kotani H, Masutani H, Oishi S, Toda Y, Fujii N, et al. Thioredoxin-interacting protein suppresses bladder carcinogenesis. Carcinogenesis. 2011;32:1459–1466. doi: 10.1093/carcin/bgr137. [DOI] [PubMed] [Google Scholar]
- Masaki S, Masutani H, Yoshihara E, Yodoi J. Deficiency of thioredoxin binding protein-2 (TBP-2) enhances TGF-beta signaling and promotes epithelial to mesenchymal transition. PLoS ONE. 2012;7:e39900. doi: 10.1371/journal.pone.0039900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Shi Y, Hou Y, Ren Y, Du C, Zhang L, Li Y, Duan H. Knockdown of thioredoxin-interacting protein ameliorates high glucose-induced epithelial to mesenchymal transition in renal tubular epithelial cells. Cell Signal. 2013;25:2788–2796. doi: 10.1016/j.cellsig.2013.09.009. [DOI] [PubMed] [Google Scholar]
- Kaimul AM, Nakamura H, Masutani H, Yodoi J. Thioredoxin and thioredoxin-binding protein-2 in cancer and metabolic syndrome. Free Radic Biol Med. 2007;43:861–868. doi: 10.1016/j.freeradbiomed.2007.05.032. [DOI] [PubMed] [Google Scholar]
- Glasauer A, Chandel NS. Targeting antioxidants for cancer therapy. Biochem Pharmacol. 2014;92:90–101. doi: 10.1016/j.bcp.2014.07.017. [DOI] [PubMed] [Google Scholar]
- Patwari P, Lee RT. An expanded family of arrestins regulate metabolism. Trends Endocrinol Metab. 2012;23:216–222. doi: 10.1016/j.tem.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alla V, Kowtharapu BS, Engelmann D, Emmrich S, Schmitz U, Steder M, Putzer BM. E2F1 confers anticancer drug resistance by targeting ABC transporter family members and Bcl-2 via the p73/DNp73-miR-205 circuitry. Cell Cycle. 2012;11:3067–3078. doi: 10.4161/cc.21476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll S, Furst K, Thomas S, Villanueva Baselga S, Stoll A, Schaefer S, Putzer BM. Dissection of cell context-dependent interactions between HBx and p53 family members in regulation of apoptosis: a role for HBV-induced HCC. Cell Cycle. 2011;10:3554–3565. doi: 10.4161/cc.10.20.17856. [DOI] [PubMed] [Google Scholar]
- Racek T, Buhlmann S, Rust F, Knoll S, Alla V, Putzer BM. Transcriptional repression of the prosurvival endoplasmic reticulum chaperone GRP78/BIP by E2F1. J Biol Chem. 2008;283:34305–34314. doi: 10.1074/jbc.M803925200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Table S1
Supplementary Table S2
Review Process File