

Introduction
I was born in Mexico City in 1942 into a family immersed in literature and art, but with no proclivity toward the sciences. My father, Bernard Ortiz de Montellano, was a poet of some standing in Mexico, an academic, and the editor of a magazine called Contemporaneos, which reported on current literary and artistic trends. In this last capacity, he was well related to the cadre of artists and writers, such as Diego Rivera and Rufino Tamayo, who were bringing Mexico into the world art forum. My mother was an adventurous American from Missouri who had ventured into Mexico in wild and woolly post-revolutionary days to earn a master's degree in Spanish and stayed to marry one of her professors. I was the second of three children. My brother, who preceded me by four years, was named Bernard Juan Ramon in honor of the Nobel Laureate and poet Juan Ramón Jiménez. My sister, Ana Luisa, was born one year after I was. Although in our childhood tiffs I usually allied with my sister against my older brother, in the long term, it was my brother who exerted the greatest influence on my life and career.
The death of my father in 1949 when I was six years old led my mother to move our family to the American Smelting and Refining complex in San Luis Potosí. There she became the sole teacher at the school established by the company for the children of senior employees who wanted them to have an American education. For four years, through the eighth grade, my sister and I had what amounted to an extended at-home education. However, when the time came for my sister and me to go to high school, my mother took the family back to the United States. That move in 1956 placed us in San Antonio, Texas, where my mother became one of the Spanish teachers at Alamo Heights High School, the same school that my sister and I attended. My interest in things mechanical and scientific developed during my high school years, although this was also the period, during one summer, that I avidly pursued the goal of reading a new historical novel every day. When graduation came in 1960, my sister emerged as the class valedictorian with a scholarship to Smith College and I with a McDermott scholarship to the Massachusetts Institute of Technology. This was a godsend, as a high school teacher's salary in Texas was woefully inadequate to support two students at East Coast universities. In my application, I had indicated an interest in pursuing chemistry as a major largely based on the fact that my brother was starting his Ph.D. work in chemistry at the University of Texas. This was not a particularly rational basis for a career choice, but fortunately, chemistry turned out to be the right place for me.
My four years at the Massachusetts Institute of Technology were challenging, stimulating, exhausting, and rewarding. I lived in Student House, a cooperative group where we reduced housing and food costs by doing most of the housework ourselves. I loved my courses in chemistry, mathematics, and the humanities, but had a more difficult time with physics. My first experience with research came during my senior year, when I took up a senior thesis project supervised by Professor Klaus Biemann on the mechanism of a mass spectrometric rearrangement. I was delighted in the same year to be accepted by the chemistry department of Harvard University for graduate studies. This was doubly fortunate, as Kirby Adams, whom I had started dating in high school, had followed my sister to Smith College and still had a year to go for her degree. In the event, we were married in 1965 after my first year of graduate school.
My real immersion in research began when I joined the group of E. J. Corey as a graduate student. My initial project was to try to trap cyclobutadiene in a spectroscopically detectable state by photolysis of 2-pyrone in a transparent frozen matrix. This project was set aside, however, when I joined Bill Russey, a graduate student in his final months at Harvard, in an effort to determine whether 2,3-oxidosqualene was a physiological intermediate in the conversion of squalene to lanosterol. The truly spectacular transformation of squalene to lanosterol involves addition of an oxygen atom, assembly of the four rings of the sterol skeleton, and migration of a methyl group, all with the stereospecific introduction of seven chiral centers. This transformation was generally viewed as a reaction triggered by oxygen addition to the internal carbon of the terminal double bond of squalene with concomitant generation of a cation at the other carbon of the double bond. However, in 1966, we were able to establish that the reaction proceeded in two distinct steps catalyzed by two distinct enzymes. In the first step, an enzyme regio- and stereospecifically produced the 2,3-epoxide of squalene (1). A second enzyme then catalyzed opening of the epoxide to give a cation that initiated the ring cyclizations and other reactions leading to lanosterol. A collaboration with Peter D. G. Dean, a postdoctoral fellow in the laboratory of Konrad Bloch, led to the identification, purification, and initial characterization of this second enzyme, which we named 2,3-oxidosqualene cyclase (2). As part of this effort, I was introduced to the joys of the slaughterhouse, where a liver taken from a freshly slaughtered pig hanging on a hook was directly dumped into my ice bucket. My subsequent work in the Corey laboratory focused on both the dissection of the mechanism of lanosterol formation by modifying the 2,3-oxidosqualene structure and the development of inhibitors of the enzyme. Competition is always a good stimulus, and our efforts were spurred on by the fact that the laboratory of E. E. van Tamelen at Stanford University was investigating the same process. It was during this period that I gave a seminar at the Massachusetts General Hospital in Boston and was asked during the question period whether the epoxidation of squalene was mediated by a cytochrome P450 enzyme. As we had not focused on the epoxidation reaction, I did not know (it is not), but in truth, this extended to the fact that I had never heard of cytochrome P450. Thus, I was glancingly introduced to what eventually became a central theme of my research career.
While I was a graduate student at Harvard, I first experienced the satisfaction of independently conceiving, planning, and carrying out a study. If 2,3-oxidosqualene was the precursor of lanosterol in animals, might it not also be the precursor of complex triterpenes in plants? Working on weekends, I was able to establish that 2,3-oxidosqualene was indeed an intermediate in the conversion of squalene to β-amyrin in etiolated pea seedlings. I was ecstatic to go into Professor Corey's office one day with a nearly complete story and a draft of the communication that was published in 1967 in the Journal of the American Chemical Society (3). Subsequent work by many laboratories has established the universal role of 2,3-oxidosqualene as the precursor of polycyclic triterpenes with a 3-hydroxyl group.
I was keen to do postdoctoral work in Europe and was delighted when I was accepted by Duilio Arigoni of the Eidgenössische Technische Hochschule (ETH) in Zürich and doubly so when I was awarded a NATO postdoctoral fellowship to support my stay in his laboratory. Thus, in the summer of 1968, a time when the world was in turmoil over the Vietnam war, I submitted my doctoral dissertation and flew to London, where my wife and I took delivery of a red Volvo that was to serve us as transportation (and sometimes home). The first stop was Bad Reichenhall in Bavaria, where we spent six weeks at the Goethe-Institut with the hope that an immersion crash course in German would help ease our way into German-speaking Switzerland. The rudiments of the German language that we acquired there provided a good basis for further study of the language, but our naïveté was exposed when we discovered that German and Swiss German were quite different and that the daily conversational traffic in Zürich was mostly in Swiss German.
I was able to spend only one year as a postdoctoral fellow at ETH because I had already accepted a position with Syntex in Mexico City with the understanding that I would take up the position after a year of postdoctoral work. In Zürich, I worked on the biosynthesis of pleuromutilin, an antibacterial sesquiterpene. But a year passed quickly and I had to leave midway through the project. I did, however, modestly improve my German, learn to ski, develop a love for opera, and discover the delights of Fasching celebrations in Munich.
One of the reasons Syntex was founded in Mexico City was its proximity to the source of cabeza de negro, a plant that was the source of diosgenin, a precursor of most steroidal drugs. However, by 1969, the bulk of Syntex research had moved to Palo Alto, California, leaving in Mexico City some manufacturing facilities and a small cadre of experienced synthetic chemists under the direction of Pierre Crabbé. Two international postdoctoral fellows were also brought in each year, one of whom was assigned to me in my initial role as a synthetic chemist. However, as Pierre Crabbé was interested in returning to an academic position in Europe, there was a clear possibility that I could inherit his role as director of the group on his departure. Unfortunately, the late sixties and early seventies were a time of political and social turmoil, and I soon realized that I would not find personal fulfillment in Mexico City. Therefore, a year into my appointment with Syntex, I began searching for an academic position in the United States, and as a step in that direction, I accepted a transfer to the Palo Alto site in 1971. As fate would have it, one of the scientists who interviewed for a position in Mexico was from the University of Washington. I had mentioned to him my interest in an academic post, and he was kind enough to write to inform me that the University of Washington had just hired a faculty member from the School of Pharmacy at the University of California, San Francisco (UCSF), creating a vacancy at that institution. I immediately applied and within weeks was offered the position with an almost immediate starting date. Thus began my long association with UCSF.
Mechanism-based Inactivation of Cytochrome P450
In shifting from an industrial setting, where research goals were largely set by management, to starting my own laboratory, I suddenly had to decide where to begin my independent research career. The first project I chose was to study the biosynthesis of squalene from farnesyl pyrophosphate, with the goal of making mechanism-based inhibitors of the enzyme squalene synthase. This was a practical choice because it made use of my specific graduate training, but focused on a different step in the cholesterol biosynthetic pathway. A practical outcome of this choice was that my first application to the National Institutes of Health, which focused on this work, was funded. Shortly afterward, I started a second project with Amrit Boparai, one of my first two graduate students. The goal of this project was to determine whether the hydrolysis of epoxides by epoxide hydrolase could be intercepted by placing a nucleophilic group on the compound, such as a hydroxyl, at an appropriate distance from the epoxide. The hypothesis was that the biosynthesis of furans in many natural products could arise by such an intramolecular cyclization. This possibility was attractive because it was thought that epoxide hydrolase involved backside addition of a water molecule to the epoxide as it was protonated by the enzyme to promote epoxide ring opening. However, the only products we obtained were the conventional diols (4). The explanation for this failure was provided many years later, when Richard Armstrong at Vanderbilt University and Bruce Hammock at the University of California, Davis, independently showed that epoxide hydrolase has a more complex mechanism in which a protein carboxyl group first adds to the epoxide to give a covalent intermediate that is subsequently hydrolyzed to release the diol (5, 6). It is amusing that an epoxide hydrolase that catalyzes a cyclization reaction such as what we were looking for was reported recently (7). In any case, our failure led to reformulation of the hypothesis and, serendipitously, to the first major discovery of my laboratory.
If epoxide hydrolysis could not be intercepted to produce cyclic products, perhaps the cytochrome P450-catalyzed epoxidation reaction itself could be interrupted by intramolecular trapping of the transition state by an appropriately placed nucleophile. In researching the relevant literature, I stumbled across references to the biological effects of 2-isopropyl-4-pentenamide (commonly known as allylisopropylacetamide). This sedative-hypnotic agent was converted metabolically to both a diol and a cyclized lactone product, the first one being the expected epoxide hydrolysis product and the latter the expected product from intramolecular trapping of the incipient epoxide by the amide group. Intriguingly, its metabolism was known to be associated with loss of both cytochrome P450 activity and heme and with the concomitant hepatic accumulation of a mysterious green pigment. As part of our study of this process (Fig. 1), we gradually pared down the 2-isopropyl-4-pentenamide structure and, to our surprise, found that ethylene gas itself caused P450 loss and accumulation of green pigments (8)! Further work established that the terminal carbon of the olefin was attached to one of the heme nitrogen atoms (Fig. 2) in a reaction that introduced a hydroxyl at the other carbon of the olefin (9). Monosubstituted acetylenes, such as those present in the ethinyl sterols used in contraceptive pills, inactivated P450 enzymes by a similar mechanism, except that the internal carbon of the unsaturated bond that was oxidized appeared as a ketone rather than as an alcohol (10). The iron atom was readily lost from N-alkylporphyrins, giving rise to N-alkylprotoporphyrin IX adducts that corresponded to the green pigments that accumulated in rodents treated with agents such as these. Thus, what started out in 1978 as an investigation of the possible intramolecular interactions of two functionalities in a substrate during its catalytic oxidation evolved a year later into a fundamental study of the nature and mechanism of the inactivation of cytochrome P450 enzymes by terminally unsaturated agents.
FIGURE 1.

Students and postdoctoral fellows in my laboratory when we entered the cytochrome P450 and heme world. Seated from left to right: Kathryn Prickett, Paul Ortiz de Montellano, and Dianne Jassawalla. Standing from left to right: Wayne Vinson, Kent Kunze, Bruce Mico, Gary Yost, and Stephen Dinizo. Kent, Bruce, Gary, and Stephen were the founding members of the P450 team. Kathryn, Dianne, and Wayne worked on squalene biogenesis.
FIGURE 2.
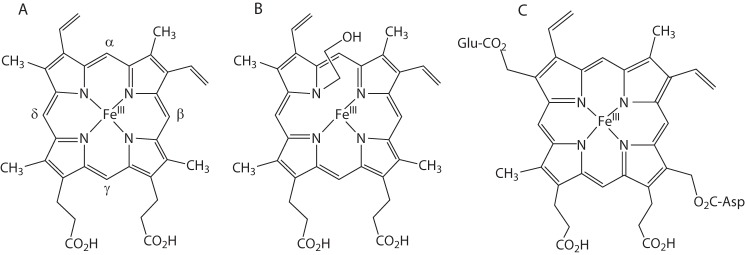
A, structure of ferric heme b, with the four meso-positions labeled. B, structure of the N-alkylheme adduct formed in the inactivation of cytochrome P450 by ethylene. C, structure of the ester links to the prosthetic heme group found in all mammalian peroxidases.
The discovery that the heme group of P450 enzymes could be alkylated by metabolically generated reactive species was an open invitation, particularly for someone with a chemistry background, to search for other functionalities that would similarly inactivate P450 enzymes. One of the earliest and most satisfying outcomes of this search stemmed from my attendance at a meeting in Brighton, England, in 1970 while I was working with Syntex in Mexico City. At that meeting, I attended a lecture by Charles W. Rees on the chemistry of 1-aminobenzotriazoles, which described their conversion to benzynes by inorganic oxidizing agents. In view of the oxidative nature of cytochrome P450, we examined the reaction of 1-aminobenzotriazole with liver microsomal P450 enzymes and determined that a benzyne-like intermediate was formed that bound covalently across two of the porphyrin nitrogens of the heme prosthetic group to give an ortho-disubstituted phenyl-bridged adduct (Fig. 3) (11). Indeed, 1-aminobenzotriazole inactivates most drug-metabolizing P450 enzymes both in vitro and in vivo, without inactivating the biosynthetic P450 isoforms. It has proven to be of practical utility and is widely used in industry and academia to test in vivo for the involvement of P450 enzymes in metabolism. Well then, if benzyne, a true chemist's molecule, was active, why not cyclobutadiene? As a graduate student, I was not able to trap photochemically generated cyclobutadiene in a frozen matrix. However, P450-catalyzed oxidation of 2,3-bis(carbethoxy)-2,3-diazabicyclo[2.2.0]hex-5-ene, anitrogen-containing cyclobutadiene precursor, satisfyingly resulted in addition of a cyclobutenyl function to one of the heme nitrogen atoms (12). Among the numerous other functions we found that catalytically inactivated cytochrome P450 enzymes were 1,2,3-benzothiadiazoles (13), arylhydrazines (14), and 3-substituted sydnones (15). Furthermore, through the efforts of several laboratories, including mine, it was established that functions that bind covalently to the heme group upon catalytic oxidation, such as terminal olefins and acetylenes, can also, through the competitive formation of electrophilic metabolites, result in covalent binding to the protein itself.
FIGURE 3.

The porphyrin formed from the prosthetic heme group in the cytochrome P450-catalyzed oxidation of 1-aminobenzotriazole to benzyne. The methyl, vinyl, and propionic acid substituents on the heme periphery are not shown. Reprinted with permission of the Biochemical Society from Reference 11.
Heme N-Alkylation and Porphyria
It was known in the 1970s that administration of many drugs could trigger episodes of porphyria in genetically susceptible individuals. When administered to normal rodents, some of these agents could engender biochemical states comparable to those characteristic of the human porphyrias. The porphyrias are disorders in which the flux through the heme biosynthetic pathway exceeds the catalytic capacity of one or more enzymes in the pathway, resulting in the accumulation of intermediates that are neuro- or phototoxic (16). In the human porphyrias, the activity of a specific enzyme is genetically depressed to the point that the normal throughput of the heme biosynthetic pathway exceeds the capacity of the enzyme or does so after heme biosynthesis is stimulated through induction of the pathway by heme depletion. One mechanism for heme depletion, of course, is to destroy the heme group of cytochrome P450 enzymes. This mechanism is often coupled with the fact that many lipophilic compounds are inducers of cytochrome P450 enzymes, which itself increases the demand for heme. However, an even more powerful mechanism was found to operate with agents such as 3,5-dicarbethoxy-2,4,6-trimethyl-1,4-dihydropyridine. In work dating back as early as the 1950s, this compound had been identified as a potent agent for the induction of porphyria in rodents by Francesco De Matteis, then at the Medical Research Council Toxicology Research Unit in Carshalton, United Kingdom, Gerald Marks at Queen's University in Canada, and other investigators. In 1981, we demonstrated that cytochrome P450 oxidation of this agent resulted in N-methylation of the prosthetic group of the enzyme, with eventual loss of the iron to produce N-methylprotoporphyrin IX (17). N-Methylprotoporphyrin turns out to be a highly potent inhibitor of ferrochelatase, the enzyme responsible for the final step of heme synthesis, in which an iron atom is inserted into the protoporphyrin IX skeleton. This results in blockage of the heme biosynthetic pathway, triggering a porphyria-like state, even in normal rodents.
In many cases, heme N-alkylation within a hemoprotein appears to involve catalytic formation of a carbon radical that binds to the ferrous iron atom and then, on oxidation of the iron to the ferric state, shifts to form a covalent bond with one of the pyrrole nitrogens of the porphyrin skeleton. The clearest demonstration of this is provided by the reactions of myoglobin and cytochrome P450 with aryldiazenes (ArNH=NH) and arylhydrazines (ArNH2NH2). As shown by NMR spectroscopy and x-ray crystallography for myoglobin, phenyldiazene (from phenylhydrazine) produces a stable phenyl-iron adduct in which the phenyl group is bound perpendicular to the heme plane by attachment of one of its carbons to the heme iron atom (18, 19). Upon aerobic denaturation of the protein under acidic conditions, the phenyl shifts to one of the porphyrin nitrogens to form N-phenylprotoporphyrin IX. Similar reactions are observed with cytochrome P450 enzymes (14), although the aryl-iron complexes of these proteins are less stable and sometimes rearrange without a requirement for prior denaturation of the protein. We were able to use the regiospecificity of the shift of the aryl group to the four distinct nitrogens of the porphyrin ring to probe the gross topologies of the distal substrate-binding sites of P450 enzymes (21). We worked with the assumption that the phenyl group would shift to the porphyrin nitrogens that were least sterically hindered by the protein structure.
Heme Modification and Peroxidases
A different type of heme reactivity is seen in the reactions of peroxidases with alkyl- and arylhydrazines, which do not form detectable aryl- or alkyl-iron complexes and N-alkyl- or N-arylporphyrin adducts. Instead, in 1987, we showed that they undergo addition of a catalytically generated substrate radical to one of the meso-carbons of the heme (22). This reactivity and its specific predilection for regiospecific addition of the alkyl or aryl group to the heme δ-meso-carbon led me to propose in 1997 that peroxidases differ critically from P450 enzymes in that access to the region above the heme is hindered in the peroxidases (23). Consequently, the oxidation of substrates by peroxidases involves electron transfer to the heme edge of the oxidatively activated heme complex; in P450 enzymes, the substrate binds within the heme crevice and directly reacts with the activated iron-bound oxygen atom. The edge reactivity of peroxidases means that aryl radical formation from aryldiazenes occurs at the δ-meso edge, resulting in addition of the aryl radical to the heme δ-meso-carbon. As crystal structures of the peroxidases have become available (e.g. Ref. 24), this general principle has been validated, although in some instances, oxidized protein residues mediate the electron abstraction from substrates.
A feature that distinguishes the mammalian peroxidases from the plant and fungal enzymes is that oxidation of certain substrates, notably the halides under acidic conditions by the plant and fungal enzymes, results in halide addition not only to the meso-carbons but also to the vinyl groups of the heme to give halohydrin, halovinyl, or dihaloalkyl adducts (25). The mammalian enzymes, for which halide oxidation is a physiologically important reaction, are much more resistant than the plant enzymes to this type of modification (26). This difference correlates with the presence of covalent bonds in the mammalian peroxidases between one or two of the heme methyl groups and protein residues. In lactoperoxidase, myeloperoxidase, eosinophil peroxidase, and probably thyroid peroxidase, two of the methyl groups of the heme are attached to protein carboxyl groups via ester links (Fig. 2) (27). In addition, in myeloperoxidase, one of the vinyl groups is covalently attached to a methionine sulfur atom via a vinyl sulfonium link. One function of these covalent links in the mammalian peroxidases is to shield the heme from reaction with catalytically generated radicals and hypohalide (HOX) species.
The covalent links between the heme and the protein in peroxidases could be forged by dedicated enzymes or could be the result of an autocatalytic maturation process. For example, the cysteine-to-vinyl links in the heme c prosthetic group of cytochrome c and other proteins are created by specific cross-linking enzymes (28). However, our results have established that the ester bonds in the mammalian peroxidases are the result of an autocatalytic maturation process. One strong piece of evidence for this conclusion is provided by the demonstration that introduction of a carboxylic acid group by site-specific mutagenesis into the active site of horseradish peroxidase, an enzyme that normally has no covalent heme-protein bonds, results in an almost quantitative formation of an ester bond between the carboxylic acid and one of the heme methyl groups upon exposure to hydrogen peroxide (29). The link that is formed is identical to that found in the mammalian peroxidases. The mechanism we advanced for this process in 1997 involves oxidation of the carboxylic acid by the enzyme Compound I ferryl species to a carboxylate radical, which abstracts a hydrogen from one of the heme methyl groups (78). Intramolecular electron transfer from the resulting carbon radical to the Fe(IV)=O species reduces the iron to the ferric state and simultaneously generates a methylene carbocation. This carbocation is finally trapped by reaction with the carboxylate group to give the ester link. In our horseradish peroxidase model, this happens only once, but in lactoperoxidase and the other mammalian peroxidases, this reaction sequence must occur twice to form ester links to two different methyl groups.
As we were elucidating this process, we were surprised to discover that the heme of most members of the CYP4 family of cytochrome P450 enzymes is also linked to the protein via an ester bond to one of the heme methyl groups (30). The combined work of my group and that of Allan Rettie at the University of Washington established that this bond is also formed autocatalytically. The members of the CYP4 family of P450 enzymes have the unique ability to oxidize the terminal methyl group of fatty acids (ω-hydroxylation), a thermodynamically disfavored reaction compared with hydroxylation of the adjacent secondary carbon ((ω-1)-hydroxylation). Some evidence suggests that the covalent ester link to the protein may increase the ω-hydroxylation regiospecificity (31), although ω-hydroxylation is favored in some CYP4 proteins that lack the covalent heme-protein link. A fully satisfactory explanation for the unique incorporation of a covalent heme-protein bond in many of the CYP4 P450 enzymes remains elusive.
Heme Oxygenase
As summarized above, hemoproteins undergo autocatalytic reactions at virtually every carbon to which a hydrogen is attached, in addition to reactions with the iron atom and the porphyrin nitrogen atoms. Given our interest in heme meso-carbon reactions, we undertook an investigation of heme degradation catalyzed by heme oxygenases. When we began our studies in 1992, the general outline of this process had already been delineated through work with microsomal enzyme preparations. Thus, in a reaction supported by molecular oxygen and NADPH-cytochrome P450 reductase, human heme oxygenase was thought to oxidize heme to the hypothetical intermediate α-meso-hydroxyheme. In a second catalytic turnover of the enzyme, this intermediate loses the α-meso-carbon as a molecule of carbon monoxide, forming verdoheme, in which an oxygen replaces the porphyrin ring carbon lost as carbon monoxide. Finally, a third catalytic turnover opens the oxygen-substituted ring to produce ferric biliverdin, in which the iron is lost after it is reduced to the ferrous state. Early work had shown that the two new oxygen atoms in the biliverdin product derived from different moleculesof molecular oxygen. After developing a recombinant expression system for heme oxygenase that allowed us to work with the purified enzyme (32), we provided direct evidence for the formation of α-meso-hydroxyheme and showed that H2O2 could substitute for the NADPH-cytochrome P450 reductase and that molecular oxygen was normally required for the reaction (33). Interestingly, reaction with meta-chloroperbenzoic acid produced a ferryl species similar to that formed with other hemoproteins, but this intermediate did not lead to normal heme cleavage and was not an intermediate in the reaction pathway (32). However, if ethyl hydroperoxide was used in place of H2O2, the reaction formed α-meso-ethoxyheme, indicating that the reaction involved either electrophilic attack of the iron-bound ethylhydroperoxy intermediate (Fe-OOEt) on the porphyrin ring or a free radical process in which homolytic fragmentation of this intermediate produced an ethoxy radical that added to the ring (34). This result excluded nucleophilic attack on the porphyrin ring by the iron-bound peroxide formed with either O2/NADPH-cytochrome P450 reductase or H2O2.
The oxidation of α-meso-phenylheme, a modified heme substrate, provided direct evidence for a postulated, but undetected, isoporphyrin intermediate occurring between the heme and α-meso-hydroxyheme intermediate. With this substrate, addition of the oxygen to the meso-carbon produces a tetrahedral intermediate, which, in the case of heme itself, rapidly loses a proton to give the α-meso-hydroxyheme. However, with a phenyl replacing the α-meso-hydrogen, no proton loss is possible, allowing detection of the tetrahedral intermediate. Further catalytic turnover of the enzyme produces the normal verdoheme with elimination of the carbon as benzoic acid rather than as carbon monoxide (35). The tetrahedral intermediate also decays non-enzymatically to a biliverdin analog with the α-meso-carbon retained as an aldehyde substituent. Interestingly, recent work by Masao Ikeda-Saito at Tohoku University in Japan and co-workers has shown that the heme oxygenase of Mycobacterium tuberculosis produces just such a formyl-substituted biliverdin (36), thus avoiding release of CO, a molecule that can bind to the mycobacterial DosS and DosT receptors (see below) to trigger dormancy.
The proximal histidine iron ligand in heme oxygenase is critical for catalysis. Mutation of the proximal histidine to an alanine yields an inactive protein whose catalytic activity can be rescued by adding imidazole. Spectroscopic studies showed that the exogenous imidazole occupies the proximal cavity of the mutant with one of its nitrogens coordinated to the iron atom (37). Site-specific mutagenesis replacement of the proximal histidine with either a cysteine or tyrosine transforms the enzyme into an NADPH oxidase (38), whereas replacement of the histidine with a highly electron-donating selenocysteine results in the NADPH-cytochrome P450 reductase-dependent formation of a covalent bond between the protein and one of the heme vinyl groups (39).
A long-standing collaboration with the laboratory of Tom Poulos at the University of California, Irvine (Fig. 4), provided the first crystal structure of a heme oxygenase in 1999 (40). This structure was followed by numerous other structures, both from our continued work with Tom Poulos and from other laboratories. These structures (a) clarified the role of steric effects in the regiochemistry of heme hydroxylation, (b) identified a flexible helix in the active site that is involved in substrate binding and release, and (c) provided evidence for a hydrogen bond network that facilitates catalysis. The best characterization of the catalytic hydrogen bond network was provided by NMR experiments carried out with Gerd La Mar at the University of California, Davis (Fig. 4), another long-term collaborator (e.g. Ref. 41).
FIGURE 4.

Gerd La Mar (left; NMR) and Tom Poulos (right; crystallography), long-term collaborators in our studies of heme oxygenase and other hemoproteins.
M. tuberculosis Hemoproteins
In the 1990s, I had the pleasure of working with a team at UCSF involved in studies of the AIDS virus and the development of therapeutic approaches to HIV (42). This project gradually evolved in my laboratory into one that focused on tuberculosis, a leading cause of death in HIV. Our interest in this topic was stimulated by the landmark publication of the genome of M. tuberculosis (43). Not surprisingly, my group focused on several classes of hemoproteins: (a) KatG, the catalase-peroxidase responsible for converting the prodrug isoniazid to its activated form; (b) EtaA, a protein we thought might be a hemoprotein but proved instead to be a flavoprotein monooxygenase that oxidatively activates ethionamide and related prodrugs; (c) DosS and DosT, the two heme-dependent sensors responsible for initiating the dormancy program that shifts mycobacteria into the latent state; and (d) the family of twenty cytochrome P450 enzymes encoded in the M. tuberculosis genome.
Isoniazid, even now one of the principal antituberculosis drugs, is oxidized by KatG to an acyl radical that attaches itself covalently to the heterocyclic ring of NAD+. The resulting adduct is a potent inhibitor of InhA, an enoyl-CoA reductase that is essential for mycobacterial cell wall synthesis (44). The most remarkable structural feature of KatG is the presence of a Met-Tyr-Trp cross-linked tripeptide within the active site that is essential for the peroxidative activity of all catalase-peroxidase enzymes. We demonstrated in 2005 that the cross-linked tripeptide was formed through an autocatalytic sequence that first linked the Tyr and Trp residues and then attached the methionine to the oxidized dipeptide (45). In this instance, a heme-dependent maturation process results in cross-linking of three protein residues rather than in cross-linking of the heme to the protein. Nevertheless, it emphasizes the importance of autocatalytic maturation processes in hemoprotein function.
Ethionamide, a second-line antituberculosis prodrug with significant toxicity issues, requires activation via a different mechanism, but it ultimately forms an NAD+ adduct similar to that formed with isoniazid to similarly inhibit cell wall synthesis (46). Ethionamide and related drugs, such as thiacetazone, contain a thioamide function known to be oxidized by hepatic cytochrome P450 enzymes to reactive toxic species. We therefore speculated that the activating enzyme might be a cytochrome P450. However, analysis of resistance mutants implicated a protein (EtaA) as the activating enzyme (47, 48). This was confirmed when we expressed and characterized EtaA as a flavoprotein monooxygenase that effectively oxidized ethionamide to its active form (49, 50).
DosT and DosS (also known as DevS) are sensor proteins with 60% sequence identity that respond to low oxygen tension or exposure to NO or CO by autophosphorylating a histidine in their own sequence. This phosphate group is then transferred to a carboxyl group on DosR, the protein partner in this two-component sensor system. Either by binding as an iron ligand or by causing iron autoxidation to the ferric state, molecular oxygen prevents the autophosphorylation of DosT and DosS. Phosphorylation of the transcription factor DosR causes it to bind to DNA, where it turns on a dormancy regulon of ∼50 proteins (51, 52). Much of the difficulty in sterilizing the infection in patients with M. tuberculosis is due to the ability of the mycobacteria to go into a dormant and drug-resistant state. A potential therapeutic approach can be envisioned in which entry into the dormant state is inhibited or reversed, forcing the mycobacteria into their active state, in which their sensitivity to standard drugs is much higher.
Both DosS and DosT are composed of a heme-containing GAF-A domain, a heme-free GAF-B domain, a histidine kinase domain, and an H-ATPase domain (53). In these proteins, the heme in the N-terminal GAF-A domain is the gas and/or redox sensor that, under appropriate conditions, triggers autophosphorylation of a histidine. We determined the structure of the truncated GAF-A heme domain of DosT (54), and the group of Beom Sik Kang at Kyungpook National University in Korea determined that of the corresponding DosS heme domain (55). In vitro assays with full-length DosT indicate that the O2-bound protein has a low ability to initiate phosphorylation, whereas phosphorylation readily occurs with the ferrous NO and CO complexes (56). The ferric protein is inactive, but the ferrous protein retains partial phosphorylation activity. The same responses are seen with DosS, although there is disagreement as to whether DosS is a gas sensor, like DosT, or a redox sensor. This disagreement arises from the fact that some laboratories have been unable to obtain a reasonably stable ferrous dioxygen complex (55, 57), which suggests that the protein responds to oxygen as a redox sensor that undergoes autoxidation to the ferric state. However, we (58) and others (56) have successfully obtained the dioxygen complex and shown that it is reasonably stable in the absence of metal ions in the medium. Site-specific mutagenesis has shown that there is a hydrogen bond system above the heme that governs the differential responses to O2, NO, and CO. Thus, mutation of a tyrosine located directly above the iron-bound ligand to a phenylalanine, which suppresses a hydrogen bond to the iron ligand, gives a protein that no longer is activated by NO or CO but still retains partial activity in the ligand-free ferrous state (59). At this time, the biggest mystery concerning this sensor system is how the on-off signal is transmitted from the heme to the kinase domain.
The M. tuberculosis genome encodes a total of 20 putative cytochrome P450 enzymes. Crystal structures have been obtained for at least seven of these enzymes, and the specific biological functions of four or five of them appear reasonably secure, but the functions of the remaining fifteen or so P450 enzymes remain undefined. Two of the enzymes, CYP125A1 and CYP142A1, are definitively involved in degradation of the cholesterol side chain, which serves both as a source of energy and as critical building blocks for the synthesis of cell-surface lipid molecules that contribute to virulence (60). A third enzyme, CYP124A1, also can oxidize the cholesterol side chain, but it is much more effective in the oxidation of terminally branched linear hydrocarbons, such as phytanic acid (61). The crystal structures (Fig. 5) of complexes of these enzymes with cholesterol and/or cholest-4-en-3-one have rationalized the specificities of these three enzymes and their role in catalyzing a three-step oxidation of the terminal methyl group to a carboxylic acid (62–64). Independent evidence has shown that cholesterol is important for the ability of M. tuberculosis to infect macrophages and to survive in its hostile environment (65). Furthermore, mass spectrometric analysis of the lipids after feeding terminally heptadeuterated cholesterol to intact mycobacteria confirmed that the terminal carbons of the cholesterol side chain are incorporated into phthiocerol dimycocerosates and other surface lipids (63). Inhibition of cholesterol utilization has emerged as a potential target for the development of new antimycobacterial agents (60).
FIGURE 5.
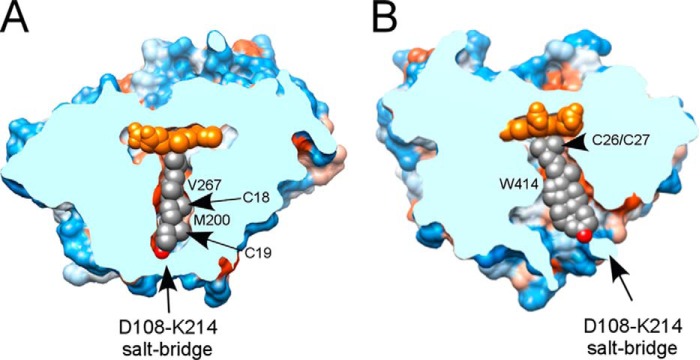
Binding of cholest-4-en-3-one in the active site of M. tuberculosis CYP125A1 as viewed after clipping the site at a plane orthogonal (A) and parallel (B) to the plane of the tetracyclic sterol framework. The heme is shown in yellow and the sterol in gray. Reprinted with permission from Reference 63.
Cytochrome P450 Catalysis
Despite four decades of effort (Fig. 6), our understanding of the mechanisms of cytochrome P450-catalyzed oxidations continues to be refined. Detailed mechanistic studies began with a landmark communication in 1978 by John Groves, Gary McClusky, Ron White, and Jud Coon from the University of Michigan, who reported partial inversion of stereochemistry in the hydroxylation of tetradeuterated norbornane by a microsomal cytochrome P450 enzyme (66). The implication that the reaction proceeded via an intermediate, probably a radical, was rationalized by a mechanism that, despite challenges over the years, has gained broad acceptance. The work of many investigators on the nature of the activated cytochrome P450 ferryl species culminated recently both in its biophysical characterization and direct monitoring of its reactions with substrates (67, 68). Many laboratories have contributed to our understanding of the mechanisms by which this activated oxygen species performs the hydroxylations, epoxidations, and other reactions characteristic of cytochrome P450 enzymes. Among the contributions of my group was the introduction of radical clock probes into studies of the cytochrome P450 mechanism, which provided strong support for a carbon radical intermediate in hydrocarbon hydroxylations (69, 70). Indirectly, the radical clock data also provided evidence for the two-state hypothesis of Shaik et al. (71), as this hypothesis elegantly rationalized the impossibly fast radical recombination rates observed with ultrafast radical clocks by Martin Newcomb, then at the University of Illinois at Chicago (72). The two-state hypothesis postulates that the catalytic heme iron-oxygen complex exists in two electronic spin states, only one of which has an energy barrier for recombination of the carbon radical intermediate with the iron-bound oxygen and therefore is the only one timed by the radical clocks. In an early experiment, we demonstrated that cytochrome P450 oxidized quadricyclane, a strained saturated hydrocarbon, to rearranged products via an electron abstraction mechanism (73). Abstraction of a single electron from the nitrogen atom of 4-alkyl-1,4-dihydropyridines, followed by extrusion of the 4-alkyl group as a free radical that alkylated the heme (74), indicated that one-electron oxidation also occurs in nitrogen oxidation. Given that epoxides were explicitly shown not to be responsible for heme adduct formation (8), the formation of N-alkylheme adducts in the oxidation of terminal olefins provided evidence that olefin epoxidation did not exclusively proceed via a concerted oxygen transfer to the double bond to form the epoxide. Furthermore, we established that the oxidation of a π-bond to give either a metabolite or an N-alkylheme adduct diverged very early in the oxidative sequence. This was most clearly illustrated by stark differences in the deuterium isotope and substituent electronic effects on the formation of ketene metabolites compared with N-alkylation of the prosthetic heme group in the oxidation of terminal acetylenes (75, 76).
FIGURE 6.

The increase in the size and content of the book Cytochrome P450: Structure, Mechanism, and Biochemistry, currently in its fourth edition, illustrates the growth of the field over the past four decades.
The Evolutionary Optimization of Heme
Heme is a highly versatile cofactor not only because it has three accessible oxidation states of the iron atom, but also because the overlap of the π-cloud of the porphyrin with the iron orbitals can be exploited to expand the redox properties and versatility of the catalytic center. The iron properties can be further modulated by varying the nature of the ligands to the iron at the positions not occupied by the four nitrogens of the porphyrin framework. Furthermore, heme provides an excellent illustration of the way in which evolution can tailor a physiological molecule to accomplish specific biochemical goals. Reactions at virtually every atom in the iron protoporphyrin IX framework have been used by evolution to tune its function. In addition to the reactions already described involving the tetrapyrrole nitrogen atoms, the meso-carbons, the methyl and vinyl substituents, and the iron atom, modifications are known to involve the propionic acid substituents. Thus, in heme d, a five-membered spirocyclic ring is formed by intramolecular addition of the propionic carboxylic acid group to the carbon to which the propionic acid is attached (77). With a few exceptions, most of these modifications, such as formation of the cysteine-to-vinyl links of heme c (20), appear to be the result of autocatalytic events supported by the normal function of the protein. For example, the heme modifications can alter the oxidation potential of the iron center, as in myeloperoxidase, or protect the heme from catalytically generated reactive metabolites, as in lactoperoxidase and the other mammalian peroxidases. In some instances, including the covalent binding that links a methyl of the heme to a carboxyl of the protein in cytochrome P450, the role of the covalent link remains uncertain. Beyond the tailoring of the heme group achieved through evolution, the chemical sensitivities of the heme group also expose an “Achilles' heel” that can result in inactivation of hemoproteins through substrate-dependent modification of the heme prosthetic group. It is a tribute to the versatility of the catalytic ferryl species of hemoproteins that it cannot only promote transformations that tune it or its protein environment, but can also catalyze an impressive diversity of reactions with the normal substrates of the enzymes.
My research career has followed a curious boomerang path, starting with studies of cholesterol biosynthesis and eventually returning to studies of cholesterol degradation and function in mycobacteria. Between these points of departure and return, my working hours have largely been devoted to the contemplation and pursuit of heme in its various guises. Who would have thought!
References
- 1. Corey E. J., Russey W. E., Ortiz de Montellano P. R. (1966) 2,3-Oxidosqualene, an intermediate in the biological synthesis of sterols from squalene. J. Am. Chem. Soc. 88, 4750–4751 [DOI] [PubMed] [Google Scholar]
- 2. Dean P. D. G., Ortiz de Montellano P. R., Corey E. J., Bloch K. (1967) A soluble 2,3-oxidosqualene sterol cyclase. J. Biol. Chem. 242, 3014–3015 [PubMed] [Google Scholar]
- 3. Corey E. J., Ortiz de Montellano P. R. (1967) Enzymic synthesis of β-amyrin from 2,3-oxidosqualene. J. Am. Chem. Soc. 89, 3362–3363 [DOI] [PubMed] [Google Scholar]
- 4. Ortiz de Montellano P. R., Boparai A. S. (1978) Aliphatic 3,4-epoxyalcohols. Metabolism by epoxide hydrase and mutagenic activity. Biochim. Biophys. Acta 544, 504–513 [DOI] [PubMed] [Google Scholar]
- 5. Lacourciere G. M., Armstrong R. N. (1993) The catalytic mechanism of microsomal epoxide hydrolase involves an ester intermediate. J. Am. Chem. Soc. 115, 10466–10467 [Google Scholar]
- 6. Borhan B., Jones A. D., Pinot F., Grant D. F., Kurth M. J., Hammock B. D. (1995) Mechanism of soluble epoxide hydrolase. Formation of an α-hydroxy ester-enzyme intermediate through Asp-333. J. Biol. Chem. 270, 26923–26930 [DOI] [PubMed] [Google Scholar]
- 7. Wong F. T., Hotta K., Chen X., Fang M., Watanabe K., Kim C.-Y. (2015) Epoxide hydrolase-lasalocid A structure provides mechanistic insight into polyether natural product biosynthesis. J. Am. Chem. Soc. 137, 86–89 [DOI] [PubMed] [Google Scholar]
- 8. Ortiz de Montellano P. R., Mico B. A. (1980) Destruction of cytochrome P-450 by ethylene and other olefins. Mol. Pharmacol. 18, 128–135 [PubMed] [Google Scholar]
- 9. Ortiz de Montellano P. R., Beilan H. S., Kunze K. L., Mico B. A. (1981) Destruction of cytochrome P-450 by ethylene. Structure of the resulting prosthetic heme adduct. J. Biol. Chem. 256, 4395–4399 [PubMed] [Google Scholar]
- 10. Ortiz de Montellano P. R., Kunze K. L., Yost G. S., Mico B. A. (1979) Self-catalyzed destruction of cytochrome P-450: covalent binding of ethynyl sterols to prosthetic heme. Proc. Natl. Acad. Sci. U.S.A. 76, 746–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ortiz de Montellano P. R., Mathews J. M. (1981) Autocatalytic alkylation of the cytochrome P-450 prosthetic heme by 1-aminobenzotriazole. Biochem. J. 195, 761–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stearns R. A., Ortiz de Montellano P. R. (1985) Inactivation of cytochrome P-450 during catalytic turnover of a cyclobutadiene precursor. J. Am. Chem. Soc. 107, 234–240 [Google Scholar]
- 13. Ortiz de Montellano P. R., Mathews J. M. (1981) Inactivation of hepatic cytochrome P-450 by a 1,2,3-benzothiadiazole insecticide synergist. Biochem. Pharmacol. 30, 1138–1141 [DOI] [PubMed] [Google Scholar]
- 14. Raag R., Swanson B. A., Poulos T. L., Ortiz de Montellano P. R. (1990) Formation, crystal structure, and rearrangement of a cytochrome P450cam iron-phenyl complex. Biochemistry 29, 8119–8126 [DOI] [PubMed] [Google Scholar]
- 15. Ortiz de Montellano P. R., Grab L. A. (1986) Inactivation of cytochrome P-450 during catalytic oxidation of a 3-(arylthioethyl)sydnone: N-vinyl heme formation via insertion into the Fe-N bond. J. Am. Chem. Soc. 108, 5584–5589 [Google Scholar]
- 16. Puy H., Gouya L., Deybach J.-C. (2010) Porphyrias. Lancet 375, 924–937 [DOI] [PubMed] [Google Scholar]
- 17. Ortiz de Montellano P. R., Beilan H. S., Kunze K. L. (1981) N-Methylprotoporphyrin IX: chemical synthesis and identification as the green pigment produced by 3,5-diethoxycarbonyl-1,4-dihydrocollidine treatment. Proc. Natl. Acad. Sci. U.S.A. 78, 1490–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kunze K. L., Ortiz de Montellano P. R. (1983) Formation of a sigma-bonded aryl-iron complex in the reaction of arylhydrazines with hemoglobin and myoglobin. J. Am. Chem. Soc. 105, 1380–1381 [Google Scholar]
- 19. Ringe D., Petsko G. A., Kerr D. E., Ortiz de Montellano P. R. (1984) Reaction of myoglobin with phenylhydrazine: a molecular doorstop. Biochemistry 23, 2–4 [DOI] [PubMed] [Google Scholar]
- 20. Kranz R., Lill R., Goldman B., Bonnard G., Merchant S. (1998) Molecular mechanisms of cytochrome c biogenesis: three distinct systems. Mol. Microbiol. 29, 383–396 [DOI] [PubMed] [Google Scholar]
- 21. Swanson B. A., Dutton D. R., Lunetta J. M., Yang C. S., Ortiz de Montellano P. R. (1991) The active sites of cytochromes P450 IA1, IIB1, IIB2, and IIE1. Topological analysis by in situ rearrangement of phenyl-iron complexes. J. Biol. Chem. 266, 19258–19264 [PubMed] [Google Scholar]
- 22. Ator M. A., Ortiz de Montellano P. R. (1987) Protein control of prosthetic heme reactivity. Reaction of substrates with the heme edge of horseradish peroxidase. J. Biol. Chem. 262, 1542–1551 [PubMed] [Google Scholar]
- 23. Ortiz de Montellano P. R. (1997) Control of the catalytic activity of prosthetic heme by the structure of hemoproteins. Acc. Chem. Res. 20, 289–294 [Google Scholar]
- 24. Gajhede M., Schuller D. J., Henriksen A., Smith A. T., Poulos T. L. (1997) Crystal structure of horseradish peroxidase C at 2.15 Å resolution. Nat. Struct. Biol. 4, 1032–1038 [DOI] [PubMed] [Google Scholar]
- 25. Huang L., Wojciechowski G., Ortiz de Montellano P. R. (2005) Prosthetic heme modification during halide ion oxidation. Demonstration of chloride oxidation by horseradish peroxidase. J. Am. Chem. Soc. 127, 5345–5353 [DOI] [PubMed] [Google Scholar]
- 26. Wojciechowski G., Huang L., Ortiz de Montellano P. R. (2005) Autocatalytic modification of the prosthetic heme of horseradish but not lactoperoxidase by thiocyanate oxidation products. A role for heme-protein covalent cross-linking. J. Am. Chem. Soc. 127, 15871–15879 [DOI] [PubMed] [Google Scholar]
- 27. Ortiz de Montellano P.R. (2013) Porphyrin modifications in the autocatalytic maturation of hemoproteins. In Handbook of Porphyrin Science (Ferreira G., ed), Vol. 30, pp. 1–30, World Scientific, Singapore [Google Scholar]
- 28. Stevens J. M., Daltrop O., Allen J. W. A., Ferguson S. J. (2004) C-type cytochrome formation: chemical and biological enigmas. Acc. Chem. Res. 37, 999–1007 [DOI] [PubMed] [Google Scholar]
- 29. Colas C., Ortiz de Montellano P. R. (2004) Horseradish peroxidase mutants that autocatalytically modify their prosthetic heme group. J. Biol. Chem. 279, 24131–24140 [DOI] [PubMed] [Google Scholar]
- 30. Hoch U., Ortiz de Montellano P. R. (2001) Covalently linked heme in cytochrome P4504A fatty acid hydroxylases. J. Biol. Chem. 276, 11339–11346 [DOI] [PubMed] [Google Scholar]
- 31. LeBrun L. A., Hoch U., Ortiz de Montellano P. R. (2002) Autocatalytic mechanisms and consequences of covalent heme attachment in the cytochrome P4504A family. J. Biol. Chem. 277, 12755–12761 [DOI] [PubMed] [Google Scholar]
- 32. Wilks A., Ortiz de Montellano P. R. (1993) Rat liver heme oxygenase. High level expression of a truncated soluble form and nature of the meso-hydroxylating species. J. Biol. Chem. 268, 22357–22362 [PubMed] [Google Scholar]
- 33. Liu Y., Moënne-Loccoz P., Loehr T. M., Ortiz de Montellano P. R. (1997) Heme oxygenase-1, intermediates in verdoheme formation and the requirement for reduction equivalents. J. Biol. Chem. 272, 6909–6917 [DOI] [PubMed] [Google Scholar]
- 34. Wilks A., Torpey J., Ortiz de Montellano P. R. (1994) Heme oxygenase (HO-1). Evidence for electrophilic oxygen addition to the porphyrin ring in the formation of α-meso-hydroxyheme. J. Biol. Chem. 269, 29553–29556 [PubMed] [Google Scholar]
- 35. Evans J. P., Niemevz F., Buldain G., Ortiz de Montellano P. R. (2008) Isoporphyrin intermediate in heme oxygenase catalysis. Oxidation of α-meso-phenylheme. J. Biol. Chem. 283, 19530–19539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nambu S., Matsui T., Goulding C. W., Takahashi S., Ikeda-Saito M. (2013) A new way to degrade heme. The Mycobacterium tuberculosis enzyme MhuD catalyzes heme degradation without generating CO. J. Biol. Chem. 288, 10101–10109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wilks A., Sun J., Loehr T. M., Ortiz de Montellano P. R. (1995) Heme oxygenase His25Ala mutant: replacement of the proximal histidine iron ligand by exogenous bases restores catalytic activity. J. Am. Chem. Soc. 117, 2925–2926 [Google Scholar]
- 38. Liu Y., Moënne-Loccoz P., Hildebrand D. P., Wilks A., Loehr T. M., Mauk A. G., Ortiz de Montellano P. R. (1999) Replacement of the proximal histidine iron ligand by a cysteine or tyrosine converts heme oxygenase to an oxidase. Biochemistry 38, 3733–3743 [DOI] [PubMed] [Google Scholar]
- 39. Jiang Y., Trnka M. J., Medzihradszky K. F., Ouellet H., Wang Y., Ortiz de Montellano P. R. (2009) Covalent heme attachment to the protein in human heme oxygenase-1 with selenocysteine replacing the His25 proximal iron ligand. J. Inorg. Biochem. 103, 316–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schuller D. J., Wilks A., Ortiz de Montellano P. R., Poulos T. L. (1999) Crystal structure of human heme oxygenase-1. Nat. Struct. Biol. 6, 860–867 [DOI] [PubMed] [Google Scholar]
- 41. Peng D., Ogura H., Zhu W., Ma L.-H., Evans J. P., Ortiz de Montellano P. R., La Mar G. N. (2009) Coupling of the distal hydrogen bond network to the exogenous ligand in substrate-bound, resting state human heme oxygenase. Biochemistry 48, 11231–11242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. DesJarlais R. L., Seibel G. L., Kuntz I. D., Furth P. S., Alvarez J. C., Ortiz de Montellano P. R., DeCamp D. L., Babé L. M., Craik C. S. (1990) Structure-based design of non-peptide inhibitors specific for the human immunodeficiency virus-1 protease. Proc. Natl. Acad. Sci. U.S.A. 87, 6644–6648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., Barry C. E. 3rd, Tekaia F., Badcock K., Basham D., Brown D., Chillingworth T., Conner R., Davies R., Devlin K., Feltwell T., Gentles S., Hamlin N., Holroyd S., Hornsby T., Jagels K., Krogh A., McLean J., Moule S., Murphy L., Oliver K., Osborne J., Quail M. A., Rajandream M.-A., Rogers J., Rutter S., Seeger K., Skelton J., Squares R., Squares S., Sulston J. E., Taylor K., Whitehead S., Barrell B. G. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 [DOI] [PubMed] [Google Scholar]
- 44. Rozwarski D. A., Grant G. A., Barton D. H. R., Jacobs W. R. Jr., Sacchettini J. C. (1998) Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science 279, 98–102 [DOI] [PubMed] [Google Scholar]
- 45. Ghiladi R. A., Knudsen G. M., Medzihradszky K. F., Ortiz de Montellano P. R. (2005) The Met-Tyr-Trp cross-link in Mycobacterium tuberculosis catalase-peroxidase (KatG). J. Biol. Chem. 280, 22651–22663 [DOI] [PubMed] [Google Scholar]
- 46. Wang F., Langley R., Gulten G., Dover L. G., Besra G. S., Jacobs W. R. Jr., Sacchettini J. C. (2007) Mechanism of thioamide drug action against tuberculosis and leprosy. J. Exp. Med. 204, 73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. DeBarber A. E., Mdluli K., Bosman M., Bekker L.-G., Barry C. E. 3rd (2000) Ethionamide activation and sensitivity in multidrug-resistant Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 97, 9677–9682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Baulard A. R., Betts J. C., Engohang-Ndong J., Quan S., McAdam R. A., Brennan P. J., Locht C., Besra G. S. (2000) Activation of the pro-drug ethionamide is regulated in mycobacteria. J. Biol. Chem. 275, 28326–28331 [DOI] [PubMed] [Google Scholar]
- 49. Vannelli T. A., Dykman A., Ortiz de Montellano P. R. (2002) The antituberculosis drug ethionamide is activated by a flavoprotein monooxygenase. J. Biol. Chem. 277, 12824–12829 [DOI] [PubMed] [Google Scholar]
- 50. Nishida C. R., Ortiz de Montellano P. R. (2011) Bioactivation of antituberculosis thioamide and thiourea prodrugs by bacterial and mammalian flavin monooxygenases. Chem. Biol. Interact. 192, 21–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bhat S. A., Singh N., Trivedi A., Kansal P., Gupta P., Kumar A. (2012) The mechanism of redox sensing in Mycobacterium tuberculosis. Free Radic. Biol. Med. 53, 1625–1641 [DOI] [PubMed] [Google Scholar]
- 52. Sivaramakrishnan S., Ortiz de Montellano P. R. (2013) The DosS-DosT/DosR mycobacterial sensor system. Biosensors 3, 259–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Saini D. K., Malhotra V., Dey D., Pant N., Das T. K., Tyagi J. S. (2004) DevR-DevS is a bona fide two-component system of Mycobacterium tuberculosis that is hypoxia-responsive in the absence of the DNA-binding domain of DevR. Microbiology 150, 865–875 [DOI] [PubMed] [Google Scholar]
- 54. Podust L. M., Ioanoviciu A., Ortiz de Montellano P. R. (2008) 2.3 Å X-ray structure of the heme-bound GAF domain of sensory histidine kinase DosT of Mycobacterium tuberculosis. Biochemistry 47, 12523–12531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cho H. Y., Cho H. J., Kim Y. M., Oh J. I., Kang B. S. (2009) Structural insight into the heme-based redox sensing by DosS from Mycobacterium tuberculosis. J. Biol. Chem. 284, 13057–13067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sousa E. H., Tuckerman J. R., Gonzalez G., Gilles-Gonzalez M. A. (2007) DosT and DevS are oxygen-switched kinases in Mycobacterium tuberculosis. Protein Sci. 16, 1708–1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kumar A., Toledo J. C., Patel R. P., Lancaster J. R. Jr., Steyn A. J. (2007) Mycobacterium tuberculosis DosS is a redox sensor and DosT is a hypoxia sensor. Proc. Natl. Acad. Sci. U.S.A. 104, 11568–11573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ioanoviciu A., Meharenna Y. T., Poulos T. L., Ortiz de Montellano P. R. (2009) DevS oxy complex stability identifies this heme protein as a gas sensor in Mycobacterium tuberculosis dormancy. Biochemistry 48, 5839–5848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yukl E. T., Ioanoviciu A., Nakano M. M., Ortiz de Montellano P. R., Moënne-Loccoz P. (2008) A distal tyrosine residue is required for ligand discrimination in DevS from Mycobacterium tuberculosis. Biochemistry 47, 12532–12539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ouellet H., Johnston J. B., Ortiz de Montellano P. R. (2011) Cholesterol catabolism as a therapeutic target in Mycobacterium tuberculosis. Trends Microbiol. 19, 530–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Johnston J. B., Kells P. M., Podust L. M., Ortiz de Montellano P. R. (2009) Biochemical and structural characterization of CYP124, a methyl-branched lipid ω-hydroxylase from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 106, 20687–20692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. McLean K. J., Lafite P., Levy C., Cheesman M. R., Mast N., Pikuleva I. A., Leys D., Munro A. W. (2009) The structure of Mycobacterium tuberculosis CYP125. Molecular basis for cholesterol binding in a P450 needed for host infection. J. Biol. Chem. 284, 35524–35533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ouellet H., Guan S., Johnston J. B., Chow E. D., Kells P. M., Burlingame A. L., Cox J. S., Podust L. M., Ortiz de Montellano P. R. (2010) Mycobacterium tuberculosis CYP125A1, a steroid C27 monooxygenase that detoxifies intracellularly generated cholest-4-en-3-one. Mol. Microbiol. 77, 730–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. García-Fernández E., Frank D. J., Galán B., Kells P. M., Podust L. M., García J. L., Ortiz de Montellano P. R. (2013) A highly conserved mycobacterial cholesterol catabolic pathway. Environ. Microbiol. 15, 2342–2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gatfield J., Pieters J. (2000) Essential role for cholesterol in entry of mycobacterium into macrophages. Science 288, 1647–1650 [DOI] [PubMed] [Google Scholar]
- 66. Groves J. T., McClusky G. A., White R. E., Coon M. J. (1978) Aliphatic hydroxylation by highly purified liver microsomal cytochrome P450. Evidence for a carbon radical intermediate. Biochem. Biophys. Res. Commun. 81, 154–160 [DOI] [PubMed] [Google Scholar]
- 67. Rittle J., Green M. T. (2010) Cytochrome P450 compound I: capture, characterization, and C-H bond activation kinetics. Science 330, 933–937 [DOI] [PubMed] [Google Scholar]
- 68. Yosca T. H., Rittle J., Krest C. M., Onderko E. L., Silakov A., Calixto J. C., Behan R. K., Green M. T. (2013) Iron(IV)hydroxide pKa and the role of thiolate ligation in C-H bond activation by cytochrome P450. Science 342, 825–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ortiz de Montellano P. R., Stearns R. A. (1987) Timing of the radical recombination step in cytochrome P-450 catalysis with ring-strained probes. J. Am. Chem. Soc. 109, 3415–3420 [Google Scholar]
- 70. He X., Ortiz de Montellano P. R. (2004) Radical rebound mechanism in cytochrome P450-catalyzed hydroxylation of the multifaceted radical clocks α- and β-thujone. J. Biol. Chem. 279, 39479–39484 [DOI] [PubMed] [Google Scholar]
- 71. Shaik S., Cohen S., Wang Y., Chen H., Kumar D., Thiel W. (2010) P450 enzymes: their structure, reactivity, and selectivity—modeled by QM/MM calculations. Chem. Rev. 110, 949–1017 [DOI] [PubMed] [Google Scholar]
- 72. Newcomb M., Chandrasena R. E. P. (2005) Highly reactive electrophilic oxidants in cytochrome P450 catalysis. Biochem. Biophys. Res. Commun. 338, 394–403 [DOI] [PubMed] [Google Scholar]
- 73. Stearns R. A., Ortiz de Montellano P. R. (1985) Cytochrome P-450-catalyzed oxidation of quadricyclane. Evidence for a radical cation intermediate. J. Am. Chem. Soc. 107, 4081–4082 [Google Scholar]
- 74. Lee J. S., Jacobsen N. E., Ortiz de Montellano P. R. (1988) Mechanisms of 4-alkyl radical extrusion in the cytochrome P450-catalyzed oxidations of 4-alkyl-1,4-dihydropyridines. Biochemistry 27, 7703–7710 [DOI] [PubMed] [Google Scholar]
- 75. Ortiz de Montellano P. R., Komives E. A. (1985) Branchpoint for heme alkylation and metabolite formation in the oxidation of arylacetylenes by cytochrome P-450. J. Biol. Chem. 260, 3330–3336 [PubMed] [Google Scholar]
- 76. Komives E. A., Ortiz de Montellano P. R. (1987) Mechanism of oxidation of π-bonds by cytochrome P-450. Electronic requirements of the transition state in the turnover of phenylacetylenes. J. Biol. Chem. 262, 9793–9802 [PubMed] [Google Scholar]
- 77. Loewen P. C., Switala J., von Ossowski I., Hillar A., Christie A., Tattrie B., Nicholls P. (1993) Catalase HPII of Escherichia coli catalyzes the conversion of protoheme to cis-heme d. Biochemistry 32, 10159–10164 [DOI] [PubMed] [Google Scholar]
- 78. Depillis G. D., Ozaki S., Kuo J. M., Maltby D. A., Ortiz de Montellano P. R. (1997) Autocatalytic processing of heme by lactoperoxidase produces the native protein-bound prosthetic group. J. Biol. Chem. 272, 8857–8860 [DOI] [PubMed] [Google Scholar]