

ABSTRACT
The pathogenicity of the Gram-negative plant-pathogenic bacterium Xanthomonas campestris pv. vesicatoria depends on a type III secretion (T3S) system, which spans both bacterial membranes and translocates effector proteins into plant cells. The assembly of the T3S system presumably involves the predicted lytic transglycosylase (LT) HpaH, which is encoded adjacent to the T3S gene cluster. Bacterial LTs degrade peptidoglycan and often promote the formation of membrane-spanning macromolecular protein complexes. In the present study, we show that HpaH localizes to the bacterial periplasm and binds to peptidoglycan as well as to components of the T3S system, including the predicted periplasmic inner rod proteins HrpB1 and HrpB2 as well as the pilus protein HrpE. In vivo translocation assays revealed that HpaH promotes the translocation of various effector proteins and of early substrates of the T3S system, suggesting a general contribution of HpaH to type III-dependent protein export. Mutant studies and the analysis of reporter fusions showed that the N-terminal region of HpaH contributes to protein function and is proteolytically cleaved. The N-terminally truncated HpaH cleavage product is secreted into the extracellular milieu by a yet-unknown transport pathway, which is independent of the T3S system.
KEYWORDS: Xanthomonas, peptidoglycan, Slt70, type III secretion, lytic transglycosylase, effector proteins
INTRODUCTION
The pathogenicity of many Gram-negative plant- and animal-pathogenic bacteria depends on a type III secretion (T3S) system, which translocates bacterial effector proteins into eukaryotic cells (1). Type III effector proteins manipulate host cellular pathways to the benefit of the pathogen and thus promote bacterial proliferation (2). T3S systems are highly complex nanomachines that span both bacterial membranes and are associated with a pilus-like appendage, which serves as a transport channel for secreted proteins (3). Protein translocation into eukaryotic cells depends on the channel-like T3S translocon, which inserts into the host plasma membrane (3, 4).
T3S systems usually consist of 20 to 25 proteins, nine of which are conserved in plant- and animal-pathogenic bacteria and constitute the core elements of the secretion apparatus. These include ring structures in the bacterial inner membrane (IM) and outer membrane (OM), which are associated with a predicted periplasmic inner rod (3). The IM ring interacts with components of the export apparatus, which is assembled by members of the conserved Sct (secretion and cellular translocation) R, S, T, U, and V families of transmembrane proteins (3). The letters refer to the Ysc proteins of the T3S system from Yersinia spp. (5, 6). Components of the export apparatus insert into the IM and interact with cytoplasmic parts of the T3S system, including the predicted cytoplasmic (C) ring and the ATPase complex, which is presumably involved in T3S substrate recognition and unfolding (7–10). The recognition of T3S substrates often depends on a secretion signal in the N-terminal protein region, which is not conserved on the amino acid level (3). In many cases, specific T3S chaperones bind to secreted proteins and might facilitate their recognition by components of the T3S system (3).
The assembly of T3S systems presumably involves the contribution of lytic transglycosylases (LTs), which cleave the glycan backbone of peptidoglycan in the bacterial periplasm and often promote the assembly of membrane-spanning macromolecular protein complexes (11–13). The deletion of single LT genes, however, often does not result in a loss of T3S because the assembly of the secretion apparatus can also take place at natural pores or breaks in the peptidoglycan layer (12). To date, a virulence function has been described for putative LTs from enterohemorrhagic Escherichia coli (EHEC), enteropathogenic Escherichia coli (EPEC), and plant-pathogenic bacteria, including pathovars of Pseudomonas syringae and Xanthomonas spp. (14–21). In most cases, however, the enzymatic activity of these proteins and their contribution to the assembly of the T3S system have not yet been experimentally confirmed.
T3S is currently being studied in several plant- and animal-pathogenic model organisms, including Xanthomonas campestris pv. vesicatoria (reclassified as Xanthomonas euvesicatoria) (22), which causes bacterial spot disease on tomato and pepper plants. The T3S system from X. campestris pv. vesicatoria is encoded by the chromosomal hrp (hypersensitive response and pathogenicity) gene cluster and translocates approximately 30 effector proteins into plant cells (23). T3S in X. campestris pv. vesicatoria is controlled by several Hpa (Hrp-associated) proteins, which contribute to, but are not essential for, pathogenicity. Previously identified Hpa proteins include the T3S substrate specificity switch (T3S4) protein HpaC, the T3S chaperone HpaB, and the predicted LT HpaH (15, 24, 25). HpaC switches the T3S substrate specificity from the secretion of the predicted inner rod protein HrpB2 to the secretion of translocon and effector proteins, whereas HpaB promotes the efficient secretion of effector proteins (24–26). The predicted LT HpaH was previously shown to contribute to virulence and to the secretion and translocation of the effector proteins XopJ and XopF1 (15, 27). In the present study, we show that HpaH from X. campestris pv. vesicatoria localizes to the bacterial periplasm and binds to peptidoglycan as well as to periplasmic components of the T3S system. The N-terminal region of HpaH contains a predicted Sec signal, which is cleaved off and contributes to the virulence function of HpaH and its transport into the periplasm. Notably, the HpaH cleavage product is itself secreted into the extracellular milieu, albeit independently of the T3S system. In vivo reporter assays revealed that HpaH promotes the type III-dependent translocation of effector and noneffector proteins, which is in agreement with the predicted contribution of HpaH to the assembly of the T3S system.
RESULTS
Translation of hpaH is presumably initiated upstream of the annotated start codon.
HpaH from X. campestris pv. vesicatoria strain 85-10 (XCV0441, GenBank accession number CAJ22072) is encoded in the flanking region of the T3S gene cluster and annotated as a protein of 157 amino acids with a predicted N-terminal Sec signal (prediction by SignalP 4.1; the predicted cleavage site is between amino acids 34 and 35 [http://www.cbs.dtu.dk/services/SignalP/]). The translation start site has not yet been experimentally determined for HpaH and homologous proteins, and comparative sequence analyses revealed that the N-terminal regions of these proteins vary in length and are not highly conserved (see Fig. S1 in the supplemental material). Given the presence of an ATG codon 90 bp upstream of the annotated start codon of hpaH (Fig. 1A), we investigated a possible alternative translation initiation of hpaH. For this, we generated expression constructs encoding HpaH or a derivative thereof with 30 additional N-terminal amino acids (designated HpaHAS for “alternative start”) under the control of the lac or the native promoter (pnat) (Fig. 1B). HpaH derivatives were analyzed in X. campestris pv. vesicatoria strain 85-10hpaHoof, which is a derivative of the wild-type (wt) strain 85-10 and contains a frameshift mutation in hpaH (15). Unfortunately, due to low expression levels of hpaH derivatives, we could not purify sufficient amounts of HpaHAS(pnat)–c-Myc from X. campestris pv. vesicatoria to determine the N-terminal amino acid sequence by mass spectrometry (see Materials and Methods).
FIG 1.

Translation of hpaH presumably is initiated upstream of the annotated translation start site. (A) Nucleotide sequence and encoded amino acids of the 5′ region of hpaH from X. campestris pv. vesicatoria strain 85-10. The annotated hpaH sequence and the upstream sequence are shown in red and black, respectively. The annotated start site of hpaH and the ATG codon in the upstream region are underlined. A black triangle indicates the predicted cleavage site. (B) Overview of the hpaH expression constructs used in this study. The red arrows refer to the annotated hpaH gene, the white box represents the upstream region, and the light red box indicates the predicted Sec signal. The lac promoter (plac) and the native hpaH promoter (pnat) are indicated by gray and green arrows, respectively. The blue arrow represents the first 33 codons of hrcC. (C) Complementation studies with HpaH derivatives. X. campestris pv. vesicatoria strains 85-10 (wt) and 85-10hpaHoof (hpaHoof) without an expression construct (−) or containing HpaH–c-Myc and derivatives thereof as indicated were inoculated into leaves of resistant ECW-10R pepper plants at a bacterial density of 108 CFU ml−1 (left panel) or 107 CFU ml−1 (right panel). Leaves were destained in ethanol at 1 dpi (left panel) or 2 dpi (right panel). Dashed lines mark the infiltrated areas. For protein analysis, equal amounts of cell extracts (adjusted according to the cell density) were analyzed by immunoblotting using a c-Myc epitope-specific antibody. As a control for equal loading, the blot was stained with Ponceau stain.
Immunoblot analyses of bacterial cell extracts revealed the presence of two HpaH–c-Myc-specific proteins of approximately 20 and 18 kDa, which likely correspond to HpaH–c-Myc and an N-terminally truncated derivative thereof lacking the predicted Sec signal (Fig. 1C). HpaHAS–c-Myc and HpaHAS(pnat)–c-Myc migrated at a higher molecular size than HpaH–c-Myc, suggesting that translation was initiated upstream of the annotated start codon (Fig. 1C). Although no Sec signal was predicted for HpaHAS, N-terminally truncated cleavage products at a size similar to that of the HpaH cleavage product were detected for all derivatives (Fig. 1C). An additional hpaH expression construct, in which the annotated start codon was mutated to CTG (Fig. 1B), migrated at a size similar to that of HpaHAS(pnat), suggesting that the annotated ATG start codon was dispensable for translation initiation (Fig. 1C).
The N-terminal 35 amino acids of HpaH are essential for protein function.
For complementation studies, bacteria were inoculated into leaves of resistant Early Cal Wonder 10R (ECW-10R) pepper plants. ECW-10R plants contain the R protein Bs1 and trigger defense responses upon recognition of the effector protein AvrBs1, which is translocated by strain 85-10 (28). AvrBs1-induced defense reactions are macroscopically visible as the hypersensitive response (HR), which is a fast cell death reaction at the infection site and restricts bacterial multiplication (29) (Fig. 1C). As expected, strain 85-10hpaHoof led to a reduced HR compared with that for the wild-type strain 85-10 (Fig. 1C) (15). The wild-type phenotype was restored by HpaH–c-Myc, HpaHAS–c-Myc, HpaHAS(pnat)–c-Myc, and HpaHAS/CTG(pnat), suggesting that these HpaH derivatives were functional (Fig. 1C). To investigate a potential contribution of the N-terminal protein region to HpaH function, we generated additional expression constructs encoding HpaH without the N-terminal 34 or 35 amino acids under the control of the lac promoter (numbers refer to amino acid positions in the annotated HpaH protein). HpaHΔ2–34–c-Myc and HpaHΔ2–35–c-Myc migrated at a size similar to that of the HpaH-specific cleavage product (Fig. 1C). HpaHΔ2–34–c-Myc partially complemented the hpaH mutant phenotype when the infected pepper leaves were inspected at 1.5 days postinoculation (dpi) (see Fig. S2 in the supplemental material) or 2 dpi (Fig. 1C), suggesting that HpaH was partially functional in the absence of a predicted Sec signal. In contrast, no complementation was observed for HpaHΔ2–35–c-Myc (Fig. 1C). The lack of complementation was presumably not caused by a dominant negative effect because ectopic expression of hpaHΔ2–34–c-myc or hpaHΔ2–35–c-myc in strain 85-10 did not alter the wild-type phenotype (Fig. S2). These findings suggest that the N-terminal 35 amino acids of the annotated HpaH protein are required for cleavage and protein function. We also generated an expression construct encoding HpaHΔ2–34 in fusion with the predicted Sec signal of the OM secretin HrcC, which is located in the N-terminal 34 amino acids. HrcC1–34-HpaHΔ2–34 complemented the hpaH mutant phenotype, indicating that the Sec signal of HrcC did not interfere with HpaH function (Fig. 1C).
HpaH binds to peptidoglycan in vitro.
HpaH shares predicted structural similarity with the catalytic region of the LT Slt70 from Escherichia coli (prediction by PHYRE; http://www.sbg.bio.ic.ac.uk/phyre) (30) (Fig. 2A). Given the putative function of HpaH as an LT, we investigated a possible interaction with peptidoglycan. For this, hpaH was in vitro transcribed and translated and the protein was incubated in the presence or absence of insoluble peptidoglycan. As positive and negative controls, we used purified lysozyme and bovine serum albumin (BSA), respectively. After centrifugation, proteins in the pellet and the supernatant fraction were analyzed by SDS-PAGE, Coomassie blue staining, or immunoblotting. In the absence of peptidoglycan, all proteins were detected mainly in the supernatant fraction (Fig. 2B). When incubated with peptidoglycan, BSA was present in the supernatant fraction whereas lysozyme and HpaH were also present in the pellet fraction, suggesting that they can bind to peptidoglycan (Fig. 2B).
FIG 2.

HpaH localizes to the bacterial periplasm and interacts with peptidoglycan. (A) HpaH shares predicted structural similarity with the LT Slt70 of E. coli. The structure of HpaH was predicted by PHYRE (http://www.sbg.bio.ic.ac.uk/phyre) and aligned with the structure of Slt70 from E. coli (UniProtKB/Swiss-Prot accession number P0AGC3.1) using the PyMOL software (http://www.pymol.org/). HpaH is shown in red, the C-terminal domain of Slt70, which contains the active site (30), in yellow, and additional Slt70 protein regions in green. The N and C termini of HpaH are indicated. (B) HpaH binds to peptidoglycan in vitro. HpaH, lysozyme, and BSA were incubated in the absence (−) or presence (+) of peptidoglycan (PG). Total cell extracts (TE) as well as proteins of the supernatant (SN) and the pellet (P) fractions were analyzed by SDS-PAGE and Coomassie blue staining or immunoblotting using antibodies specific for HpaH. (C) Mutation of the predicted catalytic glutamate residue at position 58 abolishes HpaH function. Strains 85-10 (wt) and 85-10hpaHoof (hpaHoof) containing the vector pBRM (EV) or HpaH derivatives as indicated were infiltrated at a density of 5 × 107 CFU ml−1 into leaves of resistant ECW-10R pepper plants. Leaves were destained in ethanol at 2 dpi. Dashed lines mark the infiltrated areas. For protein analysis, equal amounts of cell extracts (adjusted according to the cell density) were analyzed by immunoblotting using a c-Myc epitope-specific antibody. (D) Fractionation studies with HpaH. Strain 85*hpaHoof containing HpaH–c-Myc was incubated in secretion medium. Fractions enriched in the cytoplasm (C), the inner membrane (IM), the periplasm (P), and the outer membrane (OM) were separated by ultracentrifugation and analyzed by immunoblotting using a c-Myc epitope-specific antibody. Blots were reprobed with antibodies specific for HrpB1 and HrpB2. (E) HpaH-PhoAΔ2–120 displays phosphatase activity. Strain 85-10ΔphoA without an expression construct or containing HpaH-PhoAΔ2–120 fusions as indicated was incubated in secretion medium. Phosphatase activities were determined using pNPP as the substrate. All fusion proteins were stably synthesized, as shown in Fig. S3 in the supplemental material. Values represent the means of three measurements of one strain. Error bars represent standard deviations, and asterisks denote statistically significant differences according to the Student t test (P > 0.001).
HpaH contains a predicted catalytic glutamate residue at position 58 (E58), which is conserved in homologous LTs and was shown to be required for the activity of the LT EtgA from EPEC (16, 31) (Fig. S1). The exchange of E58 for alanine (E58A mutation) abolished the ability of HpaH from X. campestris pv. vesicatoria to complement the hpaH mutant phenotype, suggesting that the predicted enzymatic activity of HpaH is required for protein function (Fig. 2C).
HpaH localizes to the bacterial periplasm.
To analyze the subcellular localization of HpaH, we performed fractionation assays with X. campestris pv. vesicatoria strain 85-10hrpG*hpaHoof (85*hpaHoof) containing HpaH–c-Myc. Strain 85*hpaHoof is a derivative of the hrpG wild-type strain 85-10hpaHoof and encodes HrpG*, a constitutively active version of the key regulator HrpG. HrpG* activates the expression of T3S genes even under noninducing conditions, which allows the immunological detection of T3S components such as the predicted inner rod proteins HrpB1 and HrpB2 (32, 33). Bacteria were incubated under T3S-permissive conditions, and fractions enriched in cytoplasm, IM, periplasm, and OM were separated by ultracentrifugation and analyzed by immunoblotting. The cleavage product of HpaH–c-Myc was detected predominantly in the periplasm-enriched fraction, which is in agreement with the predicted localization of LTs (Fig. 2D). As described previously, a similar localization was observed for HrpB1 and HrpB2 (34, 35) (Fig. 2D).
To confirm the results of the fractionation studies, we used an N-terminal deletion derivative of the alkaline phosphatase PhoA from E. coli (PhoAΔ2–120), which lacks the native signal peptide, as reporter (36). PhoA is active only when located in the periplasm and is used as reporter to study protein topology (36–39). PhoA activities were analyzed in X. campestris pv. vesicatoria strains which lacked the predicted native phoA gene (XCV2913). HpaH-PhoAΔ2–120 led to a significant increase in PhoA activity in strain 85-10ΔphoA, suggesting that HpaH-PhoAΔ2–120 was located in the periplasm (Fig. 2E). Significantly reduced activity was measured for HpaHΔ2–34-PhoAΔ2–120 whereas HpaHΔ2–35-PhoAΔ2–120 was not active (Fig. 2E). Similar results were obtained with strain 85*ΔphoA (data not shown). The lack of activity of HpaHΔ2–35-PhoAΔ2–120 was presumably not caused by protein misfolding because all fusion proteins were active when analyzed in E. coli, which was incubated on ice (see Fig. S3 in the supplemental material). According to a previous publication, the incubation of E. coli cells on ice leads to a residual folding of cytoplasmic PhoA proteins (40). Immunoblot analysis of bacterial cell extracts revealed that all PhoAΔ2–120 fusion proteins were stably synthesized (Fig. S3). Taken together, the results of the subfractionation and the PhoA activity assays suggest that HpaH localizes to the periplasm and that the periplasmic localization of HpaH depends on the N-terminal protein region.
HpaH interacts with periplasmic components of the T3S system.
To analyze a possible interaction between HpaH and components of the T3S system, we performed in vitro glutathione S-transferase (GST) pulldown assays. For this, immobilized GST or GST fusions of the predicted inner rod proteins HrpB1 and HrpB2, the IM ring protein HrcD, the pilus protein HrpE, the OM secretin HrcC, the cytoplasmic regions of the IM proteins HrcU and HrcV, and the cytoplasmic T3S chaperone HpaB were incubated with an E. coli lysate containing the N-terminally truncated HpaHΔ2–35–c-Myc derivative. All GST fusion proteins were stably synthesized (see Fig. S4 in the supplemental material). Immunoblot analysis of the eluted proteins revealed that HpaHΔ2–35–c-Myc coeluted with GST-HrpB1, GST-HrpB2, GST-HrcD, and GST-HrpE (Fig. 3A). No interaction was observed with GST, GST-HrcC, GST-HrcU255–357, GST-HrcV324–645, and GST-HpaB (Fig. 3A), suggesting that HpaH does not unspecifically bind to GST or T3S system components.
FIG 3.

HpaH interacts with periplasmic components of the T3S system. (A) In vitro GST pulldown assays with HpaHΔ2–35–c-Myc. GST and GST fusions of HrpB1, HrpB2, HrcD, HrpE, HrcC, HrcU255–357, HrcV324–645, and HpaB as indicated were immobilized on glutathione-Sepharose and incubated with a bacterial lysate containing HpaHΔ2–35–c-Myc. Total cell extracts (TE) and eluates were analyzed by immunoblotting using a c-Myc epitope-specific antibody. All GST fusion proteins were stably synthesized, as shown in Fig. S4 in the supplemental material. It was previously shown that HrpB1, HrpB2, HrcD, and HrpE do not unspecifically interact with T3S-associated proteins (24, 35). (B) Coimmunoprecipitation experiments with X. campestris pv. vesicatoria. Strain 85*hpaHoof with or without HpaH–c-Myc was grown in secretion medium. Bacterial lysates were incubated in the presence (+) or absence (−) of c-Myc epitope-specific antibodies coupled to protein G-Sepharose. Bacterial total cell extracts (TE) and precipitated proteins (eluate) were analyzed by immunoblotting using antibodies specific for HrpB1, HrpB2, and HrcC.
To confirm the results of the in vitro GST pulldown assays, we performed in vivo coimmunoprecipitation experiments. For this, strain 85*hpaHoof without an expression construct or containing HpaH–c-Myc was grown under T3S-permissive conditions, and bacterial lysates were incubated in the presence or absence of a c-Myc epitope-specific antibody which was coupled to protein G-agarose. The predicted inner rod proteins HrpB1 and HrpB2 as well as the OM secretin HrcC were immunoprecipitated in the presence of HpaH–c-Myc (Fig. 3B). The observed interactions were specific, because HrpB1, HrpB2, and HrcC were not precipitated in the absence of HpaH–c-Myc or the c-Myc epitope-specific antibody (Fig. 3B). Given that the interaction between HpaH and HrcC was not detected by the GST pulldown assay, we assume that HrcC interacts indirectly with HpaH.
The cleavage product of HpaH is secreted in a T3S-independent manner.
Next, we investigated a possible secretion of HpaH–c-Myc. For this, we incubated strain 85*hpaHoof containing HpaH–c-Myc, HpaHAS(pnat)–c-Myc, or HpaHAS/CTG(pnat)–c-Myc in T3S-inducing medium. The HpaH–c-Myc-specific cleavage products but not the full-length proteins were detected in the culture supernatant, suggesting that they were secreted after cleavage of the signal peptide (Fig. 4A). Secretion of HpaH presumably depends on the predicted Sec signal, because HpaHΔ2–34–c-Myc and HpaHΔ2–35–c-Myc were not detected in the culture supernatant (Fig. 4B). To investigate whether HpaH secretion depends on the T3S system, we performed additional secretion assays with the T3S-deficient strain 85E*ΔhrcC, which lacks the OM secretin HrcC. The HpaH–c-Myc-specific cleavage product was present in the culture supernatant of the hrcC deletion mutant, suggesting that it was secreted independently of the T3S system (Fig. 4C). Similar findings were observed for the cleavage products of HpaHAS–c-Myc and HpaHAS(pnat)–c-Myc (see Fig. S5 in the supplemental material). Furthermore, the cleavage product of HpaH–c-Myc was also secreted by the hrpG wild-type strain 85-10hpaHoof, which does not constitutively express the T3S genes (Fig. S5). The detection of proteins in the culture supernatants was not caused by cell lysis, because the IM ring component HrcJ and the predicted inner rod protein HrpB1 were detected only in the cell extracts, as expected (Fig. 4 and S5). We therefore conclude that the cleavage product of HpaH is secreted independently of the T3S system.
FIG 4.

HpaH is secreted independently of the T3S system. (A) The HpaH-specific cleavage product is secreted. X. campestris pv. vesicatoria strains 85* (wt) and 85*hpaHoof (hpaHoof) containing vector pBRM (EV), HpaH–c-Myc, or derivatives thereof as indicated were incubated in secretion medium. Total cell extracts (TE) and culture supernatants (SN) were analyzed by immunoblotting using a c-Myc epitope-specific antibody. To ensure that no cell lysis had occurred, blots were reprobed with antibodies specific for the periplasmic HrpB1 protein and the IM protein HrcJ. To detect T3S, blots were reacted with antibodies specific for the secreted translocon protein HrpF. Note that HpaH derivatives were still detectable when the immunoblot was reprobed with HrpB1-specific antibodies. HrpB1, HpaH cleavage products, HrcJ, and HrpF are indicated by arrows. The asterisk refers to uncleaved HpaH–c-Myc or HpaHAS–c-Myc. (B) Secretion of HpaH depends on the N-terminal protein region. Strains 85* (wt) and 85*hpaHoof (hpaHoof) without an expression construct or encoding HpaH–c-Myc, HpaHΔ2–34–c-Myc, or HpaHΔ2–35–c-Myc as indicated were incubated in secretion medium. TE and SN were analyzed as described for panel A, using c-Myc epitope-, HrpB1-, and HrcJ-specific antibodies. (C) HpaH is secreted independently of the T3S system. X. campestris pv. vesicatoria strains 85* (wt), 85*hpaHoof (hpaHoof), and 85E*ΔhrcC containing vector pBRM (EV) or expression constructs encoding HpaH–c-Myc as indicated were incubated in secretion medium. TE and SN were analyzed as described for panel A.
Secretion of HpaH is independent of OMVs and the T2S systems.
Given the finding that secretion of HpaH was independent of the T3S system, we investigated a possible contribution of both type II secretion (T2S) systems to HpaH secretion. T2S systems are Sec-dependent protein transport systems which often secrete virulence factors across the outer bacterial membrane (41, 42). We previously reported that the Xps T2S system contributes to virulence and secretes degradative enzymes (43, 44). For secretion assays with HpaH–c-Myc, we used the wild-type strain 85-10 and the T2S-deficient strain 85-10ΔEEE, which lacks three putative ATPase-encoding genes of both T2S systems (43). When bacteria were incubated under T2S-permissive conditions, secretion of HpaH–c-Myc was unaffected in strain 85-10ΔEEE (Fig. 5A). We also investigated a possible contribution of outer membrane vesicles (OMVs) to the secretion of HpaH. OMVs from X. campestris pv. vesicatoria were previously shown to contain predicted virulence factors, including T2S substrates, suggesting that periplasmic proteins can be targeted to different transport routes (44). For the analysis of OMV-mediated protein secretion, strain 85-10hpaHoof containing HpaH–c-Myc was cultivated in nutrient-yeast-glycerol (NYG) medium, and bacterial culture supernatants were incubated in the absence or presence of proteinase K. OMV cargo proteins are protected from degradation by proteinase K, as is observed for the T2S substrate XCV4360–c-Myc (Fig. 5B) (44). However, this was not the case for HpaH–c-Myc, suggesting that HpaH is secreted independently of OMVs by an alternative transport pathway.
FIG 5.
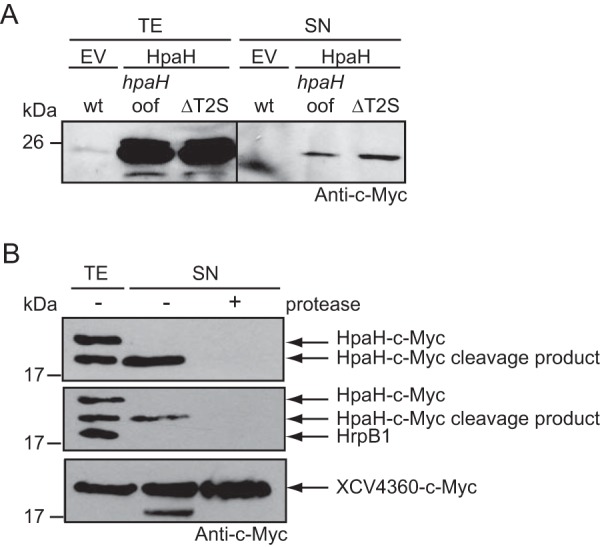
Secretion of HpaH is independent of the T2S systems and OMVs. (A) HpaH is secreted in the absence of functional T2S systems. Strains 85-10 (wt), 85-10hpaHoof (hpaHoof), and 85-10ΔEEE (ΔT2S), which is deficient in T2S, containing vector pBRM (EV) or encoding HpaH–c-Myc (HpaH) were incubated in NYG medium. Total cell extracts (TE) and culture supernatants (SN) were analyzed by immunoblotting using a c-Myc epitope-specific antibody. (B) The secreted cleavage product of HpaH is degraded by proteinase K. Strain 85* encoding HpaH–c-Myc was incubated in secretion medium. TE and SN were incubated in the presence (+) or absence (−) of proteinase K as indicated and analyzed by immunoblotting using antibodies specific for the c-Myc epitope and the periplasmic HrpB1 protein. As a control, the experiment was performed with strain 85-10 encoding the T2S substrate XCV4360–c-Myc, which was previously localized in OMVs. For the analysis of secreted XCV4360–c-Myc, bacteria were grown in NYG medium.
HpaH promotes the translocation of effector proteins.
HpaH was shown to contribute to the elicitation of plant reactions on susceptible and resistant pepper plants, suggesting that it promotes effector protein translocation (45) (Fig. 1C). In agreement with this hypothesis, strain 85-10hpaHoof induced a reduced HR on leaves of ECW-10R, ECW-20R, ECW-30R, and ECW plants, which trigger defense reactions upon recognition of the effector proteins AvrBs1, AvrBs2, AvrBs3, and AvrBsT, respectively (28, 46). To analyze the translocation of additional effector proteins, we performed translocation assays with the reporter protein AvrBs3Δ2, which is an export-deficient N-terminal deletion derivative of the effector protein AvrBs3 and is translocated into plant cells only as a fusion partner of a functional T3S and translocation signal (45, 47). Notably, we previously observed that the translocation of XopC1–200-AvrBs3Δ2 was unaffected in the absence of HpaH when the bacteria were inoculated at a density of 5 × 107 CFU ml−1. It was therefore suggested that the translocation of the effector protein XopC is HpaH independent (27). However, at lower inoculation densities, HR induction by XopC1–200-AvrBs3Δ2 was reduced in strain 85-10hpaHoof (Fig. 6B). In addition to XopC1–200-AvrBs3Δ2, we analyzed translocation of AvrBs3Δ2 fusions containing the N-terminal regions of the effector proteins HpaA and XopL (48, 49). HR induction by both proteins was significantly reduced in strain 85-10hpaHoof (Fig. 6C). Similar findings were observed for the AvrBs3Δ2 fusion of the noneffector proteins HrpB2 and HrpF (Fig. 6C). As reported previously, translocation of HrpF and HrpB2 is suppressed by the T3S chaperone HpaB and the T3S4 protein HpaC, respectively, suggesting that they are translocated as early substrates prior to effector protein translocation (25, 50). Translocation assays with HrpF1–200- and HrpB21–76-AvrBs3Δ2 fusion proteins were therefore performed in hpaB and hpaC deletion mutants, respectively. The observed influence of HpaH on type III-dependent protein translocation is in agreement with its predicted contribution to the assembly of the secretion apparatus in the periplasm. Notably, HpaH does not exert a general influence on protein secretion, because extracellular activities of proteases, cellulases, and amylases were unaltered in the absence of HpaH (Fig. 6D).
FIG 6.
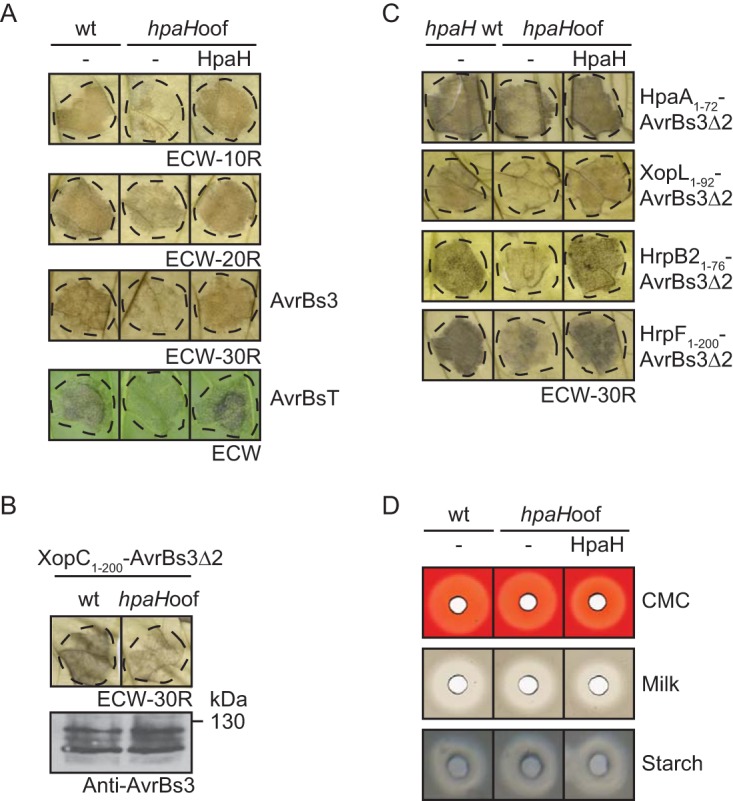
HpaH contributes to type III-dependent protein translocation. (A) Efficient HR induction by the effector proteins AvrBs1, AvrBs2, AvrBs3, and AvrBsT depends on HpaH. Strains 85-10 (wt) and 85-10hpaHoof (hpaHoof) without an expression construct or encoding HpaH–c-Myc (HpaH) as indicated were infiltrated into AvrBs1-responsive ECW-10R (at densities of 108 CFU ml−1), AvrBs2-responsive ECW-20R (at densities of 5 × 107 CFU ml−1), AvrBs3-responsive ECW-30R (at densities of 2 × 107 CFU ml−1), and AvrBsT-responsive ECW pepper plants (at densities of 4 × 108 CFU ml−1). avrBs3 and avrBsT were ectopically expressed from corresponding expression constructs. Dashed lines indicate the infiltrated areas. Leaves of ECW-10R and ECW-20R plants were destained in ethanol at 1 dpi, and leaves of ECW-30R plants were destained 2 dpi. HR phenotypes on leaves of ECW plants were photographed at 5 dpi. (B) Translocation of XopC1–200-AvrBs3Δ2 is reduced in the absence of HpaH when bacteria are infiltrated at low densities. Strains 85-10 (wt) and 85-10hpaHoof (hpaHoof) encoding XopC1–200-AvrBs3Δ2 were infiltrated at densities of 1 × 107 CFU ml−1 into leaves of AvrBs3-responsive ECW-30R pepper plants. Leaves were destained in ethanol at 2 dpi. Equal amounts of cell extracts (according to the density) were analyzed by immunoblotting using an AvrBs3-specific antibody. (C) HpaH contributes to the efficient translocation of HpaA-, XopL-, HrpB2-, and HrpF-AvrBs3Δ2 fusion proteins. X. campestris pv. vesicatoria strains 85-10 (hpaH wt) and 85-10hpaHoof (hpaHoof) without expression constructs or encoding AvrBs3Δ2 fusion proteins as indicated were inoculated into leaves of ECW-30R at densities of 4 × 108 CFU ml−1 (HpaA1–72- and HrpB21–76-AvrBs3Δ2) or 1 × 108 CFU ml−1 (XopL1–92-AvrBs3Δ2 and HrpF1–200-AvrBs3Δ2). Translocation of HrpB21–76-AvrBs3Δ2 was analyzed in strains 85*ΔhpaC (hpaH wt) and 85*ΔhpaChpaHoof (hpaHoof) lacking the T3S4 gene hpaC (50). HrpF1–200-AvrBs3Δ2 is translocated in the absence of the T3S chaperone gene hpaB (25) and was analyzed in strains 85*ΔhpaB (hpaH wt) and 85*ΔhpaBhpaHoof (hpaHoof). For better visualization of the HR, leaves were bleached in ethanol at 2 dpi. Dashed lines mark the infiltrated areas. All effector proteins were stably synthesized, as shown in Fig. S6 in the supplemental material. (D) Extracellular enzyme activities are unaffected in the absence of HpaH. Strains 85-10 (wt) and 85-10hpaHoof (hpaHoof) were incubated on NYG agar plates containing CMC, milk proteins, or starch as indicated. Halos were photographed at 2 dpi.
HpaH does not share functional redundancy with the putative LT HpaJ.
The results of the translocation and infection assays described above and in previous publications suggest that HpaH contributes to but is not essential for type III-dependent protein translocation (15, 27) (Fig. 1 and 6). Genome sequence analysis and expression studies revealed that X. campestris pv. vesicatoria strain 85-10 contains an additional predicted LT gene, hpaJ (XCV2440), which is coregulated with T3S genes and might share functional redundancy with hpaH (15, 45, 51). To analyze a potential influence of HpaJ on bacterial virulence, hpaJ wild-type and deletion mutant strains were inoculated into leaves of susceptible ECW plants. As expected, the wild-type X. campestris pv. vesicatoria strain induced disease symptoms in the form of water-soaked lesions, which became necrotic at 5 to 7 dpi (visible as a brown coloring of the infected area). Plant reactions were reduced in the absence of HpaH; however, deletion of hpaJ in strains 85-10 and 85-10hpaHoof did not lead to phenotypic changes or a reduction in bacterial in planta growth (Fig. 7A; see Fig. S7 in the supplemental material). We also tested a possible effect of the ectopic expression of hpaJ on the virulence of strain 85-10hpaHoof. Plant reactions were unaltered upon overexpression of hpaJ or hpaJ–c-myc under the control of the lac promoter in strain 85-10hpaHoof, suggesting that enhanced levels of HpaJ cannot compensate for the loss of HpaH (Fig. 7B). We therefore assume that HpaJ is dispensable for bacterial virulence and does not share functional redundancy with HpaH.
FIG 7.
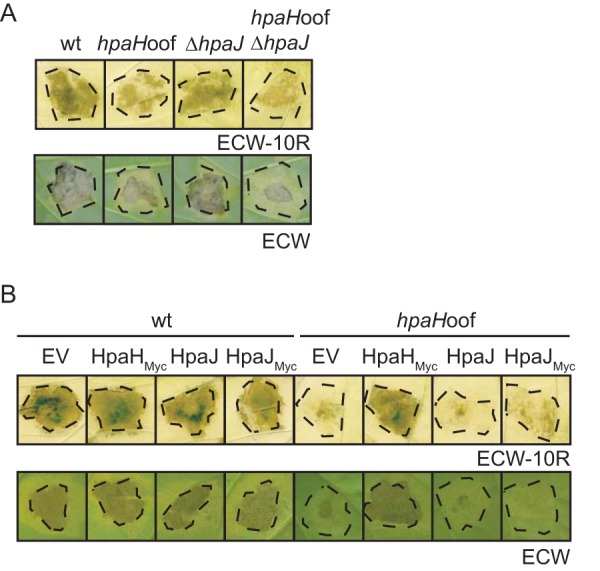
The putative LT HpaJ does not contribute to bacterial virulence. (A) Infection studies with hpaH and hpaJ mutants. X. campestris pv. vesicatoria strains 85-10 (wt), 85-10hpaHoof (hpaHoof), 85-10ΔhpaJ (ΔhpaJ), and 85-10ΔhpaHoofΔhpaJ (hpaHoofΔhpaJ) were inoculated into leaves of resistant ECW-10R and susceptible ECW pepper plants. Disease symptoms were photographed at 8 dpi. For better visualization of the HR, leaves were destained in ethanol at 2 dpi. Dashed lines mark the infiltrated areas. (B) Ectopic expression of hpaJ does not restore the wild-type phenotype in hpaH mutants. X. campestris pv. vesicatoria strains 85-10 (wt) and 85-10hpaHoof (hpaHoof) containing vector pBRM (EV) or expression constructs encoding HpaH–c-Myc (HpaHMyc), HpaJ, or HpaJ–c-Myc (HpaJMyc) as indicated were inoculated into leaves of resistant ECW-10R and susceptible ECW pepper plants. Disease symptoms were photographed at 8 dpi. For better visualization of the HR, leaves were bleached in ethanol at 2 dpi. Dashed lines mark the infiltrated areas.
DISCUSSION
In the present study, we characterized the predicted LT HpaH, which is a known virulence factor from X. campestris pv. vesicatoria (15, 27). A role in virulence was also reported for HpaH homologs (also designated Hpa2) from Xanthomonas oryzae pv. oryzae, X. oryzae pv. oryzicola, and Xanthomonas axonopodis pv. glycines (18, 19, 52). HpaH and homologs were predicted to promote the assembly of the T3S system by cleaving peptidoglycan in the periplasm. In agreement with this hypothesis, Hpa2 from X. oryzae pv. oryzae lyses bacterial cell walls (52). In the present study, the results of fractionation studies and the analysis of PhoA fusion proteins suggest that HpaH from X. campestris pv. vesicatoria is transported to the periplasm and interacts with peptidoglycan (Fig. 2). Given that the mutation of the predicted catalytic glutamate residue at position 58 abolished HpaH function, the predicted LT activity is presumably required for the contribution of HpaH to pathogenicity (Fig. 2). The predicted LT activity of HpaH, however, remains to be investigated because we could not yet obtain sufficient amounts of HpaH for enzyme activity assays.
The annotated HpaH protein contains a putative N-terminal Sec signal and therefore is likely transported via the Sec system across the IM. The predicted cleavage site is located after amino acid 34 of HpaH; however, a cleavage product of a similar size was observed for HpaHAS, which lacks a predicted Sec signal (Fig. 1). It remains to be investigated whether the cleavage of HpaH is linked to its predicted transport via the Sec system. Complementation studies showed that the predicted Sec signal of HpaH contributes to protein function and can be functionally replaced by the signal peptide of the OM secretin HrcC (Fig. 1). Unexpectedly, however, HpaHΔ2–34, which lacks the predicted Sec signal, was partially functional whereas deletion of the N-terminal 35 amino acids abolished HpaH function (Fig. 1). Given the predicted function of HpaH as a periplasmic LT, it is possible that small amounts of HpaHΔ2–34 are still transported into the periplasm, possibly by a Sec-independent mechanism. In agreement with this hypothesis, the activity of an HpaHΔ2–34-PhoAΔ2–120 fusion protein was significantly reduced but not abolished (Fig. 2). Yet, it cannot be excluded that the PhoAΔ2–120 fusion partner or the endogenous hpaH gene of the phoA deletion mutant, which was used for this assay, interfered with the transport of HpaHΔ2–34 into the periplasm. This will be addressed in future experiments.
In agreement with the predicted role of HpaH as periplasmic LT, we detected interactions between HpaH and periplasmic components of the T3S systems. Thus, the results of in vitro interaction studies suggest that HpaH interacts with the pilus protein HrpE, the IM protein HrcD, and the predicted inner rod proteins HrpB1 and HrpB2 (Fig. 3). The association of HpaH with HrpB1 and HrpB2 was confirmed by the results of coimmunoprecipitation experiments with X. campestris pv. vesicatoria lysates (Fig. 3). HpaH was also found in a complex with the OM secretin HrcC, which was not identified as an HpaH interaction partner by GST pulldown assays (Fig. 3). HrcC might, therefore, indirectly interact with HpaH. An interaction between the periplasmic domain of the OM secretin HrcC was previously reported for the predicted LT VrpA from Xanthomonas citri subsp. citri (21). Similarly to HpaH, VrpA localizes to the bacterial periplasm and contributes to T3S (21).
In X. campestris pv. vesicatoria, the interaction of HpaH with HrcC might occur via the predicted inner rod structure, which consists of HrpB1 and HrpB2. HrpB1 was previously reported to associate with HrcC as well as with HrpB2 and the pilus protein HrpE, suggesting that the predicted inner rod is connected to the pilus of the T3S system in the periplasm (34, 35). Furthermore, HrpB1 interacts with peptidoglycan and shares predicted structural similarity with the putative peptidoglycan-binding domain of the LT Slt70 from E. coli (35). It was therefore proposed that HrpB1 facilitates the stable anchoring of the T3S system to peptidoglycan. The interaction of HpaH with HrpB1 is reminiscent of the previous finding that the LT EtgA from EPEC interacts with the putative inner rod protein EscI (31, 53). The binding of LTs to periplasmic components of the T3S system might help to control LT activity and to ensure that the degradation of peptidoglycan is locally restricted. In agreement with this hypothesis, the interaction of the putative inner rod protein EscI with the LT EtgA from EPEC leads to an increase in EtgA activity (31).
The mechanisms that control the transport of HpaH into the periplasm and its association with components of the T3S system remain to be investigated. The periplasm is presumably not the final destination of HpaH, suggesting that its association with peptidoglycan and components of the T3S system is only transient. As was reported for other LTs, HpaH is released into the extracellular milieu, possibly to avoid a periplasmic accumulation of HpaH and thus deleterious effects resulting from enhanced local degradation of peptidoglycan (17, 18). Secreted putative LTs include HrpH and HopP1 from P. syringae, which are translocated into the plant cell (17). Furthermore, type III-dependent secretion was reported for the HpaH homolog Hpa2 from X. oryzae pv. oryzicola, which lacks a predicted Sec signal (18). Interestingly, efficient secretion of Hpa2 depends on the T3S chaperone HpaB, which promotes effector protein secretion (18). In contrast to Hpa2, HpaH from X. campestris pv. vesicatoria is secreted in a T3S-independent manner (Fig. 4). The transport pathway which mediates secretion of HpaH across the OM remains to be identified. We did not detect a contribution of T2S systems or OMVs to the secretion of HpaH (Fig. 5). In future studies, therefore, we will investigate a possible influence of additional protein secretion systems, including type I and type IV secretion systems, to the transport of HpaH across the OM. X. campestris pv. vesicatoria contains predicted components of type I to type VI secretion systems, including two type IV secretion systems, which have not yet been studied (51).
In summary, we conclude from our data that the predicted LT HpaH from X. campestris pv. vesicatoria is transported to the bacterial periplasm, possibly via the Sec system, and interacts with periplasmic components of the T3S system and peptidoglycan. The predicted lytic activity of HpaH presumably induces a local degradation of peptidoglycan, which might facilitate the assembly of the T3S system and thus the translocation of T3S substrates into plant cells (Fig. 8). In agreement with this hypothesis, HpaH promotes the T3S-dependent translocation of different effector proteins as well as of the translocon protein HrpF and the predicted inner rod protein HrpB2 (Fig. 6). The subsequent secretion of HpaH into the extracellular milieu presumably helps to control the local degradation of peptidoglycan and depends on a yet-unknown transport pathway, which will be the subject of future studies.
FIG 8.
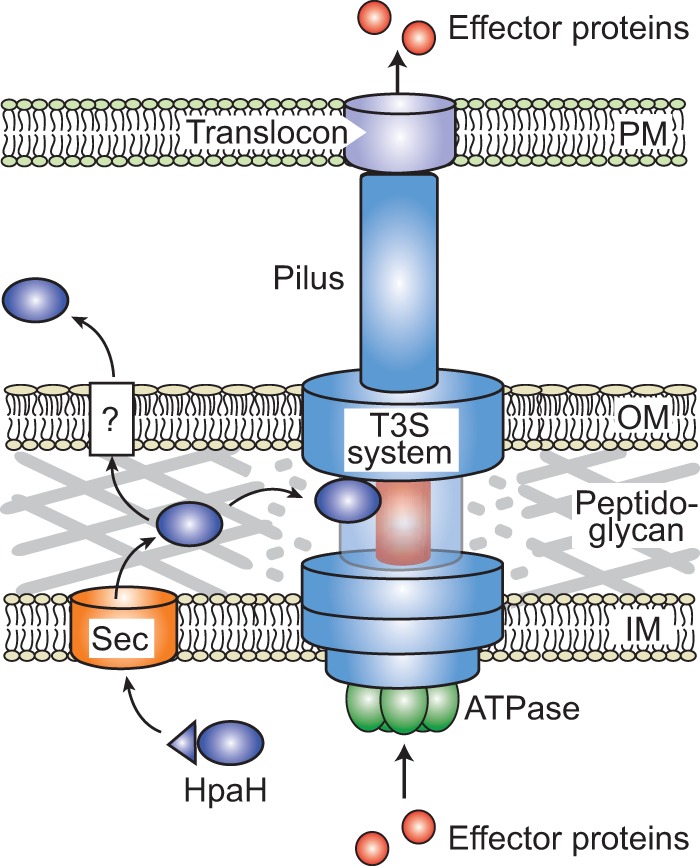
Model of HpaH function during T3S. HpaH contains a predicted Sec signal (indicated as a triangle), which is cleaved off during the transport of HpaH into the periplasm. HpaH associates with periplasmic components of the membrane-spanning T3S system, including components of the putative inner rod (depicted in red), and presumably leads to the local degradation of peptidoglycan. HpaH might thus promote the assembly of the T3S system and the type III-dependent translocation of effector proteins. The association of HpaH with the T3S system is presumably transient, because HpaH is also secreted into the extracellular milieu via a yet-unknown mechanism (indicated by a question mark). IM, inner membrane; OM, outer membrane; PM, plasma membrane.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains and plasmids that were used in this study are listed in Table 1. Escherichia coli cells were cultivated at 37°C in lysogeny broth (LB) and X. campestris pv. vesicatoria strains at 30°C in nutrient-yeast-glycerol (NYG) medium (54) or in minimal medium A (55) supplemented with sucrose (10 mM) and Casamino Acids (0.3%).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristicsa | Reference(s) or source |
|---|---|---|
| Strains | ||
| X. campestris pv. vesicatoria | ||
| 85-10 | Pepper race 2; wild type; Rifr | 56, 66 |
| 85-10ΔEEE | Derivative of strain 85-10 deleted in the predicted ATPase-encoding genes xpsE, xcsE, and XCV4312 of the Xps and Xcs T2S systems | 43 |
| 85* | 85-10 derivative containing the hrpG* mutation | 33 |
| 85*ΔhpaB | Derivative of strain 85* deleted in the T3S chaperone gene hpaB | 25 |
| 85*ΔhpaC | Derivative of strain 85* deleted in the T3S4 gene hpaC | 26 |
| 85-10hpaHoof | 85-10 derivative containing a frameshift mutation at codon 38 in hpaH | 15 |
| 85*hpaHoof | 85* derivative containing a frameshift mutation in hpaH | 15 |
| 85*ΔhpaBhpaHoof | Derivative of strain 85*ΔhpaB containing a frameshift mutation in hpaH | This study |
| 85*ΔhpaChpaHoof | Derivative of strain 85*ΔhpaC containing a frameshift mutation in hpaH | This study |
| 85-10ΔhpaJ | 85-10 derivative deleted in codons 2 to 425 of hpaJ | This study |
| 85-10hpaHoofΔhpaJ | 85-10 derivative containing a frameshift mutation in hpaH and a deletion in hpaJ | This study |
| 85-10ΔphoA | 85-10 derivative containing a 1,431-bp in-frame deletion in the phoA gene | This study |
| 85*ΔphoA | 85* derivative containing a 1,431-bp in-frame deletion in the phoA gene | This study |
| 85*ΔhrcV | 85* derivative deleted in codons 324 to 642 of hrcV | 63 |
| 85E*ΔhrcC | 85* derivative with a mutation in the eps gene cluster and deleted in hrcC | 67 |
| E. coli | ||
| BL21(DE3) | F− ompT hsdSB (rB− mB−) gal dcm (DE3) | Stratagene |
| DH5α λpir | F− recA hsdR17(rK− mK+) ϕ80dlacZΔM15 (λpir) | 68 |
| Top10 | F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 recA1 araΔ139 Δ(ara-leu)7697 galU galK rpsL endA1 nupG | Invitrogen |
| Plasmids | ||
| pBRM | Golden Gate-compatible derivative of pBBR1MCS-5 containing BsaI sites flanked by the lac promoter and a 3× c-Myc epitope-encoding sequence, respectively; Gmr | 43 |
| pBRM-Stop | Derivative of pBRM containing a stop codon upstream of the c-Myc epitope-encoding sequence | This study |
| pBRM-P | Derivative of pBRM lacking the lac promoter upstream of the BsaI recognition site; Gmr | 43 |
| pBRMhpaH | Derivative of pBRM encoding HpaH–c-Myc | This study |
| pBRMhpaHΔ2–34 | Derivative of pBRM encoding HpaHΔ2–34–c-Myc | This study |
| pBRMhpaHΔ2–35 | Derivative of pBRM encoding HpaHΔ2–35–c-Myc | This study |
| pBRMhpaHE58A | Derivative of pBRM encoding HpaHE58A–c-Myc | This study |
| pBRMhrcC1–33-hpaHΔ2–34 | Derivative of pBRM encoding HrcC1–33-HpaHΔ2–34–c-Myc | This study |
| pBRMhpaHAS | Derivative of pBRM encoding HpaHAS–c-Myc | This study |
| pBRM-PhpaHAS | Derivative of pBRM-P containing hpaH and 321-bp upstream region | This study |
| pBRM-PhpaHAS/CTG | Derivative of pBRM-P containing hpaH with 321-bp upstream region and a mutation of the annotated start codon to CTG | This study |
| pBRMhpaH-phoAΔ2-120STOP | Derivative of pBRM encoding HpaH-PhoAΔ2–120 | This study |
| pBRMhpaHΔ2–34-phoAΔ2-120STOP | Derivative of pBRM encoding HpaHΔ2–34-PhoAΔ2–120 | This study |
| pBRMhpaHΔ2–35-phoAΔ2-120STOP | Derivative of pBRM encoding HpaHΔ2–35-PhoAΔ2–120 | This study |
| pBRMhpaJ | Derivative of pBRM encoding HpaJ–c-Myc | This study |
| pBRMhpaJSTOP | Derivative of pBRM encoding HpaJ | This study |
| pBRM4360 | Derivative of pBRM encoding XCV4360–c-Myc | 44 |
| pDEST17 | Gateway-compatible expression vector, contains the T7 promoter and a 6× His-encoding sequence; Ampr | Invitrogen |
| pDEST17hpaH | Derivative of pDEST17 encoding His6-HpaH | This study |
| pDSK602 | Broad-host-range vector; contains triple lacUV5 promoter; Smr | 69 |
| pDSK604 | Derivative of pDSK602 containing an alternative polylinker | 46 |
| pDSF300 | Derivative of pDSK602 encoding AvrBs3-FLAG | 70 |
| pDSM400 | Derivative of pDSK602 encoding AvrBsT–c-Myc | 46 |
| pEGG | Golden Gate-compatible pENTR/D; Kmr | U. Bonas et al., unpublished |
| pEGGhpaH | Derivative of pEGG containing hpaH | This study |
| pGEX-2TKM | GST expression vector; ptac GST, lacIq, pBR322 ori; Apr; derivative of pGEX-2TK with polylinker of pDSK604 | 46, Stratagene |
| pGhpaB | Derivative of pGEX-2TKM encoding GST-HpaB | 26 |
| pGhrcC | Derivative of pGEX-2TKM encoding GST-HrcC | 71 |
| pGhrcD | Derivative of pGEX-2TKM encoding GST-HrcD | 35 |
| pGhrcU255–357 | Derivative of pGEX-2TKM encoding GST-HrcU255–357 | 24 |
| pGhrcV324–645 | Derivative of pGEX-2TKM encoding GST-HrcV324–645 | 71 |
| pGhrpB1 | Derivative of pGEX-2TKM encoding GST-HrpB1 | 35 |
| pGhrpB2 | Derivative of pGEX-2TKM encoding GST-HrpB2 | 72 |
| pGhrpE | Derivative of pGEX-2TKM encoding GST-HrpE | 61 |
| pOGG2 | Golden Gate-compatible derivative of pOK1; Smr | 73 |
| pOGG2hpaJ | Derivative of pOGG2 containing the flanking regions of hpaJ | This study |
| pOGG2phoA | Derivative of pOGG2 containing the flanking regions of phoA | This study |
| pRK2013 | ColE1 replicon, TraRK+ Mob+; Kmr | 74 |
| pUC57ΔBsaI | Derivative of pUC57 with mutated BsaI site | 75 |
| pUC57ΔBsaIhpaH | Derivative of pUC57ΔBsaI containing hpaH | This study |
| pL6hpaAN356 | Derivative of pLAFR6 encoding HpaA1–72-AvrBs3Δ2 | 48 |
| pL6xopL(1-92)-356 | Derivative of pLAFR6 encoding XopL1–92-AvrBs3Δ2 | 49 |
| pL6xopC356 | Derivative of pLAFR6 encoding XopC1–200-AvrBs3Δ2 | 45 |
Ap, ampicillin; Km, kanamycin; Rif, rifampin; Sm, spectinomycin; Gm, gentamicin; r, resistant.
Plant material and plant inoculations.
X. campestris pv. vesicatoria strains were inoculated into leaves of the near-isogenic pepper cultivars Early Cal Wonder (ECW), ECW-10R, ECW-20R, and ECW-30R at a concentration of 1 × 108 CFU ml−1 in 10 mM MgCl2 if not stated otherwise (28, 56, 57). After infection, plants were incubated in an incubation chamber for 16 h of light at 28°C and 65% humidity and 8 h of darkness at 22°C and 65% humidity. The appearance of disease symptoms and the hypersensitive response (HR) were scored over a period of 1 to 8 dpi. For the better visualization of the HR, leaves were destained in 70% ethanol. Experiments were repeated at least twice. In planta bacterial growth curves were determined as described previously (57).
Generation of expression constructs.
For the generation of expression constructs, hpaJ, hpaH, and derivatives thereof were amplified with or without stop codons by PCR from X. campestris pv. vesicatoria strain 85-10 and cloned into the Golden Gate-compatible expression vector pBRM, which contains a C-terminal c-Myc epitope-encoding sequence downstream and a lac promoter upstream of the cloning site (43, 58). Alternatively, PCR amplicons were cloned into vector pBRM-Stop, which was generated by PCR amplification of pBRM with phosphorylated primers that introduced a stop codon upstream of the c-Myc epitope-encoding sequence. Ligation of the PCR amplicon resulted in vector pBRM-Stop. To generate an expression construct encoding HpaHE58A, pBRMhpaH was amplified with primers containing BsaI sites and the mutation leading to the E58A exchange. The PCR amplicons were digested with BsaI and ligated, resulting in pBRMhpaHE58A. To express hpaH under the control of the native promoter, hpaH with 321 bp upstream region was amplified by PCR and cloned into pBRM-P, which is a derivative of pBRM and lacks the lac promoter. The mutation leading to the exchange of the annotated start codon to CTG was introduced by primer sequences. To obtain constructs encoding N- or C-terminally His6-tagged derivatives of HpaH and HpaHΔ2–35, respectively, the His6 epitope-encoding sequence was introduced by primers. For in vitro transcription/translation, hpaH was amplified by PCR and first inserted into the SmaI site of vector pUC57ΔBsaI in a restriction-ligation reaction (59). hpaH was subsequently cloned into the Golden Gate-compatible vector pEGG and then recombined into plasmid pDEST17, resulting in pDEST17hpaH.
For the generation of an alkaline phosphatase (phoA) expression construct, a fragment spanning nucleotides 361 to 1690 and including the stop codon of phoA from E. coli (GenBank accession number M29668) was amplified. The amplicon, which encodes an N-terminal derivative of PhoA lacking the signal peptide (PhoAΔ2–120) (36), was ligated with hpaH, hpaHΔ2–34, or hpaHΔ2–35 into the BsaI sites of the Golden Gate-compatible expression vector pBRM in a single restriction/ligation reaction. All plasmids and constructs are summarized in Table 1, and primer sequences are listed in Table S1 in the supplemental material.
Expression studies with hpaH derivatives.
To express hpaH and derivatives thereof in E. coli BL21(DE3), bacteria were transformed with constructs encoding N- or C-terminally His6-tagged or C-terminally c-Myc epitope-tagged HpaH derivatives and cultivated in LB medium at 37°C until the optical density at 600 nm (OD600) had reached a value of 0.4. After addition of isopropyl-β-d-thiogalactopyranoside at a final concentration of 2 mM, bacteria were incubated for 2 h at 37°C or 30°C in the presence of 3% (vol/vol) ethanol. Cell extracts were analyzed by SDS-PAGE and immunoblotting using antibodies specific for the His6 or the c-Myc epitope (data not shown). HpaH was stably synthesized only when present as a deletion derivative lacking the predicted Sec signal. In contrast, full-length HpaH derivatives were not detected in E. coli cell extracts and could not be purified in sufficient amounts from X. campestris pv. vesicatoria (data not shown).
Coupled in vitro transcription and translation.
For in vitro transcription and translation of hpaH, we used construct pDEST17hpaH and the TNT Quick coupled transcription/translation system (Promega GmbH). Reactions were performed according to the manufacturer's instructions.
Generation of X. campestris pv. vesicatoria deletion mutants.
To generate a 1,272-bp in-frame deletion of codons 2 to 425 of hpaJ (total length, 1,278 bp), 750-bp fragments downstream of codon 2 and upstream of codon 425 of hpaJ were amplified by PCR and cloned into the Golden Gate-compatible suicide vector pOGG2. The resulting construct, pOGG2hpaJ, was conjugated into X. campestris pv. vesicatoria strains 85-10 and 85-10hpaHoof. Double crossovers resulted in strains 85-10ΔhpaJ and 85-10hpaHoofΔhpaJ, respectively, which were selected as described previously (60).
For the generation of a phoA deletion mutant, a 702-bp fragment upstream of nucleotide 303 and a 651-bp fragment downstream of nucleotide 1734 of phoA (XCV2913; total length, 1,734 bp) were amplified by PCR and cloned into the Golden Gate-compatible suicide vector pOGG2. The resulting vector, pOGG2phoA, was conjugated into strains 85-10 and 85*. Double crossovers led to the generation of strain 85-10ΔphoA, which contains a 1,431-bp in-frame deletion in the phoA gene.
Generation of polyclonal HpaH antibodies.
For the generation of HpaH antibodies, rabbits were immunized with an HpaH-specific peptide (VGAYHSETPGERDK, amino acids 127 to 140) (Biogenes). The serum after the second booster injection was used for immunoblot analysis.
In vitro secretion assays and immunoblot analyses.
In vitro secretion assays were performed as described previously (32, 43). Equal amounts of bacterial total cell extracts and culture supernatants were analyzed by SDS-PAGE and immunoblotting. Immunoblot analyses were performed using antibodies specific for the c-Myc epitope (Roche Applied Science), the predicted periplasmic inner rod proteins HrpB1 and HrpB2, the translocon protein HrpF, the inner membrane protein HrcJ, the putative LT HpaH and the effector protein AvrBs3 (61–63). Horseradish peroxidase-labeled anti-rabbit or anti-mouse antibodies (GE Healthcare) were used as secondary antibodies. For the analysis of protein secretion via outer membrane vesicles, bacteria were incubated on a rotary shaker overnight at 30°C and supernatants were incubated with proteinase K as described previously (44). Experiments were performed three times with similar results.
Analysis of extracellular enzyme activities.
For the analysis of extracellular protease, cellulase, and amylase activities, X. campestris pv. vesicatoria strains were incubated at a density of 109 CFU ml−1 on NYG plates containing 1% agar and 1% skim milk, 1% carboxymethyl cellulose (CMC), or 1% starch. Plates were incubated at 30°C for 2 days, and bacteria were removed before documentation of the halos.
Subcellular fractionation studies.
Subcellular fractionation studies were performed according to a previously described protocol (64). For this, X. campestris pv. vesicatoria strains grown overnight in minimal medium were incubated in secretion medium for several hours until the culture had reached an OD600 of approximately 0.8. After centrifugation, bacterial cells were resuspended in 10 ml resuspension buffer (0.2 M Tris-HCl [pH 8.0], 1 M sucrose, 1 mM EDTA, and lysozyme at a final concentration of 1 mg/ml) and incubated for 5 min at room temperature. The suspension was mixed with 40 ml deionized water and incubated for 30 min on ice, and cells were centrifuged at 200,000 × g for 45 min at 4°C. The supernatant contained the fraction enriched in the periplasm. The pellet was resuspended in 7.5 ml 10 mM Tris-HCl (pH 7.5), 5 mM EDTA, 0.2 mM dithiothreitol (DTT), and DNase at a final concentration of 1 mg/ml with an Ultra-Turrax. Cells were broken with a French press, and unbroken cells were removed by centrifugation (1,789 × g for 10 min at 4°C). The supernatant was subsequently centrifuged for 2 to 3 h at 300,000 × g at 4°C. The supernatant after the last centrifugation step contained the cytoplasm-enriched fraction. The pellet corresponds to the membrane-enriched fraction and was resuspended in 9 ml 50 mM Tris-HCl (pH 8.0), 2% (wt/vol) Triton X-100, and 10 mM MgCl2. After centrifugation (85,000 × g for 30 min at 4°C), the supernatant contained the IM-enriched fraction whereas the pellet corresponded to the OM-enriched fraction and was washed in 1 ml 50 mM Tris-HCl (pH 8.0), 2% (wt/vol) Triton X-100, and 10 mM MgCl2. The sample was centrifuged as described above, and the pellet was washed three times in 500 μl deionized water before it was resuspended in 1 ml Laemmli buffer. Proteins in the periplasm-, cytoplasm-, and IM-enriched fractions were precipitated using trichloroacetic acid (TCA) and sodium-deoxycholate (DOC) as described previously (34). Fifteen microliters of each fraction was analyzed by SDS-PAGE and immunoblotting. The experiment was repeated three times with similar results.
Peptidoglycan-binding assay.
To analyze a putative binding of in vitro-synthesized HpaH to peptidoglycan, a peptidoglycan-binding assay was performed as described previously (35). Briefly, in vitro-transcribed and translated HpaH, 150 μg lysozyme (Applichem), and 150 μg bovine serum albumin (BSA) (Roth) were preincubated in phosphate-buffered saline (PBS) and centrifuged at 21,130 × g for 30 min at 4°C to precipitate insoluble proteins. The supernatants were subsequently incubated with 150 μg insoluble peptidoglycan from E. coli K-12 (Invivogen) in 1 ml PBS for 1 h at 4°C. After centrifugation at 21,130 × g for 20 min at 4°C, precipitated peptidoglycan was washed three times with 1 ml PBS to remove unbound proteins. Bound proteins were eluted with 100 μl Laemmli buffer, and boiled samples were analyzed by SDS-PAGE, Coomassie blue staining, or immunoblotting using antibodies specific for HpaH. The experiment was performed three times with similar results.
GST pulldown assays.
For the analysis of protein-protein interactions, bacterial cells from 50-ml cultures were resuspended in PBS and broken with a French press. After centrifugation, GST and GST fusion proteins were immobilized on a glutathione-Sepharose matrix according to the manufacturer's instructions. After several washing steps, the matrix was incubated with 600 μl E. coli cell lysate containing the putative interaction partner for 1 to 2 h at 4°C. The matrix was washed four times with PBS, and bound proteins were eluted with 10 mM reduced glutathione at room temperature for 2 h. Total protein lysates and eluted proteins were analyzed by SDS-PAGE and immunoblotting using antibodies specific for the c-Myc epitope and GST, respectively. Experiments were performed at least three times with similar results.
Coimmunoprecipitation studies.
For coimmunoprecipitation studies, 100-ml bacterial cultures were grown in secretion medium (minimal medium A [pH 5.3] supplemented with BSA [50 mg/ml] and thiamine). Cells were harvested by centrifugation, resuspended in 1 ml coimmunoprecipitation buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 3 mM MgCl2, 1 mM CaCl2, 2.5% glycerol), and broken with a French press. The cell lysate was incubated with 0.1% Nonidet P-40 for 15 min on ice. Cell debris was removed by centrifugation, and the supernatant was incubated with 20 μl protein G-agarose beads for 1 h at 4°C. The mixture was subsequently centrifuged for 90 s at 500 × g, and 450 μl of the supernatant was incubated with 60 μl protein G-agarose in the presence or absence of 4 ng anti-c-Myc antibody overnight at 4°C. The protein G-agarose was washed three times with coimmunoprecipitation buffer and resuspended in 50 μl Laemmli buffer. Cell lysates and unbound and immunoprecipitated proteins were analyzed by immunoblotting.
PhoA activity assays.
The PhoA activity assays were performed according to a protocol described previously (65). Briefly, bacteria grown overnight in MA medium were resuspended in secretion medium at an OD600 of 0.3. After 3 h of incubation at 30°C, the OD600 was measured, and cells of 500 μl of each culture (in triplicates) were collected by centrifugation and resuspended in resuspension buffer (0.1 M Tris-HCl [pH 7.8], 5 mM EDTA). The cells were incubated for 15 min at 30°C, permeabilized by addition of chloroform and SDS as described previously (65), and incubated as described above. Five hundred microliters of para-nitrophenylphosphate (pNPP) buffer (20 mM pNPP, 1 M Tris-HCl [pH 9.0], 10 mM MgCl2, 10 mM CaCl2) was added, and the samples were incubated at room temperature until a color change was observed. The reaction was stopped by addition of 500 μl 2 M NaOH, cell debris was pelleted by centrifugation, and the optical density of the culture supernatant was measured at 420 nm. PhoA activity was calculated as OD420 × 1,000/time (min) × OD600 × 18 (mmol−1 × cm−1) × 1 cm as units/liter (18 mmol−1 × cm−1 is the extinction coefficient of para-nitrophenol). To investigate a potential activity and thus a correct folding of PhoA fusion proteins in the absence of a periplasmic localization, proteins were analyzed in lysates of E. coli DH5α λpir cells. For this, cells were resuspended as described above and incubated for 24 h on ice before the PhoA activity was measured (40). Derivatives of PhoA were detected by immunoblotting using PhoA-specific antibodies (Thermo Fisher).
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to U. Bonas for comments on the manuscript.
This work was supported by grants from the Deutsche Forschungsgemeinschaft (BU 2145/1-2, BU 2145/5-1, and CRC 648 “Molecular mechanisms of information processing in plants” project A8) to D.B.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00788-16.
REFERENCES
- 1.Ghosh P. 2004. Process of protein transport by the type III secretion system. Microbiol Mol Biol Rev 68:771–795. doi: 10.1128/MMBR.68.4.771-795.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dean P. 2011. Functional domains and motifs of bacterial type III effector proteins and their roles in infection. FEMS Microbiol Rev 35:1100–1125. doi: 10.1111/j.1574-6976.2011.00271.x. [DOI] [PubMed] [Google Scholar]
- 3.Büttner D. 2012. Protein export according to schedule—architecture, assembly and regulation of type III secretion systems from plant and animal pathogenic bacteria. Microbiol Mol Biol Rev 76:262–310. doi: 10.1128/MMBR.05017-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mattei PJ, Faudry E, Job V, Izore T, Attree I, Dessen A. 2011. Membrane targeting and pore formation by the type III secretion system translocon. FEBS J 278:414–426. doi: 10.1111/j.1742-4658.2010.07974.x. [DOI] [PubMed] [Google Scholar]
- 5.Hueck CJ. 1998. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev 62:379–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He SY, Nomura K, Whittam TS. 2004. Type III protein secretion mechanism in mammalian and plant pathogens. Biochim Biophys Acta 1694:181–206. doi: 10.1016/j.bbamcr.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 7.Lorenz C, Hausner J, Büttner D. 2012. HrcQ provides a docking site for early and late type III secretion substrates from Xanthomonas. PLoS One 7:e51063. doi: 10.1371/journal.pone.0051063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morita-Ishihara T, Ogawa M, Sagara H, Yoshida M, Katayama E, Sasakawa C. 2006. Shigella Spa33 is an essential C-ring component of type III secretion machinery. J Biol Chem 281:599–607. doi: 10.1074/jbc.M509644200. [DOI] [PubMed] [Google Scholar]
- 9.Akeda Y, Galan JE. 2005. Chaperone release and unfolding of substrates in type III secretion. Nature 437:911–915. doi: 10.1038/nature03992. [DOI] [PubMed] [Google Scholar]
- 10.Lorenz C, Büttner D. 2009. Functional characterization of the type III secretion ATPase HrcN from the plant pathogen Xanthomonas campestris pv. vesicatoria. J Bacteriol 191:1414–1428. doi: 10.1128/JB.01446-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holtje JV. 1998. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol Mol Biol Rev 62:181–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scheurwater EM, Burrows LL. 2011. Maintaining network security: how macromolecular structures cross the peptidoglycan layer. FEMS Microbiol Lett 318:1–9. doi: 10.1111/j.1574-6968.2011.02228.x. [DOI] [PubMed] [Google Scholar]
- 13.Scheurwater E, Reid CW, Clarke AJ. 2008. Lytic transglycosylases: bacterial space-making autolysins. Int J Biochem Cell Biol 40:586–591. doi: 10.1016/j.biocel.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 14.Yu YC, Lin CN, Wang SH, Ng SC, Hu WS, Syu WJ. 2010. A putative lytic transglycosylase tightly regulated and critical for the EHEC type three secretion. J Biomed Sci 17:52. doi: 10.1186/1423-0127-17-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noël L, Thieme F, Nennstiel D, Bonas U. 2002. Two novel type III system-secreted proteins of Xanthomonas campestris pv. vesicatoria are encoded within the hrp pathogenicity island. J Bacteriol 184:1340–1348. doi: 10.1128/JB.184.5.1340-1348.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia-Gomez E, Espinosa N, de la Mora J, Dreyfus G, Gonzalez-Pedrajo B. 2011. The muramidase EtgA from enteropathogenic Escherichia coli is required for efficient type III secretion. Microbiology 157:1145–1160. doi: 10.1099/mic.0.045617-0. [DOI] [PubMed] [Google Scholar]
- 17.Oh HS, Kvitko BH, Morello JE, Collmer A. 2007. Pseudomonas syringae lytic transglycosylases coregulated with the type III secretion system contribute to the translocation of effector proteins into plant cells. J Bacteriol 189:8277–8289. doi: 10.1128/JB.00998-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li YR, Che YZ, Zou HS, Cui YP, Guo W, Zou LF, Biddle EM, Yang CH, Chen GY. 2011. Hpa2 required by HrpF to translocate Xanthomonas oryzae transcriptional activator-like effectors into rice for pathogenicity. Appl Environ Microbiol 77:3809–3818. doi: 10.1128/AEM.02849-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim JG, Park BK, Yoo CH, Jeon E, Oh J, Hwang I. 2003. Characterization of the Xanthomonas axonopodis pv. glycines Hrp pathogenicity island. J Bacteriol 185:3155–3166. doi: 10.1128/JB.185.10.3155-3166.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferreira RM, Moreira LM, Ferro JA, Soares MR, Laia ML, Varani AM, de Oliveira JC, Ferro MI. 2016. Unravelling potential virulence factor candidates in Xanthomonas citri subsp. citri by secretome analysis. Peer J 4:e1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou X, Hu X, Li J, Wang N. 2015. A novel periplasmic protein, VrpA, contributes to efficient protein secretion by the type III secretion system in Xanthomonas spp. Mol Plant Microbe Interact 28:143–153. doi: 10.1094/MPMI-10-14-0309-R. [DOI] [PubMed] [Google Scholar]
- 22.Jones JB, Lacy GH, Bouzar H, Stall RE, Schaad NW. 2004. Reclassification of the xanthomonads associated with bacterial spot disease of tomato and pepper. Syst Appl Microbiol 27:755–762. doi: 10.1078/0723202042369884. [DOI] [PubMed] [Google Scholar]
- 23.Büttner D, Bonas U. 2010. Regulation and secretion of Xanthomonas virulence factors. FEMS Microbiol Rev 34:107–133. doi: 10.1111/j.1574-6976.2009.00192.x. [DOI] [PubMed] [Google Scholar]
- 24.Lorenz C, Schulz S, Wolsch T, Rossier O, Bonas U, Büttner D. 2008. HpaC controls substrate specificity of the Xanthomonas type III secretion system. PLoS Pathog 4:e1000094. doi: 10.1371/journal.ppat.1000094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Büttner D, Gürlebeck D, Noël LD, Bonas U. 2004. HpaB from Xanthomonas campestris pv. vesicatoria acts as an exit control protein in type III-dependent protein secretion. Mol Microbiol 54:755–768. doi: 10.1111/j.1365-2958.2004.04302.x. [DOI] [PubMed] [Google Scholar]
- 26.Büttner D, Lorenz C, Weber E, Bonas U. 2006. Targeting of two effector protein classes to the type III secretion system by a HpaC- and HpaB-dependent protein complex from Xanthomonas campestris pv. vesicatoria. Mol Microbiol 59:513–527. doi: 10.1111/j.1365-2958.2005.04924.x. [DOI] [PubMed] [Google Scholar]
- 27.Büttner D, Noël L, Stuttmann J, Bonas U. 2007. Characterization of the non-conserved hpaB-hrpF region in the hrp pathogenicity island from Xanthomonas campestris pv. vesicatoria. Mol Plant Microbe Interact 20:1063–1074. doi: 10.1094/MPMI-20-9-1063. [DOI] [PubMed] [Google Scholar]
- 28.Minsavage GV, Dahlbeck D, Whalen MC, Kearny B, Bonas U, Staskawicz BJ, Stall RE. 1990. Gene-for-gene relationships specifying disease resistance in Xanthomonas campestris pv. vesicatoria-pepper interactions. Mol Plant Microbe Interact 3:41–47. doi: 10.1094/MPMI-3-041. [DOI] [Google Scholar]
- 29.Jones JD, Dangl JL. 2006. The plant immune system. Nature 444:323–329. doi: 10.1038/nature05286. [DOI] [PubMed] [Google Scholar]
- 30.van Asselt EJ, Thunnissen AM, Dijkstra BW. 1999. High resolution crystal structures of the Escherichia coli lytic transglycosylase Slt70 and its complex with a peptidoglycan fragment. J Mol Biol 291:877–898. doi: 10.1006/jmbi.1999.3013. [DOI] [PubMed] [Google Scholar]
- 31.Burkinshaw BJ, Deng W, Lameignere E, Wasney GA, Zhu H, Worrall LJ, Finlay BB, Strynadka NC. 2015. Structural analysis of a specialized type III secretion system peptidoglycan-cleaving enzyme. J Biol Chem 290:10406–10417. doi: 10.1074/jbc.M115.639013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rossier O, Wengelnik K, Hahn K, Bonas U. 1999. The Xanthomonas Hrp type III system secretes proteins from plant and mammalian pathogens. Proc Natl Acad Sci U S A 96:9368–9373. doi: 10.1073/pnas.96.16.9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wengelnik K, Rossier O, Bonas U. 1999. Mutations in the regulatory gene hrpG of Xanthomonas campestris pv. vesicatoria result in constitutive expression of all hrp genes. J Bacteriol 181:6828–6831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartmann N, Schulz S, Lorenz C, Fraas S, Hause G, Büttner D. 2012. Characterization of HrpB2 from Xanthomonas campestris pv. vesicatoria identifies protein regions that are essential for type III secretion pilus formation. Microbiology 158:1334–1349. doi: 10.1099/mic.0.057604-0. [DOI] [PubMed] [Google Scholar]
- 35.Hausner J, Hartmann N, Lorenz C, Büttner D. 2013. The periplasmic HrpB1 protein from Xanthomonas spp. binds to peptidoglycan and to components of the type III secretion system. Appl Environ Microbiol 79:6312–6324. doi: 10.1128/AEM.01226-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alexeyev MF, Winkler HH. 1999. Membrane topology of the Rickettsia prowazekii ATP/ADP translocase revealed by novel dual pho-lac reporters. J Mol Biol 285:1503–1513. doi: 10.1006/jmbi.1998.2412. [DOI] [PubMed] [Google Scholar]
- 37.Berger C, Robin GP, Bonas U, Koebnik R. 2010. Membrane topology of conserved components of the type III secretion system from the plant pathogen Xanthomonas campestris pv. vesicatoria. Microbiology 156:1963–1974. doi: 10.1099/mic.0.039248-0. [DOI] [PubMed] [Google Scholar]
- 38.Manoil C, Beckwith J. 1986. A genetic approach to analyzing membrane protein topology. Science 233:1403–1408. doi: 10.1126/science.3529391. [DOI] [PubMed] [Google Scholar]
- 39.van Geest M, Lolkema JS. 2000. Membrane topology and insertion of membrane proteins: search for topogenic signals. Microbiol Mol Biol Rev 64:13–33. doi: 10.1128/MMBR.64.1.13-33.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Derman AI, Beckwith J. 1995. Escherichia coli alkaline phosphatase localized to the cytoplasm slowly acquires enzymatic activity in cells whose growth has been suspended: a caution for gene fusion studies. J Bacteriol 177:3764–3770. doi: 10.1128/jb.177.13.3764-3770.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Natale P, Bruser T, Driessen AJ. 2008. Sec- and Tat-mediated protein secretion across the bacterial cytoplasmic membrane-distinct translocases and mechanisms. Biochim Biophys Acta 1778:1735–1756. doi: 10.1016/j.bbamem.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 42.Korotkov KV, Sandkvist M, Hol WG. 2012. The type II secretion system: biogenesis, molecular architecture and mechanism. Nat Rev Microbiol 10:336–351. doi: 10.1038/nrmicro2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Szczesny R, Jordan M, Schramm C, Schulz S, Cogez V, Bonas U, Büttner D. 2010. Functional characterization of the Xps and Xcs type II secretion systems from the plant pathogenic bacterium Xanthomonas campestris pv. vesicatoria. New Phytol 187:983–1002. doi: 10.1111/j.1469-8137.2010.03312.x. [DOI] [PubMed] [Google Scholar]
- 44.Solé M, Scheibner F, Hoffmeister AK, Hartmann N, Hause G, Rother A, Jordan M, Lautier M, Arlat M, Büttner D. 2015. Xanthomonas campestris pv. vesicatoria secretes proteases and xylanases via the Xps type II secretion system and outer membrane vesicles. J Bacteriol 197:2879–2893. doi: 10.1128/JB.00322-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Noël L, Thieme F, Gäbler J, Büttner D, Bonas U. 2003. XopC and XopJ, two novel type III effector proteins from Xanthomonas campestris pv. vesicatoria. J Bacteriol 185:7092–7102. doi: 10.1128/JB.185.24.7092-7102.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Escolar L, Van den Ackerveken G, Pieplow S, Rossier O, Bonas U. 2001. Type III secretion and in planta recognition of the Xanthomonas avirulence proteins AvrBs1 and AvrBsT. Mol Plant Pathol 2:287–296. doi: 10.1046/j.1464-6722.2001.00077.x. [DOI] [PubMed] [Google Scholar]
- 47.Szurek B, Rossier O, Hause G, Bonas U. 2002. Type III-dependent translocation of the Xanthomonas AvrBs3 protein into the plant cell. Mol Microbiol 46:13–23. doi: 10.1046/j.1365-2958.2002.03139.x. [DOI] [PubMed] [Google Scholar]
- 48.Lorenz C, Kirchner O, Egler M, Stuttmann J, Bonas U, Büttner D. 2008. HpaA from Xanthomonas is a regulator of type III secretion. Mol Microbiol 69:344–360. doi: 10.1111/j.1365-2958.2008.06280.x. [DOI] [PubMed] [Google Scholar]
- 49.Singer AU, Schulze S, Skarina T, Xu X, Cui H, Eschen-Lippold L, Egler M, Srikumar T, Raught B, Lee J, Scheel D, Savchenko A, Bonas U. 2013. A pathogen type III effector with a novel E3 ubiquitin ligase architecture. PLoS Pathog 9:e1003121. doi: 10.1371/journal.ppat.1003121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hausner J, Büttner D. 2014. The YscU/FlhB homolog HrcU from Xanthomonas controls type III secretion and translocation of early and late substrates. Microbiology 160:576–588. doi: 10.1099/mic.0.075176-0. [DOI] [PubMed] [Google Scholar]
- 51.Thieme F, Koebnik R, Bekel T, Berger C, Boch J, Büttner D, Caldana C, Gaigalat L, Goesmann A, Kay S, Kirchner O, Lanz C, Linke B, McHardy AC, Meyer F, Mittenhuber G, Nies DH, Niesbach-Klosgen U, Patschkowski T, Ruckert C, Rupp O, Schneiker S, Schuster SC, Vorholter FJ, Weber E, Pühler A, Bonas U, Bartels D, Kaiser O. 2005. Insights into genome plasticity and pathogenicity of the plant pathogenic bacterium Xanthomonas campestris pv. vesicatoria revealed by the complete genome sequence. J Bacteriol 187:7254–7266. doi: 10.1128/JB.187.21.7254-7266.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang J, Wang X, Zhang Y, Zhang G, Wang J. 2008. A conserved Hpa2 protein has lytic activity against the bacterial cell wall in phytopathogenic Xanthomonas oryzae. Appl Microbiol Biotechnol 79:605–616. doi: 10.1007/s00253-008-1457-7. [DOI] [PubMed] [Google Scholar]
- 53.Sal-Man N, Deng W, Finlay BB. 2012. EscI: a crucial component of the type III secretion system forms the inner rod structure in enteropathogenic Escherichia coli. Biochem J 442:119–125. doi: 10.1042/BJ20111620. [DOI] [PubMed] [Google Scholar]
- 54.Daniels MJ, Barber CE, Turner PC, Cleary WG, Sawczyc MK. 1984. Isolation of mutants of Xanthomonas campestris pathovar campestris showing altered pathogenicity. J Gen Microbiol 130:2447–2455. [Google Scholar]
- 55.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (ed). 1996. Current protocols in molecular biology. John Wiley & Sons, Inc., New York, NY. [Google Scholar]
- 56.Kousik CS, Ritchie DF. 1998. Response of bell pepper cultivars to bacterial spot pathogen races that individually overcome major resistance genes. Plant Disease 82:181–186. doi: 10.1094/PDIS.1998.82.2.181. [DOI] [PubMed] [Google Scholar]
- 57.Bonas U, Schulte R, Fenselau S, Minsavage GV, Staskawicz BJ, Stall RE. 1991. Isolation of a gene-cluster from Xanthomonas campestris pv. vesicatoria that determines pathogenicity and the hypersensitive response on pepper and tomato. Mol Plant Microbe Interact 4:81–88. doi: 10.1094/MPMI-4-081. [DOI] [Google Scholar]
- 58.Engler C, Kandzia R, Marillonnet S. 2008. A one pot, one step, precision cloning method with high throughput capability. PLoS One 3:e3647. doi: 10.1371/journal.pone.0003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bolchi A, Ottonello S, Petrucco S. 2005. A general one-step method for the cloning of PCR products. Biotechnol Appl Biochem 42:205–209. doi: 10.1042/BA20050050. [DOI] [PubMed] [Google Scholar]
- 60.Huguet E, Hahn K, Wengelnik K, Bonas U. 1998. hpaA mutants of Xanthomonas campestris pv. vesicatoria are affected in pathogenicity but retain the ability to induce host-specific hypersensitive reaction. Mol Microbiol 29:1379–1390. doi: 10.1046/j.1365-2958.1998.01019.x. [DOI] [PubMed] [Google Scholar]
- 61.Büttner D, Nennstiel D, Klüsener B, Bonas U. 2002. Functional analysis of HrpF, a putative type III translocon protein from Xanthomonas campestris pv. vesicatoria. J Bacteriol 184:2389–2398. doi: 10.1128/JB.184.9.2389-2398.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Knoop V, Staskawicz B, Bonas U. 1991. Expression of the avirulence gene avrBs3 from Xanthomonas campestris pv. vesicatoria is not under the control of hrp genes and is independent of plant factors. J Bacteriol 173:7142–7150. doi: 10.1128/jb.173.22.7142-7150.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rossier O, Van den Ackerveken G, Bonas U. 2000. HrpB2 and HrpF from Xanthomonas are type III-secreted proteins and essential for pathogenicity and recognition by the host plant. Mol Microbiol 38:828–838. doi: 10.1046/j.1365-2958.2000.02173.x. [DOI] [PubMed] [Google Scholar]
- 64.Thein M, Sauer G, Paramasivam N, Grin I, Linke D. 2010. Efficient subfractionation of gram-negative bacteria for proteomics studies. J Proteome Res 9:6135–6147. doi: 10.1021/pr1002438. [DOI] [PubMed] [Google Scholar]
- 65.Diepold A, Wiesand U, Amstutz M, Cornelis GR. 2012. Assembly of the Yersinia injectisome: the missing pieces. Mol Microbiol 85:878–892. doi: 10.1111/j.1365-2958.2012.08146.x. [DOI] [PubMed] [Google Scholar]
- 66.Canteros BI. 1990. Diversity of plasmids and plasmid-encoded phenotypic traits in Xanthomonas campestris pv. vesicatoria. Ph.D. thesis. University of Florida, Gainesville, FL. [Google Scholar]
- 67.Weber E, Ojanen-Reuhs T, Huguet E, Hause G, Romantschuk M, Korhonen TK, Bonas U, Koebnik R. 2005. The type III-dependent Hrp pilus is required for productive interaction of Xanthomonas campestris pv. vesicatoria with pepper host plants. J Bacteriol 187:2458–2468. doi: 10.1128/JB.187.7.2458-2468.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ménard R, Sansonetti PJ, Parsot C. 1993. Nonpolar mutagenesis of the ipa genes defines IpaB, IpaC, and IpaD as effectors of Shigella flexneri entry into epithelial cells. J Bacteriol 175:5899–5906. doi: 10.1128/jb.175.18.5899-5906.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Murillo J, Shen H, Gerhold D, Sharma A, Cooksey DA, Keen NT. 1994. Characterization of pPT23B, the plasmid involved in syringolide production by Pseudomonas syringae pv. tomato PT23. Plasmid 31:275–287. doi: 10.1006/plas.1994.1029. [DOI] [PubMed] [Google Scholar]
- 70.Van den Ackerveken G, Marois E, Bonas U. 1996. Recognition of the bacterial avirulence protein AvrBs3 occurs inside the host plant cell. Cell 87:1307–1316. doi: 10.1016/S0092-8674(00)81825-5. [DOI] [PubMed] [Google Scholar]
- 71.Hartmann N, Büttner D. 2013. The inner membrane protein HrcV from Xanthomonas is involved in substrate docking during type III secretion. Mol Plant Microbe Interact 26:1176–1189. doi: 10.1094/MPMI-01-13-0019-R. [DOI] [PubMed] [Google Scholar]
- 72.Schulz S, Büttner D. 2011. Functional characterization of the type III secretion substrate specificity switch protein HpaC from Xanthomonas. Infect Immun 79:2998–3011. doi: 10.1128/IAI.00180-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schulze S, Kay S, Büttner D, Egler M, Eschen-Lippold L, Hause G, Krüger A, Lee J, Müller O, Scheel D, Szczesny R, Thieme F, Bonas U. 2012. Analyses of new type III effectors from Xanthomonas uncover XopB and XopS as suppressors of plant immunity. New Phytol 195:894–911. doi: 10.1111/j.1469-8137.2012.04210.x. [DOI] [PubMed] [Google Scholar]
- 74.Figurski D, Helinski DR. 1979. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc Natl Acad Sci U S A 76:1648–1652. doi: 10.1073/pnas.76.4.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morbitzer R, Elsaesser J, Hausner J, Lahaye T. 2011. Assembly of custom TALE-type DNA binding domains by modular cloning. Nucleic Acids Res 39:5790–5799. doi: 10.1093/nar/gkr151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.