

Abstract
Although core mechanisms and machinery of premRNA splicing are conserved from yeast to human, the details of intron recognition often differ, even between closely related organisms. For example, genes from the pufferfish Fugu rubripes generally contain one or more introns that are not properly spliced in mouse cells. Exploiting available genome sequence data, a battery of sequence analysis techniques was used to reach several conclusions about the organization and evolution of splicing regulatory elements in vertebrate genes. The classical splice site and putative branch site signals are completely conserved across the vertebrates studied (human, mouse, pufferfish, and zebrafish), and exonic splicing enhancers also appear broadly conserved in vertebrates. However, another class of splicing regulatory elements, the intronic splicing enhancers, appears to differ substantially between mammals and fish, with G triples (GGG) very abundant in mammalian introns but comparatively rare in fish. Conversely, short repeats of AC and GT are predicted to function as intronic splicing enhancers in fish but are not enriched in mammalian introns. Consistent with this pattern, exonic splicing enhancer-binding SR proteins are highly conserved across all vertebrates, whereas heterogeneous nuclear ribonucleoproteins, which bind many intronic sequences, vary in domain structure and even presence/absence between mammals and fish. Exploiting differences in intronic sequence composition, a statistical model was developed to predict the splicing phenotype of Fugu introns in mammalian systems and was used to engineer the spliceability of a Fugu intron in human cells by insertion of specific sequences, thereby rescuing splicing in human cells.
Keywords: Fugu, zebrafish, G triplets, exonic splicing enhancers, intronic splicing enhancers
The pufferfish, Fugu rubripes, with its 7-fold-smaller genome than human, has proven to be an excellent resource for comparative genomics (1). The Fugu genome also has great potential for applications in genetics. The compactness of Fugu genes makes them ideal candidates for use in transgenesis, with the advantage over cDNA-derived constructs that they would be capable of producing all of the isoforms of a particular gene under appropriate regulatory control. However, the potential for using Fugu genes as natural minigenes for the production of transgenic mice has not been realized because initial efforts to express Fugu transgenes in mouse cells have failed because of incorrect transcript processing by the murine splicing machinery (2, 3). However, the Fugu genes studied to date are spliced and translated correctly in zebrafish, a fish whose genome size and gene organization are more similar to mammals than to Fugu.
These somewhat surprising results imply that substantial differences exist between fish and mammalian systems in exon–intron sequences and/or splicing factors. The relatively low information contents of the classical splice site signals in higher eukaryotes argues that additional transcript features are likely to be involved in recognition and splicing of many, if not all introns (4). Exonic splicing enhancers (ESEs), intronic splicing enhancers (ISEs), and exonic or intronic splicing silencers enhance or repress the use of 5′ splice sites (5′ss) or 3′ splice sites (3′ss), depending on their site and mode of action (5–8). ESEs have been the subject of many studies, and most are known to be recognized by members of the serine–arginine-rich (SR) protein family (9, 10). SR proteins bind to ESEs through their RNA-binding domains and promote splicing by recruiting spliceosomal components through protein–protein interactions by means of their arginine–serine-rich (RS) domains (9–13). The trans factors that bind to intronic splicing regulatory elements have not been characterized as thoroughly, and both SR proteins and heterogeneous nuclear ribonucleoproteins (hnRNPs) have been implicated in interactions with intronic cis elements.
By using the human (14), mouse (15) and Fugu (16) genome sequences, we applied and adapted the rescue approach for identification of splicing regulatory sequences (17) and developed methods to analyze similarities and differences in the sequences and organization of splicing regulatory elements in mammalian and fish genes. These methods revealed significant differences in predicted ISEs between mammalian and fish introns that appear to explain why certain Fugu introns are not faithfully processed by the mammalian splicing machinery.
Materials and Methods
Frequency Difference (FD) Plots. The difference between the observed frequency of a pattern (enumerated as in Table 2, which is published as supporting information on the PNAS web site) occurring in 10-bp windows (for exons of >60 bp) or 30-bp windows (for intronic regions) and the mean frequency of the same pattern in 10 random permutations (shuffles) of the sequence in the window were determined as follows, with an offset of 3 bp between successive windows. The observed frequency of a pattern of length m bp in a window of size W bp at position j in sequence i is defined as fobserved,i,j = xi,j/(W/m), where xi,j is the number of nonoverlapping occurrences of the motif whose first positions fall within the window (i.e., excluding occurrences that overlap previously counted occurrences). The average shuffled frequency of the motif of s total shuffles of the same window is defined as 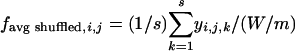 , where yi,j,k is the number of nonoverlapping occurrences of the motif in the kth shuffled version of the same window of size W bp, at position j in sequence i. Therefore, the FD of the motif at position j in sequence i is defined as FDi,j = fobserved,i,j - favg shuffled,i,j. The mean FD value μj and variance
, where yi,j,k is the number of nonoverlapping occurrences of the motif in the kth shuffled version of the same window of size W bp, at position j in sequence i. Therefore, the FD of the motif at position j in sequence i is defined as FDi,j = fobserved,i,j - favg shuffled,i,j. The mean FD value μj and variance 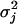 in a window of size W bp starting at position j over N sequences are calculated as
in a window of size W bp starting at position j over N sequences are calculated as 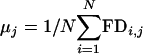 and the SEM, ε, is derived as
and the SEM, ε, is derived as 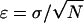 , where
, where 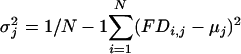 .
.
Linear Discriminant Analysis (LDA) and Intron Classification. Linear discriminant functions g1 and g2 for n1 Fugu introns and n2 mouse introns, respectively, were defined as 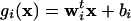 , where wi = Σ-1 μi and
, where wi = Σ-1 μi and 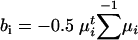 , and x is the vector of overlapping 3-mer counts computed from +5 to +65 and from -71 to -11 of the intron. Σ is the pooled covariance matrix from the individual covariance matrices: Σ = ((n1 - 1)Σ1 + (n2 - 1) Σ2))/(n1 + n2 -2). The LDA output (18), y, is defined as y(x) = g1(x) - g2(x). The intron length score, slen, was defined as slen(l) = log(fFugu(l)/fmouse(l)), where l is the length of the intron, and fFugu and fmouse are the estimated frequencies of introns falling into the relevant intron length bins in the respective organisms (Fig. 6, which is published as supporting information on the PNAS web site). Scores were generated which combine the intron length scores and the LDA outputs for Fugu and mouse introns in the following way: z(x, l) = y(x) + slen(l), where x represents a 128-long vector of 3-mer counts from an intron, and l is the intron length.
, and x is the vector of overlapping 3-mer counts computed from +5 to +65 and from -71 to -11 of the intron. Σ is the pooled covariance matrix from the individual covariance matrices: Σ = ((n1 - 1)Σ1 + (n2 - 1) Σ2))/(n1 + n2 -2). The LDA output (18), y, is defined as y(x) = g1(x) - g2(x). The intron length score, slen, was defined as slen(l) = log(fFugu(l)/fmouse(l)), where l is the length of the intron, and fFugu and fmouse are the estimated frequencies of introns falling into the relevant intron length bins in the respective organisms (Fig. 6, which is published as supporting information on the PNAS web site). Scores were generated which combine the intron length scores and the LDA outputs for Fugu and mouse introns in the following way: z(x, l) = y(x) + slen(l), where x represents a 128-long vector of 3-mer counts from an intron, and l is the intron length.
Results
Splice Site Signals and Predicted ESEs Are Conserved in Vertebrates. To identify potential splicing differences between different vertebrate organisms, three major classes of cis-acting elements were systematically analyzed: the canonical splice site/branch site motifs and two classes of splicing enhancers. By using large datasets of annotated exon–intron structures, we found that the extended consensus sequences of the classical 5′ss and 3′ss sequence motifs are essentially the same in human, mouse, zebrafish, and Fugu (these data are shown in Fig. 7A, which is published as supporting information on the PNAS web site). Putative branch point sequences identified by using a motif-finding algorithm also appear similar in sequence and are positionally conserved in orthologous mouse, human, and Fugu introns, occurring most commonly 20–40 bp upstream of the 3′ss (Fig. 7B). These data suggest that neither the branch point motif nor the 5′ss or 3′ss differ significantly between fish and mammals in the features required for recognition by the splicing machinery and that the observed differences in splicing between these systems must lie elsewhere.
Both constitutive and alternative splicing events are often modulated by elements in exons known as ESEs. To assess potential differences in ESE sequences between organisms, we applied the rescue-ese approach that was used previously to identify ESEs in human genes (17) to large datasets of annotated mouse and Fugu genes [Table 3, which is published as supporting information on the PNAS web site; access to rescue-ese hexamers for each of these organisms are available at http://genes.mit.edu/burgelab/rescue-ese (19)]. Sets of candidate ESE sequences that satisfy the two rescue-ese criteria of significant enrichment in exons relative to introns and significant enrichment in exons with weak (nonconsensus) 5′ss or 3′ss sequences relative to exons with strong splice sites were identified. Previously, predicted human ESE hexamers were clustered into 10 groups on the basis of sequence similarity (17) and then aligned to produce 10 distinct ESE motifs (Fig. 1A). Comparing the candidate mouse and Fugu ESE hexamers with those identified in human exons, a great deal of overlap was observed, with many of the same hexamers identified independently in different organisms. For example, 90 of the 100 hexamers comprising the purine-rich human 5C3D class were also predicted as ESEs in mouse, and 54 of these 100 hexamers were predicted as ESEs in Fugu exons (Fig. 1A). Of the 10 clusters of human ESEs identified, only the smallest (cluster 5E) was not represented in mouse. Furthermore, 7 of the 10 human clusters were represented in Fugu, the exceptions being 3 of the most sparsely populated human ESE clusters. Thus, rescue-ese analysis supports the presence in all three vertebrates of all of the large classes of ESEs identified in humans.
Fig. 1.

Conservation of rescue-ese sequences and distribution in vertebrates. (A) rescue-ese (17) motifs and the number of predicted ESE hexamers in mouse and Fugu that overlap with rescue-ese hexamers in human and the distribution of human rescue-ese hexamers in sets of orthologous human, mouse, and Fugu exons. The symbols + or - refer to significant increasing or decreasing, respectively, of FD gradient toward the respective splice site. No gradient (computed similarly as described in Table 4) is represented by 0. *, Conservation only in human and mouse; otherwise sign of gradient was conserved in all three organisms. (B) As an example, the FD plots for hexamers of rescue-ese class 5C3D are shown as a function of distance from the 3′ss (Left) or 5′ss (Right) of orthologous exons in human, mouse, and Fugu. Each point represents the start of a 10-bp window. Values are plotted at 3-bp intervals. Black bars show SEM (see Materials and Methods).
To further explore potential ESE-related differences between organisms, we analyzed the FD plots of rescue-ese hexamers along exons from each of the three vertebrates in sliding windows of 10 bp in width. As shown for the 5C3D cluster (Fig. 1B), most clusters of rescue-ese hexamers exhibit a concave (“smiley”) distribution, with increased FD values in the vicinity of both the 5′ss and 3′ss. This distribution is likely to result from increased selection to conserve ESEs near splice sites, which would be consistent with previous studies showing that ESEs located closer to the 3′ss of exons have higher activity than those located more distally (20) and that ESE-disrupting single-nucleotide polymorphisms are under-represented in exons near splice sites (21). For the majority of ESE classes, the shapes of the FD plots were similar in human, mouse, and Fugu (Fig. 1A and Fig. 8, which is published as supporting information on the PNAS web site). The conservation of the splice site-biased distributions of many classes of predicted ESEs between human, mouse, and Fugu argues for their functional importance in all three vertebrates.
Predicted ISEs Differ Between Mammals and Fish. In addition to exon sequences, such as ESEs, intronic elements also commonly play a role in alternative and constitutive splicing (22). To identify putative ISEs in vertebrate introns, we developed an approach called rescue-ise (Supporting Text, which is published as supporting information on the PNAS web site). By following a similar rationale to that used in our previous rescue-ese method (17), rescue-ise predicts as ISEs hexamers that share two properties: significant enrichment in introns relative to exons and significant enrichment in introns with weak (nonconsensus) 5′ss or 3′ss relative to introns with strong splice sites. Applying this method to large datasets of human and mouse introns identified the triplet motif GGG and a C-rich motif, respectively, in both mammals (Fig. 2). The GGG and C-rich hexamer clusters together comprised 96% (127 hexamers) of rescue-ise-predicted ISE hexamers in introns downstream of human 5′ss and 89% (266 hexamers) of predicted ISE hexamers in introns upstream of human 3′ss. Similar clusters comprised comparably large proportions of rescue-ise hexamers in mouse; the few remaining hexamers did not cluster into motifs that were similar between human and mouse.
Fig. 2.

rescue-predicted mammalian and Fugu ISE motifs. GGG and C-rich motifs were predicted as ISEs in human and mouse introns at both splice sites. f5A–f5E are motifs enriched in Fugu introns near the 5′ss, and f3A-f3C are enriched near the 3′ss.
Curiously, when the rescue-ise approach was applied to datasets of Fugu introns, a very different set of ISE motifs was predicted (Fig. 2), including motifs containing repetitions of CA and GT dinucleotides, but no motifs similar to the GGG or C-rich elements identified in mammals. To further explore this difference, a more detailed analysis of the predicted ISE motifs was undertaken in mammalian and fish introns by using the sea-squirt Ciona as an outgroup. From analysis of FD plots (Fig. 3), two trends were clear: (i) for GGG, an established mammalian ISE (23), there were pronounced peaks in the FD distribution in the vicinity of the 5′ss and 3′ss in both human and mouse introns; and (ii) these peaks were much more dramatic in introns with weak (nonconsensus) 5′ss or 3′ss than they were in introns with strong splice sites (Fig. 3, red curves versus blue curves). These two features can be explained if the location of the peak reflects an optimal interaction distance between hypothetical splicing regulatory factors that bind to ISEs and components of the splicing machinery bound at the splice sites and if ISEs in weak splice site introns are under increased selection to ensure efficient and accurate splicing (22). We propose that these two features comprise a sequence signature that is characteristic for ISEs.
Fig. 3.

Enrichment of predicted ISEs in introns near weak splice sites. (A) FD of GGG downstream of strong 5′ss and weak 5′ss, relative to locally permuted sequence 30-bp windows, starting from intron position +11. (B) FD of GGG upstream of strong 3′ss and weak 3′ss, starting from intron position -41. (C)FDof ACAC downstream of strong 5′ss and weak 5′ss, starting from intron position +11. (D) FD of GTGT upstream of strong 3′ss and weak 3′ss, starting from intron position -41. Black bars show SEM (see Materials and Methods). Values are plotted at 6-bp intervals.
Consistent with the differences seen in terms of predicted ISE motifs, the FD plots for Fugu introns were substantially different from those for mammalian introns (Fig. 2). Specifically, GGG was not enriched at any distance relative to the 5′ or 3′ss of Fugu introns (all FD values near zero) and had a nearly flat distribution, consistent with the absence of function in splicing. Instead, the predicted Fugu ISE motifs ACAC and GTGT showed pronounced FD peaks near the 5′ and 3′ss of Fugu introns, respectively, which were comparable in magnitude with those seen for GGG in mammalian introns. Consistent with this pattern, the peaks were more dramatic in introns with weak 5′ss and 3′ss. By contrast, the distributions of ACAC and GTGT near the 5′ss and 3′ss of mammalian introns were essentially flat, with no discernable peaks and little difference between weak and strong introns. The introns of the nonvertebrate chordate Ciona intestinalis showed modest peaks of GGG near the 5′ss and 3′ss but no clear peaks for ACAC or GTGT, and the GGG peaks in Ciona were higher for strong splice site introns rather than for weak splice site introns.
Exon and Intron Definition Mechanisms May Differ Between Mammals and Fish. The “exon definition” model of splicing postulates that the exon is the primary unit initially recognized by the splicing machinery, typically involving a complex formed across the exon containing factors that recognize the 3′ss, one or more ESEs and the 5′ss of an exon (24). This mode of splicing appears to predominate in transcripts containing small or medium-sized exons flanked by long introns (25). On the other hand, in splicing by the “intron definition” model, the intron is the primary unit initially recognized by the splicing machinery, with formation of a complex of factors recognizing the 5′ss, ISE(s), and the 3′ss of an intron (24). This mode of splicing tends to predominate in transcripts containing short introns flanked by medium or large exons (25). To analyze the effects of flanking intron length on the distribution of putative ESEs and ISEs in vertebrates, introns were categorized by length as either short (<125 bp), intermediate (125–1,000 bp), or long (>1,000 bp) (see Fig. 9, which is published as supporting information on the PNAS web site, for intron length distributions).
In human and mouse, exons flanked by longer introns contained a significantly higher abundance of most classes of rescue-ese hexamers than those flanked by intermediate-length introns, which, in turn, generally contained more such ESEs than exons flanked by short introns (Table 4 and Fig. 10, which are published as supporting information on the PNAS web site). Furthermore, short mammalian introns had higher relative frequencies of the candidate ISEs GGG and CCC near their splice sites than intermediate or long introns (Fig. 11, which is published as supporting information on the PNAS web site). Surprisingly, the relationship between ESE density and intron length was different in Fugu genes. In Fugu, there was no tendency for exons flanked by long introns to have higher densities of rescue-ese hexamers; in fact, the opposite tendency was observed for several ESE classes (Table 4). Furthermore, predicted ISE motifs ACAC and GTGT were more highly enriched in intermediate and long introns than in short introns (Fig. 11). Our proposed model is summarized in Fig. 4.
Fig. 4.

Model of association between intron length and distribution of splicing regulatory elements in mammals (A) and Fugu (B). Green triangles represent the enrichment of rescue-predicted ESEs near the splice sites in human, mouse, and Fugu exons. Red triangles represent the enrichment of rescue-predicted ISEs near the splice sites in human, mouse, and Fugu introns. The height of the triangles illustrates the relative magnitude of enrichment of rescue-ese ESEs and rescue-ise ISEs. Intron sizes in base pairs are indicated above the introns.
Differing Conservation of SR Protein and hnRNP Genes Between Mammals and Fish. Conservation of cis-regulatory elements between organisms is expected to correlate with patterns of conservation of the corresponding trans factors. To explore these relationships with respect to splicing in vertebrates, lists of human splicing factors identified previously through proteomic analysis by Zhou et al. (26) were used to identify mouse and Fugu orthologs from the EnsMart database by using reciprocal best blast hits. Domains were then predicted by using the Pfam database (27), and the results are shown in Table 1 and Tables 5–8, which are published as supporting information on the PNAS web site. Core spliceosomal components, such as small nuclear RNAs and proteins of the U1 small nuclear ribonucleoprotein (snRNP), U2 snRNP, and U4/U5/U6 tri-snRNP, are highly conserved between mammals and fish (Table 5 and data not shown). Additionally, clear orthologs with identical domain organization could be found in mouse and Fugu for all human SR proteins (Table 6), nearly all of which are known to recognize ESEs, consistent with our analysis indicating that the major rescue-ese classes are conserved between human, mouse, and Fugu. However, greater variability was seen in the domain organization and even presence/absence of H-complex hnRNP proteins, many of which are known to bind ISEs or other intronic elements (Tables 7 and 8). For example, Fugu and zebrafish orthologs for hnRNP A2/B1 (28, 29) and hnRNP F were not identified, and fish orthologs for hnRNP H and hnRNP K were missing one or more RNA recognition motifs and/or K homology domains, compared with human and mouse orthologs. In addition, Fugu orthologs for hnRNP RALY were not found, and hnRNP I/polypyrimidine tract binding protein was missing an RNA recognition motif. Given that the Fugu and zebrafish genomes are not yet complete (95% covered in Fugu and 5.7-fold coverage in zebrafish) and genome annotations are still evolving, absence of a detectable ortholog from current assemblies does not necessarily imply that an orthologous gene does not exist. Nevertheless, current data suggests greater variability in hnRNP proteins between mammals and fish than was seen for SR proteins.
Table 1. Conservation of splicing factors between human, mouse, and Fugu.
| Trans factors | Mouse | Fugu |
|---|---|---|
| SR Proteins | ||
| Domains same as human | 10/10 | 10/10 |
| Domains changed | 0/10 | 0/10 |
| Missing | 0/10 | 0/10 |
| hnRNPs | ||
| Domains same as human | 13/14 | 7/14 |
| Domains changed | 1/14 | 4/14 |
| Missing | 0/14 | 3/14 |
Domains refer to predicted RNA recognition motifs and K homology domains. Accession numbers and Ensemble identifiers for all-trans factors analyzed are provided in Tables 5-8.
Discrimination of Mammalian and Fugu Introns. The results reported above suggest that the critical differences in splicing between Fugu and mammalian introns may reside primarily in the abundance and locations of specific short oligonucleotides with ISE activity, with intron length-dependent effects also playing a role. To explore this idea, a model based on LDA was developed that utilizes intron length and nonoverlapping 3-mer counts (including GGG and CCC) as features to predict whether a given Fugu intron will be correctly spliced in mammalian cells (Fig. 6). Introns of the Fugu RCN1, HD, and ARP3 genes (2, 3, 30) were scored with this model (Fig. 5). By comparing the scores of Fugu introns to their splicing phenotypes in mammalian cells, a correlation was observed, with the highest-scoring (most Fugu-like) introns generally failing to splice in mammalian cells and introns with scores in the range observed for natural mouse introns almost always splicing correctly (Fig. 5). Thus, our method recognizes intronic features that differ between Fugu and mammalian introns and appears able to predict the spliceability of Fugu introns in mammalian cells. Independently of rescue-ise, this method ranks G triples, C-rich motifs, and AC repeats as critical features that distinguish fish and mammalian introns.
Fig. 5.

Classification of vertebrate introns. Distribution of model scores for independent sets of orthologous mouse and Fugu introns and splicing phenotypes for introns 1–5 of the Fugu RCN1 gene (3), introns 1–7 of the Fugu HD gene (2), and introns 1–11 of the Fugu ARP3 gene. Full details given in Fig. 6C.
Rescuing Splicing of Fugu Introns in Mammalian Systems. Our experience with the LDA model suggested that changing the sequence composition of a Fugu intron that was misspliced in mammalian cells by adding sequences that function as ISEs in mammalian introns might rescue the splicing phenotype. To test this idea, a Fugu ARP3 construct (Fig. 12, which is published as supporting information on the PNAS web site) was transfected into human 293T cells and into a fish (minnow) cell line, PLHC-1 (Supporting Text). After being spliced, cDNA was synthesized by reverse transcription; PCR with primers targeting exon 1 and exon 12 revealed 1.2-kb products in both cell lines. To assess the pattern of splicing, both 1.2-kb transcripts were cloned into pGEM-T vectors and sequenced. The presence of aberrant splicing was confirmed in the 293T cell line, whereas the transcript from the PLHC-1 cell line was spliced correctly. In 293T cells, introns 4 and 9 were retained, exon 7 was skipped, and exon 5 was truncated by use of a cryptic 5′ss. Based on the LDA model, we attempted to rescue splicing of Fugu ARP3 intron 4 by inserting sequences similar to the G1 and G2 G triples from intron 2 of the human alpha globin gene into the Fugu intron (23). Insertion of these sequences reduces the score of the intron substantially to a score range in which tested Fugu introns have generally spliced correctly (Fig. 5). The 88-bp wild-type intron was mutated by using site-directed mutagenesis to generate two mutants with a single and double G-triplet located near the 5′ss, resulting in mutant introns 99 and 107 bp long, respectively. These two mutant constructs were transfected into human 293T cells, and cDNA was synthesized under the same conditions as before. A PCR with primers flanking the intron was used to assess the degree of splicing. A single G2 insert was sufficient to partially rescue splicing of intron 4 (Fig. 13, which is published as supporting information on the PNAS web site). Insertion of both G1 and G2 increased the level of splicing to approximately that seen in the PLHC-1 cell line. Thus, changing the ISE composition of a misspliced Fugu intron as guided by the LDA model restored levels of correct splicing in mammalian cells comparable with that seen in fish.
Discussion
Core components of the spliceosome are universally conserved in higher eukaryotes, but less is known about the conservation of the sequences and factors that regulate splicing. The observation that some Fugu introns are not properly spliced in mammalian cells suggests that substantive differences in splicing exist between mammals and fish. Here, we conducted a large-scale bioinformatic study of cis elements and trans factors that are important in splicing, comparing mammalian and fish genomes to identify similarities and differences between organisms.
Sequence motifs at the 5′ss and 3′ss were not significantly different between mammalian and fish genes, and predicted branch site motifs are also quite similar. Applying the rescue-ese approach to identify candidate ESEs in human, mouse, and Fugu exons, substantial overlap in the sets of predicted ESE hexamers was found (Fig. 1A). Previously, the ESE activity of representatives of 10 candidate human ESE motifs predicted by rescue-ese were confirmed by using an in vivo splicing reporter assay, demonstrating high predictive accuracy for this method (17). The validity of the cross-species rescue-ese predictions are further supported by a recent study that found that the hexamers predicted here as ESEs in multiple vertebrates are significantly less likely to be disrupted by single-nucleotide polymorphisms in human than those restricted to a single species (21). Additional evidence of conserved function comes from FD plots, which document similar positional biases in rescue-ese motifs along human, mouse, and Fugu exons (Fig. 1B and Fig. 8). High conservation of splice site and predicted ESE motifs across vertebrates was mirrored in patterns of splicing factor conservation. Orthologs for all 10 human SR proteins were identified in mouse and Fugu, and domain structure was preserved.
To explore potential differences in ISEs, we introduced rescue-ise, a computational method to predict ISEs. rescue-ise and FD plot analysis identified GGG, a known mammalian ISE conserved in human and mouse (8), but did not identify any related motifs in Fugu or zebrafish introns (Fig. 2). In addition to GGG, a C-rich motif is also overrepresented in introns near splice sites in human and mouse but not in Fugu or zebrafish (Fig. 14, which is published as supporting information on the PNAS web site). Enrichment of CCC and GGG in human introns has also been observed previously (e.g., refs. 31 and 32 and references therein). McCullough and Berget (8) showed that GGG elements in human introns can base pair to nucleotides 8–10 of U1 small nuclear RNA, recruiting U1 small nuclear ribonucleoproteins to the vicinity of the 5′ss. Other splicing factors have also been implicated in binding to G-rich regions and influencing splicing, including hnRNPs A1, F, and H and other members of the hnRNP H family (33–36). H complex hnRNP proteins, which often bind to exonic splicing silencers and intronic regulatory sequences, were less conserved between mammals and fish. Orthologs of hnRNP A1 and H were identified in all three vertebrates, but an ortholog for hnRNP F was not detected in the Fugu genome. Furthermore, the fish orthologs of hnRNP H appears to lack an RNA recognition motif present in both mammalian proteins. Other differences in hnRNP genes were also observed, including the apparent absence of hnRNPs A2/B1 and RALY from the Fugu genome. Two of these genes (hnRNP F and A2/B1) appear to be absent from the zebrafish genome as well, suggesting that these represent true gene losses in the fish lineage rather than genes missed because of the incompleteness of current genome assemblies or annotations. These differences in intron-binding factors between mammals and fish may explain why certain mammalian ISEs appear absent from fish.
Applying rescue-ise to a dataset of Fugu introns identified short repeats of CA and GT dinucleotides as candidate ISEs in this organism (Fig. 2, motifs f3A and f5A). FD plots support a role for ACAC and GTGT sequences as enhancers of introns with weak 5′ss and weak 3′ss, respectively, in both Fugu and zebrafish (Fig. 3 C and D). These elements have not been identified as ISEs involved in constitutive splicing in mammals. However, a recent study showed that hnRNP L binds specifically to CA repeats to enhance alternative splicing of an upstream exon in the human endothelial nitric oxide synthase gene (37) and an ortholog of hnRNP L is present in Fugu. GU repeat sequences were also recently shown to function as ISEs involved in tissue-specific alternative splicing of the human cardiac sodium calcium exchanger gene (38). ETR-3 and the neuroblastoma apoptosis-related RNA-binding protein (NAPOR), an isoform expressed from the CUGBP2 gene, bind to GU-rich sequences in certain mammalian introns and enhance alternative splicing (39, 40). Orthologs of both genes are also present in Fugu. A search of the literature identified known mammalian splicing regulatory elements similar to candidate Fugu motifs f5D (TAG) (41) and f5E (T-rich) (42). However, our search did not identify known elements similar to motif f5C, with consensus [A/T]TAC[A/T], whose potential role in splicing will require experimental tests. These observations suggest a model in which certain repetitive motifs used primarily to regulate alternative splicing in mammals have evolved a more prominent role in constitutive splicing in fish, despite substantial reduction in repeat content in the Fugu genome.
In addition to the differences in the sequences of putative splicing regulatory elements described above, the organization of these elements also appears to differ between mammalian and fish genes. In mammalian genes, there is a compensatory relationship between ISEs and ESEs. Exons flanked by long introns are enriched in ESEs and deficient in nearby ISEs, whereas exons flanked by short introns are deficient in ESEs and enriched in nearby ISEs (Figs. 4. and 10). These observations are consistent with current splicing models for human transcripts, in which exons flanked by long introns are spliced by exon definition, which generally depends on ESEs, and short introns are recognized by an intron definition mechanism (25). Sterner et al. (25) observed that expanded human exons were efficiently included if flanking introns were at most 500 bp long but were skipped if the introns were expanded, implying an upper boundary of 500 bp for intron definition in mammals. The compaction of the Fugu genome has resulted in ≈80% of introns being <500 bp in length, presumably leading to a massive increase in intron definition. In contrast to what is seen in mammals, long Fugu introns have increased frequencies of putative ISE motifs relative to short Fugu introns, suggesting that even long Fugu introns may often be spliced by intron definition.
Our observations that putative ISE sequences differ substantially between mammalian and fish introns suggested that addition of mammalian ISEs to improperly spliced Fugu introns could rescue splicing in mammalian systems. LDA was used to combine the sequence and architectural features that distinguish mammalian and fish introns. As an application, we inserted GGG sequences into intron 4 of the Fugu ARP3 gene. This modification was predicted by the LDA analysis to rescue splicing in mammals (Fig. 5), and, indeed, this modified intron was spliced in human cell lines at a comparable level with that of the wild-type intron in a fish cell line (Fig. 13). Thus, our computational analysis has implications for effective transfer of genetic information between vertebrates. This study also represents a paradigm for analyzing the evolution of gene expression regulation. Comparative genomic approaches similar to those described here should be applicable to other steps in gene expression, including transcription and translation, that are modulated by widespread cis-regulatory elements.
Supplementary Material
Acknowledgments
We thank Paula Grabowski for critical reading of the manuscript and Tomaso Poggio and Phillip Sharp for advice. This material is based on work supported by National Science Foundation Grant 0218506 (to C.B.B.), and by a grant from the Burroughs Wellcome Fund (to C.B.B.). G.Y. was supported by the Lee Kuan Yew Fellowship of Singapore. S.H and B.V. were supported by Singapore's Agency for Science, Technology, and Research.
Author Contributions: G.Y. and C.B.B. designed research; G.Y., S.H., and B.V. performed research; G.Y. contributed new reagents/analytical tools; G.Y., S.H., B.V., and C.B.B. analyzed data; and G.Y. and C.B.B. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ESE, exonic splicing enhancer; ISE, intronic splicing enhancer; 5′ss, 5′ splice site; 3′ss, 3′ splice site; LDA, linear discriminant analysis; FD, frequency difference; hnRNP, heterogeneous nuclear ribonucleoprotein.
References
- 1.Hedges, S. B. & Kumar, S. (2002) Science 297, 1283-1285. [DOI] [PubMed] [Google Scholar]
- 2.Sathasivam, K., Baxendale, S., Mangiarini, L., Bertaux, F., Hetherington, C., Kanazawa, I., Lehrach, H. & Bates, G. P. (1997) Hum. Mol. Genet. 6, 2141-2149. [DOI] [PubMed] [Google Scholar]
- 3.Miles, C. G., Rankin, L., Smith, S. I., Niksic, M., Elgar, G. & Hastie, N. D. (2003) Nucleic Acids Res. 31, 2795-2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim, L. P. & Burge, C. B. (2001) Proc. Natl. Acad. Sci. USA 98, 11193-11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huh, G. S. & Hynes, R. O. (1993) Mol. Cell. Biol. 13, 5301-5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan, R. C. & Black, D. L. (1997) Mol. Cell. Biol. 17, 4667-4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hedjran, F., Yeakley, J. M., Huh, G. S., Hynes, R. O. & Rosenfeld, M. G. (1997) Proc. Natl. Acad. Sci. USA 94, 12343-12347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCullough, A. & Berget, S. (2000) Mol. Cell. Biol. 20, 9225-9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graveley, B. (2000) RNA 6, 1197-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blencowe, B. (2000) Trends Biochem. Sci. 25, 106-110. [DOI] [PubMed] [Google Scholar]
- 11.Graveley, B. & Maniatis, T. (1998) Mol. Cell 1, 765-771. [DOI] [PubMed] [Google Scholar]
- 12.Maniatis, T. & Tasic, B. (2002) Nature 418, 236-243. [DOI] [PubMed] [Google Scholar]
- 13.Cartegni, L., Chew, S. L. & Krainer, A. R. (2002) Nat. Rev. Genet. 3, 285-298. [DOI] [PubMed] [Google Scholar]
- 14.Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., et al. (2001) Nature 409, 860-921. [DOI] [PubMed] [Google Scholar]
- 15.Mouse Genome Sequencing Consortium (2002) Nature 420, 520-562. [DOI] [PubMed] [Google Scholar]
- 16.Aparicio, S., Chapman, J., Stupka, E., Putnam, N., Chia, J. M., Dehal, P., Christoffels, A., Rash, S., Hoon, S., Smit, A., et al. (2002) Science 297, 1301-1310. [DOI] [PubMed] [Google Scholar]
- 17.Fairbrother, W., Yeh, R.-F., Sharp, P. A. & Burge, C. B. (2002) Science 297, 1007-1013. [DOI] [PubMed] [Google Scholar]
- 18.Duda, R. O., Hart, P. E. & Stork, D. G. (2001) Pattern Classification (Wiley, New York), pp. 215-281.
- 19.Fairbrother, W. G., Yeo, G. W., Yeh, R.-F., Goldstein, P., Mawson, M., Sharp, P. A. & Burge, C. B. (2004) Nucleic Acids Res. 32, W187-W190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Graveley, B. R., Hertel, K. J. & Maniatis, T. (1998) EMBO J. 17, 6747-6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fairbrother, W. G., Holste, D., Burge, C. B. & Sharp, P. A. (2004) PLoS Biol. 2, e268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ladd, A. N. & Cooper, T. A. (2002) Genome Biol. 3, reviews0008.1–reviews0008.16. [DOI] [PMC free article] [PubMed]
- 23.McCullough, A. & Berget, S. (1997) Mol. Cell. Biol. 17, 4562-4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berget, S. (1995) J. Biol. Chem. 270, 2411-2414. [DOI] [PubMed] [Google Scholar]
- 25.Sterner, D. A., Carlo, T. & Berget, S. M. (1996) Proc. Natl. Acad. Sci. USA 93, 15081-15085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou, Z., Licklider, L., Gygi, S. & Reed, R. (2002) Nature 419, 182-185. [DOI] [PubMed] [Google Scholar]
- 27.Bateman, A., Birney, E., Cerruti, L., Durbin, R., Etwiller, L., Eddy, S. R., Griffiths-Jones, S., Howe, K. L., Marshall, M. & Sonnhammer, E. L. (2002) Nucleic Acids Res. 30, 276-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kozu, T., Henrich, B. & Schafer, K. P. (1995) Genomics 25, 365-371. [DOI] [PubMed] [Google Scholar]
- 29.Kamma, H., Horiguchi, H., Wan, L., Matsui, M., Fujiwara, M., Fujimoto, M., Yazawa, T. & Dreyfuss, G. (1999) Exp. Cell Res. 246, 399-411. [DOI] [PubMed] [Google Scholar]
- 30.Venkatesh, B. & Brenner, S. (1998) Gene 211, 169-175. [DOI] [PubMed] [Google Scholar]
- 31.Majewski, J. & Ott, J. (2002) Genome Res. 12, 1827-1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang, X. H., Heller, K. A., Hefter, I., Leslie, C. S. & Chasin, L. A. (2003) Genome Res. 13, 2637-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Min, H., Chan, R. C. & Black, D. L. (1995) Genes Dev. 9, 2659-2671. [DOI] [PubMed] [Google Scholar]
- 34.Blanchette, M. & Chabot, B. (1999) EMBO J. 18, 1939-1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hastings, M. L., Wilson, C. M. & Munroe, S. H. (2001) RNA 7, 859-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caputi, M. & Zahler, A. M. (2001) J. Biol. Chem. 276, 43850-43859. [DOI] [PubMed] [Google Scholar]
- 37.Hui, J., Stangl, K., Lane, W. & Bindereif, A. (2003) Nat. Struct. Biol. 10, 33-37. [DOI] [PubMed] [Google Scholar]
- 38.Gabellini, N. (2001) Eur. J. Biochem. 268, 1076-1083. [DOI] [PubMed] [Google Scholar]
- 39.Zhang, W., Liu, H., Han, K. & Grabowski, P. J. (2002) RNA 8, 671-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Charlet, N., Logan, P., Singh, G. & Cooper, T. A. (2002) Mol. Cell 9, 649-658. [DOI] [PubMed] [Google Scholar]
- 41.Kashima, T. & Manley, J. L. (2003) Nat. Genet. 34, 460-463. [DOI] [PubMed] [Google Scholar]
- 42.Del Gatto-Konczak, F., Bourgeois, C. F., Le Guiner, C., Kister, L., Gesnel, M. C., Stevenin, J. & Breathnach, R. (2000) Mol. Cell. Biol. 20, 6287-6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}