

Abstract
Purpose
To examine the effect of intra-tumor heterogeneity (ITH) on detection of genes within gene expression panels (GEPs), and the subsequent ability to predict prognostic risk.
Experimental Design
Multiplexed barcoded RNA analysis was used to measure the expression of 141 genes from five GEPs (Oncotype Dx, MammaPrint, PAM50, EndoPredict, and Breast Cancer Index) in breast cancer tissue sections and tumor-rich cores from 71 estrogen receptor (ER) positive node-negative tumors, on which clinical Oncotype Dx testing was previously performed. If the tumor had foci of high Ki67 (n=26), low/negative PR (n=13), or both (n=5), additional cores were obtained. In total, 181 samples were processed. Oncotype Dx recurrence scores were calculated from NanoString nCounter gene expression data.
Results
Hierarchical clustering using all GEP genes showed that majority (61/71) of tumor samples clustered by patient, indicating greater inter-patient heterogeneity (IPH) than ITH. We found a strikingly high correlation between Oncotype Dx recurrence scores obtained from whole sections versus tumor-rich cores (r=0.94). However, high Ki67 and low PR cores had slightly higher but not statistically significant recurrence scores. For 18/71 (25%) patients, scores were divergent between sections and cores and crossed the boundaries for low, intermediate and high risk.
Conclusions
Our study indicates that in patients with highly heterogeneous tumors, GEP recurrence scores from a single core could under- or over-estimate prognostic risk. Hence, it may be a useful strategy to assess multiple samples (both representative and atypical cores) to fully account for the ITH-driven variation in risk prediction.
Keywords: Intra-tumor heterogeneity, multi-gene tests, gene expression panel, prognosis prediction, breast cancer
Introduction
Breast cancer, the most common malignancy in women, is a heterogeneous disease characterized by distinct molecular subtypes.(1–3) In the past decade, gene expression profiling has enabled development of a wide variety of multi-gene prognostic signatures such as Oncotype Dx(4), MammaPrint(5), PAM50 (Prosigna)(6), EndoPredict(7), and Breast Cancer Index (BCI).(8, 9) Clinical studies on large patient cohorts have demonstrated that these gene expression panels (GEPs) may serve as tools to identify patients who are most likely to benefit from adjuvant systemic therapies, while sparing others of the unwanted side-effects and treatment-related cytotoxicity. Although there is growing recognition that GEPs are clinically relevant in breast cancer management, they have not been fully embedded into routine clinical practice.(10–12)
Among the many commercially available GEPs, Oncotype Dx and EndoPredict are the only tests that are supported by level I evidence based on the marker utility grading system.(13) Oncotype Dx (Genomic Health Inc., Redwood City, CA) measures the expression of 21 genes and calculates a recurrence score (RS) that predicts the risk of relapse in patients with ER-positive lymph node-negative early-stage breast cancer(4). Oncotype Dx is so far the most widely used GEP in clinical practice likely based upon its clinical validation(14) and approval by the National Comprehensive Cancer Network (NCCN), American Society of Clinical Oncology (ASCO), and St. Gallen European Society for Medical Oncology (ESMO).
In the past decade, there have been an increasing number of reports of intra-tumor heterogeneity (ITH) of gene expression and somatic DNA mutations.(15–20) Immunohistochemistry and fluorescence in situ hybridization studies using multiple areas of a breast tumor have shown significant ITH in ER gene expression levels and HER2 amplification.(15, 16) However, very few studies have examined the effect of ITH on measurement of GEPs and the accuracy to predict patient prognosis.(21) In a pilot study of four patients, Drury et al reported high concordance in Oncotype Dx RS scores between 0.6mm breast tumor cores and whole sections, but showed high variability in RS between individual cores resulting in prognostic misclassification.(22) Barry et al examined the influence of ITH on precision of microarray-based assays in multiple core needle biopsies, and showed high variance in recurrence risk predictions in 1 out of 18 breast cancer patients due to global variation in gene expression.(23) Another study by Gerlinger et al in a small number of renal cell carcinomas (RCCs) identified gene-expression signatures of both good and poor prognosis in different regions of the same tumor.(18) In a recent report, they reanalyzed this gene expression data in 63 regions from 10 RCCs and identified ITH in 8 of 10 tumors,(24) thus highlighting the importance of multiregion assessment in prediction of prognosis, response to therapy and risk of relapse. However, more research is needed to fully understand the degree of ITH-driven variation in risk prediction and design novel tumor sampling strategies that provide more reliable risk estimates.
In the present study, we investigated ITH of GEPs using multiplexed barcoded hybridization to measure the expression of 141 genes from five GEPs (Oncotype Dx, MammaPrint, PAM50, EndoPredict, and BCI) in whole tumor sections compared to multiple cores from 71 ER positive node-negative tumors. In addition to selecting a tumor-rich core, areas with high Ki67 and/or low PR were also used to punch cores, potentially representing aggressive areas of a tumor. In addition to examining the effect of ITH on measurement of GEPs, we also examined the effect of ITH on prediction of prognosis in the Oncotype Dx assay.
Methods
Breast tumor specimens
We previously reported inter-observer agreement amongst pathologists for hormone receptor scoring in 74 cases of ER-positive early breast cancer.(25) We used the same cohort to examine GEPs, but removed 3 cases due to inadequate tissue, for a total of 71 cases (Supplementary Table S1). All patients had clinical Oncotype Dx recurrence risk scoring performed at Genomic Health. Studies were performed with institutional review board approval PRO09100201. Immunohistochemical expression levels for ER, PR, and Ki67 were scored according to the ASCO/CAP guidelines (Supplementary Table S2).
For each patient, we selected the single FFPE block that was used for the clinical Oncotype Dx test, and cut a 5μM section, and a 0.6mm core from a tumor-rich representative part of the block. Additional cores were cut from tumors that had foci of high Ki67 (n=26), low PR (n=13), or both (n=5) to test the hypothesis that high Ki67 and/or low PR may indicate aggressive areas of a tumor. High Ki67 area in a tumor was defined as 10% or higher labeling index compared to the overall Ki67 labeling index for the whole section (Supplementary Table S2). Low PR area in a tumor was defined as >50% cells negative for PR. Five patients had all 4 types of tumor samples (including section, tumor-rich core, high Ki67 core, and low PR core), 39 patients had 3 tumor samples (section, tumor-rich core, and either high Ki67 core or low PR core) and 27 patients had 2 tumor samples (section and tumor-rich core). In total, we processed 181 samples for NanoString nCounter analysis.
RNA Isolation
The selected tumor blocks were enriched for invasive tumor and the most predominant non-invasive tissue component in the blocks was adipose tissue. For the tumor section, the whole section was scraped (without macrodissection), and the paraffin shavings were used to isolate RNA. Additionally, 1–3 cores from each sample were also used for RNA isolation. RNA was isolated using RNeasy® FFPE Kit (Qiagen, Valencia, CA) and quantified using UV spectroscopy (Nanodrop Technologies, Wilmington, DE).
Gene Expression
Barcoded-probes to measure the expression of genes comprising five GEPs and their respective housekeeping genes were manufactured by NanoString Technologies (Seattle, WA). This included Oncotype Dx (16 genes, 5 housekeeping genes), MammaPrint (66 genes), PAM50 (50 genes, 5 housekeeping genes), Endopredict (8 genes, 3 housekeeping genes), and BCI (7 genes, 4 housekeeping genes). Since there were some genes that overlapped among the five GEPs, gene expression was measured for a total of 141 unique genes (127 endogenous genes and 14 housekeeping genes) (Supplementary Table S3). The nCounter assay also included 6 positive controls and 8 negative controls. nCounter analysis was performed according to the manufacturer’s instructions using 100 ng of total RNA. Data were collected using the nCounter™ Digital Analyzer and initially processed using nSolver™ Analysis Software. QC metrics, including positive control linearity and limit of detection, were assessed using the positive and negative control probes (Supplementary Figure S1a). Raw intensities were normalized to the geometric mean of the positive controls and housekeeping genes using the R NanoStringNorm package (Supplementary Figure S1b). Data reproducibility was also assessed using technical replicates (Supplementary Figure S1c). NanoString data are available in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE79378.
Statistics
All data analyses were performed using R version 3.1.2 (http://www.r-project.org). The NanoString raw intensities were normalized to the geometric mean of positive controls and housekeeping genes using the R NanoStringNorm package. (26) Hierarchical clustering (using the Ward method on the Manhattan metric) was performed to visualize the clustering pattern of the NanoString gene expression data. To evaluate the effect of intra-tumor heterogeneity on the GEPs, we clustered all samples by genes belonging to each GEP and calculated the percentage of subjects with all samples clustered together as an indication of the robustness of the GEP to the intra-tumor heterogeneity. Since the clustering results heavily depend upon the number of clusters, we performed the clustering analysis by the number of clusters from 1 to 80.
Clinical Oncotype Dx recurrence scores were available for all patients. The Oncotype Dx score is based on an RT-PCR assay utilizing RNA isolated from macro-dissected tissue sections, which is most comparable to the whole sections used in our study. As RT-PCR and nCounter use different technologies for mRNA detection (amplification versus hybridization), the Oncotype Dx recurrence score was calculated from NanoString data (nstringRS) by fitting the following model to data from the “section” samples (1):
| (1) |
where clinRS denotes the clinical Oncotype Dx recurrence score and Xi’s represent the expression level of the 16 Oncotype Dx genes in the sections measured by Nanostring. The overall fit of the model was good (R2 = 0.87). The coefficients (β0 and βi) estimated from the linear model were then used to calculate the nstringRS for each type of tissue using (2).
| (2) |
Here the Xi’s represent the expression level of the Oncotype Dx genes measured by Nanostring for each sample.
The association of gene expression or nstringRS between two different types of tumor samples was described by the Spearman correlation coefficient. To investigate the intra-tumor variability of each gene and nstringRS, we fit a random effects model for each gene by subject. Samples from the same subject are considered to be within a cluster. From the linear mixed effect model, we derived the intra-tumor variability (1-ICC), where . (1-ICC) scores ranged between 0 and 1, and were grouped as low (0–0.2), fair (0.2–0.4), moderate (0.4–0.6), and high agreement (0.6–1.0). Kruskal-Wallis tests were used to compare gene expression levels and recurrence scores across all four sample types and Wilcoxon rank-sum tests were used for comparisons between two groups (Supplementary Table S4).
Results
Hierarchical clustering analysis of genes from five GEPs shows greater inter-patient heterogeneity than intra-tumor heterogeneity
We used nCounter analysis to measure the expression of genes from five GEPs in FFPE tumor sections compared to cores taken from tumor blocks of 71 ER-positive tumors (Figure 1A). Figure 1B shows the distribution of gene expression intensities for 141 measured genes (from 5 GEPs) and their variability across 181 tumor samples (from 71 patients). We investigated the extent of inter-tumor and intra-tumor heterogeneity in gene expression for GEPs in this patient cohort by performing hierarchical clustering using all 141 genes (Figure 1C and Supplementary Figure S2). We found that majority of tumor samples clustered by patient (61/71), indicating greater inter-patient heterogeneity (IPH) than intra-tumor heterogeneity (ITH) (Figure 1C). 10/71 (14%) patients showed discordant tumor samples, indicating ITH. A heat map shows two distinct patient clusters (rows) (Supplementray Figure S2). Based on the relative gene expression values of proliferation and survival genes, the top and bottom clusters likely represent patients with low and high recurrence risk, respectively. We also performed clustering using the genes from individual GEPs, which generally showed greater ITH compared to the combined analysis using all GEP genes (Figure 1D, Table 1 and Supplementary Figure S3a–e). MammaPrint contained the highest number of genes (n=66) among all tests and showed the least number of discordant samples (14/71) (Table 1 and Supplementary Figure S3a–e). In contrast, BCI, which had the lowest number of genes (n=7) showed the highest number of discordant samples (52/71), suggesting that the number of genes in a GEP may influence susceptibility to ITH.
Figure 1. Hierarchical clustering analysis of genes from five GEPs shows greater inter-patient heterogeneity than intra-tumor heterogeneity.
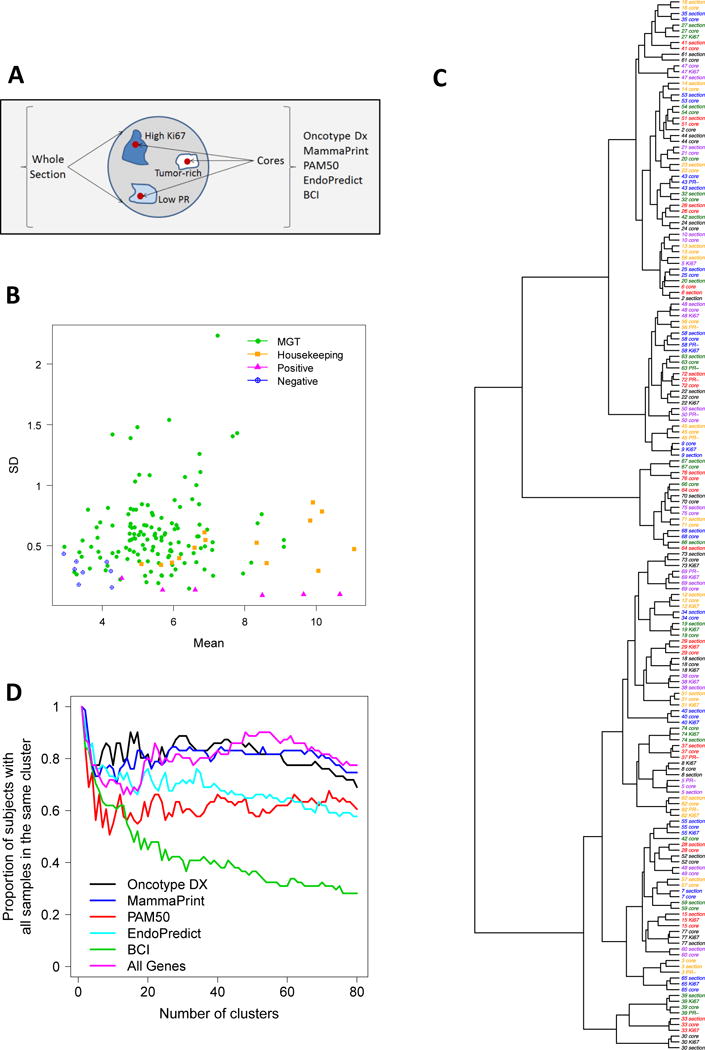
(A) nCounter analysis was used to measure the expression of genes from five GEPs in FFPE tumor sections compared to cores taken from tumor blocks for 71 ER-positive node-negative tumors. Cores were also obtained from foci of high Ki67 (n=26), low PR (n=13), or both (n=5). (B) Mean versus stand deviation plot of gene expression intensities for all measured genes (including 127 endogenous genes, 14 housekeeping genes, 6 positive controls, and 8 negative controls). Housekeeping genes show modest to high levels of gene expression with very low variation. (C–D) Hierarchical clustering by the Ward method using the Manhattan metric was performed on all GEP genes. The heatmap represents gene expression from 71 tumors (n=181 samples) profiled for 5 GEPs (127 endogenous genes). Red indicates high and green indicates low relative gene expression. Genes (columns) are clustered and tumors (rows) are clustered. (E) Clustering analysis for individual GEPs indicating the proportion of patients with all samples within the same cluster for a range of clusters (1 to 80).
Table 1.
Patients with discordant tumor samples identified by hierarchical clustering of GEP genes
| Gene Expression Panel | Number of endogenous genes | Patients with discordant tumor samples | |
|---|---|---|---|
| 1 | BCI | 7 | 52/71 |
| 2 | Endo | 8 | 30/71 |
| 3 | Oncotype Dx | 16 | 26/71 |
| 4 | PAM50 | 50 | 25/71 |
| 5 | Mammaprint | 66 | 14/71 |
| 6 | All genes | 127 | 10/71 |
Several genes involved in proliferation and invasion display high ITH
We next calculated the intra-group correlation coefficient (ICC) for each gene as a measurement of heterogeneity (Figure 2). 1-ICC values represented ITH (Figure 2A). 1-ICC scores ranging between 0 and 1 were grouped as 0–0.2 (low heterogeneity), 0.2–0.4 (fair heterogeneity), 0.4–0.6 (moderate heterogeneity) and 0.6–1.0 (high heterogeneity). Among the 127 genes that were analyzed, 73 genes showed low, 36 genes showed fair, 11 genes showed moderate, and 7 genes showed high ITH (Figure 2A and Table 2). Hence, a small proportion of genes (18/127, 14%) showed elevated variability in gene expression among the tumor samples. Figure 2B shows the distribution of 1-ICC scores for genes in the individual GEPs. 3/5 GEPs (including Oncotype Dx, MammaPrint, and PAM50) showed genes with moderate to high ITH. Supplementary Table S5 lists these heterogeneous genes and the correlation of expression values between different samples. This list includes several proliferation and invasion-related genes including MYC, FOXC1, EGFR, FGF18, CTSL2, and MMP9. When we clustered patient samples using the genes with low ITH (0–0.2 and 0.2–0.4 intra-variance scores), the majority of samples clustered by patient (Figure 2C and Supplementary Figure S4a–d). Conversely, when we clustered using the genes with 0.4–0.6 and 0.6–1.0 intra-variance scores, only a few patients had all samples clustered together, thus confirming their high ITH.
Figure 2. Intra-group correlation coefficient for GEP genes as a measurement of heterogeneity.
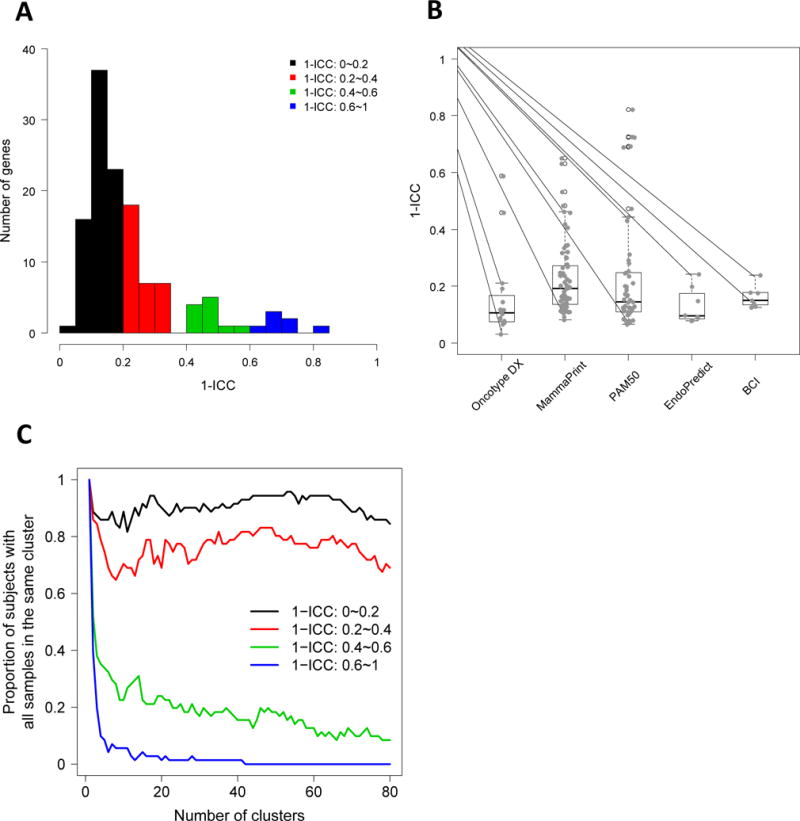
(A) Frequency distribution of 1-ICC scores representing ITH is shown for all GEP genes. 1-ICC scores range between 0 and 1 [0–0.2 (low), 0.2–0.4 (fair), 0.4–0.6 (moderate) and 0.6–1.0 (high)]. A tail to the right indicates a subgroup of the genes that are more heterogeneous among different types of tumor samples. (B) 1-ICC distribution by gene signatures. Oncotype Dx, MammaPrint, and PAM50 tests show genes with higher heterogeneity compared to Endopredict and BCI. (C) Clustering analysis was done using genes with varying ITH. Number of patients with all samples in the same cluster is plotted against the number of clusters. Lower number of patients with all samples clustered together indicates higher ITH.
Table 2.
Distribution of genes with low, fair, moderate and high intra-tumor heterogeneity
| 1-ICC
|
||||||
|---|---|---|---|---|---|---|
| 0~0.2 | 0.2~0.4 | 0.4~0.6 | 0.6~1 | Total | ||
| 1 | Oncotype | 13 | 1 | 2 | 0 | 16 |
| 2 | Mammaprint | 35 | 21 | 6 | 2 | 64 |
| 3 | PAM50 | 32 | 9 | 3 | 5 | 49 |
| 4 | Endopredict | 7 | 1 | 0 | 0 | 8 |
| 5 | BCI | 6 | 1 | 0 | 0 | 7 |
| 6 | All genes | 73 | 36 | 11 | 7 | 127 |
ITH of gene expression affects Oncotype Dx recurrence risk stratification
Oncotype Dx is the most widely used GEP for evaluating ER-positive early breast cancer prognosis. It uses a weighting algorithm to calculate the risk of recurrence score (RS), which is divided into low-risk (<18), intermediate-risk (18–30) and high-risk (≥31) categories. Based on the clinical Oncotype Dx recurrence scoring (clinRS) performed at Genomic Health, 28 patients showed low-risk with 1 case of disease recurrence; 30 patients showed intermediate-risk with 5 cases of recurrence; and 13 patients showed high-risk with 1 case of recurrence (Supplementary Table S6). To study the effect of ITH arising from sampling a tumor-enriched area or regions of high Ki67 and low PR, we estimated a predicted RS based on NanoString data (nstringRS), as described in the Statistical Methods. Supplementary figure S5a–b shows the expression of Ki67 and PR mRNA in the different types of tumor samples. In order to compare Ki67 and PR expression levels between tumor-rich cores and high Ki67/low PR cores, we selected only those patients for which both types of cores were available (n=26 and n=13, respectively) (Supplementary figure S5c–d). As expected the expression of Ki67 mRNA was significantly higher in cores from focal areas of high Ki67 and PR mRNA was significantly lower in cores from areas of low PR (Supplementary figure S5c–d). Figure 3A and Supplementary Table S6 shows nstringRS scores for the different types of samples along with the clinical Oncotype DX recurrence scores (clinRS). Overall, the nstringRS scores derived from sections and tumor-rich cores correlated well with the clinRS (Spearman’s ρ=0.92 and ρ=0.90 respectively) (Supplementary figure S6a–b). However, the Oncotype Dx-risk categories differed in 14/71 (19.7%) sections and 16/71 (22.5%) tumor-rich cores. We next looked at the agreement of nstringRS between the different types of samples (Figure 3B and Supplementary figure S6 c–f). nstringRS was strikingly similar between a whole tumor section and a tumor-rich core (Spearman’s ρ=0.94) (Figure 3B). The risk categories based on the tumor-rich cores and high Ki67 cores differed in 7/26 (27%) samples, while those derived from low PR cores differed in 3/13 (23%) samples. Although, the high Ki67 and low PR cores showed a trend towards higher median nstringRS scores compared to tumor-rich cores (Supplementary figure S6e–f), when we calculated for each patient, the difference in nstringRS scores between tumor-rich cores and high Ki67 or low PR cores, we observed no significant change in the median of this quantity (P=0.095 and P=0.675, respectively). Finally, when we compared clinRS to the nstringRS for all types of samples, we found that for majority of tumors (53/71, 75%) the different samples showed similar nstringRS scores and risk stratification. However, in 18/71 (25%) tumors the recurrence scores diverged enough to cause differential classification as the scores crossed the boundaries for low, intermediate and high risk (Figure 3C, Table 3, Supplementary Table S6, and Supplementary Table S7). 1 out of 18 tumors with discordant scores (associated with a decrease in risk due to a section) was from a patient with recurrent disease (Supplementary Table S6). Additionally, in these tumors with discordant scores, cases where at least 3 types of samples (section, tumor-rich core, high Ki67 core and/or low PR core) were available, 6 out of 10 tumors showed an increase in risk due to a high Ki67 or low PR core, 3 out of 10 tumors showed an increase in risk due to a tumor-rich core, and 1 out of 10 tumors showed an increase in risk due to both low PR core and a tumor-rich core (Supplementary Table S7).
Figure 3. Intra-tumor heterogeneity in gene expression affects Oncotype Dx recurrence risk stratification.
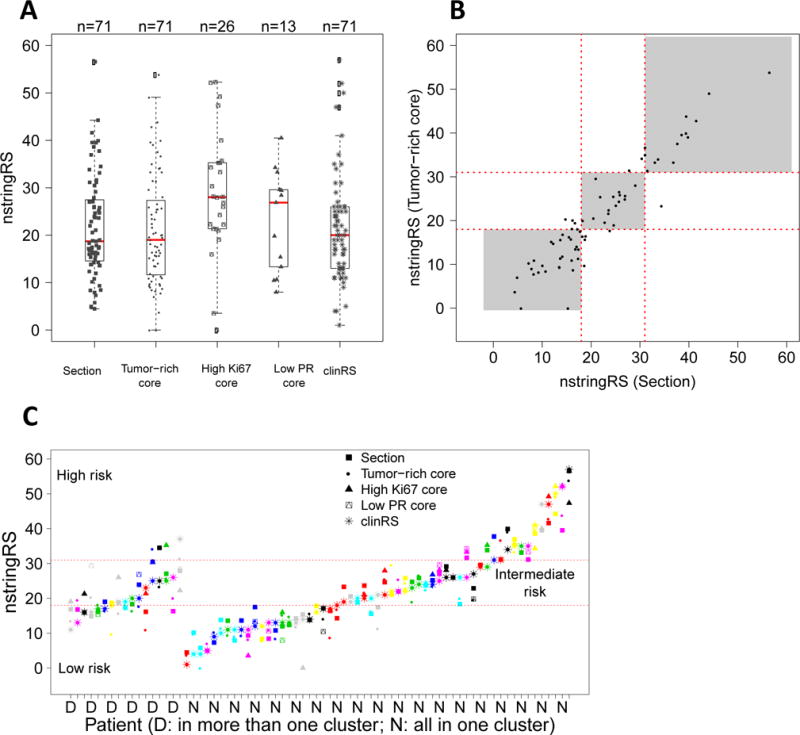
(A) NanoString-derived Oncotype Dx recurrence scores (nstringRS) are indicated for patients with different sample types; section, tumor-rich core, high Ki67 core, and low PR core, along with clinical Oncotype Dx recurrence scores (clinRS). (B) Correlation of nstringRS between whole sections and representative cores (Spearman’s ρ=0.94). (C) clinRS was compared to the nstringRS for all types of samples for changes in risk stratification. For 18/71 patients, recurrence scores crossed the boundaries for low, intermediate and high risk.
Table 3.
Differential risk stratification of patients with discordant scores based on Clinical Oncotype Dx and NanoString Oncotype Dx recurrence scores
| Number of Patients | Risk from Clinical Oncotype Dx recurrence scores (clinRS) | Risk from NanoString Oncotype Dx recurrence scores (nstringRS) |
|---|---|---|
| 7/71 (9.9%) | Low | Intermediate |
| 4/71 (5.6%) | Intermediate | High |
| 5/71 (7.0%) | Intermediate | Low |
| 1/71 (1.4%) | High | Intermediate |
Discussion
This is the first study to comprehensively examine the effect of ITH on measurement of clinically used GEPs, and their ability to predict prognostic risk in early breast cancer. The study utilized the Nanostring nCounter platform, which is ideally suited for gene expression detection in FFPE tissue. We described the ITH for each gene (n=127) in the five most commonly used GEPs. Hierachical clustering of tumors using all of the genes in the five GEPs showed relatively low ITH as individual samples from each patient clustered together for the majority of patients. However, when clustering tumors using genes in the individual GEPs, higher rates of ITH were found. An in-depth analysis of Oncotype Dx showed a strikingly high correlation between the Oncotype Dx recurrence score from a whole section (without macrodissection) and a representative tumor-rich core, suggesting little influence of the tumor microenvironment on the genes in this test. However, when measuring multiple cores within a tumor, ITH resulted in prognostic misclassification in 25% of patients.
Recent genome-wide genomic and transcriptomic studies have indicated high ITH in cancer, with some tumors having regions of both indolent and aggressive disease.(15–20) However, these studies are generally designed to identify the greatest level of ITH, as they include all transcribed genes. This is in contrast to GEPs which all use a small number of selected genes. Indeed, the effect of ITH on GEP tests seems to be a balance between the number of genes in the test, and the ITH of each of these genes. For example, when we clustered tumors based upon all of the genes that were measured, we found that most tumor samples clustered by patient, indicating greater IPH than ITH. However, selecting genes from each individual GEP resulted in lower numbers of tumors clustering per patients and higher apparent ITH. An additional level of consideration is the actual genes themselves. We show the level of ITH of each gene in each test, and find that choosing genes with low ITH results in apparent low IPH. It should be noted that none of the current GEPs used ITH as a determinant in inclusion/exclusion of genes in the test.
Our data are consistent with two previous smaller reports on the level of ITH and its effect upon risk prediction in early breast cancer. Drury et al. reported high concordance in RS scores between 0.6mm cores and whole tumor sections, similar to our data, but showed high variability in RS between the individual cores, resulting in prognostic misclassification in one out of four patients (25%).(22) Barry et al. examined the influence of ITH on the precision of microarray-based assays in multiple core needle biopsies, and showed high variance in recurrence risk predictions in 1 out of 18 patients due to global variation in gene expression.(23) In the current study, the clinical Oncotype Dx recurrence scores correlated well with the nstringRS scores derived from sections and tumor-rich cores, but, the Oncotype Dx-risk categories differed in 20% of sections and 23% of representative cores. Similarly, the risk categories based on the high Ki67 and low PR expression cores differed in 27% and 23% of patients respectively. Overall, comparing the nstringRS from sections and all three cores, we found a 25% (18/71) divergence in risk categories. In our analysis, we also found that an increase in risk was more commonly observed with a high Ki67 or low PR core, while a decrease in risk occurred more frequently due to a tumor-rich core. However, given the similarities in risk scores and differences in risk categories, the high variation observed in risk categories might be due to a limitation of the risk category cutpoints that lie in high-density areas of patient scores, even if those cutpoints have been precisely and objectively determined. Importantly, all differences in classifications were between adjacent risk groups (e.g. low to intermediate and intermediate to high) and no tumors showed divergence from low to high. Another limitation is that the Oncotype Dx and NanoString nCounter use different technologies for mRNA detection (amplification versus hybridization). Although they show a good concordance using our linear model (R2=0.87), differences in the technologies might contribute to some of the variations in the risk categories. Hence, based on the inclusion of atypical cores potentially representing aggressive areas of the tumor, the 25% variation in prognostic risk stratification provides an over-estimation of the magnitude of existing ITH. Even so, this is a clinically significant finding and underscores the need to better understand the analytic variables affecting performance of currently used GEPs. This suggests that in patients with highly heterogeneous tumors, multiple cores might be required to estimate risk prediction and that it might be a useful strategy to include both representative and atypical cores while selecting multiple samples to fully account for the ITH-driven variation in risk prediction. However, given the fact that in our study only 7/71 patients showed recurrent disease, the clinical meaning of this ITH-driven variation in prognostic risk stratification for the individual patients could not be fully addressed. Hence, future studies utilizing even larger cohorts of patients with higher number of prognostic and non-prognostic cores will be required to further confirm these findings.
The striking similarity between Oncotype Dx nstringRS scores obtained from a whole section and a tumor-rich core suggests that the tumor microenvironment may have little effect on this GEP, and likely represents the fact that the genes in this GEP are quite tumor-specific (e.g. ER, PR, ErbB2 etc). Furthermore, the tumor blocks included in this study were invasive tumor enriched and the most predominant non-invasive tissue was adipose tissue. It has been shown that a mitotically active tumor stroma(27) and a biopsy cavity with activated lymphocytes(28) can spuriously elevate the risk scores. However, the concordance between Oncotype Dx scores in a section and representative core was not due to the fact that these early breast cancers were very homogenous as histopathology showed a dynamic range of stromal and immune involvement (data not shown). Other GEPs, such as MammaPrint, contain many genes that are involved in the tumor microenvironment (e.g. VEGF), and thus may be more affected by the amount of stroma in a tumor.
In summary, a direct analysis of ITH for the five main GEPs in early breast cancer reveal intra-tumor variation, with ITH being inversely proportional to the number of genes in the test. For Oncotype Dx, the nstringRS scores strongly correlated between a tumor-rich core and whole section and suggest that a representative core may be used to determine RS. However, in instances where tumors do have ITH, the differences in gene expression can affect risk prediction. This was apparent in 25% of patients and suggests that GEPs might best be measured using knowledge and estimates of ITH from prior histopathology.
Supplementary Material
Translational Relevance.
Recent studies show tremendous transcriptomic and genomic heterogeneity not only between breast cancers, but also within a single breast cancer. This study examines the clinical importance of this heterogeneity, showing that prognostic risk scores derived from gene transcript levels deviate when taken from different regions (cores) of a breast tumor. Importantly, use of single cores can under- or over-estimate prognostic risk, and highlight the importance of understanding intra-tumor heterogeneity for breast cancer prognosis.
Acknowledgments
We thank the University of Pittsburgh Health Sciences Tissue Bank (HSTB) for collection of breast tumor specimens and clinical data.
Funding support
This work was supported in part by funds from the Breast Cancer Research Foundation (BCRF; to A.V. Lee, S. Oesterreich, and N.E. Davidson), National Cancer Institute of the National Institutes of Health award number P30CA047904, Fashion Footwear of New York (FFANY), and research support from UPMC. A.V. Lee is a recipient of a Scientific Advisory Council award from Susan G. Komen for the Cure, and is a Hillman Foundation Fellow. This project also used the University of Pittsburgh Cancer Institute (UPCI) Biostatistics Facility and Tissue and Research Pathology Services that are supported in part by award P30CA047904.
Footnotes
Conflicts of Interest
The authors have no potential conflicts of interest.
Authors’ contributions
RG carried out NanoString reproducibility experiments, acquisition of gene expression data, statistical analysis and drafted the manuscript. H-ML carried out the analysis of gene expression data under the supervision of YL. KC and DN contributed to the analysis and interpretation of data. MF performed the RNA isolation and NanoString analysis. WC carried out the NanoString analysis. AB, SO, and ND participated in study design, execution, and drafting of the manuscript. RB cored specimens and provided tissue, and DD provided clinical Oncotype Dx recurrence scores. AL designed and supervised the research and drafted the manuscript. All authors have read and approved the final manuscript.
References
- 1.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 2.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003;100:8418–23. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817–26. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 5.van ’t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 6.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27:1160–7. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Filipits M, Rudas M, Jakesz R, Dubsky P, Fitzal F, Singer CF, et al. A new molecular predictor of distant recurrence in ER-positive, HER2-negative breast cancer adds independent information to conventional clinical risk factors. Clin Cancer Res. 2011;17:6012–20. doi: 10.1158/1078-0432.CCR-11-0926. [DOI] [PubMed] [Google Scholar]
- 8.Ma XJ, Salunga R, Dahiya S, Wang W, Carney E, Durbecq V, et al. A five-gene molecular grade index and HOXB13:IL17BR are complementary prognostic factors in early stage breast cancer. Clin Cancer Res. 2008;14:2601–8. doi: 10.1158/1078-0432.CCR-07-5026. [DOI] [PubMed] [Google Scholar]
- 9.Goetz MP, Suman VJ, Ingle JN, Nibbe AM, Visscher DW, Reynolds CA, et al. A two-gene expression ratio of homeobox 13 and interleukin-17B receptor for prediction of recurrence and survival in women receiving adjuvant tamoxifen. Clin Cancer Res. 2006;12:2080–7. doi: 10.1158/1078-0432.CCR-05-1263. [DOI] [PubMed] [Google Scholar]
- 10.Zelnak AB, O’Regan RM. Genomic subtypes in choosing adjuvant therapy for breast cancer. Oncology (Williston Park) 2013;27:204–10. [PubMed] [Google Scholar]
- 11.Weigelt B, Reis-Filho JS, Swanton C. Genomic analyses to select patients for adjuvant chemotherapy: trials and tribulations. Ann Oncol. 2012;23(Suppl 10):x211–8. doi: 10.1093/annonc/mds323. [DOI] [PubMed] [Google Scholar]
- 12.Gyorffy B, Hatzis C, Sanft T, Hofstatter E, Aktas B, Pusztai L. Multigene prognostic tests in breast cancer: past, present, future. Breast Cancer Res. 2015;17:11. doi: 10.1186/s13058-015-0514-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simon RM, Paik S, Hayes DF. Use of archived specimens in evaluation of prognostic and predictive biomarkers. J Natl Cancer Inst. 2009;101:1446–52. doi: 10.1093/jnci/djp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mamounas EP, Tang G, Fisher B, Paik S, Shak S, Costantino JP, et al. Association between the 21-gene recurrence score assay and risk of locoregional recurrence in node-negative, estrogen receptor-positive breast cancer: results from NSABP B-14 and NSABP B-20. J Clin Oncol. 2010;28:1677–83. doi: 10.1200/JCO.2009.23.7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pertschuk LP, Axiotis CA, Feldman JG, Kim YD, Karavattayhayyil SJ, Braithwaite L. Marked Intratumoral Heterogeneity of the Proto-Oncogene Her-2/neu Determined by Three Different Detection Systems. Breast J. 1999;5:369–74. doi: 10.1046/j.1524-4741.1999.97088.x. [DOI] [PubMed] [Google Scholar]
- 16.Chung GG, Zerkowski MP, Ghosh S, Camp RL, Rimm DL. Quantitative analysis of estrogen receptor heterogeneity in breast cancer. Lab Invest. 2007;87:662–9. doi: 10.1038/labinvest.3700543. [DOI] [PubMed] [Google Scholar]
- 17.Navin N, Krasnitz A, Rodgers L, Cook K, Meth J, Kendall J, et al. Inferring tumor progression from genomic heterogeneity. Genome Research. 2010;20:68–80. doi: 10.1101/gr.099622.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szerlip NJ, Pedraza A, Chakravarty D, Azim M, McGuire J, Fang Y, et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:3041–6. doi: 10.1073/pnas.1114033109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marusyk A, Tabassum DP, Altrock PM, Almendro V, Michor F, Polyak K. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature. 2014;514:54–8. doi: 10.1038/nature13556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jankowitz RC, Lee AV. The evolving role of multi-gene tests in breast cancer management. Oncology (Williston Park) 2013;27:210–2, 4. [PubMed] [Google Scholar]
- 22.Drury S, Salter J, Baehner FL, Shak S, Dowsett M. Feasibility of using tissue microarray cores of paraffin-embedded breast cancer tissue for measurement of gene expression: a proof-of-concept study. J Clin Pathol. 2010;63:513–7. doi: 10.1136/jcp.2010.075754. [DOI] [PubMed] [Google Scholar]
- 23.Barry WT, Kernagis DN, Dressman HK, Griffis RJ, Hunter JVD, Olson JA, et al. Intratumor Heterogeneity and Precision of Microarray-Based Predictors of Breast Cancer Biology and Clinical Outcome. Journal of Clinical Oncology. 2010;28:2198–206. doi: 10.1200/JCO.2009.26.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gulati S, Martinez P, Joshi T, Birkbak NJ, Santos CR, Rowan AJ, et al. Systematic evaluation of the prognostic impact and intratumour heterogeneity of clear cell renal cell carcinoma biomarkers. Eur Urol. 2014;66:936–48. doi: 10.1016/j.eururo.2014.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cohen DA, Dabbs DJ, Cooper KL, Amin M, Jones TE, Jones MW, et al. Interobserver agreement among pathologists for semiquantitative hormone receptor scoring in breast carcinoma. American journal of clinical pathology. 2012;138:796–802. doi: 10.1309/AJCP6DKRND5CKVDD. [DOI] [PubMed] [Google Scholar]
- 26.Waggott D, Chu K, Yin S, Wouters BG, Liu FF, Boutros PC. NanoStringNorm: an extensible R package for the pre-processing of NanoString mRNA and miRNA data. Bioinformatics. 2012;28:1546–8. doi: 10.1093/bioinformatics/bts188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Acs G, Esposito NN, Kiluk J, Loftus L, Laronga C. A mitotically active, cellular tumor stroma and/or inflammatory cells associated with tumor cells may contribute to intermediate or high Oncotype DX Recurrence Scores in low-grade invasive breast carcinomas. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2012;25:556–66. doi: 10.1038/modpathol.2011.194. [DOI] [PubMed] [Google Scholar]
- 28.Baehner F, Quale C, Pomeroy C, Cherbavaz C, Shak S. Biopsy cavities in breast cancer specimens: their impact on quantitative RT-PCR gene expression profiles and recurrence risk assessment. Mod Pathol. 2009:28A–9A. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.