

Abstract
We analyze patterns of genetic variability of populations in the presence of a large seedbank with the help of a new coalescent structure called the seedbank coalescent. This ancestral process appears naturally as a scaling limit of the genealogy of large populations that sustain seedbanks, if the seedbank size and individual dormancy times are of the same order as those of the active population. Mutations appear as Poisson processes on the active lineages and potentially at reduced rate also on the dormant lineages. The presence of “dormant” lineages leads to qualitatively altered times to the most recent common ancestor and nonclassical patterns of genetic diversity. To illustrate this we provide a Wright–Fisher model with a seedbank component and mutation, motivated from recent models of microbial dormancy, whose genealogy can be described by the seedbank coalescent. Based on our coalescent model, we derive recursions for the expectation and variance of the time to most recent common ancestor, number of segregating sites, pairwise differences, and singletons. Estimates (obtained by simulations) of the distributions of commonly employed distance statistics, in the presence and absence of a seedbank, are compared. The effect of a seedbank on the expected site-frequency spectrum is also investigated using simulations. Our results indicate that the presence of a large seedbank considerably alters the distribution of some distance statistics, as well as the site-frequency spectrum. Thus, one should be able to detect from genetic data the presence of a large seedbank in natural populations.
Keywords: Wright–Fisher model, seedbank coalescent, dormancy, site-frequency spectrum, distance statistics
MANY microorganisms can enter reversible dormant states of low [respectively (resp.) zero] metabolic activity, for example when faced with unfavorable environmental conditions; see, e.g., Lennon and Jones (2011) for a recent overview of this phenomenon. Such dormant forms may stay inactive for extended periods of time and thus create a seedbank that should significantly affect the interplay of evolutionary forces driving the genetic variability of the microbial population. In fact, in many ecosystems, the percentage of dormant cells compared to the total population size is substantial and sometimes even dominant (for example, ∼20% in human gut, 40% in marine water, and 80% in soil; cf. Lennon and Jones 2011, box 1, table a). This abundance of dormant forms, which can be short-lived as well as stay inactive for significant periods of time (decades- or century-old spores are not uncommon), thus creates a seedbank that buffers against environmental change, but potentially also against classical evolutionary forces such as genetic drift, mutation, and selection.
In this article, we investigate the effect of large seedbanks (that is, comparable to the size of the active population) on the patterns of genetic variability in populations over macroscopic timescales. In particular, we extend a recently introduced mathematical model for the ancestral relationships in a Wright–Fisherian population of size N with geometric seedbank age distribution (cf. Blath et al. 2015b) to accommodate different mutation rates for “active” and “dormant” individuals, as well as a positive death rate in the seedbank. The resulting genealogy, measured over timescales of order N, can then be described by a new universal coalescent structure, the “seedbank coalescent with mutation,” if the individual initiation and resuscitation rates between active and dormant states as well as the individual mutation rates are of order . Measuring times in units of N and mutation rates in units of is of course the classical scaling regime in population genetic modeling; in particular, the classical Wright–Fisher model has a genealogy that converges in precisely this setup to the usual Kingman coalescent with mutation (Kingman 1982a,b,c; see Wakeley 2009 for an overview).
We provide a precise description of these (seedbank) coalescents and corresponding population models, in part motivated by recent research in microbial dormancy (Jones and Lennon 2010; Lennon and Jones 2011), in the next section. We argue that our seedbank coalescent is universal in the sense that it is robust to the specifics of the associated population model, as long as certain basic features are captured.
Our explicit seedbank coalescent model then allows us to derive expressions for several important population genetic quantities. In particular, we provide recursions for the expectation (and variance) of the time to the most recent common ancestor , the total number of segregating sites, average pairwise differences, and number of singletons in a sample (under the infinitely many sites model assumptions). We then use these recursions, and additional simulations based on the seedbank coalescent with mutation, to analyze Tajima’s D and related distance statistics in the presence of seedbanks and also the observed site-frequency spectrum.
We hope that this basic analysis triggers further research on the effect of seedbanks in population genetics, for example concerning statistical methods that allow one to infer the presence and size of seedbanks from data, to allow model selection [e.g., seedbank coalescent vs. (time-changed) Kingman coalescent], and finally to estimate evolutionary parameters such as the mutation rate in dormant individuals or the inactivation and reactivation rates between the dormant and active states.
It is important to note that our approach is different from a previously introduced mathematical seedbank model in Kaj et al. (2001). There, the authors consider a population of constant size N where each individual chooses its parent a random amount of generations in the past and copies its genetic type from there. The number of generations that separate each parent and offspring can be interpreted as the time (in generations) that the offspring stays dormant. The authors show that if the maximal time spent in the seedbank is restricted to finitely many , where m is fixed, then the ancestral process induced by the seedbank model converges, after the usual scaling of time by a factor N, to a time-changed (delayed) Kingman coalescent. Thus, typical patterns of genetic diversity, in particular the normalized site-frequency spectrum, will stay (qualitatively) unchanged. Of course, the point here is that the expected seedbank age distribution is not on the order of N, but uniformly bounded by m, so that for the coalescent approximation to hold one necessarily needs that m is small compared to N, which results in a “weak” seedbank effect. This model has been applied in Tellier et al. (2011) in the analysis of seedbanks in certain species of wild tomatoes. A related model was considered in Vitalis et al. (2004), which shares the feature that the time spent in the seedbank is bounded by a fixed number independent of the population size. For a more detailed mathematical discussion of such models, including previous work in Blath et al. (2015a), see Blath et al. (2015b). The choice of the adequate coalescent model [seedbank coalescent vs. (time-changed) Kingman coalescent] will thus also be an important question for study design, and the development of corresponding model selection rules will be part of future research.
Coalescent Models and Seedbanks
Before we discuss the seedbank coalescent, we briefly recall the classical Kingman coalescent for reference—this will ease the comparison of the underlying assumptions of both models.
The Kingman coalescent with mutation
The Kingman coalescent (Kingman, 1982a,b,c) describes the ancestral process of a large class of neutral exchangeable population models including the Wright–Fisher model (Fisher 1930; Wright 1931), the Moran model (Moran 1958), and many Cannings models (Cannings 1974). See, e.g., Wakeley (2009) for an overview. If we trace the ancestral lines (that is, the sequence of genetic ancestors at a locus) of a sample of size n backward in time, we obtain a binary tree, in which we see pairwise coalescences of branches until the most recent common ancestor is reached. Kingman proved that the probability law of this random tree can be described as follows: Each pair of lineages [there are many] has the same chance to coalesce, and the successive coalescence times are exponentially distributed with parameters , , until the last remaining pair of lines has coalesced. This elegant structure allows one to easily determine the expected time to the most recent common ancestor of a sample of size n, which is well known to be
| (1) |
Not surprisingly, we will essentially recover (1) for the seedbank coalescent defined below if the relative seedbank size becomes small compared to the active population size.
As usual, mutations are placed upon the resulting coalescent tree according to a Poisson process with rate , for some appropriate , so that the expected number of mutations of a sample of size 2 is just θ.
The underlying assumptions about the population for a Kingman coalescent approximation of its genealogy to be justified are simple but far reaching, namely that the different genetic types in the population are selectively neutral (i.e., do not exhibit significant fitness differences) and that the population size of the underlying population is essentially constant in time. If the population can be described by the (haploid) Wright–Fisher model (of constant size, say N), then, to arrive at the described limiting genealogy, it is standard to measure time in units of N, the coalescent timescale, and to assume that the individual mutation rates per generation are of order . The exact time scaling usually depends on the reproductive mechanism and other particularities of the underlying model (it differs already among variants of the Moran model), but the Kingman coalescent is still a universally valid limit for many a priori different population models (including, e.g., all reproductive mechanisms with bounded offspring variance, dioecy, age structure, partial selfing, and to some degree geographic structure), when these particularities exert their influence over timescales much shorter than the coalescent timescale; cf., e.g., Wakeley (2013). This is also the reason why the Kingman coalescent still appears as limiting genealogy of the weak seedbank model of Kaj et al. (2001) mentioned in the Introduction.
This robustness has turned the Kingman coalescent into an extremely useful tool in population genetics. In fact, it can be considered the standard null model for neutral populations. Its success is also based on the fact that it allows a simple derivation of many population genetic quantities of interest, such as a formula for the expected number of segregating sites
| (2) |
or the expected average number of pairwise differences π (Tajima 1983), the expected values of the site-frequency spectrum (cf. Fu 1995), when one assumes the infinite-sites model of Watterson (1975). This analytic tractability has allowed the construction of a sophisticated statistical machinery for the inference of evolutionary parameters. We investigate the corresponding quantities for the seedbank coalescent below.
The seedbank coalescent with mutation
Similar to the Kingman coalescent, the seedbank coalescent, mathematically introduced in Blath et al. (2015b), describes the ancestral lines of a sample taken from a population with a seedbank component. Here, we distinguish whether an ancestral line belongs to an active or dormant individual for any given point backward in time. The main difference from the Kingman coalescent is that as long as an ancestral line corresponds to a dormant individual (in the seedbank), it cannot coalesce with other lines, since reproduction and thus finding a common ancestor are possible only for active individuals.
The dynamics are now easily described as follows: If there are currently n active and m dormant lineages at some point in the past, each “active pair” may coalesce with the same probability, after an exponential time with rate , entirely similar to a classical Kingman coalescent with currently n lineages. However, each active line becomes dormant at a positive rate (corresponding to an ancestor who emerged from the seedbank), and each dormant line resuscitates, at a rate , for some The parameter K reflects the relative size of the seedbank compared to the active population and is explained below in terms of an explicit underlying population model. Since dormant lines are prevented from merging, they significantly delay the time to the most recent common ancestor. This mechanism is reminiscent of a structured coalescent with two islands (Notohara 1990; Herbots 1997), where lineages may merge only if they are in the same colony. Of course, if one samples a seedbank coalescent backward in time, one need specify not only the sample size, but also the number of sampled individuals from the active population (say n) and from the dormant population (say m).
In this article, we also consider mutations along the ancestral lines. As in the Kingman case we place them along the active line segments according to a Poisson process with rate and along the dormant segments at a rate Depending on the concrete situation, one may want to choose To determine the mutation rate in dormant individuals will be an interesting inference question. In Figure 1, we illustrate a realization of the seedbank coalescent with mutations: Ancestral lineages residing in the seedbank are represented by dotted lines and do not take part in coalescences.
Figure 1.
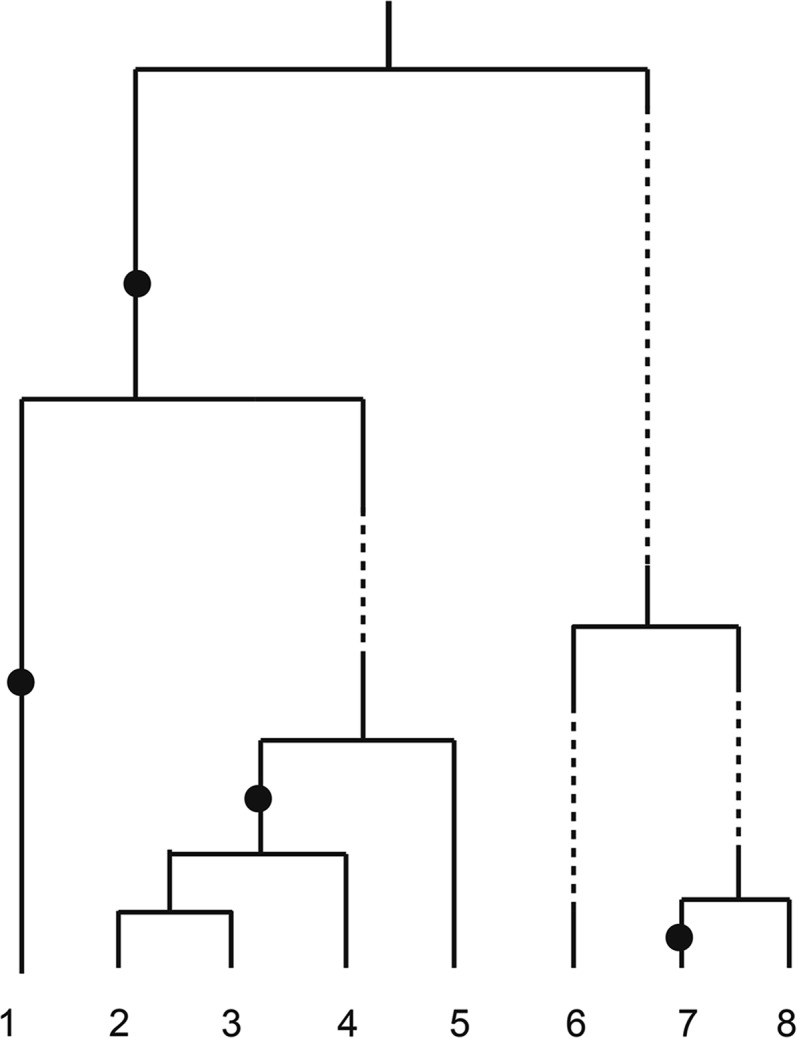
Realization of a seedbank coalescent with all sampled lineages assumed active. Mutations are, in this example, allowed to occur only on active ancestral lineages represented by solid lines; ancestral lineages residing in the seedbank are represented by dotted lines. Coalescences are allowed only among active lineages.
A formal mathematical definition of this process as a partition-valued Markov chain can be found in Blath et al. (2015b); it is straightforward to extend their framework to include mutations.
The parameters c and K can be understood as follows: c describes the proportion of individuals that enter the seedbank per (macroscopic) coalescent time unit. It is thus the rate at which individuals become dormant. If the ratio of the size of the active population and the dormant population in the underlying population is (that is, the active population is K times the size of the dormant population), and absolute (and thus also relative) population sizes are assumed to stay constant, then, for the relative amount of active and dormant individuals to stay balanced, the rate at which dormant individuals resuscitate and return to the active population is necessarily of the form (see also Figure 2). It is important to note that in this setup, the average coalescent time that an inactive individual stays dormant is of the order . We later also include a positive mortality rate for dormant individuals; this will lead to a reduced “effective” relative seedbank .
Figure 2.
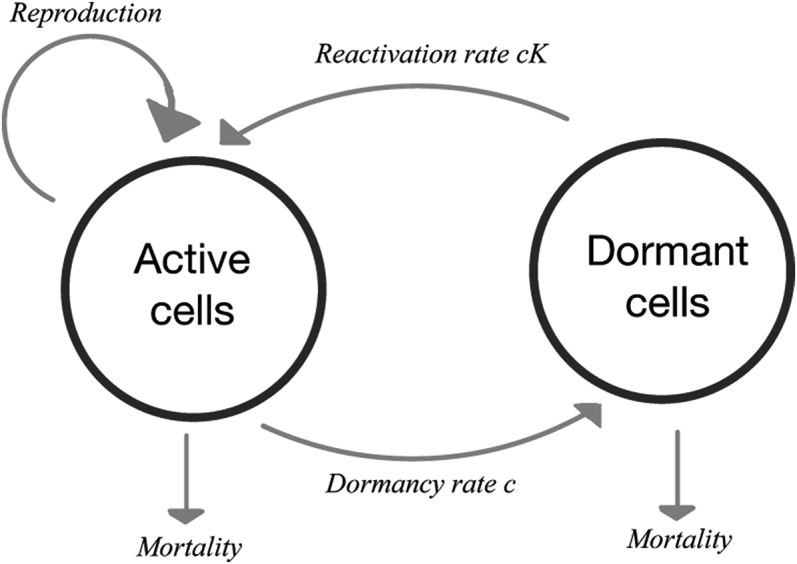
Dynamics of reversible microbial dormancy, according to Jones and Lennon (2010).
Robustness and underlying assumptions of the seedbank coalescent
As for the Kingman coalescent, it is important to understand the underlying assumptions that make the seedbank coalescent a reasonable model for the genealogy of a population: Again, we assume the types in the population to be selectively neutral, so that there are no significant fitness differences. Further, we assume the population size N and the seedbank size M to be constant and to be of the same order; that is, there exists a so that i.e., the ratio between active and dormant individuals is constant equal to Finally, the rate at which an active individual becomes dormant should be c (on the macroscopic coalescent scale), so that necessarily the average time (in coalescent time units) that an individual stays dormant before being resuscitated becomes . If one includes a positive mortality rate in the seedbank, this will lead to a modified parameter see below.
We provide below an example of a concrete seedbank population model, the “Wright–Fisher model with geometric seedbank component,” including mutation and mortality in the seedbank, for which it can be proved that the seedbank coalescent with mutation governs the genealogy if the population size N (and thus necessarily also seedbank size M) gets large, and coalescent time is measured in units of the population size N. This is the same scaling regime as in the case of the Kingman coalescent corresponding to genealogy of the classical Wright–Fisher model.
The seedbank coalescent with mutation should be robust against small alterations—such as in the transition or reproduction mechanism or in the population or seedbank size—of the underlying population, similar to the robustness of the Kingman coalescent, especially if these alterations occur on timescales that are much shorter than the coalescent timescale (which is N for the haploid Wright–Fisher model). For example, one can still obtain this coalescent in a Moran model with seedbank component, as long as the seedbank is on the same order as the active population and if the migration rates between seedbank and active population scale suitably (as well as the mutation rate) with the coalescent timescale. As mentioned above, this is an important difference from the model considered by Kaj et al. (2001), where the time an individual stays in the seedbank is negligible compared to the coalescent timescale, thus resulting merely in a (time change) of a Kingman coalescent—a weak seedbank effect.
A Wright–Fisher Model with Geometric Seedbank Distribution
We now introduce a Wright–Fisher-type population model with mutation and seedbank in which individuals stay dormant for geometrically distributed amounts of time. The model is very much in line with classical probabilistic population genetics thinking (in particular, assuming constant population size), but also captures several features of microbial seedbanks described in Lennon and Jones (2011), in particular reversible states of dormancy and mortality in the seedbank. We assume that the following (idealized) aspects of (microbial) dormancy can be observed:
Dormancy generates a seedbank consisting of a reservoir of dormant individuals.
The size of the seedbank is comparable to the order of the total population size, say in a constant ratio for some
The size of the active population N and of the seedbank stays constant in time; combining this with ii we get .
The model is selectively neutral so that reproduction is entirely symmetric for all individuals; for concreteness we assume reproduction according to the Wright–Fisher mechanism in fixed generations. That means the joint offspring distribution of the parents in each generation is symmetric multinomial. We interpret zero offspring as the death of the parent, one offspring as mere survival of the parent, and two or more offspring as successful reproduction leading to new individuals created by the parent.
Mutations may happen in the active population, at constant probability of the order , but potentially also in the dormant population [at the same, a reduced, or a vanishing probability ].
There is bidirectional and potentially repeated switching from active to dormant states, which appears essentially independently among individuals (“spontaneous switching”). The individual initiation probability of dormancy per generation is of the order , for
Dormant individuals may die in the seedbank (due to maintenance and energy costs). If mortality is assumed to be positive, the individual probability of death per generation is of order .
For each new generation, all these mechanisms occur independently of the previous generations.
We schematically visualize this mechanism in Figure 2, which is similar to figure 1 in Jones and Lennon (2010). Whether these assumptions are met of course needs to be determined for the concrete underlying real population. In this theoretical article, we use the above assumptions to construct an explicit mathematical model that leads, measuring time in units of N, to a seedbank coalescent with mutation. Still, we emphasize that, as discussed in the previous section, the seedbank coalescent is robust as long as certain basic assumptions are met.
We now turn the above features into a formal mathematical model that can be rigorously analyzed, extending the Wright–Fisher model with a geometric seedbank component in Blath et al. (2015b) by additionally including mortality in the seedbank and potentially different mutation rates in the active and dormant populations.
Definition 1 (seedbank model with mutation and mortality)
Let , and let and The seedbank model with mutation is obtained by iterating the following dynamics for each discrete generation (with the convention that all occurring numbers are integers; if not, one may enforce this using appropriate Gauss brackets):
The N active individuals from generation produce active individuals in generation by multinomial sampling with equal weights.
Additionally, c dormant individuals, sampled uniformly at random without replacement from the seedbank of size in generation 0, reactivate; that is, they turn into exactly one active individual in generation each and leave the seedbank.
The active individuals from generation 0 are thus replaced by these new active individuals, forming the active population in the next generation
In the seedbank, d individuals, sampled uniformly at random without replacement from generation , die.
To replace the vacancies in the seedbank, the N active individuals from generation 0 produce seeds by multinomial sampling with equal weights, filling the vacant slots of the seeds that were activated.
The remaining seeds from generation 0 remain inactive and stay in the seedbank.
During reproduction, each newly created individual copies its genetic type from its parent.
In each generation, each active individual is affected by a mutation with probability , and each dormant individual mutates with probability (where may be 0).
This model is an extension of the model in Blath et al. (2015b) to additionally include mortality in the seedbank and incorporate (potentially distinct) mutation rates in the active and dormant population. It appears to be a rather natural extension of the classical Wright–Fisher model. Note that the model has a geometric seedbank age distribution, since every dormant individual in each generation has the same probability to become active, resp. die, in the next generation, so that the time that an individual is in the dormant state is geometrically distributed. The parameter of this geometric distribution is given by
in the absence, resp. presence, of mortality in the seedbank. With mathematical arguments similar to those applied in Blath et al. (2015b), it is now standard to show that the ancestral process of a sample taken from the above population model converges, on the coalescent timescale N, to the seedbank coalescent with parameters c and K, resp.
and mutation rates . It is interesting to see that mortality leads to a decrease of the relative seedbank size in a way that depends on the initiation rate c, which is of course rather intuitive. In this sense gives the effective relative seedbank size.
The type frequencies in the biallelic seedbank population model
In this article, we mostly consider the infinite-sites model (Watterson 1975), where it is assumed that each mutation generates an entirely new type. However, before turning to the infinite-sites model, we briefly discuss the biallelic case, say with types Given initial type configurations and , denote by
the genetic type configuration of the active individuals (ξ) and the dormant individuals (η) in generation k (obtained from the above mechanism). We assume that each mutation causes a transition from a to A or from A to a. Let
| (3) |
We call the discrete-time Markov chain the Wright–Fisher frequency process with mutation and seedbank component. It can be seen from a generator computation that under our assumption with time measured in units of the active population size N it converges as to the two-dimensional diffusion that is the solution to the system of stochastic differential equations
| (4) |
Here, denotes standard one-dimensional Brownian motion. An alternative way to represent this stochastic process is via its Kolmogorov backward generator (cf., e.g., Karlin and Taylor 1981), which is given by
for functions . Existence and uniqueness of the stationary distribution of this process follow from compactness of the state space and strictly positive mutation rate in the active population. See Chap. 4.9 in Ethier and Kurtz (2005) for more detailed arguments. Note that ℒ is reminiscent of the backward generator of the structured coalescent with two islands (Notohara 1990; Herbots 1997); however, its qualitative behavior is very different. Its relation to the structured coalescent with two islands will be investigated in future research. Observe that the solution of (4) is driven by only one Brownian motion.
Population Genetics with the Seedbank Coalescent
In contrast to Lennon and Jones (2011), who use a deterministic population dynamics approach to study seedbanks, we are interested in probabilistic effects of seedbanks on genetic variability. We therefore use a coalescent approach to study the (random) gene genealogy of a sample. To better understand how seedbanks shape genealogies, we consider genealogical properties, such as the time to the most recent common ancestor, the total tree size, and the length of external branches.
Genealogical tree properties
First we discuss some classical population genetic properties of the seedbank coalescent when viewed as a random tree without mutations. For the results that we derive below, it will usually be sufficient to consider the block-counting process , of our coalescent, where gives the number of lines in our coalescent that are active and denotes the number of dormant lines t time units in the past. Then, is the continuous-time Markov chain started in with transitions
| (5) |
Again, introducing mutation can be done in the usual way, by superimposing independent Poisson processes with rate on the active lines and at rate on the dormant lines. If the block-counting process is currently in state , then a mutation in an active line happens at rate and a mutation in a dormant line at rate . The total jump rate from state of the backward process with mutation is thus given by
| (6) |
Time to the most recent common ancestor
It has been shown in Blath et al. (2015b, theorem 4.6) that the expected time to the most recent common ancestor for the seedbank coalescent, if started in a sample of active individuals of size n, is , in stark contrast to the corresponding quantity for the classical Kingman coalescent, which is bounded by 2, uniformly in n; cf. (1). This already indicates that one should expect elevated levels of (old) genetic variability under the seedbank coalescent, since more (old) mutations can be accumulated. While the above result shows the asymptotic behavior of the for large n, it does not give precise information for the exact absolute value, in particular for “small to medium” n. Here, we provide recursions for its expected value and variance that can be computed efficiently. First, we introduce some notation.
We define the time to the most recent common ancestor of the seedbank coalescent formally to be
If the sample consists of active and dormant individuals, for some , then the expected time to the most recent common ancestor is (Blath et al. 2015b). Here, it is interesting to note that the time to the most recent common ancestor of the Bolhausen–Sznitman coalescent is also (Goldschmidt and Martin 2005). The Bolthausen–Sznitman coalescent is often used as a model for selection; cf., e.g., Neher and Hallatschek (2013).
One can compute the expected time to most recent common ancestor recursively as follows. For let
| (7) |
where denotes expectation when started in , i.e., with n active lines and m dormant ones. Observe that we need to consider both types of lines to calculate . Write
| (8) |
and abbreviate
| (9) |
Then we have the recursive representation
| (10) |
with initial conditions The proof of (10) and a recursion for the variance of is given in Supporting Information, File S1. Since the process is nonincreasing in t, these recursions can be solved iteratively. In fact,
| (11) |
which in the case without mortality reduces to
| (12) |
Notably, is constant for sample size 2 (see Equation 11) as c varies (Table 1) if and in particular does not converge for to the Kingman case. This effect is similar to the corresponding behavior of the structured coalescent with two islands if the migration rate goes to 0; cf. Nath and Griffiths (1993). However, the Kingman coalescent values are recovered as the seedbank size decreases (e.g., for in Table 1).
Table 1. The expected time to the most recent common ancestor of the seedbank coalescent, obtained from (10), with seedbank size K, sample size n, dormancy initiation rates c as shown, and .
| Sample size n | |||
|---|---|---|---|
| c | 2 | 10 | 100 |
| , | |||
| 0.01 | 1.02 | 2.868 | 5.185 |
| 0.1 | 1.02 | 2.731 | 4.487 |
| 1 | 1.02 | 2.187 | 2.666 |
| 10 | 1.02 | 1.878 | 2.085 |
| 100 | 1.02 | 1.84 | 2.026 |
| c | 2 | 10 | 100 |
| 0.01 | 4 | 10.21 | 17.18 |
| 0.1 | 4 | 9.671 | 14.97 |
| 1 | 4 | 8.071 | 10.02 |
| 10 | 4 | 7.317 | 8.221 |
| 100 | 4 | 7.212 | 7.954 |
| c | 2 | 10 | 100 |
| 0.01 | 1.02 | 1.846 | 2.052 |
| 0.1 | 1.02 | 1.838 | 2.026 |
| 1 | 1.02 | 1.836 | 2.02 |
| 10 | 1.02 | 1.836 | 2.02 |
| 100 | 1.02 | 1.836 | 2.02 |
| 1 | 1.80 | 1.98 | |
All sampled lines are from the active population [sample configuration ]. For comparison, when associated with the Kingman coalescent . The multiplication applies only to the first section with
The fact that for can be understood heuristically if c is large: In that situation, transitions between active and dormant states happen very fast; thus at any given time the probability that a line is active is ∼1/2, and therefore the probability that both lines of a given pair are active (and thus able to merge) is ∼1/4. We can therefore conjecture that for , and the genealogy of a sample is given by a time change by a factor 4 of Kingman’s coalescent.
Table 1 and Table 2 show values of obtained from (10) for various parameter choices and sample sizes. The relative size of the seedbank (K) has a significant effect on ; a large seedbank (K small) increases , while the effect of c is to dampen the increase in with sample size (Table 1). The effect of the seedbank death rate d on is to dampen the effect of the relative size of the seedbank (Table 2).
Table 2. The expected time to most recent common ancestor of the seed-bank coalescent, obtained from (10), with all sampled lines assumed active and c, K, and d as shown.
| , , parameter d | |||||
|---|---|---|---|---|---|
| K | 0.01 | 0.1 | 1 | 10 | 100 |
| 0.01 | 2.614e+04 | 2.208e+04 | 6814 | 270.7 | 9.91 |
| 0.1 | 315.6 | 270.7 | 96.2 | 9.04 | 2.442 |
| 1 | 9.91 | 9.04 | 5.201 | 2.4 | 2.02 |
| 10 | 2.442 | 2.4 | 2.197 | 2.017 | 1.984 |
| 100 | 2.02 | 2.017 | 2 | 1.984 | 1.98 |
| , , parameter d | |||||
| c | 0.01 | 0.1 | 1 | 10 | 100 |
| 0.01 | 8.281 | 2.893 | 2.051 | 1.985 | 1.98 |
| 0.1 | 13.39 | 7.215 | 2.617 | 2.025 | 1.984 |
| 1 | 9.91 | 9.04 | 5.201 | 2.4 | 2.02 |
| 10 | 8.213 | 8.138 | 7.477 | 4.556 | 2.361 |
| 100 | 7.953 | 7.946 | 7.875 | 7.245 | 4.466 |
For comparison, (1.98 for ) when associated with the Kingman coalescent.
Total tree length and length of external branches
To investigate the genetic variability of a sample, in terms, e.g., of the number of segregating sites and the number of singletons, it is useful to have information about the total tree length and the total length of external branches. Let denote the total length of all branches while they are active and denote the total length of all branches while they are dormant. Their expectations
| (13) |
may be calculated using the following recursions for , and with given by (8),
| (14) |
| (15) |
Observe that Equations 14 and 15 differ in the factor (n resp. m) that multiplies . Similar recursions hold for their variances as well as for the corresponding values of the total length of external branches, which can be found in File S2 together with the respective proofs. From (14) and (15) one readily obtains
| (16) |
We observe that and given in (16) are independent of c if as also seen for cf. (11). We use (16) to obtain closed-form expressions for expected average number of pairwise differences.
The numerical solutions of (14) and (15) indicate that for ,
| (17) |
Hence, the expected total lengths of the active and the dormant parts of the tree are proportional, and the ratio is given by the effective relative seedbank size.
Recursions for the expected total length of external branches are given in Proposition S1.3 in File S1. Let and denote the expected total lengths of active and dormant external branches, respectively, when started with n active and m dormant lines. The numerical solutions of the recursions indicate that the ratio of expected values and is also given by (17).
Recursions for expected branch lengths associated with any other class than singletons are more complicated to derive, and we postpone those for further study. Simulation results (not shown) suggest that the result (17) we obtained for relative expected total length of active branches, and active external branches, holds for all branch length classes; if denotes the total length of active (dormant) branches subtending leaves, then, if all our sampled lines are active, we claim that is given by (17). Table S1 shows values of , i.e., the relative expected total length of external branches when our sample consists of 10 active lines and 10 dormant ones. In contrast to the case when all sampled lines are active, c clearly affects when d is small. In line with previous results, d reduces the effect of the relative size of the seedbank.
Table S2 shows the expected total lengths of active and dormant external branches and for values of c, K, and d as shown. When the seedbank is large (K small), and can be much longer than the expected length of 2 when associated with the Kingman coalescent (Fu 1995). However, as noted before, the effect of K depends on d. The effect of c also depends on d; changes in c have a bigger effect when d is large.
One can gain insight into the effects of a seedbank on the site-frequency spectrum by studying the effects of a seedbank on relative branch lengths. Let denote the relative total length of active branches subtending i leaves , relative to the total length of active branches , and we consider only the case when all n sampled lines are active. Thus, if one assumes that the mutation rate in the seedbank is negligible compared to the mutation rate in the active population, should be a good indicator of the relative number of singletons, relative to the total number of segregating sites. In addition, we investigate to learn whether and how the presence of a seedbank affects genetic variation, even if no mutations occur in the seedbank. Figure S1 shows estimates of (obtained by simulations) for values of c, K, and d as shown (all sampled lines are assumed active). The main conclusion is that a large seedbank reduces the relative length of external branches and increases the relative magnitude of the right tail of the branch length spectrum. Thus, one would expect to see a similar pattern in neutral genetic variation: a reduced relative number of singletons (relative to the total number of polymorphic sites) and a relative increase in the number of polymorphic sites in high count.
Neutral genetic variation
In this subsection we derive and study several recursions for common measures of DNA sequence variation in the infinite-sites model (ISM) of Watterson (1975). Samples are assumed to be drawn from the stationary distribution. We also investigate how these quantities differ from the corresponding values under the Kingman coalescent, in an effort to understand how seedbank parameters affect genetic variability.
Segregating sites:
First we consider the number of segregating sites S in a sample, which, assuming the ISM, is the total number of mutations that occur in the genealogy of the sample until the time of its most recent common ancestor. In addition to being of interest on its own, S is a key ingredient in commonly employed distance statistics such as those of Tajima (1989) and Fu and Li (1993). We let mutations occur on active branch lengths according to independent Poisson processes each with rate and on dormant branches with rate . The expected value of S can be expressed in terms of the expected total tree lengths as
| (18) |
An alternative recursion for the expectation and the variance of the number of segregating sites can be found in File S1 (Proposition S1.5).
Table 3 shows the expected number of segregating sites in a sample of size n taken from the active population for values of c and K as shown. The size of the seedbank K strongly influences the number of segregating sites. If there is no mutation in the seedbank, it roughly doubles for and approaches the normal value of the Kingman coalescent for small seedbanks . The parameter K seems to have a more significant influence than the parameter c.
Table 3. The expected total number of segregating sites , with values of K, c, and d as shown and sample size (all lines from the active population), with and .
| Values of K, | |||||
|---|---|---|---|---|---|
| c | 0.01 | 0.1 | 1 | 10 | 100 |
| 0.01 | 1035 | 112.8 | 20.6 | 11.38 | 10.46 |
| 0.1 | 958.3 | 105.3 | 19.98 | 11.36 | 10.46 |
| 1 | 790.6 | 90.37 | 19.4 | 11.38 | 10.46 |
| 10 | 884 | 99.92 | 20.16 | 11.39 | 10.46 |
| 100 | 1010 | 110.8 | 20.61 | 11.39 | 10.46 |
| Values of K, , | |||||
| c | 0.01 | 0.1 | 1 | 10 | 100 |
| 0.01 | 10.46 | 10.37 | 10.36 | 10.35 | 10.35 |
| 0.1 | 11.36 | 10.46 | 10.37 | 10.36 | 10.35 |
| 1 | 19.32 | 11.37 | 10.46 | 10.37 | 10.36 |
| 10 | 91.97 | 19.3 | 11.29 | 10.45 | 10.36 |
| 100 | 510.3 | 60.83 | 15.51 | 10.87 | 10.41 |
When associated with the Kingman coalescent, and
Average pairwise differences:
Average pairwise differences are a key ingredient in the distance statistics of Tajima (1983) and Fay and Wu (2000). Expected value and variance for average pairwise differences in the Kingman coalescent were first derived in Tajima (1983). Here, we give an expression for the expectation in terms of the expected total tree lengths. Denote by π the average number of pairwise differences
| (19) |
where is the total number of pairwise differences, with denoting the number of differences observed in the pair of DNA sequences indexed by . We abbreviate and obtain
which can be calculated using
| (20) |
where and are defined in (13).
Hence, given a sample configuration , i.e., our n sampled lines are all active, (20), together with (16), gives
| (21) |
If now , the dependence on c disappears again, since we have
which is obviously highly elevated compared to if the seedbank is large (K small). For comparison, when associated with the usual Kingman coalescent, which we recover in the absence of a seedbank in (21).
The site-frequency spectrum:
The site-frequency spectrum (SFS) is one of the most important summary statistics of population genetic data in the infinite-sites model. Suppose that we can distinguish between mutant and wild type, e.g., with the help of an outgroup. As before, we distinguish between the number of samples taken from the active population (say n) and the dormant population (say m). Then, the SFS of an sample is given by
| (22) |
where the , denote the number of sites at which variants appear i times in our sample of size . For the Kingman coalescent, the expected values, variances, and covariances of the SFS have been derived by Fu (1995). Expected values and covariances can in principle be computed by extending the theory in Fu (1995) and resp. Griffiths and Tavaré (1998). However, the computations would be far more involved than the previous recursions and will be treated in future research. We derive recursions for the expected number of singletons and investigate the whole SFS by simulation.
Number of singletons:
The number of singletons in a sample is often taken as an indicator of the kind of historical processes that have acted on the population. By “singletons” we mean the number of derived (or new) mutations that appear only once in the sample, which in the infinite-sites model is equal to the number of mutations occurring on external branches. Thus we can relate the expected number of singletons, denoted by , to the total length of external branches in the same way as we related the number of segregating sites to the total tree length. Let denote the expected total length of external branches when our sample consists of n active external lines, active internal lines, m dormant external lines, and dormant internal lines. Define similarly as the expected total length of dormant external branches. Recursions for and are given in File S2. For we have that the expected number of singletons is given by
Thus, one can compute the expected number of singletons by solving the recursions for external branch lengths. By way of example, Table S2 gives values of and for a sample of 10 active lines (, ).
The whole site-frequency spectrum:
Figure 3 shows estimates of the normalized expected frequency spectrum , where denotes the total number of segregating sites. Figure 3 shows that if the relative size of the seedbank is small (say, ), then the SFS is almost unaffected by dormancy, in line with intuition. If the seedbank is large (say ) and the transition rate is comparable to the mutation rate , then the spectrum differs significantly; in particular, the number of singletons is reduced by about one-half, which should be significant, and the right tail is much heavier.
Figure 3.

Estimates of the normalized site-frequency spectrum with all sampled lines assumed active and values of c and K as shown . The mutation rate in the active population is fixed, , and there is no mutation in the dormant states . All estimates based on replicates.
This can be understood as follows: If the seedbank leads to an extended time to the most recent common ancestor, then the proportion of old mutations should increase, and these should be visible in many sampled individuals, strengthening the right tail of the spectrum.
It is interesting to see that even in the presence of a large seedbank (say ), large transition rates (say ) do not seem to affect the normalized spectrum. Again, this can be understood intuitively, since by the arguments presented in the discussion after (12) large c should lead to a constant time change of the Kingman coalescent (with a time change depending on K). Such a time change does not affect the normalized spectrum.
One reason for considering the SFS is naturally that one wants to be able to use the SFS in inference, to determine, say, if a seedbank is present and how large it is. If one has expressions for the expected SFS under some coalescent model, one can use the normalized expected SFS in an approximate-likelihood inference (see, e.g., Eldon et al. 2015). The normalized spectrum is also appealing since it is quite robust to changes in the mutation rate (Eldon et al. 2015). For comparison, Figure S2 shows estimates of the expected normalized spectrum where and shows a similar pattern as for the normalized expected spectrum in Figure 3.
Distance statistics
Rigorous inference work is beyond the scope of this article. However, we can still consider (by simulation) estimates of the distribution of various commonly employed distance statistics. Distance statistics for the site-frequency spectrum are often employed to make inference about historical processes acting on genetic variation in natural populations. Commonly used statistics include the ones of Tajima (1989) (), Fu and Li (1993) (), and Fay and Wu (2000) (). These statistics contrast different parts of the site-frequency spectrum (cf., e.g., Zeng et al. 2006).
The distance:
Arguably the most natural distance statistic to consider is the distance (or sum of squares) of the whole SFS (or some lumped version thereof) between the observed SFS and an expected SFS based on some coalescent model. The statistic (n denotes sample size) is given by
| (23) |
where, in our case, expectation and variance are taken with respect to the Kingman coalescent (Fu 1995). Estimates of the distribution of are shown in Figure 4. As the size of the seedbank increases (K decreases), one observes worse fit of the site-frequency spectrum with the expected SFS associated with the Kingman coalescent.
Figure 4.

Estimates of the distribution of the statistic (Equation 23), with all sampled lines assumed active, c and K as shown, , and The vertical dashed lines are the and quantiles and the solid square (■) denotes the mean. The entries are normalized to have unit mass 1. All estimates are based on replicates.
Tajima’s D:
Tajima’s statistic for a sample of size n, with , is defined as
| (24) |
(Tajima 1989), where the variance depends on the mutation rate θ that is usually estimated from the data. Under the Kingman coalescent, Deviations from the Kingman coalescent model become significant at the 5% level if they are either >2 or < Negative values of should appear if there is an excess of either low- or high-frequency polymorphisms and a deficiency of middle-frequency polymorphisms (see, e.g., Wakeley 2009 for further details). Positive values of are to be expected if variation is common with moderate frequencies, for example in the presence of a recent population bottleneck or balancing selection.
The empirical distribution of was investigated by simulation for different seedbank parameters (Figure 5 and Figure S3), assuming that mutations do not occur in the seedbank . If the seedbank is large , then the median of D becomes significantly positive. For , there is very little deviation from the Kingman coalescent. Again D seems to be more sensitive to small values of K than changes in c. This is in line with our results on the , with highly elevated times for small K. In the latter case, old variation will dominate, thus resembling a population bottleneck, producing positive values of .
Figure 5.

Estimates of the distribution of Tajima’s (Equation 24) with all sampled lines assumed active, , and The vertical dashed lines are the and quantiles and the solid square (■) denotes the mean. The entries are normalized to have unit mass 1. The histograms are drawn on the same horizontal scale. Estimates are based on replicates.
In conclusion, might not be a very good statistic to detect seedbanks.
Fu and Li’s D:
Fu and Li (1993) statistic is defined as
| (25) |
with S being the total number of segregating sites, the total number of singletons, , and and as in Fu and Li (1993). As with , under the Kingman coalescent.
Figure 6 shows estimates of the distribution of assuming When the seedbank is large (K small), the distribution of becomes highly skewed, with most genealogies resulting in a low number of singletons compared with the total number of polymorphisms, resulting in positive . This is in line with our observations about the relative number of singletons associated with a large seedbank (Figure 3 and Figure S2) and the relative length of external branches (Figure S1).
Figure 6.

Estimates of the distribution of Fu and Li’s (Equation 25) with all sampled lines assumed active, , and The vertical dashed lines are the and quantiles and the solid square (■) denotes the mean. The entries are normalized to have unit mass 1. The histograms are drawn on the same horizontal scale. Estimates are based on replicates.
Fay and Wu’s H:
The distance statistics of Fay and Wu (2000) are defined as
| (26) |
where
| (27) |
and π is the average number of pairwise differences. A formula for the variance of was obtained by Zeng et al. (2006). Figure 7 holds estimates of the distribution of with , , and c and K as shown. As the seedbank size increases (K decreases), high-frequency variants, as captured by H, become dominant over the middle-frequency variants captured by π. In conclusion, Fu and Li’s or Fay and Wu’s may be preferable over Tajima’s statistic to detect the presence of a seedbank. A rigorous comparison of different statistics (including the E statistic of Zeng et al. 2006) and their power to distinguish between absence and presence of a seedbank must be the subject of future research.
Figure 7.

Estimates of the distribution of Fay and Wu’s (Equation 26) with all sampled lines assumed active, , and The vertical dashed lines are the and quantiles and the solid square (■) denotes the mean. The entries are normalized to have unit mass 1. The histograms are drawn on the same horizontal scale. Estimates are based on replicates.
The C code written for the computations is available at http://page.math.tu-berlin.de/∼eldon/programs.html.
Discussion
In the previous sections, we have presented and analyzed an idealized model of a population sustaining a large seedbank, as well as the resulting patterns of genetic variability, with the help of a new coalescent structure, called the seedbank coalescent (with mutation). This ancestral process appeared naturally as a scaling limit of the genealogy of large populations producing dormant forms, in a similar way to the classical Kingman coalescent that arises in conventional models, under the following assumptions: The seedbank size is of the same order as the size of the active population, the population and seedbank size are constant over time, and individuals enter the dormant state by spontaneous switching independently of each other, in a way that individual dormancy times are comparable to the active population size. We begin with a discussion of these modeling assumptions.
The assumption that the seedbank is of comparable size to the active population is based on Lennon and Jones (2011), where it is shown in box 1, table a, that this is often the case in microbial populations.
Assuming constant population size is a very common simplification in population genetics and can be explained with constant environmental conditions, we claim that “weak” fluctuations (of smaller order than the active population size) still lead to the seedbank coalescent model, as is the case for the Kingman coalescent. However, seedbanks are often seen as a bet-hedging strategy against drastic environmental changes, which is not yet covered by our models. We see this as an important task for future research, which will require serious mathematical analysis. In the case of weak seedbank effects, fluctuating population size has been considered in Živković and Tellier (2012), where the presence of the seedbank was observed to lead to an increase of the effective population size.
Assuming spontaneous switching of single individuals between the active and the dormant state is also based on Lennon and Jones (2011) (pp. 122–124). This somewhat restricts the scope of the model because it will not capture major environmental changes that may trigger a simultaneous change of state of a large proportion of individuals (e.g., due to sudden lack of nutrients). This effect is closely related to drastic changes in population size and again may lead to serious alterations of our predictions. Hence, including such large switching events will also be an important part of our future work (and will again require substantial mathematical work). In Vitalis et al. (2004) a whole proportion of the dormant population becomes active in every generation, but this should be seen in conjunction with the fact that dormancy is of limited duration, which excludes drastic alterations on a long timescale.
Assuming that the time spent in the seedbank is of the order of the population size is one of the main features that distinguishes our model from previous models of weak seedbank effects as previously investigated in Kaj et al. (2001) and Vitalis et al. (2004). Statistical inference will be needed to support or reject this assumption and to distinguish between weak and strong seedbanks. One distinguishing feature of weak and strong seedbanks is the behavior of the normalized site-frequency spectrum. Since weak seedbank effects lead to a genealogy that is a constant time change of Kingman’s coalescent (Kaj et al. 2001; Blath et al. 2013), the normalized frequency spectrum of weak seedbanks will be similar to those corresponding to the Kingman coalescent, while under our model we observe (at least for large seedbanks) a reduction in the number of singletons (Figure S2). The model of Kaj et al. (2001) was used in Tellier et al. (2011), where Tajima’s D was used to detect seedbanks.
We now discuss our results for the behavior of classical quantities describing genetic variability under our modeling assumptions, that is, when the genealogy of a sample can be described by the seedbank coalescent. In particular, we used it to derive recursions for quantities such as the time to the most recent common ancestor, the total tree length, or the length of external branches. We investigated statistics of interest to genetic variability such as the number of segregating sites, the site-frequency spectrum, Tajima’s D, Fu and Li’s D, and Fay and Wu’s H by numerical solution of our recursions and by simulation. It turns out that the seedbank size K leads to significant changes, for example in the site frequency spectrum, producing a positive Tajima’s D, indicating the presence of old genetic variability, in line with intuition. Interestingly, the influence of c seems to be less pronounced. For we observe convergence toward the Kingman coalescent regime, while seems to lead to a constant time change of Kingman’s coalescent.
We are confident that our results so far have the potential to open up many interesting research questions, both on modeling and on statistical inference, as well as in data analysis. For example, it should be interesting to derive a test to distinguish between the presence of strong vs. weak (resp. negligible) seedbanks. Another important task in future research will be to infer parameters of the model. While the relative seedbank size K can in principle be directly observed by cell counting (Lennon and Jones 2011), the parameter c seems to be difficult to observe, in particular because we have seen that many statistics we calculated are independent of or at least not very sensitive with respect to c. On the other hand, this shows that our results are fairly robust under alterations of c, such that estimations or tests may be applied to some extent without prior knowledge on c. The mortality rate d may for many practical purposes be included into the parameter K or measuring the “effective” relative seedbank size.
Estimating the mutation rates and is another goal for the future. In particular, in view of an ongoing debate on the possibility of mutations in dormant individuals (Maughan 2007), it would be important to devise a test to determine whether
Supplementary Material
Acknowledgments
J.B., B.E., and N.K. acknowledge support by Deutsche Forschungsgemeinschaft (DFG) as part of SPP Priority Programme 1590 “Probabilistic Structures in Evolution”: J.B. and B.E. through DFG grant BL 1105/3-1 and J.B. and N.K. through DFG grant BL 1105/4-1. A.G.C. is supported by DFG grant RTG 1845, “Stochastic Analysis and Applications in Biology, Finance, and Physics”; the Berlin Mathematical School (BMS); and the Mexican Council of Science in collaboration with the German Academic Exchange Service. M.W.B. is supported by DFG grant RTG 1845 and the BMS.
Footnotes
Communicating editor: W. Stephan
Supporting information is available online at www.genetics.org/lookup/suppl/doi:10.1534/genetics.115.176818/-/DC1.
Literature Cited
- Blath J., González Casanova A., Kurt N., Spanò D., 2013. The ancestral process of long-range seed bank models. J. Appl. Probab. 50: 741–759. [Google Scholar]
- Blath, J., B. Eldon, A. Casanova, and N. Kurt, 2015a Genealogy of a Wright Fisher model with strong seed bank component, in XI Symposium on Probability and Stochastic Processes, Progress in Probability, edited by R. Mena, J. C. Pardo, V. Rivero, and G. Uribe Bravo. Birkhäuser, Basel, Switzerland (in press). [Google Scholar]
- Blath J., Gonzalez-Casanova A., Kurt N., Wilke-Berenguer M., 2015b The seed-bank coalescent. Ann. Appl. Probab. (in press). [Google Scholar]
- Cannings C., 1974. The latent roots of certain Markov chains arising in genetics: a new approach, I. Haploid models. Adv. Appl. Probab. 6: 260–290. [Google Scholar]
- Eldon B., Birkner M., Blath J., Freund F., 2015. Can the site-frequency spectrum distinguish exponential population growth from multiple-merger coalescents? Genetics 199: 841–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ethier S. N., Kurtz T. G., 2005. Markov Processes: Characterization and Convergence. Wiley, Hoboken, NJ. [Google Scholar]
- Fay J. C., Wu C., 2000. Hitchhiking under positive Darwinian selection. Genetics 155: 1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher R. A., 1930. The Genetical Theory of Natural Selection. Oxford University Press, Oxford. [Google Scholar]
- Fu Y.-X., 1995. Statistical properties of segregating sites. Theor. Popul. Biol. 48: 172–197. [DOI] [PubMed] [Google Scholar]
- Fu Y.-X., Li W.-H., 1993. Statistical tests of neutrality of mutations. Genetics 133: 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt C., Martin J. B., 2005. Random recursive trees and the Bolthausen-Sznitman coalescent. Electron. J. Probab. 10: 718–745. [Google Scholar]
- Griffiths R. C., Tavaré S., 1998. The age of a mutation in a general coalescent tree. Special issue in honor of Marcel F. Neuts. Comm. Stat. Stoch. Models 14: 273–295. [Google Scholar]
- Herbots, H. M., 1997 The structured coalescent, pp. 231–255 in Progress of Population Genetics and Human Evolution, edited by P. Donnelly and S. Tavaré. Springer-Verlag, Berlin/Heidelberg, Germany/New York. [Google Scholar]
- Jones S. E., Lennon J. T., 2010. Dormancy contributes to the maintenance of microbial diversity. Proc. Natl. Acad. Sci. USA 107: 5881–5886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaj I., Krone S. M., Lascoux M., 2001. Coalescent theory for seed bank models. J. Appl. Probab. 38: 285–300. [Google Scholar]
- Karlin S., Taylor H., 1981. A Second Course in Stochastic Processes. Academic Press, New York. [Google Scholar]
- Kingman J. F. C., 1982a The coalescent. Stoch. Proc. Appl. 13: 235–248. [Google Scholar]
- Kingman J. F. C., 1982b Exchangeability and the evolution of large populations, pp. 97–112 in Exchangeability in Probability and Statistics, edited by Koch G., Spizzichino F. North-Holland, Amsterdam. [Google Scholar]
- Kingman J. F. C., 1982c On the genealogy of large populations. J. Appl. Probab. 19A: 27–43. [Google Scholar]
- Lennon J. T., Jones S. E., 2011. Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat. Rev. Microbiol. 9: 119–130. [DOI] [PubMed] [Google Scholar]
- Maughan H., 2007. Rates of molecular evolution in bacteria are relatively constant despite spore dormancy. Evolution 61: 280–288. [DOI] [PubMed] [Google Scholar]
- Moran P., 1958. Random processes in genetics. Proc. Camb. Philos. Soc. 54: 60–71. [Google Scholar]
- Nath H., Griffiths R., 1993. The coalescent in two colonies with symmetric migration. J. Math. Biol. 31: 841–852. [DOI] [PubMed] [Google Scholar]
- Neher R. A., Hallatschek O., 2013. Genealogies of rapidly adapting populations. Proc. Natl. Acad. Sci. USA 110: 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notohara M., 1990. The coalescent and the genealogical process in geographically structured population. J. Math. Biol. 29: 59–75. [DOI] [PubMed] [Google Scholar]
- Tajima F., 1983. Evolutionary relationships of DNA sequences in finite populations. Genetics 105: 437–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellier A., Laurent S. J., Lainer H., Pavlidis P., Stephan W., 2011. Inference of seed bank parameters in two wild tomato species using ecological and genetic data. Proc. Natl. Acad. Sci. USA 108: 17052–17057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitalis R., Glémin S., Olivieri I., 2004. When genes got to sleep: the population genetic consequences of seed dormacy and monocarpic perenniality. Am. Nat. 163: 295–311. [DOI] [PubMed] [Google Scholar]
- Wakeley, J., 2009 Coalescent Theory: An introduction, Vol. 1. Roberts & Company Publishers, Greenwood Village, CO. [Google Scholar]
- Wakeley J., 2013. Coalescent theory has many new branches. Theor. Popul. Biol. 87: 1–4. [DOI] [PubMed] [Google Scholar]
- Watterson G., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7: 256–276. [DOI] [PubMed] [Google Scholar]
- Wright S., 1931. Evolution in Mendelian populations. Genetics 16: 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng K., Fu Y., Shi S., Wu C., 2006. Statistical tests for detecting positive selection by utilizing high-frequency variants. Genetics 174: 1431–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Živković D., Tellier A., 2012. Germ banks affect the inference of past demographic events. Mol. Ecol. 21: 5434–5446. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.