

Abstract
Genome assembly depends critically on read length. Two recent technologies, from Pacific Biosciences (PacBio) and Oxford Nanopore, produce read lengths >20 kb, which yield de novo genome assemblies with vastly greater contiguity than those based on Sanger, Illumina, or other technologies. However, the very high error rates of these two new technologies (∼15% per base) makes assembly imprecise at repeats longer than the read length and computationally expensive. Here we show that the contiguity and quality of the assembly of these noisy long reads can be significantly improved at a minimal cost, by leveraging on the low error rate and low cost of Illumina short reads. Namely, k-mers from the PacBio raw reads that are not present in Illumina reads (which account for ∼95% of the distinct k-mers) are deemed sequencing errors and ignored at the seed alignment step. By focusing on the ∼5% of k-mers that are error free, read overlap sensitivity is dramatically increased. Of equal importance, the validation procedure can be extended to exclude repetitive k-mers, which prevents read miscorrection at repeats and further improves the resulting assemblies. We tested the k-mer validation procedure using one long-read technology (PacBio) and one assembler (MHAP/Celera Assembler), but it is very likely to yield analogous improvements with alternative long-read technologies and assemblers, such as Oxford Nanopore and BLASR/DALIGNER/Falcon, respectively.
Genome assembly quality depends on sequencing coverage depth, read accuracy, and read length (Nagarajan and Pop 2013; Myers 2016). Nowadays, the cost per sequenced base is small, so in many cases depth of coverage is no longer a major limiting factor, 100-fold coverage being routine in many projects. Such high coverage also reduces the importance of read accuracy, since errors can be effectively reduced by consensus while building contigs from the reads. Read length remains a critical factor. Its importance stems from repeated sequences, which in many cases cannot be properly assembled unless the repeated regions are shorter than the reads. For example, two identical copies of a 7-kb retrotransposable element would require reads longer than the element length for full assembly; shorter reads would produce a fragmented assembly. This limitation can be circumnavigated, but only partially, by mate-pair reads and other methods (Weber and Myers 1997; Nagarajan and Pop 2013; McCoy et al. 2014). These requirements of genome assembly are nicely encapsulated in Gene Myers’ 140-character theorem: “Thm: Perfect assembly possible iff a) errors random b) sampling is Poisson c) reads long enough 2 solve repeats” (https://dazzlerblog.wordpress.com/2014/05/15/on-perfect-assembly/).
Sanger sequencing, the first practical technology for large-scale projects, produces reads between 500 bp to1 kb, which are accurate (error rate <0.1%) but expensive, the price tag for a Drosophila-like genome being in the $1 million range (http://flybase.org/static_pages/news/whitepapers/DrosBoardWP2001.pdf). Second-generation sequencing technologies (“SGS”) such as Illumina produce reads that are inexpensive, accurate (error rate ∼0.1%), but short (<500 bp). Their low cost (Drosophila genome price tag is about $4000) allowed for an explosion of genome projects. However, due to their short read length, they produce very fragmented assemblies. Both Sanger and SGS require a huge investment of money, labor, and time if a “finished” genome is the target.
Two recently developed or improved technologies, from Pacific Biosciences (PacBio) and Oxford Nanopore, produce read lengths >20 kb, which can yield genome assemblies that are vastly superior in contiguity to those based on Sanger or short reads (Goodwin et al. 2015; Koren and Phillippy 2015; Loman et al. 2015). However, reads produced by both technologies have very high error rates (PacBio: ∼15%; Oxford Nanopore: ∼20%) and cannot be directly handled by current genome assemblers (for an exception, see Li 2016). Instead, a “hierarchical assembly process” is used: First the raw reads are error-corrected by aligning them either to Illumina reads (“hybrid assembly”) (Koren et al. 2012), or among themselves (“self-correction”) (Chin et al. 2013) and by implementing some sort of consensus algorithm, which reduces the error rate to <5%. The corrected reads are then assembled by normal “overlap-layout-consensus” assemblers (“OLC”; designed for Sanger reads). Self-correction produces better assemblies (Koren et al. 2013) and is the current state of the art, but it is computationally intensive because the all-by-all alignment must be performed with rather high sensitivity and specificity in order to detect real overlaps among the noisy reads. In practice, bacterial genomes are easily assembled, but large genomes such as those of mammals still have high computational costs (around 100,000 CPU hours) (Berlin et al. 2015). It is also unclear how far can accurate assembly can go into highly repeated regions such as heterochromatin (e.g., telomeres and centromeres), segmental duplications, and tandem gene arrays (e.g., histone and rDNA clusters).
A very efficient approach to analyze DNA sequences is to decompose them into overlapping stretches of a fixed length of k bases, called k-mers. For example, candidate overlapping reads can be detected because they share k-mers above a certain cut-off (Berlin et al. 2015). k-mers provide interesting insights into the relationship between read accuracy and computational cost. Sequencing errors introduce into the reads a large number of rare k-mers, a problem that is particularly acute for long reads due to their very high error rate. In PacBio, assuming a typical k-mer size of 16, only ∼5% of the distinct k-mers from the reads are error free (Chaisson and Tesler 2012; see also Results). The relevance of this number (5% error-free k-mers) becomes apparent when one considers that all genome assembly algorithms are based on k-mer decomposition and comparison and that at least at some steps they must track all distinct k-mers. This means that at some steps 95% of the computational resources such as memory and CPU time are wasted with k-mers that cannot indicate real read overlaps because they contain at least one wrong base.
The three main alignment algorithms for PacBio reads deal with the above problem somewhat differently. BLASR, the first developed, originally aimed to align PacBio reads to a reference genome but can also do the all-by-all alignment. It uses all k-mers and employs successive refinements of the alignment in order to detect true overlaps (Chaisson and Tesler 2012). The main limitation of BLASR is its low speed: It works well for bacteria and yeast genomes (4–12 Mbp) but is impractical for genomes such as Drosophila (180 Mbp; it used 610,000 CPU hours in the all-by-all step) (Berlin et al. 2015). DALIGNER employs highly optimized code to perform similar tasks (Myers 2014). It is computationally intensive and, in practice, requires a large computer cluster to assemble Drosophila-like genomes. The third aligner, MHAP, reduces memory usage and computational time by sampling a random subset of k-mers (“sketch”) to detect candidate overlaps (technically, the sequences are transformed into a reduced representation by applying multiple hash functions, defined by sketch size, to all k-mers in a sequence, and selecting the minimum value from each hash function). Larger sketch size results in more sensitivity, but at a higher computational cost. Typical sketch sizes range from ∼500 to 1200 k-mers, with resulting sensitivities in the range of 60%–90% (Berlin et al. 2015). MHAP is the default aligner used in the PBcR pipeline, which, after correcting the reads, feeds them into the Celera Assembler. Currently PBcR (and its recent substitute, Canu) (Koren et al. 2016) is the pipeline that requires the smaller computational infrastructure: Microbial genomes can be assembled with an eight-core desktop computer in a few hours or less, Drosophila-sized genomes are assembled in small servers (e.g., 24 cores, 64 Gb of RAM) in 3 d, and mammalian size genomes require a large cluster.
Whatever the details of the overlapper algorithm, they all have to cope with a “needle-in-a-haystack” problem (i.e., to find true overlaps amid a lot of sequencing noise) and, in principle, would work much better if the large number of “error k-mers” of the long noisy reads could be identified at the outset and ignored. We propose a simple solution to achieve this: (1) Use Illumina reads (which are accurate and inexpensive) to produce a list of error-free k-mers; (2) k-mers from the PacBio raw reads that are not present in the Illumina-derived k-mer list (which account for ∼95% of the distinct k-mers) are deemed sequencing errors and ignored at the seed alignment step.
Results
k-mer frequency distributions in PacBio and Illumina reads
In order to make clear the proposed solution, we investigated in detail the data from Escherichia coli. The genome of E. coli strain K-12 MG1655 has been sequenced and finished to high quality using Sanger reads (Blattner et al. 1997). More recently, it has been sequenced using Illumina and PacBio technologies at high coverage (77× and 94× respectively) (Kim et al. 2014; https://basespace.illumina.com). The genome is 4.64 Mbp in length and hence contains approximately 4.64 million distinct k-mers, the vast majority of them occurring only once, since bacterial genomes have few repetitive regions (throughout this manuscript we set k = 16, which is a typical value). The PacBio reads contain 436 million k-mers in total (4.64 million k-mers times 94-fold coverage); if there were no sequencing errors, these k-mers would correspond to 4.64 million distinct k-mers, each one occurring on average 94 times. However, these reads actually contain 292,687,635 distinct k-mers; among these, 4,513,248 (1.5%) are correct (i.e., present in the finished E. coli genome sequence), and the remaining 288,174,387 (98.5%) are sequencing errors (“error k-mers”; see Methods). As expected, the correct k-mers show up repeatedly, and their proportion among the total k-mers is 16.6%. On the other hand, most error k-mers are unique, because the chance that random errors create the same 16-mer sequence twice (or a pre-existing 16-mer) is small. Figure 1 shows the k-mer frequency spectrum of the PacBio and Illumina reads. First, consider the Illumina reads (Fig. 1, left panels): The huge peak on the left contains rare k-mers that mostly result from sequencing errors; the next peak, located approximately at the sequencing coverage, corresponds to single-copy sequences in the genome; and finally, smaller peaks on the right correspond to repetitive DNA (they are much more pronounced in repeat-rich genomes such as Drosophila and mammals). A similar pattern occurs with PacBio reads (Fig. 1, right panels), except that the error peak is much larger (note the y-axis scale) and that the single-copy peak is strongly shifted toward the left (because so many k-mers were “lost” due to sequencing errors). Roughly similar values were obtained for other genomes; more typically, ∼5% of the distinct k-mers in the PacBio reads are correct (Supplemental Table S1). As we commented before, this low accuracy implies a high cost: At some steps of genome assembly, 95% of the computational resources are wasted with k-mers that cannot indicate real read overlaps because they contain sequencing errors.
Figure 1.

k-mer frequency distributions for Illumina and PacBio E. coli reads. (A) Illumina, all k-mers (k = 16 in all panels). (B) Illumina, with correct k-mers shown in red and error k-mers in blue. Note that most error k-mers have very low frequency. The peak at k-mer frequency about 70 corresponds to genomic single copy k-mers. (C,D) PacBio reads. Note the huge number of error k-mers. The reference list of valid k-mers came from the finished genome (see Methods).
Note that particularly in the case of Illumina reads the large peak on the left contains nearly no correct k-mer (Fig. 1), whereas nearly all correct k-mers are located to the right. This suggests an interesting possibility: In the absence of a finished genome, an accurate list of “valid k-mers” can be obtained from the Illumina reads by taking those k-mers that occur at least, say, 10 times (single-copy k-mers are expected to occur about 70 times in this data set). In the E. coli example, if we use the Illumina-based list to validate the k-mers of the PacBio reads, we would miss only 0.003% of the correct k-mers (145 out of 4,513,248) and would incorrectly validate 0.1 % of the error k-mers (32,456 k-mers out of 288,174,387). Such an Illumina “valid k-mer list” is inexpensive to produce and may improve long-read assembly by identifying in the long reads the k-mers that should be ignored.
We implemented this k-mer validation procedure in the MHAP overlapper, as detailed in Methods, and in the next sections we tested its performance in overlap detection, read error correction, and genome assembly. We used data from genomes of five model organisms; in four of them, PacBio and Illumina reads from the same strain are available: bacteria (E. coli strain K-12 MG1655; genome size of 4.64 Mbp), yeast (Saccharomyces cerevisae strain W303; 12.1 Mbp), worm (Caenorhabditis elegans strain Bristol N2; 103 Mbp), and flies (Drosophila melanogaster strain ISO1; ∼180 Mbp). We also included the plant Arabidopsis thaliana (strain Ler-0; 135 Mbp), although in this case most of the Illumina reads came from a different strain (Ler-1) and were shallower (Supplemental Table S2). Finally, as a proof of principle, we applied the k-mer validation to three difficult regions (segmental duplications) of the human genome and to human chromosomes 15 and 17. We operationally defined as valid k-mers all those with a frequency bigger than one seventh of the single-copy peak from Illumina reads (Supplemental Fig. S1; Supplemental Table S2). This cut-off was chosen after a limited exploration (Supplemental Results).
k-mer validation increases the sensitivity and the specificity of overlap detection
The MHAP program compares pairs of uncorrected PacBio reads, aiming to detect real overlaps while keeping false positives at a minimum. We compared the performance of the modified MHAP against the standard version (1.5b1) following the procedures of the original publication (Berlin et al. 2015). Namely, artificial PacBio reads were generated by applying the typical PacBio error rates (insertion: 10%; deletion: 2%; substitutions: 1%) to 10-kb segments of known genomes (we tested E. coli, yeast, C. elegans, and Drosophila, and also random DNA sequences). These segments were arranged as pairs with a 2-kb overlap; members of different pairs do not have any real overlap but may contain similar sequences due to repetitive DNA. We measured sensitivity as the proportion of true overlaps that were detected (i.e., among members of the same pair). Overlaps between members of different pairs estimate the false-positive rate (i.e., the specificity); this is more reliably done with random DNA sequences, because biological sequences almost always contain repeats that will inflate the false-positive rate. We varied sketch size (the “num-hashes” parameter) (Berlin et al. 2015) between 64 and 2048; this parameter is very important because it controls the trade-off of computational cost (CPU time plus memory usage) versus sensitivity. All other parameters were kept fixed at their default values (k-mer size = 16; num-min-matches = 3; threshold = 0.04). As shown in Figure 2, k-mer validation caused a huge increase in sensitivity with E. coli data: At the typical sketch size of 512 (Berlin et al. 2015), the standard MHAP detected 24% of the true overlaps (61 out 250), whereas with k-mer validation, we got 95% (238 out 250). Other genomes and also random DNA sequences produce similar results (Supplemental Fig. S2). Finally, the improvement using the Illumina-derived list of valid k-mers is very similar to the one using the true k-mer list derived from the finished genome (Supplemental Fig. S3), which suggests that the former is a good proxy for the latter.
Figure 2.
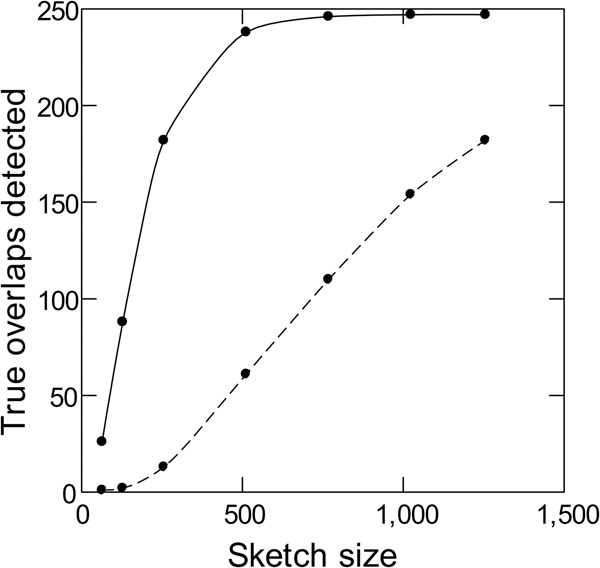
Sensitivity of read overlap detection with and without k-mer validation. Simulated PacBio reads from E. coli (250 pairs of 10-kb sequences with 2-kb overlaps) were subjected to standard MHAP (dashed line) or MHAP with masking of low-frequency k-mers (solid line) for overlap detection. The reference list of valid k-mers came from Illumina reads.
It is interesting also to look at the false-positive rate, which estimates the specificity. The observation that false positives are absent in random DNA sequences and seem to be more frequent in repeat-rich genomes (Supplemental Fig. S4) strongly suggests that repetitive DNA is the culprit, and indeed, we found transposable elements and other repeats when we checked some of them. These spurious alignments are undesirable, and k-mer validation offers a simple and effective way to nearly eliminate them: We just have to remove from the valid k-mer list all k-mers that seem to occur more than once in the genome (we used as a cut-off 1.5-fold of the Illumina single-copy peak; 105 in the E. coli case) (see Supplemental Results). As shown in Figure 3, this procedure causes minimal losses in sensitivity, while suppressing most of the “false positives.” In the next sections, we will always compare the performance of standard MHAP (“M”) with the two types of k-mer validation: masking only low-frequency k-mers (“L”) or masking both low-frequency and high frequency k-mers (“LH”). Illumina reads allow a precise k-mer classification; given enough coverage, two-copy k-mers (e.g., from a segmental duplication) can be reliably separated from single-copy ones (Supplemental Fig. S5). For the purpose of read correction and assembly, ideally only k-mers that are single copy in the genome should be used as seeds in overlap detection; as we will see below, using Illumina reads and the modified MHAP one gets close to this.
Figure 3.
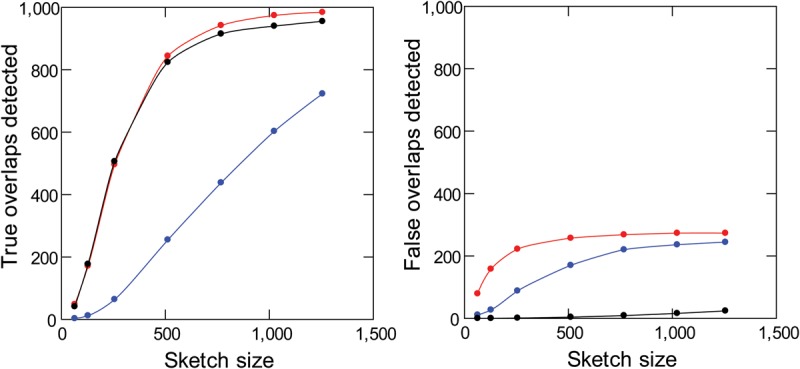
Sensitivity and specificity of read overlap detection with masking of repetitive k-mers. Simulated PacBio reads from D. melanogaster (1000 pairs of 10-kb sequences with 2-kb overlaps) were subjected to standard MHAP (blue), MHAP with masking of low-frequency k-mers (red), or MHAP with masking of low-frequency and high-frequency k-mers (black). Note that masking of low- and high-frequency k-mers cause a huge improvement in specificity (right) with minimal losses in sensitivity (left). The reference list of valid k-mers came from Illumina reads.
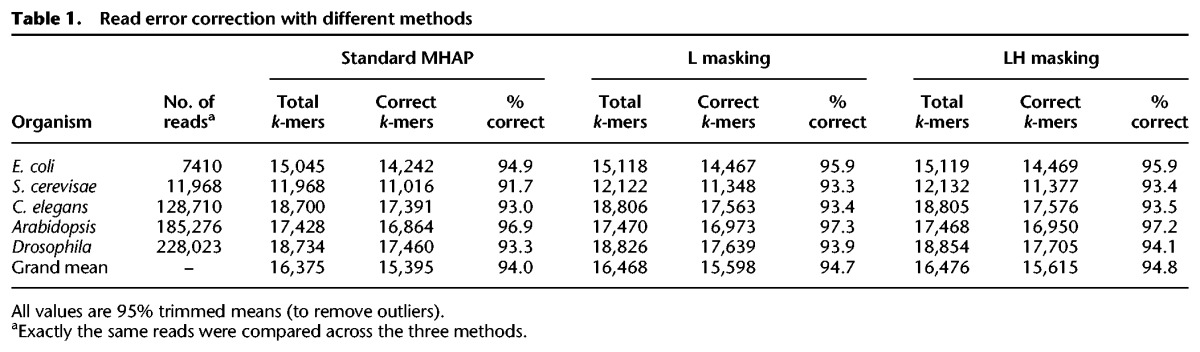
k-mer validation improves the error correction of long-reads
We assessed the performance of read error correction by counting for each read the number of correct k-mers among the total k-mers (Supplemental Methods). Uncorrected PacBio reads from different organisms contain between 15% to 38% correct k-mers (Supplemental Table S1). During read correction in all assembly pipelines, the raw reads were aligned, the regions with poor alignment were trimmed, and the discrepant bases were deemed as sequencing errors and were corrected by a consensus algorithm (Chin et al. 2013; Berlin et al. 2015). Looking first at the sequencing errors (Table 1, columns 5, 8, and 11), the standard MHAP overlapper (coupled with the default falconsense correction algorithm) brings the reads from 15%–38% to 94.0% correct k-mers (range across different organisms: 92%–97%), and k-mer validation further improves this to 94.7% (L masking) and 94.8% (LH masking). Second, there are also gains in the total amount of sequence recovered (Table 1, columns 3, 6, and 9), presumably due to improved alignment and reduction of unnecessary trimming. The combined effect of these two factors is that reads corrected with LH masking have on average 220 additional correct k-mers (i.e., 15,615 minus 15,395) compared with the standard MHAP. So k-mer validation indeed improves the correction of long-reads in both trimming and error correction. The effect differs between organisms, which is expected, since it will depend on the quality of PacBio and Illumina sequencing and on the specificities of each genome (e.g., amount and composition of repetitive DNA). In particular, the smallest improvement occurred in Arabidopsis, possibly because it has the worst Illumina data set (Supplemental Table S2; Supplemental Fig. S1). It is interesting also to note that most of the improvement in error correction seems to be due to masking of low-frequency k-mers (L-masking); LH-masking (i.e., simultaneous masking of low-frequency and high-frequency k-mers) adds little in most genomes. We will return to this point later.
Table 1.
Read error correction with different methods
During read correction (and assembly), we always used an Illumina-derived list of valid k-mers, but in E. coli and C. elegans (which have completely finished genomes), we also tested the genome-derived list of valid k-mers to guide the read alignment. The effect in read correction is negligible (Supplemental Table S3), indicating, as seen in the previous section (Supplemental Fig. S3), that Illumina-derived lists are good proxies for the real k-mer lists.
Finally, the effect of k-mer validation looks small (e.g., 220 additional k-mers in 15,395, or 1.4%), but we should note that these are average values. Most assembly breaks occur at repetitive regions, and as we will see below (see Assembly of a “Model Genome”), at these difficult regions k-mer validation has a strong effect on read correction.
k-mer validation results in more contiguous assemblies
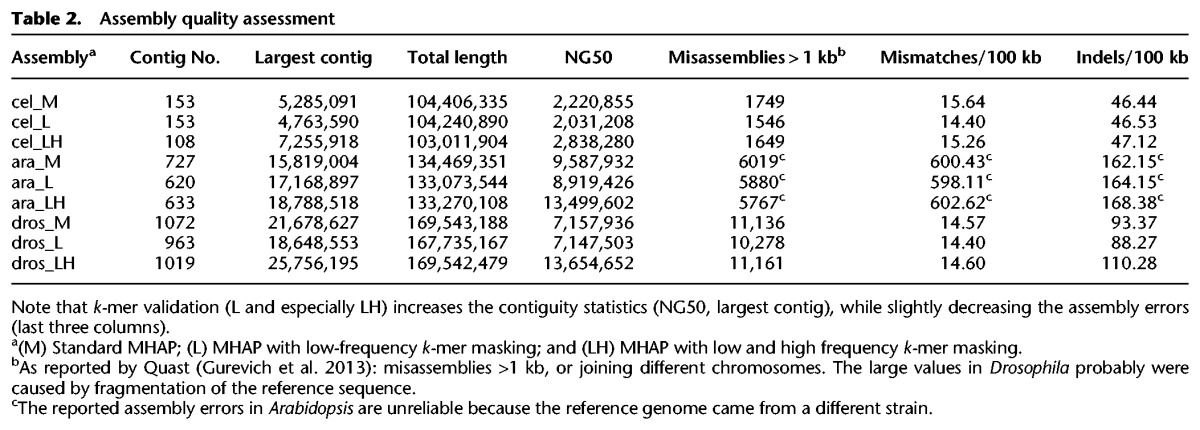
We assembled the five complete genomes with the three assembly methods (standard MHAP, L-masking, and LH-masking) and used the Quast package (Gurevich et al. 2013) to compare them for metrics such as contiguity (NG50) and misassembly frequency (Supplemental Methods). When tested with the simple genomes of E. coli (4.64 Mbp) and yeast (12.1 Mbp), all three assembly methods yield similar results (Supplemental Table S4). In E. coli, all three approaches yield one contig spanning the complete genome, with high identity to the reference sequence. In yeast, the NG50 from MHAP and LH assemblies are the same (818 kb), whereas L-masking yields a bit smaller value (751 kb). The yeast PacBio data came from W303 strain, for which there is no available finished sequence for comparison; however, the NG50 of the three assemblies approached the NG50 of the finished reference yeast strain (924 kb), so it seems that they are close to completeness. Hence both E. coli and yeast provide a nice demonstration of the power of long-reads, which, however, leaves little room for comparison among assembly methods. However, the difference between the three assembly methods becomes visible in these simple genomes when we use more challenging conditions such as low coverage data or small sketch size: In both cases, k-mer validation leads to huge improvements in assembly contiguity (Fig. 4A).
Figure 4.

Contiguity of assemblies produced with different methods. (M) standard MHAP; (L) MHAP with low-frequency k-mer masking; (LH) MHAP with low and high frequency k-mer masking. (A) Assembly of simple genomes under the challenging conditions of low coverage (E. coli; coverage reduced from 94× to 30×) or small sketch size (yeast; MHAP sketch size reduced from 512 to 128). (B) Assembly of three complex genomes (C. elegans, A. thaliana, and D. melanogaster).
When we tested the k-mer validation procedure with three complex genomes (C. elegans, A. thaliana, and D. melanogaster), we found that in all three cases it produced significantly more contiguous assemblies: In C. elegans, the NG50 rose from 2221 kb to 2838 kb; in Arabidopsis, from 9588 kb to 13,500 kb; and in Drosophila, from 7158 kb to 13,655 kb (all values MHAP vs. LH-masking) (Table 2; Fig. 4B). The improvement in contiguity is also seen in the largest contig size (Table 2). Statistics such as NG50 focus only on the largest contigs (e.g., in Drosophila only the four or five largest, all euchromatic) and can change drastically from only a few contig joins; however, the NGx plots, which capture the full continuity of the assemblies, indicate robust contiguity improvements across all size ranges (Supplemental Fig. S6). Aggressive assembly algorithms can spuriously increase statistics such as NG50 at the expense of increasing misassemblies; this was not the case of k-mer validation, which actually in most cases yield smaller numbers of misassemblies, mismatches, and indels, compared with the standard MHAP (Table 2). We also checked all assemblies with mummerplot (Kurtz et al. 2004) for the presence of gross misassemblies (e.g., contigs with spurious junctions between different chromosomes) that might inflate the NG50 of LH over MHAP assemblies, and found none (Supplemental Fig. S14). We did find in Arabidopsis a case of gross misassembly, but it occurred in all methods (M, L, and LH) (Supplemental Results, see PBcR Assemblies with Different Memory Parameters; Supplemental Fig. S7). Although we have not tested even more complex genomes such as mammals and large plant genomes, it is very likely that k-mer validation will lead to improved assemblies in these cases as well.
Table 2.
Assembly quality assessment
Three points are worth mentioning here. First, improvements in assembly caused by k-mer validation are similar when we use the Illumina- or the genome-derived list of valid k-mers (Supplemental Table S5). This shows that in terms of assembly, Illumina-derived lists are good proxies for the real k-mer lists, as seen before for overlap detection (Supplemental Fig. S3) and read correction (Supplemental Table S3).
Second, it may be argued that since Illumina sequencing has coverage bias against GC-rich and AT-rich regions (Ross et al. 2013), the use of k-mer validation might propagate such bias into the assembly of PacBio reads (which are believed to be almost bias free). We addressed this question by simulating Illumina reads with a coverage bias stronger than the reported cases (Ross et al. 2013) and then measuring its effect on the assembly of simulated PacBio reads of the same region. We found that k-mer validation assembly is insensitive to the normally encountered bias in Illumina coverage (Supplemental Results, see “k-mer Validation Assembly in the Presence of Coverage Bias of Illumina Reads; Supplemental Table S6). This counterintuitive result probably is explained by the fact that PacBio reads are much longer than the Illumina coverage gaps (which span a few hundred base pairs at most), so most read overlaps will still be detected because the k-mers outside the coverage gaps provide enough alignment seeds.
Finally, overlap detection (Fig. 3; Supplemental Fig. S4) and read error correction (Table 1) are essentially the same with L- and LH-masking, but most or all the assembly contiguity gains in complex genomes occur with LH-masking (Fig. 4B). The next section suggests an explanation for this discrepancy: Assembly contiguity gains probably are due to improved overlap specificity and read correction in a small subset of the reads and sites (i.e., at the Sequence Family Variant sites of repeated regions), which effect is imperceptible in the aggregate statistics reported in Table 1.
Assembly of a ‘model genome’
The causal events underlying the improvements in read correction (Table 1) and assembly (Fig. 4) probably are complex and scattered in many regions of the genome, making a detailed analysis impractical (e.g., how exactly does LH masking improve contiguity?). In order to better understand the reasons for the observed improvements, we isolated a small and difficult region and used it as a model: a 44-kb segmental duplication (98% identity between the two copies), which is part of a much larger segmental duplication complex located in the 10q11 region of the human genome. The finished sequence of both copies was obtained by painstaking BAC cloning and sequencing (Chaisson et al. 2015). As detailed in Supplemental Methods, we used the finished sequence to simulate PacBio reads from both copies of the 44-kb segmental duplication, along with ∼300 kb of flanking sequence; we used simulated reads because we want to know which segmental duplication copy they came from. We then assembled the reads with the three methods (standard MHAP, L-masking, and LH-masking). In the case of L- and LH-masking, we obtained the valid k-mer lists from the finished sequence. The perfect assembly of this “model genome” should yield two contigs (“left” and “right”), each representing one copy of the segmental duplication and the correct flanking sequences. Standard MHAP (“M”) assembly resulted in 11 contigs; L-masking, three contigs; and LH-masking, the expected two contigs (Supplemental Table S7). The majority of the assembly breaks in the M and L assemblies occurred within or close to the segmental duplication region, and particularly in the M assembly, there is a large amount of sequence duplication (19%), caused by partially overlapping contigs in this region (Supplemental Table S7).
Since the three assemblies differ only in the initial alignment of the uncorrected reads, all assembly differences must ultimately trace to it. When we investigated the read alignment, we found that both the standard MHAP and MHAP with L-masking fail to sort the two copies of the segmental duplication in most cases (i.e., in most reads ∼50% of the detected overlaps are between reads from different copies) (Supplemental Fig. S8), whereas with LH-masking, 92% of the detected overlaps are correct.
The next step in the assembly pipeline is the read correction by a consensus algorithm, using the overlaps obtained above. Since we know the origin of reads, we can score for each site of each corrected read if it has the right base, a wrong base, or a gap. We should distinguish three types of sites here: (1) outside the segmental duplication (NSD sites), (2) within the segmental duplication at positions that are variable between the two copies (SFV sites, for “sequence family variant”) (Dennis et al. 2012; Hughes and Rozen 2012), and (3) within the segmental duplication at positions that are conserved between the two copies (SDC sites). Note that at SFV sites, there will be conflicting sequence information in the overlaps produced by standard MHAP and by L-masking (but not by LH-masking), because as seen above these two methods mix almost indistinctly reads from the two copies of the segmental duplication. At the NSD and SDC sites, there is no such conflicting information, because at these sites either the two contigs do not align at all (NSD) or have the same sequence (SDC). As shown in Figure 5, the three methods work equally well for NSD and SDC sites: In the corrected reads, 98% of the bases at these sites are right. However, at SFV sites there is a huge difference: Whereas with LH-masking, read correction still works very well (97% right bases), with the standard MHAP and L methods, only ∼80% of the bases are right; in most cases, the SFV site is deleted (substituted by a gap). This 80% value is the average for the whole segmental duplication; sites closer to the border actually have almost perfect correction, whereas those in the middle of segmental duplication can get below 50% correct bases (Supplemental Fig. S9). This heterogeneity in error correction makes sense: Close to the border of the segmental duplication, the flanking sequence ensures the correct read overlap (and proper read correction). In the same vein, the SFV sites around a 1.5-kb indel in the middle of the segmental duplication were “protected” from miscorrection (Supplemental Fig. S9). The above results employed falconsense as the consensus algorithm; the more precise (and slower) pbdagcon yields essentially the same result (Supplemental Fig. S10), the main difference being the type of miscorrection at SFV sites: Whereas falconsense almost always introduces a gap, pbdagcon either does this or introduces a wrong base. The bottom line is that in both cases the SFV information is destroyed.
Figure 5.
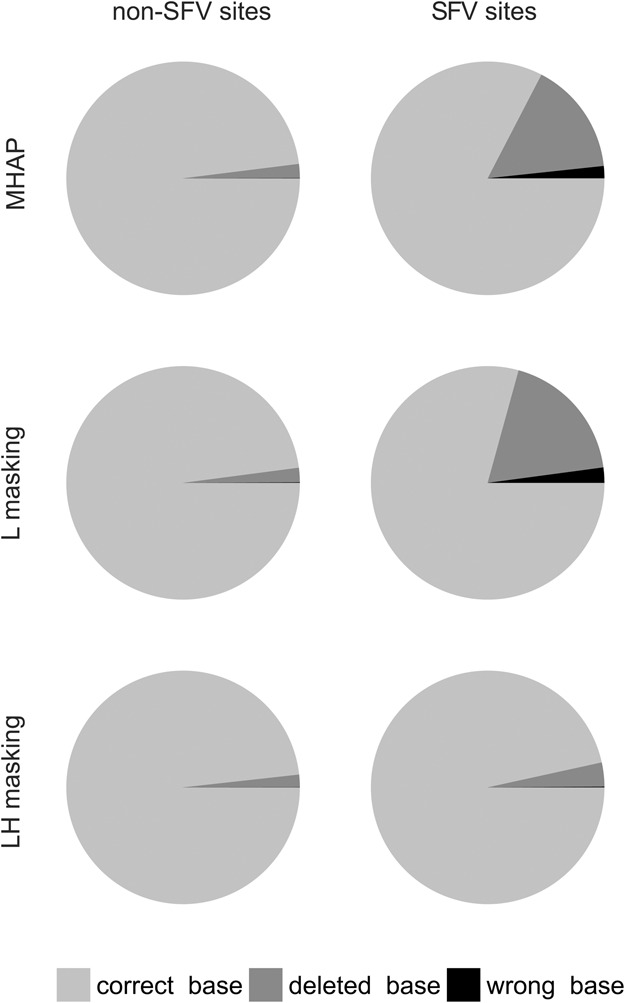
Read correction accuracy within a segmental duplication (human 10q11 region). Corrected reads were aligned with the original sequence, and each base of each read was scored as “correct” (light gray), “wrong” (black), or “deleted” (dark gray). “SFV sites” (for “sequence family variant”) are located within the segmental duplication, at positions where the two copies are different. “Non-SFV sites” are sites located within the segmental duplication and identical between the two copies or located outside the segmental duplication (they produce identical results and were lumped in the figure). Note that standard MHAP and MHAP with L-masking frequently fail at SFV sites, whereas LH-masking correctly handles them. Data from 450 sites of each type; reads were corrected with the default falconsense algorithm (for the pbdagcon correction, see Supplemental Fig. S10).
So it seems that the “model genome” provided a quite complete answer for the question “how exactly does LH-masking improve contiguity?” The increase in overlap detection efficiency due to masking of error k-mers helps. But even more important is the stringent masking of repetitive k-mers (defined as all k-mers that are not single-copy in the genome): The different copies of a repeat can be very similar (in our example, 98% identical), and without this stringent masking, the signal from SFV sites is swamped by the signal from conserved sites at the aligner step, leading first to indiscriminate overlaps (Supplemental Fig. S8), then to rampant read miscorrection at the SFV sites (Fig. 5; Supplemental Fig. S9), and finally to assembly breaks (Supplemental Table S7).
The assembly breaks are a direct consequence of the destruction of the SFV information: When a repeat is longer than the vast majority of the reads, it can only be correctly traversed by a tiling path of SFVs. Ultimately, failure to correctly handle repeats during overlap detection and read correction leads to fragmentation and other assembly errors. The problems posed by repeats in genome assembly have been recognized a long time ago (Myers 1995; Phillippy et al. 2008; Nagarajan and Pop 2009; Koren et al. 2012), and long reads have a dual relationship with them: When they fully span the repeat, they solve the problem, but when the repeat is longer than the reads, the problem becomes harder because the overlap detection in principle could not be stringent. In a sense, LH-masking implements stringent overlap detection in noisy reads.
While the above results are encouraging, it is reasonable to question their generality since they are based on one example and what resolves duplications in some cases may fragment others. So we tested additional duplications in the human genome, which were longer and harder: two segmental duplications in tandem, with sizes of 130 and 100 kb, both with 97%–98% identity. They are located in the same 10q11 region of the human genome studied by Chaisson et al. (2015). As detailed in the Supplemental Results (see Assembly of Additional Segmental Duplications), both MHAP and L-masking severely misassembled these duplications, whereas LH-masking yields a perfect assembly of them (Supplemental Table S8). Finally, it would be interesting to test k-mer validation with human data sets larger than individual segmental duplications. So, as detailed in the Supplemental Results, we applied the k-mer validation to sorted reads from human chromosomes 15 and 17; we found again that LH-masking produced more contiguous assemblies (Supplemental Fig. S11), with less misassemblies (Supplemental Table S9). Thus, the assembly improvement brought by k-mer validation seems to be a general phenomenon.
Throughout this work we have used MHAP assemblies as a baseline, and one may argue that this is somewhat unfair, since k-mer validation makes use of additional data (the Illumina reads, which provide the valid k-mer lists); under this view, a hybrid assembly would be a more appropriate baseline. The “model genome” used before is ideal for such comparisons because it is at the same time computationally tractable and a challenging assembly problem. As shown in Supplemental Results (see “Hybrid Assemblies of the Model Genome”), k-mer validation outperforms hybrid assemblies in terms of both assembly breaks (i.e., number of contigs produced) and sequence duplications (Supplemental Table S10). Indeed, LH k-mer validation has zero misassemblies of both types, whereas the best hybrid assembly introduces three assembly breaks and duplicates 7.2% of the sequence of the model genome. The above results show that LH k-mer validation uses more efficiently the information provided by the short reads; using the valid k-mers (extracted from the short reads) to guide the alignment of the long reads in the self-correction is most beneficial compared with direct correction of the long reads with the short reads.
It seems reasonable to conclude from the results presented in this section, and also from the genomes of model organisms (Fig. 4), that k-mer validation with LH masking robustly produce better assemblies.
Sampling bias in the Drosophila PacBio data
Given previous work that showed that PacBio sequencing solved two difficult repetitive regions of the Drosophila Y Chromosome (Carvalho et al. 2015; Krsticevic et al. 2015), we were surprised to find that Y-linked single-copy genes were lacking on average ∼50% of their sequence in the assemblies (Fig. 6; Supplemental Table S11). We initially thought that this was due to a combination of the lower coverage of the Y (about 45×; the Drosophila reads came from male DNA, and hence coverage of the sex-chromosomes should be half of the autosomes) and assembly parameters optimized for the approximately 95× coverage of the autosomes. However, the coding regions of 20 X-linked genes are complete (Supplemental Table S11), which excludes the above explanation. When we examined the raw PacBio reads, we found that sequencing depth was very irregular in several Y-linked genes, reaching nearly zero in large parts of kl-3, kl-5, and other genes, whereas the sequencing depth of X-linked genes is fairly constant and centered around 45×, as expected (Supplemental Fig. S12). This finding is important and may have general significance because it violates one of the conditions of Gene Myers’ “140 char theorem” (“sampling is Poisson”); such violations of random sampling may be an obstacle to perfect assemblies using PacBio technology.
Figure 6.
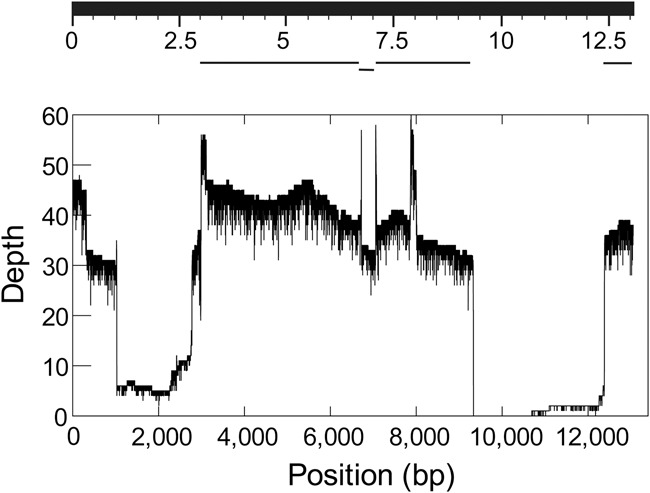
Sampling bias in the Drosophila PacBio data. (Top) BlastN search using the Y-linked kl-3 gene CDS as the query against a database of the MHAP-assembled Drosophila genome (Berlin et al. 2015). Note the large assembly gaps. (Bottom) Coverage depth of the same gene in the raw PacBio reads. Note that most of assembly gaps in the kl-3 gene actually were caused by low or absent coverage in the PacBio reads. The expected coverage depth is 45×. The Illumina coverage of the same region is fairly homogeneous (Supplemental Fig. S12).
The strong sequencing bias described above is surprising, given the success of PacBio data in assembling AT-rich or GC-rich genomes (Shin et al. 2013; Paredes et al. 2015) and previous reports of fairly uniform coverage across genomes (Ross et al. 2013). We hypothesize that this bias is related to the peculiar organization of some Drosophila Y-linked genes, which have mega base pair-sized introns composed of simple satellite DNA (e.g., (AT)n; the location of these satellite blocks is not precisely known) (Bonaccorsi and Lohe 1991; Kurek et al. 2000; Reugels et al. 2000). This will not cause problems in the assembly of exons with Sanger or short-read technologies, because the DNA is sheared in short pieces before sequencing. Indeed, the Illumina coverage is fairly constant across all Y-linked genes (except for occasional exon duplications) (Supplemental Fig. S12). However, DNA used for PacBio sequencing has a high molecular weight, in the ∼100-kb range when extracted and then sheared to ∼20 kb–40 kb; this means that some Y-linked exons will always be surrounded by a large chunk of simple repeats. Indeed, when we looked at the exon with the lowest coverage in the kl-5 gene, we found that it is surrounded by at least ∼10 kb of nearly pure (AT)n sequence on one side and a very AT-rich sequence on the other side (Supplemental Fig. S13).
How might these repeats have adversely affected PacBio sequencing? A benign hypothesis would be at the sample preparation step: Kim et al. (2014) reported the use of cesium chloride centrifugation for the Drosophila sample, which may have selected against AT-rich regions such as exons flanked by massive AT-rich satellite blocks (they will have a smaller buoyant density). A more worrisome possibility is that PacBio sequencing has some intrinsic, strong bias (e.g., against regions with very strong AT-bias or with contrasting AT-rich and GC-rich blocks). One way to solve the question would be to sequence again D. melanogaster, without the use of cesium chloride centrifugation for sample preparation. It is ironic that we failed to improve the assembly of single-copy Y-linked genes from Drosophila, since this was the original motivation of the present work.
Discussion
Single-molecule sequencing is revolutionizing genome assembly: The long reads can yield mega base pair-sized contigs that span complete chromosomes (or nearly so) of prokaryotes and simple eukaryotes, as well as the euchromatic parts of more complex genomes such as Drosophila (Berlin et al. 2015; Koren and Phillippy 2015). Their major limitation is the low accuracy. Specifically, the high error rate generates a huge number of k-mers that are not present in the original genome, and the aligners (e.g., MHAP) must sift through them in order to find shared, real k-mers that indicate true read overlaps. These problems currently are addressed by sequencing at high depth (ideally 100×), aligning the reads with improved, fast software (Myers 2014; Berlin et al. 2015) and implementing a consensus algorithm to correct the reads prior to normal assembly (Chin et al. 2013). These procedures in principle are straightforward, although the computational cost is high in the case of large genomes (e.g., mammals). A less appreciated problem is the risk of miscorrection of the reads from repetitive regions: As the initial alignment must be loose in order to detect real overlaps among the noisy reads, reads from paralogous regions (e.g., different copies of tandem rDNA genes, long transposons, or segmental duplications) will easily be lumped together (Supplemental Fig. S8); once this happens, the error correction algorithm miscorrects the reads at the “Sequence Family Variant” sites (Fig. 5), which in later assembly steps tend to cause assembly breaks.
In this article, we propose a simple and inexpensive procedure that addresses both problems: to enforce that only correct, single-copy k-mers are used as seeds for the read alignment. The enforcing of “correct k-mers” solves the “needle in a haystack problem” by making the aligner ignore the error k-mers, which are the vast majority. This by itself dramatically increases the sensitivity in overlap detection of the MHAP aligner (Fig. 2). The enforcing of k-mers that are single copy in the genome increases the specificity in read overlapping (Fig. 3; Supplemental Fig. S8) and essentially abolishes read miscorrection at “Sequence Family Variant” sites (Fig. 5). This procedure requires a list of all k-mers from the genome. Whereas a perfect list can only be obtained from a completely finished genome (i.e., when a new assembly is nonsensical), we showed that k-mers from Illumina reads provide a very good approximation to it. In contrast to the direct correction of PacBio reads with Illumina reads (“hybrid assemblies”), we used them only as a source of the list of correct single-copy k-mers. This list is used to inform the aligner of which k-mers should be ignored, thus guiding the alignment of PacBio reads for their self-correction; all sequence information came from the PacBio reads themselves. We showed that this k-mer validation procedure outperforms hybrid assemblies (Supplemental Table S10). Its use significantly improves overlap detection (Fig. 2), the accuracy of read correction (Table 1; Fig. 5; Supplemental Fig. S9), and the contiguity and accuracy of genome assembly (Fig. 4; Table 2). Gains in contiguity as measured by NG50 ranged from 28% (in C. elegans) to 91% (i.e., almost doubled, in D. melanogaster). We believe that these gains justify by themselves the use of k-mer validation, and larger gains are possible (see Supplemental Discussion). Finally, note that the additional cost of Illumina sequencing is negligible or even absent, since in nearly all cases in which a PacBio data set is available, there is also an Illumina data set from the same strain. In cases where one needs to do the Illumina sequencing, Supplemental Figure S5 suggests that an approximately 100× coverage is enough for a good separation between single-copy and repetitive k-mers, although higher coverages are beneficial.
How far can we go?
Assembly quality is a function of coverage, error rate, and read length (Phillippy et al. 2008; Nagarajan and Pop 2009; Myers 2016). Second-generation technology (e.g., Illumina) provided a good solution for this equation when fragmentation (and correct repeat reconstruction) is not a concern, for example, for sequencing genes or to identify SNP variants by comparison to a reference genome (The 1000 Genomes Project Consortium 2010). Long read sequencing provided a different solution: It yields unfragmented, nearly finished assemblies of regions with moderate repeat content, such as prokaryotic genomes and (to a large extent) the euchromatic portion of complex eukaryotic genomes, at a higher cost. Sequencing costs of new technologies tend to drop quickly, and the maturation of other long read technologies (e.g., Oxford Nanopore) brings the promise of further cost reductions. Hence, the major challenge that remains is how to correctly assemble repetitive DNA, which currently cause large assembly gaps (e.g., the histone and rDNA clusters of Drosophila; nearly all centromeres), massive fragmentation in the heterochromatin, and scattered breaks in the euchromatin (e.g., human segmental duplications). As Myers (2016) stressed, this is an open question: “… work on the assembly problem has failed to really address the issue of how to resolve repetitive sequences except in fairly superficial ways.” In a sense, technology development is pushing forward what is a repeat in assembly terms (“reads long enough 2 solve repeats”): Retrotransposons (∼7 kb long) are a major obstacle for contig building with Sanger and Illumina sequencing and are almost harmless to PacBio. So brute force, in the form of very long reads (say, average length in the 100 kb range), would solve the majority of the currently intractable regions mentioned above: Once the “golden threshold” of reads-longer-than-repeats is crossed, genome assembly became much simpler (see Fig. 1 in work by Koren and Phillippy 2015).
But “perfect assembly” is possible even when reads are not long enough to cross a repeat, as SFVs may provide a unique tiling path across it. For example, no read used in the assembly of our “model genome” spans the 44-kb segmental duplication, and yet we could assemble it in an essentially perfect form; the same happened with the 100- and 130-kb segmental duplications (Supplemental Tables S7, S8). As the results from our “model genome” show, the key is to preserve the SFV sites by not miscorrecting them, which is achieved by not swamping the overlap detection with the flood of repetitive (i.e., non-single-copy) k-mers. The k-mer validation procedure we presented here seems to be an effective implementation of this principle. Ultimately the ability to cross a repeat longer than the read length will depend on the number of SFVs per read. A tiling path requires an absolute minimum of two SFVs per read, and our model genome data had roughly 141 SFVs per read (450 SFV sites in a 44-kb segmental duplication; average length of corrected reads: 13,767 bp). It remains to be seen which read length will provide enough SFVs to cross large regions such as the histone or rDNA clusters in Drosophila (500 kb and 2 Mbp, respectively), which currently are inaccessible (both are severely fragmented even in our best Drosophila assembly). Another limit, admittedly secondary, is the assembly of simple repeats such as the intronic (AT)n blocks of Drosophila Y-inked genes, because the repeat periodicity (2–10 bp) overlaps with the error frequency of the uncorrected long reads. Finally, k-mer validation (with LH-masking) is useful even when repeats are smaller than the read length for it protects the reads from miscorrection at repeats and, hence, reduces assembly errors in these regions.
As sequencing technology and assembly software move forward, the question posed by the title of this section keeps returning (Weber and Myers 1997; Carvalho et al. 2003; Koren and Phillippy 2015; Myers 2016). But as clearly stated by Myers (https://dazzlerblog.wordpress.com/2014/05/15/on-perfect-assembly/) and Koren and Phillippy (2015) (“one chromosome, one contig”), perfect assemblies are on the verge of becoming reality, and we may now be close to the final answer.
Methods
Sequence reads
The sources of PacBio and Illumina reads for all six organisms are shown in Supplemental Table S12. All nonhuman PacBio reads came from Kim et al. (2014) and PacBio DevNet (http://www.pacb.com/); we downloaded them from the Amazon S3 repositories (listed in the Supplemental Information of Kim et al. 2014) or from the Amazon Elastic Block Storage (EBS) snapshot described by Berlin et al. (2015). These data have also been deposited at NCBI Short Read Archive (except for C. elegans), but the reads there are unfiltered (Kim et al. 2014). The sorted PacBio reads from human chromosomes 15 and 17 were kindly provided by an anonymous reviewer and came from Zook et al. (2016). The sources of Illumina reads follows: E. coli, Illumina BaseSpace (https://basespace.illumina.com); S. cerevisae, Saccharomyces Genome Database (http://www.yeastgenome.org/); A. thaliana (Cao et al. 2011; Gan et al. 2011); C. elegans (van Schendel et al. 2015); D. melanogaster (Gutzwiller et al. 2015); and H. sapiens (Zook et al. 2016).
Implementation of k-mer validation
We implemented the k-mer validation in the MHAP overlapper as follows. The standard MHAP algorithm converts each k-mer to a number (using a hash function) and saves from each read only the lowest value (called “min-mer”). The process is repeated, say, 500 times with different hash functions to generate a “sketch” of size 500, which is stored in the memory; overlapping reads were detected because their sketches share min-mers above a user-specified cut-off (for details, see Berlin et al. 2015). We implemented the k-mer validation by adding a simple step in the MHAP code: If the read k-mer is present in the valid k-mer list, it is converted to a number as described above. If it is not there (and hence probably is an error k-mer), it is converted to a very large number (technically, to Long.MAX_VALUE), effectively forcing the program to ignore it. The list of valid k-mers was previously obtained from Illumina reads with the jellyfish program (Marçais and Kingsford 2011), saved as a text file, and read by the modified MHAP code, which efficiently stores it as an array of bits (called BitSet in java language) before reading the PacBio reads. Note that these procedures implement a whitelist, whereas most aligners and overlappers use a blacklist of undesirable k-mers (either supplied by the user or produced by the program itself), which is used to remove highly repetitive k-mers, in order to reduce the computational load (e.g., the “filter-threshold” parameter in MHAP). Furthermore, their identification of repetitive k-mers is much less precise because k-mer counts are obtained from the raw PacBio reads. Illumina reads allow a much finer k-mer classification; given enough coverage, even two-copy k-mers (e.g., from a segmental duplication) can be well separated from single-copy ones (Supplemental Fig. S5). The frequency cut-off values used to build the valid k-mer lists are presented in Supplemental Table S2 and Supplemental Figure S1 and are further discussed in the Supplemental Results. When run without a valid k-mer list, the modified MHAP produces an output that is identical to the original MHAP code. The same thing happens if we use a “valid k-mer list” containing all k-mers of the PacBio reads.
The same Illumina-derived list of valid k-mers mentioned above was also used to sort “correct” and “error” k-mers in reads. For example, in Figure 1 we used a custom script that loaded the list in the memory (as an associative array) and used it to classify each read k-mer as correct (match) or error (not match).
Genome assemblies
All work was performed in three Linux servers (24 cores/64 Gb RAM, 24 cores/144 Gb RAM, and 128 cores/1 Tb RAM); assemblies used the Celera Assembler version 8.3 (PBcR pipeline). Unless otherwise noted, default PBcR parameters were used for all assemblies, including the falconsense read correction algorithm. We used the same MHAP memory parameter (ovlMemory; set to 96 Gb) in all assemblies because we found that it has a fairly strong effect on the result, including the introduction of gross misassemblies (Supplemental Results, see “PBcR Assemblies with Different Memory Parameters”; Supplemental Table S13; Supplemental Fig. S7; Supplemental Table S14). Our main purpose was to compare the Standard MHAP overlapper with the modified version (i.e., with k-mer validation) and to save time we opted to not polish the assemblies with Quiver (Chin et al. 2013).
Data access
The modified MHAP (source and compiled jar file) and the modified PBcR script from this study are available at the Supplemental_Data_S1.zip file and also at https://github.com/bernardo1963/kmer_validation. The same links provide a README file, with instructions on how to install and run the modified files. The resulting genome assemblies of C. elegans, Arabidopsis, and Drosophila are available at Supplemental_Data_S2.zip.
Supplementary Material
Acknowledgments
We thank R. Hoskins, S. Celniker, and C. Bergman for first calling our attention to PacBio sequencing; Carlos Martins and Paulo Abdon for java programming; and R. Hoskins, B. Lemos, L. Koerich, M. Vibranovski, J. Chin, A. Phillippy, S. Koren, K. Berlin, M. Chaisson, M. Sammeth, E. Ramos, C. Congrains, our lab members, and three anonymous reviewers for many valuable suggestions in the manuscript and help. We thank R. Brito (UFSCAR), C. Martins (UNESP), R. Corrêa, and G. Sachetto (UFRJ) for granting us access to their Linux servers. This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico-CNPq and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro-FAPERJ.
Author contributions: A.B.C. conceived the work, performed the research, analyzed the data, and wrote the manuscript; E.G.D. and G.G. performed the research and analyzed the data.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.209247.116.
References
- The 1000 Genomes Project Consortium. 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin K, Koren S, Chin CS, Drake JP, Landolin JM, Phillippy AM. 2015. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat Biotechnol 33: 623–630. [DOI] [PubMed] [Google Scholar]
- Blattner FR, Plunkett G III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. 1997. The complete genome sequence of Escherichia coli K-12. Science 277: 1453–1462. [DOI] [PubMed] [Google Scholar]
- Bonaccorsi S, Lohe A. 1991. Fine mapping of satellite DNA-sequences along the Y-chromosome of Drosophila melanogaster: relationships between satellite sequences and fertility factors. Genetics 129: 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Schneeberger K, Ossowski S, Gunther T, Bender S, Fitz J, Koenig D, Lanz C, Stegle O, Lippert C, et al. 2011. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet 43: 956–963. [DOI] [PubMed] [Google Scholar]
- Carvalho AB, Vibranovski MD, Carlson JW, Celniker SE, Hoskins RA, Rubin GM, Sutton G, Adams M, Myers EW, Clark AG. 2003. Y chromosome and other heterochromatic sequences of the Drosophila melanogaster genome: How far can we go? Genetica 117: 227–237. [DOI] [PubMed] [Google Scholar]
- Carvalho AB, Vicoso B, Russo CA, Swenor B, Clark AG. 2015. Birth of a new gene on the Y chromosome of Drosophila melanogaster. Proc Natl Acad Sci 112: 12450–12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, Tesler G. 2012. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, Huddleston J, Dennis MY, Sudmant PH, Malig M, Hormozdiari F, Antonacci F, Surti U, Sandstrom R, Boitano M, et al. 2015. Resolving the complexity of the human genome using single-molecule sequencing. Nature 517: 608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, et al. 2013. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10: 563–569. [DOI] [PubMed] [Google Scholar]
- Dennis MY, Nuttle X, Sudmant PH, Antonacci F, Graves TA, Nefedov M, Rosenfeld JA, Sajjadian S, Malig M, Kotkiewicz H, et al. 2012. Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication. Cell 149: 912–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan X, Stegle O, Behr J, Steffen JG, Drewe P, Hildebrand KL, Lyngsoe R, Schultheiss SJ, Osborne EJ, Sreedharan VT, et al. 2011. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature 477: 419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin S, Gurtowski J, Ethe-Sayers S, Deshpande P, Schatz MC, McCombie WR. 2015. Oxford Nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome Res 25: 1750–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29: 1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutzwiller F, Carmo CR, Miller DE, Rice DW, Newton IL, Hawley RS, Teixeira L, Bergman CM. 2015. Dynamics of Wolbachia pipientis gene expression across the Drosophila melanogaster life cycle. G3 5: 2843–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, Rozen S. 2012. Genomics and genetics of human and primate Y chromosomes. Annu Rev Genomics Hum Genet 13: 83–108. [DOI] [PubMed] [Google Scholar]
- Kim KE, Peluso P, Babayan P, Yeadon PJ, Yu C, Fisher WW, Chin C-S, Rapicavoli NA, Rank DR, Li J. 2014. Long-read, whole-genome shotgun sequence data for five model organisms. Sci Data 1: 140045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Phillippy AM. 2015. One chromosome, one contig: complete microbial genomes from long-read sequencing and assembly. Curr Opin Microbiol 23: 110–120. [DOI] [PubMed] [Google Scholar]
- Koren S, Schatz MC, Walenz BP, Martin J, Howard JT, Ganapathy G, Wang Z, Rasko DA, McCombie WR, Jarvis ED, et al. 2012. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol 30: 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Harhay GP, Smith TP, Bono JL, Harhay DM, McVey SD, Radune D, Bergman NH, Phillippy AM. 2013. Reducing assembly complexity of microbial genomes with single-molecule sequencing. Genome Biol 14: R101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Phillippy AM. 2016. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. bioRxiv 10.1101/071282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krsticevic FJ, Schrago CG, Carvalho AB. 2015. Long-read single molecule sequencing to resolve tandem gene copies: the Mst77Y region on the Drosophila melanogaster Y chromosome. G3 5: 1145–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurek R, Reugels AM, Lammermann U, Bunemann H. 2000. Molecular aspects of intron evolution in dynein encoding mega-genes on the heterochromatic Y chromosome of Drosophila sp. Genetica 109: 113–123. [DOI] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol 5: R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2016. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32: 2103–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman NJ, Quick J, Simpson JT. 2015. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods 12: 733–735. [DOI] [PubMed] [Google Scholar]
- Marçais G, Kingsford C. 2011. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27: 764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy RC, Taylor RW, Blauwkamp TA, Kelley JL, Kertesz M, Pushkarev D, Petrov DA, Fiston-Lavier AS. 2014. Illumina TruSeq synthetic long-reads empower de novo assembly and resolve complex, highly-repetitive transposable elements. PLoS One 9: e106689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers EW. 1995. Toward simplifying and accurately formulating fragment assembly. J Comput Biol 2: 275–290. [DOI] [PubMed] [Google Scholar]
- Myers EW. 2014. Efficient local alignment discovery amongst noisy long reads. In Algorithms in bioinformatics (ed. Brown D, Morgenstern B), Vol. 8701, pp. 52–67. Springer, Berlin. [Google Scholar]
- Myers EW. 2016. A history of DNA sequence assembly. Inf Technol 58: 126–132. [Google Scholar]
- Nagarajan N, Pop M. 2009. Parametric complexity of sequence assembly: theory and applications to next generation sequencing. J Comput Biol 16: 897–908. [DOI] [PubMed] [Google Scholar]
- Nagarajan N, Pop M. 2013. Sequence assembly demystified. Nat Rev Genet 14: 157–167. [DOI] [PubMed] [Google Scholar]
- Paredes JC, Herren JK, Schupfer F, Marin R, Claverol S, Kuo CH, Lemaitre B, Beven L. 2015. Genome sequence of the Drosophila melanogaster male-killing Spiroplasma strain MSRO endosymbiont. mBio 6: e02437-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillippy AM, Schatz MC, Pop M. 2008. Genome assembly forensics: finding the elusive mis-assembly. Genome Biol 9: R55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reugels AM, Kurek R, Lammermann U, Bunemann H. 2000. Mega-introns in the dynein gene DhDhc7(Y) on the heterochromatic Y chromosome give rise to the giant Threads loops in primary spermatocytes of Drosophila hydei. Genetics 154: 759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross MG, Russ C, Costello M, Hollinger A, Lennon NJ, Hegarty R, Nusbaum C, Jaffe DB. 2013. Characterizing and measuring bias in sequence data. Genome Biol 14: R51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SC, Ahn do H, Kim SJ, Lee H, Oh TJ, Lee JE, Park H. 2013. Advantages of single-molecule real-time sequencing in high-GC content genomes. PLoS One 8: e68824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Schendel R, Roerink SF, Portegijs V, van den Heuvel S, Tijsterman M. 2015. Polymerase Θ is a key driver of genome evolution and of CRISPR/Cas9-mediated mutagenesis. Nat Commun 6: 7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JL, Myers EW. 1997. Human whole-genome shotgun sequencing. Genome Res 7: 401–409. [DOI] [PubMed] [Google Scholar]
- Zook JM, Catoe D, McDaniel J, Vang L, Spies N, Sidow A, Weng Z, Liu Y, Mason CE, Alexander N, et al. 2016. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci Data 3: 160025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.