

ABSTRACT
Newly emerging human viruses such as Ebola virus, severe acute respiratory syndrome (SARS) virus, and HIV likely originate within an extant population of viruses in nonhuman hosts and acquire the ability to infect and cause disease in humans. Although several mechanisms preventing viral infection of particular hosts have been described, the mechanisms and constraints on viral host expansion are ill defined. We describe here mycobacteriophage Patience, a newly isolated phage recovered using Mycobacterium smegmatis mc2155 as a host. Patience has genomic features distinct from its M. smegmatis host, including a much lower GC content (50.3% versus 67.4%) and an abundance of codons that are rarely used in M. smegmatis. Nonetheless, it propagates well in M. smegmatis, and we demonstrate the use of mass spectrometry to show expression of over 75% of the predicted proteins, to identify new genes, to refine the genome annotation, and to estimate protein abundance. We propose that Patience evolved primarily among lower-GC hosts and that the disparities between its genomic profile and that of M. smegmatis presented only a minimal barrier to host expansion. Rapid adaptions to its new host include recent acquisition of higher-GC genes, expression of out-of-frame proteins within predicted genes, and codon selection among highly expressed genes toward the translational apparatus of its new host.
IMPORTANCE
The mycobacteriophage Patience genome has a notably lower GC content (50.3%) than its Mycobacterium smegmatis host (67.4%) and has markedly different codon usage biases. The viral genome has an abundance of codons that are rare in the host and are decoded by wobble tRNA pairing, although the phage grows well and expression of most of the genes is detected by mass spectrometry. Patience thus has the genomic profile of a virus that evolved primarily in one type of host genetic landscape (moderate-GC bacteria) but has found its way into a distinctly different high-GC environment. Although Patience genes are ill matched to the host expression apparatus, this is of little functional consequence and has not evidently imposed a barrier to migration across the microbial landscape. Interestingly, comparison of expression levels and codon usage profiles reveals evidence of codon selection as the genome evolves and adapts to its new environment.
INTRODUCTION
Mycobacteriophages are viruses that infect mycobacterial hosts (1). More than 300 completely sequenced mycobacteriophage genomes are available in GenBank, all of which—with the exception of DS6A—were either isolated on or are known to infect Mycobacterium smegmatis mc2155 (2–4). The genomic diversity of these phages is high, and there are many groups (“clusters”) that share little or no nucleotide sequence information (1, 5). Currently, 20 clusters have been described (clusters A to T), as well as nine singletons, i.e., phages for which relatives have yet to be identified (2, 3). Genomes within a cluster generally share nucleotide sequence identity spanning greater than 50% of genome length, but there is considerable diversity within most of the clusters, and many can be readily divided into subclusters based on relative nucleotide sequence similarity (5); the largest group, cluster A, currently is divided into 11 subclusters (2).
There is considerable variation in percent GC of the mycobacteriophages, ranging from 50.3% to 70% GC. In contrast, the mycobacterial hosts M. smegmatis mc2155 and Mycobacterium tuberculosis H37Rv have high GC contents—with 67.4% and 65.6% GC, respectively—typical of the genus Mycobacterium (Mycobacterium leprae is atypically low with 57.8%) (6). However, other species within the family Mycobacteriaceae span a broader percent GC range, including Corynebacterium pseudotuberculosis, with 52.2% GC, and Corynebacterium variabile, having 67.2% GC. The percent GC correlates closely with cluster designation (1), but those with the lowest percent GC are relatively rarely isolated, including the singleton Patience (50.3%) and the five cluster H phages (average, 57.3% GC). The genome diversity and percent GC range of the mycobacteriophages support a model for phage genome evolution in which phages migrate much more rapidly across a diverse bacterial landscape than their genomes ameliorate toward that of any one host (4). Thus, mycobacteriophages at the lower end of the percent GC spectrum are predicted to have infected lower-GC hosts in their recent evolutionary pasts (4). We note that there are other phage-host systems with mismatched GC contents, such as T4 and the right arm of phage lambda (35.3% and 44.4%, respectively), relative to their Escherichia coli host (50.8%).
There is also substantial variation in tRNA content of the mycobacteriophages. Many have no tRNA genes, and some have only one or a small number, while others—such as the members of clusters C and M—have more than 20 (7, 8). The rationale for carriage of these is unclear, and there is no obvious correlation between tRNA content and percent GC that might reflect tRNA acquisition to augment gene expression in newly acquired hosts. It has been noted that there is no close correlation between the tRNA specificities encoded by D29 and infrequently used codons (9) or between Bxz1 and phage codon preferences of putative high-expression genes (10). Analysis of the codon usage of 32 mycobacteriophage genomes showed that there is variation in codon usage preferences (11). In some phage genomes, the tRNAs may counteract host measures to protect themselves from infection by tRNA destruction (12).
Here, we describe mycobacteriophage Patience, a newly isolated phage of M. smegmatis mc2155 that is a singleton with no close relatives and has a GC content of 50.3%, representing the extreme low end of the percent GC spectrum for mycobacteriophages. Of the predicted 109 Patience protein-coding genes, 61% are “orphams” with no close mycobacteriophage relatives in the Phamerator_285 database, and most of the 48 genes with homologues in other mycobacteriophages are most closely related to those in clusters H, R, and D, which also have below-average GC contents. However, Patience has a distinctly different codon usage profile from both its host and other mycobacteriophages and an abundance of codons that are rarely used in the host. Proteomic analysis using mass spectrometry provides evidence for expression of at least 83 Patience proteins, two of which are from cryptic open reading frames (ORFs) embedded within annotated genes. We propose that Patience is a relatively recent visitor to the Mycobacterium neighborhood, having evolved primarily in lower-GC hosts within the Actinomycetales, and is in the process of adapting to growth in its new high-GC genetic environment.
RESULTS
Isolation and genome sequencing of mycobacteriophage Patience.
Mycobacteriophage Patience was isolated by direct plating of an environmental sample taken from near the Nelson Mandela School of Medicine at the University of KwaZulu-Natal (UKZN), Durban, South Africa, using M. smegmatis mc2155 as a host (3). Isolation and purification of Patience were components of a 2-week workshop on mycobacterial genetics offered in July 2009; the genome was sequenced at the University of Pittsburgh and annotated in a second 2-week workshop at UKZN in July 2011. Patience forms normal-size hazy (~1-mm-diameter) plaques on M. smegmatis mc2155 at 37°C under standard conditions, although we have been unsuccessful in recovering stable lysogens. It can easily be propagated on solid media to titers greater than 1010 PFU/ml. The genome is 70,506 bp long, circularly permuted, and presumably terminally redundant. For linear presentation, coordinate 1 is designated the beginning of an open reading frame upstream of the large terminase subunit consistent with the organization of the cluster H phages that contain homologues of the first open reading frame at their left ends (7). The GenBank submission (JN412589) has been reported previously (3). Genome annotation identified 109 putative open reading frames (ORFs) and one tRNA gene (Table 1); one additional ORF was annotated using mass spectrometry analysis (see below).
TABLE 1 .
Genometrics of phage Patience
| Gene | Start | Stop | Molecular mass (kDa)a | Functionb |
|---|---|---|---|---|
| 1 | 1 | 351 | 13.0 | Virion protein |
| 2 | 398 | 616 | 8.1 | NDM |
| 3 | 668 | 2029 | 50.8 | NDM |
| 4 | 2042 | 2350 | 10.7 | Virion protein |
| 5 | 2590 | 3042 | 17.5 | HNH |
| 6 | 3035 | 4702 | 63.7 | Terminase large subunit |
| 7 | 4712 | 5242 | 20.5 | Endo VII |
| 8 | 5239 | 6870 | 61.1 | Portal |
| 111 | 6881 | 6994 | 4.4 | Virion protein |
| 9 | 6991 | 7086 | 3.9 | Virion protein |
| 10 | 7102 | 7299 | 7.9 | Virion protein |
| 11 | 7300 | 7722 | 15.6 | Virion protein |
| 12 | 7723 | 7944 | 8.6 | Virion protein |
| 13 | 7977 | 8486 | 18.9 | Virion protein |
| 14 | 8486 | 9031 | 19.7 | Virion protein |
| 15 | 9031 | 9918 | 32.0 | Virion protein |
| 16 | 9978 | 10127 | 5.8 | Virion protein |
| 17 | 10120 | 10470 | 12.2 | Virion protein |
| 18 | 10470 | 11075 | 22.1 | Virion protein |
| 19 | 11079 | 11519 | 17.1 | Protease |
| 20 | 11512 | 12426 | 34.1 | MuF-like protein |
| 21 | 12449 | 13234 | 29.1 | Virion protein |
| 22 | 13312 | 14010 | 23.9 | Virion protein |
| 23 | 14060 | 15262 | 44.0 | Capsid |
| 24 | 15371 | 16057 | 25.0 | Virion protein |
| 25 | 16060 | 16389 | 11.6 | Virion protein |
| 26 | 16390 | 16761 | 13.8 | Virion protein |
| 27 | 16939 | 17307 | 13.9 | Virion protein |
| 28 | 17304 | 17705 | 14.8 | Virion protein |
| 29 | 17715 | 18134 | 13.8 | Virion protein |
| 30 | 18140 | 18358 | 8.4 | NDM |
| 31 | 18355 | 19203 | 30.5 | Major tail subunit |
| 32 | 19282 | 19842 | 20.8 | Tail assembly chaperone |
| 33 | 19880 | 20101 | 8.2 | Tail assembly chaperone |
| 34 | 20117 | 26764 | 239.4 | Tape measure protein |
| 35 | 26761 | 27651 | 33.8 | Minor tail subunit |
| 36 | 27651 | 29294 | 61.1 | Minor tail subunit |
| 37 | 29291 | 32593 | 118.6 | Minor tail subunit |
| 38 | 32593 | 33633 | 34.8 | Minor tail subunit |
| 39 | 33643 | 34008 | 12.5 | Minor tail subunit |
| 40 | 34097 | 34399 | 11.7 | Mycobacterium Hyp. |
| 41 | 34399 | 35790 | 52.4 | Lysin A |
| 42 | 35792 | 36817 | 38.2 | Lysin B |
| 43 | 36817 | 37158 | 12.3 | NDM |
| 44 | 37159 | 37653 | 18.5 | Holin? |
| 45 | 37646 | 38032 | 14.8 | NDM |
| 46 | 38032 | 38349 | 11.8 | NDM |
| 47 | 38753 | 39469 | 31.6 | NDM |
| 48 | 39702 | 40082 | 14.2 | NDM |
| 49 | 40079 | 40213 | 5.0 | NDM |
| 50 | 40236 | 40433 | 7.2 | NDM |
| 51 | 40426 | 40668 | 9.2 | Gordonia Hyp. |
| 52 | 40665 | 41027 | 13.9 | Mycobacterium Hyp. |
| 53 | 41121 | 41441 | 12.6 | NDM |
| 54 | 41450 | 41854 | 15.5 | NDM |
| 55 | 41857 | 42051 | 7.3 | NDM |
| 56 | 42103 | 42534 | 16.2 | NDM |
| 57 | 42611 | 42934 | 12.0 | NDM |
| 58 | 42937 | 43206 | 10.2 | Rhodococcus phage Hyp. |
| 59 | 43203 | 43586 | 14.3 | Rhodococcus Hyp. |
| 60 | 43583 | 43951 | 14.1 | NDM |
| 61 | 44078 | 45382 | 49.0 | Rhodococcus phage Hyp. |
| 62 | 45456 | 45602 | 5.8 | NDM |
| 63 | 45606 | 45728 | 4.6 | NDM |
| 64 | 45725 | 45862 | 5.3 | NDM |
| 65 | 45846 | 46085 | 8.7 | NDM |
| 66 | 46082 | 46231 | 5.7 | NDM |
| 67 | 46228 | 46635 | 14.6 | Excaliber Ca-binding domain |
| 68 | 46625 | 46792 | 6.4 | NDM |
| 69 | 46792 | 46995 | 7.9 | NDM |
| 70 | 47011 | 47343 | 12.4 | Enterococcus Hyp. |
| 71 | 47574 | 47897 | 11.0 | NDM |
| 72 | 48058 | 48321 | 9.5 | NDM |
| 73 | 48322 | 48591 | 10.2 | DNM |
| 74 | 48588 | 49178 | 22.4 | NDM |
| 75 | 49192 | 49716 | 20.9 | NDM |
| 76 | 49716 | 51749 | 77.0 | Helicase |
| 77 | 51746 | 52165 | 16.6 | NDM |
| 78 | 52173 | 52523 | 13.9 | NDM |
| 79 | 52672 | 53226 | 20.2 | NDM |
| 80 | 53329 | 53565 | 9.2 | NDM |
| 81 | 53552 | 54784 | 47.8 | Helicase/nuclease |
| 82 | 54781 | 54972 | 7.0 | NDM |
| 83 | 54950 | 55153 | 7.8 | NDM |
| 84 | 55153 | 55449 | 11.7 | NDM |
| 85 | 55493 | 56551 | 39.5 | RecA |
| 86 | 56615 | 57301 | 24.8 | NDM |
| 87 | 57360 | 57432 | 0.0 | tRNA-Gln |
| 88 | 57445 | 57675 | 8.3 | NDM |
| 89 | 57623 | 57922 | 8.8 | NDM |
| 90 | 57924 | 58265 | 12.7 | NDM |
| 91 | 58279 | 61512 | 122.6 | DNA Pol III alpha |
| 92 | 61521 | 61976 | 17.2 | NDM |
| 93 | 61969 | 62667 | 26.3 | RuvC |
| 94 | 62743 | 62928 | 6.9 | NDM |
| 95 | 62925 | 63299 | 14.1 | NDM |
| 96 | 63328 | 63657 | 13.2 | NDM |
| 97 | 63657 | 64004 | 13.1 | NDM |
| 98 | 64021 | 64287 | 10.1 | NDM |
| 99 | 64284 | 64436 | 5.6 | NDM |
| 100 | 64429 | 64641 | 8.1 | NDM |
| 101 | 64577 | 64987 | 12.2 | NDM |
| 102 | 64987 | 65295 | 11.6 | NDM |
| 103 | 65328 | 65792 | 17.2 | MazG-like |
| 104 | 65789 | 66052 | 10.0 | NDM |
| 105 | 66049 | 66372 | 12.4 | NDM |
| 106 | 66376 | 66765 | 14.7 | NDM |
| 107 | 66758 | 67078 | 12.2 | NDM |
| 108 | 67146 | 67319 | 6.5 | NDM |
| 109 | 67316 | 70024 | 102.1 | Primase/polymerase |
| 110 | 70021 | 70275 | 9.5 | NDM |
Predicted molecular mass of product in kilodaltons.
Function if known or predicted from BLASTP or HHPred analyses. NDM, no database match, other than to other mycobacteriophage proteins. Hyp., database match to a hypothetical protein of unknown function. Virion proteins were identified by mass spectrometry as shown in Table S1 in the supplemental material.
Virion morphology and virion PAGE analysis.
Mycobacteriophage Patience is morphologically a member of the Siphoviridae with an isometric head and a long flexible tail (Fig. 1A). The diameter of the heads averages 58 nm, and the tails average 358 nm in length, among the longest of the mycobacteriophages described to date, similar to those of the cluster M phages (348 nm [8]); other phages with notably long tails are those in cluster H (approximately 290 nm) and cluster R (approximately 288 nm) (13). SDS-PAGE analysis of Patience particles shows three abundant proteins and at least 12 other lower-abundance proteins between the sizes of 15 and 150 kDa (Fig. 1B).
FIG 1 .
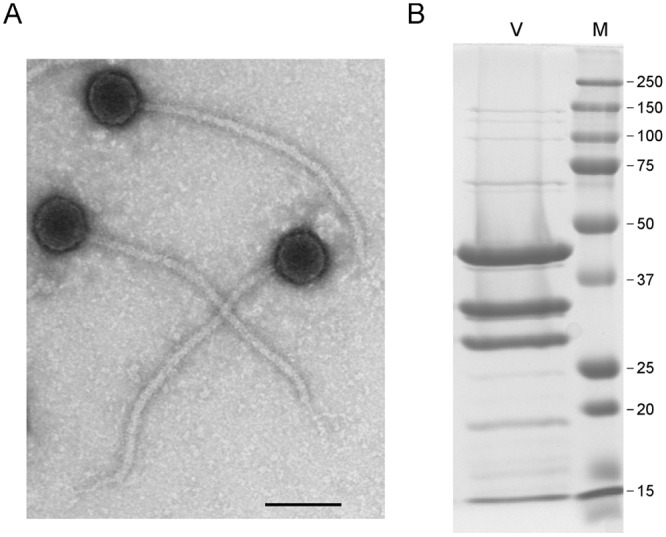
Mycobacteriophage Patience virions. (A) Electron micrograph of Patience virions. Bar, 100 nm. (B) SDS-PAGE of Patience virions (V) and marker proteins (M). The three most abundant proteins likely correspond to the major tail subunit (gp31), the capsid subunit (gp23), and gp15, with predicted molecular masses of 30.5 kDa, 35.3 kDa (after processing), and 32 kDa, respectively. The major tail subunit may migrate slower than its predicted mass, as observed in some other phages (37). Numbers at right are molecular masses in kilodaltons.
Relationship of Patience to other mycobacteriophages.
The Patience genome is not closely related to other phage genomes, although dot plot analysis shows weak similarity to cluster H phages (Fig. 2). The related segments span only 14%, 8%, and 6% of genome lengths with Barnyard, Predator, and Konstantine genomes, respectively (Fig. 2), and the closest matching segment of Patience and Barnyard is a 1,091-bp region (73% nucleotide identity) corresponding to the capsid subunit genes. Alignment of the genome maps of Patience, Barnyard, Konstantine, and Predator in Phamerator shows their architectural relationships (Fig. 3), and although the nucleotide sequence similarity is minimal, 37% of Patience genes have homologues in cluster H phages (Fig. 3). However, more than half of the Patience ORFs are orphams (genes with no close mycobacteriophage relatives [Fig. 3; Table 1]).
FIG 2 .

Dot plot analysis of Patience and cluster H phages. Dot plot analysis was performed using Gepard (31), and the designation of subcluster H1 and H2 genomes is shown.
FIG 3 .

Alignment of genome maps of mycobacteriophages Patience, Barnyard, Konstantine, and Predator. Maps were generated using Phamerator (35) and the database Mycobacteriophage_285, containing 285 complete genome sequences, and aligned by the left ends of the tape measure gene. Maps are displayed in three tiers with pairwise nucleotide sequence similarities displayed in spectrum coloring, with violet being the most similar and red the least similar (minimal BLASTN cutoff E value is 10−4). Genes are shown as colored boxes, with colors reflecting pham assignments for each gene (gene members of a phamily have the same color). Genes shown in white are orphams and have no mycobacteriophage relatives with greater than 32.5% amino acid identity or a BLASTP E value lower than 10−50. Phamily assignments with the number of phamily members in parentheses are above each gene.
The overall GC content of the Patience genome is 50.3% and is maintained almost throughout the genome (Fig. 4A). There are two notable departures suggesting recent acquisitions by horizontal exchange. One is within Patience gene 37, where the 3′ end has 81% nucleotide identity to Rosebush gene 32 (subcluster B2) and the elevated percent GC reflects that of the B2 phages (Fig. 4B). Patience gp37 is related to tail fibers of many other mycobacteriophages that are implicated in host range determination (4). The second example is gene 47 (62% GC [Fig. 4C]), although it is an orpham with no close mycobacteriophage relatives and is of unknown function.
FIG 4 .

Distribution of percent GC in the Patience genome. (A) Percent GC scan of the Patience genome. The dotted horizontal line indicates the average GC content of 50.3%. (B) Percent GC plot of the Patience minor tail protein genes. Genes 34 to 39 encoding putative minor tail proteins have GC contents similar to those of the genome as a whole, but a segment of gene 37 with nucleotide similarity to other mycobacteriophages (coordinates 31172 to 32559; blue bar) corresponds with an increase in percent GC (55.6%). (C) Variation in percent GC about gene 47.
Patience genome organization.
Genome annotation of Patience indicates that all open reading frames and one tRNA gene are transcribed in the same direction (Fig. 5; Table 1). The overall coding capacity is 94.9%, and there are no intergenic spaces greater than 250 bp. The recognizable virion structure and assembly functions lie within the gene 4 to 39 region, and the siphoviral syntenic arrangement of terminase, portal, protease, capsid, head-tail connector proteins, major tail subunit, tail assembly chaperones, tape measure, and minor tail protein genes is observed, spanning 32 kbp of the genome (14). This is atypically long not only because of the long tape measure gene corresponding to the long phage tail but because of a dozen genes of mostly unknown function located between the terminase large subunit (gene 6) and portal (gene 8) genes, between the portal (gene 8) and protease (gene 19) genes, and between the MuF-like gene (gene 20) and the putative scaffolding gene (gene 22) (Fig. 5); most of these are orphams (i.e., they do not have a close mycobacteriophage relative), with the exceptions of genes 10 and 18 (Fig. 5). Gene 7 encodes a putative Endo VII protein and has weak similarity (<30% identity) to genes in phages Konstantine and Predator (subcluster H1), as well as Dori (singleton). The gene upstream of the terminase large subunit gene encodes a protein with similarities to putative HNH homing nucleases (Fig. 5) but may function as part of the DNA packaging machinery as described for HK97 gp74 (15). The four leftmost genes (genes 1 to 4) are of unknown function.
FIG 5 .

Genome map of mycobacteriophage Patience. The map was generated using Phamerator (35) and displayed as described for Fig. 3. Putative functional assignments are shown. Identification of the gene product by mass spectrometry in particles (P) or early (E)- or late (L)-infected samples is shown above each gene.
The lysis cassette is located downstream of the virion structural genes and contains an endolysin gene (gene 41, lysin A) and a putative mycolylarabinogalactan esterase (gene 42, lysin B) (16, 17). Genes 43 and 44 code for proteins containing two and four predicted transmembrane domains, respectively, that may act as holins or lysis chaperones (18). The lysis cassette is not closely related to that of the cluster H phages, and lysin A is most closely related to the corresponding genes of cluster M phage PegLeg gp35 and Bongo gp35 (35% identity), with an Org-U domain organization (19). In contrast, lysin B is most closely related to phages in subcluster B3 (66% identity to Gadget gp48), illustrating the mosaic nature of the lysis cassette (19). Patience gp43 has no homologues, but gp44 has distant relatives in the cluster D phages (<30% identity), where the genes are located between lysin A and lysin B.
Of the other 65 Patience ORFs, only seven can be assigned putative functions, most of which are involved in DNA metabolism (helicases, RecA, DNA polymerase III [Pol III] alpha subunit, RuvC, and a DNA primase/polymerase [Fig. 4; Table 1]). These all have homologues in a variety of mycobacteriophages as well as in phages of Gordonia and Corynebacterium. One (gene 103) encodes a MazG-like protein, a putative nucleoside triphosphate pyrophosphohydrolase that is common in a variety of phage genomes (20). Patience has a single tRNA gene, and the tRNA is predicted to be charged with glutamine and has the anticodon 5′-UUG [i.e., tRNAGln(UUG)]. It is unclear how the Patience genes are transcribed, and there are no strongly predicted SigA-like promoters, similar to other mycobacteriophages such as Giles (21), even though SigA-like promoters are active in other mycobacteriophages (22–24).
Identification of Patience proteins by HPLC-MS/MS.
To determine which Patience genes are expressed in M. smegmatis, we analyzed three samples by high-pressure liquid chromatography–tandem mass spectrometry (HPLC-MS/MS): purified Patience particles and whole-cell extracts of Patience-infected samples at 30 min and 150 min after infection (Fig. 5; see also Table S1 in the supplemental material). Using stringent criteria for peptide identification, we identified 79 of the 109 previously annotated predicted gene products (82 different gene products, including previously unannotated genes) present in at least one of the samples, including 34 of the 35 putative virion structure and assembly proteins (genes 5 to 39), 27 of which are particle associated (Fig. 5). Although gp9 was not detected, particle-associated peptides corresponding to a previously unannotated 37-codon gene between genes 8 and 9 were identified, which we designate gene 111 (Fig. 5). Particles contain four additional products, gp1, gp4, gp79, and gp86, although gp86 is abundant in lytic growth and the few particle-associated peptides could be contaminants in the phage preparation (Table S1). The capsid subunit (gp23) appears to be proteolytically cleaved between residues 82 and 83 to generate a 35.3-kDa mature protein product; that and the gp31 major tail subunit (30.5 kDa) and gp15 (32 kDa) likely correspond to the three major species observed by SDS-PAGE (Fig. 1B). It is noteworthy that—with the exception of 31-residue gp9—all of the genes corresponding to insertions within the otherwise canonically syntenic virion structure and assembly operon are present in Patience particles, although only 3 spectra of gp21 were identified and could represent contaminants from the lysate (Fig. 5; Table S1).
Of the total of 81 Patience proteins identified in infected cells, only one—gp47—was not identified in the late-infected (2.5-h-postinfection) sample. Patience gp47 was identified in the early-infected sample and is presumably not expressed late in infection but also is turned over rapidly (Fig. 5; see also Table S1 in the supplemental material). Many of the virion proteins (genes 1 to 40) are expressed at higher levels (using peptide spectral count as a surrogate for expression) in the late-infected sample, although there is also substantial expression of many of these at the early time. Of the 29 annotated proteins not identified in any of the samples, 11 are predicted to be smaller than 10 kDa and may have escaped detection if they are not expressed at high levels. Twelve are predicted to be membrane or wall associated and are likely excluded from the soluble fractions used for analysis. Peptide abundance in infected cells is expected to generally correlate with expression, although other factors such as protein size and trypsin cleavage efficiencies influence peptide detection.
Unusual expression events revealed by HPLC-MS/MS.
Identification of a large number of Patience proteins provides several new insights into gene expression and posttranslational events. First, we note that peptides at or near the N terminus confirm the previously annotated translation start site for 54 proteins but also identify seven (gp4, gp17, gp29, gp47, gp53, gp89, and gp101) for which revisions of the annotated start sites are supported. For 16 proteins, the peptide coverage is insufficient to be informative about start site usage. Interestingly, seven proteins, all of them particle associated (gp1, gp20, gp21, gp26, gp31, gp37, and gp79), are acetylated at their N terminus (following methionine loss), although the functional significance—if any—is not known. Six are acetylated at an N-terminal threonine following methionine removal (the seventh is a serine acetylation), although not all proteins with N-terminal threonine residues are acetylated.
gp21 is unusual in that the N-terminal-most peptides start at residue 35 of the annotated product following a glycine in the −1 position, indicating that they were not generated either by tryptic digestion or by translation initiation. Other processes such as posttranslational processing or intron splicing prior to translation may be involved. We also note that gp24 is present in the particles but that the peptide coverage (62 total spectra) is restricted to the N-terminal 50% of the protein, whereas in infected cells, the spectra reflect 100% coverage of the protein (149 total spectra). This could be explained by assembly-associated protein processing.
Surprisingly, we identified several peptides corresponding to translation of regions wholly embedded within annotated genes. Two of these were identified only with peptides for which we have lower confidence and will not be considered further. The evidence supporting translation of the other two is, however, quite strong. The first of these is a 372-bp open reading frame transcribed on the same strand, in a different reading frame, within gene 91, the DNA polymerase III catalytic subunit (see Fig. S1A and S2A in the supplemental material). A total of 17 instances of seven different peptides were identified, and the spectra and fragmentation tables strongly support the peptide assignments (see Text S1 in the supplemental material). Moreover, 10 peptides correspond to the extreme N terminus with the methionine present, and there is a strong ribosome binding site upstream (Fig. S2). Although the protein is seemingly expressed, the functional relevance is unclear. The predicted product has no close database relatives, and the evidence for conservation is ambiguous (Fig. S1A). Barnyard contains a similar open reading frame within its polymerase, and the products share 30% amino acid identity, whereas the corresponding segments of the polymerases share only 53% identity. This region of the polymerase is more distantly related in Konstantine (40% identity), and the second open reading frame is not conserved (Fig. S1A).
A similar scenario is seen within Patience gene 20, where two peptides corresponding to a 171-bp open reading frame on the same strand were identified with high confidence (see Fig. S1B in the supplemental material). Peptides corresponding to the predicted N terminus were not identified, although a start codon with a strong ribosome binding site is present (Fig. S2B). The corresponding segment of Patience gp20 is conserved in Barnyard gp18 and Konstantine gp17, but the second open reading frame is not. Presumably, either expression of embedded out-of-frame genes is more common than anticipated or these reflect expression artifacts resulting from the use of noncognate host transcription and translation systems.
Codon selection and adaptation for mycobacterial growth.
Because M. smegmatis has a relatively high GC content (67.4%), it is not surprising that these mutational biases (reflected in base composition) result in frequent usage of GC base pairs in third-codon positions (Fig. 6A; see also Fig. S3 and S4 in the supplemental material). Each of the most commonly used of the synonymous codons has GC in the third position, and M. smegmatis mc2155 carries tRNAs with anticodons corresponding to all codons with GC in the third position, except for CGC (Fig. S3). All of the NNU codons are present infrequently and (with the exception of CGU) are decoded by wobble pairing in the codon third position (Fig. S3 and S4); M. tuberculosis H37Rv has an almost identical tRNA profile.
FIG 6 .

Codon usage and codon selection in mycobacteriophage Patience. (A) Relative synonymous codon usage (RSCU) for Patience and its host, M. smegmatis, is plotted for the 59 degenerate codons; RSCU was calculated for all genes in the respective genomes, thereby reflecting mutational biases. (B) Relationship between codon selection of 81 Patience genes and peptide counts (normalized for gene length) reported by mass spectrometry from M. smegmatis cells infected by Patience for 150 min. (C) Relationship between codon selection in Patience and its host, M. smegmatis. Codon selection is reported as δ values for each of the 59 degenerate codons. Codons are colored as in panel A (see inset).
Mycobacteriophages such as Twister (65% GC) and KayaCho (70% GC), with nucleotide compositions similar to that of M. smegmatis, have similar synonymous codon distributions (see Fig. S3 and S4 in the supplemental material). In contrast, Patience codon usage patterns are distinctly different, with notably different distributions of codons with respect to their third-position content relative to M. smegmatis (Fig. 6A; see also Fig. S3 and S4). There are a total of nine switches to a different most commonly used synonymous codon from M. smegmatis, seven of which are NNU codons that are rarely used in the host (five with normalized codon synonymous usage [NCSU] values of <0.1 [Fig. S3]). In total, NNU codons (except CGU, for which there is a cognate host tRNA) represent over 25% of all Patience codons; in contrast, these are only 5% of M. smegmatis codons. This represents an extreme end of a trend between the usage of NNU codons and the overall GC content of the phage, where usage of NNU increases as percent GC decreases (Fig. S5).
Although codon usage shows that Patience spent a significant portion of its evolutionary past in hosts with moderate GC contents, it is not clear what its current host range is. If Patience currently exploits more GC-rich hosts, then it would experience not only mutational biases, which would increase its GC content, but selective pressure on its most highly expressed genes to use tRNA pools poised to translate GC-rich genes. This selective pressure would be reflected in a preferred usage of GC-rich codons within highly expressed genes. Patience does experience codon selection, which is shown by the robust positive correlation between codon selection (adapative codon enrichment [ACEz] [25]) (see Materials and Methods) and level of gene expression, using total numbers of peptides as identified by mass spectrometry under stringent conditions as a surrogate for expression (Fig. 6B). Codon selection is measured using δ values, or the ratio of codon frequencies in genes experiencing codon selection normalized to their frequencies in genes lacking codon selection. Preferred codons are those with δ values greater than 1, indicating more frequent use in genes experiencing codon selection, frequently those expressed to greater levels. Patience favors the use of GC-rich codons in genes experiencing codon selection (Table 2), with 24 of 33 preferred codons bearing GC base pairs in their third positions, whereas 21 of 26 nonpreferred codons end with AT base pairs. Moreover, the patterns of codon preference in Patience (which codons are preferred and which are not) are strongly correlated with that of M. smegmatis (Fig. 6C), suggesting that this host imposes codon selection congruent with that currently being experienced by Patience. In contrast, Patience codon selection is not congruent with that imposed by hosts with more moderate GC content, such as Escherichia coli (Fig. 6C, inset), although such surveys can never be conclusive.
TABLE 2 .
Selection in Patience favors GC-rich codons
| Third-position base | No. of codons |
|
|---|---|---|
| Preferred (δ > 1) | Nonpreferred (δ < 1) | |
| G | 9 | 4 |
| A | 2 | 12 |
| T | 7 | 9 |
| C | 15 | 1 |
DISCUSSION
Patience represents an intriguing example of a virus that has successfully entered the mycobacterial genetic neighborhood in its relatively recent evolutionary history. Its overall GC content and codon usage profiles are distinctly different from those of its mycobacterial host, suggesting that it primarily evolved in a moderate-GC (~50%) environment. Growth in high-GC bacteria may have required multiple events, including acquisition of part of a tail gene (gene 47) by lateral gene transfer. Nonetheless, the mismatch between viral and host genomic profiles does not appear to have been a substantial impediment to host range expansion, although the highly expressed viral genes are under codon selection for more efficient translation by the host apparatus. Interestingly, although Patience conceivably could have responded by acquisition of a tRNA repertoire to facilitate phage gene expression, this has not occurred; the only phage-carried tRNA is tRNAGln(UUG), and CAA is not a rare codon in the Patience genome (see Fig. S3 in the supplemental material).
Patience is the first phage to our knowledge for which the proteomic profile in infected cells has been examined by mass spectrometry. The approach is highly informative, providing strong evidence that many of the annotated reading frames are expressed—including 29 that are shorter than 120 codons—and providing support for many of the translational start sites, as well as revisions of start sites of several genes. Moreover, a previously unannotated gene was identified (gene 111) containing only 37 codons, and the revision of start codons indicates that two pairs of genes have significant overlaps (>60 bp). Phage genome annotation is generally more error-prone than that of other genomes because of the abundance of small open reading frames and relatively small gene size (average mycobacteriophage gene length is 640 bp). HPLC-MS/MS adds confidence to genome annotation, especially for phages such as Patience, whose coding potential does not closely match its bacterial host. It is surprising to find that at least two reading frames embedded out of frame within annotated genes are also expressed. These ORFs are generally not conserved and may not express functional products, but an intriguing possibility is that this expression is a consequence of movement into higher-GC hosts, presenting a small reservoir of new products available for selection and further adaption at little evolutionary cost. We note that ribosomal profiling of phage λ suggests that previously unannotated genes are expressed from its genome (26).
The recent evolutionary history of Patience supports a model in which the diversity of viruses of a given host is a function of rapid movement from one host to another coupled with a landscape of diverse but closely related hosts in which the viruses evolve (4). The large collection of mycobacteriophages that infect the common host M. smegmatis mc2155 encompasses considerable diversity of sequence and GC content, and it seems likely that other groups of phages have entered the high-GC environment relatively recently, with Patience representing an extreme example. We note that there are bacterial strains within the order Actinomycetales, such as those of Corynebacterium pseudotuberculosis, Corynebacterium ulcerans, and Corynebacterium diphtheriae, which have moderate GC contents (52.5%, 53.4%, and 53.5%, respectively) and may be relatives of hosts that supported Patience growth in its earlier evolutionary history. Their codon usage profiles (see Fig. S6 in the supplemental material) more closely reflect those of the lower-GC mycobacteriophages such as Patience. Patience may share gene content with phages of such hosts, but to date, few have been characterized, other than some prophages such as phage Beta of C. diphtheriae (27, 28). Deeper exploration into the phages of these hosts and other hosts is thus likely to be highly informative and provide insights into viral origins and viral evolution (29).
MATERIALS AND METHODS
DNA sequencing.
Patience was isolated using standard methods as described previously (7, 30), and the genome was sequenced using 454 technology at the University of Pittsburgh’s Genomics and Proteomics Core Laboratories; a total of ~45,000 reads were assembled to yield an average redundancy of 222. Reads were assembled using Newbler (version 1.1) and evaluated using Consed 20. Fourteen additional Sanger sequencing runs using primers on genomic DNA were used to resolve weak areas. The genome assembled as a circle, and coordinate 1 was designated based on similarity to cluster H phages. Bioinformatic analyses used DNAMaster (http://cobamide2.bio.pitt.edu/), Gepard (31), ARAGORN (32), tRNAscan (33), HHpred (34), and Phamerator (35). The Phamerator database used for genomic comparisons was Mycobacteriophage_285. Phams were built using BLASTP and/or ClustalW, with similarity cutoff E values of 10−50 and 32.5% similarity or better as described elsewhere (35).
Codon usage and codon selection.
Codon usage resulting from mutational biases was estimated using the collection of all genes from a genome. The fraction (f) of each codon for a given amino acid was calculated as the ratio of the codon count to the amino acid count. Relative synonymous codon usage (RSCU) normalizes codon frequencies so that the sum of RSCU for codons of each amino acid is equal to the number of synonymous codons for that amino acid.
To measure codon selection, a second codon usage table (fo) was tabulated, limited to genes experiencing strong codon selection. For bacterial genomes, this table was constructed from homologues of Sharp’s set of 40 genes whose products participate in translation (36). For Patience, this table was constructed in three steps. First, a codon usage table was generated from 20% of the genome using the genes with the most extreme value of codon usage bias as determined by χ2 (where expected codon usage is calculated from the nucleotide composition). Next, adaptive codon enrichment (ACEu) values (25) are calculated for all genes as described previously (25), using this table to represent codon frequencies under codon selection (fo) and the frequencies of codons among all genes in the Patience genome to represent codon frequencies expected from mutational processes alone (fN). High ACEu values are shown by genes which favor codons which are overrepresented in the fo table relative to the fN table. Those genes with the highest ACEu values were used to construct another codon table; this process was repeated 50 times, reducing the size of the table to 5,000 codons total. Codon selection was measured as δ values (25), where δ = fo/fN for each codon. Preferred codons show δ values greater than 1.0.
Bioinformatic analyses used DNAMaster (http://cobamide2.bio.pitt.edu/), Gepard (31), ARAGORN (32), tRNAscan (33), HHpred (34), and Phamerator (35).
Electron microscopy.
CsCl gradient-purified Patience particles were applied to glow-discharged Formvar- and carbon-coated copper grids (400 mesh) (Ted Pella). They were stained with 1% uranyl acetate and imaged with a Morgagni 268 transmission electron microscope fitted with a Hamamatsu Orca HR side-model digital camera and AMT540 software.
SDS-PAGE.
Patience particles were concentrated and purified via CsCl gradient and ultracentrifugation. The visible phage band was dialyzed against two changes of phage buffer; 500 µl of the dialyzed CsCl band was pelleted by a 30-min spin at 14,000 rpm in a microcentrifuge. The pellet was resuspended in 75 µl of 20 mM dithiothreitol (DTT), and then 2 µl of 0.5 M EDTA and 1 µl of 1 M MgSO4 were added. The phage was disrupted by being heated to 75°C for 2 min and then sonicated on ice six times for 30 s to disrupt the DNA. The sample was then mixed with 25 µl of 4× SDS sample buffer and heated in a boiling bath for 3 min at 95°C. The sample was electrophoresed through a 12% polyacrylamide gel containing SDS and stained with Coomassie brilliant blue in methanol.
HPLC-MS/MS.
Five milliliters of exponentially growing M. smegmatis mc2155 (optical density at 600 nm [OD600] of 0.4) in 7H9-ADC medium (30) was concentrated to a 500-µl volume via low-speed centrifugation and infected with Patience at a multiplicity of infection (MOI) of 100. Phage particles were allowed to adsorb for 15 min, and then 4.5 ml of fresh 7H9 medium was added to the culture and incubated with shaking for 3 h at 37°C; the OD600 was monitored throughout to follow cell growth and lysis. At 30 min and 150 min postadsorption, a 1-ml aliquot was removed from the culture, the cells were pelleted via centrifugation (1 min, 14,000 rpm in a microcentrifuge), and the supernatant was removed. The cell pellet was frozen at −80°C and then shipped overnight on wet ice to the University of California, Davis Proteomics Core (UCDPC) (http://proteomics.ucdavis.edu). There, the cells were lysed via a MagNA Lyser, the insoluble fraction was removed, and the soluble proteins were precipitated, digested with trypsin, and cleaned up using a MacroSpin column. The peptides were then separated using an Easy-LC II high-pressure liquid chromatography (HPLC) system and loaded into a Q Exactive Orbitrap mass spectrometer with a Proxeon nanospray source (Thermo) for tandem MS analysis. Detected spectra and fragmentation profiles were matched against a database comprised of a six-frame translation of the Patience genome, the annotated proteins of M. smegmatis mc2155, and UniProt using X! Tandem. Peptide matches were analyzed using Scaffold4. The “Relaxed” settings (as reported in Table S1 in the supplemental material) used a peptide false discovery rate (FDR) of 1% and a protein FDR of 5%; the “Stringent” settings used a peptide FDR of 0.1% and a protein FDR of 0.6%. Estimation of relative protein abundance was determined by normalizing the total number of spectra detected to the gene size.
SUPPLEMENTAL MATERIAL
Annotation details. Download
Expression of overlapping genes in mycobacteriophage Patience. (A) Patience gene 91 encodes an alpha subunit of DNA polymerase III (black arrow), and homologues are encoded in phages Barnyard and Konstantine. LC-ms/ms identifies peptides corresponding to an internal gene in a second reading frame wholly within Patience gene 91 (red arrow). Both genes are present in Barnyard (percent amino acid identities in the corresponding regions are shown), but the internal gene is not conserved in Konstantine gene 80. (B) Patience gene 20 (black arrow) also contains a wholly internal second gene (red arrow), which is not conserved in Barnyard gene 20 and Konstantine gene 20. Download
Genes expressed out of frame within annotated ORFs. (A) A part of the annotated gene 91 is shown with the translated reading frame highlighted in green. A second ORF highlighted in yellow is wholly within gene 91, and peptides identified by LC-MS/MS corresponding to each of the products are shown in bold. (B) A part of the annotated gene 20 is shown with the translated reading frame highlighted in green. A second ORF highlighted in yellow is wholly within gene 20, and peptides identified by LC-MS/MS corresponding to each of the products are shown in bold. Download
Normalized synonymous codon usage (NSCU) for M. smegmatis and five phage genomes. An NSCU value of 1.0 is assigned to the most commonly used codon for each amino acid, and the abundances of other codons are given relative to it. The most commonly used codon for each of the 18 amino acids using more than one codon is shown in green. M. smegmatis carries 47 tRNAs, including those with matching anticodons to all of these with the exception of the CGC (Arg) codon. Other codons for which M. smegmatis carries a tRNA with a corresponding anticodon are shown in blue. The NSCU values are shown for five mycobacteriophages with different GC contents (in parentheses) with switches to different NSCU assignments of 1.0 from those in M. smegmatis highlighted in red. Download
Relative synonymous codon usage (RSCU) for M. smegmatis and five phage genomes. RSCU values reflect the number of times that a codon is observed divided by the number of expected occurrences given equivalent codon usage. Download
NCSU values of NNU codons correlate with genome percent GC. The NSCU values for each of the NNU codons are plotted again genome GC content, and the corresponding positions of the frequencies for the Patience, Papyrus, Plot, Twister, KayaCho, and M. smegmatis genomes are shown by the arrows. Download
Normalized synonymous codon usage (NSCU) for three relatively low-GC species of the family Mycobacteriaceae. Download
Patience peptides identified by mass spectrometry.
ACKNOWLEDGEMENTS
This work was supported in part by a grant to the University of Pittsburgh by the Howard Hughes Medical Institute (HHMI) in support of G.F.H. under HHMI’s Professorship program; from the Howard Hughes Medical Institute to William R. Jacobs, Jr.; and by National Institutes of Health grants GM093901 to G.F.H., AI26170 to W.R.J., GM077548 to J.G.L., and GM47795 to R.W.H. We thank the KwaZulu-Natal Research Institute for Tuberculosis and HIV (K-RITH) for support of the mycobacterial genetics workshop.
Phage Patience was isolated during a 2-week mycobacterial genetics workshop at the University of KwaZulu-Natal in 2009, and the genome was annotated in the 2011 offering of the same workshop. We thank the KwaZulu-Natal Research Institute for Tuberculosis and HIV (K-RITH) and the University of KwaZulu-Natal for hosting the workshops and the participants and instructors for their efforts.
Footnotes
Citation Pope WH, Jacobs-Sera D, Russell DA, Rubin DHF, Kajee A, Msibi ZNP, Larsen MH, Jacobs WR, Jr, Lawrence JG, Hendrix RW, Hatfull GF. 2014. Genomics and proteomics of mycobacteriophage Patience, an accidental tourist in the Mycobacterium neighborhood. mBio 5(6):e02145-14. doi:10.1128/mBio.02145-14.
REFERENCES
- 1. Hatfull GF. 2012. The secret lives of mycobacteriophages. Adv. Virus Res. 82:179–288. 10.1016/B978-0-12-394621-8.00015-7. [DOI] [PubMed] [Google Scholar]
- 2. Hatfull GF, Science Education Alliance Phage Hunters Advancing Genomics and Evolutionary Science (SEA-PHAGES) Program. KwaZulu-Natal Research Institute for Tuberculosis and HIV (K-RITH) Mycobacterial Genetics Course. University of California—Los Angeles Research Immersion Laboratory in Virology. Phage Hunters Integrating Research and Education (PHIRE) Program 2013. Complete genome sequences of 63 mycobacteriophages. Genome Announc. 1:e00847-13. 10.1128/genomeA.00847-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hatfull GF, Science Education Alliance Phage Hunters Advancing Genomics and Evolutionary Science Program. KwaZulu-Natal Research Institute for Tuberculosis and HIV Mycobacterial Genetics Course Students. Phage Hunters Integrating Research and Education Program 2012. Complete genome sequences of 138 mycobacteriophages. J. Virol. 86:2382–2384. 10.1128/JVI.06870-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jacobs-Sera D, Marinelli LJ, Bowman C, Broussard GW, Guerrero Bustamante C, Boyle MM, Petrova ZO, Dedrick RM, Pope WH, Science Education Alliance Phage Hunters Advancing Genomics And Evolutionary Science Sea-Phages Program. Modlin RL, Hendrix RW, Hatfull GF. 2012. On the nature of mycobacteriophage diversity and host preference. Virology 434:187–201. 10.1016/j.virol.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hatfull GF, Pedulla ML, Jacobs-Sera D, Cichon PM, Foley A, Ford ME, Gonda RM, Houtz JM, Hryckowian AJ, Kelchner VA, Namburi S, Pajcini KV, Popovich MG, Schleicher DT, Simanek BZ, Smith AL, Zdanowicz GM, Kumar V, Peebles CL, Jacobs WR, Jr, Lawrence JG, Hendrix RW. 2006. Exploring the mycobacteriophage metaproteome: phage genomics as an educational platform. PLoS Genet. 2:e92. 10.1371/journal.pgen.0020092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, III, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544. 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 7. Pedulla ML, Ford ME, Houtz JM, Karthikeyan T, Wadsworth C, Lewis JA, Jacobs-Sera D, Falbo J, Gross J, Pannunzio NR, Brucker W, Kumar V, Kandasamy J, Keenan L, Bardarov S, Kriakov J, Lawrence JG, Jacobs WR, Hendrix RW, Hatfull GF. 2003. Origins of highly mosaic mycobacteriophage genomes. Cell 113:171–182. 10.1016/S0092-8674(03)00233-2. [DOI] [PubMed] [Google Scholar]
- 8. Pope WH, Anders KR, Baird M, Bowman CA, Boyle MM, Broussard GW, Chow T, Clase KL, Cooper S, Cornely KA, DeJong RJ, Delesalle VA, Deng L, Dunbar D, Edgington NP, Ferreira CM, Hafer KW, Hartzog GA, Hatherill JR, Hughes LE, Ipapo K, Krukonis GP, Meier CG, Monti DL, Olm MR, Page ST, Peebles CL, Rinehart CA, Rubin MR, Russell DA, Sanders ER, Schoer M, Shaffer CD, Wherley J, Vazquez E, Yuan H, Zhang D, Cresawn SG, Jacobs-Sera D, Hendrix RW, Hatfull GF. 2013. Cluster M mycobacteriophages Bongo, PegLeg, and Rey with unusually large repertoires of tRNA isotypes. J. Virol. 88:2461–2480. 10.1128/JVI.03363-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kunisawa T. 2000. Functional role of mycobacteriophage transfer RNAs. J. Theor. Biol. 205:167–170. 10.1006/jtbi.2000.2057. [DOI] [PubMed] [Google Scholar]
- 10. Sahu K, Gupta SK, Ghosh TC, Sau S. 2004. Synonymous codon usage analysis of the mycobacteriophage Bxz1 and its plating bacteria M. smegmatis: identification of highly and lowly expressed genes of Bxz1 and the possible function of its tRNA species. J. Biochem. Mol. Biol. 37:487–492. 10.5483/BMBRep.2004.37.4.487. [DOI] [PubMed] [Google Scholar]
- 11. Hassan S, Mahalingam V, Kumar V. 2009. Synonymous codon usage analysis of thirty two mycobacteriophage genomes. Adv. Bioinformatics 2009:316936. 10.1155/2009/316936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kaufmann G. 2000. Anticodon nucleases. Trends Biochem. Sci. 25:70–74. 10.1016/S0968-0004(99)01525-X. [DOI] [PubMed] [Google Scholar]
- 13. Hatfull GF. 2010. Mycobacteriophages: genes and genomes. Annu. Rev. Microbiol. 64:331–356. 10.1146/annurev.micro.112408.134233. [DOI] [PubMed] [Google Scholar]
- 14. Brüssow H, Desiere F. 2001. Comparative phage genomics and the evolution of Siphoviridae: insights from dairy phages. Mol. Microbiol. 39:213–222. 10.1046/j.1365-2958.2001.02228.x. [DOI] [PubMed] [Google Scholar]
- 15. Kala S, Cumby N, Sadowski PD, Hyder BZ, Kanelis V, Davidson AR, Maxwell KL. 2014. HNH proteins are a widespread component of phage DNA packaging machines. Proc. Natl. Acad. Sci. U. S. A. 111:6022–6027. 10.1073/pnas.1320952111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Payne K, Sun Q, Sacchettini J, Hatfull GF. 2009. Mycobacteriophage lysin B is a novel mycolylarabinogalactan esterase. Mol. Microbiol. 73:367–381. 10.1111/j.1365-2958.2009.06775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gil F, Catalao MJ, Moniz-Pereira J, Leandro P, McNeil M, Pimentel M. 2008. The lytic cassette of mycobacteriophage Ms6 encodes an enzyme with lipolytic activity. Microbiology 154:1364–1371. 10.1099/mic.0.2007/014621-0. [DOI] [PubMed] [Google Scholar]
- 18. Catalão MJ, Gil F, Moniz-Pereira J, Pimentel M. 2010. The mycobacteriophage Ms6 encodes a chaperone-like protein involved in the endolysin delivery to the peptidoglycan. Mol. Microbiol. 77:672–686. 10.1111/j.1365-2958.2010.07239.x. [DOI] [PubMed] [Google Scholar]
- 19. Payne KM, Hatfull GF. 2012. Mycobacteriophage endolysins: diverse and modular enzymes with multiple catalytic activities. PLoS One 7:e34052. 10.1371/journal.pone.0034052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bryan MJ, Burroughs NJ, Spence EM, Clokie MR, Mann NH, Bryan SJ. 2008. Evidence for the intense exchange of MazG in marine cyanophages by horizontal gene transfer. PLoS One 3:e2048. 10.1371/journal.pone.0002048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dedrick RM, Marinelli LJ, Newton GL, Pogliano K, Pogliano J, Hatfull GF. 2013. Functional requirements for bacteriophage growth: gene essentiality and expression in mycobacteriophage Giles. Mol. Microbiol. 88:577–589. 10.1111/mmi.12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brown KL, Sarkis GJ, Wadsworth C, Hatfull GF. 1997. Transcriptional silencing by the mycobacteriophage L5 repressor. EMBO J. 16:5914–5921. 10.1093/emboj/16.19.5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nesbit CE, Levin ME, Donnelly-Wu MK, Hatfull GF. 1995. Transcriptional regulation of repressor synthesis in mycobacteriophage L5. Mol. Microbiol. 17:1045–1056. 10.1111/j.1365-2958.1995.mmi_17061045.x. [DOI] [PubMed] [Google Scholar]
- 24. Oldfield LM, Hatfull GF. 2014. Mutational analysis of the mycobacteriophage BPs promoter PR reveals context-dependent sequences for mycobacterial gene expression. J. Bacteriol. 196:3589–3597. 10.1128/JB.01801-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Retchless AC, Lawrence JG. 2011. Quantification of codon selection for comparative bacterial genomics. BMC Genomics 12:374. 10.1186/1471-2164-12-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu X, Jiang H, Gu Z, Roberts JW. 2013. High-resolution view of bacteriophage lambda gene expression by ribosome profiling. Proc. Natl. Acad. Sci. U. S. A. 110:11928–11933. 10.1073/pnas.1309739110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cianciotto NP, Groman NB. 1997. Characterization of bacteriophages from tox-containing, non-toxigenic isolates of Corynebacterium diphtheriae. Microb. Pathog. 22:343–351. 10.1006/mpat.1996.0120. [DOI] [PubMed] [Google Scholar]
- 28. Cianciotto N, Groman N. 1985. A beta-related corynebacteriophage which lacks a tox allele but can acquire it by recombination with converting phage. Infect. Immun. 49:32–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bibby K. 2014. Improved bacteriophage genome data is necessary for integrating viral and bacterial ecology. Microb. Ecol. 67:242–244. 10.1007/s00248-013-0325-x. [DOI] [PubMed] [Google Scholar]
- 30. Sarkis GJ, Hatfull GF. 1998. Mycobacteriophages. Methods Mol. Biol. 101:145–173. [DOI] [PubMed] [Google Scholar]
- 31. Krumsiek J, Arnold R, Rattei T. 2007. Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 23:1026–1028. 10.1093/bioinformatics/btm039. [DOI] [PubMed] [Google Scholar]
- 32. Laslett D, Canback B. 2004. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32:11–16. 10.1093/nar/gkh152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964. 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Söding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33:W244–W248. 10.1093/nar/gki162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cresawn SG, Bogel M, Day N, Jacobs-Sera D, Hendrix RW, Hatfull GF. 2011. Phamerator: a bioinformatic tool for comparative bacteriophage genomics. BMC Bioinformatics 12:395. 10.1186/1471-2105-12-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sharp PM, Bailes E, Grocock RJ, Peden JF, Sockett RE. 2005. Variation in the strength of selected codon usage bias among bacteria. Nucleic Acids Res. 33:1141–1153. 10.1093/nar/gki242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hatfull GF, Sarkis GJ. 1993. DNA sequence, structure and gene expression of mycobacteriophage L5: a phage system for mycobacterial genetics. Mol. Microbiol. 7:395–405. 10.1111/j.1365-2958.1993.tb01131.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Annotation details. Download
Expression of overlapping genes in mycobacteriophage Patience. (A) Patience gene 91 encodes an alpha subunit of DNA polymerase III (black arrow), and homologues are encoded in phages Barnyard and Konstantine. LC-ms/ms identifies peptides corresponding to an internal gene in a second reading frame wholly within Patience gene 91 (red arrow). Both genes are present in Barnyard (percent amino acid identities in the corresponding regions are shown), but the internal gene is not conserved in Konstantine gene 80. (B) Patience gene 20 (black arrow) also contains a wholly internal second gene (red arrow), which is not conserved in Barnyard gene 20 and Konstantine gene 20. Download
Genes expressed out of frame within annotated ORFs. (A) A part of the annotated gene 91 is shown with the translated reading frame highlighted in green. A second ORF highlighted in yellow is wholly within gene 91, and peptides identified by LC-MS/MS corresponding to each of the products are shown in bold. (B) A part of the annotated gene 20 is shown with the translated reading frame highlighted in green. A second ORF highlighted in yellow is wholly within gene 20, and peptides identified by LC-MS/MS corresponding to each of the products are shown in bold. Download
Normalized synonymous codon usage (NSCU) for M. smegmatis and five phage genomes. An NSCU value of 1.0 is assigned to the most commonly used codon for each amino acid, and the abundances of other codons are given relative to it. The most commonly used codon for each of the 18 amino acids using more than one codon is shown in green. M. smegmatis carries 47 tRNAs, including those with matching anticodons to all of these with the exception of the CGC (Arg) codon. Other codons for which M. smegmatis carries a tRNA with a corresponding anticodon are shown in blue. The NSCU values are shown for five mycobacteriophages with different GC contents (in parentheses) with switches to different NSCU assignments of 1.0 from those in M. smegmatis highlighted in red. Download
Relative synonymous codon usage (RSCU) for M. smegmatis and five phage genomes. RSCU values reflect the number of times that a codon is observed divided by the number of expected occurrences given equivalent codon usage. Download
NCSU values of NNU codons correlate with genome percent GC. The NSCU values for each of the NNU codons are plotted again genome GC content, and the corresponding positions of the frequencies for the Patience, Papyrus, Plot, Twister, KayaCho, and M. smegmatis genomes are shown by the arrows. Download
Normalized synonymous codon usage (NSCU) for three relatively low-GC species of the family Mycobacteriaceae. Download
Patience peptides identified by mass spectrometry.