

Abstract
UV radiation induces photolesions that distort the DNA double helix and, if not repaired, can cause severe biological consequences, including mutagenesis or cell death. In eukaryotes, both the formation and repair of UV damage occur in the context of chromatin, in which genomic DNA is packaged with histones into nucleosomes and higher-order chromatin structures. Here, we review how chromatin impacts the formation of UV photoproducts in eukaryotic cells. We describe the initial discovery that nucleosomes and other DNA-binding proteins induce characteristic ‘photofootprints’ during the formation of UV photoproducts. We also describe recent progress in genome-wide methods for mapping UV damage, which echoes early biochemical studies, and highlights the role of nucleosomes and transcription factors in UV damage formation and repair at unprecedented resolution. Finally, we discuss our current understanding of how the distribution and repair of UV-induced DNA damage influence mutagenesis in human skin cancers.
Graphical Abstract
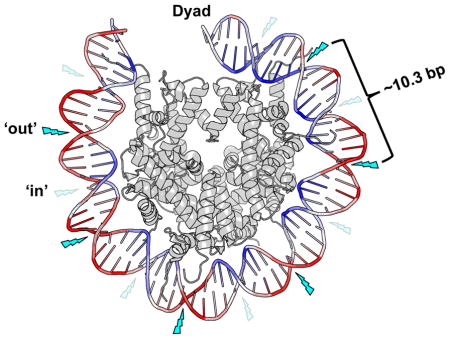
Formation of UV-induced CPD dimers occurs more frequently at ‘out’ rotational settings (indicated by bright blue lightning bolts) than at ‘in’ rotational settings (faint blue lightning bolts) in nucleosomal DNA. The relative level of local DNA mobility is represented by a color scale based on the B-factor from the 1id3 nucleosome structure, where DNA regions with a high B-factor (i.e., high mobility) are colored red, and DNA regions with a relatively low B-factor (i.e., low mobility) are colored blue.
Introduction
It has long been recognized that the target of DNA damaging agents and the ‘landscape’ of DNA repair enzymes in eukaryotes is the compact and dynamic structure of chromatin. Understanding variations in the distribution of DNA damage and repair in chromatin, together with their effects on chromatin structure, are crucial features of genomic instability. A prototype environmental agent used extensively in studies examining DNA damage and repair in chromatin has been ultraviolet (UV) radiation. UV radiation is a ubiquitous threat to the genomes of essentially all terrestrial organisms and the etiological agent underlying the development of several hereditary diseases and skin cancers. In this report, we review the historical development of our current understanding of UV-induced DNA damage in chromatin, and highlight recent developments exploring DNA damage and repair in chromatin at the genome-wide level. For brevity, we refer the reader to reviews wherever possible, which contain the seminal references for specific concepts.
Chromatin Structure Primer
In all eukaryotic cells, chromatin fibers are heterogeneous and dynamic. The basic repeating unit of chromatin fibers, the nucleosome, consists of the nucleosome core particle (NCP) and varying lengths of ‘linker DNA’ (from 20 bp to >100 bp, depending on the organism) stretching between these units (1). NCPs and linker DNA are often complexed with linker histones (H1 and associated variants), which promote compaction of the nucleosome arrays into chromatin fibers. The NCP contains an octamer of the well-conserved core histones H2A, H2B, H3, and H4, and ~147 bp of DNA coiled in ~1.7 left-handed turns around the wedge-shaped histone octamer surface (2). In the octameric complex, histones H3 and H4 form a tetramer that is flanked by two dimers of histones H2A and H2B. In addition, histone variants (primarily of histones H2A and H3) are present in subsets of nucleosomes, and play important roles in specific chromatin functions (3).
Positioning of histone octamers on DNA is greatly influenced by histone–DNA interactions in the NCP (4). These contacts are mainly electrostatic, where the minor groove of DNA interacts primarily with ‘sprocket’ arginine residues at regular ~ 10 bp intervals on each side of the pseudo-two-fold dyad axis of symmetry (5, 6). The H3–H4 tetramer strongly interacts with the central ~60 bp of NCP DNA, while the H2A–H2B dimers interact more weakly with the flanking ~30 bp of NCP DNA. The H3 αN helix specifically interacts with the ends of the NCP DNA, and thus plays an important role in regulating NCP DNA unwrapping (see below). This variation in binding strength along the DNA yields intrinsic differences in the accessibility of different DNA regions within NCPs. The reader is referred to a recent review for more detailed discussion of the nucleosome structure (2).
The stability of NCPs can be regulated in several different ways, including (a) exchange of canonical histones with histone variants (3), (b) ATP-dependent chromatin remodeling complexes (7) and (c) post-translational modifications of the core histones (8). In addition, the sequence of NCP DNA is a strong determinant of nucleosome stability (4). This latter feature of nucleosomes has been exploited for the generation of synthetic DNA sequences with strong positioning strengths (e.g., the Widom 601 positioning sequence) that bind histone octamers with high affinity (9). Use of these DNA sequences has allowed the assembly of near homogenous populations of NCPs that maintain a single rotational and translational setting on the DNA molecule. [Here, ‘rotational setting’ refers to orientations of DNA relative to the histone surface (e.g., ‘in’ rotational orientations are regions of the DNA phosphate backbone oriented toward the histone octamer and ‘out’ are regions oriented toward the solvent) and ‘translational setting’ refers to the DNA sequence positions relative to the center of symmetry (or dyad axis) of NCPs.]
Natural DNA sequences exhibit a wide range in nucleosome positioning power, where histone octamer binding affinity for different DNA sequences can vary by as much as 5000-fold (10). These differences dramatically affect the accessibility of occluded DNA sites in NCPs. Moreover, both the rotational and translational settings of DNA sites within NCPs significantly influence the accessibility of these sites. Restriction enzyme cleavage activity and Förster resonance energy transfer (FRET) have shown that regions of NCP DNA transiently unwrap from the histone surface (11). These studies revealed there is a progressive decrease in site exposure from translational settings near the ends of the DNA toward the ‘dyad center’ of NCPs (12). This dynamic equilibrium of unwrapping and rewrapping is dependent on DNA sequence. Indeed, stopped-flow FRET and fluorescence correlation spectroscopy studies have shown that the DNA unwrapping time varies by almost 10,000-fold going from the DNA ends to the dyad center (13). Thus, at the primary level of chromatin structure, are multiple, interdependent mechanisms capable of changing accessibility to DNA through the regulation of nucleosome positioning and stability.
UV Damage to DNA in Chromatin (Pre-Genomic Era)
UV Damage in Nucleosomes
Bulk Nucleosomes (Random DNA Sequences)
The location of DNA damage sites in chromatin plays an important role in their accessibility to repair enzymes. The distributions of a number of different DNA lesions in bulk (or ‘mixed sequence’) chromatin have been reported over the past four decades [e.g., see (14, 15)]. These studies helped form the basis for our understanding of the ‘efficiency’ of DNA excision repair [both nucleotide excision repair (NER) and base excision repair (BER)] in chromatin (16, 17). Early on it was clear that some classes of DNA lesions (e.g., bulky chemicals) form preferentially in nucleosome linker DNA (14). Interestingly, the major UV photoproduct in DNA [cis-syn-cyclobutane pyrimidine dimer (or CPD)] forms almost randomly between linker and core regions of chromatin (on a unit DNA basis). In contrast, the second most prevalent UV photoproduct, pyrimidine (6-4) pyrimidone dimer [or (6-4)PD], has a much stronger bias for linker DNA (14, 18).
The distribution of UV photoproducts within NCPs has also been mapped (18, 15). Using a T4 polymerase-exonuclease blockage assay, the distributions of CPDs and (6-4)PD were mapped at nucleotide resolution in NCPs from UV-irradiated cells, isolated chromatin, and isolated NCPs. These studies showed that the level of CPDs among NCPs in bulk chromatin has a striking periodic pattern, with an average periodicity of 10.3 ± 0.1 bases (Figure 1). This ‘photofootprint’ reflects the rotational setting of DNA on the histone surface, where the highest levels of CPD formation occur where the DNA phosphate backbone is farthest from the histone surface (i.e., an ‘out’ rotational setting) (19). The peak separation in the photofootprint pattern varies by as much as 1.3 bases near the nucleosome dyad relative to the average periodicity (20). In contrast, (6-4)PDs form much more randomly within nucleosome cores (Figure 1a; (21)). Thus, formation of UV photoproducts in chromatin is very sensitive to DNA-histone interactions on the octamer surface in nucleosomes.
Figure 1. UV photofootprint of nucleosome core DNA.

(a) Distributions of UV-induced (6-4)PDs and CPDs in nucleosomal DNA. Nucleosome core DNA was digested with T4 DNA polymerase-exonuclease before and after DNA photolyase treatment and the digestion profiles are shown. Lanes [CPD&(6-4)PD], no photoreversal; lanes [(6-4)PD Only], photoreversal with DNA photolyase. N, UV-irradiated nucleosome; D, UV-irradiated naked DNA. [See reference (21) for details.] (b) Laser densitometer scans of T4 DNA polymerase-exonuclease digestion profiles of UV-irradiated nucleosomes (NCP), and UV-irradiated naked DNA (DNA). NCP panel, values denote the scan absorbance (in arbitrary units) vs. distance (in bases) from the 5’ end of core DNA. Arrow represents position of the nucleosome dyad axis. DNA panel, same as NCP, except that core DNA was first isolated and then irradiated with UV light. [See reference (20) for details.]
The UV photofootprint results from the bending of DNA around histone octamers, reflecting the structural constraints imposed on DNA in nucleosomes [Figure 2, and (22)]. Moreover, the first crystal structure of a CPD-containing NCP was recently reported and indicates that CPDs do not significantly alter the DNA structure at the damage site (23). Thus, the variation in mobility of DNA on the histone surface may explain the UV photofootprint (Figure 2). After absorption of a UV photon, the [2+2] cycloaddition that occurs during the excited state of the 5–6 double bond in a pyrimidine base (24) will be more probable when adjacent pyrimidines are frequently aligned. In NCPs, the DNA mobility is highly variable, being minimal near histone–DNA contacts, where the minor groove faces toward the histone octamer, and maximal for the bases with phosphate groups on the outside of the particle (Figure 2). The increased mobility of dipyrimidines with their minor groove facing away from the histone octamer should make these regions the most favorable sites for CPD formation in NCPs.
Figure 2. Model of the nucleosome UV photofootprint.

Formation of UV-induced CPD dimers occurs more frequently at ‘out’ rotational settings (indicated by bright blue lightning bolts) than at ‘in’ rotational settings (faint blue lightning bolts) in nucleosomal DNA. The relative level of local DNA mobility is represented by a color scale based on the B-factor from the 1id3 nucleosome structure, where DNA regions with a high B-factor (i.e., high mobility) are colored red, and DNA regions with a relatively low B-factor (i.e., low mobility) are colored blue. The image was generated using Pymol to visualize the nucleosome structure 1id3 (87). Only one gyre of the nucleosome DNA is depicted.
Finally, although (6-4)PDs are less prevalent than CPDs in chromatin, the yield of these photoproducts at specific sites can be much higher than in free DNA (18). This can alter their impact on UV-induced mutagenesis at specific sites in mammalian cell chromatin (25). As these UV photoproducts are distributed more randomly than CPDs within NCPs (Figure 1a) and have a much stronger bias for linker regions (18), the distribution of (6-4)PDs in chromatin differs markedly from CPDs, possibly reflecting the different structures of these photoproducts in NCPs. Indeed, the recently reported crystal structure of a (6-4)PD-containing NCP indicates that the damaged region of NCP DNA is flexibly disordered, particularly in the solvent exposed strand (26). Thus, it is reasonable to assume that the more constrained NCP DNA is less capable of forming such structures, as compared to the linker DNA in chromatin. This feature of the (6-4)PD distribution undoubtedly plays a role in the rapid NER of these UV lesions relative to CPDs (27).
Designed Nucleosomes (Single DNA Sequences)
Reconstitution of defined DNA sequences into nucleosomes (or ‘designed nucleosomes’) allows analysis of DNA damage effects on preexisting nucleosome structures and on nucleosome formation during assembly. Most notably, designed nucleosomes allow detailed studies on repair of DNA damage in specific nucleosome locations. However, results for defined DNA sequences depend strongly on (a) sequence specificity of damage formation, (b) location of target sequences relative to the histone octamer, (c) local flexibility of DNA, and (d) stability of the designed nucleosome structure.
Several nucleosome-positioning sequences have been used to create designed nucleosomes, and many of these differ in nucleosome ‘positioning power’ (9). A favorite sequence used in early studies was the sea urchin 5S rRNA gene (28), a sequence with moderate positioning power (several-fold) over the bulk of the DNA sequences in chromatin (Table 1). On the other hand, the Widom 601 clone, having a ‘free energy of reconstitution’ (ΔΔG) of ~ 2.9 kcal/mol lower than the 5S rDNA sequence, has an ~100-fold higher positioning power than 5S rDNA (Table 1). These differences in positioning power can significantly affect the ‘occupancy time’ of DNA in preferred rotational and translational settings of designed NCPs (Figure 3) (29, 30). Thus, when comparing levels of DNA damage sites and their accessibility to repair enzymes in designed nucleosomes with different sequences, variation can arise from the differences in positioning power of these sequences.
Table 1.
Relative Affinities Reported for Different Nucleosome Positioning Sequences with or without DNA Damage1
Figure 3. Nucleosome positioning power is affected by DNA sequence [data from reference (9)].
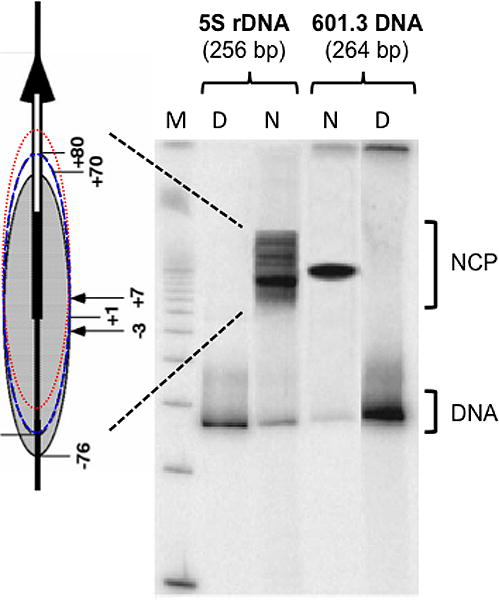
Nucleosomes reconstituted from two different nucleosome-positioning sequences (256 bp sea urchin 5S rDNA and 264 bp Widom- 601.3b sequence) are shown on a native gel. Multiple bands are shown for nucleosomes reconstituted with the relatively low affinity 5S DNA sequence. The multiplicity of bands reflects nucleosome positioning at many different positions in this sequence [schematically represented on left by 5S rDNA (thick vertical arrow) on 256 bp fragment (thin vertical line) and several NCP positions, from strongest binding translational position (shaded oval) to weaker binding positions (blue and red dashed lines)]. By contrast, nucleosome reconstitution with the high affinity 601.3b sequence shows only one dominant position. Lane M, 100 bp DNA size standard. Lanes D, naked DNA. Lanes N, reconstituted nucleosomes. [Adapted from Figure 3 of reference (9).]
Reports on the impact of nucleosome positioning on the distribution of UV-induced photoproducts in defined sequences started appearing in the 1990’s (14, 18). UV irradiation of a yeast nucleosome positioning sequence (called HISAT) yielded a strong modulation of CPD formation in NCP DNA (31). This sequence contains a 40 bp polypyrimidine stretch, including several long T-tracts, allowing measurement of CPD formation over three helical turns of DNA in a defined nucleosome. As with mixed-sequence nucleosomes, the pattern of CPD formation in HISAT nucleosomes is significantly different than in free HISAT DNA. Interestingly, the distribution of CPDs in HISAT nucleosomes only partly resembled that of random-sequence nucleosomes. Specifically, CPD levels were high at two contact points between the minor grooves of the HISAT sequence and the histone surface, a region where the random-sequence DNA predicts low levels of CPDs (Figure 1). This most likely reflects an altered T-tract structure in nucleosomes, as compared to random DNA, as these tracts have a non-B-form structure in solution (32). We note that charge neutralization of the DNA phosphates occurs essentially on one side of the helix in nucleosomes (i.e. facing the histones), which produces DNA bending around the histone octamer and formation of kinks at regular intervals in the DNA (33). Thus, the natural bending of certain DNA sequences will impact the formation of nucleosomes, being favorable or unfavorable depending on the location of these bends. Therefore, the consequence of DNA damage-induced bending on nucleosome reconstitution was studied using the 5S rDNA nucleosome (34). The results showed that damage induced by a bulky chemical carcinogen (benzo[a]pyrene diol epoxide) enhanced nucleosome formation on 5S rDNA, while UV-induced photoproducts repressed nucleosome formation (Table 1; see ΔΔG∘ values for UV and BPDE damaged 5S rDNA).
The effects of nucleosome folding on formation of UV photoproducts were also analyzed in the 5S rDNA nucleosome (35). Surprisingly, it was found that even after a fairly high amount of UV damage (~ 0.8 CPDs/NCP on average), there was no affect on either the average rotational or translational settings of the 5S rDNA. Additionally, it was found that, although nucleosome folding had little effect on CPD formation in the transcribed strand of rDNA, formation of these photoproducts was significantly repressed in the non-transcribed strand in an ~ 30 bp region around the nucleosome dyad center. These results presumably reflect constrained rotation and bending of the DNA bases, and the tight binding of the H3-H4 tetramer, near the center of NCPs (35). These authors also investigated the affect of nucleosome formation on CPD yields in a 14 base pyrimidine tract in the 5S rDNA, as it spans a complete turn of the DNA helix (35). It was found that CPD formation in the pyrimidine tract was either elevated or reduced at sites where the minor groove faced ‘out’ or ‘in’ toward the histone surface, respectively, agreeing with the CPD modulation predicted by random sequence NCPs (Figure 1).
In the late 1990’s, Kosmoski and Smerdon exploited the conformational stability of tightly positioned nucleosomes to allow incorporation of a single UV photoproduct at a specific site in NCPs (36). Using a synthetic nucleosome positioning sequence (37) to bracket a short (30 bp) DNA sequence containing a chemically synthesized CPD, these authors assembled nucleosomes specifically damaged at only one site and one structural orientation, with the DNA minor groove at the CPD site facing out from the histone surface ~5 bases from the nucleosome dyad center. Competitive gel-shift analysis showed there was only a small increase in histone binding energy required to form a NCP (ΔΔG = ~0.15 kcal/mol), which allowed complete nucleosome loading on the damaged DNA (36). Thus, it was now possible to ‘design’ specifically damaged nucleosomes for in vitro studies on how nucleosome structure influences DNA damage formation, and how DNA repair enzymes detect and repair damage that is occluded by the histone octamer. For example, it was found that NER in Xenopus oocyte extracts can effectively repair the single UV lesion in this designed nucleosome, and nucleosome reassembly occurs on the newly repaired DNA shortly after NER in these extracts (38). Many reports followed these studies using designed nucleosomes to monitor everything from the effect of rotational orientation of DNA damage to the effect of specific histone modifications on the activities of DNA repair enzymes in vitro. Although these studies are beyond the scope of the present review, the reader is referred to recent reviews that cover these topics [e.g., (16, 15, 17)].
Finally, as discussed in the Chromatin Structure Primer section, nucleosomes are dynamic and exist in a conformational equilibrium, where a portion of the NCP DNA spontaneously unwraps and rewraps around the histone octamer (Figure 4a). Förster Resonance Energy Transfer (FRET) and restriction enzyme accessibility have been used to examine how UV-induced photoproducts in nucleosome DNA affect this conformational equilibrium (39). The 147-bp Widom 601 nucleosome positioning sequence was pre-labeled with ‘donor’ and ‘accepter’ fluorophores on opposite DNA strands and used for NCP reconstitution with purified Xenopus histones (Figure 4b). This system is a sensitive detector of changes in the distance between the fluorescent dyes (Figure 4c). It was shown that UV photoproducts in the 601 sequence decreased FRET efficiency (Figure 4d) and increased restriction enzyme accessibility in the 601 NCP, indicating that UV photoproducts promote DNA unwrapping from the histone octamer (39). This increased DNA unwrapping was even detected when a UV photoproduct was inserted at a single site of the NCP. Thus, increased exposure of nucleosome DNA following DNA damage formation may play an important role in the accessibility of DNA repair enzymes in chromatin. Indeed, it was observed that rapid repair of UV-induced lesions by UV photolyase, a repair enzyme whose activity is quantitatively blocked by nucleosomes, occurs in nucleosome-loaded DNA of intact yeast cells (40). These rapid repair kinetics are consistent with the rapid kinetics of nucleosome unwrapping-rewrapping in the cell, rather than the much slower kinetics expected for histone exchange, chromatin remodeling or modification of the core histones (41).
Figure 4. The dynamic changes of nucleosomal DNA revealed by FRET.

(a) Schematic diagram of DNA transient unwrapping dynamics in the nucleosome. The green internal disk represents histone octamer. Protein binding site is indicated as dark spot (black) on DNA (purple). [Adapted from: (88)]. (b) Locations of donor (Cy3) and acceptor (Cy5) dyes on the 147 bp 601 DNA sequence used for FRET. Upper panel shows locations of dyes on the naked DNA. The distance between the Cy3 and Cy5 on the naked DNA is ~27 nm, well beyond R0 (~5 nm) for the Cy3-Cy5 pair. Lower panel shows the dye locations on 601 NCPs. The distance between the dyes is ~3 nm in NCPs, significantly less than R0, yielding efficient energy transfer from donor to acceptor. The NCP model was generated from the crystal structure in PDB (accession number 1KX5). (c) Emission spectra revealed that energy transfer was only seen in the NCPs (blue) but not in naked DNA (pink). (d) FRET analysis demonstrates enhanced nucleosome unwrapping dynamics by UV damage. The 601 DNA was irradiated with different UV doses before reconstituted into NCPs. NCP Samples were excited at 515 nm and emission spectra recorded from 550 nm to 700 nm. Less efficient energy transfer is shown by increased donor emission and decreased acceptor emission, with increased UV doses. [Panels b-d are taken from reference (39).]
UV Damage at Transcription Factor Binding Sites
The first report on protein-DNA interactions modulating UV photoproduct formation was on the lac repressor complex of Escherichia coli lac operator DNA (42). These authors used a series of chemical reactions to cleave DNA at UV photoproducts, and it was found that both increased and decreased photoproduct formation occurs at specific sites of the lac repressor binding sequence. This method was later employed to demonstrate transcription-dependent changes in the control region of the GAL1 and GAL10 genes in yeast cells (43).
These studies were followed by the use of CPD-specific endonuclease ‘T4 endo V’ (44) to detect CPD formation profiles at the single nucleotide level in protein-DNA complexes. Indeed, a number of high-resolution methods were even developed to measure UV damage at the single nucleotide level in intact cells, as the sensitivity of this assay was greatly enhanced by amplification of T4 endo V cleavage fragments in DNA and radioactive labeling (45). One such method involved amplification through ligation-mediated polymerase chain reaction, or LMPCR (46). This technique was used to show that modulation of UV photoproducts occurs in promoter regions during induction of several genes in intact human cells, including JUN, FOS, and PCNA (47). Another method used to detect DNA damage directly (i.e., not involving amplification of fragments created by strand cleavage) at the single nucleotide level excluded some potential problems with amplification methods (45). With this method, genomic DNA is digested with one or more restriction enzymes to release fragments cleaved at DNA damage sites and annealed to sequence-specific biotinylated oligonucleotides (Figure 5a). The annealed fragments can then be separated from the mix with streptavidin magnetic beads, and end-labeled by extension of the 3′ overhang of the fragment consisting of a poly(dT) tract (Figure 5a). Although less sensitive than the amplification methods, this method provides a linear representation of the actual number of DNA damage sites and has been very useful for monitoring UV damage and repair in organisms with small genomes, such as bacteria and yeast (Figure 5b). Thus, the modulation of UV photoproducts in transcription factor (TF) binding sites and positioned nucleosomes may be a general phenomenon, a premise that was recently confirmed by genome-wide mapping of UV photoproducts in intact cells (see the section on UV Damage to DNA in Chromatin (Post-Genomic Era)).
Figure 5. A method for measuring UV damage at the nucleotide level.

(a) Schematic of the high-resolution UV damage mapping technique. Cells are UV irradiated and DNA is isolated immediately after UV treatment or at different repair times. The target DNA fragment is released from genomic or plasmid DNA by restriction enzymes. CPDs are incised by T4 endo V and converted to single strand breaks. A biotinylated DNA probe containing the complementary sequence to the 3’ end of one strand is annealed to the target DNA fragment. The annealed DNA is purified with streptavidin magnetic beads, and the target DNA is end-labeled with Sequenase and [32P]-dATP, using the poly(dT) tract on the probe as the extension template. The radio-labeled products are separated on a sequencing gel. (b) Representative gel showing CPD sites and their repair in the GAL1-10 gene sequence under repressed (Glu) and activated (Gal) conditions. Yeast cells were irradiated with 50 J/m2 UV light and allowed to repair for different times. Lanes labeled as ‘U’ indicate samples without UV irradiation. The left two lanes are A+G and C+T sequence markers generated by Maxam–Gilbert sequencing procedure. [Adapted from reference (45).]
A useful model system for studying UV photoproduct formation in protein-DNA complexes in vitro has been the 5S rRNA gene sequence bound to transcription factor IIIA [TFIIIA; (48, 49)]. Binding of TFIIIA to 5S rDNA is the first step in RNA polymerase III-directed synthesis of 5S rRNA (50). This ~40-kDa protein contains nine zinc finger motifs, and binds tightly to the internal control region (ICR) of 5S rDNA, an ~50 bp sequence located inside the transcription unit (e.g., see 49–51). The dependence of the yield of UV-induced photoproducts on TFIIIA binding has been studied in the Xenopus borealis 5S rRNA gene (49, 51). TFIIIA binding was found to modulate UV photoproduct formation mainly in the template strand of 5S rDNA, where the strongest contacts with this protein are observed (52, 53). The modulation pattern was not uniform, however, and strong inhibition of CPD formation was observed at several sites in the ICR [reviewed in (15)]. Interestingly, CPD formation was also enhanced at one site in the template strand following TFIIIA binding. This region of the ICR with enhanced CPD formation binds the middle three zinc fingers of TFIIIA, and this binding differs from that of the remaining zinc fingers in TFIIIA (52, 53). The zinc finger ‘cassettes’ on the N- and the C-terminal of TFIIIA wrap along the major groove of 5S rDNA, while the three middle fingers bind nearly parallel to the helix axis in the center of the complex, and these three zinc fingers of TFIIIA may cause bending in the 5S rDNA that enhances CPD formation (51).
Finally, in addition to the modulation of UV damage in DNA by bound TFs, UV lesions themselves can alter TF binding. Tommasi et al (54) incorporated site-specific CTDs [cis-syn-cyclobutane thymine dimers] into oligonucleotides containing the recognition sequences of five different TFs (E2F, NF-Y, AP-1, NFκB, and p53). In each case, the presence of a single CTD strongly inhibited binding (11- to 60-fold) of the respective TF in vitro. More recently, Kwon and Smerdon studied the binding of TFIIIA to the ICR in 5S rDNA containing either site-specific CTDs (55) or CPDs throughout the ICR in randomly damaged 5S rDNA (51). These authors also examined the relationship between TFIIIA binding and the efficiency of DNA repair in Xenopus oocyte nuclear extracts. These studies demonstrated that (a) CTDs in the ICR of the 5 S rRNA gene can increase or decrease binding affinity of DNA for TFIIIA depending on their position, (b) modulation of TFIIIA binding correlates with the modulation in dissociation rate (koff) of the complex, (c) the decrease in binding affinity is accompanied by the loss of TFIIIA-DNA contacts near CTD sites, and (d) NER of CTDs in the TFIIIA-5S rDNA complex is extremely sensitive to changes in the dissociation rates of the complex. Thus, this simple model system has provided a detailed view of how strong protein-DNA interactions can have major effects on the yield of UV photoproducts at specific sites in the binding domain and how TF binding can be modulated by the presence of UV photoproducts.
UV Damage to DNA in Chromatin (Post-Genomic Era)
Although chromatin is uniformly comprised of nucleosomes, recent studies have highlighted the diversity of distinct types of chromatin in eukaryotic chromosomes, distinguished by differences in covalent histone modifications, histone variants, and the degree of ongoing histone exchange, nucleosome mobility, and higher-order chromatin compaction (3, 56). While much has been learned from studies of defined sequences or in bulk chromatin, it is important to elucidate how this diverse spectrum of chromatin states impact UV damage formation and subsequent repair in eukaryotic genomes. To this end, a number of different genomic approaches have been developed to map UV-induced lesions across the yeast and human genomes (57).
Genomic Approaches to Monitor UV Damage and Repair in Chromatin
Initial genomic studies adapted the chromatin immunoprecipitation-microarray (ChIP-chip) method (58, 59), using anti-CPD antibodies to immunoprecipitate lesion-containing DNA fragments, which were then detected using tiling microarrays (60–62, 57). These studies yielded insights into how DNA sequence influences UV damage formation across the genome. For example, our labs showed that UV damage formation is enriched adjacent to specific classes of repeat elements in the human genome, particularly those containing poly-T tracts (61). More recently, a microarray-based method was used to investigate how global genomic repair of CPD lesions is organized across the yeast genome by a sequence-specific DNA binding factor (i.e., Abf1) and regulated by histone acetylation (63). However, microarray-based methods can only map DNA lesions at low resolution, due to both the relatively low density of microarray probes along chromosomes and the large size of the sonicated DNA fragments [e.g., ~300–400 bp (60, 61)]. This limits their utility for characterizing how chromatin, at least at the level of individual nucleosomes, modulates UV damage formation and repair.
More recently, next generation sequencing (NGS) methods have been employed to map UV damage at higher resolution across the genome. The first of these methods was called Excision-seq (64). Excision-seq utilized DNA repair enzymes (in this case UV DNA Endonuclease [UVDE]) to generate single strand breaks (SSBs) immediately upstream of either CPDs or 6-4PDs. However, the application of Excision-seq is constrained to sequences where the SSBs are converted to DNA double strand breaks (DSBs), such as two adjacent SSBs occurring on opposing DNA strands. For this reason, very high UV doses (~10,000 J/m2) were required, in order to increase the probability that cleavable photoproducts would form in close proximity on opposing strands. Excision-seq was used to map UV damage formation across the yeast genome at single nucleotide resolution, thus revealing the expected DNA sequence preferences for CPD and 6-4PP formation (64). However, the influence of chromatin on UV damage formation was not explored in this study. A second method, known as excision repair sequencing (XR-seq), sequenced the ~25–30 nucleotide fragments that are excised during NER of UV photoproducts (65). This has proven to be a powerful method to map the repair of UV lesions across the genome (65, 66), but does not provide direct information about initial UV damage formation.
Recently, Mao et al. reported a new method named CPD-seq to map both initial CPD damage formation and subsequent NER activity in the yeast genome at single nucleotide resolution (67). CPD-seq was adapted from a method used to map ribonucleotide incorporation during DNA replication (68, 69). In CPD-seq, UV-irradiated DNA is sonicated into small DNA fragments, ligated to a double stranded DNA adapter (trP1, colored brown in Figure 6A), and treated with terminal transferase [and dideoxy-ATP (ddATP)], resulting in DNA fragments lacking free 3′-OH groups (Figure 6A). The DNA is then sequentially digested with T4 endo V and AP-endonuclease (APE1) to generate new 3′-OH groups immediately upstream of the CPD lesion (Figure 6A). These fragments are then ligated to a second adaptor DNA (A adaptor, colored orange in Figure 6A), with a biotin label on one strand to allow purification of the ligated fragments. The generated CPD-seq library is briefly amplified with primers complementary to trP1 and A adaptors and subjected for next generation DNA sequencing. Because the only 3′-OHs available for A adaptor ligation are derived from CPD cleavage by T4 endo V and APE1, the genomic location of CPD lesions is precisely determined from the corresponding DNA sequencing read, which initiates within the A adaptor (Figure 6A). Hence, using the CPD-seq strategy, one can map initial CPD formation across the genome at single nucleotide resolution. CPD maps can also be generated at different repair times to investigate the genome-wide time course of CPD removal (67).
Figure 6. Mapping UV damage in the yeast genome using the CPD-seq method.

(A) Schematic of CPD-seq [adapted from (67)]. After fragmentation of genomic DNA isolated from UV-irradiated yeast cells, the trP1 adaptor (brown), containing a 3′ dideoxy group (denoted ‘dd’), is ligated to the free DNA ends. The remaining free 3′-OHs are further blocked by terminal transferase using ddATP as the substrate. DNA is subsequently digested with T4 endo V and APE1, to generate new 3′-OHs (green) specifically at CPD sites. The A adaptor (orange) is ligated to the free 3′-OH (green) immediately upstream of the CPD site. The resulting CPD library is briefly amplified with primers complementary to the trP1 and A adaptors and subjected to next-generation sequencing. Sequencing reads are mapped to the reference yeast genome to identify genomic locations and dinucleotide sequences of the CPDs. (B) CPD formation in vivo is enhanced at outward rotational settings (positions denoted by vertical dashed lines), but repressed at inward rotational settings in strongly positioned nucleosomes across the yeast genome. The scaled ratio of CPD formation in UV irradiated yeast (in vivo) relative to UV-irradiated naked DNA is plotted for the plus and minus strands of ~10,000 strongly positioned yeast nucleosomes. Green triangles indicate the locations of previously identified peaks of CPD formation (from the 3′ side of the CPD lesion) in mammalian chromatin, based on reference (20).
Nucleosomes in the Yeast Genome Modulate CPD Formation and Repair
The high resolution of CPD-seq data provided a unique opportunity to examine how chromatin influences CPD formation across the genome. Overlaying the CPD-seq data onto a well-defined map of yeast nucleosome positions (70) revealed that yeast nucleosomes induce a strong UV photofootprint. The peaks of CPD formation (after normalizing for dipyrimidine content) coincided with outward rotational settings in the nucleosome, exhibiting a striking periodicity of ~10 bp (67), closely mirroring the UV photofootprint previously identified in mammalian chromatin [Figures 1 and 6B (19)]. Notably, the UV photofootprint was most apparent among strongly positioned nucleosomes in yeast (~10,000 nucleosomes), but was barely detectable among weakly positioned nucleosomes (~7500 nucleosomes) (67). Presumably, the lack of a uniform rotational setting among weakly positioned nucleosomes masks the UV photofooprint at these locations.
Nucleosomal DNA shows clear sequence biases: A-T rich sequences tend to adopt ‘in’ rotational settings, while G-C rich sequences tend to adopt ‘out’ rotational settings (4). For this reason, TT dinucleotides, which are most prone to forming CPD lesions, tend to be positioned at ‘in’ rotational settings [e.g., (70, 67)]. The variations of TT frequency in nucleosomal DNA are mirrored by CPD levels in UV-irradiated naked DNA, indicating that in the absence of nucleosomes, the highest CPD yields occur at locations corresponding to ‘in’ rotational settings in NCPs, and the lowest yields at locations corresponding to ‘out’ rotational settings (67). However, the exact opposite pattern occurs in vivo when DNA is packaged into nucleosomes. The impact of nucleosomes on UV damage formation is highlighted by directly comparing CPD-seq data of UV damage formation in vivo, in intact cells, with CPD-seq data of UV damage formation in vitro, in naked DNA (Figure 6B) (67). The packaging of DNA into nucleosomes suppresses CPD formation at ‘in’ rotational settings relative to naked DNA; however, CPD formation in nucleosomes is markedly higher at ‘out’ rotational settings (Figure 6B). By placing TT-rich DNA sequences at ‘in’ rotational settings, nucleosomes essentially protect (or ‘shade’) these intrinsically vulnerable sequences from UV damage. We hypothesize that this mechanism operates in all eukaryotes, and may be an important modifier of UV-induced mutagenesis (see below).
Although the rotational setting of DNA in nucleosomes modulates CPD formation, analysis of CPD removal indicates that it does not generally impact subsequent NER activity in yeast. Analysis of CPD-seq data indicates that higher CPD levels are retained at ‘out’ rotational settings during repair, even though overall CPD levels are decreased, regardless of the rotational positioning (67). Interestingly, the translational positioning significantly regulates CPD removal in strongly positioned nucleosomes. CPDs located in the distal regions of nucleosomal DNA, where spontaneous unwrapping occurs more rapidly (see the Chromatin Structure Primer section above), are repaired more efficiently than CPDs located near the nucleosome dyad (67). It is not known to what extent altering nucleosomes by incorporating histone variants or histone post-translational modifications will affect initial UV damage formation and repair on a genome-wide scale, but previous studies (e.g., (71–74)) suggest this is an important avenue for future research.
Transcription Factor Binding Significantly Alters CPD Formation
The high resolution of the CPD-seq data also permitted investigating how TF binding to DNA affects UV damage formation. It was previously reported that TF binding to DNA can modulate CPD formation (see the section on UV Damage at Transcription Factor Binding Sites), suppressing CPD formation at some locations within the binding site, and enhancing CPD formation at other locations [reviewed in (47)]. Genome-wide analysis of CPD formation at the binding sites of yeast TFs Abf1 and Reb1 demonstrated that these DNA-bound TFs generally inhibited CPD formation in vivo, although a single CPD ‘hotspot’ was detected at a specific location in Abf1 binding sites (67). TF binding appears to be critical for modulating CPD formation, as Reb1 binding sites with low Reb1 occupancy in vivo had relatively little affect on CPD formation, while high occupancy Reb1 binding sites had a striking affect on CPD formation (67). CPD formation was as much as 3-fold less frequent at Abf1 or Reb1 binding sites in vivo relative to unbound naked DNA, indicating that the magnitude (or fold-change) of the TF photofootprint is significantly greater than that of the nucleosome photofootprint. This difference in the magnitude of UV photofootprinting may be due in part to the higher intrinsic level of DNA dynamics within nucleosomes in vivo (i.e., due to DNA unwrapping, etc.) relative to TF-DNA complexes.
It will be important to investigate how different families of TFs affect UV damage formation. For example, it is possible that other families of TFs may primarily enhance CPD (or 6-4PD) formation, as has been previously reported for the TFs that bind the CCAAT-box and serum response elements in mammalian promoters (47), which could be an important contributor to mutagenesis in human cancers (see the section on Chromatin Effects on UV-induced Mutagenesis in Cancer below). Moreover, CPD-seq data could be exploited as an experimental tool to identify unknown TF binding sites across the genome based on their UV photofootprint signature.
Chromatin effects on UV-Induced Mutagenesis in Cancer
The role of UV light in inducing mutations that underlie skin cancers has long been understood (75). However, the ability to relate the genomic profile of DNA damage and lesion repair distributions to mutagenesis in human cells, model organisms, and clinical tumor samples has the potential to expand our knowledge of the important factors that govern where disease causing mutations are prone to occur (57). In the case of UV mutagenesis, the analysis of whole genome sequenced human melanomas has taken a primary focus. One of the first whole genome sequences of a human cancer was that of a melanoma cell line, where the mechanistic knowledge of Transcription Coupled Nucleotide Excision Repair (TC-NER) removal of UV lesions from the transcribed DNA strand was utilized as a means to support the observed strand bias of mutations occurring specifically within genes (76). Since then, the large number of UV-induced mutations in melanoma genomes has made this cancer type particularly well-suited for assessing how mutations are distributed across chromosomes.
Initial analyses of melanoma genomes, as well as genomes from several other cancer types, determined that mutation densities, like lesion densities (see the UV Damage to DNA in Chromatin (Post-Genomic Era) section), are highly heterogeneous, changing drastically with chromosome position (77). A comparison of the distribution of melanoma mutations to various aspects of chromosome structure detailed by the Encyclopedia of DNA Elements (ENCODE) project indicated that in addition to the expected constraints for UV-induced mutations to TC and CC dinucleotides (25), chromatin likely also impacts where these mutations occur. Specifically, melanoma mutations were found to be elevated in regions with higher nucleosome occupancy as well as in late replicating regions of the genome (78, 79). The correlation of mutation density with both of these chromosome features suggests that an impaired ability for NER to remove UV lesions within more compact chromatin prior to DNA replication is one key factor dictating the location of UV-induced mutations. The impacts of nucleosome structure on restricting NER activity have been well established both biochemically and genetically (15–17), while the realization that late replicating genome regions are generally gene poor and heterochromatic (80) suggests that neither TC-NER nor global-NER would function efficiently in these regions. Further support for chromatin accessibility impacting the location of UV-induced melanoma mutations have steadily accumulated as regions of high mutation density have been shown to be associated with histone modifications that traditionally indicate heterochromatic chromosome structure (81), while decreased mutation densities have been found within open chromatin domains marked by high DNase I accessibility (82, 83). This decreased abundance of UV-induced mutations in DNase I hypersensitive sites compared to heterochromatic regions appears to be a function of NER activity as tumors containing mutations in key NER components (83) or from XPC-deficient individuals (84) lack this phenomenon. Sancar and colleagues’ development of XR-seq (see the UV Damage to DNA in Chromatin (Post-Genomic Era) section) (65), which enables monitoring of NER activity across the genome, has provided direct evidence that globally, melanoma mutations are enriched in regions where NER occurs more slowly (66). More fine scale analyses of the distribution of NER and UV-induced mutation in active promoters indicate that DNA-binding proteins other than nucleosomes likely influence mutation positions during melanoma development (Figure 7). Specifically, TF binding sites were shown to be prone to both decreased NER activity (as measured by XR-seq) and increased mutation density (85, 86), consistent with prior biochemical work indicating that TFs can occlude lesions from NER (see the section on UV Damage at Transcription Factor Binding Sites above). However, these same analyses indicated the presence of regions adjacent to promoters that contain low relative levels of both NER activity and mutation density (Figure 7). Thus, the higher mutation densities in these regions cannot solely be explained by a difference in NER efficiency and suggests other factors, such as differences in initial UV damage levels, likely contribute to mutational heterogeneity in human tumors.
Figure 7. Effects of transcription factor binding on UV lesion formation, repair, and cancer mutations.

The genome-wide analysis of UV lesion repair in relation to transcription factor binding sites (TFBS) show potentially complex correlations with the position of melanoma mutations. A general reduction of NER as measured by XR-seq in normal human fibroblasts (NHF1 cells) (black line) occurs in TFBS. This correlates with an increased mutation density in these areas in sequenced human melanomas (orange line). However, larger regions flanking the TFBS contain both low NER activity and low melanoma mutation density, suggesting a complex relationship occurs among these parameters, which may in part be explained by differences in lesions formation. XR-seq and melanoma mutation data were obtained from reference (86).
In addition to impacting NER efficiency, chromatin could also impact melanoma mutation distributions by affecting where lesions are most likely to form. As discussed earlier, a large amount of biochemical data indicates that the structure of the nucleosome core particle, as well as TF binding to promoters, results in photofootprints after UV exposure. Despite this knowledge, most current genomic analyses of NER and mutation distributions have assumed that UV lesions form relatively evenly across chromosomes, and little information currently exists relating the formation of UV lesions to chromatin features on a genomic scale (66). The recent development of the CPD-seq method to map UV-lesions at single nucleotide resolution (see the UV Damage to DNA in Chromatin (Post-Genomic Era) section above), indicates that at least in yeast, photofootprints of the nucleosome core particle, as well as Abf1 and a Reb1 TFs, can be detected using genomic methods (67). As UV-lesion formation is a fundamental pre-requisite for both NER activity and eventual UV-induced mutations, UV photofootprints likely have a significant effect on the distribution of mutations observed in clinical melanoma samples and may help explain why certain genome regions contain both low levels of mutation and NER activity.
Conclusions
Efforts during the past four decades have led to significant progress in understanding UV damage formation and NER in the context of chromatin. Studies conducted in mammalian cells demonstrated the strong modulation of UV damage formation by DNA packaging into chromatin (19). Formation of UV damage in nucleosomes, on the other hand, alters the nucleosome unwrapping dynamics (40), which may serve as an important mechanism to recruit damage recognition factors for repair. Recent progress in mapping UV damage across the genome not only confirmed the early observations made in bulk chromatin, but also provides a unique opportunity to closely investigate the heterogeneous distribution of UV damage and variable repair dynamics in different eukaryotic chromatin domains. Considering the largely heterogeneous feature of mutation density in melanoma genomes, data generated from recent genome-wide damage maps provide new insights into mechanisms underlying variable mutation formation in the cancer genome (i.e., 82, 83). Similar mapping methods will likely be developed to map other types of DNA damage and their corresponding DNA repair pathways, which undoubtedly will strengthen our understanding of the important mechanisms that connect chromatin structure, DNA damage formation and DNA repair with mutagenesis in human cancers.
Acknowledgments
This work was supported by NIH grants ES002614 (to MJS and JJW) and ES022633 (to SAR) from the National Institute of Environmental Health Sciences (NIEHS), the Breast Cancer Research Program Breakthrough Award BC141727 from the Department of Defense (to S.A.R.), and an internal grant from the College of Veterinary Medicine at Washington State University (to JJW). Its contents are solely the responsibility of the authors.
Footnotes
This article is part of the Special Issue highlighting Dr. Aziz Sancar’s outstanding contributions to various aspects of the repair of DNA photodamage in honor of his recent Nobel Prize in Chemistry.
References
- 1.van Holde KE. Springer Series in Molecular Biology. Springer; New York,, New York, NY: 1989. Chromatin; p. 1. online resource. [Google Scholar]
- 2.McGinty RK, Tan S. Nucleosome structure and function. Chem Rev. 2015;115:2255–2273. doi: 10.1021/cr500373h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luger K, Dechassa ML, Tremethick DJ. New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat Rev Mol Cell Biol. 2012;13:436–447. doi: 10.1038/nrm3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Struhl K, Segal E. Determinants of nucleosome positioning. Nature structural & molecular biology. 2013;20:267–273. doi: 10.1038/nsmb.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.West SM, Rohs R, Mann RS, Honig B. Electrostatic interactions between arginines and the minor groove in the nucleosome. Journal of biomolecular structure & dynamics. 2010;27:861–866. doi: 10.1080/07391102.2010.10508587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hodges AJ, I, Gallegos J, Laughery MF, Meas R, Tran L, Wyrick JJ. Histone Sprocket Arginine Residues Are Important for Gene Expression, DNA Repair, and Cell Viability in Saccharomyces cerevisiae. Genetics. 2015;200:795–806. doi: 10.1534/genetics.115.175885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swygert SG, Peterson CL. Chromatin dynamics: interplay between remodeling enzymes and histone modifications. Biochimica et biophysica acta. 2014;1839:728–736. doi: 10.1016/j.bbagrm.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol. 2014;15:703–708. doi: 10.1038/nrm3890. [DOI] [PubMed] [Google Scholar]
- 9.Widom J. Role of DNA sequence in nucleosome stability and dynamics. Q Rev Biophys. 2001;34:269–324. doi: 10.1017/s0033583501003699. [DOI] [PubMed] [Google Scholar]
- 10.Segal E, Widom J. What controls nucleosome positions? Trends in genetics : TIG. 2009;25:335–343. doi: 10.1016/j.tig.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bowman GD, Poirier MG. Post-translational modifications of histones that influence nucleosome dynamics. Chem Rev. 2015;115:2274–2295. doi: 10.1021/cr500350x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson JD, Widom J. Sequence and position-dependence of the equilibrium accessibility of nucleosomal DNA target sites. Journal of molecular biology. 2000;296:979–987. doi: 10.1006/jmbi.2000.3531. [DOI] [PubMed] [Google Scholar]
- 13.Li G, Levitus M, Bustamante C, Widom J. Rapid spontaneous accessibility of nucleosomal DNA. Nature structural & molecular biology. 2005;12:46–53. doi: 10.1038/nsmb869. [DOI] [PubMed] [Google Scholar]
- 14.Smerdon MJ, Thoma F. Modulations in Chromatin Structure During DNA Damage Formation and DNA Repair. In: Nickoloff JA, Hoekstra MF, editors. DNA Damage and Repair Volume 2: DNA Repair in Higher Eukaryotes. II. Humana Press : Imprint: Humana Press; Totowa, NJ: 1998. pp. 199–222. [Google Scholar]
- 15.Liu X. In vitro chromatin templates to study nucleotide excision repair. DNA repair (Amst) 2015;36:68–76. doi: 10.1016/j.dnarep.2015.09.026. [DOI] [PubMed] [Google Scholar]
- 16.Taylor JS. Design, synthesis, and characterization of nucleosomes containing site-specific DNA damage. DNA repair (Amst) 2015;36:59–67. doi: 10.1016/j.dnarep.2015.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balliano AJ, Hayes JJ. Base excision repair in chromatin: Insights from reconstituted systems. DNA repair (Amst) 2015;36:77–85. doi: 10.1016/j.dnarep.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smerdon MJ, Conconi A. Modulation of DNA damage and DNA repair in chromatin. Prog Nucleic Acid Res Mol Biol. 1999;62:227–255. doi: 10.1016/s0079-6603(08)60509-7. [DOI] [PubMed] [Google Scholar]
- 19.Gale JM, Nissen KA, Smerdon MJ. UV-induced formation of pyrimidine dimers in nucleosome core DNA is strongly modulated with a period of 10.3 bases. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:6644–6648. doi: 10.1073/pnas.84.19.6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gale JM, Smerdon MJ. Photofootprint of nucleosome core DNA in intact chromatin having different structural states. Journal of molecular biology. 1988;204:949–958. doi: 10.1016/0022-2836(88)90054-x. [DOI] [PubMed] [Google Scholar]
- 21.Gale JM, Smerdon MJ. UV induced (6-4) photoproducts are distributed differently than cyclobutane dimers in nucleosomes. Photochemistry and photobiology. 1990;51:411–417. doi: 10.1111/j.1751-1097.1990.tb01732.x. [DOI] [PubMed] [Google Scholar]
- 22.Pehrson JR. Thymine dimer formation as a probe of the path of DNA in and between nucleosomes in intact chromatin. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:9149–9153. doi: 10.1073/pnas.86.23.9149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horikoshi N, Tachiwana H, Kagawa W, Osakabe A, Matsumoto S, Iwai S, Sugasawa K, Kurumizaka H. Crystal structure of the nucleosome containing ultraviolet light-induced cyclobutane pyrimidine dimer. Biochem Biophys Res Commun. 2016;471:117–122. doi: 10.1016/j.bbrc.2016.01.170. [DOI] [PubMed] [Google Scholar]
- 24.Cadet J, Anselmino C, Douki T, Voituriez L. Photochemistry of nucleic acids in cells. J Photochem Photobiol B. 1992;15:277–298. doi: 10.1016/1011-1344(92)85135-h. [DOI] [PubMed] [Google Scholar]
- 25.Pfeifer GP, You YH, Besaratinia A. Mutations induced by ultraviolet light. Mutation research. 2005;571:19–31. doi: 10.1016/j.mrfmmm.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 26.Osakabe A, Tachiwana H, Kagawa W, Horikoshi N, Matsumoto S, Hasegawa M, Matsumoto N, Toga T, Yamamoto J, Hanaoka F, Thoma NH, Sugasawa K, Iwai S, Kurumizaka H. Structural basis of pyrimidine-pyrimidone (6-4) photoproduct recognition by UV-DDB in the nucleosome. Scientific reports. 2015;5:16330. doi: 10.1038/srep16330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Courdavault S, Baudouin C, Charveron M, Canguilhem B, Favier A, Cadet J, Douki T. Repair of the three main types of bipyrimidine DNA photoproducts in human keratinocytes exposed to UVB and UVA radiations. DNA repair (Amst) 2005;4:836–844. doi: 10.1016/j.dnarep.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 28.Simpson RT, Stafford DW. Structural features of a phased nucleosome core particle. Proceedings of the National Academy of Sciences of the United States of America. 1983;80:51–55. doi: 10.1073/pnas.80.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lowary PT, Widom J. Nucleosome packaging and nucleosome positioning of genomic DNA. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:1183–1188. doi: 10.1073/pnas.94.4.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thastrom A, Lowary PT, Widom J. Measurement of histone-DNA interaction free energy in nucleosomes. Methods. 2004;33:33–44. doi: 10.1016/j.ymeth.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 31.Schieferstein U, Thoma F. Modulation of cyclobutane pyrimidine dimer formation in a positioned nucleosome containing poly(dA.dT) tracts. Biochemistry. 1996;35:7705–7714. doi: 10.1021/bi953011r. [DOI] [PubMed] [Google Scholar]
- 32.Losa R, Omari S, Thoma F. Poly(dA).poly(dT) rich sequences are not sufficient to exclude nucleosome formation in a constitutive yeast promoter. Nucleic acids research. 1990;18:3495–3502. doi: 10.1093/nar/18.12.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ong MS, Richmond TJ, Davey CA. DNA stretching and extreme kinking in the nucleosome core. Journal of molecular biology. 2007;368:1067–1074. doi: 10.1016/j.jmb.2007.02.062. [DOI] [PubMed] [Google Scholar]
- 34.Mann DB, Springer DL, Smerdon MJ. DNA damage can alter the stability of nucleosomes: effects are dependent on damage type. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:2215–2220. doi: 10.1073/pnas.94.6.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu X, Mann DB, Suquet C, Springer DL, Smerdon MJ. Ultraviolet damage and nucleosome folding of the 5S ribosomal RNA gene. Biochemistry. 2000;39:557–566. doi: 10.1021/bi991771m. [DOI] [PubMed] [Google Scholar]
- 36.Kosmoski JV, Smerdon MJ. Synthesis and nucleosome structure of DNA containing a UV photoproduct at a specific site. Biochemistry. 1999;38:9485–9494. doi: 10.1021/bi990297h. [DOI] [PubMed] [Google Scholar]
- 37.Shrader TE, Crothers DM. Effects of DNA sequence and histone-histone interactions on nucleosome placement. Journal of molecular biology. 1990;216:69–84. doi: 10.1016/S0022-2836(05)80061-0. [DOI] [PubMed] [Google Scholar]
- 38.Kosmoski JV, Ackerman EJ, Smerdon MJ. DNA repair of a single UV photoproduct in a designed nucleosome. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10113–10118. doi: 10.1073/pnas.181073398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duan MR, Smerdon MJ. UV damage in DNA promotes nucleosome unwrapping. The Journal of biological chemistry. 2010;285:26295–26303. doi: 10.1074/jbc.M110.140087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bucceri A, Kapitza K, Thoma F. Rapid accessibility of nucleosomal DNA in yeast on a second time scale. The EMBO journal. 2006;25:3123–3132. doi: 10.1038/sj.emboj.7601196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guintini L, Charton R, Peyresaubes F, Thoma F, Conconi A. Nucleosome positioning, nucleotide excision repair and photoreactivation in Saccharomyces cerevisiae. DNA repair (Amst) 2015;36:98–104. doi: 10.1016/j.dnarep.2015.09.012. [DOI] [PubMed] [Google Scholar]
- 42.Becker MM, Wang JC. Use of light for footprinting DNA in vivo. Nature. 1984;309:682–687. doi: 10.1038/309682a0. [DOI] [PubMed] [Google Scholar]
- 43.Selleck SB, Majors J. Photofootprinting in vivo detects transcription-dependent changes in yeast TATA boxes. Nature. 1987;325:173–177. doi: 10.1038/325173a0. [DOI] [PubMed] [Google Scholar]
- 44.Latham KA, Lloyd RS. T4 endonuclease V. Perspectives on catalysis. Ann N Y Acad Sci. 1994;726:181–196. doi: 10.1111/j.1749-6632.1994.tb52813.x. discussion 196–187. [DOI] [PubMed] [Google Scholar]
- 45.Li S, Waters R, Smerdon MJ. Low- and high-resolution mapping of DNA damage at specific sites. Methods. 2000;22:170–179. doi: 10.1006/meth.2000.1058. [DOI] [PubMed] [Google Scholar]
- 46.Pfeifer GP, Drouin R, Riggs AD, Holmquist GP. Binding of transcription factors creates hot spots for UV photoproducts in vivo. Molecular and cellular biology. 1992;12:1798–1804. doi: 10.1128/mcb.12.4.1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pfeifer GP. Formation and processing of UV photoproducts: effects of DNA sequence and chromatin environment. Photochemistry and photobiology. 1997;65:270–283. doi: 10.1111/j.1751-1097.1997.tb08560.x. [DOI] [PubMed] [Google Scholar]
- 48.Wang Z, Becker MM. Selective visualization of gene structure with ultraviolet light. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:654–658. doi: 10.1073/pnas.85.3.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu X, Conconi A, Smerdon MJ. Strand-specific modulation of UV photoproducts in 5S rDNA by TFIIIA binding and their effect on TFIIIA complex formation. Biochemistry. 1997;36:13710–13717. doi: 10.1021/bi9716736. [DOI] [PubMed] [Google Scholar]
- 50.Wolffe AP, Brown DD. Developmental regulation of two 5S ribosomal RNA genes. Science. 1988;241:1626–1632. doi: 10.1126/science.241.4873.1626. [DOI] [PubMed] [Google Scholar]
- 51.Kwon Y, Smerdon MJ. DNA repair in a protein-DNA complex: searching for the key to get in. Mutation research. 2005;577:118–130. doi: 10.1016/j.mrfmmm.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 52.Hayes JJ, Tullius TD. Structure of the TFIIIA-5 S DNA complex. Journal of molecular biology. 1992;227:407–417. doi: 10.1016/0022-2836(92)90897-s. [DOI] [PubMed] [Google Scholar]
- 53.Nolte RT, Conlin RM, Harrison SC, Brown RS. Differing roles for zinc fingers in DNA recognition: structure of a six-finger transcription factor IIIA complex. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:2938–2943. doi: 10.1073/pnas.95.6.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tommasi S, Swiderski PM, Tu Y, Kaplan BE, Pfeifer GP. Inhibition of transcription factor binding by ultraviolet-induced pyrimidine dimers. Biochemistry. 1996;35:15693–15703. doi: 10.1021/bi962117z. [DOI] [PubMed] [Google Scholar]
- 55.Kwon Y, Smerdon MJ. Binding of zinc finger protein transcription factor IIIA to its cognate DNA sequence with single UV photoproducts at specific sites and its effect on DNA repair. The Journal of biological chemistry. 2003;278:45451–45459. doi: 10.1074/jbc.M308645200. [DOI] [PubMed] [Google Scholar]
- 56.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, Ku M, Durham T, Kellis M, Bernstein BE. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wyrick JJ, Roberts SA. Genomic approaches to DNA repair and mutagenesis. DNA repair (Amst) 2015;36:146–155. doi: 10.1016/j.dnarep.2015.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, Volkert TL, Wilson CJ, Bell SP, Young RA. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- 59.Iyer VR, Horak CE, Scafe CS, Botstein D, Snyder M, Brown PO. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature. 2001;409:533–538. doi: 10.1038/35054095. [DOI] [PubMed] [Google Scholar]
- 60.Teng Y, Bennett M, Evans KE, Zhuang-Jackson H, Higgs A, Reed SH, Waters R. A novel method for the genome-wide high resolution analysis of DNA damage. Nucleic acids research. 2011;39:e10. doi: 10.1093/nar/gkq1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zavala AG, Morris RT, Wyrick JJ, Smerdon MJ. High-resolution characterization of CPD hotspot formation in human fibroblasts. Nucleic acids research. 2014;42:893–905. doi: 10.1093/nar/gkt912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Powell JR, Bennett MR, Evans KE, Yu S, Webster RM, Waters R, Skinner N, Reed SH. 3D-DIP-Chip: a microarray-based method to measure genomic DNA damage. Scientific reports. 2015;5:7975. doi: 10.1038/srep07975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu S, Evans KE, van Eijk P, Bennett M, Webster RM, Leadbitter M, Teng Y, Waters R, Jackson SP, Reed SH. Global genome nucleotide excision repair is organized into domains that promote efficient DNA repair in chromatin. Genome research. 2016;26:1376–1387. doi: 10.1101/gr.209106.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bryan DS, Ransom M, Adane B, York K, Hesselberth JR. High resolution mapping of modified DNA nucleobases using excision repair enzymes. Genome research. 2014;24:1534–1542. doi: 10.1101/gr.174052.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hu J, Adar S, Selby CP, Lieb JD, Sancar A. Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes & development. 2015;29:948–960. doi: 10.1101/gad.261271.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adar S, Hu J, Lieb JD, Sancar A. Genome-wide kinetics of DNA excision repair in relation to chromatin state and mutagenesis. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:E2124–33. doi: 10.1073/pnas.1603388113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mao P, Smerdon MJ, Roberts SA, Wyrick JJ. Chromosomal landscape of UV damage formation and repair at single-nucleotide resolution. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:9057–9062. doi: 10.1073/pnas.1606667113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reijns MA, Kemp H, Ding J, de Proce SM, Jackson AP, Taylor MS. Lagging-strand replication shapes the mutational landscape of the genome. Nature. 2015;518:502–506. doi: 10.1038/nature14183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ding J, Taylor MS, Jackson AP, Reijns MA. Genome-wide mapping of embedded ribonucleotides and other noncanonical nucleotides using emRiboSeq and EndoSeq. Nat Protoc. 2015;10:1433–1444. doi: 10.1038/nprot.2015.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brogaard K, Xi L, Wang JP, Widom J. A map of nucleosome positions in yeast at base-pair resolution. Nature. 2012;486:496–501. doi: 10.1038/nature11142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yu Y, Teng Y, Liu H, Reed SH, Waters R. UV irradiation stimulates histone acetylation and chromatin remodeling at a repressed yeast locus. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:8650–8655. doi: 10.1073/pnas.0501458102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yu Y, Deng Y, Reed SH, Millar CB, Waters R. Histone variant Htz1 promotes histone H3 acetylation to enhance nucleotide excision repair in Htz1 nucleosomes. Nucleic acids research. 2013;41:9006–9019. doi: 10.1093/nar/gkt688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.House NC, Koch MR, Freudenreich CH. Chromatin modifications and DNA repair: beyond double-strand breaks. Front Genet. 2014;5:296. doi: 10.3389/fgene.2014.00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Waters R, van Eijk P, Reed S. Histone modification and chromatin remodeling during NER. DNA repair (Amst) 2015;36:105–113. doi: 10.1016/j.dnarep.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 75.Pfeifer GP, Besaratinia A. Mutational spectra of human cancer. Human genetics. 2009;125:493–506. doi: 10.1007/s00439-009-0657-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordonez GR, Bignell GR, Ye K, Alipaz J, Bauer MJ, Beare D, Butler A, Carter RJ, Chen L, Cox AJ, Edkins S, Kokko-Gonzales PI, Gormley NA, Grocock RJ, Haudenschild CD, Hims MM, James T, Jia M, Kingsbury Z, Leroy C, Marshall J, Menzies A, Mudie LJ, Ning Z, Royce T, Schulz-Trieglaff OB, Spiridou A, Stebbings LA, Szajkowski L, Teague J, Williamson D, Chin L, Ross MT, Campbell PJ, Bentley DR, Futreal PA, Stratton MR. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, Drier Y, Zou L, Ramos AH, Pugh TJ, Stransky N, Helman E, Kim J, Sougnez C, Ambrogio L, Nickerson E, Shefler E, Cortes ML, Auclair D, Saksena G, Voet D, Noble M, DiCara D, Lin P, Lichtenstein L, Heiman DI, Fennell T, Imielinski M, Hernandez B, Hodis E, Baca S, Dulak AM, Lohr J, Landau DA, Wu CJ, Melendez-Zajgla J, Hidalgo-Miranda A, Koren A, McCarroll SA, Mora J, Lee RS, Crompton B, Onofrio R, Parkin M, Winckler W, Ardlie K, Gabriel SB, Roberts CW, Biegel JA, Stegmaier K, Bass AJ, Garraway LA, Meyerson M, Golub TR, Gordenin DA, Sunyaev S, Lander ES, Getz G. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hodgkinson A, Chen Y, Eyre-Walker A. The large-scale distribution of somatic mutations in cancer genomes. Human mutation. 2012;33:136–143. doi: 10.1002/humu.21616. [DOI] [PubMed] [Google Scholar]
- 79.Woo YH, Li WH. DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nature communications. 2012;3:1004. doi: 10.1038/ncomms1982. [DOI] [PubMed] [Google Scholar]
- 80.Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, Stamatoyannopoulos JA. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:139–144. doi: 10.1073/pnas.0912402107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schuster-Bockler B, Lehner B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012;488:504–507. doi: 10.1038/nature11273. [DOI] [PubMed] [Google Scholar]
- 82.Liu L, De S, Michor F. DNA replication timing and higher-order nuclear organization determine single-nucleotide substitution patterns in cancer genomes. Nature communications. 2013;4:1502. doi: 10.1038/ncomms2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Polak P, Lawrence MS, Haugen E, Stoletzki N, Stojanov P, Thurman RE, Garraway LA, Mirkin S, Getz G, Stamatoyannopoulos JA, Sunyaev SR. Reduced local mutation density in regulatory DNA of cancer genomes is linked to DNA repair. Nature biotechnology. 2014;32:71–75. doi: 10.1038/nbt.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zheng CL, Wang NJ, Chung J, Moslehi H, Sanborn JZ, Hur JS, Collisson EA, Vemula SS, Naujokas A, Chiotti KE, Cheng JB, Fassihi H, Blumberg AJ, Bailey CV, Fudem GM, Mihm FG, Cunningham BB, Neuhaus IM, Liao W, Oh DH, Cleaver JE, LeBoit PE, Costello JF, Lehmann AR, Gray JW, Spellman PT, Arron ST, Huh N, Purdom E, Cho RJ. Transcription restores DNA repair to heterochromatin, determining regional mutation rates in cancer genomes. Cell reports. 2014;9:1228–1234. doi: 10.1016/j.celrep.2014.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Perera D, Poulos RC, Shah A, Beck D, Pimanda JE, Wong JW. Differential DNA repair underlies mutation hotspots at active promoters in cancer genomes. Nature. 2016;532:259–263. doi: 10.1038/nature17437. [DOI] [PubMed] [Google Scholar]
- 86.Sabarinathan R, Mularoni L, Deu-Pons J, Gonzalez-Perez A, Lopez-Bigas N. Nucleotide excision repair is impaired by binding of transcription factors to DNA. Nature. 2016;532:264–267. doi: 10.1038/nature17661. [DOI] [PubMed] [Google Scholar]
- 87.White CL, Suto RK, Luger K. Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. The EMBO journal. 2001;20:5207–5218. doi: 10.1093/emboj/20.18.5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Luger K. Dynamic nucleosomes. Chromosome Res. 2006;14:5–16. doi: 10.1007/s10577-005-1026-1. [DOI] [PubMed] [Google Scholar]
- 89.Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. Journal of molecular biology. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 90.Widlund HR, Cao H, Simonsson S, Magnusson E, Simonsson T, Nielsen PE, Kahn JD, Crothers DM, Kubista M. Identification and characterization of genomic nucleosome-positioning sequences. Journal of molecular biology. 1997;267:807–817. doi: 10.1006/jmbi.1997.0916. [DOI] [PubMed] [Google Scholar]