

Abstract
Pulmonary drug disposition after inhalation is complex involving mechanisms, such as regional drug deposition, dissolution, and mucociliary clearance. This study aimed to develop a systems pharmacology approach to mechanistically describe lung disposition in rats and thereby provide an integrated understanding of the system. When drug‐ and formulation‐specific properties for the poorly soluble drug fluticasone propionate were fed into the model, it proved predictive of the pharmacokinetics and receptor occupancy after intravenous administration and nose‐only inhalation. As the model clearly distinguishes among drug‐specific, formulation‐specific, and system‐specific properties, it was possible to identify key determinants of pulmonary selectivity of receptor occupancy of inhaled drugs: slow particle dissolution and slow drug‐receptor dissociation. Hence, it enables assessment of factors for lung targeting, including molecular properties, formulation, as well as the physiology of the animal species, thereby providing a general framework for rational drug design and facilitated translation of lung targeting from animal to man.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC? ☑ Several drug and formulation properties are held as individually effective for achieving lung‐selectivity. However, simple empirical inhalation PK‐models do not allow for evaluation of the net effect of property combinations or for translation of inhaled drug pharmacology. • WHAT QUESTION DID THIS STUDY ADDRESS? ☑ What combinations of drug and formulation properties result in lung‐selective receptor occupancy, given the physiology of the test species?. • WHAT THIS STUDY ADDS TO OUR KNOWLEDGE ☑ A mechanistic inhalation PK‐model was developed and is made available. This model can guide the design of compounds and inhaled drug formulations with optimal local pharmacology and provide a logic framework for translation of inhaled drug pharmacology. Specific findings in this study include lung‐selectivity possibly being unattainable in the well‐perfused parts of the lung and that slow drug‐receptor dissociation can be a drug property providing lung‐selectivity. • HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS ☑ The model can be used in clinical studies to tailor inhaled drug formulations, target appropriate dose ranges, and interpret study results.
Inhalation is an attractive route of administration that has been used for more than 2,000 years.1 The capability of delivering drug directly to the target organ has been associated with advantages, such as a rapid onset of action and a higher and more sustained local tissue concentration.2 The latter offers an opportunity to increase the therapeutic index by achieving lung‐selectivity and thus fulfilling the aim of locally acting inhaled drugs (i.e., to obtain high concentrations at the lung target site while the systemic concentrations are kept at a minimum).3 In order to minimize the systemic exposure, and thus systemic side effects, drug discovery typically aims to develop inhaled drugs with high hepatic clearance to obtain a rapid elimination and to avoid absorption from the gastrointestinal tract.4
Nevertheless, achieving lung‐selectivity after inhalation is not a trivial task. The large surface area, good vascularization, and thin alveolar epithelium offer the potential for rapid absorption into the systemic circulation.5 Indeed, with the exception of i.v. administration, inhalation is the fastest route for systemic drug delivery of small molecules. This is particularly prominent for small lipophilic molecules, in which the absorption half‐life is ∼1–2 minutes.6 Several strategies for enhancing lung retention have therefore been explored, including increasing basicity,7 formulation approaches8 and low solubility.9
However, assessment of lung‐selectivity has so far proven to be elusive. Collection of relevant exposure measurements is recognized as a challenge both within clinical and preclinical research. Because the appearance of drug in the systemic circulation is the result of pulmonary absorption, unbound concentrations in plasma cannot be assumed to reflect the target site concentration in the lung.2 This constitutes a challenge because unbound plasma concentrations usually form the basis for establishing pharmacokinetic/pharmacodynamic (PK/PD) relationships.
In a preclinical setting, lungs can be collected by destructive sampling at several time points after intratracheal administration or dry powder inhalation. Drug concentrations are subsequently measured in lung tissue homogenates, providing a time profile of total lung concentrations in which the organ is erroneously reflected as one anatomic entity. Moreover, the homogenization process severely distorts the data interpretation by disrupting the normal compartmentalization (e.g., lysosomal trapping) and by dissolving solid drug particles.10 Indeed, the establishment of PK/PD relationships based on total lung concentrations is known to be more challenging for poorly soluble compounds.7 As receptor occupancy is driven by the unbound drug concentration at the target site, such measurements could clarify the PK after topical administration. A methodology capable of in vivo receptor occupancy measurements in the lung and in a reference organ for systemic exposure has therefore been developed.11 By applying this method, further understanding can be gained as comparison of pulmonary and systemic occupancies provides a quantitative readout of the degree of lung‐selectivity achieved by inhalation.
Even so, interpretation of data from preclinical PK studies can be challenging. Rodents are generally exposed via nose‐only inhalation, in which a substantial deposition of drug particles will occur in the nose.10 Drug deposited in the lung and the nose will both be subject to the self‐cleansing mechanism mucociliary clearance (MCC), which transports drug particles toward the pharynx where they are eventually swallowed.12 Accordingly, the resulting plasma PK is a result of parallel absorption from the lung, the nose, and the gastrointestinal tract.13, 14
Hence, despite the historical success and widespread use of locally acting inhaled drugs, pulmonary drug disposition remains poorly understood. It is recognized as complex because several important processes take place simultaneously, such as regional drug deposition, dissolution of solid drug particles and MCC. Additional complexity comes from the heterogeneous nature of the organ with distinct differences between the tracheobronchial and alveolar regions.9 An integrated understanding, which takes the mechanistic processes as well as the organ heterogeneity into account, is thus desirable.
Simulation models have previously been used to predict the systemic exposure for inhaled corticosteroids in humans. Weber and Hochhaus15 developed a compartmental simulation tool, in which the lung was divided into two subcompartments representing the central and peripheral region, respectively. The model also included features, such as MCC and drug dissolution, described by rate constants.15 Earlier simulations, using an even simpler model structure with one lung compartment and receptor binding described by a static model, showed that slow drug dissolution gives lung‐selectivity.16 Chaudhuri et al.17 used GastroPlus to predict the systemic PK of budesonide. The simulated plasma profiles of both models proved to agree well with experimental data. Nevertheless, a mechanistic model predictive of local tissue concentrations combined with measurements such as receptor occupancy for validation is currently lacking. Such a model would be necessary to elucidate the highly complex processes involved in pulmonary drug disposition.18
This study aimed to develop a physiologically based pharmacokinetic model including lung disposition for rats, which can integrate the current knowledge of the system with drug‐ and formulation‐specific properties. The model puts emphasis on lung disposition by mathematically describing both the physiology and the fate of the deposited dose. When drug‐ and formulation‐specific input parameters for the poorly soluble drug fluticasone propionate (FP) were fed into the model, it accurately predicted the PK both after i.v. administration as well as via nose‐only inhalation. It was also possible to predict target site exposure, as validated by receptor occupancy measurements. Because this approach allows for separation between drug‐ and system‐specific properties, it will aid in understanding which drug‐and/or formulation‐specific properties are associated with lung‐selectivity and thus yield favorable efficacy/safety profiles for inhaled drugs. Moreover, because the parametrization of this model is based on the physiology of the animal, it provides a framework for a facilitated translation by switching to human system‐specific properties.
METHODS
Experiments
Please refer to the Supplementary Material for experimental details about the PK and the receptor occupancy studies.
PK study
The PK of FP was studied in rats after i.v. administration of 90 and 1,000 nmol/kg (n = 2 and n = 3, respectively). Blood samples were repeatedly collected from a venous catheter for 8 hours after administration.
Receptor occupancy studies
The time course of glucocorticoid receptor occupancy and the PK was studied in rats after nose‐only inhalation (lung deposited dose [LDD]: 11.3 nmol/kg) and i.v. administration of FP (90 nmol/kg), the latter study as well as a detailed experimental protocol are described in a previous study.11 The inhalation study was performed twice using the same experimental setup.
Model development
This section gives an overview of the model. Detailed descriptions of the model, its different subcomponents, and the mathematical derivations are included in the Supplementary Material.
Model structure
A mechanistic physiologically based pharmacokinetic model (PBPK) including lung disposition was implemented in MATLAB R2013a (Mathworks, Natick, MA). The structural model is illustrated in Figure 1 a. Blood flows and volumes of the tissue compartments included are presented in Table 1.
Figure 1.
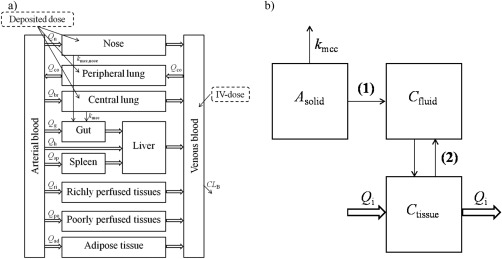
(a) Structure of the whole‐body physiologically based pharmacokinetic (PBPK) model, and (b) compartmental representation of the central lung, peripheral lung, and the nose: solid drug (Asolid), dissolved drug in the epithelial or nasal lining fluid (Cfluid), and drug in tissue (Ctissue). In the nose and the central lung, solid particles are transported by mucociliary clearance (kmcc). Drug particles are dissolved in the lining fluid (1), once dissolved the drug may permeate through the epithelial membrane to the tissue (2).
Table 1.
System‐specific input parameters for the rat.
| Tissue | Volume (fraction of BW) | Blood flow (fraction of Q CO) |
|---|---|---|
| Adipose | 0.040a | 0.009217a |
| Gut | 0.0259b | 0.14b |
| Liver | 0.04c | 0.024b |
| Lung | 0.004127d | 0.021b/1 |
| Nose | 0.000254e | 0.0015f |
| Poorly perfusedg | 1‐(the rest) | 1‐(the rest) |
| Richly perfusedh | 0.039c | 0.5096c |
| Spleen | 0.002b | 0.0715i |
| Arterial blood | 0.02c | NA |
| Venous blood | 0.04c | NA |
The lung was divided into a tracheobronchial and alveolar region; each of these was in turn divided into three separate compartments (Figure 1 b). The same compartmental representation was used for the nose: (1) solid drug (Asolid); (2) dissolved drug in the epithelial or nasal lining fluid (Cfluid); and (3) drug in tissue (Ctissue). The tracheobronchial region is perfused by the bronchial blood flow (Qbr), the alveolar region by the entire cardiac output (QCO), and the nose by the nasal blood flow (Qn).
Perfusion‐rate limited distribution was assumed to apply for all tissues. For compartment i, the rate of change of quantity within the organ can be described as24:
| (1) |
where Vi is the tissue volume, Ci is the drug tissue concentration, Qi is the blood flow to the tissue, CA is the arterial drug concentration, R is the blood/plasma ratio, and Kp,i is the tissue‐plasma partition coefficient.
Particle size distribution and regional deposition
The particle size distribution was determined using an impactor, providing a discrete distribution consisting of eight particle size classes. The mass fractions (f 1,…,f 8) are presented in Table 2 and experimental details in the Supplementary Material.
Table 2.
Drug‐ and formulation‐specific input parameters for fluticasone propionate
| Parameter | Value |
|---|---|
| Blood/plasma ratio | 0.95 |
| CLB (L/h/kg) | 11.53 |
| CLP (L/h/kg) | 10.95 |
| Cs (nM) | 4530 |
| Diffusion coefficient (m2/s) | 2.27*10−11 |
| f 1,…,f 8 a | 0.17, 0.30, 0.26, 0.18, 0.073, 0.0091, 0.0032, 0.0035 |
| F b | 0 |
| fu | 0.016 |
| fu,fluid | 1 |
| Kd (nM) | 0.015 ± 0.0045 |
| Koff (h−1) | 0.51 ± 0.17 |
| Kon (L/nmol/h) | 34 ± 20 |
| logD7.4 | 4.2 |
| Molecular weight (g/mol) | 500.6 |
| Papp (cm/s) | 46.9*10−6 |
| Particle density (nmol/dm3) | 1.430*109 |
| r 1,…,r 8 (µm) | 3.55, 2.31, 1.42, 0.887, 0.544, 0.349, 0.231, 0.118 |
| Vdss (L/kg) | 12.5 |
| Vu,lung (mL/g lung tissue) | 213.4 |
CLB = blood clearance; CLP = plasma clearance; C s = solubility; f 1,…,f 8 = mass fractions for particle size classes 1,…,8; F = oral bioavailability; fabs = fraction absorbed; fgut = fraction escaping gut metabolism; fh = fraction escaping hepatic metabolism; f u = fraction unbound in plasma; fu,fluid = fraction unbound in epithelial or nasal lining fluid; Kd = dissociation constant; Koff = dissociation rate constant; Kon = association rate constant; Papp = apparent permeability; r 1,…,r 8 = initial geometric radius for particle size classes 1,…,8; Vd,ss = volume of distribution at steady state; Vu,lung = unbound lung volume of distribution.
when all decimal places are used.
F=fabs*fgut*fh.
Inhaled drug particles can be deposited in the extrathoracic, tracheobronchial, and alveolar region. This model neglects deposition in the pharynx, hence only nasal deposition is considered in the extrathoracic region. Henceforth, the tracheobronchial and alveolar regions are referred to as the central and peripheral lung, respectively.
Several models have been developed for the prediction of regional particle deposition in rat lungs.25, 26, 27, 28 By using deposition fractions for the relevant aerodynamic diameters extracted from ref. 25 and the mass fractions for the corresponding particle size classes, the number of deposited particles of size class i in region j (Nj,i) can be calculated. As specified in the Supplementary Material, Nj,i is used for simulating drug dissolution and total amount of solid drug.
Mucociliary clearance
Because of MCC, Nj,i was modeled as an exponential decay:
| (2) |
where Nj,i(0) is the number of particles of size class i in region j at t = 0 and kmcc,j is MCC in region j. MCC in the central lung (kmcc,lung) was estimated from ref. 29 as described in the Supplementary Material and MCC in the nose (kmcc,nasal) was extracted from ref. 30; the rate constants are presented in Table 3. MCC in the peripheral lung was assumed to be negligible, since it is primarily associated with the tracheobronchial region.34 Consequently, Nj,i in the peripheral region is constant. Drug removed by MCC is transported to the gut, where the bioavailable fraction (F) subsequently can be absorbed into the systemic circulation. F is defined as:
| (3) |
Table 3.
System‐specific input parameters for the central lung, peripheral lung, and the nose
| Parameter | Central lung | Peripheral lung | Nose |
|---|---|---|---|
| Blood flow (fraction of Q CO) | 0.021a | 1 | 0.0015b |
| Surface area (dm2/kg) | 3.27c | 276.4d | 0.416e |
| Lining fluid volume (µL/kg) | 163.6a | 193.5a | 20.8a |
| Fraction of tissue volume | 0.19a | 0.81a | NA |
| k mcc (h−1) | 0.0472f | NA | 0.2079 |
kmcc, rate constant for mucociliary clearance; NA, not available; QCO, cardiac output.
That is, F accounts for the fraction absorbed from the gastrointestinal tract (fabs), the fraction that escapes the gut (fgut), and hepatic extraction (fh).35 Because of the high blood clearance (CLb), fh was set to 0.
Dissolution of drug
Drug particles are dissolved in the epithelial lining fluid or in the nasal lining fluid. The dissolution process is modeled by the Nernst–Brunner equation,36, 37 describing how the dissolution rate depends on the solubility of the drug (Cs), the concentration in the dissolution medium, the diffusion coefficient (D), and the surface area of the particle (4πrj,i(t)2). Detailed descriptions of both the dissolution and calculation of D 35 are provided in the Supplementary Material.
Detailed description of the lung and nose
The compartmental representation of the nose and the two lung regions is shown in Figure 1 b. The system‐specific input parameters for the nose and lung are summarized in Table 3. Once the drug has dissolved (Supplementary Material Eq. S33) it may permeate to the tissue according to:
| (4) |
where dJ/dt is the molar flow of drug (nmol/h), P is the permeability, Asurf is the surface area, Ci is the tissue concentration of drug, and Kp,u,i is the tissue‐to‐unbound plasma partition coefficient. A detailed description of the prediction of Kp,u‐values as well as the subsequent calculations of Kp‐values are provided in the Supplementary Material, all Kp‐values are presented in Table 4. The in vitro apparent permeability across CaCo2‐monolayers was measured and used as P (Table 2).
Table 4.
Tissue‐plasma partition coefficients (Kp) for tissues included in the model
Receptor binding
Receptor binding was included in all tissue compartments and was described as:
| (5) |
where RD is the concentration of the drug‐receptor complex, Kon is the association rate constant, Bmax is the receptor density, and Koff is the dissociation rate constant.
Bmax for the spleen was 31.5 nM,40 Bmax for the lung was set to 21 nM,11 and Bmax in the other tissue compartments was set to the mean value of Bmax over five brain regions (23 nM).41
Parameterization of the model
Drug‐specific input parameters
All drug‐specific input parameters are specified in Tables 2 and 4. The blood/plasma ratio (R) was used for calculating CLB from the plasma clearance (CLP) obtained from the PK‐study (Supplementary Material Eq. S21). As CLP was estimated from venous drug concentrations, elimination was set to occur from the venous compartment. Accordingly, CLB acts on the absorbed drug prior to entering the organs.
Parameter estimation
Cs was estimated using nonlinear least squares in the MATLAB curve‐fitting toolbox, in which the Levenberg–Marquardt algorithm minimized the difference between observed and predicted total lung concentrations (11.3 nmol/kg, LDD). The initial estimate had been selected from an exhaustive search, in which the sum of squares was initially evaluated using a broad search space (102≤Cs≤105 nM). The search space was then confined to the proximity of the best solution Cs,0 (Cs,0−200≤Cs≤ Cs,0+200 nM), in which 300 candidates were evaluated.
With the exception of Cs, all input parameters in the PBPK model are from literature sources, internal AstraZeneca data, or parameter estimates obtained from modeling of the PK and glucocorticoid receptor occupancy studies as specified.
Characterization of binding kinetics
Modeling of the binding kinetics is described in detail in the Supplementary Material.
Model validation and verification
Administrations via the i.v. route (20, 90, 150, 750, and 1,000 nmol/kg) and via nose‐only inhalation (11.3 and 100 nmol/kg, LDD) were simulated using drug‐specific input parameters for FP (Tables 2 and 4). Neither particle size distribution nor density was available for the higher LDD, it was therefore assumed to have the same formulation‐specific properties as the lower dose. The simulations were subsequently compared with experimental data obtained from this study, AstraZeneca's internal database, or ref. 11.
Evaluation of concepts for lung‐selectivity
In this article, lung‐selectivity is defined as the difference between pulmonary (ROlung) and systemic receptor occupancy (spleen, ROsp) i.e., the criteria of lung‐selectivity is met when:
| (6) |
Because the lung is divided into a central and a peripheral region, lung‐selectivity can be evaluated for each region individually. Besides, the whole lung can also be considered by using a weighted average based on the occupancy for the two regions (ROave; Supplementary Material Eq. S51). Because the experimental methodology cannot discriminate between central and peripheral occupancy, ROave corresponds to the observations.
The impact of the following drug‐specific input parameters on lung‐selectivity was evaluated: CL, F, Cs, and Koff. Furthermore, one formulation‐ and one system‐specific input parameter were each investigated: particle size and Qn, respectively. The contribution of nasal absorption was evaluated by comparing the systemic PK profile from a base‐case scenario (input parameters from Tables 1, 2, 3, 4) to a scenario in which Qn was set to 0. The particle size distribution was investigated by comparing the base‐case scenario to one in which the particles were evenly distributed between the four smallest size classes, that is fi = 0.25 for i = 5,…,8 and fi = 0 otherwise. In all simulations, LDD was fixed at 11.3 nmol/kg.
Statistics and data presentation
Monte Carlo simulations were used for receptor occupancy simulations to account for the uncertainty in the estimated binding kinetics parameters. Random parameter values for Kd and Koff were generated from a lognormal distribution based on the parameter precision obtained from the modeling. A large number of simulations (n = 1,000) were made and 90% confidence intervals were constructed.
Receptor occupancy measurements are presented as mean ± one SE. As described in the Supplementary Material, receptor occupancy was calculated using the ratio method42 and propagation of error was used for the calculation of SE.
Sensitivity analysis was performed to investigate how the dynamic behavior of the system responded to changes in selected input parameters (Supplementary Figure S5a,b).
RESULTS
Characterization of binding kinetics
Kon and Koff were estimated to be 34 ± 20 nM−1h−1 and 0.51 ± 0.17 h−1 (Supplementary Figure S1b). An exhaustive search algorithm confirmed that the global minimum had been found within the defined parameter space.
Model verification and validation
I.V. administration
Model predictions of the plasma profiles as well as the time course of occupancy (Figure 2 a,b) were consistent with experimental i.v. data (90 and 1,000 nmol/kg). The predicted occupancy profiles in the two lung regions and the spleen were identical. No occupancy measurements were available for the higher dose. Regardless of the route of administration, inclusion of the receptor‐bound concentration was essential for the predictive capability of spleen concentrations (Figure 2 c).
Figure 2.
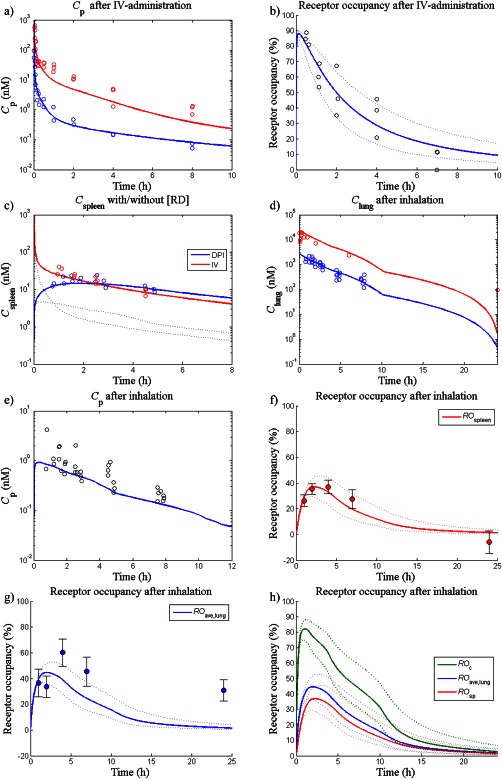
Simulations and observations of: (a) plasma concentrations (Cp) after i.v. administration of 90 (blue line) and 1,000 nmol/kg (red line), (b) receptor occupancy in the spleen/peripheral lung/central lung after i.v. administration of 90 nmol/kg (blue line), (c) spleen concentration (Cspleen) after i.v. administration of 90 nmol/kg (red line) and after inhalation of a lung deposited dose (LDD) of 11.3 nmol/kg (blue line). The dashed lines show Cspleen excluding the receptor‐bound concentration (d) total lung concentrations after an LDD of 11.3 nmol/kg (blue line) and 100 nmol/kg (red line), (e) Cp after a LDD of 11.3 nmol/kg (blue line), (f) receptor occupancy in the spleen (ROsp) after an LDD of 11.3 nmol/kg (red line), (g) whole lung receptor occupancy (ROave) after an LDD of 11.3 nmol/kg (blue line), and (h) receptor occupancy in the central lung (ROc), ROave, and ROsp after an LDD of 11.3 nmol/kg (green, blue, and red line, respectively). For each receptor occupancy simulation, a 90% confidence interval was created by a Monte Carlo simulation, which repeatedly sampled random values from a lognormal distribution of the binding kinetics parameters (n = 1,000, dashed lines). DPI, dry powder inhalation.
The model was predictive of receptor occupancy measurements made 1.5 hours after three i.v. doses of FP. Simulated occupancies were 27%, 64%, and 86% for 20, 150, and 750 nmol/kg, respectively. Simulations agreed well with observed data: 27 ± 9.7, 74 ± 5.0, and 100 ± 3.5% for 20, 150, and 750 nmol/kg, respectively.
Nose‐only inhalation
Following estimation of Cs (95% confidence interval = 4,530 [3,845–5,215] nM), total spleen, lung and plasma concentrations were well‐predicted by the model after nose‐only inhalation (Figure 2 c–e). An underprediction was noted for the last time point of the higher dose.
Model predictions of the systemic occupancy were consistent with the observations (Figure 2 f). ROave captured key trends in the data, although a tendency toward underprediction was noted (Figure 2 g). Neither plasma concentrations nor receptor occupancy had been measured following inhalation of the higher dose.
Evaluation of concepts for lung‐selectivity
It was noted that lung‐selectivity could not be achieved if the peripheral lung was considered as the pulmonary region. That is, no difference was obtained between the occupancy in the peripheral lung and the spleen in this model. Therefore, the occupancy in the central lung (RO C) is used as the pulmonary region for evaluation of lung‐selectivity.
As can be seen in Figure 3 a, a longer period of lung‐selectivity was obtained for a poorly soluble drug (Cs = 2.5 µM) than for a highly soluble drug (Cs = 50 µM). A transient concentration gradient created during dissolution of a highly soluble compound (Cs = 50 µM) could also give rise to a prolonged lung‐selectivity given a slow Koff (Figure 3 b).
Figure 3.
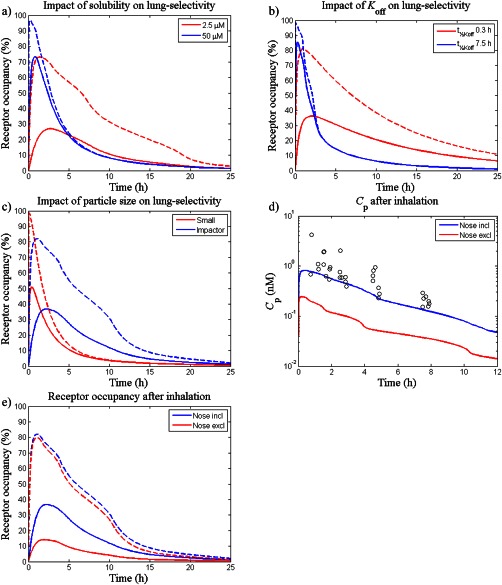
The impact of different drug‐, formulation‐, and system‐specific properties on lung‐selectivity was evaluated by varying the following parameters: (a) solubility; Cs=2.5 µM (red line) and 50 µM (blue line), (b) dissociation rate; t½, Koff=7.5 h (blue line) and 0.3 h (red line), (c) particle size distribution; f 1,…,f 8 from Table 2 (blue line) and fi = 0.25 for i = 5, …, 8 and fi = 0 otherwise (red line), (d) nasal absorption; nose included (blue line) and nose excluded (red line), and e) nasal absorption; nose included (blue line) and nose excluded (red line). Except for subfigure e) which shows predictions (lines) and observations (open circles) of plasma concentrations of fluticasone propionate (Cp), dashed lines represent receptor occupancy in the central lung and solid lines represent occupancy in a systemic reference organ.
Given a certain LDD, simulations showed that the particle size distribution had an impact on the occupancy profile as well as the degree of lung‐selectivity (Figure 3 c).
Simulations showed a negative correlation between F and lung‐selectivity, whereas a positive correlation was found for CL (simulations not shown).
According to the simulations, nasal absorption significantly contributed to the systemic exposure and decreased the degree of lung‐selectivity following nose‐only inhalation of FP (Figure 3 d,e). Noteworthy is that the nasally deposited dose was predicted to be several‐fold higher than LDD (Supplementary Figure S6).
DISCUSSION
This article presents a systems pharmacology approach for prediction of systemic and pulmonary PK for inhaled drugs, which was validated by experimental measurements of drug concentrations and receptor occupancy. By virtue of being mechanistic, this model provides a tool to theoretically explore pulmonary drug disposition and how key processes in a physiological context produce lung‐selectivity.
Data generated from i.v. dosing as well as inhalation were used for validation, which thus allows the model to be used to compare the two administration routes. Model predictions of i.v. administrations were consistent with experimental data (Figure 2 a–c), supporting a perfusion‐rate limited distribution. Notably, input parameters (CL, Vd,ss, Kon, and Koff) obtained from the modeling of one dataset (90 nmol/kg, i.v.) proved predictive of data from four other i.v. dose levels, thus offering strong support and confidence in its predictive capability to determine systemic PK and receptor occupancy. Interestingly, inclusion of receptor binding was necessary for accurate predictions of spleen concentrations (Figure 2 c), which verifies that FP has a high receptor‐bound fraction.11 This elucidates the potential pitfall of only relying on Kp‐values when predicting tissue concentrations after low doses of highly potent compounds. Under such circumstances, underpredictions are inevitable as Kp‐values do not account for receptor binding.
As the aqueous solubility of poorly soluble compounds tends to underpredict the in vivo dissolution rate,43 the single parameter Cs was estimated from observations of total drug concentrations in the lung made in one inhalation study (11.3 nmol/kg, LDD). It is worth noting that the estimate of Cs (4,530 nM) was close to the measured FaSSIF‐solubility (3,120 nM). When the optimized model was tested on another dataset (100 nmol/kg, LDD), it was shown to be predictive of the total lung concentrations with the exception of the last time point (Figure 2 d). This could be explained by either inaccurately described particle size distribution or limitations of the Nernst–Brunner equation for the alveoli where the epithelial lining fluid layer might be smaller than the particle diameter.
Model predictions of plasma concentrations and systemic occupancy after nose‐only inhalation agreed well with experimental data (Figure 2 e,f). This consistency confirms that FP has a dissolution rate‐limited absorption and emphasizes the importance of mechanistically describing the dissolution process for such compounds.
Validation of lung occupancy simulations was slightly more complex as these measurements reflect whole lung occupancy. Thus, for comparison of observations and simulations, a weighted average accounting for the relative contribution of each region was needed. Although exact determination of the tissue fractions cannot be made, the volume fraction of the central lung (fv,c) was approximated to be 0.19 (Supplementary Material). Given the uncertainty in fv,c and the slightly lower accuracy of lung occupancy measurements, a whole lung occupancy prediction that qualitatively captures key features, including lung‐selectivity and late occupancy peak (Figure 2 g), can be regarded as a good description of the data.
For validation purposes, emphasis was not put on explaining the variability in the data, which is partly caused by the use of destructive sampling (one animal/time point). Apart from interindividual differences in model input parameters, a high variation in LDD is expected from preclinical studies. The validation instead focused on how well the model captured key features in the observations, both on a quantitative and a qualitative level.
The model was used to provide mechanistic insight into pulmonary drug disposition and to identify key determinants for lung‐selectivity. As expected, simulations confirmed the current dogma that inhaled drugs benefit from a high CL and a low F. A previously unforeseen result was that lung‐selectivity could not be achieved in the peripheral lung. This is attributed to the high perfusion rate of this region, which thus rapidly equilibrates with the systemic circulation. In fact, the model predicted the tissue distribution half‐life of FP in the peripheral lung to be below 2 seconds. However, lung‐selectivity could be obtained in the central region after inhalation as its lower perfusion‐rate allows for a longer equilibration time.
It was shown that a concentration gradient, and thus lung‐selectivity, was obtained during the dissolution phase (Figure 3 a). As expected, the model described the risk of only obtaining transient lung‐selectivity for a rapidly dissolving drug without any additional mechanisms enhancing its lung‐retention (i.e., a scenario resembling i.v. administration in which no lung‐selectivity is obtained). Nevertheless, a transient concentration gradient can give rise to an extended period of lung‐selectivity provided that the drug receptor dissociation rate is relatively slow (Figure 3 b). The latter feature was unforeseen by earlier models because the receptor binding was described by a static Emax‐model.16 Accordingly, two mechanisms were found to be good strategies for achieving lung‐selectivity: (1) slow dissolution; and (2) slow drug receptor dissociation. Notably, at the other end of the spectrum are low affinity compounds for which a low Cs might disrupt the opportunity of obtaining sufficiently high target site concentrations to elicit a pharmacological response. Hence, several drug‐specific properties need to be considered during lead compound optimization and the presented model is indeed useful for such exercises.
Simulations verified that the particle size distribution also has an impact on the dissolution rate and can thus be used to partly control this process (Figure 3 c). Moreover, particle size is well‐known to be an important determinant of the regional deposition pattern. Model predictions elucidated that a high nasal deposition, possibly accompanied by significant absorption, is expected following nose‐only inhalation studies (Supplementary Figure S6). Although nasal absorption is absent for orally inhaled products in the clinic, simulations suggest that nasal uptake reduces the degree of lung‐selectivity seen in preclinical models (Figure 3 d,e). Thus, accounting for this process might be important for interpretation and translation of preclinical data, further emphasizing the need of an integrated understanding of formulation‐specific, drug‐specific, and system‐specific properties. If the technical challenges can be overcome, experiments addressing the extent of nasal absorption after nose‐only inhalation would indeed be useful. Furthermore, this example illustrates how the model lends itself for translation. As it relies on a physiological parametrization, translation from animal to man can be done by changing from nasal to oral inhalation and exchanging system‐specific (rat vs. human physiology) and formulation‐specific parameters (particle size, etc.). Human system‐specific parameters are provided in the Supplementary Material. This study demonstrated the value of mechanistically describing the underlying processes of drug disposition in the lung to understand how the delicate interactions among drug‐specific, formulation‐specific, and system‐specific properties produce the final outcome of the system. The model thereby provides a framework for rational drug design and a facilitated translation from animal to man, which will be instrumental to any drug discovery or development program targeting the lung via the inhaled route.
Study Highlights
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Several drug and formulation properties are held as individually effective for achieving lung‐selectivity. However, simple empirical inhalation PK‐models do not allow for evaluation of the net effect of property combinations or for translation of inhaled drug pharmacology.
WHAT QUESTION DID THIS STUDY ADDRESS?
The combinations of drug and formulation properties that result in lung‐selective receptor occupancy, given the physiology of the test species.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
A mechanistic inhalation PK‐model was developed and is made available. This model can guide the design of compounds and inhaled drug formulations with optimal local pharmacology and provide a logic framework for translation of inhaled drug pharmacology. Specific findings in this study include lung‐selectivity possibly being unattainable in the well‐perfused parts of the lung and that slow drug‐receptor dissociation can be a drug property providing lung‐selectivity.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS
The model can be used in clinical studies to tailor inhaled drug formulations, target appropriate dose ranges, and interpret study results.
Conflict of Interest. The authors declared no conflict of interest.
Author Contributions. E.B., N.E., M.J.C., A.L., P.E., A.W., and M.F. wrote the article. E.B., P.E., and M.F. designed the research. E.B., A.L., A.W., and M.F. performed the research. E.B., N.E., M.J.C., P.E., and M.F. analyzed the data. A.L. contributed new reagents/analytical tools.
Supporting information
Supplementary information accompanies this paper on the CPT: Pharmacometrics & Systems Pharmacology website (http://www.wileyonlinelibrary.com/psp4).
Supplementary Information Figure 1.
Supplementary Information Figure 2.
Supplementary Information Figure 3.
Supplementary Information Figure 4.
Supplementary Information Figure 5.
Supplementary Information Figure 6.
Supplementary Information Figure 7.
Supplementary Information Table 1.
Supplementary Information Table 2.
Supplementary Information Table 3.
Supplementary Information Table 4.
Supplementary Information
Supplementary Information
Acknowledgments
This work was supported by the Marie Curie FP7 People ITN European Industrial Doctorate (EID) [Project No. 316736] and the Innovative Modelling for Pharmacological Advances through Collaborative Training (IMPACT). The authors thank Susanne Arlbrandt, Britt‐Marie Fihn, Marie Johansson, and Gina Hyberg for their invaluable contributions to the in vivo studies. Sara Johansson and Annica Jarke are acknowledged for their help with formulating nanosuspensions of FP for the PK study. Furthermore, the authors would like to thank Jan Westergren and Ulrika Tehler for fruitful discussions. Finally, gratitude is also extended to Erica Bäckström for kindly providing measurements of the unbound lung volume of distribution for FP.
References
- 1. Sanders, M. Inhalation therapy: an historical review. Prim. Care Respir. J. 16, 71–81 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bäckman, P. , Adelmann, H. , Petersson, G. & Jones, C.B. Advances in inhaled technologies: understanding the therapeutic challenge, predicting clinical performance, and designing the optimal inhaled product. Clin. Pharmacol. Ther. 95, 509–520 (2014). [DOI] [PubMed] [Google Scholar]
- 3. Olsson, B. et al Pulmonary Drug Metabolism, Clearance, and Absorption Controlled Pulmonary Drug Delivery (eds. Smyth H. & Hickey A.) pp. 21–50 (Springer, New York, NY, 2011). [Google Scholar]
- 4. Winkler, J. , Hochhaus, G. & Derendorf, H. How the lung handles drugs: pharmacokinetics and pharmacodynamics of inhaled corticosteroids. Proc. Am. Thorac. Soc. 1, 356–363 (2004). [DOI] [PubMed] [Google Scholar]
- 5. Agu, R.U. , Ugwoke, M.I. , Armand, M. , Kinget, R. & Verbeke, N. The lung as a route for systemic delivery of therapeutic proteins and peptides. Respir. Res. 2, 198–209 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Patton, J.S. , Fishburn, C.S. & Weers, J.G. The lungs as a portal of entry for systemic drug delivery. Proc. Am. Thorac. Soc. 1, 338–344 (2004). [DOI] [PubMed] [Google Scholar]
- 7. Cooper, A.E. , Ferguson, D. & Grime, K. Optimisation of DMPK by the inhaled route: challenges and approaches. Curr. Drug Metab. 13, 457–473 (2012). [DOI] [PubMed] [Google Scholar]
- 8. Loira–Pastoriza, C. , Todoroff, J. & Vanbever, R. Delivery strategies for sustained drug release in the lungs. Adv. Drug Deliv. Rev. 75, 81–91 (2014). [DOI] [PubMed] [Google Scholar]
- 9. Patton, J.S. & Byron, P.R. Inhaling medicines: delivering drugs to the body through the lungs. Nat. Rev. Drug Discov. 6, 67–74 (2007). [DOI] [PubMed] [Google Scholar]
- 10. Forbes, B. et al Challenges in inhaled product development and opportunities for open innovation. Adv. Drug Deliv. Rev. 63, 69–87 (2011). [DOI] [PubMed] [Google Scholar]
- 11. Boger, E. et al A novel in vivo receptor occupancy methodology for the glucocorticoid receptor: toward an improved understanding of lung pharmacokinetic/pharmacodynamic relationships. J. Pharmacol. Exp. Ther. 353, 279–287 (2015). [DOI] [PubMed] [Google Scholar]
- 12. Edsbäcker, S. , Wollmer, P. , Selroos, O. , Borgström, L. , Olsson, B. & Ingelf, J. Do airway clearance mechanisms influence the local and systemic effects of inhaled corticosteroids? Pulm. Pharmacol. Ther. 21, 247–258 (2008). [DOI] [PubMed] [Google Scholar]
- 13. Sakagami, M. In vivo, in vitro and ex vivo models to assess pulmonary absorption and disposition of inhaled therapeutics for systemic delivery. Adv. Drug Deliv. Rev. 58, 1030–1060 (2006). [DOI] [PubMed] [Google Scholar]
- 14. Sakagami, M. , Kinoshita, W. , Sakon, K. & Makino, Y. Fractional contribution of lung, nasal and gastrointestinal absorption to the systemic level following nose‐only aerosol exposure in rats: a case study of 3.7‐ micro m fluorescein aerosols. Arch. Toxicol. 77, 321–329 (2003). [DOI] [PubMed] [Google Scholar]
- 15. Weber, B. & Hochhaus, G. A pharmacokinetic simulation tool for inhaled corticosteroids. AAPS J. 15, 159–171 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hochhaus, G. , Möllmann, H. , Derendorf, H. & Gonzalez–Rothi, R.J. Pharmacokinetic/pharmacodynamic aspects of aerosol therapy using glucocorticoids as a model. J. Clin. Pharmacol. 37, 881–892 (1997). [DOI] [PubMed] [Google Scholar]
- 17. Chaudhuri, S.R. , Lukacova, V. & Woltosz, W.S. Simulating the disposition of budesonide from dry powder inhalers (DPIs) and nebulizer. <http://abstracts.aaps.org/Verify/aaps2013/postersubmissions/T3181.pdf> (2013).
- 18. Borghardt, J.M. , Weber, B. , Staab, A. & Kloft, C. Pharmacometric models for characterizing the pharmacokinetics of orally inhaled drugs. AAPS J. 17, 853–870 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gearhart, J.M. , Jepson, G.W. , Clewell, H.J. 3rd , Andersen, M.E. & Conolly, R.B. Physiologically based pharmacokinetic and pharmacodynamic model for the inhibition of acetylcholinesterase by diisopropylfluorophosphate. Toxicol. Appl. Pharmacol. 106, 295–310 (1990). [DOI] [PubMed] [Google Scholar]
- 20. Arundel, P.A. A multi‐compartmental model generally applicable to physiologically‐based pharmacokinetics. <https://www.researchgate.net/publication/267672776_A_multi-compartmental_model_generally_applicable_to_physiologically-based_pharmacokinetics_Astra-Zeneca_UK> (1997).
- 21. Brown, R.P. , Delp, M.D. , Lindstedt, S.L. , Rhomberg, L.R. & Beliles, R.P. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol. Ind. Health 13, 407–484 (1997). [DOI] [PubMed] [Google Scholar]
- 22. Campbell, J.L. , Andersen, M.E. & Clewell, H.J. A hybrid CFD‐PBPK model for naphthalene in rat and human with IVIVE for nasal tissue metabolism and cross‐species dosimetry. Inhal. Toxicol. 26, 333–344 (2014). [DOI] [PubMed] [Google Scholar]
- 23. Chen, A. & Kaufman, S. Splenic blood flow and fluid efflux from the intravascular space in the rat. J. Physiol. 490(Pt 2), 493–499 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Peters, S.A. Physiological model for distribution In Physiologically‐Based Pharmacokinetic (PBPK) Modeling and Simulations: Principles, Methods, and Applications in the Pharmaceutical Industry (ed. Peters S.A.) p. 111 (Wiley, Hoboken, NJ, 2012). [Google Scholar]
- 25. Lee, D. & Wexler, A.S. Particle deposition in juvenile rat lungs: a model study. J. Aerosol Sci. 42, 567–579 (2011). [Google Scholar]
- 26. Anjilvel, S. & Asgharian, B. A multiple‐path model of particle deposition in the rat lung. Fundam . Appl. Toxicol. 28, 41–50 (1995). [DOI] [PubMed] [Google Scholar]
- 27. Hofmann, W. , Asgharian, B. , Bergmann, R. , Anjilvel, S. & Miller, F.J. The effect of heterogeneity of lung structure on particle deposition in the rat lung. Toxicol. Sci. 53, 430–437 (2000). [DOI] [PubMed] [Google Scholar]
- 28. Schmid, O. et al Model for the deposition of aerosol particles in the respiratory tract of the rat. I. Nonhygroscopic particle deposition. J. Aerosol Med. Pulm. Drug Deliv. 21, 291–307 (2008). [DOI] [PubMed] [Google Scholar]
- 29. Pesic, J. et al In vivo assessment of mucociliary clearance in rodents via SPECT imaging after instillation and inhalation of a 99mTc‐labelled colloid. American Thoracic Society International Conference Abstracts. Abstract #C37. COPD: What is new in imaging? A4327–A4327 (2014).
- 30. Creutzenberg, O. , Muhle, H. & Bellmann, B. Nasal retention and clearance of wood dust in rats. Ann. Occup. Hyg. 38 (Suppl), 895–901 (1994). [Google Scholar]
- 31. Yeh, H.C. , Schum, G.M. & Duggan, M.T. Anatomic models of the tracheobronchial and pulmonary regions of the rat. Anat. Rec. 195, 483–492 (1979). [DOI] [PubMed] [Google Scholar]
- 32. Jones, J.H. & Longworth, K.E. Gas exchange at rest and during exercise in mammals In Treatise on Pulmonary Toxicology, Volume I: Comparative Biology of the Normal Lung (ed. Parent R.A.) pp. 271–307 (CRC Press, Boca Raton, FL, 1992). [Google Scholar]
- 33. Schreider, J.P. Nasal airway anatomy and inhalation deposition in experimental animals and people In Nasal Tumors in Animals and Man (eds. Reznik G. & Stinson S.F.) pp. 1–26 (CRC Press, Boca Raton, FL, 1983). [Google Scholar]
- 34. Lansley, A.B. Mucociliary clearance and drug delivery via the respiratory tract. Adv. Drug Deliv. Rev. 11, 299–327 (1993). [Google Scholar]
- 35. Peters, S.A. Drug absorption and gut bioavailability In Physiologically‐Based Pharmacokinetic (PBPK) Modeling and Simulations: Principles, Methods, and Applications in the Pharmaceutical Industry (ed. Peters S.A.) p. 47 (Wiley, Hoboken, NJ, 2012). [Google Scholar]
- 36. Brunner, E. & Nernst, W. Theorie der Reaktionsgeschwindigkeit in heterogenen Systemen. Hoppe Seylers Z. Physiol. Chem. 47, 56–102 (1904). [Google Scholar]
- 37. Noyes, A. & Whitney, W.R. The rate of solution of solid substances in their own solutions. J. Am. Chem. Soc. 19, 930–934 (1897). [Google Scholar]
- 38. Rodgers, T. & Rowland, M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 95, 1238–1257 (2006). [DOI] [PubMed] [Google Scholar]
- 39. Bäckström, E. et al Development of a novel lung slice methodology for profiling of inhaled compounds. J. Pharm. Sci.; e‐pub ahead of print 2015. [DOI] [PubMed] [Google Scholar]
- 40. Miller, A.H. , Spencer, R.L. , Stein, M. & McEwen, B.S. Adrenal steroid receptor binding in spleen and thymus after stress or dexamethasone. Am. J. Physiol. 259(3 Pt 1), E405–E412 (1990). [DOI] [PubMed] [Google Scholar]
- 41. Spencer, R.L. , Young, E.A. , Choo, P.H. & McEwen, B.S. Adrenal steroid type I and type II receptor binding: estimates of in vivo receptor number, occupancy, and activation with varying level of steroid. Brain Res. 514, 37–48 (1990). [DOI] [PubMed] [Google Scholar]
- 42. Wadenberg, M.L. , Kapur, S. , Soliman, A. , Jones, C. & Vaccarino, F. Dopamine D2 receptor occupancy predicts catalepsy and the suppression of conditioned avoidance response behavior in rats. Psychopharmacology (Berl) 150, 422–429 (2000). [DOI] [PubMed] [Google Scholar]
- 43. Jones, H. & Rowland–Yeo, K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacometrics Syst. Pharmacol. 2, e63 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information accompanies this paper on the CPT: Pharmacometrics & Systems Pharmacology website (http://www.wileyonlinelibrary.com/psp4).
Supplementary Information Figure 1.
Supplementary Information Figure 2.
Supplementary Information Figure 3.
Supplementary Information Figure 4.
Supplementary Information Figure 5.
Supplementary Information Figure 6.
Supplementary Information Figure 7.
Supplementary Information Table 1.
Supplementary Information Table 2.
Supplementary Information Table 3.
Supplementary Information Table 4.
Supplementary Information
Supplementary Information