

ABSTRACT
Mycobacterium tuberculosis, the etiological agent of tuberculosis (TB), has a unique cell envelope which accounts for its unusual low permeability and contributes to resistance against common antibiotics. The main structural elements of the cell wall consist of a cross-linked network of peptidoglycan (PG) in which some of the muramic acid residues are covalently attached to a complex polysaccharide, arabinogalactan (AG), via a unique α-l-rhamnopyranose–(1→3)-α-d-GlcNAc-(1→P) linker unit. While the molecular genetics associated with PG and AG biosynthetic pathways have been largely delineated, the mechanism by which these two major pathways converge has remained elusive. In Gram-positive organisms, the LytR-CpsA-Psr (LCP) family of proteins are responsible for ligating cell wall teichoic acids to peptidoglycan, through a linker unit that bears a striking resemblance to that found in mycobacterial arabinogalactan. In this study, we have identified Rv3267 as a mycobacterial LCP homolog gene that encodes a phosphotransferase which we have named Lcp1. We demonstrate that lcp1 is an essential gene required for cell viability and show that recombinant Lcp1 is capable of ligating AG to PG in a cell-free radiolabeling assay.
IMPORTANCE
Tuberculosis is an infectious disease caused by the bacterial organism Mycobacterium tuberculosis. Survival of M. tuberculosis rests critically on the integrity of its unique cell wall; therefore, a better understanding of how the genes and enzymes involved in cell wall assembly work is fundamental for us to develop new drugs to treat this disease. In this study, we have identified Lcp1 as an essential phosphotransferase that ligates together arabinogalactan and peptidoglycan, two crucial cell wall macromolecules found within the mycobacterial cell wall. The discovery of Lcp1 sheds new light on the final stages of mycobacterial cell wall assembly and represents a key biosynthetic step that could be exploited for new anti-TB drug discovery.
INTRODUCTION
Tuberculosis (TB) remains the single most important bacterial cause of global mortality and morbidity, causing around 9.6 million new cases and 1.5 million deaths each year (1). This prevalence is largely due to the increased susceptibility of HIV-infected individuals, and the rise of multidrug-resistant (MDR), extensively drug-resistant (XDR), and, more recently, totally drug-resistant (TDR) TB strains (2). It is of paramount importance that we extend our understanding of the pathogenicity and physiology of the tubercle bacillus in the hope of priming novel therapeutic approaches against this ancient human adversary. Mycobacterial peptidoglycan (PG) forms the basal layer of the mycolyl-arabinogalactan–peptidoglycan (mAGP) complex and is composed of alternating N-acetylglucosamine (GlcNAc) and modified muramic acid (Mur) residues, linked in a β(1→4) configuration (3). Unlike archetypal PG commonly found in bacteria such as Escherichia coli, the muramic acid residues in Mycobacterium tuberculosis contains a mixture of N-acetyl and N-glycolyl derivatives, whereby the N-acetyl function has been oxidized to an N-glycolyl function to form MurNGly (4–6). In addition, approximately 10 to 12% of the muramic acid residues of PG are covalently tethered to arabinogalactan (AG) via a phosphodiester bond (7). The 6′-OH groups of these muramic acid residues serve as attachment sites forming a phosphodiester bond via a unique α-l-rhamnopyranose–(1→3)-α-d-GlcNAc-(1→P) linker unit (LU) (8). Collectively, PG and AG form a huge macropolymer positioned between the cytoplasmic membrane and the outer mycolic acid layer of the TB bacillus. Our understanding of both the structure and biosynthesis of mycobacterial AG has developed steadily over the last 2 decades, with much emphasis being placed on investigating the molecular genetics of how this complex structure is assembled (extensively reviewed in reference 9). While the cytoplasmic and extracytoplasmic intermediate biosynthetic steps of cell wall assembly have been almost fully delineated, our understanding of the latter stages of AG biosynthesis, specifically the attachment of AG to PG, remains fragmented at best. Previous studies have demonstrated that attachment of AG to the mycobacterial cell wall occurs only when the PG has been fully matured via the cross-linking action of endogenous transpeptidases (10). However, the enzyme responsible for this action remains elusive. In Gram-positive bacteria, the structure of the linker unit (d-GlcNAc-d-ManNAc) that connects wall teichoic acids (WTA) to PG via a phosphodiester bond is remarkably similar to the linker unit found within the M. tuberculosis cell wall (d-GlcNAc-l-Rhap) (11). Other commonalities also exist between mycobacterial and Gram-positive cell wall glycan biosynthetic pathways. For instance, in organisms such as Staphylococcus aureus polyribitol-phosphate (Rbo-P) is assembled upon a polyprenyl pyrophosphoryl lipid carrier which, after biosynthesis is completed, is ultimately attached to the C6-hydroxyl of MurNAc within the PG glycan strands (8, 11). Mycobacterial AG is also assembled upon a polyprenyl pyrophosphoryl lipid carrier before its final deposition onto specific muramic acid residues of PG (12). We postulated that WTA-containing Gram-positive organisms and members of the Actinomycetales might utilize similar ligases from the same protein superfamily. Kawai et al. conducted a structural and genetic study and proposed that a family of genes encoding the LytR-CpsA-Psr (LCP) proteins catalyze the ligation of the murine linkage unit of WTA to the MurNAc units of PG in Bacillus subtilis (13). More recent studies have revealed further corroborating evidence that the LCP family of proteins are responsible for attaching capsular polysaccharides to the murine sacculus of S. aureus (14) and Streptococcus pneumoniae (15). Here, we demonstrate that the open reading frame contained in Rv3267, one of three LCP ortholog genes in M. tuberculosis, encodes a peptidoglycan–arabinogalactan ligase, which we have termed Lcp1.
RESULTS
Genome comparison of the Lcp1 locus.
Using S. aureus, S. pneumoniae, and B. subtilis LCP proteins as search sequences, we performed a bioinformatic analysis of mycobacterial and corynebacterial genomes to identify putative genes that might encode the mycobacterial LCP homologs (see Fig. S1 in the supplemental material). We identified three putative LCP homologs in M. tuberculosis, with Rv3267 sharing 20% amino acid sequence identity with Cps2A from S. pneumoniae, which is in accordance with a previous phylogenetic study of bacterial LCP proteins (16). The remaining two putative LCP orthologs present in M. tuberculosis, encoded by Rv3484 and Rv0822c, share 15% and 12% amino acid identity with Cps2A from S. pneumoniae. Rv3267 shares 36% and 26% amino acid sequence identity with Rv3484 and Rv0822c, respectively. Both Rv3484 and Rv0822c are predicted to contain a single N-terminal transmembrane (TM) α-helix, similar to that of Rv3267, and are predicted to contain domains belonging to both LCP and LytR_C superfamilies. Interestingly, the genome of Mycobacterium leprae, which shows massive gene decay and is thus considered to represent a “minimal” mycobacterial genome, has retained only two LCP orthologs in the form of ML0750 and ML2247, sharing 79.1% and 32.7% amino acid sequence identity with Rv3267, respectively. Inspection of the Mycobacterium smegmatis genome reveals four putative orthologs, MSMEG_1824, MSMEG_6421, MSMEG_0107, and MSMEG_5775, each sharing amino acid sequence identities of 72%, 35%, 31%, and 24% with Rv3267, respectively. Furthermore, an alignment of the genes surrounding Rv3267 reveals genetic synteny with orthologous regions from M. smegmatis, M. leprae, and Corynebacterium glutamicum (Fig. 1A). This close arrangement of genes is highly indicative of conserved functionality. Interestingly, Rv3267 is positioned immediately upstream of rmlD and wbbL1, both of which encode enzymes responsible for the formation of the linker unit during early-stage AG biosynthesis (Fig. 1A) (17, 18). Based on these analyses, we hypothesized that Rv3267 was the primary LCP homolog responsible for ligating PG to AG in mycobacteria, which we have named Lcp1.
FIG 1 .

Comparison of the lcp1 locus and in silico analysis of Lcp1 by TMHMM and I-TASSER. (A) The locus in bacteria analyzed consists of lcp1, which in M. tuberculosis has the locus tag Rv3267 and in M. smegmatis has the locus tag MSMEG1824. For M. leprae and C. glutamicum, locus tags are ML0756 and NCgl0708, respectively. (B) The TMHMM server was used to predict the membrane topology of MtbLcp1 (48). (C) I-TASSER homology model of MtbLcp1 positioned in relation to the cytoplasmic membrane and predicted N-terminal TM α-helix (19).
In silico analysis of MtbLcp1 and identification of copurifying polyisoprenoid phosphate.
Analysis of the M. tuberculosis Lcp1 (MtbLcp1) amino acid sequence suggests that a single transmembrane-spanning α-helix occurs at the N terminus of the protein (Fig. 1B). Similarly, a single N-terminal transmembrane α-helix has also been observed in other LCP proteins, such as Cps2A from S. pneumoniae (13, 16). An alignment of the amino acid sequences of MtbLcp1 and Cps2A from S. pneumoniae indicates that these two proteins share 20% sequence identity (see Fig. S1 in the supplemental material). The crystal structure of ΔTM-Cps2A (residues 98 to 481) has been solved to a resolution of 1.69 Å with one molecule of decaprenol-1-monophosphate bound in a central hydrophobic cavity (2XXP) (13). Although we were able to express large amounts of pure, soluble His6-tagged MtbLcp1 (see below), our attempts to obtain an experimental crystal structure of MtbLcp1 have so far proven unsuccessful. Therefore, we generated an MtbLcp1 homology model using the I-TASSER server (utilizing the S. pneumoniae Cps2A structure as the template), resulting in a C-score of −2.64 (19, 20). Unlike the S. pneumoniae ΔTM-Cps2A structure, which also harbors an N-terminal accessory domain, the MtbLcp1 amino acid sequence and homology model suggests that only a single “LCP-like” domain is present (Fig. 1C) (13). Our MtbLcp1 homology model indicates that a 5-stranded β-sheet forms the core of the protein, with α-helices surrounding a central β-sheet. Our homology model also retains the polyisoprenoid-binding cavity of Cps2A (Fig. 1C) (13). Overexpression of His6-tagged MtbLcp1 in E. coli BL21(DE3) cells and subsequent purification by immobilized metal affinity chromatography (IMAC) resulted in a preparation of His6-tagged MtbLcp1 that was stable in solution at a concentration of 50 mg/ml. Since Cps2A was shown to copurify with a polyisoprenoid phosphate, we also investigated whether His6-tagged MtbLcp1 copurified with a similar lipid (13). We performed an organic solvent extraction on His6-tagged MtbLcp1 followed by thin-layer chromatography (TLC) analysis, which revealed a spot that migrated with an Rf identical to that of decaprenyl-1-monophosphate (Fig. 2A). Mass spectrometric analysis of this copurifying lipid revealed an m/z of 777, which correlates with the expected mass of decaprenyl-1-monophosphate (Fig. 2D). This evidence was further corroborated by comparison to known standards of decaprenyl-1-monophsophate and undecaprenyl-1-monophsophate, which reveal m/z values of 777 and 845, respectively (Fig. 2B and C). We also performed an identical solvent extraction on a highly purified preparation of EmbCCT (the C-terminal globular domain of the arabinofuranosyltransferase EmbC from M. tuberculosis [21]) (Fig. 2A). This negative control confirms that the product extracted from MtbLcp1 is not a contaminating impurity from the extraction process.
FIG 2 .
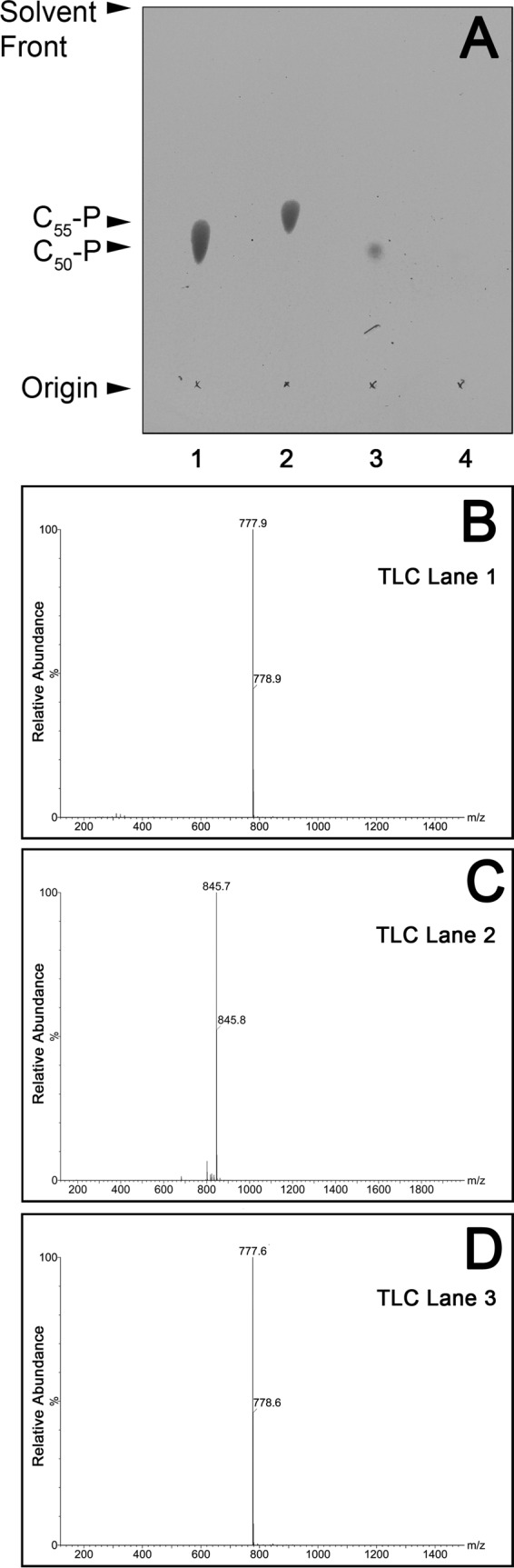
TLC and ES-MS analysis and identification of decaprenyl-1-monophosphate bound to recombinant MtbLcp1. (A) TLC analysis of organic extractable material of recombinant MtbLcp1. Standards of decaprenyl-1-monophosphate and undecaprenyl-1-monophosphate are in lanes 1 and 2, respectively. Lane 3 contains material extracted from MtbLcp1, while lane 4 contains a negative control of material extracted from EmbCCT (21). Lipids were analyzed by TLC using silica gel plates (5735 silica gel 60F254; Merck) and developed in CHCl3-CH3OH-NH4OH-H2O (65:25:0.5:3.6, vol/vol/vol/vol), and plates were sprayed with 5% ethanolic molybdophosphoric acid and charred to visualize the lipids. (B) ES-MS analysis of decaprenyl-1-monophosphate standard (TLC, lane 1). (C) ES-MS analysis of undecaprenyl-1-monophosphate standard (TLC, lane 2). (D) ES-MS organic extracted material from MtbLcp1 (TLC, lane 3).
MsLcp1 is essential for viability of M. smegmatis.
Because of its putative role in covalently attaching AG to PG in mycobacteria, we hypothesized that Mtblcp1 would likely be an essential gene. In order to test this hypothesis, we used M. smegmatis as a model system to investigate the essentiality of Lcp1 in mycobacteria using the conditional expression specialized transduction essentiality test (CESTET) (22–25). We constructed a knockout phage, phΔMslcp1, designed to replace MSMEG1824 (M. smegmatis lcp1 [Mslcp1]) with a hygromycin resistance cassette (22), but as predicted, we were unable to directly generate a null mutant due to the failure to yield any transductants. However, we were able to generate an Mslcp1 null mutant (M. smegmatis ΔMslcp1 pMV306-ACET-Mslcp1) by transducing a merodiploid strain containing a second, inducible copy of Mslcp1. Expression of this recombinant copy of Mslcp1 was induced by the addition of acetamide to the growth medium, and only when this inducer was present on agar plates were we able to generate conditional knockout mutants. In this regard, the conditional knockout mutant (M. smegmatis ΔMslcp1 pMV306-ACET-Mslcp1) exhibited an altered colony phenotype compared to the wild type, which is suggestive of a defect or lesion in the cell wall, which might not be fully recovered by complementation via the acetamide-inducible pMV306-ACET-Mslcp1 plasmid (Fig. 3A). To further investigate the essentiality of Mslcp1, we cultured M. smegmatis ΔMslcp1 pMV306-ACET-Mslcp1 in liquid medium and withdrew the inducer acetamide from the growth medium, with subsequent measurement of CFU. We observed a rapid loss of cell viability immediately after acetamide was removed from growth medium, indicating that expression of the pMV306-ACET-driven copy of Mslcp1 was required for cell growth (Fig. 3B). This confirms that Mslcp1 is indeed essential in M. smegmatis. The rapid loss in cell viability of the ΔMslcp1 mutant made any attempt to characterize the resultant phenotype (specifically cell wall lesions) technically challenging. Therefore, we turned our attention toward the biochemical characterization of recombinant MtbLcp1.
FIG 3 .

Essentiality of Mslcp1 in M. smegmatis mc21551. (A) Growth and colony morphology of M. smegmatis and ΔMslcp1 conditional mutant on tryptic soy agar with the inducer acetamide. WT, wild type. (B) CFU counts of M. smegmatis ΔMslcp1 conditional mutant cultured in tryptic soy broth in the presence and absence of the inducer acetamide (data plotted represent the mean ± standard deviation from three independent biological replicate experiments).
Carbohydrate binding of linker unit mimetics to MtbLcp1.
Our laboratory has previously synthesized neoglycolipid acceptors modeled on motifs found within mycobacterial AG, which have been successfully employed to probe both enzymatic function and ligand binding relationships of cell wall processing enzymes (21, 26, 27). In this regard, we synthesized a panel of analogues of the mycobacterial linker unit (compounds 1 to 4) (Fig. 4A; see also Text S1 in the supplemental material) in order to probe the interaction of MtbLcp1 with decaprenyl-pyrophosphoryl-GlcNAc-Rha-Galactose, its natural physiological substrate. Each of the ligands used (compounds 1 to 4) was synthesized to include an octyl (C8) group attached to the anomeric carbon. Historically, these alkyl groups are useful for performing solvent extractions from glycosyltransferase assay mixtures when used as neoglycolipid acceptors (26). In this study, their sole use was to mimic the hydrophobicity of the lipid moiety of the natural substrate, which would be accommodated by decaprenyl-pyrophosphate. Due to the chemical complexity and labile nature of the natural substrate (12, 28), using synthetic analogues offered us an alternative approach to investigate the biochemistry of this key enzymatic step. By way of intrinsic tryptophan fluorescence (ITF), we probed the binding of four potential ligands (compounds 1 to 4) to MtbLcp1, each of which is a variant of the mycobacterial linker unit (Fig. 4A). ITF is an extremely useful approach to study protein-ligand interactions (29) and has been successfully used by our laboratory to biochemically characterize several mycobacterial proteins (21, 27, 30, 31). Fitting the binding data to a single-site saturation model yielded an equilibrium dissociation constant, KD, of 57.68 µM for compound 1, the ligand which resembles the mycobacterial linker unit disaccharide l-Rha-α(1→3)-d-GlcNAc (Fig. 4B). ITF experiments repeated with a tetrasaccharide ligand that resembles an extension of the linker unit by two Galf residues afforded an ~10-fold increase in affinity, with a KD of 5.13 µM. Further elongation with a third Galf unit resulted in a calculated KD of 20.39 µM. The cytoplasmic steps of mycobacterial galactan formation proceed via a linear biosynthetic pathway; therefore, compounds 2 and 3 represent chemical mimetics of glycolipid-3 (GL-3) and glycolipid-4 (GL-4) intermediates of this pathway, respectively (12, 28), without the pyrophosphoryl-decaprenyl moiety linked to the anomeric position of the GlcNAc unit. In addition, we investigated the binding of a fourth ligand against MtbLcp1. Compound 4, a disaccharide of d-Galf-β(1→4)-l-Rhap (27), mimics the terminal component of the linker unit with a single Galf residue and displays an apparent KD of 97.61 µM. However, compound 4 achieves a Bmax of only 177, which is significantly lower than those of compounds 1 to 3 and reflects the changes in fluorescence observed upon addition of equivalent volumes of water. Therefore, compound 4 displays no significant binding affinity (above background) toward MtbLcp1 in ITF assays (Fig. 4B).
FIG 4 .

Binding of linker unit analogues to MtbLcp1. (A) Chemical structure of the mycobacterial linker unit and novel chemical scaffolds used as ligands to probe the interaction of the linker unit with MtbLcp1. (B) Saturation binding experiments using intrinsic tryptophan fluorescence of MtbLcp1 to study binding of ligands 1 to 4. Data plotted represent the mean ± standard deviation from three independent biological replicate experiments.
MtbLcp1 ligates newly synthesized galactan to peptidoglycan in a cell-free assay.
We sought to investigate whether MtbLcp1 is capable of ligating AG to PG (from decaprenyl-pyrophosphoryl-GlcNAc-Rha-galactose) by developing a cell-free biochemical functional assay. Since MtbLcp1 contains a single N-terminal TM α-helix, and attachment of AG to PG is likely to occur at or around the cytoplasmic membrane, we postulated that membranes prepared from mycobacteria should contain endogenous PG-AG ligase activity and, as such, could be investigated using an appropriate [14C]carbohydrate radiolabeling assay. We have previously developed a radiolabel assay for monitoring the synthesis of decaprenyl-pyrophosphoryl-linked intermediates of mycobacterial cell wall biosynthesis (28). In this study, we modified this assay system so that we could directly investigate the in vitro covalent attachment of AG intermediates to PG. Briefly, membranes and the P60 fraction from M. smegmatis (both rich in endogenous cell wall biosynthetic activity) were combined with substrates in the form of purified peptidoglycan (PPG) and UDP-[14C]Galp. The endogenous mutase activity within the P60 fraction of this assay mix (used to convert UDP-[14C]Galp to UDP-[14C]Galf) was sufficient to allow direct incorporation of [14C]Galf into cell wall intermediates, as previously demonstrated (12, 28). PPG is a highly purified preparation of nascent M. smegmatis PG free from AG covalently attached to the 6′-OH position of MurNAc via a phosphodiester bond (32). The purpose of PPG is both to provide a substrate for attachment of 14C-labeled linker unit-galactan intermediates and to serve as a “vehicle” for extraction from the assay mixture for subsequent radiochemical quantification. Finally, the assay mixture was initiated by addition of highly purified recombinant MtbLcp1. The analytical strategy for the products formed from this assay is illustrated as a flow chart in Fig. S4 in the supplemental material. Analysis of the fraction containing the CHCl3-CH3OH organic extraction by TLC reveals three major bands migrating toward the bottom of the TLC plate (Fig. 5A, lane 2). These bands correspond to 14C-labeled decaprenyl-P-P-GlcNAc-Rha-[14C]Galf (GL-3) and decaprenyl-P-P-GlcNAc-Rha-[14C]Galf2 (GL-4), with decaprenyl-P-P-GlcNAc-Rha-[14C]galactan remaining at the origin of the TLC plate (12, 28). Interestingly, the intensity of each of these bands diminished in a titration-dependent response as the amount of MtbLcp1 increased in the assay mix (Fig. 5A, lanes 3 and 4), which was also quantified by densitometric analysis. When we repeated the assay at the highest MtbLcp1 concentration in the absence of purified PG (PPG), we observed a product profile on the TLC with band intensities almost identical to that of the control reaction (Fig. 5A, lane 5). These data clearly demonstrate that upon addition of MtbLcp1 to the assay mixture, the relative amount of [14C]Galf-labeled material is reduced in the extracted organic fraction. We continued our analysis of the assay products by turning our attention to the insoluble fraction that contains PG and PG-linked [14C]Galf-labeled material. Complete acid hydrolysis for the purpose of [14C]sugar identification was conducted on the insoluble material (fraction 2 [see Fig. S4 in the supplemental material]) that contains nascent PG with newly synthesized AG material. Hydrolysates were then reduced and per-O-acetylated before analysis via silica TLC. Analysis of per-O-acetylated alditol acetates from this insoluble fraction revealed a major band that migrated to the position of per-O-acetylated galactitol (Fig. 5B, lane 2). Other bands that correspond to per-O-acetylated glucitol and per-O-acetylated N-acetyl glucitol were also identified (Fig. 5B, lane 2). However, we observed no statistically significant variation in the intensity of these bands across all samples and repeats and attribute this phenomenon to metabolism of UDP-[14C]Gal into other sugars under these reaction conditions (Fig. 5B, lane 2), and this phenomenon has also been previously reported (12). Overall, this background activity [14C]Gal incorporation can be attributed to endogenous MsLcp1 present in the M. smegmatis membrane preparation used in the assay mixture (Fig. 5B, lane 2). Due to the essentiality of Mslcp1, we were unable to culture M. smegmatis depleted of MsLcp1 in order to derive membranes free from MsLcp1 activity. Therefore, we adopted a strategy whereby we assayed for Lcp1 ligase activity over and above endogenous background activity (Fig. 5B, lane 2). The addition of recombinant MtbLcp1 to reaction mixtures resulted in a concomitant increase in band density that corresponds to per-O-acetylated galactitol (Fig. 5B, lanes 3 and 4), which is also quantified by densitometric analysis. However, in control reaction mixtures where PPG is removed from the assay mixture, we observed no significant incorporation of [14C]Galf-labeled material, as manifested by the disappearance of a band corresponding to per-O-acetylated galactitol (Fig. 5B, lane 5). Taken together, these data clearly demonstrate two major findings. First, we are able to directly monitor the ligation of [14C]Galf cell wall material to PG. Second, recombinant MtbLcp1 is capable of using decaprenyl-P-P-[14C]Galfn as a substrate for subsequent attachment to PG.
FIG 5 .

AG-PG ligase activity of MtbLcp1 using a radiolabeled cell-free functional assay. (A) Organic solvent-extracted fractions from 14C-labeled assay mixtures were analyzed by TLC and developed in CHCl3-CH2OH-H2O-NH4OH (65:25:3.6:0.5, vol/vol/vol/vol) on aluminum-backed silica gel plates (5735 silica gel 60F254; Merck), and products were visualized by autoradiography, exposing the TLCs to X-ray film (Kodak X-Omat). TLC is representative of three independent biological replicate experiments, and corresponding densitometric data plotted represent the mean ± standard deviation for each band. Associated P values are as follows: *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001. (B) Insoluble material from fraction 2 of the assay mixtures was hydrolyzed in 2 M TFA, reduced, and per-O-acetylated. The resulting 14C-labeled per-O-acetylated alditol acetate derivatives were analyzed by TLC using ethyl acetate-hexane (4:6, vol/vol) on aluminum-backed silica gel plates (5735 silica gel 60F254; Merck), and products were visualized by autoradiography, exposing the TLCs to X-ray film (Kodak X-Omat), and compared to known alditol-acetate standards of d-galactose (Gal), d-glucose (Glc), and d-N-acetylglucosamine (GlcNAc). TLC is representative of three independent biological replicate experiments, and corresponding densitometric data plotted represent the mean ± standard deviation for each band. Associated P values are as follows: *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
DISCUSSION
After conducting a bioinformatics analysis of organisms belonging to the Actinomycetales, we identified MtbLcp1 (Rv3267) as the primary putative LCP homolog in M. tuberculosis, sharing 20% identity with Cps2A from S. pneumoniae (see Fig. S1 in the supplemental material). Since lcp1 is positioned immediately upstream of two genes involved in linker unit biosynthesis, in the form of wbbL1 and rmlD (Fig. 1A), we decided to investigate the M. smegmatis lcp1 ortholog (MSMEG_1824) as a model system to study the molecular genetics of this gene in order to assess its essentiality and to confirm its role as a primary LCP protein involved in mycobacterial cell wall assembly.
Mycobacterial AG is a highly branched heteropolysaccharide that serves to attach PG to the outer “mycomembrane,” and the biosynthesis of this essential cell wall component has been extensively studied (reviewed in reference 9). Since we have previously used M. smegmatis to successfully study the molecular genetics of mycobacterial cell wall biosynthesis (22, 24), we attempted to generate a clean deletion of Mslcp1 from M. smegmatis. However, due to the essential nature of Mslcp1 in attaching AG to PG in the cell envelope, we anticipated that a direct deletion of Mslcp1 would result in a nonviable, lethal strain, and our hypothesis was justified by our inability to generate a null mutant. The essentiality of Mslcp1 was confirmed when we performed a CESTET essentiality experiment (Fig. 3) (22). Generating null mutants of genes in mycobacteria that are involved in essential aspects of cell envelope biosynthesis is technically challenging and can be attributed to corresponding defects in the cell wall that produce nonviable phenotypes. The M. smegmatis ΔaftC mutant represents a strain of mycobacteria for which we have succeeded in generating a significant cell wall defect via genetic manipulation (24). Due to the complete loss of α(1→3)-arabinosyltransferase activity, the M. smegmatis ΔaftC mutant produces a significantly altered AG but, crucially, still connects PG to the outer (albeit reduced) mycolate layer (24). Unsuccessful attempts by our laboratory to generate a direct knockout (KO) of any other gene(s) involved in cell wall biosynthesis upstream of this event are due to the formation of nonviable mutants which are completely unculturable. Given the essential nature of the mycobacterial cell wall toward cell viability, the generation of conditional mutants followed by phenotypic characterization via the CESTET procedure is an extremely useful tool to confirm gene essentiality. Furthermore, M. smegmatis encodes three additional Lcp orthologs in the forms of the products of MSMEG_0107, MSMEG_6421, and MSMEG_5775, none of which can complement the function of MSMEG_1824 KO. In M. tuberculosis, two additional Lcp orthologs are encoded by Rv3484 and Rv0822c. In this regard, the function of these remaining lcp genes requires further investigation.
The presence of a hydrophobic channel within the MtbLcp1 homology model is consistent with similar cavities found within LCP proteins from B. subtilis and S. pneumoniae (13, 15) (Fig. 1C). Indeed, biochemical analysis of recombinant MtbLcp1 clearly demonstrates that decaprenyl-1-monophosphate copurifies and is likely to be bound within the hydrophobic cavity of the protein (Fig. 2). In order to investigate the biochemical function of MtbLcp1, we chemically synthesized a panel of four ligands that represent chemical structures found within the mycobacterial AG proximal to the linker unit and used intrinsic ITF to study the ligand binding properties of these ligands against MtbLcp1. We observed a ligand binding relationship that reached saturation with a defined Bmax for each of the compounds 1 to 3, all of which display a binding affinity in the low-micromolar range. Compound 1 was synthesized to mimic the l-Rha-α(1→3)-d-GlcNAc disaccharide moiety of the linker unit, and we recorded a KD of 58.71 µM. However, our binding data clearly demonstrate that the ligand with highest affinity for MtbLcp1 is the tetrasaccharide Galf2-Rha-GlcNAc-O-C8 (compound 2) with a KD of 5.13 µM. Interestingly, extension of this molecule with an additional Galf residue (compound 3) caused a subtle 4-fold decrease in binding affinity (Fig. 4B). For technical reasons, we were unable to synthesize Galf-Rha-GlcNAc-O-C8 and therefore were unable to study the binding of a ligand that represents the linker unit with only a single Galf extension. In a control experiment, we investigated the ligand binding properties of Galf-β(1→4)-l-Rhap (compound 4) toward MtbLcp1. This compound displayed no recordable affinity toward the protein (over changes in background fluorescence), which suggests that the presence of d-GlcNAc in the linker unit is vital for biophysical association between protein and ligand. However, since MtbLcp1 is able to bind both the linker unit (compound 1) and the linker unit with 2 to 3 Galf residues (compounds 2 and 3, respectively), our data suggest that MtbLcp1 might catalyze the phosphotransfer and ligation of AG to PG upon encountering a true physiological substrate that contains a decaprenyl-pyrophosphate-linker unit with at least one (if not two) Galf residue at the nonreducing end of the glycan chain (Fig. 6).
FIG 6 .

Schematic diagram of the latter stages of mycobacterial cell wall biosynthesis. (1) Translocation of galactan polymer across plasma membrane. Lcp1 then recognizes and binds the linker unit (i.e., represented by compounds 2 and 3). (2) Phosphotransferase activity links AG to the 6′-OH of muramyl residues, releasing decaprenyl-1-monophosphate for recycling. (3) Newly bound AG is released by Lcp1 to go on to form mature mAGP. Additional synthesis of new PG, requiring AG addition by Lcp1, takes place simultaneously.
We extended our investigation toward the development of a cell-free functional assay to measure the ligation of AG to PG in the presence of MtbLcp1. Chemical analysis of a reaction mixture that contained 14C-radiolabeled AG supplemented with nascent PG clearly shows that MtbLcp1 is capable of ligating both macromolecules in a titratably dependent manner (Fig. 5). However, because this assay requires the use of mycobacterial membranes that contain endogenous AG-PG ligase activity, an alternative explanation is that recombinant MtbLcp1 could indirectly activate this residual membrane activity. It has been previously reported that ligation of AG to PG in mycobacteria requires concomitant synthesis of both cell wall polymers (10, 12); however, until now, the enzyme responsible for this activity has not been reported. This study now provides compelling genetic and biochemical evidence that MtbLcp1 is indeed the enzyme that reconciles two major mycobacterial cell wall biosynthetic pathways by forming a covalent phosphodiester between PG and AG (Fig. 6). The discovery of MtbLcp1 sheds new light on a key reaction involved during late-stage cell wall assembly which is likely to be critical for all members of the Actinomycetales harboring AG attached to PG via a phosphodiester linker unit.
In S. aureus, the first two genes of the WTA pathway (tagO and tagA) are nonessential and can be deleted without retarding growth of both mutant strains (33, 34). However, deletion of any of the subsequent WTA-synthesizing enzymes (TagBDFGH or TarIJL) produces a lethal phenotype, unless each corresponding mutant also contains an inactivation in tagO or tagA (34). A proposed hypothesis to explain this synthetic viable phenotype relates to the limited availability of the bactoprenol lipid carrier in the cytoplasmic membrane, which is required for not only WTA biosynthesis but also lipid II biosynthesis, which ultimately leads to PG assembly (34). Furthermore, similar effects have been demonstrated in S. aureus by chemical means, whereby targocil, an inhibitor of late-stage WTA biosynthesis, causes an accumulation of undecaprenyl-linked intermediates, thus preventing recycling of the undecaprenyl-phosphate lipid carrier, causing synthetic lethality (35). This phenomenon most likely reflects the phenotypic consequence of an Mslcp1 deletion in M. smegmatis. Upon acetamide withdrawal from growing cultures of M. smegmatis ΔMslcp1 pMV306-ACET-Mslcp1, expression of Mslcp1 diminishes, which will likely result in two major effects: (i) loss of AG-PG ligase activity and (ii) a concomitant increase in the decaprenyl-pyrophosphoryl-linked galactan intermediate(s). While the former effect induces a devastating cell wall lesion, the latter effect will cause a dramatic reduction in available decaprenyl monophosphate that is also required for PG biosynthesis, via loss of lipid II. Furthermore, Actinomycetales such as M. tuberculosis and C. glutamicum also use decaprenyl phosphate in the production of decaprenyl-phosphoarabinose (DPA) and decaprenyl-phosphomannose (DPM), which is directed toward the formation of d-arabinan and lipomannan (which is the precursor to lipoarabinomannan [LAM]), respectively (36, 37). Benzothiazinones (BTZs) are a class of compounds that are particularly potent inhibitors of mycobacterial growth. BTZs target DprE1, which is an oxidoreductase involved in the epimerization of decaprenyl-phosphoribose (DPR) to decaprenyl-phosphoarabinose (DPA), thus arresting d-arabinan biosynthesis (38, 39). However, it has recently been shown that BTZ’s potent mode of action is attributed to the blocking of decaprenyl monophosphate recycling, a process which is required to allow cell wall biosynthesis to function properly (40). In this regard, Lcp1 is a potential drug target, since its inhibition will affect late-stage cell wall maturation and reduce the availability of decaprenyl monophosphate, leading to synthetic lethality.
MATERIALS AND METHODS
Homology modeling and sequence alignment.
The amino acid sequence of M. tuberculosis Rv3267 (MtbLcp1, Swiss-Prot ID P96872_MYCTU) was submitted for three-dimensional (3D) structure prediction using the I-TASSER server (19, 20), and the conformation of the secondary structural features for the resultant MtbLcp1 model was predicted using pdbsum (41). The homology model of MtbLcp1 was analyzed in comparison to the Cps2A structures from S. pneumoniae (PDB ID 2XXP) (13).
Chemicals, reagents, and enzymes.
All chemicals and solvents were from Sigma-Aldrich (Dorset, United Kingdom), Bio-Rad (CA, USA), and Fisher Chemicals (United Kingdom) unless otherwise stated and were of analytical grade or equivalent. Plasmids were propagated during cloning in Escherichia coli Top10 cells (Invitrogen). All restriction enzymes and Phusion DNA polymerase enzyme were sourced from New England Biolabs. The Bioline quick ligation kit was used to perform ligation reactions. Oligonucleotides were from MWG Biotech Ltd., and PCR fragments were purified using the QIAquick gel extraction kit (Qiagen). Plasmid DNA was purified using the QIAprep purification kit (Qiagen). The preparation of oligosaccharides 1 to 3 using methods previously reported (42) is described in Text S1 in the supplemental material. Disaccharide 4 was prepared previously (43).
Bacterial strains and growth conditions.
E. coli Top10 cells were routinely grown in Luria-Bertani broth or Luria-Bertani agar (LB; Difco) at 37°C. E. coli BL21(DE3) cells were grown in Terrific Broth (Difco) at 37°C. M. smegmatis mc2155 was grown in tryptic soy broth (TSB; Difco) containing Tween 80 supplemented with the appropriate antibiotic(s) (25 µg/ml kanamycin [Kan] and/or 100 µg/ml hygromycin).
Construction of recombinant plasmids.
For generation of the MtbLcp1 expression vector, a 1,389-bp region of Rv3267 was cloned from M. tuberculosis H37Rv genomic DNA using the following primer pairs (restriction sites underlined): Rv3267FW (5′-GATCGATCCATATGCGGTCGTTCGAAGACGGCATCTTCCAC-3′) and Rv3267DN (5′-GATCGATCAAGCTTTCAGTTGATGCACTCCGGCGCGTCGGAG-3′). The PCR product was ligated into pET28b vector and cut with NdeI and HindIII, yielding pET28b-Mtblcp1. The plasmid was subsequently sequenced to check for construct integrity (Eurofins MWG).
For generating an integrative vector containing MSMEG1824, the open reading frame (ORF) was amplified from M. smegmatis mc2155 genomic DNA using the primers MS1824FW (5′-GATCGATCGATATCTTGATCAGGTCCATTGCTGTGGCCGCA-3′) and MS1824DN (5′-GATCGATCAAGCTTTCAGTTCACGCACTGCGGGTCGTTGGC-3′). The DNA amplicon was digested with HindIII and EcoRV and subsequently cloned into pMV306 together with the Pacetamidase promoter region from pSD26 as described previously (22), thus yielding pMV306-ACET-MSMEG1824 (pMV306-ACET-Mslcp1). The plasmid was subsequently sequenced to check for construct integrity (Eurofins MWG).
For generation of the ΔMslcp1 phasmid, approximately 1-kb sequences of the upstream and downstream regions of MSMEG1824 were PCR amplified from M. smegmatis mc2155 genomic DNA with the primer pairs MS1824LL (5′-TTTTTTTTCCATAAATTGGCGACCACCAGGGGGCGGGCATCGTCCA-3′) and MS1824LR (5′-TTTTTTTTCCATTTCTTGGACCGGGGCAGGCGGCATGGTCGGGCGC-3′) and MS1824RL (5′-TTTTTTTTCCATAGATTGGACGACGCCACCGGTGAACGCATCGAGC-3′) and MS1824RR (5′-TTTTTTTTCCATCTTTTGGGCCGACGACAGGCTGCTCGACGACTAC-3′), respectively (all primers had Van91I recognition sites incorporated at the 5′ end). The PCR fragments were digested with Van91I and directly cloned into Van91I-digested p0004S (gift from T. Hsu and W. R. Jacobs, Jr., Albert Einstein College of Medicine, NY). Recombinant plasmids obtained after transforming E. coli Top10 cells were digested with Van91I for confirmation and sequenced. One plasmid, pΔMSMEG1824, was linearized by PacI digestion and packaged into the temperature-sensitive mycobacteriophage phAE159, as previously described (44), to yield phasmid DNA of the knockout phage phΔMSMEG1824 (phΔMslcp1).
Generation of the ΔMslcp1 conditional mutant.
The M. smegmatis ΔMslcp1 conditional mutant was generated using the CESTET procedure (22). Briefly, a merodiploid strain was first generated by introducing pMV306-ACET-Mslcp1 by electroporation into M. smegmatis mc2155 (22). The merodiploid strain M. smegmatis mc2155::pMV306-ACET-Mslcp1 was then subjected to specialized transduction as previously described (22) using a temperature-sensitive, recombinant phage, phΔMslcp1, designed to replace MSMEG1824 with a hygromycin resistance marker. Transductants were selected at the nonpermissive temperature of 37°C on selective plates containing 25 µg/ml kanamycin, 100 µg/ml hygromycin B, and 0.2% (wt/vol) acetamide. Allelic exchange in hygromycin-resistant transductants was confirmed by PCR and Southern blotting.
Conditional depletion of the ΔMslcp1 conditional mutant.
The ΔMslcp1 conditional mutant was grown in tryptic soy broth (TSB; Difco) containing 0.05% Tween 80, 25 µg/ml kanamycin, 100 µg/ml hygromycin B, and 0.2% (wt/vol) acetamide and subsequently passaged twice in medium with and without acetamide. Ten-milliliter cultures were allowed to grow for a further 56 h at 37°C with shaking. Ten-microliter samples were taken at 8-h intervals and plated on TBS agar plates supplemented with 25 µg/ml kanamycin and 100 µg/ml hygromycin B with or without 0.2% (wt/vol) acetamide to measure CFU.
Expression and purification of MtbLcp1.
The plasmid pET28b-Mtblcp1 was transformed into E. coli BL21(DE3) cells, spread onto LB agar plates containing 50 mg/ml Kan, and incubated at 37°C for 18 h. Single colonies were selected to inoculate 5 ml LB broth containing 50 µg/ml Kan and were incubated at 37°C overnight. The overnight cultures were then used to inoculate 1 liter Terrific Broth containing 50 mg/ml Kan, which was incubated (37°C, 180 rpm) until an optical density at 600 nm (OD600) of 0.5 was reached. Protein production was then induced by the addition of 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside; 12 h at 16°C). Cells were harvested via centrifugation at 6,000 × g for 15 min (4°C). Supernatant was discarded, and cells were washed in 30 ml phosphate-buffered saline (PBS). Pellets were stored at −20°C until further use. A pellet was defrosted and then resuspended in 30 ml lysis buffer (50 mM KH2PO4 [pH 7.9], 300 mM NaCl, 20 mM imidazole), including a proteinase inhibitor tablet (Roche). The mixture was subsequently sonicated using a Soniprep 150 ultrasonic disintegrator (MSE) (30 s on, 30 s off, 10 cycles). The solution was then centrifuged at 27,000 × g for 40 min (4°C), and the supernatant was retained (clarified lysate). Protein was loaded onto a HisTrap 5-ml chelating HP column (GE Healthcare), charged with 0.1 M NiCl2, equilibrated in lysis buffer, and eluted via a stepwise 50 to 500 mM imidazole concentration gradient (in lysis buffer). Fractions were analyzed by 12% SDS-PAGE to check for purity, and those fractions containing purified protein were dialyzed into dialysis buffer (25 mM Tris-HCl [pH 7.9], 10 mM NaCl, and 10% glycerol). Protein concentration was measured using a Bradford assay.
Extraction of decaprenyl-1-monophosphate from MtbLcp1.
Two milliliters of recombinant MtbLcp1 (2 mg/ml dissolved in dialysis buffer) was combined with 2 ml CHCl3 and 1 ml of CH3OH and heated at 60°C for 2 h with agitation. After centrifugation at 5,000 × g for 10 min, the lower organic fraction was recovered and dried under nitrogen. The resulting extract was resuspended in CHCl3-CH3OH (100 µl, 2:1, vol/vol) and analyzed by electrospray mass spectrometry (ES-MS) in the negative mode on a Micromass LCT mass spectrometer. An identical extraction was also carried out on 2 mg/ml of EmbCCT (21), which was used as a negative control.
Ligand binding experiments.
Intrinsic tryptophan fluorescence (ITF) ligand binding experiments of MtbLcp1 with saccharide ligands were conducted on a Hitachi F7000 fluorescence spectrophotometer with the temperature thermostatically regulated to 25°C. MtbLcp1 was diluted to 0.026 µM with dialysis buffer, and fluorescence spectra were measured at an excitation wavelength of 280 nm and emission wavelength of 300 to 400 nm with an excitation and emission slit width of 5 nm using a 500-µl crystal cuvette. ITF maximum (Femissionmax) was recorded at λ = 341 nm, providing a basal Femission coordinate for the collection of subsequent ITF data. Increasing concentrations of ligands representing the linker unit (compounds 1 to 4) were added to the cuvette. The change in fluorescence emission (ΔFemission) was calculated by subtracting Femission (recorded 2 min after each ligand addition) from Femissionmax. Changes of background fluorescence observed upon addition of equivalent volumes of dialysis buffer (the solvent in which compounds 1 to 4 were dissolved) were subtracted from the overall change in fluorescence, and the data were then plotted against ligand concentration, [L] (3 independent biological experiments). A plot of ΔFemission at λ = 341 nm versus [L] was fitted to a one-site specific binding equation using GraphPad Prism 5 software: ΔFemission = Fmax × [L]/(KD + [L]).
Preparation of nascent mycobacterial peptidoglycan.
One liter of M. smegmatis mc2155 was cultured in TSB as described above and harvested by centrifugation. Pelleted M. smegmatis cells were resuspended in H2O before addition of CHCl3-CH3OH (1:1, vol/vol) to create CHCl3-CH3OH-H2O (10:10:3, vol/vol/vol) single phase. The suspension was centrifuged at 3,300 × g for 15 min, the resulting supernatant was discarded, and the pellet was resuspended in 30 to 40 ml H2O. The cell suspension was sonicated (60 s on, 90 s off, 15 cycles) before addition of 1% (vol/vol) Triton X-100. The cell suspension was incubated at room temperature for 16 h and then centrifuged at 3,300 × g for 15 min. The crude insoluble material from the above extraction was extracted three times with 2% SDS in PBS at 95°C for 1 h; washed with water, 80% (vol/vol) acetone in water, and acetone; and finally lyophilized to yield a highly purified cell wall mAGP preparation (45). Mycolic acids were removed from mAGP by treatment with 0.5% (wt/vol) KOH in methanol at 37°C for 4 days. The treated mAGP was collected by centrifugation at 27,000 × g for 20 min and then washed repeatedly with methanol and recentrifuged at 27,000 × g, and the pellet was recovered. The mycolic acids were then extracted from the treated mAGP using diethyl ether and recentrifuged at 27,000 × g, and the pellet (AGP) and soluble mycolic acid methyl esters were recovered in the diethyl ether supernatant. The extraction process using diethyl ether was repeated three times. The AGP complex was hydrolyzed with 0.2 M H2SO4 at 85°C for 30 min and neutralized with BaCO3 (32). The supernatant (which contained solubilized arabinogalactan [AG]) was removed via centrifugation at 27,000 × g for 30 min. The residual pellet (containing the highly purified peptidoglycan [PPG] fraction) was finally extensively washed with water, freeze-dried, and stored at −20°C.
Functional biochemical assay for MtbLcp1.
Membranes (containing membrane-bound enzymes involved in lipid-linked cell wall biosynthetic processes) and P60 (a Percoll-derived cell-free fraction rich in carbohydrate and enzymes associated with cell wall processes as described in reference 46) fractions from M. smegmatis were prepared as described previously (24, 26, 46) and resuspended in buffer A (50 mM morpholinepropanesulfonic acid [MOPS; pH 7.9] containing 5 mM β-mercaptoethanol and 10 mM MgCl2) to a final concentration of 15 mg/ml and 10 mg/ml for membrane and P60 fractions, respectively.
The basic assay mix consisted of buffer A, 0.5 mg membranes (33 µl), 0.5 mg of P60 (50 µl), 0.2 mM ATP, 0.2 mM NADH, 0.1 µCi of UDP-d-[U-14C]galactose (specific activity, 300 mCi/mmol; ARC Radiochemicals), and 1 mg PPG; the reaction was initiated with the addition of 0.1 mg/ml MtbLcp1 (final volume of 200 µl); and the reaction mixture was incubated for 17 h at 37°C. Reactions were quenched by the addition of 1,333 µl of CHCl3-CH3OH (1:1, vol/vol). After mixing and centrifugation at 27,000 × g for 15 min at 4°C, the supernatant was removed and dried under nitrogen and the pellet was retained for further processing. The dried supernatant was resuspended into 2 ml of CHCl3-H2O (1:1, vol/vol), and the lower organic phase (fraction 1) was removed and dried under nitrogen. Material in fraction 1 was subjected to TLC using CHCl3-CH2OH-H2O-NH4OH (65:25:3.6:0.5, vol/vol/vol/vol) on aluminum-backed silica gel plates (5735 silica gel 60F254; Merck), and products were visualized by autoradiography, exposing the TLCs to X-ray film (Kodak X-Omat). The pellet from the previous step was resuspended into 1 ml H2O-C2H5OH-(C2H5)2-pyridine-NH4OH (15:15:5:1:0.017, vol/vol/vol/vol/vol) and left at room temperature to be extracted for 2 h. Samples were again centrifuged at 27,000 × g for 15 min, and supernatant was discarded. The remaining pellet (fraction 2) was resuspended in 200 µl H2O. All fractions were quantified by liquid scintillation counting using 10% of the labeled material and 5 ml of EcoScintA (National Diagnostics, Atlanta, GA).
Material in fraction 2 was hydrolyzed in 2 M trifluoroacetic acid (TFA) and reduced with NaBH4, and the resultant alditols were per-O-acetylated as described previously (47). The resulting 14C-labeled per-O-acetylated alditol acetate derivatives were analyzed by TLC using ethyl acetate-hexane (4:6, vol/vol) on aluminum-backed silica gel plates (5735 silica gel 60F254; Merck), and products were visualized by autoradiography, exposing the TLCs to X-ray film (Kodak X-Omat) and comparing them to known standards. All experiments were performed in triplicate as biological replicates.
SUPPLEMENTAL MATERIAL
Supplemental methods. Download
Sequence alignment of Lcp1 orthologues from M. tuberculosis, M. smegmatis, and C. glutamicum and LCP homologs from B. subtilis and S. pneumoniae. Amino acid sequences were aligned using ClustalW and rendered with EsPRIPT. Secondary structure information was obtained from PDB coordinates 2XXP. Download
Reaction scheme for the synthesis of octyl α-l-rhamnopyranosyl-(1→3)-2-acetamido-2-deoxy-α-d-glucopyranoside (compound 1). Download
Reaction scheme for the synthesis of octyl-β-d-galactofuranosyl-(1→5)-β-d-galactofuranosyl-(1→4)-α-l-rhamnopyranosyl-(1→3)-2-acetamido-2-deoxy-α-d-glucopyranoside (compound 2) and octyl-β-d-galactofuranosyl-(1→6)-β-d-galactofuranosyl-(1→5)-β-d-galactofuranosyl-(1→4)-α-l-rhamnopyranosyl-(1→3)-2-acetamido-2-deoxy-α-d-glucopyranoside (compound 3). Download
Flow chart tracking radioactivity incorporated from UDP-[14C]Galp through each of the analytical steps leading to TLC analysis (Fig. 5). Download
ACKNOWLEDGMENTS
J.H. is a Medical Research Council Quota Student. T.L.L. and M.J. acknowledge support from the Alberta Glycomics Centre and the Natural Sciences and Engineering Research Council of Canada. G.S.B. acknowledges support in the form of a Personal Research Chair from James Bardrick, Royal Society Wolfson Research Merit Award, as a former Lister Institute-Jenner Research Fellow. We acknowledge funding from the MRC (grant G1100127). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
J.H., G.L., A.L., G.S.B., and L.J.A. designed the experiments. J.H., G.L., and H.W.-M. performed biochemical experiments. J.H., G.L., E.R., and A.B. performed mutational experiments. G.S.B., M.J., and T.L.L. performed chemical synthesis.
Footnotes
Citation Harrison J, Lloyd G, Joe M, Lowary TL, Reynolds E, Walters-Morgan H, Bhatt A, Lovering A, Besra GS, Alderwick LJ. 2016. Lcp1 is a phosphotransferase responsible for ligating arabinogalactan to peptidoglycan in Mycobacterium tuberculosis. mBio 7(4):e00972-16. doi:10.1128/mBio.00972-16.
REFERENCES
- 1.WHO 2015. Global tuberculosis report 2015. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Loewenberg S. 2012. India reports cases of totally drug-resistant tuberculosis. Lancet 379:205. doi: 10.1016/S0140-6736(12)60085-3. [DOI] [PubMed] [Google Scholar]
- 3.Lederer E, Adam A, Ciorbaru R, Petit J, Wietzerbin J. 1975. Cell walls of mycobacteria and related organisms; chemistry and immunostimulant properties. Mol Cell Biochem 7:87–104. doi: 10.1007/BF01792076. [DOI] [PubMed] [Google Scholar]
- 4.Mahapatra S, Crick DC, Brennan PJ. 2000. Comparison of the UDP-N-acetylmuramate:L-alanine ligase enzymes from Mycobacterium tuberculosis and Mycobacterium leprae. J Bacteriol 182:6827–6830. doi: 10.1128/JB.182.23.6827-6830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahapatra S, Scherman H, Brennan PJ, Crick DC. 2005. N glycolylation of the nucleotide precursors of peptidoglycan biosynthesis of Mycobacterium spp. is altered by drug treatment. J Bacteriol 187:2341–2347. doi: 10.1128/JB.187.7.2341-2347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raymond JB, Mahapatra S, Crick DC, Pavelka MS Jr. 2005. Identification of the namH gene, encoding the hydroxylase responsible for the N-glycolylation of the mycobacterial peptidoglycan. J Biol Chem 280:326–333. doi: 10.1074/jbc.M411006200. [DOI] [PubMed] [Google Scholar]
- 7.Amar C, Vilkas E. 1973. Isolation of arabinose phosphate from the walls of Mycobacterium tuberculosis H37Ra. C R Acad Sci Hebd Seances Acad Sci D 277:1949–1951. [PubMed] [Google Scholar]
- 8.McNeil M, Daffe M, Brennan PJ. 1990. Evidence for the nature of the link between the arabinogalactan and peptidoglycan of mycobacterial cell walls. J Biol Chem 265:18200–18206. [PubMed] [Google Scholar]
- 9.Jankute M, Grover S, Rana AK, Besra GS. 2012. Arabinogalactan and lipoarabinomannan biosynthesis: structure, biogenesis and their potential as drug targets. Future Microbiol 7:129–147. doi: 10.2217/fmb.11.123. [DOI] [PubMed] [Google Scholar]
- 10.Hancock IC, Carman S, Besra GS, Brennan PJ, Waite E. 2002. Ligation of arabinogalactan to peptidoglycan in the cell wall of Mycobacterium smegmatis requires concomitant synthesis of the two wall polymers. Microbiology 148:3059–3067. doi: 10.1099/00221287-148-10-3059. [DOI] [PubMed] [Google Scholar]
- 11.Yokoyama K, Miyashita T, Araki Y, Ito E. 1986. Structure and functions of linkage unit intermediates in the biosynthesis of ribitol teichoic acids in Staphylococcus aureus H and Bacillus subtilis W23. Eur J Biochem 161:479–489. doi: 10.1111/j.1432-1033.1986.tb10469.x. [DOI] [PubMed] [Google Scholar]
- 12.Yagi T, Mahapatra S, Mikusova K, Crick DC, Brennan PJ. 2003. Polymerization of mycobacterial arabinogalactan and ligation to peptidoglycan. J Biol Chem 278:26497–26504. doi: 10.1074/jbc.M302216200. [DOI] [PubMed] [Google Scholar]
- 13.Kawai Y, Marles-Wright J, Cleverley RM, Emmins R, Ishikawa S, Kuwano M, Heinz N, Bui NK, Hoyland CN, Ogasawara N, Lewis RJ, Vollmer W, Daniel RA, Errington J. 2011. A widespread family of bacterial cell wall assembly proteins. EMBO J 30:4931–4941. doi: 10.1038/emboj.2011.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan YG, Kim HK, Schneewind O, Missiakas D. 2014. The capsular polysaccharide of Staphylococcus aureus is attached to peptidoglycan by the LytR-CpsA-Psr (LCP) family of enzymes. J Biol Chem 289:15680–15690. doi: 10.1074/jbc.M114.567669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eberhardt A, Hoyland CN, Vollmer D, Bisle S, Cleverley RM, Johnsborg O, Håvarstein LS, Lewis RJ, Vollmer W. 2012. Attachment of capsular polysaccharide to the cell wall in Streptococcus pneumoniae. Microb Drug Resist 18:240–255. doi: 10.1089/mdr.2011.0232. [DOI] [PubMed] [Google Scholar]
- 16.Hübscher J, Lüthy L, Berger-Bächi B, Stutzmann Meier P.. 2008. Phylogenetic distribution and membrane topology of the LytR-CpsA-Psr protein family. BMC Genomics 9:617. doi: 10.1186/1471-2164-9-617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mills JA, Motichka K, Jucker M, Wu HP, Uhlik BC, Stern RJ, Scherman MS, Vissa VD, Pan F, Kundu M, Ma YF, McNeil M. 2004. Inactivation of the mycobacterial rhamnosyltransferase, which is needed for the formation of the arabinogalactan-peptidoglycan linker, leads to irreversible loss of viability. J Biol Chem 279:43540–43546. doi: 10.1074/jbc.M407782200. [DOI] [PubMed] [Google Scholar]
- 18.Ma Y, Pan F, McNeil M. 2002. Formation of dTDP-rhamnose is essential for growth of mycobacteria. J Bacteriol 184:3392–3395. doi: 10.1128/JB.184.12.3392-3395.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y. 2007. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins 69(Suppl 8):108–117. doi: 10.1002/prot.21702. [DOI] [PubMed] [Google Scholar]
- 21.Alderwick LJ, Lloyd GS, Ghadbane H, May JW, Bhatt A, Eggeling L, Fütterer K, Besra GS. 2011. The C-terminal domain of the arabinosyltransferase Mycobacterium tuberculosis EmbC is a lectin-like carbohydrate binding module. PLoS Pathog 7:e00972-16. doi: 10.1371/journal.ppat.1001299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhatt A, Jacobs WR Jr. 2009. Gene essentiality testing in Mycobacterium smegmatis using specialized transduction. Methods Mol Biol 465:325–336. doi: 10.1007/978-1-59745-207-6_22. [DOI] [PubMed] [Google Scholar]
- 23.Bhatt A, Kremer L, Dai AZ, Sacchettini JC, Jacobs WR Jr. 2005. Conditional depletion of KasA, a key enzyme of mycolic acid biosynthesis, leads to mycobacterial cell lysis. J Bacteriol 187:7596–7606. doi: 10.1128/JB.187.22.7596-7606.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Birch HL, Alderwick LJ, Bhatt A, Rittmann D, Krumbach K, Singh A, Bai Y, Lowary TL, Eggeling L, Besra GS. 2008. Biosynthesis of mycobacterial arabinogalactan: identification of a novel α(1→3) arabinofuranosyltransferase. Mol Microbiol 69:1191–1206. doi: 10.1111/j.1365-2958.2008.06354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rana AK, Singh A, Gurcha SS, Cox LR, Bhatt A, Besra GS. 2012. Ppm1-encoded polyprenyl monophosphomannose synthase activity is essential for lipoglycan synthesis and survival in mycobacteria. PLoS One 7:e00972-16. doi: 10.1371/journal.pone.0048211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee RE, Brennan PJ, Besra GS. 1997. Mycobacterial arabinan biosynthesis: the use of synthetic arabinoside acceptors in the development of an arabinosyl transfer assay. Glycobiology 7:1121–1128. doi: 10.1093/glycob/7.8.1121. [DOI] [PubMed] [Google Scholar]
- 27.Alderwick LJ, Dover LG, Veerapen N, Gurcha SS, Kremer L, Roper DL, Pathak AK, Reynolds RC, Besra GS. 2008. Expression, purification and characterisation of soluble GlfT and the identification of a novel galactofuranosyltransferase Rv3782 involved in priming GlfT-mediated galactan polymerisation in Mycobacterium tuberculosis. Protein Expr Purif 58:332–341. doi: 10.1016/j.pep.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 28.Birch HL, Alderwick LJ, Rittmann D, Krumbach K, Etterich H, Grzegorzewicz A, McNeil MR, Eggeling L, Besra GS. 2009. Identification of a terminal rhamnopyranosyltransferase (RptA) involved in Corynebacterium glutamicum cell wall biosynthesis. J Bacteriol 191:4879–4887. doi: 10.1128/JB.00296-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghisaidoobe AB, Chung SJ. 2014. Intrinsic tryptophan fluorescence in the detection and analysis of proteins: a focus on Förster resonance energy transfer techniques. Int J Mol Sci 15:22518–22538. doi: 10.3390/ijms151222518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alderwick LJ, Lloyd GS, Lloyd AJ, Lovering AL, Eggeling L, Besra GS. 2011. Biochemical characterization of the Mycobacterium tuberculosis phosphoribosyl-1-pyrophosphate synthetase. Glycobiology 21:410–425. doi: 10.1093/glycob/cwq173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alderwick LJ, Molle V, Kremer L, Cozzone AJ, Dafforn TR, Besra GS, Fütterer K. 2006. Molecular structure of EmbR, a response element of Ser/Thr kinase signaling in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 103:2558–2563. doi: 10.1073/pnas.0507766103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shenderov K, Barber DL, Mayer-Barber KD, Gurcha SS, Jankovic D, Feng CG, Oland S, Hieny S, Caspar P, Yamasaki S, Lin X, Ting JP, Trinchieri G, Besra GS, Cerundolo V, Sher A. 2013. Cord factor and peptidoglycan recapitulate the Th17-promoting adjuvant activity of mycobacteria through mincle/CARD9 signaling and the inflammasome. J Immunol 190:5722–5730. doi: 10.4049/jimmunol.1203343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.D’Elia MA, Millar KE, Beveridge TJ, Brown ED. 2006. Wall teichoic acid polymers are dispensable for cell viability in Bacillus subtilis. J Bacteriol 188:8313–8316. doi: 10.1128/JB.01336-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.D’Elia MA, Pereira MP, Chung YS, Zhao W, Chau A, Kenney TJ, Sulavik MC, Black TA, Brown ED. 2006. Lesions in teichoic acid biosynthesis in Staphylococcus aureus lead to a lethal gain of function in the otherwise dispensable pathway. J Bacteriol 188:4183–4189. doi: 10.1128/JB.00197-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Campbell J, Singh AK, Swoboda JG, Gilmore MS, Wilkinson BJ, Walker S. 2012. An antibiotic that inhibits a late step in wall teichoic acid biosynthesis induces the cell wall stress stimulon in Staphylococcus aureus. Antimicrob Agents Chemother 56:1810–1820. doi: 10.1128/AAC.05938-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolucka BA, de Hoffmann E. 1995. The presence of β-d-ribosyl-1-monophosphodecaprenol in mycobacteria. J Biol Chem 270:20151–20155. doi: 10.1074/jbc.270.34.20151. [DOI] [PubMed] [Google Scholar]
- 37.Gurcha SS, Baulard AR, Kremer L, Locht C, Moody DB, Muhlecker W, Costello CE, Crick DC, Brennan PJ, Besra GS. 2002. Ppm1, a novel polyprenol monophosphomannose synthase from Mycobacterium tuberculosis. Biochem J 365:441–450. doi: 10.1042/BJ20020107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Makarov V, Manina G, Mikusova K, Möllmann U, Ryabova O, Saint-Joanis B, Dhar N, Pasca MR, Buroni S, Lucarelli AP, Milano A, De Rossi E, Belanova M, Bobovska A, Dianiskova P, Kordulakova J, Sala C, Fullam E, Schneider P, McKinney JD, Brodin P, Christophe T, Waddell S, Butcher P, Albrethsen J, Rosenkrands I, Brosch R, Nandi V, Bharath S, Gaonkar S, Shandil RK, Balasubramanian V, Balganesh T, Tyagi S, Grosset J, Riccardi G, Cole ST. 2009. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 324:801–804. doi: 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Batt SM, Jabeen T, Bhowruth V, Quill L, Lund PA, Eggeling L, Alderwick LJ, Fütterer K, Besra GS. 2012. Structural basis of inhibition of Mycobacterium tuberculosis DprE1 by benzothiazinone inhibitors. Proc Natl Acad Sci U S A 109:11354–11359. doi: 10.1073/pnas.1205735109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grover S, Alderwick LJ, Mishra AK, Krumbach K, Marienhagen J, Eggeling L, Bhatt A, Besra GS. 2014. Benzothiazinones mediate killing of Corynebacterineae by blocking decaprenyl phosphate recycling involved in cell wall biosynthesis. J Biol Chem 289:6177–6187. doi: 10.1074/jbc.M113.522623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laskowski RA, Watson JD, Thornton JM. 2005. ProFunc: a server for predicting protein function from 3D structure. Nucleic Acids Res 33:W89–W93. doi: 10.1093/nar/gki414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Completo GC, Lowary TL. 2008. Synthesis of galactofuranose-containing acceptor substrates for mycobacterial galactofuranosyltransferases. J Org Chem 73:4513–4525. doi: 10.1021/jo800457j. [DOI] [PubMed] [Google Scholar]
- 43.Pathak AK, Besra GS, Crick D, Maddry JA, Morehouse CB, Suling WJ, Reynolds RC. 1999. Studies on β-d-Galf-(1→4)-α-l-Rhap octyl analogues as substrates for mycobacterial galactosyl transferase activity. Bioorg Med Chem 7:2407–2413. doi: 10.1016/S0968-0896(99)00199-6. [DOI] [PubMed] [Google Scholar]
- 44.Bardarov S, Bardarov S Jr, Pavelka MS Jr, Sambandamurthy V, Larsen M, Tufariello J, Chan J, Hatfull G, Jacobs WR Jr. 2002. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148:3007–3017. doi: 10.1099/00221287-148-10-3007. [DOI] [PubMed] [Google Scholar]
- 45.Besra GS, Khoo KH, McNeil MR, Dell A, Morris HR, Brennan PJ. 1995. A new interpretation of the structure of the mycolyl-arabinogalactan complex of Mycobacterium tuberculosis as revealed through characterization of oligoglycosylalditol fragments by fast-atom bombardment mass spectrometry and 1H nuclear magnetic resonance spectroscopy. Biochemistry 34:4257–4266. doi: 10.1021/bi00013a015. [DOI] [PubMed] [Google Scholar]
- 46.Besra GS, Morehouse CB, Rittner CM, Waechter CJ, Brennan PJ. 1997. Biosynthesis of mycobacterial lipoarabinomannan. J Biol Chem 272:18460–18466. doi: 10.1074/jbc.272.29.18460. [DOI] [PubMed] [Google Scholar]
- 47.Alderwick LJ, Radmacher E, Seidel M, Gande R, Hitchen PG, Morris HR, Dell A, Sahm H, Eggeling L, Besra GS. 2005. Deletion of Cg-emb in Corynebacterianeae leads to a novel truncated cell wall arabinogalactan, whereas inactivation of Cg-ubiA results in an arabinan-deficient mutant with a cell wall galactan core. J Biol Chem 280:32362–32371. doi: 10.1074/jbc.M506339200. [DOI] [PubMed] [Google Scholar]
- 48.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental methods. Download
Sequence alignment of Lcp1 orthologues from M. tuberculosis, M. smegmatis, and C. glutamicum and LCP homologs from B. subtilis and S. pneumoniae. Amino acid sequences were aligned using ClustalW and rendered with EsPRIPT. Secondary structure information was obtained from PDB coordinates 2XXP. Download
Reaction scheme for the synthesis of octyl α-l-rhamnopyranosyl-(1→3)-2-acetamido-2-deoxy-α-d-glucopyranoside (compound 1). Download
Reaction scheme for the synthesis of octyl-β-d-galactofuranosyl-(1→5)-β-d-galactofuranosyl-(1→4)-α-l-rhamnopyranosyl-(1→3)-2-acetamido-2-deoxy-α-d-glucopyranoside (compound 2) and octyl-β-d-galactofuranosyl-(1→6)-β-d-galactofuranosyl-(1→5)-β-d-galactofuranosyl-(1→4)-α-l-rhamnopyranosyl-(1→3)-2-acetamido-2-deoxy-α-d-glucopyranoside (compound 3). Download
Flow chart tracking radioactivity incorporated from UDP-[14C]Galp through each of the analytical steps leading to TLC analysis (Fig. 5). Download