

Abstract
The mucosal microbiota lives in close proximity with the intestinal epithelium and may interact more directly with the host immune system than the luminal/fecal bacteria. The availability of nutrients in the mucus layer of the epithelium is also very different from the gut lumen environment. Inferred metagenomic analysis for microbial function of the mucosal microbiota is possible by PICRUSt. We recently found that by using this approach, actively inflamed tissue of ulcerative colitis (UC) patients have mucosal communities enriched for genes involved in lipid and amino acid metabolism, and reduced for carbohydrate and nucleotide metabolism. Here, we find that the same bacterial taxa (e.g. Acinetobacter) and predicted microbial pathways enriched in actively inflamed colitis tissue are also enriched in the mucosa of subjects undergoing routine screening colonoscopies, when compared with paired samples of luminal/fecal bacteria. These results suggest that the mucosa of healthy individuals may be a reservoir of aerotolerant microbial communities expanded during colitis.
Keywords: acinetobacter, colitis, inflammation, microbiome, mucosa, proteobacteria
Introduction
The epithelial cells of the gastrointestinal tract form a physical barrier to both pathogenic and commensal bacteria, and also coordinate the mucosal immune system to maintain homeostasis.1 Commensal bacteria provide important signals through pattern recognition receptors (PRRs) that are necessary for maintaining intestinal epithelial homeostasis and repair.2 Goblet cells secreting mucus and Paneth cells secreting antimicrobial proteins (AMPs) are essential for reinforcing the barrier function of the intestinal epithelium. Hence, microbial communities that reside on the surface of the intestinal mucosa encounter an environment specialized to prevent their translocation across the epithelial barrier, which is distinct from the luminal environment.
In inflammatory bowel disease (IBD), this homeostatic relationship between the commensal bacteria and mucosal immune responses becomes disrupted in inflammatory bowel disease (IBD).3-5 Most IBD studies have focused on luminal/fecal bacteria, instead of bacterial communities adherent to the intestinal mucosa,6,7 although the mucosal microbiota are in closer proximity to immune cells.8 Although the luminal microbiota may simply be a numerical transformation of the mucosal microbiome,9 comparisons of mucosal and fecal samples have shown that these 2 intestinal compartments have significantly different microbial communities.10-12
Next generation 16S rRNA sequencing reveals alterations to bacterial taxa, but not changes to the metabolic activity and function of microbial communities. Shotgun metagenomic studies of total bacterial gene content for functional analysis are often limited by insufficient material and host DNA contamination (e.g., in biopsy samples). Recently, bacterial functional profiles can be investigated by inferring the metagenome of the closest available whole genome sequences using 16S gene sequence profiles.9,13 This approach showed that in IBD, functional differences were more pronounced than taxonomic differences.9
Using the same approach, we recently compared the microbial functional profiles of biopsies taken from regions of known inflammation states of IBD patients.14 We found that predicted microbial functions were significantly altered in inflamed tissue of ulcerative colitis (UC) patients (compared to normal tissue), with a reduction in carbohydrate and nucleotide metabolism in favor of increased lipid and amino acid metabolism. However, differences in predicted microbial function based on inflammation state were not observed in samples from Crohn's Disease (CD) patients. Since mucosal microbial function in CD did not vary as much as UC with inflammation state, we hypothesized that there is systemic perturbation of host-bacteria interactions in CD compared to more localized dysregulation in inflamed tissues for UC patients.
In this study, we inferred functional differences in paired mucosal and luminal/fecal microbiota of subjects undergoing routine screening colonoscopy. We find that the same bacterial taxa15 and predicted microbial pathways14 enriched in the mucosal microbiota of actively inflamed colitis tissue of UC patients are also enriched in the mucosa of these research subjects without gastrointestinal symptoms, when compared with luminal/fecal bacteria. Hence, the mucosal and luminal bacteria that are expanded during active inflammation in colitis are likely to be always present in smaller numbers in the mucosa of healthy individuals.
Results
Mucosal and luminal microbiota in subjects undergoing routine screening colonoscopy are significantly distinct
After filtering for singletons and samples with <1000 sequences, the final data set had 37 samples (18 biopsy samples and 19 stool samples), with a total of 267735 sequences and an average of 7236 ± 2158.514 sequences per sample. Fastq files are available upon requests. There were 17 paired stool-biopsy samples that were used for paired analyses. Fourteen subjects had polyps detected, of which 6 were reported as tubular adenoma. The microbiota from the biopsy samples with adenoma was inspected using unweighted UniFrac followed by principle coordinate analysis (PCoA) (Fig. S1) to determine if this could have yielded a heterogeneous sample set, but no visible clusters of samples were detected.
Microbial diversity for the mucosal microbiota was significantly less than that for the luminal microbiota, with reduced number of OTUs (Fig. 1A), species richness (by chao1, Fig. S2) and species evenness (by Shannon index, Fig. S2) in the mucosal microbiome. Unweighted UniFrac-based comparison (β diversity) of the mucosal and luminal microbiota (Fig. 1B) indicated that the differences in microbial composition could be clustered by sample type (R=0.5391, p=0.001). The main phyla driving these differences were Firmicutes, Bacteroidetes and Proteobacteria (Fig. 1C). Proteobacteria is the most abundant phylum in the mucosa (mean 42.2%), whereas Firmicutes was most abundant in fecal samples (65.3%), followed by Bacteroidetes (25.9%). The comparison of average UniFrac distances calculated between individual subjects and between sample types showed that the differences in bacterial communities between biopsy and fecal samples were greater than the differences in bacterial communities between individual subjects (Fig. 1D).
Figure 1.

(A) The luminal microbiome (represented by fecal samples) has higher microbial diversity than the mucosal microbiome (represented by biopsy samples) as determined by paired t-test. ***P < 0.0005. (B) PCoA plot of unweighted UniFrac distance, showing that the bacterial communities of the stool samples (blue dots) were distinct from those in the biopsy samples (red dots). (C) Relative abundance of taxonomic groups averaging across stool and biopsy samples, with the 10 most abundant phyla being represented. (D) Average UniFrac distance calculated between the luminal and mucosal microbial communities was greater than the average UniFrac distance calculated for different microbial communities between different individuals. (E) Bacterial taxa identified to be differentially abundant in the mucosal and luminal microbiome by LEfSe, at a logarithmic LDA threshold score of 2.0.
Bacterial taxa enriched in the mucosa are similar to those enriched under inflammatory conditions
Utilizing the LEfSe algorithm,16 we proceeded to identify bacterial taxa that were differentially abundant between mucosa and fecal samples (Fig. 1E). Strikingly, many of the taxa identified to be differentially abundant in this comparison were also observed to be differentially abundant in ulcerative colitis (UC) patients based on inflammation status of the mucosa.15 Most interestingly, the genus Acinetobacter from the Family Moraxellaceae, Order Pseudomonadales, Class Gammaproteobacteria and Phylum Proteobacteria was highly enriched both in the mucosa of subjects undergoing routine screening colonoscopy (relative to lumen) (Fig. 2 and S3) and in biopsy samples from UC patients with active inflammation (relative to biopsies from normal regions).15
Figure 2.

(A) Bacterial taxa that were significantly enriched in the mucosa of participants undergoing routine screening colonoscopy and were previously identified to be differentially enriched in mucosa of patients with colitis. (B) Bacterial taxa that were significantly enriched in the luminal microbiome of participants undergoing routine screening colonoscopy and were previously identified to be reduced in the mucosa of inflamed tissue of UC patients relative to uninflamed normal mucosa. Paired t-tests: *P < 0.05, ***P < 0.0005.
An increased level of Clostridiales was present in the luminal microbiota relative to the mucosal microbiota (Fig. 2B). Increased Clostridiales was also observed in normal tissue from UC patients, relative to inflamed tissue.15 Hence, many of the same bacterial taxa that are enriched or depleted during intestinal inflammation are also enriched or depleted in the mucosal microbiota, relative to the luminal microbiota.
Functional pathways enriched in the mucosal microbiota are similar to those enriched under inflammatory conditions
We used PICRUSt to predict the bacterial functional pathways that were differentially abundant in the mucosal and luminal microbiota (Fig. 3A). The functional differences between stool and biopsy samples include a switch toward lipid and amino acid metabolism in the mucosal microbiota (Fig. 3B), from carbohydrate and nucleotide metabolism in the luminal microbiota (Fig. 3C). Genetic information processing, replication and repair are also enriched in the luminal microbiota (Fig. 3A and C), perhaps indicative of greater bacterial turnover and replication compared to the mucosa. We had recently found that the same functional pathways also distinguished between inflamed tissue and normal tissue of UC patients,14 although these differences were not observed in samples from CD patients.
Figure 3.

(A) Supervised analysis with LEfSe (Log LDA > 2.00) showed functional pathways that were differentially enriched in the mucosal microbiome and the luminal microbiome. (B) Paired analyses of stool and biopsy samples from the same individuals found lipid and amino acid metabolism pathways to be enriched in the mucosal microbiome, while carbohydrate and nucleotide metabolism pathways, as well as genetic information processing pathways to be more abundant on the luminal microbiome. Paired t-tests: **P < 0.005, ***P < 0.0005.
Positive correlation between stool/biopsy differences and inflamed/normal tissue differences in microbiota profiles
The striking similarities between stool/biopsy and inflamed/normal tissue comparisons led us to hypothesize that there is a correlation between these different conditions. To test this, we calculated the differences in mucosa-adherent bacteria abundance and luminal bacteria abundance, as well as the differences in bacteria abundance of inflamed tissue and that of normal tissue. Both sets of values were subjected to Spearman correlation analysis. The significant positive correlation of these 2 sets of values (R = 0.5784, p = 0.007 for differences at phyla level; R = 0.5191, p<0.0001 for differences at genus level) suggests that the differences between the mucosa-adherent bacteria and the luminal bacteria are indeed similar with the changes in bacterial taxa that occurred during inflammation in colitis (Fig. 4A and B). This same correlation analysis was repeated for relative abundance values of predicted bacterial metabolic pathways and a significant Spearman correlation (R = 0.9930, p < 0.0001) was obtained as well (Fig. 4C). Hence, there is a significant correlation between the abundance of bacterial taxa and microbial pathways in actively inflamed colitis tissue with the mucosa of subjects without gastrointestinal symptoms, when compared with paired samples of luminal/fecal bacteria. In order to validate the enrichment of the Acinetobacter genus in the mucosal microbiota, we performed quantitative PCR (qPCR) analysis with genus specific primers and confirmed a very significant difference in the relative abundance of Acinetobacter genus between biopsy and stool samples (Fig. 4D).
Figure 4.
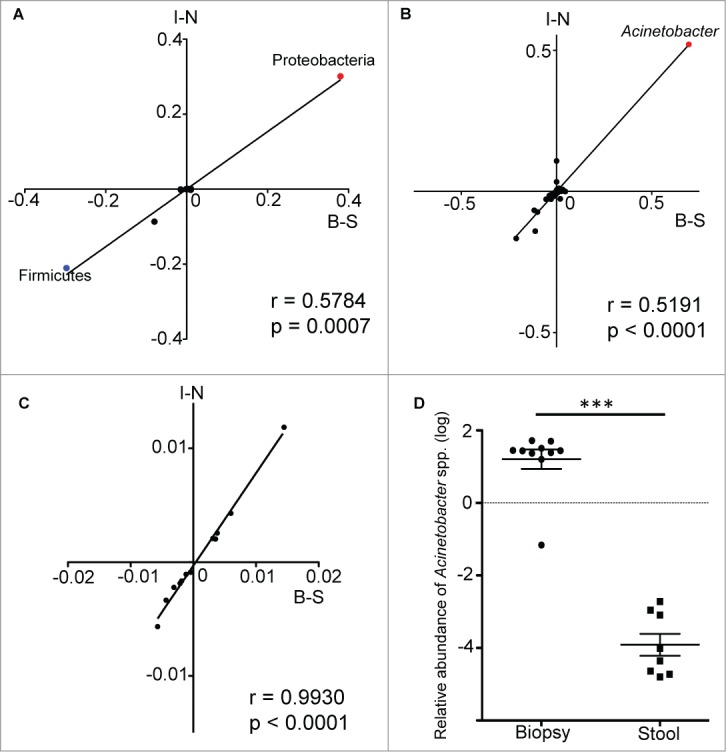
Spearman correlation analysis of the differences in relative abundance values of bacterial taxa from both data sets. The values along the B-S axis represented the differences in relative abundance of bacteria between biopsy and stool samples, while the values along the I-N axis represented the differences in relative abundance of bacteria between inflamed and normal biopsy tissues. Each data point is representative of a single bacteria taxa. The correlation analysis was performed at both the phylum (A) and genus (B) levels, with the data points for the phyla Proteobacteria and Firmicutes, as well as the genus Acinetobacter being highlighted. The same analysis was repeated with relative abundance values of predicted metabolic pathways (C). (D) The enrichment of Acinetobacter genus in mucosal biopsy was validated by performing qPCR on biopsy and stool samples. Mann-Whitney test: ***P < 0.0005.
Discussion
In this study, we find that the bacterial taxa and functional pathways that are enriched in the stool of ulcerative colitis patients,17 or in their mucosal tissues during active inflammation,14,15 are also enriched in the mucosal microbiota of subjects undergoing routine screening colonoscopy when compared to their luminal/fecal microbiota. We propose a model (Fig. 5A) whereby the intestinal mucosa of most individuals in a steady state are colonized with mucosal microbial communities kept in check by the luminal bacteria and intestinal immune response and barrier function. During colitis, this relationship may break down and specific components of the mucosal microbial communities may expand in numbers, spilling over into the luminal contents, as well as translocating across the intestinal epithelial barrier surface and triggering a strong anti-bacterial response (Fig. 5B). A recent study observed similar differences in Proteobacteria in the mucosal associated microbiota of healthy human subjects and tied these differences to the oxygen gradient of the intestine.18 We are particularly attracted to the model proposed by the authors whereby the oxidative nature of the host inflammatory response favors the expansion of aerotolerant organisms. Alterations in the oxygen gradient (Fig. 5) may expand low level keystone communities in the mucosal microbiota leading to dysbiosis that positively feeds back into the inflammatory response.
Figure 5.
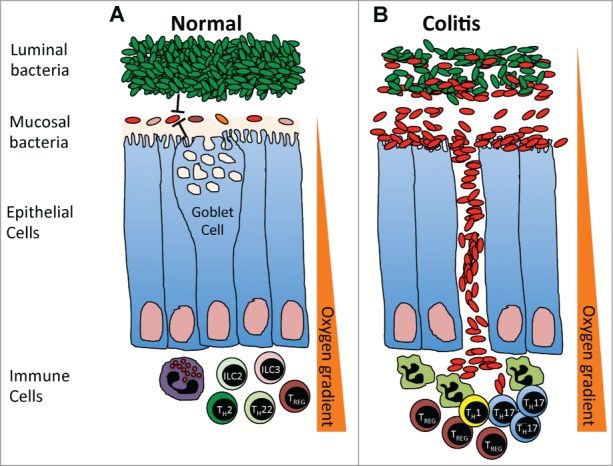
(A) In a steady-state, the mucosal microbial communities are kept in check by the luminal bacteria, intestinal immune response and barrier function. An oxygen gradient exists within the intestinal environment, with the mucosal interface being largely aerobic and the intestinal lumen largely anaerobic. (B) During colitis, the intestinal barrier breaks down, increasing the oxygen content within the intestinal lumen and leading to the expansion of aerotolerant mucosal microbial communities, which spill over into the intestinal lumen and translocate across the intestinal epithelial barrier surface to trigger a strong anti-bacterial response.
The overlap of bacterial taxa and microbial functional pathways observed between biopsy vs. stool, and between active inflammation vs. normal mucosal tissue is striking. This observation may provide some support to the proposed oxygen-gradient model.18 Acinetobacter in particular was enriched during active colitis (vs. normal tissue) and in the mucosal microbiota of individuals without gastrointestinal problems (vs. luminal bacteria). The strictly aerobic Acinetobacter genus has conventionally been studied in the context of nosocomial infections and antibiotic resistance.19 Its role in the largely anaerobic intestinal environment is unclear. Recently, a steep oxygen gradient was described within the intestine, extending from the more aerobic mucosal interface toward the largely anaerobic intestinal lumen.18 The mucosa may thus favor the colonization of aerotolerant bacterial communities, particularly from the phylum Proteobacteria.18 The breakdown of the mucosal barrier during colitis may increase host-gut microbiota oxygen exchange (e.g. through disrupted blood vessels and neutrophil activation), supporting the expansion of the genus Acinetobacter in biopsy tissues with active inflammation. In contrast, Clostridiales and Ruminococcaceae are more abundant in the luminal microbiome and these taxa are also enriched in normal uninflamed tissue, when compared to samples with active colitis.20 Hence, the source of bacteria expanded during colitis could be the mucosal microbiota present in healthy individuals. Notably, in a recent large study of treatment naïve new-onset CD patients, Proteobacteria was also increased and Clostridiales was decreased relative to non-IBD controls.17 While it is clear that the aerotolerant Acinetobacter genus are enriched in the mucosa, these results are correlative and require functional confirmation to determine if there is a causal relationship between Acinetobacter and intestinal inflammation.
Recent efforts in molecular systems ecology have been focused on characterizing host-specific microbial communities in terms of microbial functional traits, rather than specific bacterial taxa.21 A previous study found that differences in metabolic pathways were more significant between the microbiota of IBD patients and non-IBD controls than were reflected in differences between bacterial taxa alone.9 We also found previously that lipid and amino acid metabolic pathways were enriched at the expense of a reduction in carbohydrate and nucleotide metabolism for the mucosal microbiota of ulcerative colitis patients,14 although these alterations were not observed in CD patients. Strikingly, the same metabolic pathways were identified in this study to be enriched for the mucosal microbiota compared to the luminal/fecal microbiota. These results suggest to us that the mucosa-adherent bacterial communities present in healthy individuals are further expanded in the mucosa during active inflammation in UC, and can spill over into the luminal/fecal compartment during active disease. Alterations to the intestinal environment during active inflammation favor the expansion of mucosal communities enriched for genes involved in lipid and amino acid metabolism. The luminal environment favors communities encoding genes involved in carbohydrate and nucleotide metabolism and these communities compete more successfully in the mucosa when inflammation is absent.
Lipids and xenobiotics metabolism pathways, which we describe here to be enriched in the mucosal microbiota, have been found to be of higher abundance in mice with colitis, as compared to mice that were in treatment-induced remission.22 This may be related to alterations in phospholipid turnover that has previously been associated with the inflamed mucosa of IBD patients.23 The importance of bacterial xenobiotics metabolism in the gut was first highlighted when it was found that xenobiotics exposure induced expression of bacterial genes involved in drug resistance and metabolism. In a more recent study of mouse model colitis, xenobiotics metabolism pathways, particularly benzoate biodegradation, were implicated to promote the growth of Enterobacteriaceae, a family of bacteria enriched during colitis.22 Similarly, at a higher level of KEGG annotation of the predicted functional pathways, we found that the mucosa-adherent bacterial communities were enriched with benzoate metabolism pathways (Fig. S4). The functional pathways related to metabolism of terpenoids and polyketides, which we have predicted to be enriched in the mucosal microbiome, have also been previously implicated in the pathogenesis of intestinal inflammation and colitis-associated colorectal cancer.24 It was suggested that colonic inflammation favored the growth of bacteria with the polyketide synthase gene, which was genotoxic and promoted tumorigenesis in colitis.24
The mucosal and luminal microbiota have distinct profiles of biodiversity and bacterial taxa. The intestinal mucosa and lumen are 2 distinct microbial habitats. Lower microbial diversity at the intestinal mucosa is expected since this is an environment specialized to prevent their translocation across the epithelial barrier, with goblet cells secreting mucus and Paneth cells secreting antimicrobial proteins (AMPs).25 In conclusion, the mucosal microbiota may be a reservoir for keystone species that contributes to disease activity in colitis. In future studies, germ-free mice could be colonized with bacterial cultures from mucosal biopsies to determine if mucosal-adherent bacteria induce more intestinal inflammation than luminal/fecal bacteria.
Methods
Ethical considerations
The study was approved by the New York Harbor Veterans Affairs Hospital Institutional Review Board prior to research subject recruitment. Written informed consent was received from all participants.
Research subjects
Twenty male research subjects over the age of 50 and with no medical co-morbidities were recruited at the Manhattan campus of the Veteran Affairs Hospital after being referred to the Gastroenterology clinic for average risk screening colonoscopies. The median age was 60.5. Ethnicity was 30% (n = 6) Caucasian, 25% (n = 5) African American, 30% (n = 6) Hispanic and 15% (n = 3) other. Average BMI was 28.56 ± 5.73 (SD). Six study participants had a normal colonoscopy while 14 had polyps detected. All visualized polyps were resected except for one participant whose polyp was unable to be retrieved after resection. Biopsies from 7 of the participants were reported as normal mucosa or hyperplastic polyps, while 6 were reported as tubular adenoma.
Sample acquisition and DNA extraction
Patients underwent standard screening colonoscopies as clinically indicated. Endoscopic evaluations were performed using Olympus CF-Q160 or CF-H180 videoendoscopes. Polyps were measured using the open-forceps technique and then removed as clinically indicated. Mucosal pinch biopsies were taken 20 cm from the anal verge. Stool samples were collected from the patients prior to starting bowel preparation for their colonoscopy and was immediately stored at −80°C until DNA extraction. DNA extraction was performed using the MoBio PowerSoil DNA Isolation Kit (Carlsbad, CA) according to the manufacturer's instructions and without any modifications.
Amplification and sequencing of variable 4 (V4) region of 16S rRNA (rRNA) gene
Amplification of the V4 region of the bacterial 16S rRNA gene was carried out using the protocol modified from Caporaso et al.26 The forward primer construct contained the 5′ Illumina adapter, the forward primer pad, a 2-base linker (‘GT’) and the 515F primer (5′- AAT GAT ACG GCG ACC GAG ATC TAC ACT ATG GTA ATT GTG TGC CAG CMG CCG CGG TAA -3′). The reverse primer construct contained the 3′ Illumina adapter, a unique 12-base error-correcting Golay barcode, the reverse primer pad, a 2-base linker sequence (‘CC’) and the 806R primer (5′- CAA GCA GAA GAC GGC ATA CGA GAT NNN NNN NNN NNN AGT CAG TCA GCC GGA CTA CHV GGG TWT CTA AT -3′). Cycling protocol consisted of 94°C for 3min, 35 cycles of 94°C for 45s, 50°C for 60s and 72°C for 90s, with a final extension of 72°C for 10 min.
Amplification was carried out in triplicates and replicate amplicons were pooled before DNA concentration quantification with Quant-iT PicoGreen dsDNA kit (Invitrogen, Grand Island NY, USA). The amplicons were then pooled in equimolar ratio and purified using QIAquick PCR purification kit (Qiagen Inc., Chatsworth, CA, USA). The final concentration of purified amplicon was determined using Qubit PicoGreen dsDNA BR assay kit (Invitrogen, Grand Island, NY, USA). The pooled amplicon was sequenced on the Illumina MiSeq with a 2 × 150 cycle run (Illumina, San Diego CA, USA).
Sequencing reads demultiplexing and OTU picking
The paired-end sequencing was not effective and the resulting sequencing reads were single-ended. The single-end reads were demultiplexed using the 12-base Golay barcodes. Unmatched barcodes were first removed and excluded from demultiplexing. The sequencing reads were then assigned to their respective samples using the filtered set of barcodes and the split_library.py function from the Quantitative Insights In Microbial Ecology (QIIME) suite tools.27 Demultiplexing was done using the default parameters to filter for quality reads: minimum quality score of 25, minimum/maximum length of 200/1000, no ambiguous bases allowed and no mismatches allowed in the primer sequence.
We utilized a combination of closed reference and de novo sequence clustering methods (pick_open_reference_otus.py workflow in QIIME) to identify Operational Taxonomic Units (OTUs) for biodiversity analyses. Briefly, the quality-filtered sequences were first aligned to the Greengenes collection of reference sequences (gg_otus_13_5.tar.gz, http://greengenes.secondgenome.com/downloads/database/13_5). 0.1% of the sequences that failed to align were then subjected to de novo clustering with the UCLUST consensus taxonomy assigner, using a 97% similarity threshold.28 Sequences within the centroid de novo clusters were used for a second round of closed reference alignment and 0.1% of the sequences that failed to align were subjected to a second round of de novo clustering with 97% similarity threshold. The Ribosomal Database Project (RDP) classifier was used to assign taxonomy for the de novo sequences.29 The final outputs of the workflow were a biom-formatted matrix of OTU abundances in each sample and a phylogenetic tree constructed using the PyNAST alignment algorithm.30 The resulting OTU table was filtered for singletons and samples with low read number (<1000 sequences per sample) before being used for downstream diversity analyses.
Diversity analysis using QIIME
Relative abundance of all OTUs in all samples were calculated and averaged across the stool and biopsy samples. Alpha diversity analysis was done using the metrics observed OTUs, Shannon index31 and chao1.32 The QIIME α rarefaction workflow (α_rarefaction.py) was used on an OTU table that was first rarefied to a depth of 2000 sequences per sample. Rarefaction analysis between a depth of 10 sequences per sample and 2000 sequences per sample was iterated over 10 steps. To test for statistical significance of the difference in α diversity between the stool and biopsy samples, (unpaired) non-parametric 2-sample t-test and paired t-test were done on the α diversity metrics obtained at the rarefaction depth of 2000 sequences per sample for both groups of samples. The non-parametric t-test was done using the compare_alpha_diversity.py function in QIIME, while paired t-test was done using Prism 6.0 (GraphPad software, La Jolla, California).
Beta diversity analysis was calculated using unweighted UniFrac distance performed on an OTU table rarefied at 2000 sequences per sample.33,34 Principle Coordinate Analyses (PCoA) was performed on the UniFrac distance matrices and the resulting PCoA plot visualized using the Emperor graphics program.35 The statistical significance of the differences in β diversity between the mucosal and luminal microbiome was tested with the non-parametric ANOSIM test, using the QIIME compare_categories.py function and the unweighted UniFrac distance matrix as an input. The R statistic computed by the ANOSIM test indicated the similarity of the samples, with an R value of 0 indicating no significant differences between the groups and an R value of 1 indicating significant differences between the groups. Statistical significance of the R value was determined through 999 permutations.
Differential analysis using LEfSe
To identify for bacterial genus that were differentially abundant in the mucosal and luminal microbiome, Linear Discriminant Analysis Effect Size (LEfSe) analysis was performed on the OTU table (genus level) using the online Galaxy interface.16 Using the LEfSe algorithm, bacterial genus that were differentially abundant in the mucosal and luminal microbiome were first identified and tested using the Kruskal Wallis test. The identified genus were then subjected to the Linear Discriminant Analysis (LDA) model and ranked by their LDA scores respectively. The threshold logarithmic LDA score was set at 2.0, in order to identify bacterial genus that were truly significant in both groups of samples.
Inferred metagenomics and predicted functional analysis using PICRUSt
We utilized closed reference sequence clustering to identify OTUs for metagenome prediction and functional analysis. The quality-filtered, demultiplexed sequences were clustered by 97% similarity. Representative sequences were picked and aligned to the sequences from the Greengenes reference collection (gg_otus_13_5.tar.gz) to assign taxonomy.
The resulting OTU table was then used to generate inferred metagenome data using the PICRUSt workflow on the online Galaxy interface. The abundance values of each OTU were first normalized to their respective predicted 16S rRNA copy numbers. The normalized OTU abundance values were then multiplied by the respective predicted gene counts for metagenome prediction. Predicted functional pathways were annotated using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database at hierarchy levels 2 and 3. LEfSe analysis was used to identify bacterial functional pathways that were differentially abundant in the mucosal and luminal microbiome.16 Paired t-tests of functional pathway abundance were done using Prism 6.0.
Combined analysis of subjects undergoing routine screening colonoscopy and IBD subjects
To better demonstrate that the differences between the mucosal microbiome and the luminal microbiome of healthy subjects overlapped with the differences between inflamed tissue microbiome and normal tissue microbiome of the IBD patients from the previous data set, we calculated the differences in relative abundance values of the bacterial taxa (at phyla and genus taxonomic levels) for both sets of comparisons and performed correlation analysis of both sets of values. The Spearman correlation was calculated and tested for statistical significance. The same correlation analysis was also repeated for the relative abundance values of predicted bacterial metabolic pathways in both data sets.
Acinetobacter genus real time quantitative PCR
We quantified the abundance of Acinetobacter genus by real time quantitative PCR (qPCR) using 10 biopsy samples and 8 stool samples. The biopsy samples were selected from the same cohort of individuals who participated in this study and for which the samples had sufficient amount of nuclei acid material remaining after performing 16S microbial sequencing. The stool samples used for qPCR were collected fresh from 8 individuals undergoing routine screening colonoscopy. DNA isolation was done as previously described. The Acinetobacter genus specific primer pairs used were Ac436f (5′ TTT AAG CGA GGA GGA GG 3′) and Ac676r (5′ ATT CTA CCA TCC TCT CCC 3′).36 The primer pair amplified for a region of the Acinetobacter 16S rRNA gene and was of 100% homology between members of the Acinetobacter genus.36 Acinetobacter 16S rRNA was normalized to total 16S using the universal primer pairs 5′ ACT CCT ACG GGA GGC AGC AGT 3′ and 5′ ATT ACC GCG GCT GCT GGC 3′.37 The 20 μl reaction volume consisted of 10 μl SYBR Green PCR Mix (2X), 0.25 μl forward primer, 0.25 μl reverse primer, 1 μl DNA and 8μl RNase-free water. Cycling was done on a BioRad C1000 Thermal Cycler, using a protocol that consisted of 95°C for 5 minutes, 60 cycles of 95°C for 15s, 58°C for 30s and plate read, followed by a standard melt curve.
Statistical analyses
Statistical analysis was performed using Prism 6.0 (GraphPad Software, La Jolla, CA). Paired t-test was used whenever possible to assess statistical significance between paired mucosa and stool samples. The unpaired t-test or Mann-Whitney test was used to assess statistical significance between samples that are not paired, depending on the number of samples compared and whether they fall in a normal distribution.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by funds from the Kevin and Marsha Keating Family Foundation (PL), and UM/MoHE High Impact Research (H-20001–00-E000061) grant from University of Malaya (YALL and PL), the Diane Belfer Program in Human Microbial Ecology (IC). PL is also supported by grants from the National Institutes of Health (AI093811 and AI094166) and the Broad Medical Research Program. SCL is supported by a University of Malaya Student Grant (PV024/2011B). MST is supported by the Grants-in-Aid of Research (G2012161472) from Sigma Xi, The Scientific Research Society. JP is supported by the AGA-Eli and Edythe Broad Student Research Fellowship. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev 2014; 14:141-53; PMID:24566914; http://dx.doi.org/ 10.1038/nri3608 [DOI] [PubMed] [Google Scholar]
- 2. Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004; 118:229-41; PMID:15260992; http://dx.doi.org/ 10.1016/j.cell.2004.07.002 [DOI] [PubMed] [Google Scholar]
- 3. Abraham C, Medzhitov R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology 2011; 140:1729-37; PMID:21530739; http://dx.doi.org/ 10.1053/j.gastro.2011.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Honda K, Littman D. The microbiome in infectious disease and inflammation. Annu Rev Immunol 2012; 30:759-95; PMID:22224764; http://dx.doi.org/ 10.1146/annurev-immunol-020711-074937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Manichanh C, Borruel N, Casellas F, Guarner F. The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol 2012; 9:599-608; PMID:22907164; http://dx.doi.org/ 10.1038/nrgastro.2012.152 [DOI] [PubMed] [Google Scholar]
- 6. Frank D, St Amand A, Feldman R, Boedeker E, Harpaz N, Pace N. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A 2007; 104:13780-5; PMID:17699621; http://dx.doi.org/ 10.1073/pnas.0706625104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, et al. Reduced diversity of faecal microbiota in crohn's disease revealed by a metagenomic approach. Gut 2006; 55:205-11; PMID:16188921; http://dx.doi.org/ 10.1136/gut.2005.073817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Packey C, Sartor R. Interplay of commensal and pathogenic bacteria, genetic mutations, and immunoregulatory defects in the pathogenesis of inflammatory bowel diseases. J Intern Med 2008; 263:597-606; PMID:18479259; http://dx.doi.org/ 10.1111/j.1365-2796.2008.01962.x [DOI] [PubMed] [Google Scholar]
- 9. Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 2012; 13:R79; PMID:23013615; http://dx.doi.org/ 10.1186/gb-2012-13-9-r79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eckburg P, Bik E, Bernstein C, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science 2005; 308:1635-8; PMID:15831718; http://dx.doi.org/ 10.1126/science.1110591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Momozawa Y, Deffontaine V, Louis E, Medrano J. Characterization of bacteria in biopsies of colon and stools by high throughput sequencing of the V2 region of bacterial 16S rRNA gene in human. PLoS One 2011; 6:e16952; PMID:21347324; http://dx.doi.org/ 10.1371/journal.pone.0016952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zoetendal E, von Wright A, Vilpponen-Salmela T, Ben-Amor K, Akkermans A, de Vos W. Mucosa-associated bacteria in the human gastrointestinal tract are uniformly distributed along the colon and differ from the community recovered from feces. Appl Environ Microbiol 2002; 68:3401-7; PMID:12089021; http://dx.doi.org/ 10.1128/AEM.68.7.3401-3407.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 2013; 31:814-21; PMID:23975157; http://dx.doi.org/ 10.1038/nbt.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Davenport M, Poles J, Leung J, Wolff M, Abidi W, Ullman T, et al. Metabolic alterations to the mucosal microbiota in inflammatory bowel disease. Inflamm Bowel Dis 2014; 20:721-31; PMID:24583479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leung JM, Davenport M, Wolff MJ, Wiens KE, Abidi WM, Poles MA, et al. IL-22-producing CD4+ cells are depleted in actively inflamed colitis tissue. Mucosal Immunol 2013; 7:124-33; PMID:23695510; http://dx.doi.org/ 10.1038/mi.2013.31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol 2011; 12:R60; PMID:21702898; http://dx.doi.org/ 10.1186/gb-2011-12-6-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, Ren B, et al. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe 2014; 15:382-92; PMID:24629344; http://dx.doi.org/ 10.1016/j.chom.2014.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Albenberg L, Esipova TV, Judge CP, Bittinger K, Chen J, Laughlin A, et al. Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology 2014; 147:1055-63; PMID:25046162; http://dx.doi.org/ 10.1053/j.gastro.2014.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peleg AY, Seifert H, Paterson DL. Acinetobacter baumannii: emergence of a successful pathogen. Clinical Microbiol Rev 2008; 21:538-82; PMID:18625687; http://dx.doi.org/ 10.1128/CMR.00058-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leung J, Davenport M, Wolff M, Wiens K, Abidi W, Poles M, et al. IL-22-producing CD4+ cells are depleted in actively inflamed colitis tissue. Mucosal Immunol 2014; 7:124-33; PMID:23695510; http://dx.doi.org/ 10.1038/mi.2013.31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shafquat A, Joice R, Simmons SL, Huttenhower C. Functional and phylogenetic assembly of microbial communities in the human microbiome. Trends Microbiol 2014; 22:261-6; PMID:24618403; http://dx.doi.org/ 10.1016/j.tim.2014.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rooks M, Veiga P, Wardwell-Scott L, Tickle T, Segata N, Michaud M, et al. Gut microbiome composition and function in experimental colitis during active disease and treatment-induced remission. ISME J 2014; 8:1403-17; PMID:24500617; http://dx.doi.org/ 10.1038/ismej.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morita H, Nakanishi K, Dohi T, Yasugi E, Oshima M. Phospholipid turnover in the inflamed intestinal mucosa: arachidonic acid-rich phosphatidyl/plasmenyl-ethanolamine in the mucosa in inflammatory bowel disease. J Gastroenterol 1999; 34:46-53; PMID:10204610; http://dx.doi.org/ 10.1007/s005350050215 [DOI] [PubMed] [Google Scholar]
- 24. Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012; 338:120-3; PMID:22903521; http://dx.doi.org/ 10.1126/science.1224820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maynard C, Elson C, Hatton R, Weaver C. Reciprocal interactions of the intestinal microbiota and immune system. Nature 2012; 489:231-41; PMID:22972296; http://dx.doi.org/ 10.1038/nature11551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 2011; 108 1:4516-22; PMID:20534432; http://dx.doi.org/ 10.1073/pnas.1000080107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010; 7:335-6; PMID:20383131; http://dx.doi.org/ 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010; 26:2460-1; PMID:20709691; http://dx.doi.org/ 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- 29. Wang Q, Garrity GM, Tiedje JM, Cole JR, Naïve bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 2007; 73:5261-7; PMID:17586664; http://dx.doi.org/ 10.1128/AEM.00062-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 2010; 26:266-7; PMID:19914921; http://dx.doi.org/ 10.1093/bioinformatics/btp636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shannon CE. A mathematical theory of communication. Bell System Tech J 1948; 27:379-423; 623-56; http://dx.doi.org/ 10.1002/j.1538-7305.1948.tb01338.x [DOI] [Google Scholar]
- 32. Chao A. Nonparametric estimation of the number of classes in a population. Scandinavian J Statistics 1984:265-70; PMID:6422541.6422541 [Google Scholar]
- 33. Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: an effective distance metric for microbial community comparison. ISME J 2011; 5:169-72; PMID:20827291; http://dx.doi.org/ 10.1038/ismej.2010.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen J, Bittinger K, Charlson ES, Hoffmann C, Lewis J, Wu GD, et al. Associating microbiome composition with environmental covariates using generalized unifrac distances. Bioinformatics 2012; 28:2106-13; PMID:22711789; http://dx.doi.org/ 10.1093/bioinformatics/bts342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vazquez-Baeza Y, Pirrung M, Gonzalez A, Knight R. EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience 2013; 2:16; PMID:24280061; http://dx.doi.org/ 10.1186/2047-217X-2-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vanbroekhoven K, Ryngaert A, Wattiau P, Mot R, Springael D. Acinetobacter diversity in environmental samples assessed by 16S rRNA gene PCR-DGGE fingerprinting. FEMS Microbiol Ecol 2004; 50:37-50; PMID:19712375; http://dx.doi.org/ 10.1016/j.femsec.2004.05.007 [DOI] [PubMed] [Google Scholar]
- 37. Ramanan D, Tang MS, Bowcutt R, Loke P, Cadwell K. Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal bacteroides vulgatus. Immunity 2014; 41:311-24; PMID:25088769; http://dx.doi.org/ 10.1016/j.immuni.2014.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.