

Abstract
Assaying in vivo accrual of DNA damage and DNA mutations by stem cells and pinpointing sources of damage and mutations would further our understanding of aging and carcinogenesis. Two main hurdles must be overcome. First, in vivo mutation rates are orders of magnitude lower than raw sequencing error rates. Second, stem cells are vastly outnumbered by differentiated cells, which have a higher mutation rate—quantification of stem cell DNA damage and DNA mutations is thus best performed from small, well-defined cell populations. Here we report a mutation detection technique, based on the “duplex sequencing” principle, with an error rate below ∼10−10 and that can start from as little as 50 pg DNA. We validate this technique, which we call SIP-HAVA-seq, by characterizing Caenorhabditis elegans germline stem cell mutation accrual and asking how mating affects that accrual. We find that a moderate mating-induced increase in cell cycling correlates with a dramatic increase in accrual of mutations. Intriguingly, these mutations consist chiefly of deletions in nonexpressed genes. This contrasts with results derived from mutation accumulation lines and suggests that mutation spectrum and genome distribution change with replicative age, chronological age, cell differentiation state, and/or overall worm physiological state. We also identify single-stranded gaps as plausible deletion precursors, providing a starting point to identify the molecular mechanisms of mutagenesis that are most active. SIP-HAVA-seq provides the first direct, genome-wide measurements of in vivo mutation accrual in stem cells and will enable further characterization of underlying mechanisms and their dependence on age and cell state.
Mutations are thought to be a major cause of aging and to underlie oncogenic transformation and acquisition of drug resistance by cancer cells (Helleday et al. 2014). Yet much remains to be established about the mutagenic process, at both the cell and tissue levels. Multiple sorts of uncertainties exist at the cell level. First, the relative contributions to mutation accrual of errors in DNA replication and of exogenous damage are unclear, and these contributions might change, e.g., depending on physiological or external stresses to which cells are exposed and on the fidelity of the proofreading and repair mechanisms that are most active at a given time. Second, it is not always clear which kinds of errors or damage are most relevant to mutation accrual, since damage that occurs frequently can have a minimal contribution if it is efficiently repaired in an error-free fashion. Third, little is known about the extent to which the mutation rate and spectrum change with chronological or replicative age or with acquired mutations. At the tissue level, fundamental questions about clonal dynamics remain unaddressed. Tissues may be designed following strategies to minimize the opportunity for mutations to persist and spread as a result of cell proliferation (Cairns 1975; Chiang et al. 2015), but few techniques allow direct, genome-wide measurement of this spreading to test theories. Addressing these fundamental unknowns will be critical, e.g., to identify the factors that most strongly influence the likelihood of carcinogenesis (Tomasetti and Vogelstein 2015; Wu et al. 2016) or to devise effective therapies (e.g., Akhmetzhanov and Hochberg 2015).
Unknowns in the mutagenic process stem from severe technical limitations in the detection of rare mutations in relevant cell populations. Detection of mutations present at high frequency can be readily performed, for example by DNA sequencing, when using large quantities of starting material. But this approach is unsuitable for the study of well-defined populations of progenitor cells that may be present in small numbers. Even when large numbers of cells are available, as can be the case for cancer samples, subclonality of the mutations can make them difficult to detect (Wang et al. 2013); despite strong technical limitations that obscure rare mutations, current data already make it clear that a substantial number of mutations represent a low fraction of the reads (Nik-Zainal et al. 2012), which is due not just to contamination by “normal” cells but also in large part to diversity within the cancerous cell population. Data on non-rare mutations only address a late stage of the mutation accumulation process, since they leave out mutations that have not achieved sufficiently high frequency through clonal expansion, and thus do not provide a direct window into the clonal dynamics that shape tissue-level mutation accumulation. Techniques that bypass the limitations of naïve sequencing approaches in detecting rare mutations rely, e.g., on in vitro setups (Gundry et al. 2012) or on bulk tissue detection of mutation accrual at specific loci, for example, using rescue of inserted bacterial plasmids (Gossen et al. 1989) or tracking of mutations in genes such as APC in the intestine (Lynch 2010). These techniques share a key limitation with naïve sequencing approaches in that they do not distinguish between cell subpopulations. Stem cell mutation accrual is a critical source of tissue-wide mutation accumulation, yet using detection of bulk tissue mutations, the characteristics of this accrual can only be inferred indirectly—using models that make assumptions about the number of stem cells, their cell cycle kinetics, and selection for or against mutations after they have been acquired, all of which can change widely as our understanding of stem cell populations progresses. Importantly, one often makes the additional assumptions that all mutations are replication dependent, which is likely not the case in practice (Dollé et al. 2000; Beerman et al. 2014), and that mutation rates and spectra do not vary with age, with acquired mutator mutations, or with cell differentiation status—assumptions that are also invalid (Fox et al. 2013). It would thus be particularly advantageous to add to the limited direct data that are available (e.g., Dollé et al. 2000) and derive a genome-wide view of the damage accrual and mutagenesis process in progenitor populations.
A number of programs exist that can examine sequencing data to identify “rare variants,” i.e., mutations that are present in only a small fraction of the sampled cells (e.g., Koboldt et al. 2012; Cibulskis et al. 2013). These programs rely in part on statistical models that play a critical role in distinguishing the rare variants from sequencing errors. An important limitation of these models is that they do not account for errors that occur at an early step of in vitro amplification; a substantial number of mutated reads can derive from a common ancestral in vitro error and be mistakenly assigned to a rare variant in the original sample. As we show in the following, this can lead to a large underestimation of the false-positive rate in called mutations.
A recent breakthrough has substantially increased the practicality of Illumina sequencing to detect mutations in large cell populations, overcoming errors occurring during in vitro amplification of the DNA to be sequenced (Schmitt et al. 2012). Building on this technique, called “duplex sequencing,” we report a new method that allows sequencing of samples up to 2000-fold smaller than previously reported, at error rates lower than 10−10, i.e., orders of magnitude lower than for other techniques, including those that only target predetermined loci (10−5–10−8 reported error rates) (e.g., Dollé et al. 2000; Bielas and Loeb 2005), and also substantially lower than demonstrated in previous duplex sequencing reports (10−7 in study by Schmitt et al. 2012). We illustrate our technique by measuring endogenous mutation accrual in the Caenorhabditis elegans germ line—using libraries created from microdissected mitotic zones (MZs) comprising ∼50 pg DNA—and by asking how mutation accrual changes with cell cycling.
Results
Duplex sequencing principle
The fundamental issue with Illumina sequencing–based mutation detection is that sample preparation requires amplification both to prepare the on-chip cluster, and to overcome the high error rate of ∼0.1% by reading each genome position multiple times. However, even the best polymerases used in vitro have error rates orders of magnitude higher than in vivo error rates (McInerney et al. 2014). Errors that occur during the first rounds of in vitro amplification are particularly damaging because they may represent a large fraction of the reads and may even be stochastically largely overamplified compared to correct variants (Kanagawa 2003); these errors thus cannot be easily rooted out by great sequencing depth—even in the case of single-cell sequencing, which additionally suffers from the related problem of allele dropout (Piyamongkol et al. 2003). Schmitt et al. (2012) reported a simple but powerful idea to prevent bona fide mutations from being drowned out by technical errors. While applying this idea reduces error rates considerably, previous implementations required a high quantity of input genomic DNA that made studies of low-abundance cells problematic—a problem to which we return in the following. The principle is to tag purified DNA using Y-shaped adapters containing variable barcodes before any amplification is performed, in such a way that amplification products can be grouped based on the individual original DNA molecule from which they were derived, and such that it can be determined whether they were amplified from the “top” or “bottom” strand of that original molecule (Fig. 1A); we call this principle “strand identity preservation.” Because the original top and bottom strands are replicated independently, a substantial reduction in technical error rate can be achieved by accepting only candidate mutations that are detected both in DNA amplified from the original top strand and in DNA amplified from the original bottom strand. This scheme was referred to as “duplex sequencing” (Schmitt et al. 2012).
Figure 1.

Preparation of sequencing libraries, de novo mutation calling, and overall experimental scheme of SIP-HAVA-seq. (A) DNA to sequence is sheared, end-repaired, and, before any amplification is performed, ligated to hairpin adapters comprising a variable barcode. After linearization using NEB's “USER” enzyme, a first PCR is performed to amplify the material, and a second to add Illumina sequencing adapters. Illumina reads that correspond to the same “duplex” are grouped using the custom program “Mutinack” that relies on mapping position and variable barcode sequence. Mutation candidates are only reported if they are supported by both top and bottom strand amplification products. Mutinack also reports disagreements between the top and bottom strands, which can be indicative of a mismatch or damage-induced covalent modification in the original piece of DNA (since covalent modification can cause incorrect base-pairing during replication) or of an in vitro amplification error that occurred at an early step of the PCR; this step of the analysis does not distinguish between the two potential sources of disagreement. (BC) Illumina multiplexing barcode; (P5, P7) flow cell attachment sites. (B) EdU continuous labeling time course in hermaphrodites, taken at ages spanning the interval between onset of adulthood and time of collection for sequencing. (C) Experimental scheme. Each library is created from a single dissected worm germline MZ (red oval), comprising ∼250 cells. Mutations detected in one MZ but not the sister MZ from the same worm must have arisen de novo (stars in diagram). The speed of MZ cell cycling is increased by mating, and one can thus compare mutation rates in germ cells cycling at different speeds.
Adaptation of duplex sequencing protocol
We lowered the quantity of input DNA required for library preparation following the original duplex sequencing protocol by switching from dA-tailing to dT-tailing and using hairpin adapters that minimize adapter self-ligation. This lowered the required quantity from 0.3- to 1-µg input DNA to ∼50 pg (Methods; Supplemental Text section I; Supplemental Fig. S1; Supplemental Tables S15, S16). Our adapted protocol, which we call SIP-HAVA-seq (for Strand-Identity-Preserving Hairpin-Variable-Adapter sequencing), relies on adapters that have a shorter variable barcode that is not unique across molecules. To group sequencing reads into duplexes, we thus developed a new program dubbed “Mutinack” that relies on both short variable barcodes and read mapping position in the reference genome (Supplemental Text section II.A). Mutinack identifies mutation candidates and rates them as “Q1” or “Q2” depending on how well they are supported by the sequencing data (see Supplemental Text section II; for details of quality thresholds that are applied, see Supplemental Table S1); Q1 candidates are not output as bona fide mutations, but they cause any identical Q2 candidate detected in the “sister sample” from the same individual to be discarded, which helps ensure that mutations that are part of the individual's genetic background are ignored with high probability even if they are heterozygous.
Validation on the C. elegans germline
The nematode worm C. elegans has two sexes: hermaphrodite and male. Mating with males provides hermaphrodites with extra sperm and stimulates oocyte production and germ cell progenitor cycling (Cinquin et al. 2016); mating thus provides a physiological way of increasing activity of the hermaphrodite reproductive system. The gonad of hermaphrodites, on which we focus here, consists of two arms, each of which contains a distal MZ with stem cells that ensure growth during development and renewal throughout reproductive life; an MZ comprises ∼250 cells. Germ cells are derived from two precursors set aside early during development; the two arms grow independently as the precursors and their descendants proliferate, and the cell lineages of the two arms are thus separated early on. Germ cell cycling is high up to the second day of adulthood (last larval stage L4 + 2 d), as assayed by uptake of the thymidine analog EdU (5-ethynyl-2′-deoxyuridine) that marks S-phase cells (Fig. 1B) and drops at day 3 only in unmated hermaphrodites. Based on these data and measurements of cell cycle length (Chiang et al. 2015), we estimate that germ cells of mated hermaphrodites have undergone approximately one more cycle by L4 + 4 d than germ cells of unmated hermaphrodites, an increase of ∼5% (see Supplemental Text section III.A).
To ask how mutation accrual is affected by mating, we measured mutation rates in mated or unmated hermaphrodites prepared at L4 + 4 d (Fig. 1C). We prepared separate libraries from the MZ of each arm (Fig. 1C), using four mated and three unmated hermaphrodites for a total of 14 MZs, which we sequenced to an average depth of 70-fold—yielding an average of ∼16 duplexes per genome position after discarding repetitive regions to avoid spurious mutation detection and applying other Q1 thresholds (Supplemental Text sections II.C and III.B; Supplemental Figs. S2–S4).
High technical error rates close to ligation sites
Library preparation requires end-repair of sample DNA after sonication, which may be error-prone and thereby introduce spurious mutation candidates. These spurious candidates are expected to localize for the most part to the very end of repaired molecules, but proofreading activity of the end-repair polymerase may allow cycles of degradation and resynthesis to extend further inside the molecule. To address this problem, we plotted the frequency of mutation candidate detection as a function of distance to the closest adapter ligation site. Mutation candidates are indeed found at a higher than expected frequency close to ligation sites (Supplemental Fig. S5a), as are duplex disagreements (Supplemental Fig. S5b); in contrast, Phred scores are overall higher closer to the end of the reads (Supplemental Fig. S5c), suggesting that sequencer errors are not the problem. The trend of decreased mutation candidate enrichment as distance to the ligation site increases disappeared by position 35, which we thus chose as a threshold for Q2 mutation candidates. Our conclusions are robust against changes in this threshold (Supplemental Table S2).
Duplex strand disagreements provide an upper bound on technical error rate in mutation detection
The average duplex disagreement rate across all samples was 2.3 × 10−5 (mostly contributed by substitutions) (Supplemental Tables S3, S4; Supplemental Fig. S6a,b). Since the top and bottom strands in the original sample are replicated independently, the technical error rate can be conservatively estimated as (2.3 × 10−5)2/3 = 1.8 × 10−10, which is an estimate of the rate at which in vitro replication errors lead by chance to the same spurious mutation appearing for top and bottom strand amplification products. Since bona fide top/bottom strand mismatches in the original sample contribute to the disagreements in addition to errors introduced during in vitro replication, this estimate is an upper bound (i.e., the technical error rate may be even lower). As expected, the rate of duplex disagreements in which both the “top” and “bottom” bases differed from the wild type was low (1.5 × 10−9); we nonetheless excluded such disagreements to avoid contributions of reference genome errors to our analysis.
Differences in disagreement spectra further support validity of mutation detection
As a test of SIP-HAVA-seq, we compared the duplex disagreement spectrum to spectra of raw mismatches derived directly from the alignment of each individual read to the reference genome and to the spectrum of Q2 mutations (Supplemental Fig. S7). The duplex disagreement spectrum should be dominated by in vitro replication errors—some of which may be due to covalent modifications of bases in the starting material—and by bona fide mismatches in the starting material. The raw mismatch spectra should be dominated by sequencer errors but may also receive a contribution from in vitro replication errors occurring during the later cycles of PCR and from reference genome errors. Finally, the mutation spectrum is determined by the combination of DNA damage and repair events.
We found that raw mismatches are mostly T → A and duplex disagreements are G → T or C → A (Supplemental Text section II.D; Supplemental Fig. S6c,d), while Q2 mutations are mostly deletions (see below). The mutation spectrum (Supplemental Fig. S8) is thus highly distinct from the other spectra, showing that the mutations we identified are not “bleed-through” of in vitro amplification or sequencer errors.
Mating alters duplex disagreement distribution
To begin asking what information duplex disagreements may hold about the in vivo presence of mismatches or of covalent modifications that lead to in vitro misreplication, we compared disagreements in MZ DNA extracted from mated worms to those from unmated worms (Fig. 2). The main difference we identified is that median deletion disagreement length increases upon mating (Supplemental Text section II.D). This increase prompted us to refine our analysis, breaking down disagreements by length. We chose a threshold of 3 bp based on deletion disagreement lengths (Fig. 2B) and found that deletion and insertion disagreements longer than 3 bp are increased upon mating (P < 0.001 and P < 0.026, respectively; sixfold increase for deletion disagreements), mostly due to a 10-fold rate increase in gene regions (although that increase does not itself reach statistical significance: P > 0.052 after correction for multiple hypothesis testing) (Fig. 2E). This trend was reversed when considering disagreements not exceeding 3 bp (Fig. 2F).
Figure 2.

Characterization of duplex disagreements and comparison to deletion lengths. (A) Duplex disagreement rates broken down by genome location. (B,C) Distribution of duplex insertion and deletion disagreement lengths. (D) Duplex disagreement rates broken down by coding or template strand type. (E) Duplex disagreement rates broken down by genome location for disagreements longer than 3 bp. (F) Duplex disagreement rates broken down by genome location, for disagreements not exceeding 3 bp. (G) Deletion lengths. (A, D–F) Error bars, SEM; asterisks denote significance of pairwise differences computed using bootstrapped confidence intervals ([*] P < 0.05; [**] P < 0.01; [***] P < 0.001), with P-values corrected using Hochberg's step-up procedure with a 5% false-discovery rate.
That mating exerts opposite effects on short and long disagreement rates suggests that both kinds of disagreement reflect relevant properties of the DNA samples and do not stem solely from technical artifacts. Although various DNA covalent modifications may also result in duplex deletion disagreements (see Discussion), a straightforward source of these disagreements would be the presence of single-stranded gaps in input DNA. Such single-stranded gaps may lead to deletions if unrepaired before DNA replication or if repaired in an error-prone fashion (see Discussion). Mating increases the frequency of disagreements longer than 3 bp, and although shorter deletion disagreements are much more frequent (both in mated and unmated cases; Fig. 2B), the majority of the deletions that we detected were longer than 3 bp (n = 9/12) (Fig 2G; next section). In addition, rates in gene regions of both deletion disagreements longer than 3 bp and deletions longer than 3 bp are increased by mating (the former show a 10-fold increase and the latter go from a rate of zero to a rate of 1.5 × 10−8). This further suggests that deletion disagreements longer than 3 bp reflect at least in part single-stranded gaps in input DNA and that these longer gaps are a source of mutations—not because they are more prevalent than shorter gaps but perhaps because they are repaired less efficiently or through error-prone pathways, or because they occur concomitantly with other kinds of damage such as interstrand crosslinks (Sarkar et al. 2006).
We next refined our analysis of duplex substitution disagreements by computing a breakdown of disagreements over the different kinds of substitutions and over gene and nongene regions as well as worm mating status (Fig. 3; Supplemental Fig. S9). Following Schmitt et al. (2012), we used the ratio of the rate of each kind of substitution to that of its reciprocal (e.g., G → T/C → A). That ratio is expected to be one if there are no biases in disagreement-generating mechanisms; biases may be introduced by preferential covalent modification of a base, e.g., oxidation of guanine to 8-oxoguanine that frequently leads to mispairing with A during replication and thus G:C → T:A transversions (Shibutani et al. 1991). Of note, duplex disagreement ratios differ greatly from raw Q2 mismatch ratios (Supplemental Fig. S9), indicating that the former do not stem from “bleed-through” of sequencer errors. The relative prevalence of G → T substitution disagreements was slightly lower in mated worms compared with unmated worms (1.5 vs. 1.7, P < 0.0076; n = 14), which is attributable to a prevalence decrease in nongene regions upon mating (1.6 vs. 2.4, P < 0.0006; n = 14), and lower in gene than in nongene regions when only considering unmated worms (1.6 vs. 2.4, respectively, P < 0.0038; n = 14) (Fig. 3A).
Figure 3.

Duplex substitution disagreement ratios. (A,B) Ratios between each kind of duplex substitution disagreement and its reciprocal broken down by genome location (A) or coding or template strand type (B). Error bars and confidence intervals computed as in Figure 2.
Increased G → T disagreements in noncycling cells could have been explained by increased oxidative stress in noncycling cells increasing levels of 8-oxoguanine, which may be less efficiently repaired in compact chromatin regions (Lukas et al. 2011). To test this idea, we stained mated and unmated MZs and found that mated MZs show in fact a higher prevalence of 8-oxoguanine (38% of scored MZ cells show foci vs. 22% for unmated; n = 15 and n = 18 MZs; P < 0.036) (Supplemental Fig. S10, Supplemental Text section III.C). Increased G → T disagreements in unmated worms must thus have another source than increased 8-oxoguanine levels. One possibility is decreased efficiency of DNA mismatch repair in noncycling cells, consistent with the idea that mismatch repair is most active during S phase (Schroering et al. 2007). Further experimentation will be required to explore this idea.
Overall, although mechanistic details would benefit from further study, the fact that some disagreement characteristics change according to mating status shows that they reflect at least partially in vivo mechanisms. Further supporting the idea that duplex disagreements identify intermediate states in the mutagenesis process, the substantial increase upon mating of deletion disagreements longer than 3 bp in gene regions is matched by an increase in the rate of deletions of similar characteristics—as detailed in the next section.
Mating increases deletion rate
Having characterized duplex disagreements, we next turn to mutations. We retrieved a total of 12 deletions and five substitutions (Tables 1, 2), making for an average mutation rate of 1.9 × 10−8 per base pair. The average mutation rate of DNA from mated worms was 1.9-fold higher than that from unmated worms (2.3 × 10−8 vs. 1.2 × 10−8, respectively) (Fig. 4A), but the difference did not reach statistical significance (95% bootstrapped confidence interval for difference of the means: −4.2 × 10−8—0.4 × 10−8). Interestingly, however, whereas the substitution rates were highly similar for mated and unmated worms (5.5 × 10−9 vs. 5.7 × 10−9; P > 0.8; n = 14), the deletion rates were significantly higher in mated worms (1.8 × 10−8 vs. 6.5 × 10−9; P < 0.03; n = 14) (Fig. 4A; Supplemental Text section II.D). Mated worm deletions had a median length of 7 bp (Table 2); although our analysis—which relies on gaps in read alignments—does not detect deletions >49 bp, below that threshold read length does not appear to substantially bias our estimate of median deletion length (Supplemental Fig. S11). We could not identify any conspicuous genome context at deletion sites (Supplemental Text section II.E; Supplemental Tables S5–S10). Further breaking down the analysis by genome region, we found that gene regions had a significantly higher mutation rate in mated worms than in unmated worms (2.3 × 10−8 vs. 3.5 × 10−9; Bonferroni-corrected P < 0.007 × 2 = 0.014) (Fig. 4B), which stemmed from an increased deletion rate (1.7 × 10−8 vs. 0; Bonferroni-corrected P < 0.001 × 4 = 0.004); the difference in gene-region substitution rates between mated and nonmated worms did not reach statistical significance (5.3 × 10−9 vs. 3.5 × 10−9; P > 0.9). Nongene regions showed a minimal difference in mutation rates. In summary, we conclude that mating increases the deletion rate in gene regions.
Table 1.
Mutation sites and gene expression at those sites
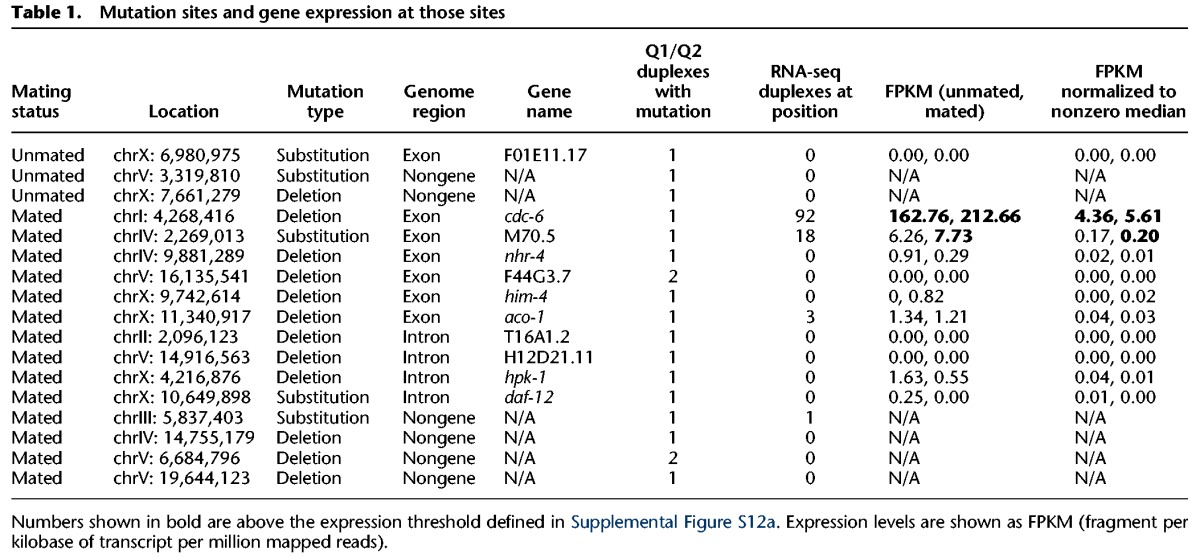
Table 2.
Genome context of mutations
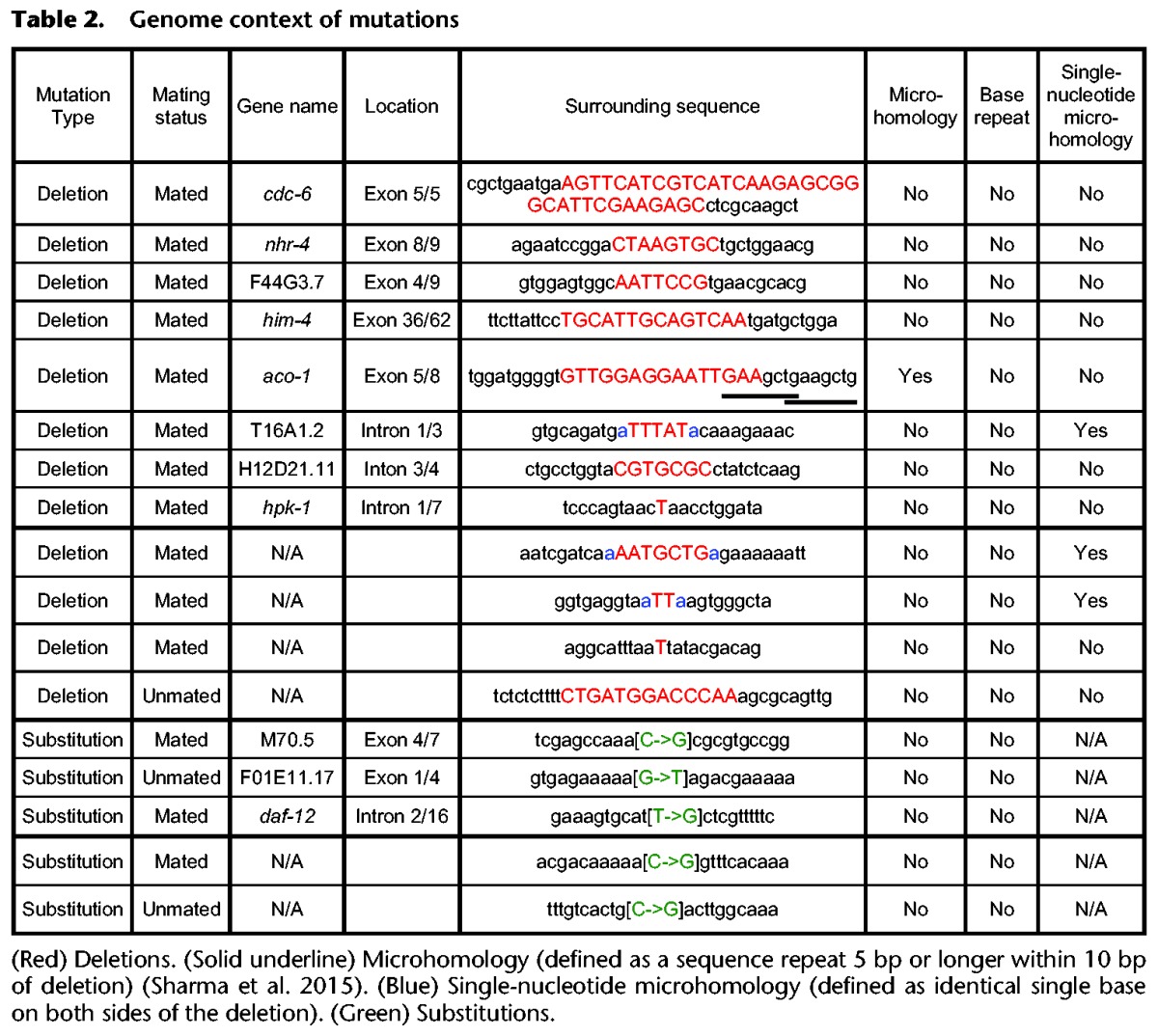
Figure 4.
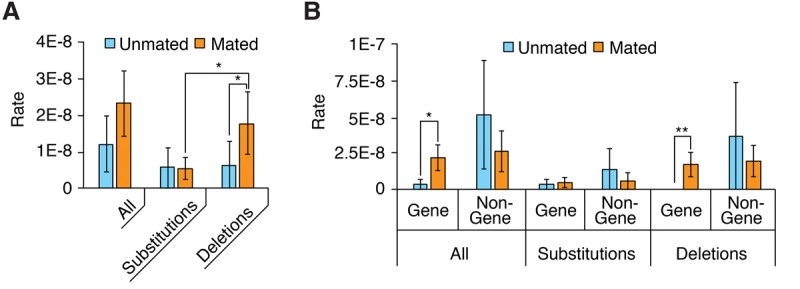
Characterization of mutations. (A,B) Distribution of mutations broken down by mutation type (A) or genome region (B). Error bars, SEM; asterisks denote the significance of pairwise differences computed using bootstrapped confidence intervals ([*] P < 0.05; [**] P < 0.01), with P-values corrected using the Bonferroni procedure.
Nonexpressed regions have higher mutation rate
Because extra mutations induced by mating were substantially enriched in gene regions, we next asked if the occurrence of mutations correlates with gene expression. To measure gene expression levels, we performed RNA-seq on MZs of mated and unmated worms dissected at L4 + 4 d (see Supplemental Text section III.B). As expected from previous reports (Strome et al. 2014), little expression was detected in either sample type from Chromosome X (Fig. 5), the chromosome on which the highest number of mutations was detected. We first compared disagreement rates between expressed and nonexpressed regions (as defined in Supplemental Fig. S12). We found that duplex insertion disagreements are less frequent in nonexpressed gene regions of mated samples than in expressed regions of the same samples or nonexpressed regions of unmated samples (2.7 × 10−7 vs. 5.5 × 10−7 or 5.7 × 10−7; P < 0.016 and P < 0.001, respectively; n = 14) (Supplemental Fig. S12); deletion or substitution disagreements are unaffected by expression status (Supplemental Fig. S12c).
Figure 5.
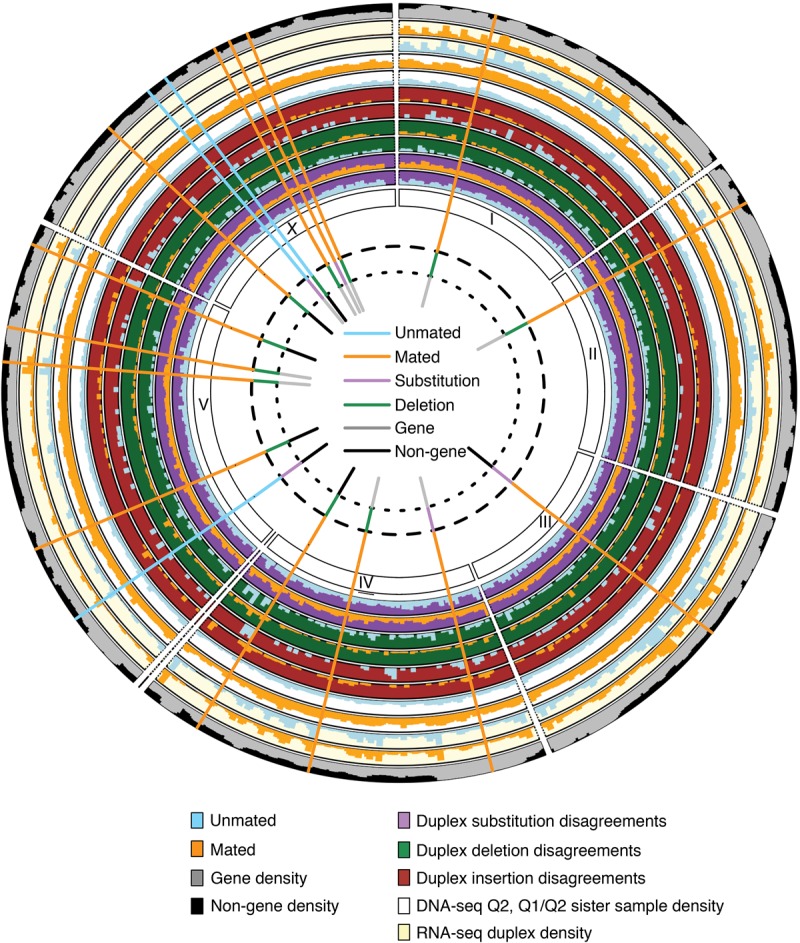
Graphical overview of sequencing results and detected mutations. Densities of DNA-seq Q2 candidates with at least one Q1/Q2 candidate at a matching position in the sister sample, RNA-seq duplex coverage, gene regions, and duplex disagreements are shown on outer tracks. Detected mutations, color-coded by kind, sample type (mated or unmated), and location type (gene or nongene) are shown as bars.
Second, we compared mutation rates between mated and unmated worms, considering expressed and nonexpressed genes. Nonexpressed genes showed a significantly higher mutation rate in mated worms, while expressed genes did not show a clear difference (Supplemental Fig. S13; Supplemental Table S11; see also Supplemental Table S12; Supplemental Figs. S14, S15), although that may be because of their smaller number. It thus appears that the significant deletion increase associated with extra cell cycling may be preferentially contributed by nonexpressed gene regions.
Comparison with other approaches
To help situate SIP-HAVA-seq on the fast-evolving landscape of mutation detection techniques, we compared it to two alternative approaches. A common kind of approach to dealing with small quantities of DNA is to perform multiple displacement amplification (MDA) prior to sequencing library preparation. We verified that using MDA prior to SIP-HAVA-seq is impractical because of the high technical error rate it introduces (Supplemental Text section IV.A; Supplemental Fig. S16). Another kind of approach is to prepare libraries with a small amount of amplification and to identify rare variants using software that attempts to distinguish sequencing errors from bona fide mutations.
To quantitatively assay the advantage afforded by SIP-HAVA-seq over rare-variant-calling software, we compared the output of Mutinack to the output of two popular programs, VarScan 2 (Koboldt et al. 2012) and MuTect (Cibulskis et al. 2013), which have been shown to be top-performing tools (Wang et al. 2013). When run on our data, these programs displayed an extremely high false-positive rate (Supplemental Text section IV.B,C; Supplemental Figs. S17–S22; Supplemental Tables S13, S14). We thus conclude that—at least for the kind of sample used in the present study, which requires substantial amplification before sequencing because of the low quantity of starting material—Mutinack enables an analysis that would not be possible using preexisting tools.
Discussion
Technical improvements to duplex sequencing principle are of general applicability
Before addressing the biological significance of our findings, we first briefly discuss their technical aspect. Compared to the original “duplex sequencing” and subsequent reports, SIP-HAVA-seq contributes four broad improvements. First, by the use of hairpin adapters, it enables a substantial reduction—of a few orders of magnitude—in the quantity of DNA used as starting material. Here we have applied this technique to microdissected tissue, but we expect it will work just as well on rare stem cell populations purified by any other means such as cell flow sorting. It should, in principle, be possible to extend it to single-cell libraries (although given the lack of preamplification that may come at the cost of severely reduced breadth of genome coverage: Duplexes that fail to ligate to adapters prior to amplification are lost from the library). As a second improvement, the estimated error rate of SIP-HAVA-seq is substantially lower than reported in previous duplex sequencing reports. An important germane discovery, which is likely relevant irrespective of the particular duplex sequencing protocol that is followed, is that technical error rates are higher in a relatively large region abutting duplex ends; a crucial feature of our software is that it prevents these regions from providing Q2 candidates, taking into account the distance of each read base to both duplex ends. This suggests that the cost efficiency of duplex sequencing in general will benefit from improvements to Illumina sequencing that increase read length. As a third improvement, our software allows for straightforward analysis of all duplex disagreement types over desired genome regions, the utility of which is illustrated, e.g., by the different behavior of gene and nongene regions. Fourth, and finally, the variable barcode regions we have used are shorter than in previous reports, because we rely both on mapping position and on the variable barcode to group reads into duplexes. This has the important advantage of simplifying adapter preparation, but samples with substantially larger numbers of input duplexes would require increasing variable barcode length to allow proper duplex separation. Overall, we expect that the improvements afforded by SIP-HAVA-seq will apply in a straightforward fashion to other sample types.
Comparison to mutation accumulation lines suggests age and/or mating status dependence in mutation rates and spectra
SIP-HAVA-seq provides an unprecedented view of mutation accrual in a small progenitor population and of its relationship with cell cycling. Mutation rates and spectra have been measured in the C. elegans germ line using mutation accumulation lines (Denver et al. 2004, 2006, 2009; Meier et al. 2014). Such experiments detect mutations that have occurred at some point in the life cycle of the worm; they do not distinguish between mutations that occurred in young vs. old individuals or in MZ cells vs. cells undergoing meiosis. The former distinction is important because it is not clear whether mutation rates (and not just accumulated mutations) increase in aging individuals; the latter because meiosis may be a step at which a substantial proportion of mutations are incurred, as suggested, e.g., in the case of human spermatogenesis (Grégoire et al. 2013).
How do the mutations we characterize compare to those from previous worm germline studies? The average mutation rate that we computed from DNA of unmated worms, 1.2 × 10−8, is somewhat higher than in previous reports (3.1 × 10−9 [Denver et al. 2009] or 10−8 [Meier et al. 2014] per site per worm generation), but the main difference that we observe is in the mutation spectrum. One early publication reported a high frequency of insertions (Denver et al. 2004), while later reports found mainly (Meier et al. 2014) or exclusively (Denver et al. 2009) substitutions (the reasons for these differences are unclear, but our comparison between mated and unmated worms suggests that a factor could perhaps be replicative age of the gametes that formed new individuals chosen as founders of each generation). In contrast to previous reports, we find balanced substitutions and deletions (based on a limited number of mutations in unmated worms) or a majority of deletions (in mated worms).
Further experiments will be required to explain the difference between the mutation rates and spectra derived from our experiments and those derived from mutation accumulation lines. Two broad, nonmutually exclusive possibilities are that there is a difference in mutation accrual between the two kinds of experiments or that there is a difference in selective forces that apply to those mutations. We discuss these two possibilities in turn. Mutation accrual may change with chronological and/or replicative age, with cell state, or with worm mating status to favor deletions. Compatible with age-dependent changes, only two mutations we detected were present in more than one duplex, which suggests that observed mutations arose late, since none of them had a chance to stochastically expand in the cell population. Importantly, the germ cell progenitors that we dissected at the older L4 + 4 d stage only infrequently contribute fertilized gametes in the case of worms passaged without mating; age-dependent changes in mutation spectrum could thus explain the difference between our spectra and those observed with mutation accumulation lines (Denver et al. 2004, 2006, 2009; Meier et al. 2014). Compatible with cell state–dependent changes, it has been suggested that a substantial proportion of mammalian mutations are acquired during meiosis (Grégoire et al. 2013), i.e., after germ cells leave the mitotic state; if mutations acquired during or after meiosis are enriched for substitutions and insertions, this could explain why accumulation lines show such mutations in larger proportions. Finally, compatible with mating-specific changes, seminal fluid introduced by mating in all likelihood induces organism-wide changes in worms as it does in Drosophila (Avila et al. 2011); indeed, seminal fluid is likely responsible for shortening of C. elegans hermaphrodite lifespan even in the absence of germ cells (Shi and Murphy 2014). It may thus be that mating affects mutation accrual indirectly, e.g., by causing oxidative stress in various tissues including the MZ, and causes a shift in mutation spectrum, which does not occur with mutation accumulation lines because their propagation relies on hermaphrodite self-fertilization rather than on mating with males.
Another possibility to explain mutation spectrum differences derives from the fact that a high proportion of meiotic cells (in excess of 50%) undergo apoptosis (MZ cells do not appear to undergo apoptosis) (Gumienny et al. 1999; Jaramillo-Lambert et al. 2007; Chiang et al. 2015). Little is known about the upstream controls of the apoptosis decision, but it is possible that apoptosis preferentially targets cells (Bhalla and Dernburg 2005; Jaramillo-Lambert et al. 2010) that have incurred specific kinds of damage, which may substantially reshape the mutation spectrum; such a mechanism may rely on epigenetic memory of DNA damage (Polo et al. 2006; Li et al. 2015). It is also possible that there is selection against certain kinds of mutations; for example, deletions, which we see more frequently than in mutation accumulation lines, may be more detrimental than substitutions because of the frameshifts they often introduce. This selection may act at the meiotic step and perhaps also at later stages for mutations that cause, e.g., embryonic lethality.
Overall, the low number of mutations detected in more than one copy qualitatively argues that an age-dependent increase in mutation rate takes place in the MZ; changes in mutation spectra may also occur. Irrespective of the combination of possibilities discussed above that apply, SIP-HAVA-seq reveals mutation characteristics in proliferating germ cells that are not detected using mutation accumulation lines and opens new avenues to tease out various sources of mutation accrual.
Duplex deletion disagreements identify single-stranded DNA gaps as potential mutation precursors
One unique aspect of our study is that we identify not only de novo mutations but also marks of molecular intermediates that may lead to these mutations, under the form of duplex disagreements. These disagreements can be generated in multiple ways, some of which (mismatches in starting material and covalent modifications of that material) are relevant and others (accrual during in vitro amplification) are not. Although we cannot distinguish between these possibilities for any disagreement taken in isolation, we can and do detect different biases in different genome regions and in different sample types (mated and unmated), strongly supporting the biological relevance of these biases. Although substitution disagreements can have a relatively straightforward interpretation, especially in the case of G → T, a transversion expected as a result of guanine oxidation to 8-oxoguanine (Shibutani et al. 1991), we find that a more complex picture emerges in that 8-oxoguanine levels in fact do not positively correlate with G → T disagreement levels.
Deletion disagreements are also complex, because multiple mechanisms may lead to a molecular state where a sequence is deleted in one strand of a molecule but not in the other strand of the same molecule. DNA damage can lead to deletions upon replication (Shibutani and Grollman 1993), and deletion disagreements may thus stem indirectly from some forms of DNA damage that lead to deletion-inducing misreplication of one of the duplex strands early in the amplification process; this possibility will require further investigation. Another possibility is that deletion disagreements stem from single-stranded gaps: The DNA ligase used in the process of library preparation can seal such single-stranded gaps of up to 100 bases (Nilsson and Magnusson 1982), which should allow gap-carrying duplexes to make it into the sequencing library.
How would single-stranded gaps be formed in DNA? We first note that gaps are associated with single-strand breaks, which may be the form of DNA damage that is incurred at the highest rate (Vilenchik and Knudson 2003), and that they can persist from one phase of the cell cycle to another before being repaired (Diamant et al. 2012); embryonic stem cells display such gaps at high frequency, possibly because of a short G1 phase (Ahuja et al. 2016). A high frequency of single-strand gaps is thus not surprising. A major source may be both nucleotide excision repair and base excision repair (BER), both of which are active in the worm germ line (Lans and Vermeulen 2011) and rely on the formation of single-strand gaps as intermediates that require repair synthesis. Interestingly, in the case of long-patch BER there does not appear to be a mechanism to directly “hand off” excision-generated gaps to complexes that fill those gaps (Prasad et al. 2011), which may contribute to gap perdurance. The reported length of long-patch-BER–generated gaps, approximately two to 12 bases (Sattler et al. 2003), is largely compatible with the length of observed deletion disagreements (Fig. 2B). Other contributors to gap accrual may be oxidative damage–induced covalent modifications that can hamper hand-off and possibly lead to DNA end trimming (Çağlayan and Wilson 2015), or error-prone translesion synthesis across damaged regions (e.g., Bassett et al. 2002).
Effects of cell cycling on mutation accrual
Another unique aspect of our study of germ cell mutation accrual is that we assay highly similar tissues (mated and unmated MZs) that differ in their cell cycle rate. We modulated cell cycle rates in a physiological way: by exposure of wild-type hermaphrodites to males. Hermaphrodite reproduction is such that mating mainly affects cell cycle rate over the last 2 d before sample collection at L4 + 4. The period over which detected mutations could theoretically have been accrued covers the time of onset of germ cell proliferation (during larval development) to L4 + 4, but in practice, most mutations appear to have arisen late in worm life (see above). Therefore, although we estimate that mating causes an increase in total cell cycle number of only ∼5% over the period of germ cell proliferation, cell cycle activity may be more substantially different over the period during which most detected mutations are accrued, i.e., late in life (Fig. 1B). We note that, as discussed above, changes in mutation accrual upon mating may not be due solely to extra cell cycling but perhaps also, e.g., to global changes induced by seminal fluid. Furthermore, mutation accrual due to extra cell cycling may not be solely due to replication errors but perhaps also, at least in part, to oxidative stress.
Effects of mating on mutation accrual are surprising: We do not detect any substitution rate increase (possibly in large part because mating only modestly increases total cell cycle number, as discussed above), but we do detect a substantial deletion rate increase in nonexpressed gene regions. How does this deletion rate increase come about? That deletions but not substitutions increase in frequency upon mating strongly suggests that the deletion rate increase is not simply a result of increased DNA replication rate with errors occurring at a constant rate per cell division. That, in addition, duplex deletion disagreements longer than 3 bp also increase in frequency upon mating, in the same genome regions as deletions, suggests that these disagreements may identify single-stranded gaps that can be converted to deletions. We note that, unlike deletions, about a third of deletion disagreements longer than 3 bp show microhomology; it may thus be that there are multiple relevant subcategories of long deletion disagreements, some of which give rise to deletions at a higher rate. Gaps longer than 3 bp perhaps increase in frequency after mating because cell cycling requires high metabolic activity that causes oxidative damage repaired, e.g., through long-patch BER, and are perhaps more frequent in nonexpressed gene regions because of a combination of accessibility of DNA to damaging agents and slower repair kinetics than in actively transcribed regions because of, e.g., chromatin structure differences (Misteli and Soutoglou 2009). Although gaps may be repaired in an error-free fashion, through translesion synthesis or homologous recombination (HR) (Ma et al. 2013), they may alternatively generate deletions either as a result of DNA replication or through error-prone HR (Guirouilh-Barbat et al. 2014) or as a result of replication-fork-mediated conversion of gaps to double-strand breaks (Kuzminov 2001). Although our data are compatible with nonhomologous end joining playing a role in generating the deletions we detected, they do not currently allow us to finely distinguish different double strand break repair pathways (Supplemental Text section II.F).
Further experiments will be necessary to test directly whether single-stranded gaps are a major source of deletions, to ask why longer gaps would be more mutagenic than shorter gaps, to ask how gene expression and coding/template strand status affect gap accrual (Supplemental Text section II.G; Supplemental Fig. S23), and to identify the molecular mechanisms at play in the generation of the gaps and their processing to deletions. A fruitful approach would be to perform the same experiments in mutants that selectively inactivate specific pathways (many such C. elegans mutants are already available) (see, e.g., Meier et al. 2014) and to perform time courses to compare the accumulation of gaps and deletions with age. In addition, more ways exist to modulate cell cycle activity physiologically (Cinquin et al. 2016), which will provide further means to probe the relationship among cell cycling, DNA damage accrual, and error-free or error-prone repair.
Despite the numerous unknowns that remain, our results already paint a nuanced picture of the relationship between cell cycling and mutation accumulation. It may be that with respect to mutation accumulation, cycling is beneficial in some respects but detrimental in others (Bielas and Heddle 2000; Beerman et al. 2014), a question that SIP-HAVA-seq will make possible to resolve in more detail. This question will also be particularly interesting within the context of clonal dynamics: Mutation accumulation may provide competitive advantages to particular clones expanding by proliferation, both in cancerous tissue and in normal self-renewing organs such as the human testis (Goriely et al. 2003). Two of our samples had a given mutation that was present in more than one duplex, suggesting that mutation copy number increased as a result of cell cycling; with deeper sequencing of samples collected at an older age, SIP-HAVA-seq should make it possible to identify selection for or against particular mutations that are preferentially accrued in cycling or in resting states.
Methods
Optimized library preparation protocol
End repair and adapter ligation
The NEBNext ultra DNA library prep kit was used, scaling down volumes 5.5-fold to accommodate 10 µL starting material and using our custom variable barcode adapters (Supplemental Table S15). Samples were brought up to 50 µL with water and purified with 0.8× Ampure XP beads (Agencourt), as per NEB protocol for a 200-bp insert size library (used instead of 300-bp selection to increase recovery), and eluted in 11.5 µL of EB (Qiagen).
Library amplification
The following primers were used for the first amplification step (PCR 1 in Fig. 1A): Amp1F, ACACTCTTTCCCTACACGACGCTCTTCCGATCT; Amp1R, GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT. The PCR mix was 12.5 µL NEBNext high-fidelity 2× PCR master mix, 0.5 µL Amp1F (100 µM), 0.5 µL Amp1R (100 µM), and 11.5 µL library. The PCR program was 17 cycles of 10 sec at 98°C, 30 sec at 55°C, and 30 sec at 72°C. Sample was brought up to 50 µL with water and purified and size-selected with 0.8× Ampure XP beads, with elution in 11.9 µL EB (Qiagen). For the second amplification step (PCR 2 in Fig. 1A) the following primers were used: Amp2F, AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGAC; Amp2R, CAAGCAGAAGACGGCATACGAGATXXXXXXGTGACTGGAGTTCAGACGTGTGC (where XXXXXX represents a fixed multiplexing barcode). The PCR mix was 12.5 µL NEBNext high-fidelity 2× PCR master mix, 0.1 µL Amp2F (100 µM), 0.5 µL Amp2R (20 µM), and 11.9 µL of PCR 1 product. PCR 1 followed the same program as PCR 2, but with only six cycles. For final purification, samples were brought up to 50 µL with water, purified and size-selected with 0.8× Ampure XP beads, and eluted in 15 µL EB (Qiagen). Libraries from MDA subsamples were sonicated to 500 bp, allowing for purification with the GeneRead size selection kit (Qiagen).
Library quality control
Library fragment size distribution was evaluated using the Bioanalyzer high sensitivity DNA analysis kit (Agilent Technologies), to verify the lack of a distinct self-ligated adapter peak at 150 bp and the presence of a broad 300- to 500-bp peak; concentrations were quantified using the GeneRead library quant kit (Qiagen). Coverage was verified by qPCR with a 1:50 dilution of the library using nine primer sets (Supplemental Table S16) covering a total of three genes on chromosomes I, IV, and X. Only libraries positive for all loci were sequenced.
SIP-HAVA-seq adapter details
SIP-HAVA-seq adapter sequences were as follows: phosphate 5′-GAN1N2N3AGATCGGAAGAGCACACGTCTGAACTCCAGTCUA CACTCTTTCCCTACACGACGCTCTTCCGATCTN′3N′2N′1TCT-3′. The regions highlighted in bold, which correspond to the standard Illumina adapter sequence, are complementary and designed to provide adapter self-annealing into a double-stranded stem, while the underlined region is designed to form a single-stranded loop that can be cleaved at the uracil residue by addition of the USER enzyme (New England BioLabs) (Hendrickson 2012). A list of the 10 variable barcodes we used (N1N2N3 and reverse complement N′3N ′2N ′1 in sequence above) is given in Supplemental Table S15. Each adapter was synthesized independently (Invitrogen), resuspended at 15 µM in 10 mM Tris-HCl (pH 8.0), incubated for 5 min at 95°C, and immediately placed on ice to promote hairpin formation. All adapters were then mixed to form an equimolar solution of 0.15 µM each in Tris-HCl (pH 8.0).
Sequencing
Paired-end sequencing was performed on either an Illumina HiSeq 2500 (∼5% of our sequence data, spread across samples; read length 100 bases) or NextSeq 500 (remaining 95%; read length 150 bases). Sequence data were analyzed as detailed in Supplemental Text section II.
Data access
DNA-seq and RNA-seq data from this study have been submitted to the European Nucleotide Archive (http://www.ebi.ac.uk/ena) and ArrayExpress (https://www.ebi.ac.uk/arrayexpress/) under accession numbers PRJEB10953 and E-MTAB-4774, respectively. Source code for Mutinack is available as Supplemental File 1 and at https://github.com/cinquin/mutinack/ under an open source, free software license that allows for commercial usage.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants R21-AG042125, R01-GM102635, and P50-GM076516 and by University of California's Cancer Research Coordinating Committee award CRC-15-380484. We thank the anonymous reviewers for helpful comments; Ali Mortazavi for access to sequencing equipment; Nicole Che El-Ali and Ricardo Ramirez for assistance with that equipment; Arthur Lander for access to a qPCR machine; the Genomic High Throughput Facility shared resource of the Cancer Center (supported by National Institutes of Health grants CA-62203, S10RR025496, and S10OD010794) for access to a HiSeq 2500 sequencer; and UC Irvine's Optical Biology Core facility for access to microscopes. Some computations were performed on UC Irvine's high-performance computing cluster.
Author contributions: P.H.T. generated libraries and sequence data, tested software written by O.C., and used it to analyze sequence data. A.C. performed cell cycle and oxidative damage analyses. P.H.T. and O.C. wrote the manuscript, which was edited and approved by all authors.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.200501.115.
References
- Ahuja AK, Jodkowska K, Teloni F, Bizard AH, Zellweger R, Herrador R, Ortega S, Hickson ID, Altmeyer M, Méndez J, et al. 2016. A short G1 phase imposes constitutive replication stress and fork remodelling in mouse embryonic stem cells. Nat Commun 7: 10660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhmetzhanov AR, Hochberg ME. 2015. Dynamics of preventive vs post-diagnostic cancer control using low-impact measures. eLife 4: e06266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila FW, Sirot LK, LaFlamme BA, Rubinstein CD, Wolfner MF. 2011. Insect seminal fluid proteins: identification and function. Annu Rev Entomol 56: 21–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett E, Vaisman A, Tropea KA, McCall CM, Masutani C, Hanaoka F, Chaney SG. 2002. Frameshifts and deletions during in vitro translesion synthesis past Pt-DNA adducts by DNA polymerases β and η. DNA Repair (Amst) 1: 1003–1016. [DOI] [PubMed] [Google Scholar]
- Beerman I, Seita J, Inlay MA, Weissman IL, Rossi DJ. 2014. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 15: 37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla N, Dernburg AF. 2005. A conserved checkpoint monitors meiotic chromosome synapsis in Caenorhabditis elegans. Science 310: 1683–1686. [DOI] [PubMed] [Google Scholar]
- Bielas JH, Heddle JA. 2000. Proliferation is necessary for both repair and mutation in transgenic mouse cells. Proc Natl Acad Sci 97: 11391–11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielas JH, Loeb LA. 2005. Quantification of random genomic mutations. Nat Methods 2: 285–290. [DOI] [PubMed] [Google Scholar]
- Çağlayan M, Wilson SH. 2015. Reprint of “Oxidant and environmental toxicant-induced effects compromise DNA ligation during base excision DNA repair”. DNA Repair (Amst) 36: 86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns J. 1975. Mutation selection and the natural history of cancer. Nature 255: 197–200. [DOI] [PubMed] [Google Scholar]
- Chiang M, Cinquin A, Paz A, Meeds E, Price CA, Welling M, Cinquin O. 2015. Control of C. elegans germline stem cell cycling speed meets requirements of design to minimize mutation accumulation. BMC Biol 13: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. 2013. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31: 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinquin A, Chiang M, Paz A, Hallman S, Yuan O, Vysniauskaite I, Fowlkes CC, Cinquin O. 2016. Intermittent stem cell cycling balances self-renewal and senescence of the C. elegans germ line. PLoS Genet 12: e1005985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver DR, Morris K, Lynch M, Thomas WK. 2004. High mutation rate and predominance of insertions in the Caenorhabditis elegans nuclear genome. Nature 430: 679–682. [DOI] [PubMed] [Google Scholar]
- Denver D, Feinberg S, Steding C, Durbin M, Lynch M. 2006. The relative roles of three DNA repair pathways in preventing Caenorhabditis elegans mutation accumulation. Genetics 174: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver DR, Dolan PC, Wilhelm LJ, Sung W, Lucas-Lledó JI, Howe DK, Lewis SC, Okamoto K, Thomas WK, Lynch M, et al. 2009. A genome-wide view of Caenorhabditis elegans base-substitution mutation processes. Proc Natl Acad Sci 106: 16310–16314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamant N, Hendel A, Vered I, Carell T, Reissner T, de Wind N, Geacinov N, Livneh Z. 2012. DNA damage bypass operates in the S and G2 phases of the cell cycle and exhibits differential mutagenicity. Nucleic Acids Res 40: 170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dollé ME, Snyder WK, Gossen JA, Lohman PH, Vijg J. 2000. Distinct spectra of somatic mutations accumulated with age in mouse heart and small intestine. Proc Natl Acad Sci 97: 8403–8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox EJ, Prindle MJ, Loeb LA. 2013. Do mutator mutations fuel tumorigenesis? Cancer Metastasis Rev 32: 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goriely A, McVean GAT, Röjmyr M, Ingemarsson B, Wilkie AOM. 2003. Evidence for selective advantage of pathogenic FGFR2 mutations in the male germ line. Science 301: 643–646. [DOI] [PubMed] [Google Scholar]
- Gossen JA, de Leeuw WJ, Tan CH, Zwarthoff EC, Berends F, Lohman PH, Knook DL, Vijg J. 1989. Efficient rescue of integrated shuttle vectors from transgenic mice: a model for studying mutations in vivo. Proc Natl Acad Sci 86: 7971–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grégoire MC, Massonneau J, Simard O, Gouraud A, Brazeau MA, Arguin M, Leduc F, Boissonneault G. 2013. Male-driven de novo mutations in haploid germ cells. Mol Hum Reprod 19: 495–499. [DOI] [PubMed] [Google Scholar]
- Guirouilh-Barbat J, Lambert S, Bertrand P, Lopez BS. 2014. Is homologous recombination really an error-free process? Front Genet 5: 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumienny TL, Lambie E, Hartwieg E, Horvitz HR, Hengartner MO. 1999. Genetic control of programmed cell death in the Caenorhabditis elegans hermaphrodite germline. Development 126: 1011–1022. [DOI] [PubMed] [Google Scholar]
- Gundry M, Li W, Maqbool SB, Vijg J. 2012. Direct, genome-wide assessment of DNA mutations in single cells. Nucleic Acids Res 40: 2032–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleday T, Eshtad S, Nik-Zainal S. 2014. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet 15: 585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson C. 2012. Oligonucleotide adapters: compositions and methods of use. U.S. patent no. 13/513,726. http://www.google.com/patents/US20120238738.
- Jaramillo-Lambert A, Ellefson M, Villeneuve AM, Engebrecht J. 2007. Differential timing of S phases, X chromosome replication, and meiotic prophase in the C. elegans germ line. Dev Biol 308: 206–221. [DOI] [PubMed] [Google Scholar]
- Jaramillo-Lambert A, Harigaya Y, Vitt J, Villeneuve A, Engebrecht J. 2010. Meiotic errors activate checkpoints that improve gamete quality without triggering apoptosis in male germ cells. Curr Biol 20: 2078–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanagawa T. 2003. Bias and artifacts in multitemplate polymerase chain reactions (PCR). J Biosci Bioeng 96: 317–323. [DOI] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. 2012. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22: 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzminov A. 2001. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc Natl Acad Sci 98: 8241–8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lans H, Vermeulen W. 2011. Nucleotide excision repair in Caenorhabditis elegans. Mol Biol Int 2011: 542795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Calder RB, Mar JC, Vijg J. 2015. Single-cell transcriptogenomics reveals transcriptional exclusion of ENU-mutated alleles. Mutat Res 772: 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas J, Lukas C, Bartek J. 2011. More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol 13: 1161–1169. [DOI] [PubMed] [Google Scholar]
- Lynch M. 2010. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci 107: 961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Westmoreland JW, Resnick MA. 2013. Homologous recombination rescues ssDNA gaps generated by nucleotide excision repair and reduced translesion DNA synthesis in yeast G2 cells. Proc Natl Acad Sci 110: E2895–E2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInerney P, Adams P, Hadi MZ. 2014. Error rate comparison during polymerase chain reaction by DNA polymerase. Mol Biol Int 2014: 287430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier B, Cooke SL, Weiss J, Bailly AP, Alexandrov LB, Marshall J, Raine K, Maddison M, Anderson E, Stratton MR, et al. 2014. C. elegans whole-genome sequencing reveals mutational signatures related to carcinogens and DNA repair deficiency. Genome Res 24: 1624–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T, Soutoglou E. 2009. The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat Rev Mol Cell Biol 10: 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, Raine K, Jones D, Marshall J, Ramakrishna M, et al. 2012. The life history of 21 breast cancers. Cell 149: 994–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson SV, Magnusson G. 1982. Sealing of gaps in duplex DNA by T4 DNA ligase. Nucleic Acids Res 10: 1425–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piyamongkol W, Bermúdez MG, Harper JC, Wells D. 2003. Detailed investigation of factors influencing amplification efficiency and allele drop-out in single cell PCR: implications for preimplantation genetic diagnosis. Mol Hum Reprod 9: 411–420. [DOI] [PubMed] [Google Scholar]
- Polo SE, Roche D, Almouzni G. 2006. New histone incorporation marks sites of UV repair in human cells. Cell 127: 481–493. [DOI] [PubMed] [Google Scholar]
- Prasad R, Beard WA, Batra VK, Liu Y, Shock DD, Wilson SH. 2011. A review of recent experiments on step-to-step “hand-off” of the DNA intermediates in mammalian base excision repair pathways. Mol Biol (Mosk) 45: 586–600. [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Davies AA, Ulrich HD, McHugh PJ. 2006. DNA interstrand crosslink repair during G1 involves nucleotide excision repair and DNA polymerase zeta. EMBO J 25: 1285–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler U, Frit P, Salles B, Calsou P. 2003. Long-patch DNA repair synthesis during base excision repair in mammalian cells. EMBO Rep 4: 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, Loeb LA. 2012. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci 109: 14508–14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroering AG, Edelbrock MA, Richards TJ, Williams KJ. 2007. The cell cycle and DNA mismatch repair. Exp Cell Res 313: 292–304. [DOI] [PubMed] [Google Scholar]
- Sharma S, Javadekar SM, Pandey M, Srivastava M, Kumari R, Raghavan SC. 2015. Homology and enzymatic requirements of microhomology-dependent alternative end joining. Cell Death Dis 6: e1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Murphy CT. 2014. Mating induces shrinking and death in Caenorhabditis mothers. Science 343: 536–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibutani S, Grollman AP. 1993. On the mechanism of frameshift (deletion) mutagenesis in vitro. J Biol Chem 268: 11703–11710. [PubMed] [Google Scholar]
- Shibutani S, Takeshita M, Grollman AP. 1991. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 349: 431–434. [DOI] [PubMed] [Google Scholar]
- Strome S, Kelly WG, Ercan S, Lieb JD. 2014. Regulation of the X chromosomes in Caenorhabditis elegans. Cold Spring Harb Perspect Biol 6: a018366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasetti C, Vogelstein B. 2015. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347: 78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilenchik MM, Knudson AG. 2003. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc Natl Acad Sci 100: 12871–12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Jia P, Li F, Chen H, Ji H, Hucks D, Dahlman KB, Pao W, Zhao Z. 2013. Detecting somatic point mutations in cancer genome sequencing data: a comparison of mutation callers. Genome Med 5: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Powers S, Zhu W, Hannun YA. 2016. Substantial contribution of extrinsic risk factors to cancer development. Nature 529: 43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.