

Abstract
Vibrio cholerae encodes six resistance-nodulation-division (RND) efflux systems which function in antimicrobial resistance, virulence factor production, and intestinal colonization. Among the six RND efflux systems, VexAB exhibited broad substrate specificity and played a predominant role in intrinsic antimicrobial resistance. The VexAB system was encoded in an apparent three gene operon that included vexR; which encodes an uncharacterized TetR family regulator. In this work we examined the role of vexR in vexRAB expression. We found that VexR bound to the vexRAB promoter and vexR deletion resulted in decreased vexRAB expression and increased susceptibility to VexAB antimicrobial substrates. Substrate-dependent induction of vexRAB was dependent on vexR and episomal vexR expression provided a growth advantage in the presence of the VexAB substrate deoxycholate. The expression of vexRAB increased, in a vexR-dependent manner, in response to the loss of RND efflux activity. This suggested that VexAB may function to export intracellular metabolites. Support for this hypothesis was provided by data showing that vexRAB was upregulated in several metabolic mutants including tryptophan biosynthetic mutants that were predicted to accumulate indole. In addition, vexRAB was found to be upregulated in response to exogenous indole and to contribute to indole resistance. The collective results indicate that vexR is required for vexRAB expression in response to VexAB substrates and that the VexAB RND efflux system modulates the intracellular levels of metabolites that could otherwise accumulate to toxic levels.
Introduction
Vibrio cholerae is a noninvasive gram negative bacterial pathogen that causes the disease cholera. Cholera is a severe acute diarrheal disease that affects an estimated 3–5 million people per year [1]. Untreated cholera can rapidly lead to dehydration, hypotensive shock, and death. Cholera is contracted by ingesting V. cholerae contaminated food or water [2]. Following ingestion, V. cholerae colonizes the small intestine via a process that is dependent upon the induction of genes which are required for intestinal colonization and disease development. These in vivo expressed genes contribute to V. cholerae pathogenesis in diverse ways and range from traditional virulence factors (e.g. cholera toxin and the toxin co-regulated pilus) to genes that facilitate V. cholerae survival in the gastrointestinal (GI) tract [3]. Persistence in the intestine is dependent upon V. cholerae’s ability to overcome antibacterial barriers intrinsic to the GI tract, including the presence of high concentrations of toxic small molecules (such as bile acids and other detergent-like molecules), antimicrobial products generated by resident flora, and products of the innate immune system. In response to these toxic compounds, V. cholerae activates genes which function to protect the cell by modulating its outer membrane (OM) permeability barrier and by activating efflux transporters [4–6]. For example, in response to bile acids V. cholerae alters the porin composition of the OM to effectively reduce the rate of bile salt diffusion, and presumably the diffusion of other soluble toxic molecules, across the OM [7–10]. In conjunction with reduced OM permeability, V. cholerae expresses RND-family transport systems that function to efflux bile salts, and multiple other antimicrobial compounds, from within the cell envelope to the external environment [4–6,11]. Together, the activated RND efflux systems and reduced OM permeability function synergistically to provide V. cholerae with high-level resistance to lethal antimicrobial compounds present in the host. The importance of these responses in the pathobiology of this organism is highlighted by the fact that V. cholerae exhibits a greatly diminished ability to colonize the intestinal tract in the absence of these adaptive responses [5,11,12].
The RND efflux systems have been a focal point in bacterial antimicrobial resistance research due to the ability of individual RND systems to provide resistance to a broad range of chemically unrelated substrates that include antibiotics, detergents, dyes, and antimicrobial peptides [13]. The RND efflux systems are found in most gram negative bacteria and function as proton-substrate antiporters [14]. Individual RND efflux systems are composed of three components: an outer membrane pore protein that is homologous to Escherichia coli TolC, an integral cytoplasmic membrane pump protein belonging to the RND superfamily, and a periplasmic membrane fusion protein that links the outer membrane pore protein to the RND pump protein [15–18]. Together these three components form a transport apparatus that spans the cell envelope and functions to efflux substrates from within the cell envelope into the external environment. Although the RND transport apparatus is responsible for the efflux of antimicrobials, phylogenetic analysis suggests that the RND efflux systems evolved independent of xenobiotic selection [19,20]. Indeed, there is mounting evidence that the RND efflux systems are involved in diverse functions (reviewed in [21]) such as biofilm formation, iron acquisition, plant-bacteria interactions, lipid transport, bacterial virulence, divalent cation resistance, and the removal of metabolic byproducts from within the cell.
The V. cholerae genome encodes six RND efflux systems [22]. Inhibition of the RND efflux systems renders V. cholerae hypersensitive to multiple antimicrobial compounds and attenuates the expression of virulence factors including cholera toxin (CT) and the toxin co-regulated pilus (TCP) [4,23,24]. Among the six RND efflux systems, the VexAB system was shown to be the primary system that contributes to intrinsic antimicrobial resistance in vitro [4,5]. Several studies have suggested that the V. cholerae VexAB RND efflux system is important to V. cholerae pathogenesis. This included finding that vexAB was induced in vivo in humans and animals [25], that vexAB expression was enhanced by the Cpx system [26], and that VexAB was required for high-level virulence factor production [5]. While there is ample evidence to suggest that VexAB is important in pathogenesis, the regulatory mechanisms controlling its expression are unknown.
In many bacteria regulation of the RND efflux systems is mediated by a linked TetR family regulator [27]. The TetR family of regulatory proteins function in diverse phenotypes including antibiotic resistance, metabolism, stress responses, and pathogenicity [28]. TetR proteins contain two functional domains: a conserved N-terminal DNA-binding domain and a variant C-terminal ligand binding domain [28]. The ligand binding domain is capable of binding to effector molecules that modulate the interaction of the DNA binding domain with its target sequences. In the case of RND efflux systems, the activity of the RND efflux system’s TetR regulators is often modulated by the binding of efflux substrates of the linked RND system. In many cases, TetR proteins appear to be capable of binding a diverse set of ligands that correspond to the multiple substrates of linked RND efflux systems [28]. The vast majority of reported TetR family regulators function as repressors that bind the promoter region and repress transcription in the absence of bound ligands [29,30]. In addition to regulating their specific target genes, many TetR regulators also regulate their own expression [27,28].
In this work we tested the hypothesis that the TetR family protein VexR regulated the expression of the vexAB RND efflux system [22,31,32]. VexR, which has not been characterized, was encoded by the first gene in a three gene operon that included vexA and vexB; a genetic organization that was distinct from most RND efflux systems [28,29]. Our results confirmed that VexR functioned in the regulation of the vexRAB operon, but in a manner that was opposite to most RND efflux system associated TetR regulators. Our results indicated that VexR was required for activation of the vexRAB operon, whereas most RND-linked TetR regulators function as repressors. We further found that the vexRAB promoter was upregulated, in a vexR-dependent manner, in response to the loss of RND efflux activity. This suggested that endogenous metabolites may serve as activators of the vexRAB efflux operon and could be VexAB substrates. Consistent with this idea, vexRAB was upregulated in several metabolic mutants, including tryptophan biosynthetic mutants. Furthermore indole, an intermediate in tryptophan biosynthesis, induced vexRAB expression. Taken together our results suggested that VexR was required for activation of the vexRAB operon. We further posit that a native role of the VexAB RND efflux system is to remove excess cellular metabolites from within the cell that could otherwise accumulate to toxic levels.
Materials and Methods
Bacterial strains and culture conditions
The bacterial strains used in this study are listed in Table 1. Escherichia coli EC100Dpir+, SM10λpir, and ER2566 were used for cloning, plasmid mobilization, and protein purification, respectively. Vibrio cholerae strains used in this study were derivatives of O1 El Tor strains N16961 and C6706 [22,33,34]. V. cholerae strain JB58 (N16961-ΔlacZ SmR) or JB804 (C6706-lacZ+ SmR) were used as wild-type (WT) control strains as indicated. The C6706 transposon mutants [33] were graciously supplied by Dr. John Mekalanos (Harvard Medical School). Bacterial strains were grown at 37°C in Luria-Bertani (LB) broth or on LB agar. AKI broth was used for virulence inducing conditions as described previously [5]. Bacterial stocks were maintained at -80°C in LB broth containing 25% glycerol. Growth media was supplemented with carbenicillin (Cb) and streptomycin (Sm) at 100 μg/mL, kanamycin (Km) at 50 μg/mL, or chloramphenicol (Cm) at 1μg/mL (for V. cholerae) and 20 μg/mL (for E. coli) as required. Bacterial growth media was purchased from Difco (Lawrence, KS) and chemicals were purchased from Sigma-Aldrich (St Louis, MO).
Table 1. Strains, plasmids and oligonucleotides.
| Strain: | Genotype: | Source: |
|---|---|---|
| Vibrio cholerae | ||
| JB804 | 01 El Tor strain C6706, Smr | [34] |
| JB3 | 01 El Tor strain N16961, Smr | [4] |
| JB58 | 01 El Tor strain N16961 ΔlacZ, Smr | [4] |
| JB114 | JB58 ΔvexM | [5] |
| JB116 | JB58 ΔvexH | [5] |
| JB432 | JB58 ΔvexF | [5] |
| JB464 | JB58 ΔvexD ΔvexF ΔvexH ΔvexK ΔvexM | [5] |
| JB485 | JB58 ΔvexB ΔvexD ΔvexF ΔvexH ΔvexK ΔvexM | [5] |
| JB495 | JB58 ΔvexB | [4] |
| JB528 | JB58 ΔvexK | [5] |
| JB692 | JB58 ΔvexD | [4] |
| JB694 | JB58 ΔvexB ΔvexD | [4] |
| JB718 | JB58 ΔvexR ΔvexD | This study |
| XBV218 | JB58 ΔvexR | This study |
| XBV220 | JB58 ΔvexR ΔvexB ΔvexD ΔvexF ΔvexH ΔvexK ΔvexM | This study |
| MKW589 | ΔcpxR lacZ::cpxP-lacZ | [26] |
| DT1458 | JB58 lacZ::cpxP-lacZ | [26] |
| DT1572 | JB58ΔvexB lacZ::cpxP-lacZ | [26] |
| DT1460 | JB485 lacZ::cpxP-lacZ | [26] |
| DT1693 | JB495 lacZ::cpxP-lacZ | This study |
| VA367 | JB58 ΔvexR ΔcpxR lacZ::cpxP-lacZ | This study |
| EC8508 | C6706 Tn::VC0027(ilvA) | [33] |
| EC20412 | C6706 Tn::VC0051(purK) | [33] |
| EC1769 | C6706 Tn::VC0052(purE) | [33] |
| EC23411 | C6706 Tn::VC0164(vexB) | [33] |
| EC24273 | C6706 Tn::VC0374(pgi) | [33] |
| EC14462 | C6706 Tn::VC0384(cysJ) | [33] |
| EC5082 | C6706 Tn::VC0385(cysI) | [33] |
| EC9587 | C6706 Tn::VC0386(cysH) | [33] |
| EC19978 | C6706 Tn::VC0537(cysM) | [33] |
| EC11960 | C6706 Tn::VC0767(guaB) | [33] |
| EC4709 | C6706 Tn::VC0774 | [33] |
| EC11507 | C6706 Tn::VC0819(aldA-1) | [33] |
| EC8862 | C6706 Tn::VC0923 | [33] |
| EC11848 | C6706 Tn::VC0968(cysK) | [33] |
| EC10232 | C6706 Tn::VC1061 | [33] |
| EC5818 | C6706 Tn::VC1169(trpA) | [33] |
| EC24412 | C6706 Tn::VC1170(trpB) | [33] |
| EC12331 | C6706 Tn::VC1171(trpC/F) | [33] |
| EC11883 | C6706 Tn::VC1172(trpD) | [33] |
| EC11883 | C6706 Tn::VC1173(trpG) | [33] |
| EC11131 | C6706 Tn::VC1174(trpE) | [33] |
| EC1872 | C6706 Tn::VC1579 | [33] |
| EC389 | C6706 Tn::VC1732(aroA) | [33] |
| EC14803 | C6706 Tn::VC1819(aldA-2) | [33] |
| EC12803 | C6706 Tn::VC2013(ptsG) | [33] |
| EC7541 | C6706 Tn::VC2092(gltA) | [33] |
| EC18511 | C6706 Tn::VC2209(vibF) | [33] |
| EC10553 | C6706 Tn::VC2348(deoB) | [33] |
| EC24541 | C6706 Tn::VC2362(thrC) | [33] |
| EC19558 | C6706 Tn::VC2363(thrB) | [33] |
| EC13310 | C6706 Tn::VC2364(thrA) | [33] |
| EC1335 | C6706 Tn::VC2558(cysC) | [33] |
| EC13560 | C6706 Tn::VC2559(cysN) | [33] |
| EC21282 | C6706 Tn::VC2560(cysD) | [33] |
| EC9914 | C6706 Tn::VC2649(cysE) | [33] |
| EC20144 | C6706 Tn::VCA0013(malP) | [33] |
| EC2460 | C6706 Tn::VCA0014(malQ) | [33] |
| EC4499 | C6706 Tn::VCA0765(ybjU) | [33] |
| EC9834 | C6706 Tn::VCA0886(kbl) | [33] |
| EC3123 | C6706 Tn::VCA0896(zwf) | [33] |
| EC14445 | C6706 Tn::VCA0987(ppsA) | [33] |
| EC21873 | C6706 Tn::VCA1046(mtlD) | [33] |
| Escherichia coli | ||
| EC100Dpir+ | F- mcrA Δ(mrr-hsdRMS-mcrBC) Φ80dlacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara, leu)7697 galU galK λ- rpsL (SmR) nupG pir+ | Epicentre |
| SM10λpir | thi-1 thr leu tonA lacY supE recA::RP4-2-4-Tc::Mu KmR (λ pirR6K) | [62] |
| ER2566 | F- glnV44(AS) galK2(Oc) rpsL704(strR) xylA5 mtl-1 argE3(Oc) thiE1 tfr-3 λ DE3 = λ sBamHIo ∆EcoRI-B int::(lacI::PlacUV5::T7 gene1) i21 Δnin5 | New England BioLabs |
| Plasmids: | Description: | |
| pBAD18 | Expression plasmid, CbR, pBR322 origin of replication | [63] |
| pCM10 | Vector for construction of luxCDABE transcriptional fusions, KmR, ori101 | [64] |
| pDT1076 | pCM10 containing the vexR promoter region from N16961 | This study |
| pDT1146 | pMMB66EH::vexR | This study |
| pDT1777 | pDT1076 with Cm-mark cassette inserted into the vector, CmR | This study |
| pJB703 | pBAD18::vexR | This study |
| pMAL-c2 | Expression plasmid for fusion of proteins to MBP and cytoplasmic expression, CbR, pBR322 origin of replication | New England BioLabs |
| pMMB66EH | Expression plasmid, CbR, oriV/T | [7] |
| pSC137 | Vector for transposon mutagenesis of bacteria, CmR, oriR6K | |
| pMAL-c2::vexR | vexR cloned into pMAL-c2 | This study |
| pTL61T | Vector for construction of lacZ transcriptional fusions, CbR, oriRK2 | [12] |
| pWM91 | Suicide plasmid vector used for allelic exchange, CbR, oriR6K/fl | [65] |
| pWM91::ΔvexR | pWM91::ΔvexR | This study |
| pXB233 | pTL61T containing the vexR promoter region from N16961 | This study |
| pXB228 | pTL61T containing the vexEF promoter region | [26] |
| pXB229 | pTL61T containing the vexGH promoter region | [26] |
| pXB230 | pTL61T containing the vexIJK promoter region | [26] |
| pXB231 | pTL61T containing the vexCD promoter region | [26] |
| pXB232 | pTL61T containing the vexLM promoter region | [26] |
| pΔR | cpxR::Km allelic exchange vector | [37] |
| pJL1P’Z | Allelic exchange vector for placing cpxP-lacZ into the V. cholerae genome | [37] |
| Oligonucleotides: | DNA sequence (5’–3’):: | |
| 166b-F-XhoI | AACTCGAGGCAGAGAAATGTGATGT | |
| 166b-R-XbaI | AATCTAGAGCCAAACAGCAGGATCG | |
| 166c-F-XhoI | TTCTCGAGGGGTCCGGAGACGTACT | |
| 166c-R-XbaI | CGTCTAGAGGAGCTGTTTATCGCCG | |
| Biotin | GCGGGAGTCGGCAGCG | |
| MCS4.VexA.R | CCGGATCCCATTCTGGTGCGAACTCCAAATTAGTGTTG | |
| VC0166-SacI-F | CTGAGCTCAAGGGTTCATATGCA | |
| VC0166-XbaI-R | TTTCTAGATTAGTGTTGAGTAATTGCA | |
| VC0166-F1 | CAGGATCCACTTTAGCACCGTTACTCAG | |
| VC0166-F2 | TCATTGCATCCTGTTTATCGCCGTACACTATTTC | |
| VC0166-R1 | ACCTCGAGTATTGGCCAGTATGACCTTG | |
| VC0166-R2 | CGATAAACAGGATGCAATGATTCAAGCCAGTTGG | |
| VC0166-F-pMAL-SmaI | GGCCCGGGTTGCAGAGAAATGTGATGTCTGAAATAGTG | |
| VC0166-R-pMal-EcoR1 | GGAATTCTTAGTGTTGAGTAATTGCATCC | |
| vexR-F1 | GCGGGAGTCGGCAGCGATAATAATCCGCTCACCGAG | |
| vexR-R1 | GCGGGAGTCGGCAGCGCCCCTGTTTTGCAATACACTTG | |
| vexR-F2 | GCGGGAGTCGGCAGCGTGCAAAACAGGGGGTATTAG | |
| vexR-R2 | GCGGGAGTCGGCAGCGGCCGTACACTATTTCAGACA | |
| XWL-BRL-F | CGCAGGGTTTTCCCAGTCACGAC | |
Growth curves were generated as follows. The indicated strains were grown overnight in LB broth and normalized to an OD600 of 1.0. The cultures were then diluted 1:20,000 into LB broth with or without arabinose and/or deoxycholate as indicated. Aliquots of 100μL of each strain were then distributed in triplicate into the wells of a flat bottom 96-well microtiter plate. The 96-well plate was then incubated in a BioTek ELx808 plate reader at 37°C with intermittent shaking and growth was monitored by measuring the absorbance at 600 nm every 20 min for 14h. The data was then averaged for the triplicate samples at each time point.
Plasmid and mutant construction
Plasmids and oligonucleotides used in this study are listed in Table 1. Enzymes for cloning experiments were purchased from New England Biolabs (Beverly, MA). pXB233 (PvexRAB-lacZ) was constructed as follows. The 166c-F-XhoI and 166c-R-XbaI PCR primers were used to amplify the vexRAB promoter from N16961 genomic DNA. The resulting PCR amplicon was then restricted with XhoI and XbaI endonucleases before being ligated into the XhoI/XbaI site of pTL61T to generate pXB233 (PvexRAB-lacZ). pDT1076 (PvexRAB-lux) was generated by cloning the vexRAB promoter from pXB233 into pCM10 as follows. The vexRAB promoter was PCR amplified from pXB233 using the XWL-BRL-F and166c-F-XhoI PCR primers. The resulting PCR amplicon was then made blunt-ended before being restricted with BamHI. The resulting fragment was then cloned into pCM10 linearized with EcoRI, and then blunt-ended, before being restricted with BamHI. A Cm-marked version of pDT1076 was generated by transposing the Cm-marked mariner transposon from pSC137 into pDT1076 to yield pDT1777. All plasmids were verified by DNA sequencing.
The vexR expression plasmids were constructed as follows. pJB703 was generated by amplifying vexR from N16961 using the VC0166F-SacI and VC0166-XbaI-R PCR primers. The resulting amplicon was restricted with SacI and XbaI endonucleases before being ligated with similarly digested pBAD18. pDT1146 (pMMB66EH::vexR) was generated by amplifying the vexR gene from N16961 using the VC0166F-SacI and MCS4.VexA.R primers. The resulting amplicon was blunt-ended, digested with BamHI, and then ligated with similarly treated pMMB66EH to generate pDT1146. The sequence of vexR in both plasmids was confirmed by DNA sequencing. pMAL-c2-vexR was constructed by amplifying the vexR gene from N16961 using the VC0166-F-pMAL-SmaI and VC0166-R-pMal-EcoRI PCR primers. The resulting PCR amplicon was then digested with EcoRI and SmaI endonucleases and cloned into the same sites of pMAL-c2 (New England Biolabs).
The allelic exchange vector pWM91::ΔvexR was generated by crossover PCR as previously described [4,5,35,36]. Briefly, vexR-specific VC0166-F1/-R2 and VC0166-F2/-R1 PCR primer pairs (Table 1) were used in separate PCR reactions with N16961 genomic DNA as a template. The resulting ~1.1 Kb PCR products were gel purified, pooled, and then used as the template for a second PCR reaction using the flanking VC0166-F1/-R1 PCR primers to generate the ~2.2 Kb vexR deletion construct. The resulting ~2.2 Kb PCR amplicon was then purified, restricted with XhoI and BamHI endonucleases before being ligated with similarly digested pWM91 to generate pWM91::ΔvexR. The resulting plasmid was then used to delete vexR. Briefly, pWM91::ΔvexR was conjugated into the V. cholerae and cointegrants were selected for Cb/Sm resistance. Several Cb/Sm resistant colonies were then streaked for single colonies onto LB agar (without NaCl) containing 5% sucrose to select for loss of the integrated plasmid. Several sucrose-resistant colonies were screened for Cb sensitivity to verify plasmid loss before the vexR deletion was confirmed by PCR using flanking primers.
Deletion of cpxR was accomplished by allelic exchange using pΔR as previously described [26,37]. The cpxP-lacZ chromosomal reporter was introduced into the lacZ locus using pJL1P’Z as previously described [26,37].
Reporter assays
The β-galactosidase assays were performed as follows. Test strains were grown in LB broth or under AKI conditions and culture aliquots were taken in triplicate at the indicated time points post-inoculation to quantify β-galactosidase activity as previously described [7]. All experiments were performed at least three times and the results averaged. Statistical significance was determined using ANOVA with indicated post-test.
The bioluminescence reporter assays were performed as follows. E. coli EC100Dpir+ containing pDT1041 and pDT1124 (or pBAD18) were grown overnight in LB broth before being diluted 1:200 into fresh LB broth with or without 0.2% arabinose. Aliquots (100μL) of the diluted cultures were then distributed in triplicate into the wells of white 96-well microtiter plates with clear bottoms (Corning) and incubated with shaking at 37°C for the duration of the assay. Luminescence and the OD600 at indicated time points were determined using a BioTek Synergy HT plate reader. The reported results are the average relative light units (RLU) for each test sample divided by the optical density.
Quantification of vexRAB expression in the C6706 transposon mutants was accomplished using pDT1777 (vexRAB-lux) as follows. Strains grown in LB broth were diluted 1:200 from the overnight culture into fresh LB broth with Cm and grown under the same conditions as listed above. Strains grown under AKI conditions were diluted 1:10,000 into AKI broth with Cm. Aliquots (370μL) of the diluted cultures were then distributed in triplicate as described above. Plates were incubated statically at 37°C for 4h, at which point 215μL of culture was removed from each well for a final volume of 155μL/well. Plates were then grown with shaking at 37°C for the remainder of the assay. Luminescence production was determined as described above. The results are the average RLUs for each test sample divided by the optical density. Two-way ANOVA with Dunnet’s post-hoc test was used to determine significance relative to WT.
Antimicrobial susceptibility assays
Antimicrobial susceptibility tests were performed using gradient agar plates as previously described [5]. Each 9 cm x 9 cm gradient plate was inoculated with six V. cholerae strains, including WT which served as an internal control. The plates were then incubated overnight at 37°C before growth was measured. The minimum inhibitory concentration (MIC) of each strain was calculated by the percent growth across the plate multiplied by the antimicrobial concentration used in the plate. One-Way ANOVA with Tukey’s multiple comparison post-test or with Dunnet’s post-hoc test, as indicated, was used to calculate the level of significance for the tested strains.
Purification of VexR
Proteins for the gel mobility shift assay were purified as follows. E. coli ER2566 carrying pMAL-c2 or pMAL-c2-vexR were grown overnight at 37°C with aeration. The cultures were then diluted 100-fold into LB broth with Cb and incubated at 37°C with shaking until reaching an OD600 of ~0.5 when isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 0.3 mM and the cultures were incubated for an additional 2 h. The cells were then harvested by centrifugation, the supernatant removed, and the pellet resuspended in column buffer (20 mM Tris-HCl, 200 mM NaCl, 1 mM EDTA) plus 1 mM phenylmethylsulfonyl fluoride (PMSF). The cells were then lysed using a M-11P Microfluidizer processor according to the manufacturer’s instructions (Microfluidics). The resulting lysates were cleared of particulate matter by centrifugation at 15,000 x g for 20 min at 4°C. The clarified supernatants (i.e. VexR-MBP or MBP) were diluted 1:6 with column buffer and loaded onto a 0.8 x 7 cm column containing 1 mL of amylose resin (New England Biolabs). The column was washed with 12 column volumes of column buffer before the bound proteins were eluted with elution buffer (20 mM Tris-HCl, 200 mM NaCl, 1 mM EDTA, 10 mM maltose). Protein concentrations were determined using the Coomassie Plus Assay kit according to manufacturer’s instructions (Thermo Scientific). The purity of the eluted proteins were assessed by SDS–PAGE with Coomassie Brilliant Blue R-250 staining.
Electrophoretic Mobility Shift Assay (EMSA)
DNA fragments designated vexR1 (the nucleotide sequence between -129 and -46 of the vexR promoter region) and vexR2 (-59 to +21 of the vexR promoter region) were amplified from N16961 using the primers vexR-F1/vexR-R1 and vexR-F2/vexR-R2 primers, respectively. The fragments were then used as a template for a second PCR reaction with the flanking biotin primer. The biotin primer was biotinylated by the manufacturer (IDT) to end label the fragments. The resulting biotinylated probes (2.5 nM) were incubated with purified VexR-MBP or MBP in amounts ranging from 0 to 250 nM in 10 μl of binding buffer containing 10 mM Tris (pH 7.4), 150 mM KCl, 0.1 mM DTT, 0.1 mM EDTA (pH 8), and 200 μg/mL sheared salmon sperm DNA. The binding reactions were incubated at room temperature for 20 min before being subjected to electrophoresis on a nondenaturing 5% polyacrylamide TBE gel in 0.25x TBE buffer at 200V for 45 min. The DNA in the gel was transferred to a nylon membrane in 0.5x TBE buffer at 380 mA for 1 h, UV cross-linked, before the biotinylated probes were detected using the Chemiluminescent Nucleic Acid Detection Module (Thermo Scientific) and documented using a FluorChem E imaging system (Protein Simple).
Results
Genetic organization of the vexRAB operon
In many gram negative bacteria the RND efflux systems encode a linked TetR family regulatory protein that functions as a repressor of the linked RND efflux system [27,28]. The V. cholerae genome encodes six RND efflux systems; among these six, only the VexAB RND system contained a linked TetR-family regulator which was previously named vexR (VC0166) [4,22]. The vexR gene was present as the first gene in the vexRAB operon (Fig. 1A). Relative to most other RND efflux systems, the genetic arrangement of vexR within the vexRAB operon is unusual [27–29]. In most RND efflux systems that encode a linked TetR regulator, the regulatory gene is expressed from a divergently expressed promoter that is upstream of and overlaps with the RND efflux system promoter. This arrangement is demonstrated by the archetypical acrAB RND efflux system in E. coli where acrR is encoded upstream and divergently from the acrAB efflux system (Fig. 1B) [28]. The AcrR protein functions to regulate the expression of the acrAB RND system in response to its antimicrobial substrates while also regulating its own expression [38].
Fig 1. Genetic organization of RND efflux systems.
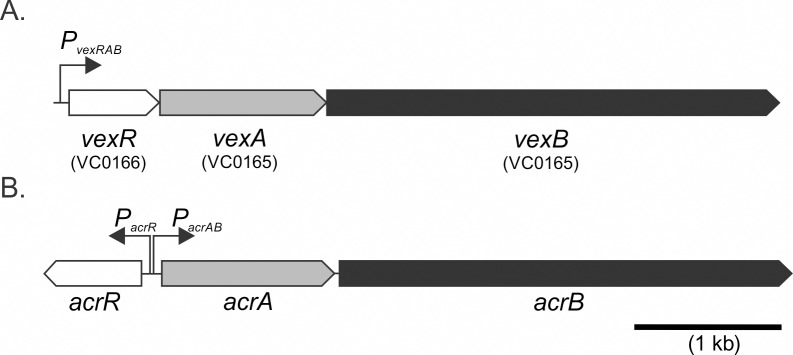
(A) Schematic of the V. cholerae vexRAB operon. (B) Schematic of the E. coli acrR-acrAB locus. Genes encoding a TetR-family (white), membrane fusion family (grey), and RND-family (black) proteins are shown. Putative promoters for each respective operon are indicated by the thin black arrows.
Expression of vexRAB is induced in response to VexAB efflux substrates
The role of the RND efflux pumps is to export their substrates out of the cell. As such, their expression is typically regulated by a feedback loop in response to the presence of their efflux substrates [28]. Since bile salts were shown to be major substrates of VexAB [4,5], we hypothesized that vexRAB expression may be regulated in response to bile salts. To test this we cultured V. cholerae JB58 (ΔlacZ) under virulence gene inducing conditions in AKI broth containing sub-lethal concentrations of deoxycholate and measured vexRAB expression using a vexRAB-lacZ reporter (i.e. pXB233). AKI growth conditions were selected because previous studies had indicated a linkage between RND efflux and virulence gene expression [5]. We also tested erythromycin, an antibiotic that is a substrate of VexAB. The results showed a concentration-dependent induction of vexRAB expression in response to both erythromycin (Fig. 2A) and deoxycholate (Fig. 2B). Deoxycholate at 0.2% resulted in a 2-fold increase in vexRAB expression while the presence of erythromycin at 0.2 μg/mL resulted in a nearly 3-fold increase in vexRAB expression. Based on these results we concluded that the vexRAB operon is regulated in response to deoxycholate and erythromycin, two of its known efflux substrates.
Fig 2. VexAB efflux substrates affect vexRAB expression.

V. cholerae strain JB58 (ΔlacZ SmR) containing pXB233 (vexRAB-lacZ) was grown under AKI conditions for 5 h with the indicated concentrations of (A) erythromycin or (B) deoxycholate when vexRAB expression was quantified as described in the methods. The reported data are in Miller Units (MU) and are the mean ± SD of three independent experiments. Statistical significance was determined relative to the media control by one-way ANOVA with Dunnet’s post-hoc test. * = P<0.05.
VexR is required for expression of the vexRAB operon
The finding that vexRAB expression was induced in response to VexAB efflux substrates indicated the possibility that the vexRAB system was regulated in a fashion that was similar to other RND efflux systems. Given that most TetR-family regulators function as repressors in the absence of their efflux substrates, we hypothesized that VexR would function to repress vexRAB expression. If this was true, then deletion of vexR should result in increased vexRAB expression. We therefore compared vexRAB-lacZ expression in JB58 and an isogenic ΔvexR mutant strain. The strains were grown in LB broth to an OD600 of 0.8 when vexRAB-lacZ expression was quantified. The results showed that vexRAB expression was significantly decreased in the ΔvexR strain relative to WT (Fig. 3A). This finding suggested that VexR was a positive, rather than a negative regulator of the vexRAB operon.
Fig 3. VexR is required for vexRAB expression.

(A) N16961 WT and ΔvexR containing pXB233 (vexRAB-lacZ) were grown in LB broth. At 3.5h deoxycholate (DOC) was added to a final concentration of 0.2% and the cultures were incubated for an additional 30 min before vexRAB expression was determined as described in the methods. Data is the mean of three independent experiments ± SD. Significance was determine by one-way ANOVA with Tukey-Kramer multiple comparison test. Unless otherwise indicated, asterisks are significance relative to WT (*p<0.05; **p<0.001). (B) E. coli containing pDT1076(vexRAB-lux) and either pJB703(pBAD18::vexR) or pBAD18 was grown in LB broth with or without 0.2% arabinose and bioluminescence was assayed at 0h, 1h, and 2h. Error bars indicate the mean ± SD of three replicates. The results are representative of three independent experiments. (C) Gel mobility shift assay showing the binding of VexR to the vexRAB promoter. The promoter was split into two fragments (vexR1) -129 to -46 and (vexR2) -59 to +21 relative to the ATG start site. Biotin labeled DNA (2.5nM) from vexR1 (lane 1–4) or vexR2 (lane 5–12) fragments was incubated with either purified VexR-MBP or MBP as indicated at 0 nM (lane 1), 25 nM (lane 2), 100 nM (lane 3), or 250 nM (lane 4) prior to electrophoresis. Specific binding reaction, detection, and visualization are discussed in the Material and Methods.
We examined whether the substrate-dependent induction is contingent on the presence of vexR because vexRAB expression was increased in response to the VexAB substrates (Fig. 2). We chose to focus on deoxycholate as an inducer because it is the biologically most relevant vexAB substrate with regards to V. cholerae pathogenesis [5]. Growth of WT in the presence of 0.2% deoxycholate resulted in a ~5.5-fold increase in vexRAB-lacZ expression (Fig. 3A). Deletion of vexR greatly reduced the deoxycholate-dependent induction of vexRAB expression. This result further supported the conclusion that vexR was required for vexRAB expression. While VexR appeared to be required for robust vexRAB expression, deoxycholate still induced vexRAB expression by ~2-fold in the ΔvexR strain relative to the LB broth control, although the expression level was greatly reduced compared to JB58 grown in LB broth (Fig. 3A). This suggested that there may be other factors involved in regulating vexRAB expression; a scenario similar to the E. coli acrAB operon where multiple factors contribute to its expression. Taken together these findings show that vexR contributes to the positive regulation of vexRAB.
The VexAB RND efflux system is the only RND efflux system that encodes a linked regulatory system. While the VexCD RND efflux system has been shown to be under negative control by BreR [6], the regulatory mechanisms controlling the expression of the other five RND efflux systems were unknown. We therefore tested the hypothesis that VexR may regulate the expression of the other five RND efflux systems. We introduced plasmid based lacZ reporters for each of the other five RND efflux systems (i.e. vexCD, vexEF, vexGH, vexIJK, and vexLM) into JB58 and its isogenic vexR deletion strain. The resulting strains were then cultured in LB broth in the presence or absence of 0.02% deoxycholate for two hours when β-galactosidase activity was quantified. The results showed that there was no significant difference in the expression of the five tested RND efflux systems in WT relative to the ΔvexR mutant under either growth condition (S1 Fig.). From this we concluded that vexR was specific for the vexRAB operon.
To further confirm that VexR contributes to activation of the vexRAB operon we tested whether recombinant vexR expression would activate transcription of the vexRAB promoter in E. coli. We therefore transformed E. coli bearing pDT1076 (vexRAB-lux) with pBAD18 (empty vector control) or pBAD18::vexR. The resulting strains were then grown in LB broth plus or minus 0.2% arabinose (to induce vexR expression from the arabinose inducible promoter in pBAD18) and luminescence production was measured at zero, one and two hours. The results showed that the empty vector control (i.e. pBAD18) did not affect vexRAB expression (Fig. 3B). This indicated that neither pBAD18, nor arabinose, affected the expression of the vexRAB-lux reporter. In contrast, there was a marked increase in luminescence production in the strain containing pBAD18::vexR. The presence of pBAD18::vexR, even in the absence of arabinose, was sufficient to induce vexRAB expression as evidenced by the increase in luminescence production at one and two hours (Fig. 3B); this may reflect leaky vexR expression from pBAD18::vexR. The addition of arabinose to the broth further increased vexRAB expression. Based on these results, plus the results presented in Fig. 3A, we concluded that vexR contributes to positive regulation of the vexRAB operon.
VexR binds to the vexRAB promoter directly
The above experiments suggested that VexR may act directly at the vexRAB promoter. To test this hypothesis we performed gel mobility shift assays using VexR-MBP with the vexRAB promoter. The 5’ upstream region of vexRAB was arbitrarily split into two fragments that covered from -129 to -46 (vexR1) and -59 to +21 (vexR2) relative to the ATG start site (Fig. 3C). The results of the gel shift assays showed that purified VexR-MBP was able to shift the vexR2 promoter fragment, but not the vexR1 promoter fragment. Incubation of the vexR2 fragment with MBP did not result in a shift confirming that the results were due to VexR (Fig. 3C). These results validated that VexR directly binds to the vexRAB promoter and supports the conclusion that VexR directly regulates vexRAB expression.
VexR contributes to antimicrobial resistance
If VexR contributes to positive regulation of the vexRAB operon, we hypothesized that the deletion of vexR should increase V. cholerae susceptibility to antimicrobial compounds that are substrates of VexAB. To test this we determined the MIC of deoxycholate, erythromycin, and Triton X-100 for WT strain JB58 and its isogenic ΔvexR mutant. While deoxycholate, erythromycin, and Triton X-100 are all substrates of VexAB, other RND efflux systems possess redundant efflux activity for deoxycholate and Triton X-100. The results showed a 2.7-fold decrease in the erythromycin MIC in the ΔvexR mutant (Table 2). In contrast, there was no change in the susceptibility of the ΔvexR mutant to deoxycholate or Triton X-100. These results contrast the resistance profile of a ΔvexB mutant. For example, the erythromycin MIC for the vexR mutant was 1.65 μg/mL compared to 0.07 μg/mL for the vexB mutant. Likewise, the Triton X-100 MIC for the vexR mutant was >3% while the vexB mutant MIC was 0.0017%. This indicated that the vexR mutant exhibited a phenotype that was intermediate between WT and the ΔvexB strain. Since VexB is the only RND efflux pump that contributes to erythromycin resistance [4], these results suggest that vexAB is expressed at a low basal level in the ΔvexR mutant.
Table 2. Antimicrobial susceptibility of vexR mutants.
| MIC (s.d.) 1 | ||||
|---|---|---|---|---|
| Strain | Genotype | Em (μg/mL) | Doc (%) | TX-100 (%) |
| JB58 | WT | 4.40 (2.1) | >3 (0) | >3 (0) |
| XBV218 | ΔvexR | 1.65 (0.9) 2 , 3 | >3 (0) | >3 (0) |
| JB495 | ΔvexB | 0.07 (0.005) 2 | >3 (0) | 0.0017 (0.0011) 2 |
| JB692 | ΔvexD | ND | >3 (0) | ND |
| JB718 | ΔvexRΔvexD | ND | 0.020 (0.001) 2 , 4 | ND |
| JB694 | ΔvexBΔvexD | ND | 0.007 (0.003) 2 | ND |
(1)Minimum Inhibitory Concentration (MIC) for erythromycin (Em), deoxycholate (Doc), and Triton X-100 (TX-100) for the indicated N16961 strains with standard deviations in parenthesis.
(2) P< 0.05 relative to WT.
(3) P<0.05 relative to ΔvexB.
(4) P<0.05 relative to ΔvexBD. ND = not determined.
The fact that vexR deletion did not affect deoxycholate resistance was expected as previous studies have shown that the VexCD RND efflux system was redundant with VexAB for bile salt resistance [4]. Because of overlapping specificity for deoxycholate, mutation of both vexB and vexD are required to produce a bile salt hypersensitive phenotype [4]. Therefore, to address whether vexR contributed to deoxycholate resistance we examined the effect of vexR deletion in a ΔvexD background. The results showed that deletion of vexR in a ΔvexD background reduced the deoxycholate MIC to 0.019%; a MIC that was 2.7-fold greater than the deoxycholate MIC observed with the ΔvexBΔvexD mutant (Table 2). The intermediate resistance phenotype of the ΔvexRΔvexD mutant (relative to the ΔvexBΔvexD mutant) further supported the conclusion that the vexRAB efflux system is expressed at a low basal level in the vexR mutant.
VexAB is the primary RND efflux system involved in Triton X-100 resistance [4]. Consistent with this, a >1,700-fold increase in Triton X-100 susceptibility was observed in a ΔvexB mutant (Table 2). However, deletion of vexR did not affect Triton X-100 susceptibility (Table 2). This suggests that the low-level vexAB expression in the vexR mutant was sufficient to provide WT-level Triton X-100 resistance. Alternatively, it is possible that deletion of vexR resulted in the induction of other resistance traits that contributed to Triton X-100 resistance and thus compensated for the reduction in vexRAB expression in the vexR mutant.
The collective MIC data indicated that VexR was required for maximum resistance to erythromycin and deoxycholate under the tested conditions. When vexR was deleted, the contribution of VexAB to antimicrobial resistance was significantly reduced, but not to a level that was equal to a vexB null mutant. This suggested that vexAB was expressed at a low basal level in the ΔvexR mutant.
We recently reported that activation of the Cpx membrane stress response system can enhance vexRAB expression in V. cholerae [26]. We also showed that vexB deletion resulted in activation of the V. cholerae Cpx system which suggested that vexRAB and the Cpx system were reciprocally regulated. Based on this we hypothesize that if vexR was required for vexAB expression, then mutation of vexR should also result in activation of the Cpx system. To test this hypothesis we assayed cpxP expression. The expression of cpxP is regulated by CpxR and has been used as a reporter for the activation state of the cpx system [26,37]. We introduced a cpxP-lacZ chromosomal reporter into ΔvexR and ΔvexRΔcpxR mutants. We then compared cpxP-lacZ expression on LB-X-gal agar plates. In these assays we also included WT, ΔvexB, ΔvexBDFHKM and ΔcpxR mutants as controls. The results showed that WT produced white colonies on LB agar (S2 Fig.). This indicated that the Cpx system was inactive under standard growth conditions as previously reported [37]. Growth of the same strain in the presence of CuCl2, which is an inducer of the Cpx system, resulted in the formation of dark blue colonies. This confirmed that the Cpx reporter system was functioning as expected. In contrast to WT, the ΔvexR mutant produced light blue colonies on LB agar that were similar in appearance to the ΔvexB mutant. This indicated that deletion of vexR, like deletion of vexB, resulted in activation of the Cpx system.
Since deletion of vexR activated the Cpx system, we tested whether the Cpx system affected vexRAB expression in the ΔvexR background. If this was true we predicted that deletion of cpxR in the ΔvexR background would result in decreased vexRAB expression and increased sensitivity to VexAB efflux substrates. We first determined the effect of individual and combined deletions of vexR and cpxR on erythromycin resistance. The results showed that the erythromycin MIC for the ΔvexRΔcpxR mutant was not different from the ΔvexR mutant (S2 Table) suggesting that the Cpx system is not contributing to enhance resistance to VexAB substrates in the vexR mutant. Consistent with this result, deletion of cpxR in a ΔvexR mutant did not affect vexRAB expression (S3 Fig.). Taken together these results confirmed that vexR deletion activated the Cpx system, but that the Cpx system did not affect the basal expression of vexRAB in the absence of vexR. One potential explanation for this observation is that CpxR-dependent enhancement of vexRAB expression may require vexR.
Overexpression of vexR enhances resistance to deoxycholate
We hypothesized that if vexR was required for vexRAB upregulation and antimicrobial resistance, then vexR overexpression should complement a ΔvexR mutant for growth in the presence of deoxycholate and erythromycin. We tested this hypothesis by determining the deoxycholate and erythromycin MIC for WT, ΔvexR and ΔvexRΔvexD strains that expressed vexR from the arabinose promoter in pBAD18. In these experiments the respective strains containing pBAD18 or pBAD18::vexR were grown on antimicrobial gradient agar plates that contained a range of different arabinose concentrations. However, we were unable to identify an arabinose concentration which resulted in complementation of the tested mutants for deoxycholate or erythromycin resistance (data not shown). We considered that this result could have been an artifact of the pBAD18 expression system, and therefore we cloned vexR into the low copy number IPTG-inducible pMMB66EH expression vector and repeated the complementation experiments and obtained identical results.
The reason that we were unable to complement the vexR mutant is not clear. The inability to complement the vexR deletion likely did not result from the introduction of a secondary mutation during the construction of the vexR mutant since DNA sequencing confirmed the integrity of the vexR deletion construct (i.e. pWM91::ΔvexR) and the vexRAB locus in the vexR deletion strain. Further, the ΔvexR and ΔvexRΔvexD mutants were independently created from different parental strains, making it unlikely that the complementation defect was due to an unlinked spontaneous mutation. The lack of complementation was also not due to mutations introduced into the complementing plasmids as we confirmed the DNA sequence of the complementing plasmids. This latter conclusion is further supported by the observation that the presence of pBAD18::vexR activated vexRAB-lacZ expression in E. coli (Fig. 3B).
Since the vexR deletion mutant could not be complemented by ectopic vexR expression, we sought another method to confirm that VexR contributed to antimicrobial resistance. We hypothesized that if vexR was indeed an activator of the vexRAB operon, then episomal expression of vexR in WT should result in enhanced growth in the presence of sub-lethal concentrations of VexAB substrates. To test this hypothesis we generated growth curves for WT(pBAD18::vexR) and WT(pBAD18) in LB broth containing a sub-lethal concentration of deoxycholate. The results of this analysis showed that in the absence of deoxycholate, WT(pBAD18::vexR) grew equally well during logarithmic phase of growth as WT(pBAD18) through six hours (Fig. 4A). Thereafter WT(pBAD18) growth continued to increase whereas the strain bearing pBAD18::vexR peaked and the cell density declined through 12 hours. This indicated that over-expression of vexR is detrimental under standard laboratory growth conditions. We speculated that this was a result of increased vexAB expression which has been suggested to be growth inhibitory under non-selective conditions [27,39]. When the same strains were cultured in LB broth containing deoxycholate, WT (pBAD18::vexR) exhibited a growth advantage over the empty vector control regardless of the presence or absence of arabinose (Fig. 4B). The addition of arabinose did not significantly affect the growth of WT(pBAD18::vexR) relative to the same culture grown in the absence of arabinose (Fig. 4B). The complementation results were reminiscent of the findings observed for vexR activation of vexRAB in E. coli (Fig. 3B) where vexR enhanced vexRAB-lux expression even in the absence of arabinose, and further enhanced vexRAB-lux expression with the addition of arabinose. Taken together these results provide additional support for the hypothesis that vexR contributes to vexRAB activation.
Fig 4. VexR contributes to V.

cholerae survival under inhibitory antimicrobial conditions. V. cholerae N16961 WT (A & B) and ΔvexB (C & D) containing pJB703(pBAD18::vexR) or pBAD18 were grown in triplicate wells of microtiter plates containing LB broth (A & C) or LB broth plus 0.015% deoxycholate (B & D). Expression of vexR was induced by adding 0.1% arabinose to the growth media as indicated. Cell growth was monitored as the change in the optical density at 600 nm and plotted versus time as the mean ±SEM. The results are representative of three independent experiments.
The above results suggested that episomal vexR expression enhanced deoxycholate resistance, but did not allow us to discriminate if the resistance phenotype was mediated by VexAB or some other factor. If enhanced growth was due to VexR activation of VexAB production, then deletion of vexB should alleviate the vexR-dependent growth enhancement in the presence of deoxycholate. We therefore repeated the above growth experiments in a vexB deletion strain. The results showed that the growth of the ΔvexB(pBAD18) control strain was slightly attenuated relative to ΔvexB(pBAD18::vexR) in LB broth (Fig. 4C). This suggested that vexR may impart a growth advantage in the vexB mutant during growth in LB broth. The growth of the ΔvexB strain was significantly inhibited in the presence of deoxycholate (Fig. 4D). Unlike what was observed in the WT background, vexR expression in the ΔvexB mutant did not significantly affect cell growth (Fig. 4D). This suggested that the enhanced growth observed with vexR in WT cells grown in the presence of deoxycholate may have resulted from vexR-dependent activation of the vexRAB operon (Fig. 4B). We also tested if pBAD18::vexR would provide a growth advantage to a vexR deletion strain under the same growth conditions, but the results were congruent with the gradient agar plate results and showed that pBAD18::vexR did not complement the ΔvexR mutant (S4 Fig.). Taken together the collective results are consistent with the idea that vexR overexpression enhanced V. cholerae growth in the presence of deoxycholate via the VexAB RND efflux system.
Deletion of the RND efflux systems induces vexRAB expression
Recent studies in E. coli suggested that the RND systems may have evolved to remove potentially toxic metabolites from the cell cytoplasm [40–42]. If this was true, we hypothesized that vexRAB expression would be upregulated in RND negative cells due to the accumulation of metabolites that were substrates of the RND efflux systems. Consistent with this hypothesis vexRAB has been reported to be upregulated in RND negative strain JB485 [26]. To further expand upon this and to determine whether vexR contributed to this phenotype we introduced the vexRAB-lacZ reporter into WT, RND deficient strain JB485, and JB485ΔvexR. The resulting strains were then cultured under AKI conditions in AKI broth for 5 h when vexRAB expression was assessed (Fig. 5). AKI growth conditions were selected for these studies because previous work had indicated a linkage between RND efflux and virulence gene expression [5]. The results showed a ~4.5-fold increase in vexRAB expression in JB485 confirming previous results [26]. Deletion of vexR in JB485 reduced vexRAB expression to near background levels which suggested that the increase in vexRAB expression in JB485 was dependent upon vexR (Fig. 5).
Fig 5. Expression of vexRAB is induced in V.

cholerae RND efflux mutants. The indicated N16961 strains containing pXB233 (vexRAB-lacZ) were grown for 5 h under AKI conditions when vexRAB expression was quantified as described in the methods. Strain JB485 is RND negative and lacks all six RND efflux pump proteins. Error bars indicate ± SD of three independent experiments. One-way ANOVA with Dunnet’s post-hoc test was used to determine significant differences relative to WT. * = P<0.05; ** = P<0.01; ***P = <0.0001.
We next tested whether increased vexRAB expression in JB485 was due to loss of vexB or a result of the deletion of all six RND efflux systems. We first quantified vexRAB expression in a ΔvexB mutant. The results showed that vexRAB expression increased in the vexB mutant (~3-fold), but to a level that was less than observed in JB485 (Fig. 5). This suggested that at least one of the other RND systems can partially compensate for the loss of vexB. The expression of vexRAB in a ΔvexDFHKM (vexB+) was not significantly different from WT. This latter finding was consistent with previous work showing that vexB, due to its broad substrate specificity, was able to complement for the loss of the other five RND efflux systems [5,24]. Taken together these results indicated that vexRAB expression was responsive to the RND efflux status of the cell and that deletion of vexB resulted in vexRAB expression being elevated. The higher level of vexRAB expression in JB485 relative to the vexB mutant suggested that one or more of the other RND efflux systems were able to efflux the inducing factor(s). Based on these results we inferred that V. cholerae compensates for reductions in RND efflux activity by upregulating vexRAB expression. Exactly how V. cholerae senses efflux activity was unclear, but we speculated this resulted from the intracellular accumulation of natural substrates of the RND efflux systems in the absence of RND efflux activity.
The expression of vexRAB is altered in metabolic mutants
We tested whether V. cholerae upregulated vexRAB expression in response to the accumulation of metabolic byproducts. Our approach for these experiments was based on the assumption that the mutation of metabolic genes would disrupt biosynthetic pathways and result in the intracellular accumulation of chemical intermediates of the targeted biochemical pathway which would then activate vexRAB expression. To conduct these studies we obtained a number of metabolic mutants from a defined transposon mutant library that was constructed in V. cholerae El Tor strain C6706 [33]. We selected mutants that targeted a number of different metabolic pathways including some that have been shown to affect E. coli acrAB expression [42]. This included mutants that affected purine metabolism, cysteine metabolism, sulfur metabolism, vibriobactin biosynthesis, glycolysis, gluconeogenesis, pyruvate metabolism, amino acid metabolism, and the citric acid cycle (S1 Table).
The metabolic mutants were transformed with pDT1777 (vexRAB-lux) and cultured in AKI broth for five hours when vexRAB expression was quantified. While we were most interested in the mutants that activated vexRAB expression, we noted several mutations reduced vexRAB expression. Based on the idea that vexRAB expression is modulated by metabolite accumulation, one explanation for reduced vexRAB expression is the loss of down-stream activators. However, we cannot exclude that the observed reduction of vexRAB expression was an artifact resulting from cellular byproducts inhibiting the luciferase reporter [43]. Three mutants were identified (VC1172, VC1579, and VCA1046) that resulted in a >2-fold increase in vexRAB expression (S1 Table). VC1172 encodes TrpD which functions in tryptophan biosynthesis; VC1579 encodes AlmE which is a lipid A modification enzyme [44]; and VCA1046 encodes mannitol-1-phosphate 5-dehydrogenase and is involved in mannitol metabolism. These results confirmed that vexRAB expression was upregulated in response to interruption of at least three metabolic pathways. To further investigate this phenomenon we focused on tryptophan biosynthesis due to the availability of mutants and two chemical intermediates in the tryptophan biosynthetic pathway.
Disruption of the tryptophan biosynthetic pathway affects vexRAB expression
Mutation of trpD (VC1172) resulted in the strongest induction of vexRAB expression suggesting that tryptophan biosynthesis intermediates may function as inducers of the vexRAB operon. To investigate this we examined vexRAB expression in six different tryptophan biosynthetic transposon mutants (Fig. 6A). The results showed that vexRAB expression increased 3-fold in the trpB mutant, and ~2-fold in the trpA and trpD mutants (Fig. 6B). Mutation of trpB (VC1170) is predicted to result in indole accumulation while mutation of trpA (VC1169) and trpD (VC1172) would result in indole-3-glycerol phosphate and anthranilate accumulation, respectively. This suggests that indole, indole-3-glycerol phosphate and anthranilate could contribute to vexRAB induction.
Fig 6. Indole activates vexRAB expression.

(A) Schematic of the V. cholerae tryptophan biosynthetic pathway. (B) The indicated C6706 strains bearing pDT1777 (vexRAB-lux) were grown in 96-well plates under AKI conditions for 5 h when luminescence (RLU) and OD600 were measured. Data are the mean +/- SEM of three independent assays. Two-way Anova with Dunnet’s post-hoc test compared to WT was used to determine significance. * = P<0.01; ** = P<0.0001. (C) V. cholerae N16961 containing pXB233 (vexRAB-lacZ) was grown with or without indole, L-tryptophan, or anthranilic acid at the indicated concentrations under AKI conditions for 5 h before vexRAB expression was determined as described in the methods. Bars indicate the mean +/- SD of three independent experiments. One-way ANOVA with Dunnet’s post-hoc test was used to determine significant changes relative to growth in media alone. * = P<0.001.
If intermediates of tryptophan biosynthesis were vexRAB inducers, we hypothesized that adding these compounds to the growth media would activate vexRAB expression. As anthranilate and indole were available from commercial sources, we tested if the addition of these compounds affected vexRAB-lacZ expression in WT. The results showed that indole at 1.5 mM increased vexRAB expression by ~1.5 fold (Fig. 6C). This finding, combined with the observation that vexRAB was induced in the trpB mutant (Fig. 6B), suggested that indole may contribute to vexRAB activation. The addition of anthranilate or tryptophan did not have a significant effect on vexRAB expression (Fig. 6C). There are at least two explanations for this result. First, it is possible that mutation of trpD may have pleiotropic effects on other metabolic pathways which could be responsible for the effects on vexRAB expression. Second, it is also possible that V. cholerae lacks an anthranilate transport system which would limit the effects of exogenous anthranilate.
Indole is a substrate of the VexAB RND efflux system
Indole is an intermediate in the biosynthesis of tryptophan and is also produced by V. cholerae as a byproduct of tryptophan degradation. Indole has also been shown to function as a signaling molecule in biofilm formation [45,46]. At high concentrations indole is toxic, which suggested that it may be a potential substrate of the V. cholerae RND efflux systems. We therefore tested if the RND efflux systems contributed to indole resistance by comparing the growth of WT and the RND deficient strain JB485 (ΔvexBDFHKM) on indole gradient agar plates. The results showed that the WT MIC was 2.1 mM while the JB485 MIC was 1.6 mM (Table 3). This suggested that the V. cholerae RND efflux systems contributed to indole resistance. In an effort to determine which RND efflux systems contributed to indole resistance we examined mutants lacking each of the six RND efflux systems pumps (i.e. vexB, vexD, vexF, vexH, vexK, vexM). Only the vexB mutant had a small but significant decrease in the indole MIC (1.8 mM), which was higher than the MIC exhibited by JB485 (Table 3). Although small, the change in the vexB MIC was consistent with the idea that indole is a substrate of the VexAB RND efflux system. The discrepancy between the vexB and JB485 MICs suggest that at least one of the other RND efflux systems function in indole resistance. All together this data indicates that indole may be a substrate of the RND efflux systems and that indole may serve as an inducer of the vexRAB operon.
Table 3. Minimum inhibitory concentration of indole for RND mutants.
| Strain | Genotype | Indole MIC (s.d) 1 |
|---|---|---|
| JB58 | WT | 2.1 (0.1) |
| XBV218 | ΔvexR | 2.0 (0.2) |
| JB495 | ΔvexB | 1.8 (0.2) 2 |
| JB692 | ΔvexD | 2.0 (0.2) |
| JB432 | ΔvexF | 2.0 (0.1) |
| JB116 | ΔvexH | 2.1 (0.1) |
| JB528 | ΔvexK | 2.1 (0.1) |
| JB114 | ΔvexM | 2.0 (0.1) |
| JB485 | ΔvexBDFHKM | 1.6 (0.1) 3 |
(1)Minimum Inhibitory Concentration (MIC) of Indole (mM) for the indicated N16961 strains. One-way ANOVA with Dunnet’s post-hoc test was used to determine statistical difference relative to WT.
(2) P< 0.05 relative to WT;
(3) P<0.001 relative to WT.
Discussion
The VexAB RND efflux system was the only V. cholerae RND system that was associated with a linked TetR family regulator (Fig. 1). In contrast to what is observed with most RND loci, VexR was encoded as the first gene in the vexRAB operon. VexR contributed to vexRAB expression as evidenced by the finding that vexR deletion decreased vexRAB expression (Fig. 3A) and resulted in increased susceptibility to the VexAB substrate erythromycin (Table 2). These results indicated that vexR contributed to the positive regulation of the vexRAB operon. This conclusion was supported by subsequent experiments showing that VexR directly bound the vexRAB promoter (Fig. 3C), that VexR was able to increase vexRAB expression in E. coli (Fig. 3B), and that vexR overexpression in V. cholerae resulted in a vexB-dependent growth advantage in the presence of sub-lethal concentrations of deoxycholate (Fig. 4).
The finding that VexR was required for vexRAB expression was unexpected. The vast majority of TetR-family regulators behave as transcriptional repressors [30]. Thus our findings highlight differences in the regulation of vexRAB relative to its orthologous Enterobacteriaceae system (i.e. acrR-acrAB). In E. coli acrR functioned as a negative regulator of acrAB while global regulatory systems such as the Mar operon functioned to positively regulate acrAB expression in response to antimicrobial exposure (Fig. 1) [28,29]. Although V. cholerae lacks the Mar operon, VexR could function in a similar role as the Mar operon by contributing to vexRAB expression in response to antimicrobial exposure. It is worth noting that the vexRAB and vexGH RND efflux systems and the Cpx system are reciprocally regulated in V. cholerae [26]. Recent studies showed that mutation of vexB or vexH resulted in activation of the Cpx system while activation of the Cpx system resulted in upregulation of vexRAB and vexGH. Herein our results show that deletion of vexR resulted in activation of the Cpx system (S2 Fig.), but that Cpx activation did not affect vexRAB expression (S3 Fig.) or VexAB mediated antimicrobial resistance (S2 Table). This result may suggest that the Cpx-dependent upregulation of the vexRAB operon is dependent on vexR. The fact that the Cpx system does not respond to antimicrobial substrates of the V. cholerae RND efflux systems further indicates that vexRAB expression is under the influence regulatory systems that respond to distinct stimuli. This could explain the inability to transcomplement the vexR deletion strain. It is possible that VexR-dependent regulation of the vexRAB operon is dependent on VexR interaction with other regulatory elements. Alternatively, cis-acting sequencing within the vexR open reading frame could be required for vexRAB upregulation.
The expression of vexRAB was highly elevated upon loss of RND-mediated efflux (Fig. 5). This upregulation was abolished when vexR was deleted (Fig. 5), indicating that vexR was required for vexRAB expression in the RND efflux mutants. This suggested the possibility that metabolites accumulated within the cell and induced vexRAB expression in the absence of RND-mediated efflux. This idea is consistent with recent reports in E. coli which have linked efflux to cell metabolism [40–42,47]. The finding that vexRAB expression was upregulated in three of the 36 tested metabolic mutants provided evidence to support this hypothesis (S1 Table). Altogether, this suggests the VexAB RND efflux system plays a role in removing excess metabolites from the cell. While vexRAB was induced in three of the metabolic mutants, it is also possible that other efflux systems may function in a similar or redundant role to remove other metabolites from the cell. Precedence for this was found in E. coli where the acrEF, yfiK and aaeAB efflux systems contributed to indole, cysteine-cystine, and p-hydroxybenzoate export, respectively [48–50]. The fact that vexRAB expression in the ΔvexB was lower than what was observed in RND negative strain JB485 supports the conclusion that other V. cholerae RND efflux systems also participate in the removal of excess metabolites from within the cell (Fig. 5).
The mutations that activated vexRAB expression disrupted three different metabolic pathways. This suggested that vexRAB expression is modulated in response to multiple different metabolites (S1 Table and Fig. 6B). The mutation of trpB resulted in the greatest increase in vexRAB expression. Since TrpB catalyzes the conversion of indole to tryptophan (Fig. 6A), this suggested that indole likely contributed to vexRAB induction in the trpB mutant (Fig. 6). Activation of vexRAB expression by exogenous indole supported this conclusion (Fig. 6C). In addition, both JB485 and the ΔvexB mutant exhibited increased susceptibility to indole. This suggested that indole was a substrate for VexAB and likely additional RND efflux systems (Table 3). Based on the fact that indole both contributed to induction, and likely functioned as a substrate of vexRAB, we propose that the VexAB efflux system functions to remove excess indole from within the cell before it reaches concentrations that are detrimental to cell growth. Given the broad substrate specificity of the VexAB system, combined with the observation that vexRAB was induced in two other metabolic mutants; we suggest that the function of VexAB in metabolic relief may extend beyond indole.
In conclusion, we have shown that vexR contributed to the positive regulation of vexRAB. VexR appears to be necessary for activation of the vexRAB operon in response to efflux substrates of the VexAB RND efflux system. The finding that vexR was necessary for positive regulation was unusual. To the best of our knowledge only ten TetR family regulators from the more than 20,000 distinct TetR family proteins have been characterized as activators [29,51–61]. However, it is possible that TetR activators may be more common than currently thought as only a fraction of the annotated TetR proteins have been characterized. We have also established a novel role for VexAB in responding to the metabolic state and efflux status of the cell. We suggest that that vexRAB expression is modulated in response to metabolic products and that VexAB relieves stress by preventing the accumulation of metabolic byproducts to toxic levels. Our results were similar to what has been observed in E. coli where metabolic mutants and metabolic products were found to activate the acrAB RND system [40–42,47]. Therefore, the ability of the RND efflux systems to alleviate stress due to excess metabolites appears to be an evolutionarily conserved mechanism.
Supporting Information
Overnight cultures of Vibrio cholerae strain JB58 (N16961 ΔlacZ SmR) or the isogenic ΔvexR mutant bearing the transcriptional fusion reporters for the indicated RND efflux system were diluted 1:100 into LB broth and grown with shaking for 3.5 h at 37°C when deoxycholate (DOC) was added to 0.02%. The culture was incubated with shaking at 37°C for an additional 30 min when aliquots were collected in triplicate and β-galactosidase activity was quantified as described in the methods. (A.) vexCD; (B.) vexEF; (C.) vexGH; (D.) vexIJK; (E.) vexLM. The reported results are in Miller Units (MU) and are the mean ± SD of three independent experiments.
(PDF)
The indicated strains containing a chromosomal cpxP-lacZ transcriptional reporter were cultured overnight in LB broth before being diluted 1:100 into fresh LB broth. The cultures were incubated an additional hour and diluted 1,000-fold into PBS. Aliquots of the diluted cultures were then inoculated onto the surface of LB agar plates containing X-gal (160 μg/mL) plus and minus 500 μM CuCl2 (to induce expression of the Cpx system). The plates were then incubated overnight at 37°C before being photographed.
(PDF)
The indicated V. cholerae N16961 strains containing a vexRAB-lacZ transcriptional reporter were cultured to middle logarithmic phase in LB broth when vexRAB-lacZ expression was quantified by a β-galactosidase assay as described in the methods. The results are the average ±SD of five independent experiments.
(PDF)
V. cholerae N16961 ΔvexR containing pJB703 (pBAD18::vexR) or pBAD18 were grown in triplicate wells of microtiter plates containing (A) LB broth or (B) LB broth plus 0.015% deoxycholate. Arabinose (0.1%) was added to the media as indicated to induce expression of vexR from the arabinose regulated promoter in pBAD18. Cell growth was then monitored as the change in the optical density at 600 nm and plotted versus time as the mean ±SEM.
(PDF)
(PDF)
(PDF)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by National Institutes of Health (NIH) award R01AI091845. DLT and MFH were supported by NIH training grant T32AI049820. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ali M, Lopez AL, You YA, Kim YE, Sah B, et al. (2012) The global burden of cholera. Bulletin of the World Health Organization 90: 209–218A. 10.2471/BLT.11.093427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kaper JB, Morris JG Jr., Levine MM (1995) Cholera. Clinical microbiology reviews 8: 48–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Childers BM, Klose KE (2007) Regulation of virulence in Vibrio cholerae: the ToxR regulon. Future microbiology 2: 335–344. [DOI] [PubMed] [Google Scholar]
- 4. Bina JE, Provenzano D, Wang C, Bina XR, Mekalanos JJ (2006) Characterization of the Vibrio cholerae vexAB and vexCD efflux systems. Archives of microbiology 186: 171–181. [DOI] [PubMed] [Google Scholar]
- 5. Bina XR, Provenzano D, Nguyen N, Bina JE (2008) Vibrio cholerae RND family efflux systems are required for antimicrobial resistance, optimal virulence factor production, and colonization of the infant mouse small intestine. Infection and immunity 76: 3595–3605. 10.1128/IAI.01620-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cerda-Maira FA, Ringelberg CS, Taylor RK (2008) The bile response repressor BreR regulates expression of the Vibrio cholerae breAB efflux system operon. Journal of bacteriology 190: 7441–7452. 10.1128/JB.00584-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Provenzano D, Schuhmacher DA, Barker JL, Klose KE (2000) The virulence regulatory protein ToxR mediates enhanced bile resistance in Vibrio cholerae and other pathogenic Vibrio species. Infect Immun 68: 1491–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goulart CL, dos Santos GG, Barbosa LC, Lery LM, Bisch PM, et al. (2010) A ToxR-dependent role for the putative phosphoporin VCA1008 in bile salt resistance in Vibrio cholerae El Tor N16961. Microbiology 156: 3011–3020. 10.1099/mic.0.043117-0 [DOI] [PubMed] [Google Scholar]
- 9. Mathur J, Waldor MK (2004) The Vibrio cholerae ToxR-regulated porin OmpU confers resistance to antimicrobial peptides. Infect Immun 72: 3577–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Merrell DS, Bailey C, Kaper JB, Camilli A (2001) The ToxR-mediated organic acid tolerance response of Vibrio cholerae requires OmpU. Journal of bacteriology 183: 2746–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Taylor DL, Bina XR, Bina JE (2012) Vibrio cholerae VexH encodes a multiple drug efflux pump that contributes to the production of cholera toxin and the toxin co-regulated pilus. PLoS One 7: e38208 10.1371/journal.pone.0038208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Provenzano D, Klose KE (2000) Altered expression of the ToxR-regulated porins OmpU and OmpT diminishes Vibrio cholerae bile resistance, virulence factor expression, and intestinal colonization. Proceedings of the National Academy of Sciences of the United States of America 97: 10220–10224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Van Bambeke F, Glupczynski Y, Plesiat P, Pechere JC, Tulkens PM (2003) Antibiotic efflux pumps in prokaryotic cells: occurrence, impact on resistance and strategies for the future of antimicrobial therapy. J Antimicrob Chemother 51: 1055–1065. [DOI] [PubMed] [Google Scholar]
- 14. Delmar JA, Su CC, Yu EW (2014) Bacterial multidrug efflux transporters. Annual review of biophysics 43: 93–117. 10.1146/annurev-biophys-051013-022855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eswaran J, Koronakis E, Higgins MK, Hughes C, Koronakis V (2004) Three's company: component structures bring a closer view of tripartite drug efflux pumps. Current Opinion in Structural Biology 14: 741–747. [DOI] [PubMed] [Google Scholar]
- 16. Koronakis V, Sharff A, Koronakis E, Luisi B, Hughes C (2000) Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature 405: 914–919. [DOI] [PubMed] [Google Scholar]
- 17. Akama H, Matsuura T, Kashiwagi S, Yoneyama H, Narita S, et al. (2004) Crystal structure of the membrane fusion protein, MexA, of the multidrug transporter in Pseudomonas aeruginosa. The Journal of biological chemistry 279: 25939–25942. [DOI] [PubMed] [Google Scholar]
- 18. Murakami S, Nakashima R, Yamashita E, Yamaguchi A (2002) Crystal structure of bacterial multidrug efflux transporter AcrB. Nature 419: 587–593. [DOI] [PubMed] [Google Scholar]
- 19. Martinez JL (2009) The role of natural environments in the evolution of resistance traits in pathogenic bacteria. Proceedings Biological sciences / The Royal Society 276: 2521–2530. 10.1098/rspb.2009.0320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alonso A, Sanchez P, Martinez JL (2001) Environmental selection of antibiotic resistance genes. Environ Microbiol 3: 1–9. [DOI] [PubMed] [Google Scholar]
- 21. Alvarez-Ortega C, Olivares J, Martinez JL (2013) RND multidrug efflux pumps: what are they good for? Front Microbiol 4: 7 10.3389/fmicb.2013.00007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, et al. (2000) DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406: 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bina XR, Philippart JA, Bina JE (2009) Effect of the efflux inhibitors 1-(1-naphthylmethyl)-piperazine and phenyl-arginine-beta-naphthylamide on antimicrobial susceptibility and virulence factor production in Vibrio cholerae. J Antimicrob Chemother 63: 103–108. 10.1093/jac/dkn466 [DOI] [PubMed] [Google Scholar]
- 24. Taylor DL, Bina X. R., Bina J.E (2012) Vibrio cholerae vexH Encodes a Multiple Drug Efflux Pump that Contributes to the Production of Cholera Toxin and the Toxin Co-Regulated Pilus. Plos One 7: e38208 10.1371/journal.pone.0038208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bina J, Zhu J, Dziejman M, Faruque S, Calderwood S, et al. (2003) ToxR regulon of Vibrio cholerae and its expression in vibrios shed by cholera patients. Proceedings of the National Academy of Sciences of the United States of America 100: 2801–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor DL, Bina XR, Slamti L, Waldor MK, Bina JE (2014) Reciprocal regulation of RND efflux systems and the Cpx two-component system in Vibrio cholerae. Infect Immun IAI.00025-14 [pii] 10.1128/IAI.00025-14. [DOI] [PMC free article] [PubMed]
- 27. Grkovic S, Brown MH, Skurray RA (2002) Regulation of bacterial drug export systems. Microbiol Mol Biol Rev 66: 671–701, table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ramos JL, Martinez-Bueno M, Molina-Henares AJ, Teran W, Watanabe K, et al. (2005) The TetR family of transcriptional repressors. Microbiol Mol Biol Rev 69: 326–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ahn SK, Cuthbertson L, Nodwell JR (2012) Genome context as a predictive tool for identifying regulatory targets of the TetR family transcriptional regulators. Plos One 7: e50562 10.1371/journal.pone.0050562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cuthbertson L, Nodwell JR (2013) The TetR family of regulators. Microbiol Mol Biol Rev 77: 440–475. 10.1128/MMBR.00018-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M (2012) KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40: D109–114. 10.1093/nar/gkr988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanabe M, Kanehisa M (2012) Using the KEGG database resource. Curr Protoc Bioinformatics Chapter 1: Unit1 12. [DOI] [PubMed]
- 33. Cameron DE, Urbach JM, Mekalanos JJ (2008) A defined transposon mutant library and its use in identifying motility genes in Vibrio cholerae. Proceedings of the National Academy of Sciences of the United States of America 105: 8736–8741. 10.1073/pnas.0803281105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thelin KH, Taylor RK (1996) Toxin-coregulated pilus, but not mannose-sensitive hemagglutinin, is required for colonization by Vibrio cholerae O1 El Tor biotype and O139 strains. Infect Immun 64: 2853–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bina JE, Mekalanos JJ (2001) Vibrio cholerae tolC is required for bile resistance and colonization. Infect Immun 69: 4681–4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Imai Y, Matsushima Y, Sugimura T, Terada M (1991) A simple and rapid method for generating a deletion by PCR. Nucleic Acids Res 19: 2785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Slamti L, Waldor MK (2009) Genetic analysis of activation of the Vibrio cholerae Cpx pathway. Journal of bacteriology 191: 5044–5056. 10.1128/JB.00406-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ma D, Alberti M, Lynch C, Nikaido H, Hearst JE (1996) The local repressor AcrR plays a modulating role in the regulation of acrAB genes of Escherichia coli by global stress signals. Mol Microbiol 19: 101–112. [DOI] [PubMed] [Google Scholar]
- 39. Li XZ, Zhang L, Nikaido H (2004) Efflux pump-mediated intrinsic drug resistance in Mycobacterium smegmatis. Antimicrob Agents Chemother 48: 2415–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rosner JL, Martin RG (2009) An excretory function for the Escherichia coli outer membrane pore TolC: upregulation of marA and soxS transcription and Rob activity due to metabolites accumulated in tolC mutants. Journal of bacteriology 191: 5283–5292. 10.1128/JB.00507-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Helling RB, Janes BK, Kimball H, Tran T, Bundesmann M, et al. (2002) Toxic waste disposal in Escherichia coli. Journal of bacteriology 184: 3699–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruiz C, Levy SB (2013) Regulation of acrAB expression by cellular metabolites in Escherichia coli. J Antimicrob Chemother dkt352 [pii]10.1093/jac/dkt352. [DOI] [PMC free article] [PubMed]
- 43. Hatzios SK, Ringgaard S, Davis BM, Waldor MK (2012) Studies of dynamic protein-protein interactions in bacteria using Renilla luciferase complementation are undermined by nonspecific enzyme inhibition. Plos One 7: e43175 10.1371/journal.pone.0043175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hankins JV, Madsen JA, Giles DK, Brodbelt JS, Trent MS (2012) Amino acid addition to Vibrio cholerae LPS establishes a link between surface remodeling in gram-positive and gram-negative bacteria. Proceedings of the National Academy of Sciences of the United States of America 109: 8722–8727. 10.1073/pnas.1201313109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mueller RS, Beyhan S, Saini SG, Yildiz FH, Bartlett DH (2009) Indole acts as an extracellular cue regulating gene expression in Vibrio cholerae. Journal of bacteriology 191: 3504–3516. 10.1128/JB.01240-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mueller RS, McDougald D, Cusumano D, Sodhi N, Kjelleberg S, et al. (2007) Vibrio cholerae strains possess multiple strategies for abiotic and biotic surface colonization. Journal of bacteriology 189: 5348–5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rosner JL, Martin RG (2013) Reduction of cellular stress by TolC-dependent efflux pumps in Escherichia coli indicated by BaeSR and CpxARP activation of spy in efflux mutants. Journal of bacteriology 195: 1042–1050. 10.1128/JB.01996-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kawamura-Sato K, Shibayama K, Horii T, Iimuma Y, Arakawa Y, et al. (1999) Role of multiple efflux pumps in Escherichia coli in indole expulsion. FEMS Microbiol Lett 179: 345–352. [DOI] [PubMed] [Google Scholar]
- 49. Franke I, Resch A, Dassler T, Maier T, Bock A (2003) YfiK from Escherichia coli promotes export of O-acetylserine and cysteine. Journal of bacteriology 185: 1161–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Van Dyk TK, Templeton LJ, Cantera KA, Sharpe PL, Sariaslani FS (2004) Characterization of the Escherichia coli AaeAB efflux pump: a metabolic relief valve? Journal of bacteriology 186: 7196–7204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sahota G, Stormo GD (2010) Novel sequence-based method for identifying transcription factor binding sites in prokaryotic genomes. Bioinformatics 26: 2672–2677. 10.1093/bioinformatics/btq501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Christen S, Srinivas A, Bahler P, Zeller A, Pridmore D, et al. (2006) Regulation of the Dha operon of Lactococcus lactis: a deviation from the rule followed by the Tetr family of transcription regulators. The Journal of biological chemistry 281: 23129–23137. [DOI] [PubMed] [Google Scholar]
- 53. Hu B, Lidstrom M (2012) CcrR, a TetR family transcriptional regulator, activates the transcription of a gene of the Ethylmalonyl coenzyme A pathway in Methylobacterium extorquens AM1. Journal of bacteriology 194: 2802–2808. 10.1128/JB.00061-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. de Eugenio LI, Galan B, Escapa IF, Maestro B, Sanz JM, et al. (2010) The PhaD regulator controls the simultaneous expression of the pha genes involved in polyhydroxyalkanoate metabolism and turnover in Pseudomonas putida KT2442. Environ Microbiol 12: 1591–1603. 10.1111/j.1462-2920.2010.02199.x [DOI] [PubMed] [Google Scholar]
- 55. Quinones B, Pujol CJ, Lindow SE (2004) Regulation of AHL production and its contribution to epiphytic fitness in Pseudomonas syringae. Mol Plant Microbe Interact 17: 521–531. [DOI] [PubMed] [Google Scholar]
- 56. Chatterjee A, Cui Y, Hasegawa H, Chatterjee AK (2007) PsrA, the Pseudomonas sigma regulator, controls regulators of epiphytic fitness, quorum-sensing signals, and plant interactions in Pseudomonas syringae pv. tomato strain DC3000. Appl Environ Microbiol 73: 3684–3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hirano S, Tanaka K, Ohnishi Y, Horinouchi S (2008) Conditionally positive effect of the TetR-family transcriptional regulator AtrA on streptomycin production by Streptomyces griseus. Microbiology 154: 905–914. 10.1099/mic.0.2007/014381-0 [DOI] [PubMed] [Google Scholar]
- 58. Park H, Ro YT, Kim YM (2011) MdoR is a novel positive transcriptional regulator for the oxidation of methanol in Mycobacterium sp. strain JC1. Journal of bacteriology 193: 6288–6294. 10.1128/JB.05649-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Uguru GC, Stephens KE, Stead JA, Towle JE, Baumberg S, et al. (2005) Transcriptional activation of the pathway-specific regulator of the actinorhodin biosynthetic genes in Streptomyces coelicolor. Mol Microbiol 58: 131–150. [DOI] [PubMed] [Google Scholar]
- 60. Pompeani AJ, Irgon JJ, Berger MF, Bulyk ML, Wingreen NS, et al. (2008) The Vibrio harveyi master quorum-sensing regulator, LuxR, a TetR-type protein is both an activator and a repressor: DNA recognition and binding specificity at target promoters. Mol Microbiol 70: 76–88. 10.1111/j.1365-2958.2008.06389.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chattoraj P, Mohapatra SS, Rao JL, Biswas I (2011) Regulation of transcription by SMU.1349, a TetR family regulator, in Streptococcus mutans. Journal of bacteriology 193: 6605–6613. 10.1128/JB.06122-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Miller VL, and Mekalanos J.J. (1988) A novel suicide vector and its use in construction of insertion mutations: Osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR . Journal of bacteriology 170: 2575–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Guzman LM, Belin D, Carson MJ, Beckwith J (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. Journal of bacteriology 177: 4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Morin CE, Kaper JB (2009) Use of stabilized luciferase-expressing plasmids to examine in vivo-induced promoters in the Vibrio cholerae vaccine strain CVD 103-HgR. FEMS Immunol Med Microbiol 57: 69–79. 10.1111/j.1574-695X.2009.00580.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schild S, Tamayo R, Nelson EJ, Qadri F, Calderwood SB, et al. (2007) Genes induced late in infection increase fitness of Vibrio cholerae after release into the environment. Cell Host Microbe 2: 264–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Overnight cultures of Vibrio cholerae strain JB58 (N16961 ΔlacZ SmR) or the isogenic ΔvexR mutant bearing the transcriptional fusion reporters for the indicated RND efflux system were diluted 1:100 into LB broth and grown with shaking for 3.5 h at 37°C when deoxycholate (DOC) was added to 0.02%. The culture was incubated with shaking at 37°C for an additional 30 min when aliquots were collected in triplicate and β-galactosidase activity was quantified as described in the methods. (A.) vexCD; (B.) vexEF; (C.) vexGH; (D.) vexIJK; (E.) vexLM. The reported results are in Miller Units (MU) and are the mean ± SD of three independent experiments.
(PDF)
The indicated strains containing a chromosomal cpxP-lacZ transcriptional reporter were cultured overnight in LB broth before being diluted 1:100 into fresh LB broth. The cultures were incubated an additional hour and diluted 1,000-fold into PBS. Aliquots of the diluted cultures were then inoculated onto the surface of LB agar plates containing X-gal (160 μg/mL) plus and minus 500 μM CuCl2 (to induce expression of the Cpx system). The plates were then incubated overnight at 37°C before being photographed.
(PDF)
The indicated V. cholerae N16961 strains containing a vexRAB-lacZ transcriptional reporter were cultured to middle logarithmic phase in LB broth when vexRAB-lacZ expression was quantified by a β-galactosidase assay as described in the methods. The results are the average ±SD of five independent experiments.
(PDF)
V. cholerae N16961 ΔvexR containing pJB703 (pBAD18::vexR) or pBAD18 were grown in triplicate wells of microtiter plates containing (A) LB broth or (B) LB broth plus 0.015% deoxycholate. Arabinose (0.1%) was added to the media as indicated to induce expression of vexR from the arabinose regulated promoter in pBAD18. Cell growth was then monitored as the change in the optical density at 600 nm and plotted versus time as the mean ±SEM.
(PDF)
(PDF)
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.