

Abstract
Temperature-dependent hydrogen-deuterium (H/D) exchange of the thermophilic alcohol dehydrogenase (htADH) has been studied by using liquid chromatography-coupled mass spectrometry. Analysis of the changes in H/D exchange patterns for the protein-derived peptides suggests that some regions of htADH are in a rigid conformational substate at reduced temperatures with limited cooperative protein motion. The enzyme undergoes two discrete transitions at ≈30 and 45°C to attain a more dynamic conformational substate. Four of the five peptides exhibiting the transition above 40°C are in direct contact with the cofactor, and the NAD+-binding affinity is also altered in this temperature range, implicating a change in the mobility of the cofactor-binding domain >45°C. By contrast, the five peptides exhibiting the transition at 30°C reside in the substrate-binding domain. This transition coincides with a change in the activation energy of kcat for hydride transfer, leading to a linear correlation between kcat and the weighted average exchange rate constant kHX(WA) for the five peptides. These observations indicate a direct coupling between hydride transfer and protein mobility in htADH, and that an increased mobility is at least partially responsible for the reduced Eact at high temperature. The data provide support for the hypothesis that protein dynamics play a key role in controlling hydrogen tunneling at enzyme active sites.
There has been increasing speculation that protein dynamics may play an active role in facilitating catalysis in enzymes. NMR studies of a number of enzymes such as HIV protease (1), dihydrofolate reductase (2), cyclophilin A (3), ribonuclease A (4), and ribonuclease binase (5) show flexible regions of enzyme that may be correlated with catalysis. However, the demonstration of a causal relationship between the observed protein motions and the rate of the elementary step of an enzyme-catalyzed process remains a formidable experimental challenge. Due to the multidimensional and quantum-tunneling nature of hydrogen transfer reactions, the rate of hydrogen transfer is highly sensitive to protein motions that modulate the height and width of the reaction barrier (6–8). Therefore, enzymes catalyzing proton, hydride, or hydrogen atom transfer reaction are particularly valuable for understanding the role of protein motion in catalysis.
Recently, experimental studies of some enzyme systems that include alcohol dehydrogenase (9), soybean lipoxygenase (10), dihydrofolate reductase (11), methylamine dehydrogenase (12), and morphinone reductase (7) have produced anomalous kinetic data that can be rationalized only by an environmentally coupled tunneling model in which heavy atom motion controls the probability of hydrogen transfer. Among these intriguing systems is the thermophilic alcohol dehydrogenase (htADH) isolated from Bacillus stearothermophilus strain LLD-R. htADH catalyzes the reversible hydride transfer between the cofactor NAD+/NADH and the substrate alcohol/aldehyde with different activation energies below and above 30°C (Fig. 1). Temperature-dependent kinetic isotope effect studies have further shown that the tunneling properties become more conspicuous at elevated temperatures (>30°C) (9), a highly counterintuitive result, because it is normally expected that tunneling will diminish with increasing temperature. These results were rationalized within an environmentally coupled tunneling model, whereby hydrogen transfer is enhanced at elevated temperatures by a sampling of the conformational space that is inaccessible at low temperature. This has the effect of reducing the reaction barrier width and enhancing the vibrational/electronic coupling between reactant and product states (9). Indeed, theoretical modeling studies of several enzyme-catalyzed hydrogen transfer reactions have generated evidence supporting this view (13, 14).
Fig. 1.

Arrhenius plots for the oxidation of protonated (•) and deuterated (▪) benzyl alcohol by htADH (adapted from ref. 9).
The lack of a generic tool for probing protein motion makes it difficult to test directly the environmentally coupled tunneling model. Dynamic NMR techniques are not suitable for most of the systems mentioned above due to their large sizes. In a previous study, the flexibility of htADH was compared to a mesophilic ADH from yeast by using Fourier transform infrared spectroscopy to probe for global changes in protein flexibility (15). Herein, we present results from a temperature-dependent amide hydrogen-deuterium (H/D) exchange study of htADH using liquid chromatography-coupled mass spectrometry (16). By analyzing the H/D exchange patterns for protein-derived peptides, it is possible to detect changes in protein mobility that reveal a one-to-one correlation with temperature-dependent changes in catalytic rate. This behavior is restricted to a small subset of peptides that surround the substrate-binding pocket, implying a direct link between regional protein dynamics and the rate of hydride transfer.
Materials and Methods
Cloning, Isolation, and Purification of htADH. The htADH gene was amplified directly by PCR from the genomic DNA of B. stearothermophilus strain LLD-R (NCIMB 12403; www.ncimb.co.uk) with the primers 5′-cgtcatgaaagccgctgtagttg-3′ and 5′-atgagacagccctagaatgccggat-3′. The gene was first cloned into the vector pCR 2.1-TOPO by TA cloning with the TOPO TA cloning kit from Invitrogen, and the plasmid was propagated and sequenced to confirm the insertion of the gene. With the ADH gene containing plasmid as template, a second round of PCR with the primers 5′-gcgcgctagcatgaaagccgctgtagttgaacaatt-3′ and 5′-tttaattgcccgcgcatcaaa attgcaatcttctatttattggatccc-3′ generated a ≈1.1-kb PCR product with two restriction sites incorporated for ligation. The PCR product was double digested with NheI and BamHI and ligated into the expression vector pET-24b(+) to give the plasmid pET-ADH. The plasmid was subsequently transformed into Escherichia coli expression system BL21(DE3). For large-scale expression and purification of the enzyme, 6 × 1.6 liters of LB medium containing 30 μg/ml kanamacyin were inoculated. The culture was incubated at 37°C with shaking until the OD550 reached 0.6, then isopropyl β-D-thiogalactoside (0.6 mM) was added to induce protein expression. After growth for another 16 h, the cells were harvested and lysed following standard procedures. The supernatant containing cell contents was incubated at 60°C for 15 min, and the debris was removed by centrifugation. The remaining supernatant was first passed through a fast-flow DEAE-Sepharose column, and the fractions with ADH activity were pooled, concentrated, and passed through a sephacryl S-300-HR column. The protein was further purified by using an affinity column (Reactive Blue 2, Sigma) and eluted with a salt gradient [final yield: ≈9 mg/liter media, specific activity: 380 units/mg (60°C)]. The homogeneity of the enzyme was confirmed by SDS/PAGE gel under reducing and nonreducing conditions.
H/D Exchange. The exchange reactions were initiated by adding 10 μl of newly thawed protein solution (≈0.5 mg/ml, 50 mM KPi, pH 7.4/250 mM KCl/2.5 mM DTT) to 90 μl of D2O. After incubation, the exchange reaction was rapidly quenched by lowering the pH to 2.4 and temperature to 0°C and was followed by pepsin digestion of the protein. The resulting peptides were analyzed on a reversed-phase capillary column (POROS 20 R1, PerSeptive Diagnostics, Cambridge, MA) coupled to a QStar Pulsar quadrupole time-of-flight mass spectrometer with normal electrospray interface (PE Biosystems). The maximal time from the initiation of digestion to the elution of any peptide was 22 min. Zero time-point controls (the “artifactual in-exchange” control) were performed by adding quench buffer to the protein solution before exposure to D2O. Measured peptide masses were corrected for artifactual in-exchange at t = 0, normalized to 100% D2O, and corrected for back exchange following the method described by Resing et al. (17). Typically, 12 data points were collected at each temperature, with the shortest incubation time of 7 s and the longest incubation time of 130 min. Kinetics of exchange using corrected peptide masses were fit by nonlinear least squares to the equation: Y = N - Ae-k1t - Be-k2t - Ce-k3t, where N is the total number of amides exchanging over the observed time course for each individual peptide, and A, B, and C correspond to the number of amides exchanging with rate constants k1, k2, and k3, respectively. The number of nonexchanging amide Hs is calculated by subtracting the number of exchanging amides (A+B+C) from the total number of backbone amides (NH) in the peptide, excluding proline residues.
Fluorescence Measurement. The quenching of protein fluorescence by titration with NAD+ was performed on a FluoroMax-3 fluorometer. Excitation wavelength was set at 295 nm to selectively excite tryptophan. The buffer conditions are the same as described in the H/D exchange experiments. All of the protein and NAD+ solutions were degassed and filtered before measurement. Aliquots of NAD+ solution were titrated into a 1 × 1-cm cell containing the free enzyme and the intensity of the fluorescence maximum was recorded. From the quenching titration curves, the dissociation constants (Kd) at different temperatures for NAD+ have been obtained with a one-site binding model.
Results and Discussion
Temperature-Dependent H/D Exchange. Amide H/D exchange was carried out at 10, 20, 40, 55, and 65°C with apo-enzyme. Independent activity assays confirmed that the enzyme is fully active after incubation for 130 min under the exchange conditions. Twenty-one nonoverlapping peptides generated by pepsin digestion and identified for H/D exchange analysis are shown in Fig. 2. For each peptide, the number of incorporated deuterons or exchanging Hs was obtained by calculating the difference between the weighted average mass of the peptide before and after H/D exchange. The plots of exchanging Hs vs. time were fitted with a three-exponential model to give the exchange rate constants k1, k2, and k3 and the number of corresponding exchanging Hs (see Tables 1–8 and Fig. 7, which are published as supporting information on the PNAS web site). Based on their exchanging rates, the amide Hs are categorized into four groups: fast-exchanging Hs (kex > 1 min-1), intermediate-exchanging Hs (0.1 min-1 < kex < 1 min-1), slow-exchanging Hs (0.001 min-1 < kex < 0.1 min-1), and nonexchanging Hs (kex < 0.001 min-1).
Fig. 2.

Primary sequence of htADH. A total of 21 peptides generated by pepsin digestion are numbered starting from the N terminus. Peptides located in the cofactor-binding domain are in green, and peptides located in the substrate-binding domain are in red.
An examination of the H/D exchange of the 21 peptides reveals that the peptides show strikingly different patterns and can be categorized into three distinct groups based on their temperature-dependent H/D exchange patterns. The first group, including 11 peptides, is represented by the six peptides shown in Fig. 3. For these peptides, the number of the exchangeable Hs appears to increase successively with temperature. The H/D exchange kinetic traces consist of both slow and fast kinetic components at low temperatures, whereas the exchange is dominated by fast kinetic components at high temperatures. In sharp contrast, the second group, including peptides 8–10, 17, and 21, displays a clear transition between 40°C and 55°C in the temperature-dependent H/D exchange (Fig. 4 a–e). At low temperatures (10, 20, and 40°C), the exchange shows weak temperature-dependent behavior and is dominated by fast kinetic components. At higher temperatures, there are significantly more slow-exchanging Hs, as indicated by the emergence of slow kinetic components in the plots. Although these five peptides are distributed in both the cofactor-binding domain (peptides 10 and 17) and the substrate-binding domain (peptides 8, 9, and 21), they all have some residues involved in NAD+ binding except peptide 8 (Fig. 4g). The third group, which includes peptides 1–4 and 7, exhibits a discontinuity or transition at lower temperature (between 20°C and 40°C) in the temperature-dependent H/D exchange (Fig. 5 a–e). Below the transition temperature, the exchange is fast and reaches an equilibrium within the first 20 min, and both the number of exchangeable Hs and the exchange rate show small changes with temperature. Above the transition temperature, there are significantly more slow and intermediate-exchangeable Hs, as evidenced by the appearance of slow kinetic components. Interestingly, all five of the peptides are located in the substrate-binding domain and are intertwined with each other to form the core of this domain (Fig. 5g). Note that the residues responsible for substrate binding are also located on the five peptides in the third group.
Fig. 3.

H/D exchange time course plots for peptides 11, 13–15, 18, and 19 at 10°C (•), 20°C (○), 40°C (▾), 55°C (▿), and 65°C (▪).
Fig. 4.

Temperature-dependent H/D exchange of the five peptides that show a transition between 40°C and 55°C. (a–e) H/D exchange time course plots for peptides 8–10, 17, and 21 at 10°C (•), 20°C (○), 40°C (▾), 55°C (▿), and 65°C (▪). (f) Plot of ln(kHX(WA)) vs. 1/T. The weight average H/D exchange rate constant kHX(WA) is defined as Ak1 + Bk2 + Ck3)/NH. (g) Structure of htADH monomer. The two zinc ions and the modeled-in cofactor NAD+ are shown in yellow. The cofactor-binding domain is on the left, and the substrate-binding domain is on the right. The five peptides exhibiting a transition above 40°C in the temperature-dependent H/D exchange are mapped onto the structure and colored.
Fig. 5.

Temperature-dependent H/D exchange of the five peptides that show a transition between 20°C and 40°C. (a–e) H/D exchange time course plots for peptides 1–4 and 7 at 10°C (•), 20°C(○), 40°C(▾), 55°C(▿), and 65°C (▪). (f) Plot of ln(kHX(WA)) vs. 1/T. The weight average H/D exchange rate constant kHX(WA) is defined as Ak1 + Bk2 + Ck3)/NH. (g) Structure of htADH monomer. The five peptides exhibiting a transition at 30°C in the temperature-dependent H/D exchange are mapped onto the structure and colored.
To analyze the exchange data in a more quantitative way, we introduce a weighted average hydrogen exchange rate constant defined as:
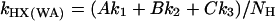 |
[1] |
where NH is the total number of amide Hs for each individual peptide, and A, B, and C are the number of amide Hs exchanging with rate constant k1, k2, and k3, respectively. In this manner, kHX(WA) represents the average rate constant for all amide Hs within a given peptide. Although kHX(WA) is not a traditional rate constant, it is an approximated measure of the extent of H/D exchange, because it takes into account the number of exchangeable Hs as well as the rate of exchange. The plots of the kHX(WA) vs. 1/T are nonlinear, and the lines break at ≈45°C for the peptides in group two (Fig. 4f) and at ≈30°C for the peptides in group three (Fig. 5f).
H/D Exchange Mechanism for Native Protein. Only single envelopes of isotope peaks and no bimodal isotope patterns have been observed in the mass spectra (16), indicating that htADH is structurally homogeneous and in its native state under exchange temperatures. For the amide hydrogens of a native protein to exchange with D2O, transient structural fluctuations are necessary to break intrinsic protein H bonds and to bring exchangeable amide Hs into H bonding contact with the catalyst OD- (18). The structural fluctuations can be either fast local fluctuations or relatively slower fluctuations involving cooperative motions or large subglobal unfoldings (19, 20).
It is generally assumed that exchange can be modeled as a two-state process involving the opening (kopen) and closing (kclose) of protein followed by reaction of the exposed amide with OD- (kex). Further, under native conditions and neutral pH, the closing step is generally much faster than the exchange step (kclose >> kex), and the H/D exchange falls into the EX2 limit, Eq. 2. Under this condition, the overall exchange rate is proportional to the equilibrium constant Keq for the interconversion between the closed and open states of protein and kHX is expressed as the product of kex and Keq.
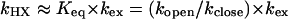 |
[2] |
Even under native conditions, a small fraction of the population of any protein will assume high energy conformational substates as described by the Boltzmann distribution (21), and raising the temperature is expected to populate these high-energy conformational substates. Because kex is also temperature dependent, the expectation is that the rate of exchange will increase with temperature in an Arrhenius fashion (see Fig. 3). In contrast, a number of the peptides from htADH exhibit a discontinuous change in the rate and the number of deuterons incorporated (Figs. 4 and 5). In these instances, we apply Eq. 3, which invokes a transition between two states as temperature is raised above a threshold value:
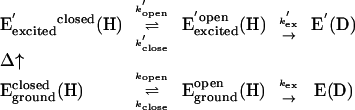 |
[3] |
Physical Origin of the Temperature-Dependent Transitions in H/D Exchange. It has been shown that an E. coli ADH that is highly homologous to htADH undergoes a conformational change when it binds NAD+ (22). The conformational change involves a rigid body rotation of the catalytic domain relative to the NAD+-binding domain, although the extent of rotation (≈2.5°) is significantly smaller than that for dimeric ADHs such as horse liver ADH (22). It was feasible that the transition in temperature-dependent H/D exchange shown in Figs. 4 and 5 resulted from a switch to a more open cofactor-free conformation at high temperatures, leading to the solvent exposure of a larger number of amide hydrogens. This appears highly unlikely, however, because the x-ray structure of htADH shows that the protein is already in an open form at reduced temperatures (23). If a structural change occurred as the temperature was raised into the range that is optimal for catalysis, it might be expected to bring the protein into a form that is better able to support catalysis, i.e., a more closed configuration. The strongest evidence against a temperature-dependent domain closure as the origin of the observations in H/D exchange comes from H/D exchange experiments in the presence of a high concentration of NAD+ (10 mM), which clearly show a reduction in exchange for some peptides together with no effect on the extent of deuteration and exchange rates for peptides 1–4, 7, and 9 (23).
The H/D exchange of the 10 peptides displaying a temperature-dependent transition reaches equilibrium quickly at low temperatures (Figs. 4 and 5), consistent with the exchangeable Hs within these peptides being made available to solvent mainly through fast local protein fluctuation characterized by small activation energy. The exchange patterns also indicate that the remainder of the amide hydrogens are well protected from the solvent (compactness) and that large-scale protein motion is limited (rigidity), supporting the suggestion that thermophilic proteins are less flexible in comparison with their mesophilic and psychrophilic counterparts at reduced temperature (24, 25). At elevated temperatures, some nonexchangeable Hs that are protected by hydrophobic packing and hydrogen bonding start to exchange with D2O, as evidenced by the emergence of intermediate and slow-exchanging Hs. The changes in the H/D exchange pattern, together with the appearance of the intermediate and slow-exchanging Hs, strongly suggest an initiation of large cooperative protein motions characterized by higher activation barriers. These cooperative motions are frozen out at low temperatures due to the absence of adequate thermal energy to overcome the kinetic barrier between the low- and high-energy conformational substates.
The transition above 40°C is accompanied by a change in NAD+-binding affinity, as measured by the quenching of protein fluorescence by NAD+. The fluorescence spectrum of free htADH exhibits a temperature-independent maximum at 336 nm when Trp residues are selectively excited. The dissociation constants (Kd) for NAD+ remain almost constant up to 45°C and increase by ≈2-fold above this temperature (see Table 7). Considering that four of the five peptides that show a transition above 40°C are in direct contact with the cofactor (Fig. 4g), the transitions in H/D exchange and binding affinity are most likely due to the same structural or dynamical changes around the cofactor-binding pocket. Peptide 17 plays a particularly important role in the binding and releasing of NAD+. A single-residue change within this fragment can have a significant impact on cofactor binding for ADHs, as suggested by the studies of horse liver ADH and the thermophilic ADH from Sulfolobus solfataricus (26, 27).
The transitions above 20°C are restricted to five peptides that reside in the substrate-binding domain (Fig. 5). One distinct feature of the substrate-binding domain is its primary composition of antiparallel β strands that are interconnected by multiple hydrogen bonds. It is probable that these observed transitions in H/D exchange are analogous to “phase-transition” processes characterized by simultaneous breakage of multiple hydrogen bonds. Additionally, we note the presence of 10 proline residues, including a cis-proline (Pro 56) located on peptide 3 (23), within the substrate-binding domain. The cis-proline at position 56, which is expected to rigidify the local structure, may undergo a cis to trans isomerization that initiates or contributes to the observed transition at 30°C.
It is clear that htADH undergoes at least two cooperative changes with increasing temperature, and that the changes are not related to domain opening/closing. The thermal activation of htADH is best viewed as a shift from a rigid substate to a preexisting high-energy substates with greater flexibility (Eq. 3). It is possible that this shift is also accompanied by some structural changes that alter the average configuration of the atoms, but a structural change without an accompanying alteration in protein dynamics cannot explain the changes in H/D exchange patterns. The possibility that there are subtle changes in the average structure of the thermally activated htADH, especially for surface loops, is currently under investigation by x-ray crystallographic studies.
Correlation Between Catalysis and Protein Mobility. One of the most compelling observations from this study is the coincidence of the transition in kHX(WA) for peptides 1–4 and 7 with kcat (see Figs. 1 and 5f). This leads to the linear correlations shown in Fig. 6. These five peptides are the only ones that show a linear relationship between kcat and kH(WA) with correlations of determination r2 > 0.9.¶ From a structural perspective, with the exception of peptide 1, peptides 2–4 and 7 contain residues located in a region of the active site that is expected to participate in substrate binding. We are led to the conclusion that an increase in mobility within the substrate-binding domain is intimately linked to the efficiency of hydride transfer. The observed correlations in Fig. 6 provide support for the view that specific and regional protein motions of htADH will evolve to enhance catalysis, presumably through promoting hydride transfer in htADH according to the environmentally coupled tunneling model (10). Although the peptides that show a transition above 40°C in H/D exchange have residues clustered around the NAD+-binding pocket, the transition in H/D exchange for these peptides occurs at a higher temperature than the transition in Arrhenius plot and any implied correlation between catalysis and flexibility changes in the regions of the protein near these peptides is more tenuous. The importance of conformational flexibility for catalysis in ADH is further supported by a comparative H/D exchange study, which shows that a highly homologous cold-adapted ADH exhibits enhanced conformational flexibility in the regions involved in substrate and cofactor binding at its physiologically relevant temperature (10°C) (Z.-X.L., I. Tsigos, T.L., V. Bouriotis, K.A.R., N.G.A., and J.P.K., unpublished work).
Fig. 6.

Correlation between kcat [obtained from the oxidation of benzyl alcohol (9)] and kHX(WA) for peptides 1 (coefficient of determination r2 = 0.98), 2 (r2 = 0.97), 3 (r2 = 0.93), 4 (r2 = 0.97), and 7 (r2 = 0.91).
Although NMR studies have suggested that the protein motion responsible for H/D exchange is likely to be on a micro- to millisecond time scale (28, 29), no information regarding the time scale of the motions responsible for exchange can be gleaned from the composite rate constant kHX(WA). It should be emphasized that the correlation between kcat and kHX(WA) in Fig. 6 does not indicate that the protein fluctuations responsible for H/D exchange are on the same time scale of protein motions as for catalysis. The catalysis-related protein vibrational modes are likely to be on a considerably shorter time scale than the cooperative protein fluctuations that are activated at elevated temperatures and are responsible for the H/D exchange (10, 30). Importantly, the correlations observed in this study imply that catalysis is proportional to the population of the high-energy conformational substates that determine the ratio of protein opening and closing rates for H/D exchange. Further spectroscopic studies, as well as molecular dynamics simulations in long time regimes, may yield insight into the nature of protein motion that both allows the access of amide to OD- during H/D exchange and leads to enhancement of hydrogen transfer catalysis.
Conclusion
A thermophilic alcohol dehydrogenase (htADH) has previously been shown to undergo a temperature-dependent transition between enzyme forms that differ in their properties for the hydride transfer and its accompanying tunneling parameters (9, 15). Using H/D exchange coupled to mass spectrometry, we now show that specific regions of htADH undergo changes in dynamic properties that correlate with cofactor binding and hydride transfer. Analysis of the H/D exchange pattern for 21 protein-derived peptides between 10 and 65°C indicates that approximately half of the enzyme is frozen in a rigid conformational substrate at reduced temperature. As the temperature is raised, the enzyme undergoes transitions to attain high-energy substates with enhanced mobility. For five peptides that reside in the substrate-binding domain, statistically significant linear correlations are observed between the temperature dependence of the average rate constants for H/D exchange and that for enzymatic turnover. These results provide unprecedented experimental support for the hypothesis that specific and regional protein motions are linked to the efficiency of enzymatic hydride transfer.
Supplementary Material
Acknowledgments
We thank Drs. Walter Englander and Brian Bahnson for valuable discussions. This work was supported by National Science Foundation Grants MCB0135446 (to J.P.K.), T32 GM07135 (to T.L.), GM48521 (to N.G.A.), and CA87648 (to K.A.R.).
Abbreviations: htADH, thermophilic alcohol dehydrogenase; H/D, hydrogen-deuterium.
Footnotes
The five peptides that show a transition above 40°C show some degree of correlation with r2 in the range of 0.82–0.89. The remaining peptides show weak correlation with r2 below 0.8, with the exception of peptides 11 (r2 = 0.89) and 20 (r2 = 0.87). All values for r2 are summarized in Table 8.
References
- 1.Nicholson, L. K., Yamazaki, T., Torchia, D. A., Grzesiek, S., Bax, A., Stahl, S. J., Kaufman, J. D., Wingfield, P. T., Lam, P. Y. S. & Jadhav, P. K. (1995) Nat. Struct. Biol. 2, 274-280. [DOI] [PubMed] [Google Scholar]
- 2.Osborne, M. J., Schnell, J., Benkovic, S. J., Dyson, H. J. & Wright, P. E. (2001) Biochemistry 40, 9846-9859. [DOI] [PubMed] [Google Scholar]
- 3.Eisenmesser, E. Z., Bosco, D. A., Akke, M. & Kern, D. (2002) Science 295, 1520-1523. [DOI] [PubMed] [Google Scholar]
- 4.Cole, R. & Loria, J. P. (2002) Biochemistry 41, 6072-6081. [DOI] [PubMed] [Google Scholar]
- 5.Wang, L., Pang, Y., Holder, T., Brender, J. R., Kurochkin, A. V. & Zuiderweg, E. R. P. (2001) Proc. Natl. Acad. Sci. USA 98, 7684-7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Knapp, M. J. & Klinman, J. P. (2002) Eur. J. Biochem. 269, 3113-3121. [DOI] [PubMed] [Google Scholar]
- 7.Basran, J., Harris, R. J., Sutcliffe, M. J. & Scrutton, N. S. (2003) J. Biol. Chem. 278, 43973-43982. [DOI] [PubMed] [Google Scholar]
- 8.Benkovic, S. J. & Hammes-Schiffer, S. (2003) Science 301, 1196-1202. [DOI] [PubMed] [Google Scholar]
- 9.Kohen, A., Cannio, R., Bartolucci, S. & Klinman, J. P. (1999) Nature 399, 496-499. [DOI] [PubMed] [Google Scholar]
- 10.Knapp, M. J., Rickert, K. & Klinman, J. P. (2002) J. Am. Chem. Soc. 124, 3865-3874. [DOI] [PubMed] [Google Scholar]
- 11.Maglia, G. & Allemann, R. K. (2003) J. Am. Chem. Soc. 125, 13372-13373. [DOI] [PubMed] [Google Scholar]
- 12.Basran, J., Patel, S., Sutcliffe, M. J. & Scrutton, N. S. (2001) J. Biol. Chem. 276, 6234-6242. [DOI] [PubMed] [Google Scholar]
- 13.Hammes-Schiffer, S. (2002) Biochemistry 41, 13335-13343. [DOI] [PubMed] [Google Scholar]
- 14.Caratzoulas, S., Mincer, J. S. & Schwartz, S. D. (2002) J. Am. Chem. Soc. 124, 3270-3276. [DOI] [PubMed] [Google Scholar]
- 15.Kohen, A. & Klinman, J. P. (2000) J. Am. Chem. Soc. 122, 10738-10739. [Google Scholar]
- 16.Hoofnagle, A. N., Resing, K. A. & Ahn, N. G. (2003) Annu. Rev. Biophys. Biomol. Struct. 32, 1-25. [DOI] [PubMed] [Google Scholar]
- 17.Resing, K. A., Hoofnagle, A. N. & Ahn, N. G. (1999) J. Am. Soc. Mass Spectrom. 10, 685-702. [DOI] [PubMed] [Google Scholar]
- 18.Milne, J. S., Mayne, L., Roder, H., Wand, A. J. & Englander, S. W. (1998) Protein Sci. 7, 739-745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Englander, S. W., Sosnick, T. R., Englander, J. J. & Mayne, L. (1996) Curr. Opin. Struct. Biol. 6, 18-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maity, H., Lim, W. K., Rumbley, J. N. & Englander, S. W. (2003) Protein Sci. 12, 153-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frauenfelder, H., Sligar, S. G. & Wolynes, P. G. (1991) Science 254, 1598-1603. [DOI] [PubMed] [Google Scholar]
- 22.Karlsson, A., El-Ahmad, M., Johansson, K., Shafqat, J., Jornvall, H., Eklund, H. & Ramaswamy, S. (2003) Chem. Biol. Interact. 143–144, 239-245. [DOI] [PubMed] [Google Scholar]
- 23.Ceccarelli, C., Liang, Z. X., Strickler, M., Prehna, G., Goldstein, B. M., Klinman, J. P. & Bahnson, B. J. (2004) Biochemistry 43, 5266-5277. [DOI] [PubMed] [Google Scholar]
- 24.D'Amico, S., Marx, J. C., Gerday, C. & Feller, G. (2003) J. Biol. Chem. 278, 7891-7896. [DOI] [PubMed] [Google Scholar]
- 25.Wintrode, P. L., Zhang, D., Vaidehi, N., Arnold, F. H. & Goddard, I. & William A. (2003) J. Mol. Biol. 327, 745-757. [DOI] [PubMed] [Google Scholar]
- 26.Rubach, J. K., Ramaswamy, S. & Plapp, B. V. (2001) Biochemistry 40, 12686-12694. [DOI] [PubMed] [Google Scholar]
- 27.Giordano, A., Cannio, R., Cara, F. L., Bartolucci, S., Rossi, M. & Raia, C. A. (1999) Biochemistry 38, 3043-3054. [DOI] [PubMed] [Google Scholar]
- 28.Arrington, C. B. & Robertson, A. D. (2000) J. Mol. Biol. 296, 1307-1317. [DOI] [PubMed] [Google Scholar]
- 29.Englander, S. W. & Hiller, R. (2001) in Methods in Enzymology (Academic, New York) Part C, Vol. 334, pp. 342-350. [DOI] [PubMed] [Google Scholar]
- 30.Francisco, W. A., Knapp, M. J., Blackburn, N. J. & Klinman, J. P. (2002) J. Am. Chem. Soc. 124, 8194-8195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.