

Abstract
Background
Microsatellite markers are widely used for estimating genetic diversity within and differentiation among populations. However, it has rarely been tested whether such estimates are useful proxies for genome-wide patterns of variation and differentiation. Here, we compared microsatellite variation with genome-wide single nucleotide polymorphisms (SNPs) to assess and quantify potential marker-specific biases and derive recommendations for future studies. Overall, we genotyped 180 Arabidopsis halleri individuals from nine populations using 20 microsatellite markers. Twelve of these markers were originally developed for Arabidopsis thaliana (cross-species markers) and eight for A. halleri (species-specific markers). We further characterized 2 million SNPs across the genome with a pooled whole-genome re-sequencing approach (Pool-Seq).
Results
Our analyses revealed that estimates of genetic diversity and differentiation derived from cross-species and species-specific microsatellites differed substantially and that expected microsatellite heterozygosity (SSR-H e) was not significantly correlated with genome-wide SNP diversity estimates (SNP-H e and θ Watterson) in A. halleri. Instead, microsatellite allelic richness (A r) was a better proxy for genome-wide SNP diversity. Estimates of genetic differentiation among populations (F ST) based on both marker types were correlated, but microsatellite-based estimates were significantly larger than those from SNPs. Possible causes include the limited number of microsatellite markers used, marker ascertainment bias, as well as the high variance in microsatellite-derived estimates. In contrast, genome-wide SNP data provided unbiased estimates of genetic diversity independent of whether genome- or only exome-wide SNPs were used. Further, we inferred that a few thousand random SNPs are sufficient to reliably estimate genome-wide diversity and to distinguish among populations differing in genetic variation.
Conclusions
We recommend that future analyses of genetic diversity within and differentiation among populations use randomly selected high-throughput sequencing-based SNP data to draw conclusions on genome-wide diversity patterns. In species comparable to A. halleri, a few thousand SNPs are sufficient to achieve this goal.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3459-7) contains supplementary material, which is available to authorized users.
Keywords: Microsatellites, SSR, Arabidopsis halleri, Genetic diversity, Expected heterozygosity, SNPs, Population genomics, Whole-genome re-sequencing, Pool-Seq, Conservation units
Background
Genetic diversity is essential for organisms to adapt to changing environmental conditions and is recognised as a key component of biodiversity (e.g. [1, 2]). Microsatellite markers (also known as simple sequence repeats, SSRs) are a widely used marker system to estimate genetic diversity in population genetic studies and are often implicitly assumed to reflect the genome-wide diversity of a taxon [3]. The use of microsatellites has increased linearly since their detection in the 1980s [4], and they are nowadays extensively applied, for example in conservation genetics (e.g. [5]), forensic DNA profiling, paternity analyses, and studies of neutral genetic population structure (for reviews see [3, 6, 7]). However, the challenge of correctly interpreting microsatellite data is often strongly underrated [8], and the question whether a limited number of microsatellite markers accurately reflects genome-wide diversity remains a contentious issue (e.g. [9–12]). Single nucleotide polymorphisms (SNPs) on the basis of traditional DNA sequencing [13] have long been known, but in contrast to microsatellites, were relatively rarely used in population genetics until recently because of the difficulties associated with their characterization and genotyping in non-model organisms [14]. Moreover, their (mostly) bi-allelic state limits the information content per locus compared to the more polymorphic microsatellite markers [15–17]. In recent years, the use of SNPs has been exponentially increasing [7], mainly because newly developed high-throughput sequencing techniques can efficiently be applied to a wide range of organisms. These techniques allow for the identification of thousands to millions of unbiased SNPs, and the simultaneous estimation of SNP frequencies across the genomes of individuals, populations and species [18–20].
Microsatellites have unique properties that distinguish them from the rest of the genome, and these should be taken into consideration when analysing and interpreting them [8]. Microsatellites are codominant markers and typically consist of simple sequence repeats varying in length between one and six base pairs. Their variability originates from DNA polymerase slippage during replication, leading to the formation of shorter or longer alleles (for further details see [21–23]). In plants, microsatellite mutation rates range between 10−6 and 10−2 per locus and generation (for a review see [24]), thus varying approximately 10,000-fold, and are affected by various factors, including repeat type, repeat copy number, marker location in the genome, and taxon [23]. In contrast, spontaneous mutation rates for SNPs only vary about 100-fold [25]. Knowledge of direct estimates of SNP mutation rates is limited, but the rate has been accurately estimated e.g. in Arabidopsis thaliana to be 7 × 10−9 substitutions per site per generation [26]. Microsatellite mutation rates are therefore several orders of magnitude higher and much more variable than those of SNPs. In combination with the often small number of markers used, microsatellite-based studies typically sample a narrow fraction of the genome with unusually high mutation rate [21]. This may be aggravated when only the most polymorphic microsatellite markers are selected for further analysis after initial screening of a small subsample of individuals or populations. Estimates of genetic diversity may then suffer from ascertainment bias [15, 27]. Additionally, amplification variation of primers [28] and fragment size homoplasy [29] potentially reduce the accuracy of genetic estimates inferred from microsatellite markers. The use of microsatellite markers may thus lead to estimates of genetic diversity and differentiation that do not well reflect genome-wide patterns of variation.
Despite these potential caveats, a large number of studies has relied on microsatellite markers to estimate genetic diversity and genetic differentiation, not only within and among populations, but also among species (e.g. [3, 7, 10, 30]). In a conservation context, microsatellites are also used to identify conservation units (CUs), whose genetic variation and distinctness is potentially relevant for species survival (e.g. [5, 31, 32]). Well-known case studies are the Florida panther [33, 34] or the African elephant from Eritrea [35], for which management decisions were taken based on genetic data derived from few microsatellites.
To date, only few studies have explored in detail to what degree microsatellite variation reflects genetic variation at other nuclear loci, and which genetic diversity estimator for microsatellites provides the most accurate prediction of genome-wide diversity. Positive but sometimes weak correlations between expected microsatellite heterozygosity (SSR-H e) and SNP diversity in nuclear gene sequences have been reported at the population level in salmon [11, 36, 37] and several carnivore species [10], as well as different rice varieties and sheep breeds [38, 39]. Most of these studies, however, have investigated only a limited number of SNPs (ranging from tens to a few thousand). The outcome of the comparison of SNP versus microsatellite diversity in these studies was strongly affected by the number of SNP markers used. Studies in which low SNP numbers (<300) were compared to microsatellites found that the latter had more power to infer differences in genetic summary statistics [10, 38–46] or found similar results when approximately 400 SNPs were used [47]. In contrast, studies using larger numbers of SNPs (~3000) found that SNPs performed better than microsatellites [11, 12, 37]. Many of these studies used existing genotyping arrays for SNP detection. These may, however, cause ascertainment bias as a consequence of the overrepresentation of common SNPs [8]. To date, no unbiased whole-genome re-sequencing approach has been used for comparison. Studies based on reduced representation libraries (e.g. restriction-site associated DNA sequencing; RADseq), which sample a subset of all SNPs of the genome, showed that SNPs have more power than microsatellites, e.g. to detect heterozygosity–fitness correlations in natural populations of oldfield mice [9]. Further, demographic inferences drawn from RADseq-derived SNPs in bumble bees reflected important long-term differences in population size better than microsatellites, which instead signalled either recent demographic changes or mutational processes [48].
Because of the widespread application of microsatellite markers both in basic research and practical conservation, it is important to evaluate the tenet that microsatellite variation adequately reflects genome-wide genetic diversity, especially for situations in which only a limited number of markers are used, as is often the case in conservation genetics, where on average only 12 microsatellites are used per study [49]. It is further relevant to evaluate the power of next-generation sequencing (NGS) based genotyping approaches to infer genome-wide diversity and population structure, e.g. to estimate the number of SNPs required to achieve accurate and consistent estimates of genome-wide diversity.
We used two types of microsatellite markers (markers developed for the same species and cross-species markers) as well as genome-wide SNP variation in the meadow rock cress, Arabidopsis halleri (L.), to compare estimates of genetic diversity and differentiation. Overall, we genotyped 180 individuals of A. halleri from nine natural populations using 20 microsatellite markers, which is above the average number of microsatellites typically used in population and conservation genetic studies [49]. We compared them to a pooled whole-genome re-sequencing approach (Pool-Seq; [50, 51]) and tested whether estimates of genetic variation derived from microsatellite polymorphisms are valid and useful proxies of genome-wide genetic variation and differentiation. Specifically, we tested whether estimates from both marker types were correlated (relative comparison) and had similar absolute values (absolute comparison). Further, we used down-sampling to assess how many random and presumably unlinked SNPs are required to calculate accurate estimates of genome-wide diversity.
Methods
Study system
Arabidopsis halleri is a perennial, insect-pollinated, strictly outcrossing and functionally self-incompatible herb [52] with a wide geographic distribution from central Europe to eastern Asia [53]. It grows in diverse habitats including mountain slopes, grassy meadows, forest margins and rocky crevices [54, 55] and has been widely used as a model to study heavy metal tolerance (e.g. [55, 56]). The species is diploid with 2n = 16 [54] and has an estimated genome size of 255 Mbp [57].
Sampling and DNA extraction
Leaf tissue from 20 individuals each of nine populations of A. halleri was sampled in south-eastern Switzerland and northern Italy (Additional file 1: Table S1). The selected samples size per population should allow to accurately estimate population-specific genetic diversity and differentiation [58]. A minimal distance of two, but preferably 4 m, was maintained between collected individuals, as genetic structure and diversity may be affected by clonal growth when plants are separated by less than one meter [59]. Leaf samples were dried in silica gel, and DNA was extracted with the DNeasy Plant Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. DNA concentrations were measured with a Qubit® 1.0 Fluorometer (dsDNA BR, Carlsbad, USA), and DNA quality was examined using a NanoDrop 8000 Spectrophotometer (Thermo Scientific, Waltham, USA) as well as 1.5% agarose gels stained with GelRed (Biotium, Hayward, USA).
Microsatellite analyses
The 180 samples were genotyped using 20 microsatellite markers in three multiplex PCRs, each amplifying either six or eight microsatellite markers (Additional file 2: Table S2). The first two multiplex sets included 12 microsatellite loci that were originally developed for A. thaliana [52, 60, 61], hereafter referred to as “cross-species” microsatellites. Some of these primer sequences were adapted to A. halleri by comparing them to our own de-novo assembly of the A. halleri genome [51] using IGV 2.1 [62], identifying potential mismatches and changing the primer sequences accordingly (Additional file 2: Table S2). Further, eight microsatellite primer pairs that were specifically developed for A. halleri [63] were combined in a third multiplex set. These markers are hereafter referred to as “species-specific” microsatellites. Detailed lab protocols can be found in the Additional Methods (Additional file 3). Alleles were called using GeneMapper 4.1 (Applied Biosystems).
Estimating microsatellite-based genetic diversity
For every marker and population, we assessed the following population genetic parameters. The inbreeding coefficient F IS and its p-value, which indicate whether markers or populations deviate from Hardy–Weinberg equilibrium, were calculated with GenoDive 2.0b23 [64] using the heterozygosity-based G IS statistic with 999 permutations and applying Bonferroni correction for multiple testing. Null allele frequencies were calculated with FreeNA [28]. Pairwise values of genetic differentiation among populations, F ST, were calculated based on allele identity with Genepop 4.2.2 [65, 66], whereas allele frequencies, expected heterozygosity (SSR-H e) and mean number of alleles (allelic richness, A r) per locus were quantified with Genetix 4.05 [67]. We consistently genotyped 20 individuals per population, therefore, A r did not have to be corrected with a rarefaction approach. All population parameters were computed for three different marker sets including (i) all, (ii) the cross-species, and (iii) the species-specific microsatellite markers (Additional file 2: Table S2). To infer marker bias, we tested for quantitative differences in estimates of SSR-H e and A r estimated in each population from cross-species and species-specific markers using a paired t-test (function ‘t.test’) in R 3.2 [68]. For the relative comparison of SSR-H e derived from cross-species and species-specific markers, we used a Pearson’s correlation test (function ‘cor.test’) in R. To test whether population-specific estimates of SSR-H e obtained from the different microsatellite types differ [69], we used a pairwise Wilcoxon signed-rank test (function ‘pairwise.wilcox.test’) in R. By plotting SSR-H e medians and quantiles for each population, we inferred whether non-significant differences were caused by high variance. When the absence of significant differences between populations was obviously caused by overly high variances in the genetic diversity estimates computed per microsatellite marker and population, we interpreted this as variance bias. P-values were adjusted for multiple testing using Bonferroni correction.
Pool-Seq and Illumina read processing
Pooled next-generation sequencing (Pool-Seq) has been shown to produce accurate population-specific allele frequencies [20, 51, 70]. For NGS, individually extracted high-quality DNA was equimolarly pooled using the same 20 individuals from the nine populations as for the microsatellite genotyping presented above. These nine pools were high-throughput sequenced and further processed as described below and, in more detail, in Fischer et al. [50]. For a subset of SNPs and populations of the present dataset, the accuracy of exactly the same Pool-Seq approach had been validated [51]: differences in estimates of population-specific allele frequencies compared to those from individual genotyping were on average less than 4%. Library preparation (~170–250 bp insertion size; 100-bp paired-end reads) and sequencing on an Illumina HiSeq2000 (Illumina, San Diego, USA) were performed by GATC Biotech (Constance, Germany) and the Quantitative Genomics Facility (D-BSSE, ETH Zürich, Switzerland). Forward and reverse raw reads were trimmed for tags and adaptors with Cutadapt [71]. Phred-type quality scores Q20 were used for quality trimming with the FASTX toolkit (http://hannonlab.cshl.edu/fastx_toolkit). The separately trimmed forward and reverse reads were then re-synchronized to pairs with an in-house perl script. Only paired sequences were used for further analysis [50].
Read mapping, SNP calling and genome-wide population genetic estimates
To estimate genome-wide genetic diversity and differentiation for all nine populations of A. halleri, reads were mapped to the A. thaliana reference genome (TAIR10, from which organellar DNA was excluded [72–74]) using BWA aln, allowing for 10% mismatch, and sampe [75]. All ambiguously mapped reads were removed and the remaining high-quality reads were sorted with SAMtools 0.1.18 [76]. SNPs were called for the nine populations by producing mpileup files with SAMtools (for details see [50, 76]).
To obtain population-specific genome-wide estimates of genetic diversity, we first calculated Watterson’s theta (θ Watterson), an estimator that takes into account the number of segregating sites to estimate the population mutation rate. θ Watterson was calculated on a gene-by-gene basis (only exons) for each population using the gene and exon annotation of TAIR10 (GFF3_genes.gff; [74]). The perl script ‘Variance-at-position.pl’ of the software package PoPoolation [77] was used with the mpileup file of each population and the.gtf annotation file (transformed from GFF3_genes.gff file) to calculate exonic θ Watterson. This approach provides unbiased estimates for pooled samples as it corrects for coverage. Exon based θ Watterson is a conservative estimate of genetic diversity as it infers diversity from genomic regions that are predominantly under purifying selection, hence from slightly less diverse regions than the rest of the genome. Exon-based diversity estimates are of direct relevance for the adaptive potential of a population, because exons harbour functionally relevant polymorphisms that allow populations to adapt to changing environments. To accurately estimate allele frequencies for estimating θ Watterson, minimum counts for minor alleles were set to two to account for sequencing errors, leading to a minor allele frequency threshold of 0.05. The minimum coverage per site within populations was set to 20×, which mimics the number of individuals. To further correct for potential errors caused by repeated sequences, a maximum coverage of 400× per population was used as threshold for SNP identification. In order to be included in the genome-wide estimates of gene diversity, 50% of all SNPs within a gene had to reach the above-mentioned thresholds in all nine populations [50]. For all analyses, pool size per population was set to 40 because 20 diploid genomes were represented in each population pool.
Second, we calculated genome-wide SNP-based expected heterozygosity (SNP-H e), taking all SNPs into account, not only those located in exons. Mpileup files were synchronized and filtered for base quality (Q20) with the perl script ‘mpileup2sync.pl’ of PoPoolation2 [78]. Next, major and minor allele frequencies were calculated with the script snp-frequency-diff.pl. The coverage threshold was the same as mentioned above, except that the minor allele count was set to four, as all nine populations were jointly used to infer minor allele frequencies [50], leading to a more sensitive, but less error-prone minor allele frequency threshold of 0.011. We only used bi-allelic SNPs and calculated the average genome-wide SNP-H e as
where n is the number of SNPs, and p i is the minor allele frequency of the ith allele. This approach assumes Hardy–Weinberg equilibrium within populations.
To infer potential demographic events that could strongly influence genetic diversity within populations, we calculated exome-wide Tajima’s D using the TAIR10 gene annotation and the perl script ‘Variance-at-position.pl’ in PoPoolation [77]. It is suggested to use a coverage threshold of less than three times smaller than the pool size [79], which is in our case 13×. A negative genome-wide Tajima’s D is indicative of an expansion after a bottleneck, whereas a positive D is compatible with a scenario of a decrease in population size [80, 81]. To test whether the average of the resulting distribution of Tajima’s D was significantly different from zero, we used t-tests against random normal distributions (functions ‘t.test’ and ‘rnorm’ in R) with an average of zero and the same standard deviation as observed in the real data of each population.
Estimates of pairwise population genetic differentiation (F ST) were calculated with ‘fst-sliding.pl’ in PoPoolation2 [78, 82]. Average values of pairwise F ST were calculated using the same parameters as mentioned above for the estimates of SNP-H e as explained in detail in Fischer et al. [50].
Comparisons of genetic diversity and differentiation derived from microsatellites and genome-wide SNPs
To explore associations between estimates of genetic diversity derived from microsatellites and SNPs, we performed Pearson’s correlations (‘cor.test’ in R) of microsatellite-based allelic richness (A r) and expected heterozygosity (SSR-H e). Estimates of genome-wide SNP diversity were derived from exon sequences (θ Watterson) and genome-wide SNP expected heterozygosity (SNP-H e). Further, to account for possible confounding effects due to linkage, associations were also tested for a subset of SNPs, consisting of every 50th SNP.
We performed Mantel tests to check for correlations between values of F ST derived from genome-wide SNPs and (i) all, (ii) cross-species, (iii) and species-specific microsatellite markers. The same analysis was used to assess correlations between values of F ST derived from species-specific and cross-species microsatellite markers. All analyses were performed with 1001 permutations using Ecodist 1.2.7 [83] in R. Finally, we used paired t-tests implemented in R to quantitatively evaluate whether H e and F ST derived from microsatellite markers and SNPs significantly differ.
Estimating the number of unbiased SNPs required for accurate estimates of genetic diversity
We used a down-sampling procedure to estimate SNP-H e with the aim to infer the required number of randomly selected and unlinked SNP markers to obtain accurate estimates of genetic diversity and to reliably rank populations according to their genetic diversity (e.g. for CU identification). Thus, each population was resampled for the same k random SNP markers drawn from the pool of more than 2 million SNPs. For each value of k varying between 100 and 400,000, we created 1000 random subsamples of k SNP markers (starting from k 100 up to 10,000 we sampled k at steps of 100 and from k 10,000 to 400,000 SNPs we sampled k in steps of 1000). We then computed the mean expected heterozygosity (H e) and 95% confidence intervals for each value of k observed in each of the 1000 subsamples. Obtained results were used to draw curves representing the variation of the estimated H e as a function of genotyping effort in each population. We then identified the number of SNPs for which the upper and lower confidence intervals for expected heterozygosity (SNP-H e) fell below ±0.01, ±0.005, and ±0.001.
Results
Microsatellite diversity
Twenty microsatellite loci were initially used to characterise 180 A. halleri individuals from nine populations. We excluded marker ah59 from further analyses, because it deviated significantly from Hardy–Weinberg equilibrium and exhibited an estimated null allele frequency of 10% (Additional file 4: Table S3). The remaining 19 microsatellite markers harboured 83 alleles and only 0.3% missing data. Allelic richness per locus (A r) ranged between 2.2 and 3.1 per population, with an average of 2.71 (±0.29 SD), and expected heterozygosity (SSR-H e) ranged from 0.025 to 0.717 per microsatellite marker. Further details are given in Table 1 and Additional file 4: Table S3. Population allele frequency distributions were fairly noisy, see Additional file 5: Figure S1A. None of the nine populations showed significant deviation from Hardy–Weinberg equilibrium after Bonferroni correction (Table 1).
Table 1.
Population genetic parameters inferred from 19 microsatellites and genome-wide SNPs for nine populations of Arabidopsis halleri. Allelic richness (A r), expected heterozygosity (SSR-H e) and inbreeding coefficient F IS including its one-sided p-value (i.e. heterozygote deficiency) are given. No F IS value was significantly different from zero after Bonferroni correction. θ Watterson was calculated for 20,617 genes. Expected heterozygosity (SNP-H e) was calculated from all SNPs across the genome. Tajima’s D was calculated for 22,210 genes, p-values refer to deviations from zero (t-test)
| Population | Microsatellites | SNPs | ||||||
|---|---|---|---|---|---|---|---|---|
| A r | SSR-H e | F IS | F IS p-values | θ Watterson | SNP-H e | Tajima’s D | Tajima’s D p-values | |
| Aha09 | 2.7 | 0.392 | 0.073 | 0.051 | 0.0088 | 0.154 | −0.029 | <0.001 |
| Aha11 | 2.6 | 0.318 | 0.043 | 0.217 | 0.0081 | 0.138 | −0.114 | <0.001 |
| Aha18 | 2.6 | 0.360 | −0.015 | 0.386 | 0.0086 | 0.152 | −0.009 | 0.743 |
| Aha19 | 2.8 | 0.332 | −0.084 | 0.027 | 0.0086 | 0.150 | −0.033 | <0.001 |
| Aha21 | 2.7 | 0.404 | 0.075 | 0.075 | 0.0083 | 0.148 | −0.021 | 0.106 |
| Aha31 | 3.1 | 0.387 | 0.068 | 0.082 | 0.0093 | 0.157 | −0.119 | <0.001 |
| AhaN1 | 2.9 | 0.465 | 0.081 | 0.030 | 0.0092 | 0.155 | −0.169 | <0.001 |
| AhaN3 | 3.1 | 0.399 | 0.110 | 0.010 | 0.0089 | 0.154 | −0.151 | <0.001 |
| AhaN4 | 2.2 | 0.343 | 0.058 | 0.151 | 0.0067 | 0.119 | −0.103 | <0.001 |
| Mean | 2.7 | 0.378 | 0.045 | 0.0085 | 0.148 | −0.083 | ||
All twelve cross-species microsatellite markers, initially developed for A. thaliana, successfully amplified in A. halleri. A r at cross-species microsatellite markers was 2.2 alleles per marker and population and thus significantly lower than A r at species-specific markers (3.7 alleles, p < 0.0001, paired t-test; Fig. 1a). The same pattern (p < 0.0001, paired t-test; Fig. 1b) was observed for SSR-H e, which was 0.32 and 0.47, respectively. No significant correlation was observed among estimates of H e inferred from cross-species and species-specific microsatellite markers (Pearson’s r = 0.439, p = 0.237; Fig. 1c).
Fig. 1.
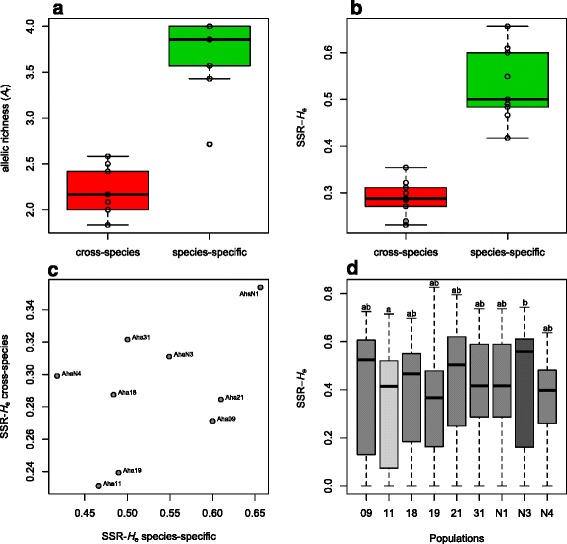
Comparison of estimates of genetic diversity derived from cross-species (developed for Arabidopsis thaliana) and species-specific microsatellite markers (developed for Arabidopsis halleri) for a allelic richness (A r, p < 0.0001, paired t-test) and b expected microsatellite heterozygosity (SSR-H e, p < 0.0001, paired t-test). c Estimates of SSR-H e inferred separately from cross-species and species-specific microsatellite markers were not significantly correlated (Pearson’s r = 0.439, p = 0.237). Dots are labelled with population codes (Additional file 1: Table S1). d No significantly different estimates of H e were observed among populations after Bonferroni correction (pairwise Wilcoxon signed-rank test). Without correction for multiple testing only population Aha11 and AhaN3 showed significantly different estimates of H e (indicated with different colouring and letters)
The variance in the estimates of SSR-H e among microsatellite markers within and among population was so high that no significant differences in genetic diversity among populations could be inferred after Bonferroni correction (pairwise Wilcoxon signed-rank test). Without Bonferroni correction, only populations Aha11 and AhaN3 differed significantly in their estimates of H e (Fig. 1d). Similar results were found when cross-species and species-specific microsatellites were analysed separately and corrections for multiple testing were performed (Additional file 6: Figure S2).
Average pairwise F ST for the 19 microsatellite loci was 0.173 (range: 0.021–0.375); species-specific (mean: 0.169; range: 0.00–0.381) and cross-species microsatellites markers (mean: 0.173; range: 0.021–0.418) did not deviate significantly from each other (p = 0.743, paired t-test).
Illumina sequencing and genome-wide diversity
The Illumina sequencing yielded a total of 1,247,939,483 100-bp paired-end reads corresponding to 249,587,896,600 nucleotides. After quality filtering and trimming, 1,197,105,373 paired-end reads were mapped to the A. thaliana reference genome (TAIR10), from which organellar DNA was excluded. The average coverage per site, after filtering with the defined thresholds, was 60.7× with a range of population-wise coverage of 52.7 to 69.3 × .
We detected 2,178,204 SNPs, which were used for the calculation of pairwise F ST, and 2,064,681 bi-allelic SNPs, which were used for the calculation of population-specific SNP-H e. All populations had even population-specific allele frequency distributions (Additional file 5: Figure S1B). Values of pairwise F ST ranged between 0.02 and 0.09, and population-specific SNP-H e was between 0.12 and 0.16. Overall, 20,617 and 22,210 genes fulfilled our thresholds of coverage for calculating θ Watterson and Tajima’s D, respectively. θ Watterson ranged from 0.0067 to 0.0093, and Tajima’s D values were all slightly negative, ranging from −0.01 to −0.17. Only two of the nine populations showed no significant deviation from zero. In other words, most populations showed weak demographic changes probably related to a bottleneck with later expansion (Table 1 and Additional file 7: Figure S3).
Comparisons of genetic diversity estimates derived from microsatellites and genome-wide SNPs
No significant correlation was observed between population-specific estimates of H e derived from microsatellites and genome-wide SNPs (Pearson’s r = 0.550, p = 0.125; Fig. 2a), independent of whether all SNPs or a subset of presumably unlinked SNPs (every 50th SNP) were used (Pearson’s r = 0.572, p = 0.108; Additional file 8: Figure S4). Estimates of SNP-H e were overall significantly lower than those of SSR-H e (paired t-test, p < 0.0001; Fig. 2a insert). SSR-H e was also not significantly correlated with θ Watterson (Pearson’s correlation: r = 0.553, p = 0.123; Fig. 2b). The correlation coefficient was higher when using only species-specific markers (Pearson’s r = 0.640, p = 0.063), but lower when only cross-species markers were used (Pearson’s r = 0.263, p = 0.494; Fig. 2b), though neither of the two correlations was significant.
Fig. 2.
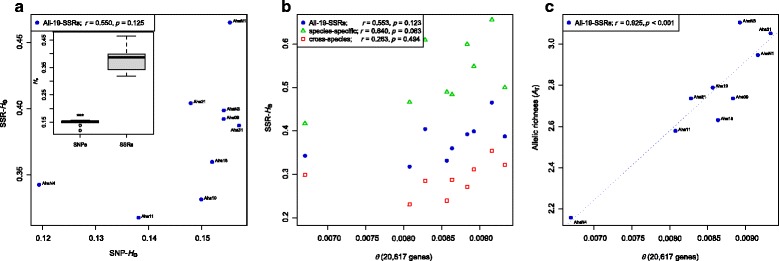
Relationships between population genetic parameters for nine populations of Arabidopsis halleri estimated on the basis of 19 microsatellites and 2 million genome-wide single nucleotide polymorphisms (SNPs). a Relationship between expected genome-wide SNP heterozygosity (SNP-H e) and expected microsatellite heterozygosity (SSR-H e). The insert illustrates differences in H e between SNPs and microsatellites across the nine A. halleri populations. b Relationship between genome-wide diversity estimated by average θ Watterson across 20,617 genes and expected heterozygosity for all 19 microsatellites (filled circles) and separately for species-specific (open triangles) and cross-species microsatellites (open squares). c Comparisons of allelic richness (A r) with genome-wide SNP variation estimated by θ Watterson
In marked contrast to heterozygosity, A r of microsatellite markers was significantly correlated with θ Watterson (Pearson’s r = 0.925, p = 0.0004; Fig. 2c). The correlation based on ranks was still significant (Spearmen’s ρ = 0.817, p = 0.0108), but slightly weaker than non-ranked comparisons.
The SNP-based genome-wide diversity estimates, θ Watterson and SNP-H e, were highly correlated (Pearson’s r = 0.979; p < 0.001; Fig. 3), even though values of θ Watterson were derived exclusively from coding regions (exons), and estimates of SNP-H e were calculated from more than two million SNPs across the entire genome. In fact, θ Watterson estimates inferred from introns and intergenic regions were highly correlated to estimates of exon-based θ Watterson (Pearson’s r = 0.988; p < 0.001; see Additional file 9: Figure S5).
Fig. 3.
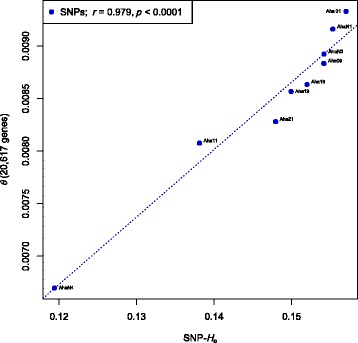
Pearson’s correlation (dashed line) between exome-wide θ Watterson and genome-wide expected SNP heterozygosity (SNP-H e) in Arabidopsis halleri
Comparison of genetic differentiation estimates derived from microsatellite versus genome-wide SNP variation
Mantel tests revealed highly significant correlations between values of pairwise F ST derived from genome-wide SNP data and microsatellite markers (Fig. 4). The best correlation was achieved when using all 19 microsatellites (r MT = 0.947, p = 0.001; Fig. 4a). However, values of F ST derived from microsatellite markers were 3.35-fold higher than those from SNPs and were significantly different (p < 0.0001, paired t-test; Fig. 4b). If we split the microsatellite markers into species-specific and cross-species, the correlations were slightly weaker for cross-species microsatellites (r MT = 0.942, p = 0.001; Fig. 4a) and for species-specific microsatellites (r MT = 0.866, p = 0.008; Fig. 4a). The correlation among species-specific and cross-species microsatellites was high (r MT = 0.829, p = 0.004; Additional file 10: Figure S6).
Fig. 4.
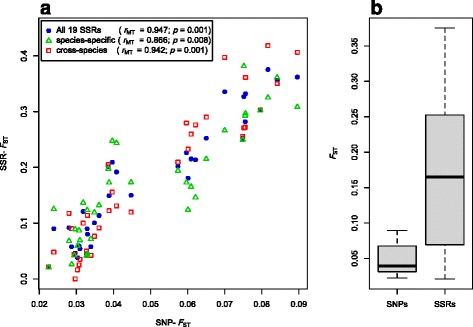
Comparison of pairwise population genetic differentiation (F ST) among nine populations of Arabidopsis halleri estimated with different genetic markers. a Comparison of F ST derived from 2,178,204 genome-wide SNPs with F ST inferred on the basis of all 19 microsatellite markers (filled circles), and separately for 12 cross-species (open triangles), and seven species-specific microsatellites (open squares). Given are correlation coefficients (r MT) and p-values from Mantel tests. b Estimates of F ST inferred from SNPs were significantly lower than those based on microsatellites (p < 0.0001, paired t-test)
Estimating the number of unbiased SNPs required for accurate estimates of genetic diversity
SNP down-sampling revealed that the upper and lower confidence intervals for expected heterozygosity (SNP-H e) fell below ±0.01, ±0.005, and ±0.001 with 1000, 4000 and 93,000 random SNPs, respectively. To accurately and consistently rank all A. halleri populations according to their genome-wide diversity (i.e. non-overlapping 95% confidence intervals), 300,000 SNPs were required (Fig. 5). However, populations Aha09 and AhaN3 could not be distinguished, as they have the same genome-wide SNP-H e (Table 1).
Fig. 5.
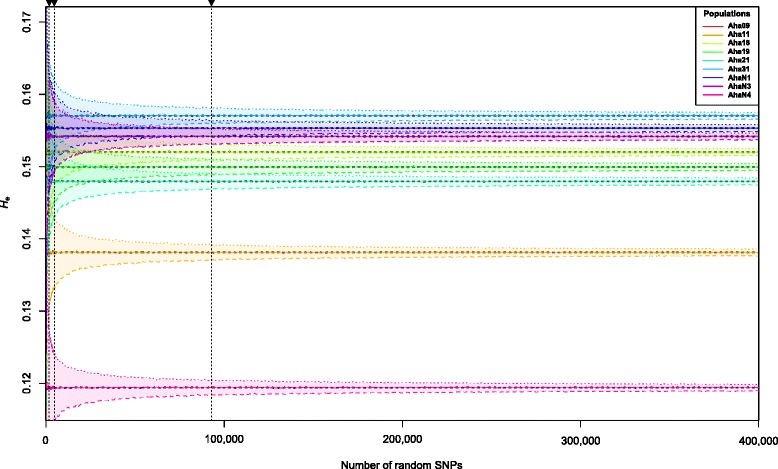
The effect of the number of randomly selected SNP markers on the estimation of H e represented for each of nine populations of Arabidopsis halleri. Light colours indicate the 95% confidence intervals as inferred from 1000 randomly drawn replicates. Dashed horizontal lines (marked by arrows) indicate the numbers of SNPs for which 95% confidence intervals for SNP-H e are below ±0.01, ±0.005, and ±0.001
Discussion
The application of microsatellite markers is widespread in population and conservation genetic studies. However, NGS-based SNP genotyping approaches are rapidly developing and can be applied to a wide diversity of model and non-model organisms. Our comparative analysis of genetic diversity estimates based on microsatellites and genome-wide SNPs revealed interesting differences. Most importantly, we found no significant correlation between expected microsatellite heterozygosity (SSR-H e), an estimator of genetic diversity that is widely used and reported in microsatellite studies, and genome-wide SNP diversity (Fig. 2a and b). This finding indicates that SSR-H e does not adequately reflect genome-wide genetic diversity in the investigated populations of A. halleri. In contrast, microsatellite allelic richness (A r) was a much better proxy for genome-wide diversity (Fig. 2c). Further, genetic differentiation in terms of F ST estimated from microsatellite variation correlated reasonably well with that based on genome-wide SNP data. However, absolute values of the different summary statistics inferred from different marker types varied considerably (Figs. 2a and 4b). Our results do not question the usefulness of microsatellites per se, but point to research questions for which SNPs may be better suited than microsatellite markers, given the availability of robust and cost-effective high-throughput sequencing-based SNP genotyping approaches.
Thanks to the massive advances in sequencing technology, thousands of SNPs can efficiently be genotyped in any given organism, and these may improve our ability to adequately estimate genetic diversity and differentiation. Previous studies found that the potential of SNPs to resolve population genetic structure strongly depends on their number. Indeed, in studies using a low number of assay-based SNPs, microsatellites performed similarly well or better than SNPs [10, 38–47]. However, in studies using larger SNP numbers, especially when they were derived from NGS-based approaches, the relative performance of SNPs clearly improved [9, 11, 12, 37, 48]. These studies show that a large number of SNPs compensates for the lower information content of these typically bi-allelic markers compared to more polymorphic microsatellite markers. Our study shows that a few thousand SNPs are enough to accurately estimate genome-wide diversity in terms of H e (Fig. 5, see below).
Evidence available from animal species [10, 36, 46] as well as from this study reveals that there is no or only weak congruence between estimates of heterozygosity derived from microsatellites and SNPs. Indeed, theory suggests that an association of heterozygosity estimates between microsatellites and genome-wide SNPs is not expected a priori. According to Ljungqvist et al. [84], this association is shaped by identity disequilibrium, i.e. the non-random association of diploid genotypes between loci. Simulations indicated that a strong positive correlation only emerges when the studied populations are characterized by substantial identity disequilibrium, as was the case in the studies on salmon and carnivores [10, 36]. The absence of this correlation in A. halleri suggests that identity disequilibrium is weak or absent, which is compatible with the existence of a strong self-incompatibility system in this species and observed non-significant values of F IS. This finding indicates that heterozygosity estimates based on microsatellites (SSR-H e) might not be good surrogates for genome-wide diversity in outbreeding species. Further, we found that the marker-specific variation of SSR-H e is too high to distinguish populations based on their levels of genetic diversity (Fig. 1d) and that even among microsatellite markers originally developed for different taxa, important discrepancies can be observed (Fig. 1c and Additional file 6: Figure S2). This variance bias is especially strong when a low number of microsatellites is used, as is the case in many population and conservation genetic studies (on average about 12 markers [49]). The larger sampling variance associated with a limited number of microsatellite markers is evidenced by their allele frequency distributions, which are much noisier than those derived from the SNP data set (Additional file 5: Figure S1).
An alternative estimator to H e is microsatellite allelic richness (A r), one of the simplest estimators of genetic diversity available. A r was significantly correlated with genome-wide SNP diversity in our study (Fig. 2c) and thus appears to be a useful proxy of genome-wide genetic diversity. Congruent results of A r and SNP diversity were also reported in other studies [10, 36, 49] and might be explained by several reasons. First, SSR-H e estimates are based on few markers with noisy allele frequency distributions (Additional file 5: Figure S1A) and represent a proportion, ranging between 0 and 1, whereas allelic richness is an infinite count. Accordingly, one additional allele, especially when it is rare and many alleles are already present, does not strongly influence SSR-H e, but will affect A r estimates, making the latter a more sensitive estimator of diversity. Moreover, especially for microsatellites with a high number of alleles, accurate estimates of population-specific heterozygosity can be problematic [85, 86], and stochasticity may have a strong impact on estimates of H e at the lower range of allelic diversity. Finally, A r is more sensitive to population bottlenecks than H e [49]. Therefore, A r better reflects the population’s demographic history and hence is a more relevant estimator of genetic diversity to predict the short-term survival of a population.
Despite the good performance of A r as a proxy of genome-wide diversity, it was not sufficient to accurately rank populations according to their genetic diversity (Fig. 2c). Consequently, the identification of conservation units (CUs) or decision taking for conservation actions based on microsatellite-derived rankings of genetic diversity may be misguiding. Nevertheless, using microsatellite-derived A r rather than SSR-H e provides more accurate estimates of genome-wide genetic diversity derived from a limited number of microsatellite markers. These considerations are strengthened by a simulation study [87], which found allelic richness to be two to four times more powerful than H e for the identification of a temporal genetic decline in a population.
A possible reason for the deviation of estimates of genetic diversity derived from microsatellite and genome-wide SNP data could be the influence of very recent demographic changes. As a consequence of their higher mutation rate [21], microsatellites respond more strongly to recent demographic events than genome-wide SNPs [48], and SNPs uncover a different and likely older demographic history. Evidence for this hypothesis has also been presented for bumble bees [48] and may indicate a fruitful application of microsatellites in analyses of populations that may have undergone very recent demographic changes. However, it is important to note that loci with high mutation rate (e.g. microsatellites) may violate demographic model assumptions, such as mutation–migration–drift equilibrium [8]. Further, for the long-term survival of populations, genetic diversity in coding and regulatory sequences is arguably more relevant than microsatellite diversity, because most microsatellites are located in non-coding regions and are mostly selectively neutral, hence of less evolutionary importance. Consequently, estimates of genome-wide SNP diversity better reflect functionally important and potentially adaptive genetic variation [88], and should therefore be used preferentially, especially in conservation genetics studies. In A. halleri the long-term demographic history inferred from genome-wide data indicates a bottleneck with later expansion for most populations, as values of Tajima’s D were slightly negative and significantly different from zero (Table 1 and Additional file 7: Figure S3).
The origin of the microsatellite markers used, i.e. whether they are species-specific or cross-species markers, may further impact estimates of genetic diversity [27]. In this study, species-specific microsatellites displayed significantly higher A r and SSR-H e than cross-species markers (Fig. 1a, b) originally developed for A. thaliana [52, 60, 61]. Further, species-specific markers resulted in more accurate estimates of genetic diversity (A r ; Fig. 2b), but less accurate estimates of divergence (F ST; Fig. 4a) among populations of A. halleri. Hence, the practicability of microsatellites for population genetic studies is limited and difficult to assess a priori [8].
High estimates of genetic diversity derived from species-specific microsatellite markers may be a consequence of ascertainment bias caused by selecting the most polymorphic markers [15, 27], whereas cross-species microsatellites are mostly chosen based on their amplification success in the study species. The consequence of this ascertainment bias is evident in this study, as estimates of SSR-H e for cross-species microsatellites are not significantly correlated with SSR-H e for species-specific microsatellites (Fig. 1c). These differences further emphasize our inference that marker choice may substantially bias estimates of genetic diversity and may invalidate comparisons between populations or species, most notably when different microsatellite loci are assessed [10].
A different pattern emerges for estimates of population genetic differentiation in terms of pairwise F ST. We found a significant positive correlation between values of pairwise F ST derived from microsatellites and genome-wide SNPs (Fig. 4). Similar findings were reported for salmons and threespine sticklebacks [36, 37, 46]. A reason for the better correlation between estimates of genetic differentiation compared to estimates of genetic diversity may be that more values are involved in pairwise comparisons, and that differences in allele frequencies of the common alleles are more important for the accurate estimation of genetic differentiation than those of rare alleles. Importantly, estimates of F ST derived from microsatellites were consistently and substantially higher than those based on genome-wide SNPs. This seems counterintuitive, because multi-allelic microsatellite markers with high mutation rates (and thus high genetic diversity) should cause lower F ST values than low-diversity markers like SNPs [89]. However, pooled whole-genome re-sequencing studies with high coverage, such as this one, also detect rare variants; these low-frequency SNPs reduce overall F ST [90]. Overall, we consider pairwise population genetic differentiation estimated from microsatellites a useful proxy for genome-wide differentiation, but only in relative and not in absolute terms (Fig. 4b). This finding has serious implications, because absolute values of F ST continue to be frequently used to infer indirect estimates of gene flow and migration, even though estimates of gene-flow should not be derived from F ST [91]. Further, this marker-specific difference in F ST estimates has a major impact on comparative studies of the divergence of quantitative traits, known as Q ST–F ST comparisons, because the inference of the role of natural selection and genetic drift as causes of population genetic differentiation in complex polygenic traits is biased [92].
While our results suggest that microsatellites should not be used for estimating genome-wide heterozygosity, we emphasize that microsatellites remain useful molecular markers for other applications. For example, microsatellites perform very well in genetic stock identification or paternity analysis owing to their high variability [15, 93–96] and may therefore continue to play an important role in molecular ecology. However, before embarking on a molecular analysis, it remains a key issue to carefully assess the inherent strengths and limitations associated with different molecular markers [8]. Only then it is possible to select the most appropriate method for a given ecological or evolutionary question [46].
In contrast to microsatellite-derived data, estimates of genome-wide diversity inferred from whole-genome re-sequencing data, e.g. exome-wide θ Watterson, intronic and intergenic θ Watterson, or genome-wide SNP-H e, were highly correlated with each other and led to the same ranking of populations (Fig. 3; Additional file 9: Figure S5). Even though values of θ Watterson were either derived exclusively from coding regions or intronic and intergenic regions, and SNP-H e was calculated from positions across the whole genome, their estimates were highly congruent. The slight variation observed among θ Watterson and SNP-H e (Fig. 3) might be explained by differences in the demographic history among populations (Tajima’s D in Table 1), because the demographic history of a population has a stronger influence on θ Watterson (the number of segregating sites) than on SNP-H e or SNP nucleotide diversity estimates [49], as rare alleles are more likely to be lost during a bottleneck than common ones. Further, we found in our whole-genome re-sequencing study that the confounding effects of genetic linkage are negligible in the highly outcrossing and self-incompatible A. halleri, most likely because the small-scale linkage effects are compensated by the large numbers of unlinked SNPs. Thus, our SNP-H e estimates based on a subset of putatively unlinked SNPs were nearly identical to the estimates inferred for all SNPs, see Fig. 2a and Additional file 8: Figure S4 . Similar to our genome re-sequencing study, approaches that use reduced representation libraries (e.g. RADseq) to sample a subset of genome-wide SNPs can accurately estimate genome-wide heterozygosity. As a consequence of the much smaller proportion of the genome surveyed with such approaches, however, care should be taken to avoid confounding effects of linkage, for example by considering only one SNP per RAD-locus [97]. For example, the inbreeding coefficient of a known pedigree in oldfield mice showed strong concordance with the inferred estimates of heterozygosity obtained from 13,198 RADseq SNPs [9]. This result indicates that, as long as a sufficiently large number of unbiased NGS-based SNPs is analysed across the genome, SNP estimates accurately reflect genome-wide diversity in natural populations. Therefore, approaches like RADseq [98], Pool-Seq [20] and whole-genome re-sequencing at low coverage [99, 100] are more appropriate than array-based SNP approaches, which may be affected by strong ascertainment bias [17, 101].
An important question to consider in many studies may be the number of SNPs that are needed to estimate genetic diversity. Our down-sampling approach indicated that the number of random SNPs that are required to resolve genetic diversity difference among populations range from 1000 (confidence intervals ± 0.01 SNP-H e) to 93,000 SNPs (±0.001 SNP-H e). This number is in the range of SNPs that can be inferred with standard RADseq protocols also in non-model organisms [98]. However, to differentiate among populations with very similar levels of genetic diversity, we required approximately 300,000 SNPs (Fig. 5). Thus, large SNP datasets that are ideally identified de novo through NGS approaches (to prevent ascertainment bias) are highly suitable to distinguish, for example, between populations differing in genetic diversity and may therefore support decision-making in conservation management. A further advantage of genome-wide SNP data is that they not only allow one to estimate neutral genetic diversity, but also to identify adaptive genetic variation (e.g. [50, 102–104]), which is considered essential for delimitating conservation units (CUs; [5, 32, 105]). The large technical advances in nucleotide sequencing technology in recent years have not only massively increased the number of nucleotides that can be sequenced per individual or population, but have also led to reduced costs per nucleotide to the extent that screening a handful of microsatellite markers may be as expensive as surveying thousands of SNPs using latest NGS-based genotyping technologies (e.g. [19, 98]).
Conclusion
This case study in the perennial and outcrossing plant A. halleri reveals that genetic diversity estimated from microsatellite markers, notably expected heterozygosity, may not adequately reflect genome-wide genetic diversity estimated from single-nucleotide polymorphisms and may therefore be a poor proxy for genome-wide estimates of genetic diversity. Possible causes include the limited number of microsatellite markers used, marker ascertainment bias, as well as the high variance in microsatellite-derived diversity estimates. Interestingly, microsatellite allelic richness (A r) was found to be a reasonable proxy for genome-wide diversity, but the absolute ranking of populations was still inconsistent. Estimates of genetic differentiation (F ST) among populations derived from microsatellites were consistently higher than SNP-based estimates but were significantly correlated with the latter.
Our results do not question the usefulness of microsatellites per se, but point to research questions for which NGS-derived SNPs may be better suited than microsatellite markers, given the availability of robust and cost-effective SNP genotyping approaches based on high-throughput sequencing. As a consequence, we recommend using genome-wide analyses of SNP diversity when the inference and comparison of genetic diversity within and among populations and species is the goal of a study. A few thousand NGS-derived SNPs are sufficient for this purpose and this number of unbiased SNPs can nowadays easily been obtained also for non-model species, for example by using a reduced representation sequencing approach such as RADseq [98].
Acknowledgments
We thank C. Beisel for supporting our Illumina sequencing efforts at the Quantitative Genomics Facility, D-BSSE, ETH Zürich. We also thank A. Bösch, E. Schnyder, J. Zimmermann, D. Zulliger, N. Quebre, and N. Aellen for sampling A. halleri populations, and C. Michel, R. Graf, and the Genetic Diversity Centre Zürich (GDC) for experimental support, A. Tedder for helpful input, as well as S. Fior and N. Zemp for bioinformatics support. We acknowledge the valuable comments made by anonymous reviewers on an earlier version of this article.
Funding
This study was funded by the Swiss National Science Foundation (project CRSI33_127155) and the Adaptation to a Changing Environment (ACE) Center of ETH Zurich.
Availability of data and materials
The datasets supporting the conclusions of this article are available in the DRYAD digital repository, (http://dx.doi.org/10.5061/dryad.11jp5; http://datadryad.org), in the European Nucleotide Archive (ENA; PRJEB18647; www.ebi.ac.uk/ena) and in the Sequence Read Archive (SRA; SRP029378; http://www.ncbi.nlm.nih.gov/sra).
Authors’ contributions
MCF, CR, and AW designed the study; FG, MCF, and AW conducted field-work; MCF, CR, ML, and MR performed the research and analyzed the data; MCF, CR, and AW wrote the manuscript with substantial contributions from ML, MR, FG, and RH. AW, KKS and RH conceived the project and obtained funding. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- Ar
Microsatellite allelic richness
- He
Expected heterozygosity
- NGS
Next-generation sequencing
- Pool-Seq
Pooled next-generation sequencing
- RADseq
Restriction-site associated DNA sequencing
- SNP
Single nucleotide polymorphism
- SSR
Simple sequence repeat
- θWatterson
Watterson’s theta
Additional files
Sampling locations of the nine study populations of Arabidopsis halleri. (PDF 31 kb)
Characteristics of 20 microsatellite markers used for Arabidopsis halleri. Source refers to the species for which a microsatellite marker was originally developed. Sequences of forward and reverse primers are shown in 5′ - 3′ direction. The fluorescent dyes used in multiplex PCRs and allele size ranges in base pairs are given for each locus. (PDF 60 kb)
Additional Methods. (PDF 23 kb)
Locus-specific summary statistics for all 20 microsatellite markers genotyped in Arabidopsis halleri. Estimated frequency of null alleles, total number of alleles found among the 180 individuals, expected heterozygosity (SSR-H e), and inbreeding coefficient F IS with one-sided p-value (significant heterozygote deficiency indicated in bold) are given. (PDF 28 kb)
(A) Population specific allele frequency distributions in nine populations of A. halleri for 19 microsatellite markers (blue). (B) Minor allele frequency distributions in the same nine populations across 2,064,681 SNPs (green). Histograms are labelled with population codes (Additional file 1: Table S1). (PDF 1421 kb)
Box-whisker plot of expected heterozygosity calculated for each microsatellite marker (SSR-H e) genotyped in Arabidopsis halleri. For cross-species microsatellites (left, red), no significantly different estimates of H e (pairwise Wilcoxon signed-rank test) were observed after Bonferroni correction, as a consequence of the high variance. Without correction for multiple testing, only population Aha11 had a significantly different H e estimates from Aha31 and AhaN1 (indicated with different colouring and letters). For species-specific markers (right, green), no significant differences in H e estimates were observed after Bonferroni correction. Without multiple corrections, only Aha21 was significantly different from AhaN4. (PDF 225 kb)
Observed (solid red lines) versus expected (dotted black lines) distributions of Tajima’s D for nine populations of Arabidopsis halleri. The expected normal distribution consists of 22,210 values with an average of zero and the same standard deviation as the real dataset. p-values indicate whether there was a significant deviation of the average from zero using a t-test. (PDF 370 kb)
Relationships between expected SNP heterozygosity (SNP-H e) and expected microsatellite heterozygosity (SSR-H e) of Arabidopsis halleri estimated on the basis of 19 microsatellites and 41,294 genetically unlinked genome-wide single nucleotide polymorphisms (SNPs). For this analysis only every 50th SNP of the more than 2 million SNPs was used. The median distance among two neighbouring SNPs is 1600 bp on the A. thaliana reference genome. (PDF 198 kb)
Pearson’s correlation (dashed line) between exome-wide θ Watterson and θ Watterson calculated from introns and intergenic regions in Arabidopsis halleri. (PDF 191 kb)
Comparison of pairwise population genetic differentiation (F ST) among nine populations of Arabidopsis halleri based on 12 cross-species microsatellite markers and seven species-specific microsatellite markers. r MT represents the correlation coefficient of the Mantel test and p the significance of the correlation. (PDF 181 kb)
Contributor Information
Martin C. Fischer, Phone: +41 44 633 93 19, Email: martin.fischer@env.ethz.ch
Christian Rellstab, Email: christian.rellstab@wsl.ch.
Marianne Leuzinger, Email: marieleu@bluewin.ch.
Marie Roumet, Email: marie.roumet@env.ethz.ch.
Felix Gugerli, Email: felix.gugerli@wsl.ch.
Kentaro K. Shimizu, Email: kentaro.shimizu@ieu.uzh.ch
Rolf Holderegger, Email: rolf.holderegger@wsl.ch.
Alex Widmer, Email: alex.widmer@env.ethz.ch.
References
- 1.Reed DH, Frankham R. Correlation between fitness and genetic diversity. Conserv Biol. 2003;17:230–237. doi: 10.1046/j.1523-1739.2003.01236.x. [DOI] [Google Scholar]
- 2.Agudo R, Carrete M, Alcaide M, Rico C, Hiraldo F, Donázar JA. Genetic diversity at neutral and adaptive loci determines individual fitness in a long-lived territorial bird. Proc R Soc Lond B. 2012;279:3241–3249. doi: 10.1098/rspb.2011.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Selkoe KA, Toonen RJ. Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecol Lett. 2006;9:615–629. doi: 10.1111/j.1461-0248.2006.00889.x. [DOI] [PubMed] [Google Scholar]
- 4.Tautz D, Renz M. Simple sequences are ubiquitous repetitive components of eukaryotic genomes. Nucleic Acids Res. 1984;12:4127–4138. doi: 10.1093/nar/12.10.4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Funk WC, McKay JK, Hohenlohe PA, Allendorf FW. Harnessing genomics for delineating conservation units. Trends Ecol Evol. 2012;27:489–496. doi: 10.1016/j.tree.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ouborg NJ, Pertoldi C, Loeschcke V, Bijlsma R, Hedrick PW. Conservation genetics in transition to conservation genomics. Trends Genet. 2010;26:177–187. doi: 10.1016/j.tig.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Guichoux E, Lagache L, Wagner S, Chaumeil P, LÉGer P, Lepais O, Lepoittevin C, Malausa T, Revardel E, Salin F, et al. Current trends in microsatellite genotyping. Mol Ecol Resour. 2011;11:591–611. doi: 10.1111/j.1755-0998.2011.03014.x. [DOI] [PubMed] [Google Scholar]
- 8.Putman AI, Carbone I. Challenges in analysis and interpretation of microsatellite data for population genetic studies. Ecol Evol. 2014;4:4399–4428. doi: 10.1002/ece3.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoffman JI, Simpson F, David P, Rijks JM, Kuiken T, Thorne MAS, Lacy RC, Dasmahapatra KK. High-throughput sequencing reveals inbreeding depression in a natural population. Proc Natl Acad Sci USA. 2014;111(10):3775–80. [DOI] [PMC free article] [PubMed]
- 10.Väli U, Einarsson A, Waits L, Ellegren H. To what extent do microsatellite markers reflect genome-wide genetic diversity in natural populations? Mol Ecol. 2008;17:3808–3817. doi: 10.1111/j.1365-294X.2008.03876.x. [DOI] [PubMed] [Google Scholar]
- 11.Glover K, Hansen M, Lien S, Als T, Hoyheim B, Skaala O. A comparison of SNP and STR loci for delineating population structure and performing individual genetic assignment. BMC Genet. 2010;11:2. doi: 10.1186/1471-2156-11-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gärke C, Ytournel F, Bed’hom B, Gut I, Lathrop M, Weigend S, Simianer H. Comparison of SNPs and microsatellites for assessing the genetic structure of chicken populations. Anim Genet. 2012;43:419–428. doi: 10.1111/j.1365-2052.2011.02284.x. [DOI] [PubMed] [Google Scholar]
- 13.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–7. [DOI] [PMC free article] [PubMed]
- 14.Helyar SJ, Hemmer-Hansen J, Bekkevold D, Taylor MI, Ogden R, Limborg MT, Cariani A, Maes GE, Diopere E, Carvalho GR, et al. Application of SNPs for population genetics of nonmodel organisms: new opportunities and challenges. Mol Ecol Resour. 2011;11:123–136. doi: 10.1111/j.1755-0998.2010.02943.x. [DOI] [PubMed] [Google Scholar]
- 15.Haasl RJ, Payseur BA. Multi-locus inference of population structure: a comparison between single nucleotide polymorphisms and microsatellites. Heredity. 2011;106:158–171. doi: 10.1038/hdy.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu N, Chen L, Wang S, Oh C, Zhao H. Comparison of single-nucleotide polymorphisms and microsatellites in inference of population structure. BMC Genet. 2005;6 Suppl 1:S26. doi: 10.1186/1471-2156-6-S1-S26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morin PA, Luikart G, Wayne RK, the SNPwg SNPs in ecology, evolution and conservation. Trends Ecol Evol. 2004;19:208–216. doi: 10.1016/j.tree.2004.01.009. [DOI] [Google Scholar]
- 18.Metzker ML. Sequencing technologies — the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 19.Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet. 2011;12:499–510. doi: 10.1038/nrg3012. [DOI] [PubMed] [Google Scholar]
- 20.Schlötterer C, Tobler R, Kofler R, Nolte V. Sequencing pools of individuals - mining genome-wide polymorphism data without big funding. Nat Rev Genet. 2014;15:749–763. doi: 10.1038/nrg3803. [DOI] [PubMed] [Google Scholar]
- 21.Ellegren H. Microsatellites: simple sequences with complex evolution. Nat Rev Genet. 2004;5:435–445. doi: 10.1038/nrg1348. [DOI] [PubMed] [Google Scholar]
- 22.Kelkar YD, Strubczewski N, Hile SE, Chiaromonte F, Eckert KA, Makova KD. What is a microsatellite: a computational and experimental definition based upon repeat mutational behavior at A/T and GT/AC repeats. Genome Biol Evol. 2010;2:620–635. doi: 10.1093/gbe/evq046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ellegren H. Microsatellite mutations in the germline: implications for evolutionary inference. Trends Genet. 2000;16:551–558. doi: 10.1016/S0168-9525(00)02139-9. [DOI] [PubMed] [Google Scholar]
- 24.Bhargava A, Fuentes FF. Mutational dynamics of microsatellites. Mol Biotechnol. 2010;44:250–266. doi: 10.1007/s12033-009-9230-4. [DOI] [PubMed] [Google Scholar]
- 25.Leffler EM, Bullaughey K, Matute DR, Meyer WK, Ségurel L, Venkat A, Andolfatto P, Przeworski M. Revisiting an old riddle: what determines genetic diversity levels within species? PLoS Biol. 2012;10:e1001388. doi: 10.1371/journal.pbio.1001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ossowski S, Schneeberger K, Lucas-Lledó JI, Warthmann N, Clark RM, Shaw RG, Weigel D, Lynch M. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science. 2010;327:92–94. doi: 10.1126/science.1180677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Queirós J, Godinho R, Lopes S, Gortazar C, de la Fuente J, Alves PC. Effect of microsatellite selection on individual and population genetic inferences: an empirical study using cross-specific and species-specific amplifications. Mol Ecol Resour. 2015;15:747–760. doi: 10.1111/1755-0998.12349. [DOI] [PubMed] [Google Scholar]
- 28.Chapuis M-P, Estoup A. Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol. 2007;24:621–631. doi: 10.1093/molbev/msl191. [DOI] [PubMed] [Google Scholar]
- 29.Estoup A, Jarne P, Cornuet J-M. Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Mol Ecol. 2002;11:1591–1604. doi: 10.1046/j.1365-294X.2002.01576.x. [DOI] [PubMed] [Google Scholar]
- 30.Balloux F, Lugon-Moulin N. The estimation of population differentiation with microsatellite markers. Mol Ecol. 2002;11:155–165. doi: 10.1046/j.0962-1083.2001.01436.x. [DOI] [PubMed] [Google Scholar]
- 31.Willi Y, Van Buskirk J, Hoffmann AA. Limits to the adaptive potential of small populations. Annu Rev Ecol Evol Syst. 2006;37:433–458. doi: 10.1146/annurev.ecolsys.37.091305.110145. [DOI] [Google Scholar]
- 32.Allendorf FW, Hohenlohe PA, Luikart G. Genomics and the future of conservation genetics. Nat Rev Genet. 2010;11:697–709. doi: 10.1038/nrg2844. [DOI] [PubMed] [Google Scholar]
- 33.Johnson WE, Onorato DP, Roelke ME, Land ED, Cunningham M, Belden RC, McBride R, Jansen D, Lotz M, Shindle D, et al. Genetic restoration of the Florida panther. Science. 2010;329:1641–1645. doi: 10.1126/science.1192891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pimm SL, Dollar L, Bass OL. The genetic rescue of the Florida panther. Anim Conserv. 2006;9:115–122. doi: 10.1111/j.1469-1795.2005.00010.x. [DOI] [Google Scholar]
- 35.Brandt AL, Hagos Y, Yacob Y, David VA, Georgiadis NJ, Shoshani J, Roca AL. The elephants of Gash-Barka, Eritrea: nuclear and mitochondrial genetic patterns. J Hered. 2014;105:82–90. doi: 10.1093/jhered/est078. [DOI] [PubMed] [Google Scholar]
- 36.Ryynänen HJ, Tonteri A, Vasemägi A, Primmer CR. A comparison of biallelic markers and microsatellites for the estimation of population and conservation genetic parameters in Atlantic salmon (Salmo salar) J Hered. 2007;98:692–704. doi: 10.1093/jhered/esm093. [DOI] [PubMed] [Google Scholar]
- 37.Ozerov M, Vasemägi A, Wennevik V, Diaz-Fernandez R, Kent M, Gilbey J, Prusov S, Niemelä E, Vähä J-P. Finding markers that make a difference: DNA pooling and SNP-arrays identify population informative markers for genetic stock identification. PLoS One. 2013;8:e82434. doi: 10.1371/journal.pone.0082434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ciani E, Cecchi F, Castellana E, D’Andrea M, Incoronato C, D’Angelo F, Albenzio M, Pilla F, Matassino D, Cianci D, et al. Poorer resolution of low-density SNP vs. STR markers in reconstructing genetic relationships among seven Italian sheep breeds. Large Anim Rev. 2013;19:236–241. [Google Scholar]
- 39.Singh N, Choudhury DR, Singh AK, Kumar S, Srinivasan K, Tyagi RK, Singh NK, Singh R. Comparison of SSR and SNP markers in estimation of genetic diversity and population structure of Indian rice varieties. PLoS One. 2013;8:e84136. doi: 10.1371/journal.pone.0084136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fernández ME, Goszczynski DE, Lirón JP, Villegas-Castagnasso EE, Carino MH, Ripoli MV, Rogberg-Muñoz A, Posik DM, Peral-García P, Giovambattista G. Comparison of the effectiveness of microsatellites and SNP panels for genetic identification, traceability and assessment of parentage in an inbred Angus herd. Genet Mol Biol. 2013;36:185–191. doi: 10.1590/S1415-47572013000200008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herráeza DL, Schäfer H, Mosner J, Fries H-R, Wink M. Comparison of microsatellite and single nucleotide polymorphism markers for the genetic analysis of a Galloway cattle population. Z Naturforsch C. 2005;60:637–643. doi: 10.1515/znc-2005-7-821. [DOI] [PubMed] [Google Scholar]
- 42.Coates BS, Sumerford DV, Miller NJ, Kim KS, Sappington TW, Siegfried BD, Lewis LC. Comparative performance of single nucleotide polymorphism and microsatellite markers for population genetic analysis. J Hered. 2009;100:556–564. doi: 10.1093/jhered/esp028. [DOI] [PubMed] [Google Scholar]
- 43.Livingstone D, III, Motamayor J, Schnell R, Cariaga K, Freeman B, Meerow A, Brown JS, Kuhn D. Development of single nucleotide polymorphism markers in Theobroma cacao and comparison to simple sequence repeat markers for genotyping of Cameroon clones. Mol Breeding. 2011;27:93–106. doi: 10.1007/s11032-010-9416-2. [DOI] [Google Scholar]
- 44.Granevitze Z, David L, Twito T, Weigend S, Feldman M, Hillel J. Phylogenetic resolution power of microsatellites and various single-nucleotide polymorphism types assessed in 10 divergent chicken populations. Anim Genet. 2014;45:87–95. doi: 10.1111/age.12088. [DOI] [PubMed] [Google Scholar]
- 45.Ross CT, Weise JA, Bonnar S, Nolin D, Satkoski Trask J, Smith DG, Ferguson B, Ha J, Kubisch HM, Vinson A, et al. An empirical comparison of short tandem repeats (STRs) and single nucleotide polymorphisms (SNPs) for relatedness estimation in Chinese rhesus macaques (Macaca mulatta) Am J Primatol. 2014;76:313–324. doi: 10.1002/ajp.22235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DeFaveri J, Viitaniemi H, Leder E, Merilä J. Characterizing genic and nongenic molecular markers: comparison of microsatellites and SNPs. Mol Ecol Resour. 2013;13:377–392. doi: 10.1111/1755-0998.12071. [DOI] [PubMed] [Google Scholar]
- 47.Miller JM, Malenfant RM, David P, Davis CS, Poissant J, Hogg JT, Festa-Bianchet M, Coltman DW. Estimating genome-wide heterozygosity: effects of demographic history and marker type. Heredity. 2014;112:240–247. doi: 10.1038/hdy.2013.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lozier JD. Revisiting comparisons of genetic diversity in stable and declining species: assessing genome-wide polymorphism in North American bumble bees using RAD sequencing. Mol Ecol. 2014;23:788–801. doi: 10.1111/mec.12636. [DOI] [PubMed] [Google Scholar]
- 49.Vilas A, Pérez-Figueroa A, Quesada H, Caballero A. Allelic diversity for neutral markers retains a higher adaptive potential for quantitative traits than expected heterozygosity. Mol Ecol. 2015;24:4419–4432. doi: 10.1111/mec.13334. [DOI] [PubMed] [Google Scholar]
- 50.Fischer MC, Rellstab C, Tedder A, Zoller S, Gugerli F, Shimizu KK, Holderegger R, Widmer A. Population genomic footprints of selection and associations with climate in natural populations of Arabidopsis halleri from the Alps. Mol Ecol. 2013;22:5594–5607. doi: 10.1111/mec.12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rellstab C, Zoller S, Tedder A, Gugerli F, Fischer MC. Validation of SNP allele frequencies determined by pooled next-generation sequencing in natural populations of a non-model plant species. PLoS One. 2013;8:e80422. doi: 10.1371/journal.pone.0080422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Llaurens V, Castric V, Austerlitz F, Vekemans X. High paternal diversity in the self-incompatible herb Arabidopsis halleri despite clonal reproduction and spatially restricted pollen dispersal. Mol Ecol. 2008;17:1577–1588. doi: 10.1111/j.1365-294X.2007.03683.x. [DOI] [PubMed] [Google Scholar]
- 53.Pauwels M, Vekemans X, Godé C, Frérot H, Castric V, Saumitou-Laprade P. Nuclear and chloroplast DNA phylogeography reveals vicariance among European populations of the model species for the study of metal tolerance, Arabidopsis halleri (Brassicaceae) New Phytol. 2012;193:916–928. doi: 10.1111/j.1469-8137.2011.04003.x. [DOI] [PubMed] [Google Scholar]
- 54.Al-Shehbaz IA, O’Kane SL. Taxonomy and phylogeny of Arabidopsis (Brassicaceae) In: Somerville CR, Meyerowitz EM, editors. The Arabidopsis Book. Rockville: American Society of Plant Biologist; 2002. pp. 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clauss MJ, Koch M. Poorly known relatives of Arabidopsis thaliana. Trends Plant Sci. 2006;11:449–459. doi: 10.1016/j.tplants.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 56.Meyer C-L, Kostecka AA, Saumitou-Laprade P, Créach A, Castric V, Pauwels M, Frérot H. Variability of zinc tolerance among and within populations of the pseudometallophyte species Arabidopsis halleri and possible role of directional selection. New Phytol. 2010;185:130–142. doi: 10.1111/j.1469-8137.2009.03062.x. [DOI] [PubMed] [Google Scholar]
- 57.Johnston JS, Pepper AE, Hall AE, Chen ZJ, Hodnett G, Drabek J, Lopez R, Price HJ. Evolution of genome size in Brassicaceae. Ann Bot. 2005;95:229–235. doi: 10.1093/aob/mci016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hale ML, Burg TM, Steeves TE. Sampling for microsatellite-based population genetic studies: 25 to 30 individuals per population is enough to accurately estimate allele frequencies. PLoS One. 2012;7:e45170. doi: 10.1371/journal.pone.0045170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Van Rossum F, Bonnin I, Fenart S, Pauwels M, Petit D, Saumitou-Laprade P. Spatial genetic structure within a metallicolous population of Arabidopsis halleri, a clonal, self-incompatible and heavy-metal-tolerant species. Mol Ecol. 2004;13:2959–2967. doi: 10.1111/j.1365-294X.2004.02314.x. [DOI] [PubMed] [Google Scholar]
- 60.Bell CJ, Ecker JR. Assignment of 30 microsatellite loci to the linkage of Arabidopsis. Genomics. 1994;19:137–144. doi: 10.1006/geno.1994.1023. [DOI] [PubMed] [Google Scholar]
- 61.Clauss MJ, Cobban H, Mitchell-Olds T. Cross-species microsatellite markers for elucidating population genetic structure in Arabidopsis and Arabis (Brassicaeae) Mol Ecol. 2002;11:591–601. doi: 10.1046/j.0962-1083.2002.01465.x. [DOI] [PubMed] [Google Scholar]
- 62.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Godé C, Decombeix I, Kostecka A, Wasowicz P, Pauwels M, Courseaux A, Saumitou-Laprade P. Nuclear microsatellite loci for Arabidopsis halleri (Brassicaceae), a model species to study plant adaptation to heavy metals. Am J Bot. 2012;99:e49–e52. doi: 10.3732/ajb.1100320. [DOI] [PubMed] [Google Scholar]
- 64.Meirmans PG, Van Tienderen PH. GenoType and GenoDive: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes. 2004;4:792–794. doi: 10.1111/j.1471-8286.2004.00770.x. [DOI] [Google Scholar]
- 65.Raymond M, Rousset F. GENEPOP: population genetics software for exact tests and ecumenicism. J Hered. 1995;86:248–249. [Google Scholar]
- 66.Rousset F. genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour. 2008;8:103–106. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- 67.GENETIX 4.05, logiciel sur Windows™ pour la génétique des populations. http://kimura.univ-montp2.fr/genetix/.
- 68.R Core Team . R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2015. [Google Scholar]
- 69.Ciosi M, Miller NJ, Kim KS, Giordano R, Estoup A, Guillemaud T. Invasion of Europe by the western corn rootworm, Diabrotica virgifera virgifera: multiple transatlantic introductions with various reductions of genetic diversity. Mol Ecol. 2008;17:3614–3627. doi: 10.1111/j.1365-294X.2008.03866.x. [DOI] [PubMed] [Google Scholar]
- 70.Gautier M, Foucaud J, Gharbi K, Cézard T, Galan M, Loiseau A, Thomson M, Pudlo P, Kerdelhué C, Estoup A. Estimation of population allele frequencies from next-generation sequencing data: pool-versus individual-based genotyping. Mol Ecol. 2013;22:3766–3779. doi: 10.1111/mec.12360. [DOI] [PubMed] [Google Scholar]
- 71.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17:10. [Google Scholar]
- 72.Lamesch P, Berardini TZ, Li D, Swarbreck D, Wilks C, Sasidharan R, Muller R, Dreher K, Alexander DL, Garcia-Hernandez M, et al. The Arabidopsis information resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 2012;40:D1202–D1210. doi: 10.1093/nar/gkr1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kaul S, Koo HL, Jenkins J, Rizzo M, Rooney T, Tallon LJ, Feldblyum T, Nierman W, Benito MI, Lin XY, et al. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- 74.TAIR; The Arabidopsis Information Resource. http://www.arabidopsis.org/.
- 75.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Subgroup GPDP The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kofler R, Orozco-terWengel P, De Maio N, Pandey RV, Nolte V, Futschik A, Kosiol C, Schlötterer C. PoPoolation: a toolbox for population genetic analysis of next generation sequencing data from pooled individuals. PLoS One. 2011;6:e15925. doi: 10.1371/journal.pone.0015925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kofler R, Pandey RV, Schlötterer C. PoPoolation2: identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq) Bioinformatics. 2011;27:3435–3436. doi: 10.1093/bioinformatics/btr589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Achaz G. Testing for neutrality in samples with sequencing errors. Genetics. 2008;179:1409–1424. doi: 10.1534/genetics.107.082198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Biswas S, Akey JM. Genomic insights into positive selection. Trends Genet. 2006;22:437–446. doi: 10.1016/j.tig.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 82.Hartl DL, Clark AG. Principles of population genetics. Sutherland: Sinauer; 2007. [Google Scholar]
- 83.Goslee SC, Urban DL. The ecodist package for dissimilarity-based analysis of ecological data. J Stat Softw. 2007;22:1–19. doi: 10.18637/jss.v022.i07. [DOI] [Google Scholar]
- 84.Ljungqvist M, ÅKesson M, Hansson B. Do microsatellites reflect genome-wide genetic diversity in natural populations? A comment on Väli et al. (2008) Mol Ecol. 2010;19:851–855. doi: 10.1111/j.1365-294X.2010.04522.x. [DOI] [PubMed] [Google Scholar]
- 85.Wang J. Does GST, underestimate genetic differentiation from marker data? Mol Ecol. 2015;24:3546–3558. doi: 10.1111/mec.13204. [DOI] [PubMed] [Google Scholar]
- 86.Jost L. G(ST) and its relatives do not measure differentiation. Mol Ecol. 2008;17:4015–4026. doi: 10.1111/j.1365-294X.2008.03887.x. [DOI] [PubMed] [Google Scholar]
- 87.Hoban S, Arntzen JA, Bruford MW, Godoy JA, Rus Hoelzel A, Segelbacher G, Vilà C, Bertorelle G. Comparative evaluation of potential indicators and temporal sampling protocols for monitoring genetic erosion. Evol Appl. 2014;7:984–998. doi: 10.1111/eva.12197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mittell EA, Nakagawa S, Hadfield JD. Are molecular markers useful predictors of adaptive potential? Ecol Lett. 2015;18:772–778. doi: 10.1111/ele.12454. [DOI] [PubMed] [Google Scholar]
- 89.Whitlock MC. G’ST and D do not replace FST. Mol Ecol. 2011;20:1083–1091. doi: 10.1111/j.1365-294X.2010.04996.x. [DOI] [PubMed] [Google Scholar]
- 90.Roesti M, Salzburger W, Berner D. Uninformative polymorphisms bias genome scans for signatures of selection. BMC Evol Biol. 2012;12:94. doi: 10.1186/1471-2148-12-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Whitlock MC, McCauley DE. Indirect measures of gene flow and migration: Fst ≠ 1/(4 Nm + 1) Heredity. 1999;82:117–125. doi: 10.1038/sj.hdy.6884960. [DOI] [PubMed] [Google Scholar]
- 92.Leinonen T, McCairns RJS, O’Hara RB, Merila J. QST-FST comparisons: evolutionary and ecological insights from genomic heterogeneity. Nat Rev Genet. 2013;14:179–190. doi: 10.1038/nrg3395. [DOI] [PubMed] [Google Scholar]
- 93.Hess JE, Matala AP, Narum SR. Comparison of SNPs and microsatellites for fine-scale application of genetic stock identification of Chinook salmon in the Columbia River Basin. Mol Ecol Resour. 2011;11:137–149. doi: 10.1111/j.1755-0998.2010.02958.x. [DOI] [PubMed] [Google Scholar]
- 94.Jennings TN, Knaus BJ, Mullins TD, Haig SM, Cronn RC. Multiplexed microsatellite recovery using massively parallel sequencing. Mol Ecol Resour. 2011;11:1060–1067. doi: 10.1111/j.1755-0998.2011.03033.x. [DOI] [PubMed] [Google Scholar]
- 95.Hamblin MT, Warburton ML, Buckler ES. Empirical comparison of simple sequence repeats and single nucleotide polymorphisms in assessment of maize diversity and relatedness. PLoS One. 2007;2:e1367. doi: 10.1371/journal.pone.0001367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Weinman LR, Solomon JW, Rubenstein DR. A comparison of single nucleotide polymorphism and microsatellite markers for analysis of parentage and kinship in a cooperatively breeding bird. Mol Ecol Resour. 2015;15:502–511. doi: 10.1111/1755-0998.12330. [DOI] [PubMed] [Google Scholar]
- 97.Lee T-H, Guo H, Wang X, Kim C, Paterson AH. SNPhylo: a pipeline to construct a phylogenetic tree from huge SNP data. BMC Genomics. 2014;15:1–6. doi: 10.1186/1471-2164-15-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Andrews KR, Good JM, Miller MR, Luikart G, Hohenlohe PA. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat Rev Genet. 2016;17:81–92. doi: 10.1038/nrg.2015.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Korneliussen TS, Albrechtsen A, Nielsen R. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics. 2014;15:1–13. doi: 10.1186/s12859-014-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Foote AD, Vijay N, Avila-Arcos MC, Baird RW, Durban JW, Fumagalli M, Gibbs RA, Hanson MB, Korneliussen TS, Martin MD, et al. Genome-culture coevolution promotes rapid divergence of killer whale ecotypes. Nat Commun. 2016;7:11693. doi: 10.1038/ncomms11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. 2011;12:363–376. doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Holderegger R, Kamm U, Gugerli F. Adaptive vs. neutral genetic diversity: implications for landscape genetics. Landscape Ecol. 2006;21:797–807. doi: 10.1007/s10980-005-5245-9. [DOI] [Google Scholar]
- 103.Turner TL, Bourne EC, Von Wettberg EJ, Hu TT, Nuzhdin SV. Population resequencing reveals local adaptation of Arabidopsis lyrata to serpentine soils. Nat Genet. 2010;42:260–263. doi: 10.1038/ng.515. [DOI] [PubMed] [Google Scholar]
- 104.Rellstab C, Gugerli F, Eckert AJ, Hancock AM, Holderegger R. A practical guide to environmental association analysis in landscape genomics. Mol Ecol. 2015;24:4348–4370. doi: 10.1111/mec.13322. [DOI] [PubMed] [Google Scholar]
- 105.Bonin A, Nicole F, Pompanon F, Miaud C, Taberlet P. Population adaptive index: a new method to help measure intraspecific genetic diversity and prioritize populations for conservation. Conserv Biol. 2007;21:697–708. doi: 10.1111/j.1523-1739.2007.00685.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the conclusions of this article are available in the DRYAD digital repository, (http://dx.doi.org/10.5061/dryad.11jp5; http://datadryad.org), in the European Nucleotide Archive (ENA; PRJEB18647; www.ebi.ac.uk/ena) and in the Sequence Read Archive (SRA; SRP029378; http://www.ncbi.nlm.nih.gov/sra).