

SUMMARY
The RAG1 endonuclease, together with its cofactor RAG2, is essential for V(D)J recombination but is a potent threat to genome stability. The sources of RAG1 mis-targeting and the mechanisms that have evolved to suppress it are poorly understood. Here, we report that RAG1 associates with chromatin at thousands of active promoters and enhancers in the genome of developing lymphocytes. The mouse and human genomes appear to have responded by reducing the abundance of “cryptic” recombination signals near RAG1 binding sites. This depletion operates specifically on the RSS heptamer, whereas nonamers are enriched at RAG1 binding sites. Reversing this RAG-driven depletion of cleavage sites by insertion of strong recombination signals creates an ectopic hub of RAG-mediated V(D)J recombination and chromosomal translocations. Our findings delineate rules governing RAG binding in the genome, identify areas at risk of RAG-mediated damage, and highlight the evolutionary struggle to accommodate programmed DNA damage in developing lymphocytes.
Graphical abstract
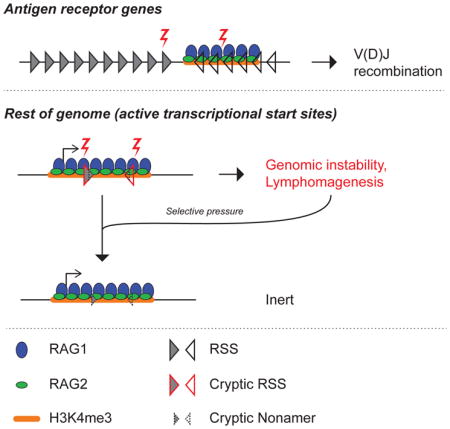
INTRODUCTION
Vertebrate lymphocytes appear to be unique in their tolerance for, and dependence on, programmed DNA breakage and rearrangement during multiple developmental stages. V(D)J recombination occurs during early B- and T-lymphocyte development, assembling the immunoglobulin (Ig) and T-cell receptor (Tcr) genes from arrays of V, D, and J gene segments. The diversity arising from this locus-specific recombination creates the basis for the immense variation in antigen recognition capacity of the mature lymphocyte population.
The proteins that initiate V(D)J recombination, RAG1 and RAG2, form an endonuclease with a preferred DNA substrate – the recombination signal sequence (RSS) – that immediately flanks V, D, and J gene segments. The RSS consists of a heptamer (consensus 5′-CACAGTG) and nonamer (consensus 5′-ACAAAAACC) separated by a degenerate spacer of either 12 or 23 base pairs (bp). The heptamer serves as the cleavage target and the nonamer provides an important binding site for RAG1 (Schatz and Swanson, 2011). Efficient DNA cleavage and recombination require synapsis of one 12-RSS and one 23-RSS. Once cleaved, the DNA ends are ligated by non-homologous end joining (NHEJ) (Rooney et al., 2004), resulting in the juxtaposition of two coding gene segments and precise joining of the RSSs.
RAG1 serves as the workhorse for DNA cleavage. It contains a DDE motif that coordinates divalent metal ions and catalyzes cleavage, as well as several regions that directly bind to the RSS (Schatz and Swanson, 2011). RAG2, in contrast, is an accessory factor that enables RAG1 cleavage activity and enhances sequence-specific recognition of the RSS by RAG1. RAG2 makes limited direct contact with the DNA and does not contribute directly to enzymatic activity (Swanson, 2004; Zhao et al., 2009). A C-terminal plant homeodomain (PHD) tethers RAG2 to tri-methylated histone H3 lysine 4 (H3K4me3) (Liu et al., 2007; Matthews et al., 2007), a ubiquitous chromatin marker of active transcription start sites (TSSs). Consequently, RAG2 localization to chromatin mirrors the deposition of H3K4me3 (Ji et al., 2010), exhibiting no particular preference for the Ig or Tcr loci over the thousands of other highly expressed genes in lymphocytes.
Potential substrates for RAG-mediated cleavage are remarkably abundant in vertebrate genomes. Such cryptic RSSs (cRSSs) occur an estimated once per 600 bp (Lewis et al., 1997). This can be attributed to the sequence variation tolerated at multiple positions in the heptamer and nonamer, along with the degeneracy of the spacers. In fact, CA repeats (which mimic the heptamer and nonamer, and exist in thousands of copies in mammalian genomes) can support RAG-mediated recombination (Agard and Lewis, 2000; Sakata et al., 2004; Stallings et al., 1991). Lapses in RAG fidelity involving cRSSs have been shown to cause oncogenic translocations and interstitial deletions (Gostissa et al., 2011; Marculescu et al., 2002; Onozawa and Aplan, 2012; Tsai and Lieber, 2010).
Given that RAG2 occupies a significant proportion of the active chromatin landscape, and that cRSSs are strewn about the genome, one might expect that the site-specificity of V(D)J recombination would be enforced through restriction of RAG1 binding to antigen receptor genes. However, here we show that RAG1 is not exclusively targeted to the Ig and Tcr genes but instead colocalizes with RAG2 at thousands of sites. This begs the question: how are these sites protected from RAG cleavage through the multiple rounds of V(D)J recombination required for lymphocyte maturation? Developing B and T cells, by necessity, perch on the edge of genomic instability, and even a single instance of off-target cleavage creates broken DNA ends that can contribute to local rearrangements or long-range translocations.
We postulated that the deleterious effects of mis-targeted RAG activity on whole-organism fitness could have applied selective pressure on vertebrate genomes, gradually suppressing functional cRSSs over evolutionary time. In support of this hypothesis, we show that the sequence environments surrounding RAG1 binding sites exhibit a reduced probability of containing cRSSs, particularly heptamer-like motifs, compared to sites in the genome that are devoid of RAG1 binding. We used genome editing to experimentally “reverse” the cRSS depletion in vivo, converting weak cRSS-like sequences into pairs of consensus RSSs. This created ectopic hotspots for local rearrangement and long-range translocation, highlighting the danger imposed by strong cRSSs in the genome. We propose that the selective pressure exerted by RAG off-target activity has re-shaped the vertebrate genome, buffering against the obligate genotoxic stress that occurs during lymphocyte development.
RESULTS
Our previous analysis of the murine Tcrα, Tcrβ, Igκ, and Igh loci using chromatin immunoprecipitation (ChIP) revealed that the RAG proteins accumulate in a “recombination center” over the J and J-proximal D gene segments (Ji et al., 2010). This analysis left largely unaddressed the questions of if and where RAG1 binds outside of antigen receptor loci. To address these issues, we performed ChIP-seq for RAG1 and RAG2 in total thymocytes and CD19+ B-lineage bone marrow cells from WT or RAG-mutant mice. “D” mice are deficient in endogenous RAG1, but harbor a bacterial artificial chromosome (BAC) expressing a RAG1 catalytic mutant (D708A) that retains its DNA binding properties. Combining “D” with a Tcrβ transgene or with a pre-rearranged B1-8i Igh knock-in allele yielded “Dβ” or “BD” mice, which were used for the analysis of thymocytes (almost exclusively CD4+CD8+ pre-T cells) or B lineage cells (almost exclusively pre-B cells), respectively (Ji et al., 2010). We also analyzed RAG1−/− and RAG2−/− mice bred to contain the Tcrβ transgene or B1-8i Igh allele, as well as core RAG1 (“cR1”) mice (expressing the catalytic core of RAG1, aa 384-1008 of 1040 aa) and “R2ΔC” mice (expressing aa 1-352 of the 527 aa RAG2 protein) that lacks the PHD (Gigi et al., 2014; Liang et al., 2002)). Both lymphocyte development and V(D)J recombination occur with reduced efficiencies in cR1 and R2ΔC mice (Dudley et al., 2003; Liang et al., 2002). Finally, we analyzed human thymocytes to investigate whether RAG binding patterns are evolutionarily conserved.
Recombination centers in high-resolution
Analysis of thymocytes from recombination-incompetent Dβ mice identified the Tcrα and Tcrβ recombination centers as broad peaks of RAG1, RAG2, and H3K4me3 over the 5′ Jα segments (Figure 1A and 1B: i, iv, vii), the Dβ1-Jβ1 segments (Figure S1A), and Dβ2-Jβ2 segments (not shown), consistent with our previous analysis (Ji et al., 2010). The Tcrδ recombination center was tightly focused over Dδ2-Jδ1 (Figure 1C). A similar analysis of B-lineage cells from BD mice confirmed the location of the Igκ (Figure 1D) and Igh (Figure S1B) recombination centers, and identified RAG recruitment hubs in Igλ, which featured strong RAG binding over the Jλ3-Jλ1 region and weaker binding at Jλ2 and Vλ3 (Figure 1E).
Figure 1. Recombination centers of antigen receptor genes.

ChIP-seq (reads per million mapped reads, in raw (unfiltered) form) for RAG1 (blue), RAG2 (green), and H3K4me3 (orange) are shown for selected antigen receptor genes in mouse lymphocytes. (A) The Tcrα/δ locus, (B) Tcrα recombination center (magnified), and (C) TCRδ recombination center (magnified) are shown for thymocytes. (D) The Igκ recombination center (magnified) and (E) Igλ locus are shown for pre-B cells. See also Figure S1.
In WT thymocytes, which are heterogeneous in developmental stage and recombination status of their antigen receptor genes, RAG1, RAG2, and H3K4me3 spread into 3′ Jα segments (Figure 1B: ii, v, viii). This alteration likely reflects the incremental extension of chromatin accessibility to downstream Jα segments as recombination progresses (Hawwari and Krangel, 2007) and the resulting cellular heterogeneity in Jα segment usage. A similar pattern of RAG binding was observed in the human TCRα/δ locus (Figure S1C).
RAG1 occupancy often extended outside of the regions containing the gene segments and their RSSs, spreading along the domain marked by H3K4me3 (Figures 1D, S1A, and S1B). This provided an initial indication that RAG1 recruitment could be dissociated from the RSS, in contrast to the conclusions of our previous study (Ji et al., 2010). Indeed, as we examined the remainder of the genome, we found the presence or absence of RSSs was a poor predictor of RAG1 localization and that most of the rules governing RAG1 binding applied to both the antigen receptor loci and the rest of the genome.
RAG1 binds to thousands of sites in the lymphocyte genome
Our previous study identified genome-wide co-localization of RAG2 and H3K4me3 in thymocytes (Ji et al., 2010). Here, we used a different α-RAG2 antibody to validate this in mouse thymocytes (Figures 1B, 2A–C, and S2A; and Table S1), mouse pre-B cells (Figures 1D–E, 2H), and human thymocytes (Figures S1C and S2C). These datasets confirmed that RAG2 binding patterns are RAG1-independent (Figures S1D: vi, vii; S2A: vi, vii), but require the RAG2 C-terminal region containing the PHD (Figures S1D: x; S2A: x). Since RAG2 exhibits no known catalytic activities, its widespread distribution to active genes seemed relatively innocuous.
Figure 2. RAG1 and RAG2 bind to thousands of sites in the lymphocyte genome.

(A) ChIP-seq for RAG1, RAG2, and H3K4me3 are shown for a region of chromosome 6 in mouse thymocytes (same color coding as in Figure 1). (B) Overlap and (C) correlation between RAG2 and H3K4me3 peaks. (D) Overlap and (E) correlation between RAG1 and H3K4me3 peaks. (F) Overlap and (G) correlation between RAG1 and RAG2 peaks. Boxplots in (D) and (F) show the H3K4me3 and RAG2 RPKM (reads per kb per million mapped reads), respectively, in sections “B” and “C” of the corresponding Venn diagrams. (H) ChIP-seq for RAG1, RAG2, and H3K4me3 are shown for a selected region of chromosome 5 in mouse pre-B cells. (I) Venn diagrams show the overlap between RAG1 and H3K4me3, and between RAG1 and RAG2 in pre-B cells. The sum of H3K4me3 peaks is not equal between (B) and (D) because one RAG peak can overlap with more than one H3K4me3 peak and vice versa. See also Figure S2 and Table S1.
Surprisingly, RAG1 co-localized with a subset of RAG2 and H3K4me3 peaks (Figures 2A, 2D, and 2F; and Table S1), affixing a potent endonuclease to ~3,500 sites in the thymocyte genome. Similarly, RAG1 occupied ~3,400 sites in mouse pre-B cells (Figure 2H, I) and ~1,800 sites in human thymocytes (Figure S2C–E), with the lower number in the latter likely reflecting the weaker ChIP-seq signals obtained. The intensity of RAG1 binding was, in some cases, comparable to that observed in recombination centers (Figure 1B, 2A), and correlated well with the density of RAG2 and H3K4me3 present (Figures 2E and 2G). Given the strong correlation between RAG1 binding and H3K4me3, it is unsurprising that the majority (81%) of RAG1 peaks overlapped with TSSs (Figure 3A). The remaining, TSS-distal peaks (>2 kb away from a TSS) showed lower levels of RAG1 binding, reduced H3K4me3 density, and overlap with active enhancers (H3K4me1+, H3K27Ac+) (Figures 3A–C). The reproducibility of peaks (Figure S1E) and extremely weak signals in our negative controls (RAG1 and RAG2 ChIP-seq in their respective knock-outs; one exception is noted in Figure S2F) (Figures 1B and 2A: iii, vi) provided strong support for the veracity of these RAG1 binding sites. Furthermore, we validated multiple TSS-proximal RAG1 peaks using ChIP-qPCR, a far less sensitive method than ChIP-seq (Figure S2B).
Figure 3. RAG1 binding sites overlap with TSSs and accessible chromatin.

(A) RAG1 and H3K4me3 peaks in mouse thymocytes were characterized by their overlap with TSSs (defined as +/− 2 kb from an annotated gene start position), gene bodies, or intergenic regions. Non-TSS peaks were further categorized as overlapping with active or poised enhancers. (B) RAG1 RPKM and (C) H3K4me3 RPKM are shown for each category of RAG1 peak. (D) Overlap between RAG1, H3K4me3, and accessible chromatin (defined by ATAC-seq) is shown for mouse pre-B cells. See also Figure S3.
The association of RAG1 with active TSSs and enhancers suggested that chromatin accessibility might be important in governing RAG1 binding. To explore this, we compared RAG1 binding sites to areas of accessible chromatin identified by ATAC-seq (Assay for Transposase-Accessible Chromatin) (Buenrostro et al., 2013). This revealed that 80% of RAG1 binding regions in pre-B cells lie within accessible areas (Figure 3D). Furthermore, 84% and 86% of RAG1 binding sites in thymocytes and pre-B cells, respectively, lie within 1 kb of a CpG island (Extended Experimental Procedures), an element associated with promoters and low nucleosome density (Fenouil et al., 2012). Together, our data suggested that high levels of H3K4me3 and chromatin accessibility define a preferred repertoire of genomic binding sites for RAG1.
RAG1 binding was reduced in magnitude and perturbed in thymocytes lacking RAG2 (Figures S1D, S2A, S3A, S3B). Furthermore, in both R2ΔC and cR1 thymocytes, RAG1 binding became less selective, expanding to thousands of additional sites characterized by relatively low levels of RAG1 and H3K4me3 (Figure S3C–H). The interpretation of these observations is uncertain because RAG1 expression is elevated in thymocytes of these genotypes (Figure S1F). RNA-sequencing and micrococcal nuclease-sequencing experiments revealed no clear evidence that widespread RAG binding influences gene expression or nucleosome positioning (Extended Experimental Procedures; Figures S3I, J; and Table S2).
Off-target recombination by RAG appears to be the exception, not the rule
Outside of one indispensable characteristic (the 5′ CAC of the heptamer) and a preference for an AT-rich central nonamer region (Swanson, 2004), RAG substrate recognition accommodates sequence variation in both the heptamer and nonamer. As a consequence, the mouse and human genomes are predicted to contain millions of cryptic RSSs with at least a low level of function (Lewis et al., 1997; Merelli et al., 2010).
Using our genomic maps of RAG1 binding as a starting point, we investigated the extent of RAG1-mediated rearrangements in the genome. The RSS Information Content (RIC) algorithm (Cowell et al., 2002), was used to query the top 100 RAG1 binding peaks in mouse thymocytes for cryptic 12- and 23-RSS pairs passing established thresholds and falling within one kb of the peak boundaries. Three examples (Rag1, Cd8a, and Ets2) were tested for local rearrangements in mouse thymocytes using PCR assays designed to detect the expected deletional or inversional recombination product (Figure 4A–C and Figure S4A). Despite the robust accumulation of RAG1, RAG2, and H3K4me3 near these cRSSs, recombination events were not detected for any of the pairs, even after assaying millions of thymocyte genomes (Extended Experimental Procedures).
Figure 4. Off-target cleavage by RAG is rare and restricted by ATM.

RAG1, RAG2, and H3K4me3 ChIP-seq data are shown for (A) Rag1, (B) Cd8a, (C) Ets2, (F) Notch1, and (G) Bcl11b in WT mouse thymocytes. The 12-cRSSs and 23-cRSSs are indicated by open and filled triangles, respectively. Small arrows represent PCR primers used to assay for RAG-dependent deletions or inversions, and thick lines represent gene bodies. (D and E) The intrinsic recombination activity of selected cRSSs was assayed using a plasmid substrate, diagrammed above each panel. Arrows, PCR primers for recombination assay; position 1, reference cRSS (either the Lmo2 12-cRSS or a modified Ttg1 23-cRSS, open triangle); position 2, test 12- or 23-cRSS (red open triangle); position 3, appropriate consensus partner RSS (filled black triangle). Recombination activity (mean +SEM) of each test cRSS is shown relative to that of the reference cRSS. RAG-mediated deletions at the mouse (H) Notch1 and (I) Bcl11b loci were detected using a nested PCR assay. The frequency of deletions (mean ±SEM) is shown for WT, Rag1−/−, Atm−/−, H2ax−/, Mdc1−/−, and P53−/− thymocytes. Each point represents data generated from one mouse. Unpaired t-test, *p ≤ 0.05, *** p ≤ 0.001, ns = not significant. See also Figure S4 and Table S3.
We used a plasmid substrate to measure the intrinsic recombination potential of the tested cRSSs when paired with a consensus partner RSS (Figures 4D, E). The cRSSs were tested in competition with a known cRSS (Figure 4), which facilitated quantitation and comparisons of activity (Agard and Lewis, 2000). Recombination was assessed by PCR after transfection into 293T cells (together with RAG expression vectors), with activity expressed relative to that of the internal reference cRSS. The cRSSs present in the Rag1 and Cd8a loci showed very low recombination activity in this assay, consistent with their lack of activity in vivo, while Ets2 contained an active 12-cRSS but a weak 23-cRSS (Figure 4D, E). The two Ets2 cRSSs showed no detectable recombination activity when paired with one another in a plasmid recombination assay (Figure S4D), consistent with their inactivity in vivo.
Prior studies of lymphoma-associated chromosomal rearrangements identified a small number of active cRSS pairs, including those located in the mouse Notch1 and Bcl11b loci (Sakata et al., 2004; Tsuji et al., 2004). The 5′ end of the Notch1 locus features low levels of RAG1 occupancy (ranked 846th among RAG1 binding sites in thymocytes), along with a pair of cRSS that span ~12 kb around the TSS (Figure 4F). Even though the RIC scores of these two cRSSs fall below those of Rag1, Cd8a, and Ets2 (Figure S4A), we detected Notch1 deletions at a frequency of ~2.5 × 10−6 in wild-type thymocytes (Figures 4H and S4B), consistent with prior studies (Tsuji et al., 2004). The Notch1 deletion junctions showed hallmarks of classical coding joints (Table S3). Repeated isolation of identical junction sequences suggested that clones bearing individual deletion events had expanded in vivo, obscuring the true deletion frequency.
At Bcl11b (ranked 101st in RAG1 binding density), a ~64 kb deletion takes place between a defined 12-cRSS and one of several 23-cRSSs that arise within a CA-rich region near the promoter (Figures 4G and S4A). We detected these events in wild type thymocytes at a frequency of ~2 × 10−5 (Figures 4I and S4C), consistent with prior reports (Sakata et al., 2004). In contrast to the pauciclonal junctions observed for Notch1 deletions, the coding joints formed at Bcl11b were highly diverse (Table S3). Analysis of the Bcl11b coding and signal joints identified multiple 23-cRSSs within the CA-rich region (Table S3), demonstrating that a CA repeat region can create multiple substrates for off-target recombination.
The intrinsic recombination activity of each Notch1 and Bcl11b cRSS was assessed in the plasmid recombination assay (Figure 4D and 4E). The Notch1 cRSSs showed weak activity while the Bcl11b cRSSs were more active, in line with their respective recombination activities in vivo. However, when paired with one another in a plasmid recombination assay, the two Bcl11b cRSSs showed no detectable activity (Figure S4D), suggesting that the ability to detect their activity in vivo may reflect biological selection. Such selection operates on the Notch1 deletion, which allows for production of a constitutively active form of the Notch1 protein (Ashworth et al., 2010).
Cryptic recombination increases in the absence of ATM
The Ataxia-Telangiectasia Mutated kinase (ATM) mediates the cellular response to DNA double-stranded breaks (DSBs) (Shiloh and Ziv, 2013). Lymphocytes lacking ATM resolve V(D)J recombination intermediates less proficiently, allowing broken ends to accumulate and participate in hybrid joint formation (Bredemeyer et al., 2006) and chromosomal translocations (Helmink and Sleckman, 2012; Zhang et al., 2012). Notably, Notch1 and Bcl11b deletion frequencies increased approximately two-fold in Atm−/− mouse thymocytes (Figures 4H and 4I). This effect depended more strongly on ATM than on H2AX and MDC1, two phosphorylation targets of ATM involved in the DSB repair response. ATM-dependent apoptosis is unlikely to counteract cRSS-mediated rearrangements, since p53−/− and WT thymocytes showed similar frequencies of Notch1 and Bcl11b deletions (Figures 4H and 4I).
To determine if ATM deficiency also sensitized thymocytes to other rare cryptic recombination events, we tested Atm−/− mouse thymocytes for RAG-mediated recombination of the cRSSs at Rag1, Cd8a, and Ets2. No events were detected, again after screening millions of genomes (Extended Experimental Procedures). Efforts to detect hybrid joint formation at Cd8a and Ets2 in Atm−/− thymocytes were equally unsuccessful (data not shown). Thus, ATM deficiency did not unmask cRSS recombination at these three loci.
Cryptic RSSs and heptamers, but not nonamers, are depleted from RAG1 binding sites
We hypothesized that the fitness disadvantage conferred by cRSS-mediated rearrangements had applied selective pressure during vertebrate evolution against the presence of cRSSs near sites of RAG1 recruitment. We first asked whether the regions encompassed by RAG1 binding peaks (outside of antigen receptor genes) had a lower incidence of cRSSs than similarly sized but randomly chosen regions of the genome. Indeed, RAG1+ regions from mouse pre-B cells and thymocytes exhibited a 25–48% lower density of 12- and 23-cRSSs than random segments of the genome (Figure 5A). This demonstrates that co-binding of RAG1 and RAG2 occurs in regions of the genome that are relatively depleted of potential cleavage sites.
Figure 5. Sites of RAG1 binding are preferentially depleted of cryptic RSSs.

(A) The density of 12- and 23-cRSSs was calculated for regions of the genome containing a RAG1 binding peak (blue) in mouse thymocytes (top) or pre-B cells (bottom) compared to randomly assigned (Shuffle, gray) genomic regions (10,000 shuffles ± standard deviation). (B) The densities of 12-cRSSs, 23-cRSSs, and CA dinucleotides were plotted across a 10 kb region centered on the TSS in mouse lymphocytes. RAG1+ regions include those bound by RAG1 in either mouse thymocytes or pre-B cells. (C) The 12-cRSS content in RAG1+ H3K4me3+ regions in mouse thymocytes (left) and pre-B cells (right) was compared to the 12-cRSS content in comparable H3K4me3+ regions of non-lymphoid tissues. Cumulative distribution plots show the fraction of regions that contain more than a threshold number (x-axis) of 12-cRSSs. A shift to the left indicates depletion; a shift to the right indicates enrichment. Similar analyses were done for (D) 23-cRSSs, (E) high scoring heptamers, and (F) high scoring nonamers. Densities of high scoring heptamers and nonamers in RAG1+ and RAG1− regions are plotted relative to TSSs for (G) mouse lymphocytes and (H) human thymocytes. (I) RAG1 and H3K4me3 ChIP-qPCR (mean + SEM, n=3) in STI-571-treated Rag1−/− v-abl cells reconstituted with a retrovirus expressing no RAG1 (empty), WT RAG1, or RAG1 NBD mutant (NBDm). See also Figure S5 and Table S4.
Is this driven by the presence of RAG, or is it simply a side effect of the types of genomic regions bound by RAG1? For instance, TSSs and their flanking promoters might have a reduced incidence of cRSSs simply because of functional constraints inherent in these regions. To control for this, we compared the density of cRSSs surrounding RAG1+ and RAG1− TSSs, which revealed that the area flanking TSSs as a whole did indeed have a lower incidence of cRSSs than TSS-distal regions (Figure 5B). Importantly, however, cRSS density was lower in RAG1+ TSSs (n=4,087; combined list from thymocytes and pre-B cells) compared to RAG1− TSSs (n=16,412), a phenomenon that also extended to CA density (Figure 5B). Hence, RAG1+ TSSs show a substantial depletion of both cRSS and CA density beyond that of other TSSs.
This analysis did not take into account certain features of RAG1+ TSSs, namely elevated CpG and GC content (Figures S5A–B). Such differences in base composition between the RAG1+ and RAG1− groups could contribute to the differences observed. To control for this, we created lists of genomic regions in non-lymphoid cell types that mimicked the RAG1+ regions from pre-B cells and thymocytes in terms of overlap with TSSs (Figure S5C), H3K4me3 levels (Figure S5D), and CpG (Figure S5E) and GC (Figure S5F) content (Extended Experimental Procedures). Our hypothesis predicted that cRSS concentration should be lower in RAG1+ regions in lymphocytes than in closely matched regions from other tissues that do not express RAG. Strikingly, the densities of both 12-cRSSs (Figure 5C) and 23-cRSSs (Figure 5D) were lower in the RAG1+ regions of thymocytes and pre-B cells than in matched control regions of six different non-lymphoid cell types. The differences were small but highly significant, particularly for the 23-cRSS (Table S4).
Given the different functions served by the heptamer (site of cleavage) and nonamer (DNA binding), we hypothesized that the two RSS components were subject to different degrees of selective pressure. We repeated the above analysis but focused on “high scoring” heptamers and nonamers (defined by position weight matrix; Extended Experimental Procedures). This revealed a highly significant reduction in the probability of finding heptamers at RAG1+ regions as compared to control regions in both pre-B cells and thymocytes (Figure 5E), and remarkably, the opposite for nonamers, which were enriched in RAG1+ regions (Figure 5F; Table S4). These phenomena were also evident in a TSS-centered analysis, where heptamers were less abundant in RAG1+ versus RAG1− regions, while nonamers were more abundant (Figure 5G). Similar results were obtained for RAG1+ versus RAG1− TSSs in human thymus (Figure 5H). This argues for selection during vertebrate evolution focused on depleting the heptamer, consistent with its role in directing DNA cleavage by RAG.
Nonamer enrichment at RAG1+ regions raised the possibility that a portion of RAG1 binding is mediated by direct RAG1-nonamer interactions. To test this, we reconstituted a Rag1−/− Abelson kinase-transformed pre-B cell line (v-abl) with WT RAG1 or a RAG1 mutant (NBDm) in which DNA contact residues in the nonamer binding domain (NBD) were altered (R391A, R393A, R407A). We found that binding of the NBDm protein was invariably decreased (though not ablated) relative to WT RAG1 (Figure 5I). This difference was most prominent at Jκ5, as expected (Ji et al., 2010). Importantly, decreased binding was observed at six different promoter regions (Figure 5I), including three genes with no high-scoring nonamers within 1 kb of the site assayed for binding. Hence, the NBD is important for the association of RAG1 with chromatin and its contribution likely derives from both sequence-specific and sequence-nonspecific interactions with DNA.
RSSs inserted into the genome create ectopic recombination hotspots
Our analyses thus far suggested that substrate availability is an important functional difference between a RAG complex occupying an antigen receptor gene and an “ectopic” RAG complex occupying a non-antigen receptor gene. This predicted that the latter can be converted into a recombination hotspot simply by the presence of a strong RSS pair. To test this, we introduced consensus 12- and 23-RSS pairs into specific sites of the lymphocyte genome (Figure 6A) in a v-abl pre-B cell line, which upregulates RAG and Igκ recombination in response to the v-abl kinase inhibitor STI-571. A site-specific TALEN (Transcription activator-like effector nuclease) and a targeting construct containing point mutations in its 5′ homology arm that converted weak RSS-like motifs into consensus RSSs (Figure 6A) were used to introduce RSSs into a target locus while minimizing additional sequence alterations (Figure S6A). Depending on the region of crossover, the modified allele harbored either a pair of 12- and 23-RSSs (designated as “12/23”), or a single 23-RSS (designated as “23”) (Figure 6A).
Figure 6. Insertion of RSSs into the genome results in ectopic V(D)J recombination.

(A) Strategy for targeted introduction of RSSs into the genome. Diagrammed are the target locus (TALEN cleavage site, red arrow) and targeting construct (blue triangles, LoxP sites; Bsr, Blasticidin resistance gene; T2A, self-cleaving peptide; TK, thymidine kinase; black arrows, PCR primers to detect recombination; open and filled triangles, consensus 12 and 23 RSSs, respectively). Depending on the region of crossover in the 5′ homology arm, indicated by the dashed blue and orange lines, homologous recombination results in either 12/23 or 23-RSSki, respectively. RAG1 binding patterns at the mouse (B) Cd79b locus and (D) Aicda E1 enhancer are shown in v-abl-transformed pre-B cells treated with STI-571 for 48 hours. RAG1, RAG2, and H3K4me3 ChIP-seq data are also shown for pre-B cells from BD mice. The RSSki alleles are diagrammed at the bottom. PCR was used to assay for recombination between the knocked-in RSSs in the (C) Cd79b locus and (E) Aidca E1 enhancer pre- and post-treatment with STI-571. The assay is specific to the RSSki allele, and amplifies a product from both the un-recombined (upper band) and recombined (lower band) loci. Two clones are shown for each RSSki cell line, along with the parental v-abl cell line (WT), where the knock-in allele is not amplified by PCR. Histograms show the percent recombination in each cell line after STI-571 treatment, as determined by a semi-quantitative PCR assay. The effects of ATM inhibition (treatment with either ATM inhibitor [black bars] or DMSO [white bars] in combination with STI-571) on recombination are shown for the (F) Cd79b and (G) Aicda E1 12/23-RSSki cell lines. Two subclones (designated “A” and “B”) are shown for each of the 12/23-RSSki cell lines. See also Figure S6 and Table S5.
We first knocked-in RSSs at Cd79b (ranked 73rd in RAG1 binding density in pre-B cells), which features substantial RAG1 and RAG2 recruitment in both v-abl cell lines and primary pre-B cells (Figure 6B). Upon treatment with STI-571, the Cd79b-12/23 cell lines showed clear recombination activity, whereas the Cd79b-23 cell lines remained inert (Figure 6C). The deletion junctions showed hallmarks of classical V(D)J recombination (Table S5). ChIP experiments confirmed RAG1 recruitment to the 12/23-modified locus (Figure S6B).
We then examined whether a site normally devoid of RAG binding (the E1 enhancer of Aicda) was also susceptible to off-target RAG1 activity. Since Aicda is not significantly expressed at the pre-B cell stage, neither the E1 enhancer nor the gene showed significant RAG recruitment or H3K4me3 deposition in v-abl cell lines or primary pre-B cells (Figure 6D). Paired RSSs placed in the E1 enhancer recombined detectably, albeit at lower levels than at Cd79b, while single 23-RSSs were again inactive (Figure 6E, S6B, and Table S5; note that the E1-12/23-1 line was modified on both E1 alleles). Though recruitment of RAG1 was not as robust at the E1-12/23 locus compared to the Cd79b-12/23 locus (Figure S6B), the presence of recombination events at E1 confirmed that even low levels of ectopic RAG recruitment can prove biologically functional. ATAC-seq revealed a peak of accessibility over E1 in pre-B cells (Figure S6C), suggesting that it is poised for activity in pre-B cells and cell lines, consistent with the finding that Aicda expression is cytokine-inducible in v-abl cell lines (Kumar et al., 2013). Together, these results support the catalytic competence of RAG complexes assembled throughout the genome and indicate that even relatively quiescent areas of the genome are not intrinsically protected from RAG activity.
The observation of increased recombination between Notch1 and Bcl11b cRSSs in the context of ATM deficiency led us to ask if ATM inhibition in the RSSki cell lines would have a similar effect on the knocked-in RSSs. This was indeed the case, as treatment with an ATM kinase inhibitor (ATMi) reproducibly increased recombination of the 12/23 RSSki insertions at the Cd79b and E1 loci (Figure 6F, G). Hence, ATM can regulate recombination of both cryptic and strong RSSs, perhaps through a feedback inhibition mechanism (Steinel et al., 2013).
Breaks at inserted RSSs undergo chromosomal translocations
Since RAG-dependent breaks in the antigen receptor genes are known to participate in chromosomal translocations (Gostissa et al., 2011), we tested the Cd79b and E1 RSSki for translocation activity. In v-abl cells, RAG-induced breaks at Igκ provide an abundant source of potential translocation partners, and ATMi strongly sensitizes the cells to RAG-mediated translocations (Zhang et al., 2012). We readily detected translocations between Jκ gene segments and the RSSki in the Cd79b-12/23 and E1-12/23 cell lines (Figures 7A, B). Detection of translocations depended on both STI-571 and ATMi, as expected (Zhang et al., 2012), and an isolated consensus 23-RSS was inactive for translocation (Figure 7B). Translocations between two coding ends, as well as those involving an RSSki coding end and the Jκ5 signal end (although, curiously, not the Jκ1 signal end), were readily detected and featured imprecise joints with nucleotide additions and deletions (Figure 7B and Table S6). Translocations involving the RSSki signal ends (to a Jκ coding or signal end) were almost never detected, which might reflect the design of the RSSki loci (Extended Experimental Procedures).
Figure 7. RAG-dependent breaks at knocked-in RSSs undergo long-range chromosome rearrangements.

(A) Three loci were tested for translocations (left to right): the RSSki, Igκ, and Hprt (targeted for cleavage by CRISPR/Cas9). Purple, 12RSS coding end; orange, 23RSS coding end; green triangle, Jκ1 23RSS; green rectangle, Jκ1 gene segment (coding end); blue triangle, Jκ5 23RSS; blue rectangle, Jκ5 gene segment (coding end); red arrow, CRISPR/Cas9 cleavage site. (B) RSSki lines were tested for translocation activity using PCR assays to detect junctions between RSSki DSBs and RAG-dependent DSBs originating from Igκ (i-viii) or RAG-independent DSBs generated at Hprt (ix). The outcome of each translocation is diagrammed. 12/23-RSSki (12/23), 23-RSSki (23), and WT cell lines were assayed with or without treatment with STI-571 (STI) and an ATM inhibitor (ATMi). Translocation products were amplified in at least two independent PCR experiments. To illustrate translocations between the CD79b RSSki and Hprt (ix), a set of 24 PCR reactions are shown for either wild-type (WT) cells or a Cd79b 12/23-RSSki cell line. See also Figure S7 and Table S6.
We also examined whether RSSki breaks could translocate to RAG-independent, CRISPR/Cas9-generated DSBs in the Hprt locus. In a Cd79b 12/23-RSSki cell line, this resulted in clear STI-571/ATMi-dependent RSSki–Hprt translocations with nucleotide additions and deletions at the junctions (Figure 7B: ix and Table S6). We were unable to detect such translocations for the E1 12/23 RSSki, presumably due to the lower frequency of RSSki breaks. Together, our data demonstrate unambiguously that a strong 12/23 RSS pair can create an ectopic hub of recombination and translocation activity, whereas an isolated RSS, even in an actively transcribed region, is far less dangerous.
The above results were confirmed by RSSki into the Cd79b locus in an Atm−/− v-abl cell line, revealing robust recombination and translocation activity for the 12/23 RSSki, while single 23 RSSki alleles were inert (Figure S7A, B). To determine whether such an assay would allow detection of recombination between cryptic RSSs, the Notch1 or the Bcl11b cRSS pairs were knocked into the Cd79b or Aicda E1 loci in the Atm−/− v-abl cell line. Despite normal levels of RAG protein expression (not shown), none of these lines yielded detectable recombination after STI-571 induction (Figure S7C). These findings emphasize the low recombination potential associated with cRSSs whose activity remains detectable in vivo.
DISCUSSION
The prior, limited analysis of RAG1 binding at non-antigen receptor genes was consistent with the model that RAG1 association with active chromatin was restricted to regions containing strong RSSs (Ji et al., 2010). This provided an appealing mechanism by which to limit the genotoxic threat of RAG. Our data, however, reveal that this model is not correct and that antigen receptor genes are not exceptional in their ability to recruit RAG1. This fundamentally alters our understanding of the threat to the genome constituted by RAG, and raises questions regarding the evolutionary response to such widespread RAG1/RAG2 binding. Our study begins to answer these questions by characterizing how the genome has adapted to the threat of RAG and the consequences of reversing this adaptation. Our data also provide an important resource for the analysis and explanation of patterns of genome instability in lymphoid malignancies.
Regulation of RAG recruitment
Our data identify a set of rules that govern the steady state association of RAG1 and RAG2 with chromatin. For RAG2, binding is independent of RAG1, correlates tightly with H3K4me3, and appears to be strongly dependent on the RAG2 C-terminal region, in which the critical domain is almost certainly the PHD.
For RAG1, chromatin accessibility appears to be an important factor, as demonstrated by strong correlations between RAG1 binding and active promoters and enhancers, H3K4me3, ATAC-seq, and CpG islands. Open chromatin allows for access to DNA, and RAG1 has substantial non-specific DNA binding activity (Schatz and Swanson, 2011; Zhao et al., 2009). Our finding that mutation of the NBD reduced RAG1 binding even at regions lacking strong nonamers argues that non-specific DNA binding accounts for a substantial portion of the RAG1 binding pattern and is consistent with structural analysis showing sequence-nonspecific interactions between the NBD and DNA (Yin et al., 2009). However, sequence-specific interactions between RAG1 and the nonamer might also enhance the association of RAG1 with chromatin, as suggested by our finding that nonamers are somewhat enriched in the vicinity of RAG1 binding regions. This is consistent with in vivo and biochemical studies demonstrating specific RAG1-nonamer interactions in the absence of a heptamer (Swanson, 2004). In addition, the perturbations in RAG1 binding observed when RAG2 is absent or truncated suggest that the RAG1-RAG2 interaction is also a significant factor in localizing RAG1 to active chromatin. This agrees with a recent modeling analysis that predicted that the RAG complex should localize efficiently to chromatin rich in H3K4me3 (Askary et al., 2014). Finally, given the ability of RAG1 to bind and ubiquitinate histone H3 (Deng et al., 2015; Grazini et al., 2010; Jones et al., 2011), it is plausible that direct RAG1-histone interactions contribute to the observed RAG1 binding pattern.
Our analyses provide a cumulative population description and it is unknown to what extent individual lymphocytes reflect the population averages described by ChIP-seq. We also note that DNA looping between bona fide protein binding sites and distal elements might explain a portion of the weak, TSS-distal RAG1 binding peaks detected.
While we were unable to identify a function for RAG binding in regulating gene expression, it is noteworthy that the catalytic function of RAG is required for the proper development of a subset of natural killer cells and perhaps other non-B/non-T hematopoietic lineages as well (Karo et al., 2014). This implies the involvement of RAG-mediated DNA breaks, and while the sites of such putative cleavage events are unknown, it is appealing to think that colocalization of RAG1 and RAG2 to open transcriptional regulatory regions might be relevant.
Strong ectopic RSSs in the genome create hotspots for genomic instability
Robust recombination and translocation activity of the Cd79b RSSki illustrate the dangers of RSS availability in a pre-existing region of RAG1 recruitment. This is consistent with results obtained using a Tcrβ minilocus inserted into the cMyc first intron, which recombined (Ranganath et al., 2008) and activated the translocation potential of formerly inert cRSSs in its vicinity (Tepsuporn et al., 2014). This insertion, however, differed from ours in its inclusion of antigen receptor gene segments and endogenous RSSs, one of which contained a TATA box and a cFos binding site that recruits RAG (Wang et al., 2008). Even the Aicda E1 enhancer, which is normally devoid of RAG binding, transformed into a recombination and translocation hotspot upon the introduction of paired RSSs. This indicates that de novo sequence-driven RAG recruitment is no less dangerous than the presence of RSSs in a pre-existing RAG binding site. In contrast, a single RSS did not produce detectable levels of local or long-range rearrangement, arguing that neither uncoupled (i.e., single RSS) cleavage nor inter-chromosomal recombination are major contributors to genomic instability, at least in this assay system. Thus, the genome appears to derive substantial protection from the requirement for simultaneous engagement of two RSS substrates for RAG-mediated DSB formation.
Preventing off-target recombination
Multiple mechanisms are likely to protect the lymphocyte genome in the face of widespread RAG binding. The first is the ability of inaccessible chromatin to prevent recombination of even strong RSSs. This is amply illustrated by the inactivity of Tcr loci in developing B cells, of Ig loci in developing T cells, and of all antigen receptor loci in fibroblasts expressing RAG (Cobb et al., 2006). In such instances, not only is RAG likely to have difficulty gaining access to RSSs, but the dearth of H3K4me3 would be predicted to significantly hamper stable RAG recruitment (Askary et al., 2014).
The second mechanism, identified here, is to restrict the availability of RSS substrates in the genome. We provide evidence consistent with selection against cRSSs and heptamer sequences near sites of RAG1 binding, and given our results with the Aicda E1 RSSki, it is plausible that such selection extends to other areas of the genome as well. The extent to which an individual cRSS represents a threat to genome integrity, and thus might be selected against, is likely to depend not only on the linear distance in the genome to a compatible partner cRSS, but also on higher order chromatin architecture and dynamics, which will determine the potential for two sites to interact and support RAG-mediated cleavage (Lucas et al., 2014). Our detection of cRSS-mediated recombination was restricted to Notch1 and Bcl11b, where rare deletion events are detected by virtue of their oncogenic potential (Onozawa and Aplan, 2012). This theme extends to human B cell acute lymphoblastic leukemias harboring an ETV6-RUNX1 translocation, which express RAG and contain a high incidence of recurrent interstitial deletions. Strikingly, the deletion breakpoints are enriched at active promoters and correlate strongly with heptamer motifs without a discernable nonamer (Papaemmanuil et al., 2014). This provides a biological rationale for our observation that evolutionary selection has operated primarily on the heptamer.
Strong cRSSs (with RIC scores comparable to those of antigen receptor gene RSSs) are relatively rare in the mouse genome, and it is even more rare to find a strong 12-cRSS in close proximity to a strong 23-cRSS (Table S4). This likely reflects the stringent requirements for such sequences, which must match the RSS consensus at many heptamer and nonamer positions. The principal danger to genome stability in developing lymphocytes probably derives from weak cRSS sequences, consistent with our finding of a relative depletion of cRSS and heptamers near RAG1 binding sites. Notably, modeling predicts that RAG will dissociate from non-specific DNA sequences more rapidly than it can introduce nicks (Askary et al., 2014), and such a mechanism might help suppress genome instability at the many heptamers that remain in open regions of the genome.
The ATM kinase contributes to a third mechanism that restricts cRSS-mediated recombination, as illustrated by the increase in Notch1 and Bcl11b deletions in Atm−/− versus WT thymocytes. Recent studies support a feedback inhibition model in which RAG-mediated DNA breaks signal through ATM to temporarily down-regulate further RAG-mediated DNA cleavage, helping to protect the genome and enforce allelic exclusion (Hewitt et al., 2009; Steinel et al., 2013). Notably, neither MDC1 nor H2AX were required for feedback inhibition (Steinel et al., 2013), consistent with our findings (Figure 4H, I). This model can also explain the increase we observe in recombination at the RSSki alleles following treatment with ATMi, since the inhibitor would prevent feedback inhibition arising from breaks at Igκ.
Evolutionary adaptation to RAG
The arrival of RAG1 and RAG2, perhaps as components of a transposable element (Thompson, 1995), and their adaptation into the recombinase underlying adaptive immunity, presented a significant evolutionary challenge to vertebrate genome integrity. One possible response could have been to tightly restrict RAG binding to antigen receptor genes, but this might be inherently difficult to accomplish for DNA binding proteins, as illustrated by the promiscuous binding of transcription factors at a multitude of sites of uncertain or no functional relevance (Biggin, 2011). Instead, we propose that an evolutionary response unfolded through gradual selection for mutations that deactivated dangerous ectopic cRSSs, though it is clear that this purifying process is not complete (Papaemmanuil et al., 2014).
EXPERIMENTAL PROCEDURES
ChIP and ChIP-seq
ChIP experiments (assayed either by qPCR or sequencing) were performed as described previously (Ji et al., 2010) using custom monoclonal rabbit antibodies for RAG1 (mAb #23) and RAG2 (mAb #39) (Coster et al., 2012) or a polyclonal rabbit antibody for H3K4me3 (Millipore, 07-475). Taqman qPCR primer and probe sequences can be found in Ji et al., 2010 or in Table S7. ChIP-qPCR signals were expressed as IP/input minus IgG background × 1000. For ChIP-seq, libraries were prepared and sequenced following Illumina protocols. Sequence tags were aligned to the mouse (GRCm38p2/mm10) or human (GRCh37/hg19) genomes using Bowtie (version 0.12.7). Peak calling was performed with MACS 1.4.2. See Extended Experimental Methods for details on data analysis.
Cell lines
v-abl-transformed pre-B cell lines – either wild type (A70.2) (Bredemeyer et al., 2006) or Atm−/− containing an I-SceI substrate integrated in chr18 (Zhang et al., 2012) – were treated with 3 μM STI-571 for 24 or 48 h. RSSki v-abl cell lines were created using TALEN expression plasmids targeting Cd79b or Aicda E1 enhancer (Kieffer-Kwon et al., 2013). The region 5′ of the TALEN cleavage site was scanned for pre-existing heptamer- and nonamer-like sequences, and mutations were designed to convert these RSS-like sequences into consensus or cryptic RSSs, along with 10 bp of coding flank. A 5′ semi-homology arm bearing the desired point mutations (synthesized by Blue Heron Biotechnology) and a 3′ homology arm were cloned into a targeting construct containing a floxed Blasticidin-T2a-TK selection cassette. The TALEN expression vectors and the targeting construct were transfected into v-abl cells by Amaxa nucleofection. Blasticidin-resistant single cell clones were isolated and transfected with a Cre expression plasmid to allow for deletion of the selection cassette. Ganciclovir-resistant single cell subclones were isolated. For descriptions of cell line genotyping, see Extended Experimental procedures.
Supplementary Material
Acknowledgments
The authors thank S. Zhou for assistance with mice, E. Corbett for antibody production, Y. Zhang and F. Alt for the Atm−/− I-SceI v-abl cell line, J. Henao-Meija for the CRISPR/Cas9 guide RNA sequence for Hprt, and S. Smale and J. Langermann for valuable advice. This work was supported in part by an NIH NRSA Institutional Postdoctoral Training Grant (T32 AI007019) and a Cancer Research Institute Irvington Postdoctoral Fellowship (G.T.), R37 AI32524 (D.G.S.), R01 GM0888847 (M.C.), by the Intramural Research Program of NIAMS, NIH (R.C.), and by a Burroughs Wellcome Fund Career Award (L.G.C.). D.G.S. is an investigator of the Howard Hughes Medical Institute. This study used high-performance computational capabilities of the NIH Biowulf Linux cluster.
Footnotes
ACCESSION NUMBERS
ChIP-, RNA-, and MNase-seq datasets are available in the GEO database (XX)
Supplemental information includes Extended Experimental Procedures, 7 figures, and 7 tables.
AUTHOR CONTRIBUTIONS
G.T., R.C., and D.G.S. designed wet lab experiments, which were performed by G.T. G.T., R.C. D.G.S., Y.M., W.R., M.K., and L.G.C. designed computational analyses. Y.M. and W.R. performed computational analyses of ChIP-seq experiments. M.K. and L.G.C. performed computational analyses of cRSS density, and Y.M. performed analyses of heptamer and nonamer density. A.Y. and K.K. generated sequencing libraries and performed sequencing runs. J.Q. created TALEN expression plasmids. M.M. and M.C. generated ATAC-seq data. Y. J. generated the mouse thymocyte H3K4me3 ChIP-seq dataset. E.M. prepared human thymocytes. G.T. and D.G.S. wrote the paper with input from all other authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agard EA, Lewis SM. Postcleavage sequence specificity in V(D)J recombination. Mol Cell Biol. 2000;20:5032–5040. doi: 10.1128/mcb.20.14.5032-5040.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth TD, Pear WS, Chiang MY, Blacklow SC, Mastio J, Xu L, Kelliher M, Kastner P, Chan S, Aster JC. Deletion-based mechanisms of Notch1 activation in T-ALL: key roles for RAG recombinase and a conserved internal translational start site in Notch1. Blood. 2010;116:5455–5464. doi: 10.1182/blood-2010-05-286328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askary A, Shimazaki N, Bayat N, Lieber MR. Modeling of the RAG reaction mechanism. Cell Rep. 2014;7:307–315. doi: 10.1016/j.celrep.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggin MD. Animal transcription networks as highly connected, quantitative continua. Dev Cell. 2011;21:611–626. doi: 10.1016/j.devcel.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC, Nuskey B, Sullivan KE, Pandita TK, Bassing CH, et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature. 2006;442:466–470. doi: 10.1038/nature04866. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb RM, Oestreich KJ, Osipovich OA, Oltz EM. Accessibility control of V(D)J recombination. Adv Immunol. 2006;91:45–109. doi: 10.1016/S0065-2776(06)91002-5. [DOI] [PubMed] [Google Scholar]
- Coster G, Gold A, Chen D, Schatz DG, Goldberg M. A dual interaction between the DNA damage response protein MDC1 and the RAG1 subunit of the V(D)J recombinase. J Biol Chem. 2012;287:36488–36498. doi: 10.1074/jbc.M112.402487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell LG, Davila M, Kepler TB, Kelsoe G. Identification and utilization of arbitrary correlations in models of recombination signal sequences. Genome Biol. 2002;3:RESEARCH0072. doi: 10.1186/gb-2002-3-12-research0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Liu H, Liu X. RAG1-mediated ubiquitylation of histone H3 is required for chromosomal V(D)J recombination. Cell Res. 2015;25:181–192. doi: 10.1038/cr.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley DD, Sekiguchi J, Zhu C, Sadofsky MJ, Whitlow S, DeVido J, Monroe RJ, Bassing CH, Alt FW. Impaired V(D)J recombination and lymphocyte development in core RAG1-expressing mice. J Exp Med. 2003;198:1439–1450. doi: 10.1084/jem.20030627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenouil R, Cauchy P, Koch F, Descostes N, Cabeza JZ, Innocenti C, Ferrier P, Spicuglia S, Gut M, Gut I, et al. CpG islands and GC content dictate nucleosome depletion in a transcription-independent manner at mammalian promoters. Genome Res. 2012;22:2399–2408. doi: 10.1101/gr.138776.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigi V, Lewis S, Shestova O, Mijuskovic M, Deriano L, Meng W, Luning Prak ET, Roth DB. RAG2 mutants alter DSB repair pathway choice in vivo and illuminate the nature of ‘alternative NHEJ’. Nucleic Acids Res. 2014;42:6352–6364. doi: 10.1093/nar/gku295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gostissa M, Alt FW, Chiarle R. Mechanisms that promote and suppress chromosomal translocations in lymphocytes. Annu Rev Immunol. 2011;29:319–350. doi: 10.1146/annurev-immunol-031210-101329. [DOI] [PubMed] [Google Scholar]
- Grazini U, Zanardi F, Citterio E, Casola S, Goding CR, McBlane F. The RING domain of RAG1 ubiquitylates histone H3: a novel activity in chromatin-mediated regulation of V(D)J joining. Mol Cell. 2010;37:282–293. doi: 10.1016/j.molcel.2009.12.035. [DOI] [PubMed] [Google Scholar]
- Hawwari A, Krangel MS. Role for rearranged variable gene segments in directing secondary T cell receptor alpha recombination. Proc Natl Acad Sci U S A. 2007;104:903–907. doi: 10.1073/pnas.0608248104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmink BA, Sleckman BP. The response to and repair of RAG-mediated DNA double-strand breaks. Annu Rev Immunol. 2012;30:175–202. doi: 10.1146/annurev-immunol-030409-101320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt SL, Yin B, Ji Y, Chaumeil J, Marszalek K, Tenthorey J, Salvagiotto G, Steinel N, Ramsey LB, Ghysdael J, et al. RAG-1 and ATM coordinate monoallelic recombination and nuclear positioning of immunoglobulin loci. Nat Immunol. 2009;10:655–664. doi: 10.1038/ni.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Resch W, Corbett E, Yamane A, Casellas R, Schatz DG. The in vivo pattern of binding of RAG1 and RAG2 to antigen receptor loci. Cell. 2010;141:419–431. doi: 10.1016/j.cell.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JM, Bhattacharyya A, Simkus C, Vallieres B, Veenstra TD, Zhou M. The RAG1 V(D)J recombinase/ubiquitin ligase promotes ubiquitylation of acetylated, phosphorylated histone 3.3. Immunol Lett. 2011;136:156–162. doi: 10.1016/j.imlet.2011.01.005. [DOI] [PubMed] [Google Scholar]
- Karo JM, Schatz DG, Sun JC. The RAG Recombinase Dictates Functional Heterogeneity and Cellular Fitness in Natural Killer Cells. Cell. 2014;159:94–107. doi: 10.1016/j.cell.2014.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer-Kwon KR, Tang Z, Mathe E, Qian J, Sung MH, Li G, Resch W, Baek S, Pruett N, Grontved L, et al. Interactome maps of mouse gene regulatory domains reveal basic principles of transcriptional regulation. Cell. 2013;155:1507–1520. doi: 10.1016/j.cell.2013.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Wuerffel R, Achour I, Lajoie B, Sen R, Dekker J, Feeney AJ, Kenter AL. Flexible ordering of antibody class switch and V(D)J joining during B-cell ontogeny. Genes Dev. 2013;27:2439–2444. doi: 10.1101/gad.227165.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SM, Agard E, Suh S, Czyzyk L. Cryptic signals and the fidelity of V(D)J joining. Mol Cell Biol. 1997;17:3125–3136. doi: 10.1128/mcb.17.6.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang HE, Hsu LY, Cado D, Cowell LG, Kelsoe G, Schlissel MS. The “dispensable” portion of RAG2 is necessary for efficient V-to-DJ rearrangement during B and T cell development. Immunity. 2002;17:639–651. doi: 10.1016/s1074-7613(02)00448-x. [DOI] [PubMed] [Google Scholar]
- Liu Y, Subrahmanyam R, Chakraborty T, Sen R, Desiderio S. A plant homeodomain in RAG-2 that binds Hypermethylated lysine 4 of histone H3 is necessary for efficient antigen-receptor-gene rearrangement. Immunity. 2007;27:561–571. doi: 10.1016/j.immuni.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas JS, Zhang Y, Dudko OK, Murre C. 3D trajectories adopted by coding and regulatory DNA elements: first-passage times for genomic interactions. Cell. 2014;158:339–352. doi: 10.1016/j.cell.2014.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marculescu R, Le T, Simon P, Jaeger U, Nadel B. V(D)J-mediated translocations in lymphoid neoplasms: a functional assessment of genomic instability by cryptic sites. J Exp Med. 2002;195:85–98. doi: 10.1084/jem.20011578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews AG, Kuo AJ, Ramon-Maiques S, Han S, Champagne KS, Ivanov D, Gallardo M, Carney D, Cheung P, Ciccone DN, et al. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature. 2007;450:1106–1110. doi: 10.1038/nature06431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merelli I, Guffanti A, Fabbri M, Cocito A, Furia L, Grazini U, Bonnal RJ, Milanesi L, McBlane F. RSSsite: a reference database and prediction tool for the identification of cryptic Recombination Signal Sequences in human and murine genomes. Nucleic Acids Res. 2010;38:W262–267. doi: 10.1093/nar/gkq391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onozawa M, Aplan PD. Illegitimate V(D)J recombination involving nonantigen receptor loci in lymphoid malignancy. Genes Chromosomes Cancer. 2012;51:525–535. doi: 10.1002/gcc.21942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J, Alexandrov LB, Van Loo P, Cooke SL, Marshall J, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet. 2014;46:116–125. doi: 10.1038/ng.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganath S, Carpenter AC, Gleason M, Shaw AC, Bassing CH, Alt FW. Productive coupling of accessible Vbeta14 segments and DJbeta complexes determines the frequency of Vbeta14 rearrangement. J Immunol. 2008;180:2339–2346. doi: 10.4049/jimmunol.180.4.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney S, Chaudhuri J, Alt FW. The role of the non-homologous end-joining pathway in lymphocyte development. Immunol Rev. 2004;200:115–131. doi: 10.1111/j.0105-2896.2004.00165.x. [DOI] [PubMed] [Google Scholar]
- Sakata J, Inoue J, Ohi H, Kosugi-Okano H, Mishima Y, Hatakeyama K, Niwa O, Kominami R. Involvement of V(D)J recombinase in the generation of intragenic deletions in the Rit1/Bcl11b tumor suppressor gene in gamma-ray-induced thymic lymphomas and in normal thymus of the mouse. Carcinogenesis. 2004;25:1069–1075. doi: 10.1093/carcin/bgh094. [DOI] [PubMed] [Google Scholar]
- Schatz DG, Swanson PC. V(D)J recombination: mechanisms of initiation. Annu Rev Genet. 2011;45:167–202. doi: 10.1146/annurev-genet-110410-132552. [DOI] [PubMed] [Google Scholar]
- Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14:197–210. [PubMed] [Google Scholar]
- Stallings RL, Ford AF, Nelson D, Torney DC, Hildebrand CE, Moyzis RK. Evolution and distribution of (GT)n repetitive sequences in mammalian genomes. Genomics. 1991;10:807–815. doi: 10.1016/0888-7543(91)90467-s. [DOI] [PubMed] [Google Scholar]
- Steinel NC, Lee BS, Tubbs AT, Bednarski JJ, Schulte E, Yang-Iott KS, Schatz DG, Sleckman BP, Bassing CH. The ataxia telangiectasia mutated kinase controls Igkappa allelic exclusion by inhibiting secondary Vkappa-to-Jkappa rearrangements. J Exp Med. 2013;210:233–239. doi: 10.1084/jem.20121605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson PC. The bounty of RAGs: recombination signal complexes and reaction outcomes. Immunol Rev. 2004;200:90–114. doi: 10.1111/j.0105-2896.2004.00159.x. [DOI] [PubMed] [Google Scholar]
- Tepsuporn S, Hu J, Gostissa M, Alt FW. Mechanisms That Can Promote Peripheral B-cell Lymphoma in ATM-Deficient Mice. Cancer Immunol Res. 2014;2:857–866. doi: 10.1158/2326-6066.CIR-14-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CB. New insights into V(D)J recombination and its role in the evolution of the immune system. Immunity. 1995;3:531–539. doi: 10.1016/1074-7613(95)90124-8. [DOI] [PubMed] [Google Scholar]
- Tsai AG, Lieber MR. Mechanisms of chromosomal rearrangement in the human genome. BMC Genomics. 2010;11(Suppl 1):S1. doi: 10.1186/1471-2164-11-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji H, Ishii-Ohba H, Katsube T, Ukai H, Aizawa S, Doi M, Hioki K, Ogiu T. Involvement of illegitimate V(D)J recombination or microhomology-mediated nonhomologous end-joining in the formation of intragenic deletions of the Notch1 gene in mouse thymic lymphomas. Cancer Res. 2004;64:8882–8890. doi: 10.1158/0008-5472.CAN-03-1163. [DOI] [PubMed] [Google Scholar]
- Wang X, Xiao G, Zhang Y, Wen X, Gao X, Okada S, Liu X. Regulation of Tcrb recombination ordering by c-Fos-dependent RAG deposition. Nat Immunol. 2008;9:794–801. doi: 10.1038/ni.1614. [DOI] [PubMed] [Google Scholar]
- Yin FF, Bailey S, Innis CA, Ciubotaru M, Kamtekar S, Steitz TA, Schatz DG. Structure of the RAG1 nonamer binding domain with DNA reveals a dimer that mediates DNA synapsis. Nat Struct Mol Biol. 2009;16:499–508. doi: 10.1038/nsmb.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, McCord RP, Ho YJ, Lajoie BR, Hildebrand DG, Simon AC, Becker MS, Alt FW, Dekker J. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 2012;148:908–921. doi: 10.1016/j.cell.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Gwyn LM, De P, Rodgers KK. A non-sequence-specific DNA binding mode of RAG1 is inhibited by RAG2. J Mol Biol. 2009;387:744–758. doi: 10.1016/j.jmb.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.