

Abstract
FHIT is a genome caretaker gene that is silenced in >50% of cancers. Loss of Fhit protein expression promotes accumulation of DNA damage, affects apoptosis and epithelial-mesenchymal transition, though molecular mechanisms underlying these alterations have not been fully elucidated. Initiation of genome instability directly follows Fhit loss and the associated reduced Thymidine Kinase 1 (TK1) protein expression. The effects on TK1 of Fhit knockdown and Fhit induction in the current study confirmed the role of Fhit in regulating TK1 expression. Changes in Fhit expression did not impact TK1 protein turnover or transcription from the TK1 promoter, nor steady-state levels of TK1 mRNA or turnover. Polysome profile analysis showed that up-regulated Fhit expression resulted in decreased TK1 RNA in non-translating messenger ribonucleoproteins and increased ribosome density on TK1 mRNA. Fhit does not bind RNA but its expression increased luciferase expression from a transgene bearing the TK1 5’-UTR. Fhit has been reported to act as a scavenger decapping enzyme, and a similar result with a mutant (H96) that binds but does not cleave nucleoside 5’,5’-triphosphates suggests the impact on TK1 translation is due to its ability to modulate the intracellular level of cap-like molecules. Consistent with this, cells expressing Fhit mutants with reduced activity toward cap-like dinucleotides exhibit DNA damage resulting from TK1 deficiency, whereas cells expressing wild-type Fhit or the H96N mutant do not. The results have implications for the mechanism by which Fhit regulates TK1 mRNA, and more broadly, for its modulation of multiple functions as tumor suppressor/genome caretaker.
Keywords: Fhit, translational control, Thymidine Kinase 1, 5’-UTR
Graphical abstract
1. Introduction
Common chromosomal fragile sites are large, unstable regions of the genome that are highly sensitive to genotoxic and replication stress [1, 2]. In 1996, the first gene spanning a chromosome fragile site was reported and named FHIT (fragile histidine triad), owing to the fragility of the gene locus and sequence homology to members of the histidine triad family of enzymes [3]. Members of this family each contain a His-X-His-X-His-XX sequence motif that forms the core of the protein active site and functions in the hydrolysis of nucleotide derivatives. The FHIT gene product, Fhit, is a diadenosine triphosphate (Ap3A) hydrolase that asymmetrically cleaves dipurine polyphosphates in vitro, producing ADP and AMP [4]. Additionally, cancer cell lines that have lost Fhit expression produce excess Ap3A [5], a favored in vitro Fhit substrate. Draganescu et al [6] showed that GpppBODIPY, an mRNA cap-like molecule that fluoresces when cleaved, could also serve as a Fhit substrate, and Fhit was recently identified as a scavenger-decapping enzyme [7]. Scavenger-decapping enzymes degrade the methylguanosine (m7GpppN) dinucleotides that are generated by 3’-5’ degradation of mRNAs [8] that might otherwise compete with eIF4E for binding the mRNA cap.
Due to the sensitivity of the FHIT gene locus to diverse inducers of replication stress, inactivating genetic or epigenetic alterations frequently occur in early preneoplastic lesions, resulting in reduced or lost FHIT/Fhit expression in over 50% of human cancers. Examples of this include epithelial-derived cancers of the lung, esophagus, breast, stomach, skin, pancreas, cervix, and kidney [9]. There is considerable phenotypic evidence that Fhit loss is not a passenger event in cancer, but rather that Fhit, a mainly cytoplasmic protein, is a tumor suppressor. Fhit knockout mice develop more spontaneous tumors and dramatically more carcinogen-induced tumors than wild-type mice [10, 11] and viral-mediated FHIT gene delivery prevents and even causes regression of carcinogen-induced tumors [12–15] by activating apoptotic pathways. In response to oxidative stress, Fhit interacts with ferredoxin reductase in mitochondria to enhance reactive oxygen species production, which initiates caspase-3 activation and apoptosis [16, 17]. These tumor suppressor activities have been shown to be independent of Fhit enzymatic activity but dependent on Fhit-substrate binding [16, 17]. Fhit mutants that show reduced binding of substrates, and thus reduced substrate hydrolysis, lack tumor suppressor abilities, while mutants that can bind but not hydrolyze substrates retain tumor suppressor function [4, 18]. Therefore, it has been hypothesized that Fhit-substrate complexes are active signaling molecules for tumor suppression [19]. Fhit has also been shown to negatively regulate the epithelial-to-mesenchymal transition, tumor invasion, and metastasis [20, 21], though a direct route for Fhit participation in these cellular transitions has not been defined.
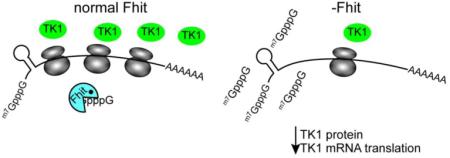
Fhit also functions as a genome caretaker; cells lacking Fhit exhibit increased double-strand DNA breaks (DSBs) and γH2AX and 53BP1 nuclear foci, due to endogenous replication stress [22]. Mechanistically, Fhit positively regulates expression of Thymidine Kinase 1 (TK1) protein, an enzyme necessary for sufficient thymidine triphosphate (TTP) pool synthesis [22]. Insufficient TK1 expression causes nucleotide imbalance in Fhit-deficient cells, leading to replication fork slowing, stalling, and DNA breaks, without checkpoint activation and cell cycle arrest. This replication stress provides an environment of ongoing DNA damage that leads to increased chromosomal instability in cells from Fhit knockout mice and human cells with silenced Fhit, including chromosome breaks, aneuploidy, and increased APOBEC-mediated point mutations [23, 24]. The current study shows that Fhit acts to maintain and/or stimulate the translation of TK1 mRNA, and suggests that this is dependent on the ability of Fhit to modulate the intracellular level of nucleoside 5’,5’-triphosphates.
2. Materials and Methods
2.1. Cell lines and reagents
H1299 lung carcinoma cells, in which the FHIT gene is silenced by CpG island methylation, were previously transfected with inducible FHIT cDNA and tightly regulated inducible clones were isolated: the D1 clone, inducible for wild-type (WT) Fhit, and the E1 clone carrying an empty vector [25]. The H96N and Y114F mutant Fhit-expressing H1299 clones were similarly prepared. Briefly, an H1299 cell line stably transfected with the ecdysone-inducible vector pVgRXR was transfected with pIND-FHIT plasmid containing either a single H96N or Y114F mutation. G418-resistant clones were selected and tested for Fhit expression with and without induction with ponasterone A. The Fhit H96N mutant binds the substrate similarly to WT Fhit, but can not cleave the in vitro cap-like substrate, Ap3A [4]. The Fhit Y114F mutant shows 5-fold reduced binding of Ap3A substrate [16, 17]. The H96N mutant suppresses tumorigenicity, while the Y114F mutant does not [16]. See Supplementary Table S1 for the kinetics of these mutants. H1299 cells were maintained in DMEM medium with 10% FBS, zeocin, gentamicin and geneticin. FHIT expression was induced by addition of ponasterone A (2 μM) (Life Technologies) to the growth medium. 293 human embryonic kidney cells transformed by Adeno-5 T antigen were cultured in DMEM with 10% FBS and 100 μg/ml gentamicin. HCT116 colon cancer cells expressing endogenous Fhit protein, were cultured in RPMI with 10% FBS and 100 μg/ml gentamicin.
2.2. Thymidine/nocodazole synchronization
293 cells were split 1:3 into 10 cm2 plates; 10 h later thymidine was added to the culture medium at a final concentration of 2 mM, and cells incubated 20 h. The culture medium was then removed, cells were washed 2x with PBS and normal growth medium added. After 4 h of incubation in normal medium, nocodazole was added at a final concentration of 100 ng/ml, and cells incubated another 12 h. The culture medium was removed, cells were washed 2x with PBS and released into normal growth medium. Cells were collected every 4 h for up to 24 h and lysed for western blot analysis.
2.3. siRNAs transfections
293 cells (60-70% confluent) were transfected with siRNAs targeting FHIT and non-specific scrambled control siRNAs (Santa Cruz Biotechnology); H1299 E1 cells (80-90% confluent) were transfected with siRNAs targeting human DcpS or a non-specific control siRNA (Santa Cruz Biotechnology), using the manufacturer’s recommended protocol. For each 60 mm dish, 1 μg of siRNA and 5 μl of Lipofectamine 2000 (Invitrogen) were diluted in Opti-MEM (Gibco) and incubated for 30 min. Cells were washed in Opti-MEM, overlaid with the siRNA/Lipofectamine solution and incubated overnight at 37°C. Verification of siRNA knockdown was performed 48-96 h later for Fhit expression and 48 h later for DcpS expression by Western blot.
2.4. Western blot analysis
Cells were lysed with RIPA buffer (Thermo Scientific) supplemented with Halt Protease Cocktail Inhibitors (Thermo Scientific). Cytoplasmic extracts specific to each assay (see below) were used for DRB, polysome and luciferase experiments, Proteins were separated by SDS gel electrophoresis, transferred to nylon membranes, and immunoblotted with antisera against human Fhit [26], human TK1 (Santa Cruz), human DcpS (Santa Cruz), human Cyclin A2 (Abcam), human α-tubulin (Sigma), or human Vinculin (Abcam).
2.5. Comet assays
The H1299 cell lines expressing Fhit WT, H96N, and Y114F proteins were used to assess comparative levels of DNA double strand breaks in order to correlate Fhit structural features [9] with genome caretaker function. Neutral comet assays were performed using the CometAssay kit (Trevigen) protocol. Images were acquired with a Zeiss AxioskopWideField40 fluorescent microscope mounted with an AxioCamHRc camera, and using an A-Plan 10×/0.25 objective lens. Images were converted to Bitmap files, and comet tail moments were scored using Tri Tek Comet score 1.6.1.22 (TriTek, autocomet.com). Immunoblots of lysates of the same cell lines with and without induction of WT or mutant Fhit expression were assessed in parallel for levels of Fhit expression.
2.6. Luciferase reporter construction
To determine if TK1 promoted transcription is affected by level of Fhit expression, the full-length TK1 promoter (NM_003258.4, 437 bp) was amplified using primers that included recognition sequences for SpeI and XhoI restriction endonucleases [27]. TK1 promoter amplicons were then sticky-end cloned into pMCS-Red firefly luciferase plasmid (ThermoFisher Scientific) following restriction digest with SpeI and XhoI restriction enzymes. For the UTR reporter constructs, the HBB 5’-UTR in CMV-glo-SPA [28] was replaced with an XhoI cloning site using the Change-IT site directed mutagenesis kit (Affymetrix). The DNA sequence encoding the entire 210 nt long 5’ UTR of TK1 (plus overhangs including XhoI restriction sites) was ordered as a single gBlock from Integrated DNA Technologies (IDT) and cloned into the newly inserted XhoI site. Finally, the restriction site was removed from UTR-less (control) and TK1-UTR constructs with subsequent site directed mutagenesis reactions as above. The FLAG tagged pRc/CMV WT FHIT plasmid has been described [4]. A FHIT cDNA with a PCR introduced single base pair mutation (H96N) without a FLAG tag was similarly cloned into the eukaryotic expression vector pRc/CMV (Invitrogen).
2.7. TK1 promoted luciferase activity in Fhit expressing and non-expressing cells
The TK1 promoted luciferase expression vector was co-transfected with pRL-TK (a Renilla luciferase reporter to normalize for transfection efficiency) into H1299 D1 and E1 cells using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s instructions. Three independent transfections were assayed in triplicate for all luciferase experiments. Luciferase output from both Firefly and Renilla luciferase were measured step-wise by luminometry on the Veritas microplate luminator 48 h post-transfection using the Dual-Glo luciferase assay system (Promega). Luminescence values for Firefly Luciferase were normalized to Renilla values to control for transfection efficiency. Relative firefly luciferase light units (RLU) in H1299 E1 or D1 cells were plotted relative to E1 cells. For UTR-containing reporter constructs, Lipofectamine 2000 was used to transiently co-transfect CMV-promoted, firefly Luciferase constructs with either no UTR or the TK1 5’-UTR, and pRL-TK into H1299 D1 and E1 cells that had been uninduced or induced to express Fhit 24 h prior to transfection. Cell extracts were harvested 24 h later and assayed for both luciferase mRNAs by qPCR and luciferase activity. In a follow-up experiment, H1299 cells were co-transfected with pCMV-empty, WT FHIT-flag, or mutant Fhit-H96N and the 5’-UTR reporters used earlier and then luciferase activity was assessed as above.
2.8. Analysis of TK1 mRNA decay
Triplicate cultures of H1299 E1 and D1 cells (0.75 x 106 cells) were seeded into 100 mm dishes. 24 h later, ponasterone A was added to a final concentration of 2 μM for 24 h followed by 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB) (Sigma-Aldrich) addition (60 μM). After 0, 2, 4, 6, 8, and 10 h of DRB treatment, cells were washed twice with PBS, scraped from the dish and pelleted by centrifugation at 2500 xg for 5 min. Cytoplasmic extract was prepared by lysing pellets in 5 pellet volumes of ice cold cytoplasmic lysis buffer (50 mM Tris pH 7.5, 10 mM KCl, 10 mM MgCl2, 150 mM NaCl, 0.2 % NP-40, 2 mM DTT, 0.5 mM PMSF, 1 mM sodium orthovanadate, supplemented with 5 μl/ml RNAseOUT (Life Technologies), 25 μl/ml protease inhibitor cocktail (Sigma), 10 μl/ml each phosphatase inhibitor cocktails 2 and 3 (Sigma)). Lysates were incubated on ice for 10 min with mild agitation every two min. Nuclei and other debris were pelleted with a 10 min centrifugation at 16,100 xg and pellets were discarded. RNA was extracted from the lysates using Direct-Zol (Zymo) according to the manufacturer’s Reaction Clean-Up instructions. For each sample, 2.5 μg of cytoplasmic RNA was spiked with luciferase control RNA (0.5 ng/sample, Promega) and analyzed by RT-qPCR.
2.9. Reverse Transcription qPCR
cDNA was synthesized from cytoplasmic RNA using Superscript III (Life Technologies) according to the manufacturer’s instructions for random hexamer priming (with a 55°C extension temperature). qPCR was performed with SensiFAST-SYBR No-ROX (Bioline) using a CFX Connect Real-Time System (BioRad). For the DRB experiments, samples were analyzed in triplicate and normalized to both the spiked Luciferase mRNA and 18S rRNA (an internal, DRB-unresponsive control). Gene specific primers for TK1 (Hs.PT.58.39947559), 18S rRNA: 18S_For 5'-CTGAGAAACGGCTACCACATC-3’ 18S_Rev 5'-GCCTCGAAAGAGTCCTGTATTG-3’ Luciferase: Pro_Luc_For 5'-CTTATGCATGCGGCCGCATCTAGAGG-3' and Pro_Luc_Rev, 5'-CAGTTGCTCTCCAGCGGTTCCATCC-3' and RLPP0 RPLP0-For 5'-GGAGAAACTGCTGCCTCATATC-3', RPLP0-Rev 5'-CAGCAGCTGGCACCTTATT-3' were purchased from IDT. The comparative ΔΔCt method was used to calculate the relative mRNA levels (compared to 0 h DRB).
2.10. Polysome gradients
Triplicate cultures of H1299 E1 and D1 cells (1.75 x 106 cells) were seeded into 150 mm dishes. 24 h later, the cells were induced with ponasterone A (2 μM) for 48 h. Cycloheximide (CHX, 100 μg/ml) was added to the medium for 10 min at 37°C. Cells were then washed twice with PBS+CHX (100 μg/ml), scraped from the dish and pelleted by centrifugation at 2500 xg for 5 min. Subsequent procedures were carried out on ice. Collected cell pellets were lysed in 5 pellet volumes of ice cold cytoplasmic lysis buffer (see section 2.8 above) supplemented with 200 μg/ml CHX and lysates were prepared as above. 75% (25% reserved for input samples and western blotting) of the supernatants were loaded onto 11 ml 10-50% linear sucrose gradients and centrifuged for 3 h at 35,000 rpm in a Sorvall TH641 rotor at 4ºC. Gradients were collected in 0.5 ml fractions with continuous monitoring of absorbance at 254 nm. 400 μl of alternating fractions were spiked with a luciferase control RNA (1 ng/fraction, Promega) and RNA was recovered using Direct-Zol (Zymo) according to the manufacturer’s Reaction Clean-Up instructions. 100% of the RNA recovered from each fraction (and paired, spiked, 15% input samples) was reverse transcribed with Superscript III as above. The comparative ΔΔCt method was used to calculate the relative mRNA levels (compared to sample-matched total gradient input).
2.11. Statistical analysis
Two-sided T-tests were used to determine significance for data with a normal distribution and equal variances. Nonparametric data was analyzed using the Mann-Whitney rank sum test for single comparisons or using the Kruskal-Wallis test for multiple comparisons. Groups with P-values less than a value of 0.05 were considered significantly different.
3. Results
3.1. Loss of Fhit protein expression abrogates S phase-specific expression of TK1
Fhit silencing causes depletion of TK1 protein expression in transformed human embryonic kidney cells, 293, as also observed in Fhit-negative H1299 lung carcinoma cells (E1) vs a sister clone carrying an inducible wild-type (WT) Fhit transgene (D1 induced) (Fig. 1, A and B). We have also previously shown decreased TK1 expression in kidney cells established from Fhit−/− mice and A549 lung cancer cells with Fhit knockdown for 9 weeks. Expression of exogenous Fhit is sufficient to restore TK1 expression to normal levels [22]. Thus, Fhit modulation of TK1 expression is a general phenomenon, necessary to prevent TTP imbalance and allow unperturbed DNA synthesis. To identify the level(s) at which Fhit regulates TK1 expression, we first silenced Fhit in 293 cells and observed the effect on the synthesis and degradation of TK1 protein after treatment with the proteasome inhibitor MG132. In this experiment, steady-state levels of TK1 should increase with prolonged proteasome inhibition, with the rate of TK1 level increase a function of the rate of protein synthesis. MG132 treatment led to a robust increase in TK1 expression only in Fhit-positive 293 cells (Fig. 1C). Using the reverse strategy by blocking protein synthesis with cycloheximide (CHX), steady-state levels will decrease with prolonged inhibition, with the rate of TK1 decrease being a function of the rate of protein degradation. Under these conditions, siCtrl and siFHIT 293 cells showed similar rates of TK1 protein degradation (Fig. 1D), indicating that Fhit promotes TK1 protein expression by stimulating protein production rather than protecting TK1 protein from degradation. The tight regulation of TK1 protein expression to the S and G2 phases of the cell cycle prevents aberrant accumulation of TTP levels when they are not needed for DNA synthesis or repair [29]. To determine if this positive regulation of TK1 by Fhit was S-phase specific, we synchronized cells to mitosis using a thymidine/nocodazole synchronization protocol and followed TK1 protein expression for 24 h after synchronization release. As cells cycled through S-phase, TK1 protein expression increased only in control cells, peaking during G2. S phase up-regulation was not observed in the Fhit-deficient cells and TK1 was only slightly expressed in G2 (Fig. 1E), indicating that Fhit is important for S-phase induction of TK1 protein expression.
Fig. 1. S-phase specific TK1 protein synthesis in Fhit-positive and negative cells.

A. Immunoblot of Vinculin, TK1, and Fhit in 293 cells 72 h post transfection with siCtrl and siFHIT. B. Immunoblot of Vinculin, TK1, and Fhit in H1299 E1 and D1 induced cells. Cells were harvested 48 h after treatment with ponasterone A. C. Immunoblot of TK1 and GAPDH in siRNA transfected 293 cells treated for indicated times with 10 μM MG132 (left panel). Relative TK1 protein expression determined by densitometric analysis of the immunoblots (right panel). D. Immunoblot of TK1 and GAPDH in siRNA transfected 293 cells treated for indicated times with 100 μg/ml cycloheximide (CHX) (left panel). Protein lysates from siFHIT cells were loaded at 2:1 ratio compared to siCtrl lysates to more accurately measure rate of degradation. Relative TK1 protein expression determined by densitometric analysis of immunoblots (right panel). E. Immunoblot of TK1 and Cyclin A2 in siRNA transfected 293 cells synchronized by thymidine/nocodazole block and released for up to 24 h. Cells pass through S and G2 phases between 16-24 h after release as determined by Cyclin A2 expression. The initial ‘0’ lanes on the two blots show siFHIT and siCtrl lanes for direct comparison to the juxtaposed ‘0’ lanes for the siCtrl and siFHIT time-course immunoblots of TK1 and Cyclin A2 expression. Error bars indicate standard deviation.
3.2. TK1 transcription, steady-state levels of TK1 mRNA and TK1 mRNA decay are unaffected by Fhit
To determine if Fhit affects TK1 transcription we cloned the full-length TK1 promoter regulatory region upstream of the firefly luciferase gene and co-transfected this with a plasmid expressing Renilla luciferase into H1299 E1 or D1 induced cells. A representative Western blot of Fhit induction is shown in the inset in Fig. 2C. Luciferase expression remained unchanged regardless of Fhit expression (Fig. 2A), indicating a post-transcriptional process was likely responsible for Fhit regulation of TK1 expression. The Fhit-negative H1299 lung cancer cell lines in Fig. 1B were used to examine the impact of Fhit on steady-state levels of TK1 mRNA. Results in Fig. 2B show there is no statistically significant difference in the steady-state level of TK1 mRNA in Fhit-deficient versus Fhit-expressing cells. An internal control of RPLP0 mRNA was similarly unaffected by Fhit. We next examined the impact of Fhit on the turnover of TK1 mRNA. In Fig. 2C triplicate cultures of D1 induced and E1 cells were treated with DRB and TK1 mRNA was quantified over the next 10 h by RT-qPCR and the results were compared to a spiked luciferase mRNA and 18S rRNA, a DRB-unresponsive internal control. TK1 mRNA had a half-life in excess of 10 h which was unaffected by Fhit. Together, results in Fig. 2 show that Fhit has no impact on the accumulation or decay of TK1 mRNA.
Fig. 2. Fhit does not regulate the transcription or stability of TK1 mRNA.

A. H1299 E1 and D1 induced cells were co-transfected with plasmids expressing firefly luciferase under control of the TK1 promoter and Renilla luciferase. Results are presented as the ratio of D1/E1 for relative light units of firefly luciferase normalized to Renilla luciferase and represent the mean ± standard deviation (n=3). B. RT-qPCR analysis of relative TK1 mRNA expression or RPLP0, an internal control, in H1299 E1 and D1 induced cells. mRNA levels, shown as fold change after induction of WT Fhit, were normalized to an internal luciferase control transcript. C. H1299 E1 and D1 induced cells were treated with DRB to block transcription. TK1 mRNA was determined by RT-qPCR and normalized to 18S rRNA and luciferase that was added as a recovery control. The insert is a Western blot of cytoplasmic extract from these cells probed with antibodies to Fhit and tubulin. While this is specific to C it is representative of Western blots done for A and B. The results in B and C represent the mean ± SEM from three independent experiments each performed in triplicate.
3.3. Fhit affects polysome binding of TK1 mRNA
To determine if Fhit affects the translation of TK1 mRNA, cytoplasmic extracts from E1 and D1 induced (Fhit expressing) cells were separated on sucrose gradients and analyzed for overall changes in polysome profile and for the distribution of TK1 mRNA. Expression of Fhit resulted in a reproducible increase in 80S monosomes (see Discussion, Fig. 3A) but had little overall impact on polysomes. RNA recovered from even-numbered fractions was spiked with a luciferase transcript as an internal control for sample recovery and analyzed by RT-qPCR for TK1 mRNA (Fig. 3B).
Fig. 3. Fhit increases polysome-bound TK1 mRNA.

A. Cytoplasmic extracts from H1299 E1 and D1 induced cells were separated on a 10-50% sucrose gradient and fractions were monitored continuously at 254 nm. A representative gradient (of 3 independent gradients) is shown for each cell line and a representative Western blot of Fhit expression is shown in the inset. B. Even-numbered fractions were spiked with 1 ng of luciferase RNA as a control for sample recovery, and recovered RNA was analyzed by RT-qPCR for TK1 and luciferase. TK1 was normalized to luciferase and plotted as the percent of mRNA in each gradient fraction relative to input. Results shown are the means ± SEM of three independent experiments each performed in triplicate.
In Fhit-expressing D1 cells the distribution of TK1 is shifted to heavier polysomes and the overall ribosome density of TK1 mRNA in D1 cells is 27% higher than that of Fhit-deficient E1 cells. Together with the corresponding ~2.5-fold decrease in TK1 mRNA in the non-translating pool at the top of the gradient these data indicate that increased translation of TK1 mRNA is responsible for the Fhit-dependent increase in expression seen in Fig. 1.
3.4. The 5’-UTR determines Fhit regulation of TK1 translation
Translation is regulated primarily at initiation, which in turn is affected by sequence and structural features of the 5’-UTR [30]. To determine if features of the TK1 5’-UTR are responsible for its increased translation in Fhit-expressing cells we inserted this sequence upstream of luciferase in a CMV promoter-driven plasmid. This or a construct lacking the TK1 5’-UTR were co-transfected with a plasmid expressing Renilla luciferase into D1 cells and luciferase activity was assessed as a function of Fhit expression (see Western blot in Fig. 4A, top). Fhit induction had no effect on expression of the control luciferase reporter (Fig. 4A); however, upon addition of the TK1 5’-UTR the luciferase reporter became susceptible to Fhit regulation. Overall, luciferase expression was 25% greater in Fhit-expressing cells compared to cells in which Fhit was not induced or cells carrying luciferase without the TK1 5’-UTR.
Fig. 4. Fhit mediates translational regulation through the TK1 5’-UTR.

A. H1299 D1 and D1 induced cells were transfected with a plasmid expressing firefly luciferase with no added 5’-UTR (control, grey bars), or the same plasmid expressing firefly luciferase with the TK1 5’-UTR (TK1, black bars). Results were normalized to co-transfected Renilla luciferase. Fhit induction is shown in the Western blot at the top. B. The experiment in A was repeated using Fhit-negative H1299 E1 cells treated with or without ponasterone A. Data shown in A and B are the means ±SEM of three independent experiments (each measured in triplicate). The asterisk (*) indicates p <0.05 as determined by two-tailed Student T test.
To confirm that the impact of Fhit on luciferase expression is linked to the TK1 5’-UTR we repeated this experiment using Fhit-negative E1 cells. In the experiment in Fig. 4B plasmids expressing the constructs used in Fig. 4A were transfected into untreated or Ponasterone A treated E1 cells, and, as in that experiment, firefly luciferase was normalized to a co-transfected Renilla luciferase plasmid. Unlike results in Fig. 4A the presence of the TK1 5’-UTR had no impact on luciferase expression, thus confirming Fhit regulation of TK1 translation is mediated by sequence or structural properties of the TK1 5’-UTR. A predicted secondary structure map of the available primate TK1 5’-UTRs is shown in Supplementary Fig. S1. In this the cap is juxtaposed to a base-paired region, raising the possibility that TK1 mRNA might be susceptible to conditions that influence cap binding by eIF4E [30].
3.5. The activity of Fhit on nucleoside 5’-5’-triphosphates correlates with its impact on TK1 translation and its action as a genome protective protein
Fhit has not been identified as an RNA-binding protein in any of the messenger ribonucleoprotein proteome studies done to date; however, it is a nucleoside 5’,5’-triphosphate hydrolase [4]. The recent identification of Fhit as a scavenger decapping enzyme [7] raised the possibility that the catalytic activity of Fhit might be responsible for the maintenance/stimulation of TK1 translation. To examine this we compared the impact on translation of wild-type Fhit with a mutant form of Fhit (H96N) that has a >106 fold lower kcat for nucleoside 5’-5’-triphosphates but nonetheless binds these with high affinity [16, 19]. In the experiment in Fig. 5A H1299 cells were transfected with empty vector, or plasmids expressing WT Fhit or H96N together with the TK1 5’-UTR luciferase reporter used in Fig. 4. We reasoned if Fhit acts by modulating the concentration of nucleoside 5’,5’-triphosphates the H96N form of the protein should affect a similar increase in luciferase expression as WT Fhit. Western blotting confirmed these proteins were expressed to a similar extent (Fig. 5A, upper panel). Importantly co-expression of either WT Fhit or H96N resulted in the same 25% increase in luciferase expression seen in Fig. 4 (Fig. 5A, lower panel), thus supporting the proposal that the activity of Fhit against nucleoside 5’,5’-triphosphates is the mechanism responsible for stimulating TK1 mRNA translation.
Fig. 5. Translational regulation of TK1 is mediated by Fhit scavenger decapping activity.

A. Parental H1299 cells were co-transfected with plasmids expressing firefly luciferase with the TK1 5’-UTR and Renilla luciferase, and either empty vector or plasmids expressing WT Fhit or the catalytically inactive H96N mutant. The impact of Fhit catalytic activity on expression driven by the TK1 5’-UTR was determined by dual luciferase assays. Data shown are the means ±SEM of three independent experiments (each measured in triplicate). The asterisk (*) indicates p <0.05 as determined by two-tailed Student T test. B. Extracts from DcpS positive HCT116 cells and H1299 cells used in this study were analyzed by Western blotting for DcpS and for Vinculin. C. H1299 E1 cells were transfected with control or DcpS siRNA and monitored by Western blotting for DcpS, Fhit, Vinculin and TK1.
Cells have two scavenger decapping enzymes, Fhit and DcpS, and if Fhit acts to reduce nucleoside 5’,5’-triphosphates (eg. cap dinucleotides) one might expect DcpS would mitigate the impact of Fhit loss. However, the H1299 lung cancer cells used in this study have significantly less DcpS protein than HCT116 colon cancer cells (Fig. 5B). DcpS expression changed little upon Fhit re-expression, indicating that catalytic activity of Fhit is associated with the observed changes in TK1 translation. The limited expression of TK1 in these cells is lower in DcpS knockdown cells (Fig. 5C), a result that is again consistent with a role for scavenger decapping activity in maintenance/stimulation of TK1. It should be noted that the gel was overloaded to visualize TK1.
3.6. The genome protective effect of Fhit depends on its ability to bind to or hydrolyze 5’-5’ nucleoside triphosphates
Finally we asked if both Fhit binding to and hydrolysis of substrates is necessary for Fhit genome caretaker function. The experiment in Fig. 6 used H1299 lung cancer cell lines stably-transfected with ponasterone-inducible transgenes expressing WT Fhit, H96N, or Fhit with tyrosine at position 114 changed to phenylalanine (Y114F), a mutation that shows 5-fold reduced binding of 5’-5’ nucleoside triphosphates [16,31]. Cells were left untreated or treated with ponasterone A to induce expression of the Fhit proteins and then assessed for genome DSBs by comet assay and for expression of Fhit and TK1 by Western blot.
Fig. 6. Assessment of DNA damage in cells expressing Fhit.

Fhit genome caretaker function requires binding and cleavage of Fhit substrate to prevent DSBs. A. Neutral comet assay of H1299 cells ± 48 h ponasterone A treatment. Box plots of tail moments: D1, n = 292; D1 induced, n = 207; H96N induced, n = 185; Y114F induced, n = 152. Scatter plots of tail moments include data from 3 separate experiments. Statistical significance was determined using the wilcoxon test. B. Representative photos of neutral comet assay fields of H1299 cells 48 h after induction of WT or mutant Fhit proteins. C. Immunoblot assay showing levels of TK1 and Fhit protein expression in the H1299 cells examined in panels A and B. Densitometry of TK1 expression relative to Vinculin loading control.
DNA damage was quantified in these cells via neutral comet assay. The neutral comet assay utilizes the faster migration of damaged DNA through agarose, resulting in DSBs visible as comet “tails” following electrophoresis. The product of tail length and the percentage of total DNA in the tail is then used to determine the tail moment in individual cells. Results demonstrated that there was an elevated level of DNA damage in uninduced H1299 D1 cells (Fig. 6A) which express a minimally detectable level of Fhit (Fig. 6C), compared to D1 induced cells that exhibit robust Fhit expression. (P<0.0001). H1299 cells expressing a Fhit mutant (Y114F), with reduced binding of nucleoside 5’-5’ triphosphates, did not protect the cells from genome instability and showed a level of DSBs similar to the D1 cells not induced to express Fhit (see Fig. 6A and B). This confirms that substrate binding (H96N mutant) or catalytic activity of Fhit is important for its genome caretaker function. Importantly, the decrease in DNA damage seen with this Fhit H96N mutant is similar to that observed with WT Fhit (p<0.0001). Collectively, these results are consistent with a relationship between the genome caretaker activity of Fhit and its ability to modulate the level of nucleoside 5’-5’ triphosphates within the cell.
4. Discussion
Since discovery of the FHIT gene and its identification as a tumor suppressor, it has been the subject of nearly two thousand publications, with some contending that its loss of expression in cancer is due to passenger gene alterations at the fragile locus. This is argued in spite of reports that FHIT gene replacement can cause loss of tumorigenicity in vitro and in vivo [12, 13]. Resolution of these opposing claims is important if losses of Fhit downstream signal pathways, which occur in some normal cells of all humans, are to be considered as cancer prevention and treatment targets. Recent reports from our laboratory have established a role for Fhit loss in the initiation of genome instability [22–24, 32], and have unambiguously characterized Fhit as a ‘genome caretaker’, a role intimated by the discovery that Fhit knockout mice develop sebaceous tumors [10], similar to those caused by loss of DNA mismatch repair enzymes [33]. Such sebaceous carcinomas in humans can also be caused either by loss of Fhit or loss of mismatch repair enzymes [34, 35].
Saldivar et al [22] also noted that loss of Fhit in various cell types in vitro led to DNA replication stress that can be caused by imbalance in production of dNTP pools, and found that the TTP level is reduced by Fhit loss through Fhit down-modulation of TK1 protein expression. Since this was the first direct effect of Fhit on expression of a specific gene, the next goal was to determine the mechanism through which Fhit controls the level of the TK1 protein, with the idea that this would lead to understanding of the capacity of Fhit to participate in control of other cellular and biological signal pathways. In keeping with results of that study, Fhit knockdown is accompanied by the parallel loss of TK1 (Fig. 1A), and this finding is complemented by results showing restoration of TK1 expression following induction of a Fhit transgene in Fhit-negative cells (Fig. 1B). Faced with the absence of an impact of Fhit on TK1 protein stability (Fig. 1, C and D), we concluded that Fhit acts to stimulate or maintain TK1 gene expression.
Fhit does not control TK1 expression through an effect on the TK1 promoter (Fig. 2A) and has no impact on the steady-state level of TK1 mRNA (Fig. 2B) or TK1 mRNA turnover (Fig. 2C). Where we did see an effect was on translation (Fig. 3), where there was a reduction in TK1 mRNA in non-translating messenger ribonucleoprotein and a corresponding increase in polysome-bound TK1 with the population of TK1 mRNA shifted to heavier polysomes. Overall polysome binding of TK1 mRNA increased by 27%, and this matched a similar Fhit-stimulated increase in luciferase when expressed as a fusion with the full-length TK1 5’-UTR (Fig. 4A). None of the studies done to date on the messenger ribonucleoprotein proteome identify Fhit as an RNA-binding protein [36]. This suggested Fhit regulation of TK1 may be an indirect consequence of its function as a scavenger decapping enzyme [7]. Support for this is seen in Fig. 5A, where overexpression of the H96N mutant protein had the same impact as wild-type Fhit on luciferase translated from mRNA with the TK1 5’-UTR. This is notable because H96N binds cap-like nucleoside 5’,5’-triphosphates with an affinity similar to WT Fhit (Supplementary Table S1). This binding activity is retained despite the fact that although H96N cripples the activity of Fhit in vitro (Kcat reduced over a million-fold) it still serves as a tumor suppressor in vivo [4]. We propose that overexpressed H96N has the potential to reduce the free intracellular pool of nucleoside 5’-5’-triphosphates in vivo. The millimolar Km of WT FHIT and H96N in vitro might seem to argue against this hypothesis; however, those values were obtained using ApppA as substrate, which is likely to differ from the Km of these proteins for m7GpppN cap dinucleotides in vivo. Further, the tumor suppressor phenotype exhibited by WT and H96N, but not Y114A FHIT in vivo supports this interpretation. Additional support was seen in Fig. 4B which shows that H1299 cells have little DcpS and levels of this scavenger decapping enzyme are unaffected by Fhit. Even though there is limited DcpS in these Fhit-deficient cells, its knockdown also reduced TK1 expression (Fig. 4C), a finding that provides additional support for the involvement of scavenger decapping activity in regulating TK1 expression.
Fig. 6 provided functional evidence that Fhit acts through its ability to reduce free nucleoside 5’,5’-triphosphates. In this experiment Fhit deficiency-associated DNA DSBs, initiated through Fhit loss and the associated TTP imbalance and quantified by the comet assay, were fully prevented by induced expression of WT Fhit. Importantly, this protective function of Fhit is also seen with the H96N form of Fhit. Additional support for this was seen when we examined the reduced substrate-binding Y114F mutant. Fhit/Y114F has 5-fold greater Km for nucleoside 5’,5’-triphosphates than WT Fhit protein. This reduced ability to bind cap-like molecules is consistent with the inability of Fhit/Y114F to protect cells from DNA damage.
In repeated experiments we observed a reproducible increase in 80S monosomes in Fhit-expressing cells compared to controls but little change in absorbance across polysome-containing fractions. Until recently 80S monosomes were thought to consist of non-functional couplings of 40S and 60S ribosome subunits. However, this changed with the demonstration in [37] that 80S monosomes are in fact engaged in translation, particularly of low abundance regulatory proteins. Although TK1 mRNA is not enriched in 80S monosomes, the increase in 80S monosomes in Fhit-expressing cells is consistent with Fhit acting upstream of the translation of regulatory proteins that are involved in its tumor suppressor/genome protective effect.
Since Fhit does not bind RNA how might that happen? H1299 cells are deficient in DcpS, so a likely answer is that the loss of Fhit is associated with an overall decrease in scavenger decapping activity. This would result in the accumulation of cap dinucleotides generated by 3’-5’ mRNA decay. Given that eIF4E binds cap dinucleotides independently of mRNA [38], an increase in their intracellular concentration might result in competition with capped ends of some mRNAs for binding by a more limited population of eIF4E. We speculate that this might be particularly relevant for mRNAs whose capped ends are not readily accessible for binding, for example by juxtaposition to highly structured 5’-UTRs. This is consistent with a predicted secondary structure map of the available primate TK1 5’-UTRs (Supplementary Figure S1). In this figure the TK1 5’-UTR sequences of 14 different primates were aligned and their common RNA structure was computationally predicted emphasizing compensatory mutations consistent with the base pairing pattern. The high degree of conservation and presence of multiple base pairings supported by compensatory mutations, while still needing to be confirmed experimentally, lends confidence to the overall picture.
Although cancer is associated with elevated levels of eIF4F complex proteins, most notably eIF4E [39], the large literature on loss of FHIT and cancer suggests a complex interrelationship between these processes. Elevated eIF4E promotes malignant transformation at least in part by increasing translation of mRNAs with specific sequence or structural elements within their 5’-UTR [40]. While elevated eIF4E might be expected to mitigate competition from cap dinucleotides, this has yet to be shown, and indeed both processes may impact cancer formation. For example, Fhit loss occurs early in preneoplastic lesions, increasing the cap dinucleotide pool, and perhaps leading to selective pressure to upregulate eIF4E. Thus, it will be important to identify additional mRNAs whose translation contribute to Fhit tumor suppressor and genome caretaker functions, and to identify the pathways in which they function as targets for synthetic lethality strategies to eliminate Fhit-deficient cells early during cancer progression.
Supplementary Material
Highlights.
Fhit loss leads to decreased TK1 protein independent of protein stability
Fhit does not affect TK1 transcription, TK1 mRNA level or decay
Fhit increases polysome binding and ribosome density on TK1 mRNA
Fhit translational control is mediated through the TK1 5’-UTR
Fhit may control TK1 through modulation of nucleoside 5’,5’-triphosphate levels
Acknowledgments
Funding
This research was supported by a multi-PI IDEA grant from the Ohio State University Comprehensive Cancer Center Pelotonia fund (to KH, DS, RB), from the National Cancer Institute (grant numbers CA120516 and CA154200, to KH), from the National Institute of General Medical Science (grant number GM084177, DRS), and the National Science Foundation (grant numbers DMR-1105458 and DMR-1410172, RB). DLK was supported by a Pelotonia postdoctoral fellowship and training grant T32 CA0093338 from the National Cancer Institute, CEW was supported by a predoctoral fellowship from the Ohio State University Medical Center and a Pelotonia PhD student fellowship, IO was supported by a scholarship for graduate research from the Egyptian Cultural and Educational Bureau, and JCS was supported by an F31 fellowship from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of Pelotonia, The Ohio State University, the National Institutes of Health or the National Science Foundation.
Abbreviations
- FHIT
fragile histidine triad gene
- Fhit
the FHIT gene protein product
- TK1
thymidine kinase gene and protein
- DSB
double-strand break
- Ap3A
diadenosine triphosphate
- TTP
thymidine triphosphate
- CHX
cycloheximide
- DRB
5,6-dichloro-1-β-D-ribofuranosylbenzimidazole
- dNTPs
deoxyribonucleotide triphosphates
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schwartz M, Zlotorynski E, Goldberg M, Ozeri E, Rahat A, le Sage C, Chen BP, Chen DJ, Agami R, Kerem B. Genes Dev. 2005;19:2715–2726. doi: 10.1101/gad.340905. 10.1101/gad.340905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koundrioukoff S, Carignon S, Técher H, Letessier A, Brison O, Debatisse M. PLoS Genet. 2013;9:e1003643. doi: 10.1371/journal.pgen.1003643. 10.1371/journal.pgen.1003643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ohta M, Inoue H, Cotticelli MG, Kastury K, Baffa R, Palazzo J, Siprashvili Z, Mori M, McCue P, Druck T, Croce CM, Huebner K. Cell. 1996;84:587–597. doi: 10.1016/s0092-8674(00)81034-x. 10.1016/S0092-8674(00)81034-X. [DOI] [PubMed] [Google Scholar]
- 4.Barnes LD, Garrison PN, Siprashvili Z, Guranowski A, Robinson AK, Ingram SW, Croce CM, Ohta M, Huebner K. Biochemistry. 1996;35:11529–11535. doi: 10.1021/bi961415t. 10.1021/bi961415t. [DOI] [PubMed] [Google Scholar]
- 5.Murphy GA, Halliday D, McLennan AG. Cancer Res. 2000;60:2342–2344. [PubMed] [Google Scholar]
- 6.Draganescu A, Hodawadekar SC, Gee KR, Brenner C. J Biol Chem. 2000;275:4555–4560. doi: 10.1074/jbc.275.7.4555. 10.1074/jbc.275.7.4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taverniti V, Seraphin B. Nucleic Acids Res. 2014;43:482–492. doi: 10.1093/nar/gku1251. 10.1093/nar/gku1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Y, Kiledjian M. Wiley Interdiscip. Rev. RNA. 2010;1:253–265. doi: 10.1002/wrna.15. 10.1002/wrna.15. [DOI] [PubMed] [Google Scholar]
- 9.Pichiorri F, Palumbo T, Suh SS, Okamura H, Trapasso F, Ishii H, Huebner K, Croce CM. Future Oncol. 2008;4:815–824. doi: 10.2217/14796694.4.6.815. 10.2217/14796694.4.6.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fong LY, Fidanza V, Zanesi N, Lock LF, Siracusa LD, Mancini R, Siprashvili Z, Ottey M, Martin SE, Druck T, McCue PA, Croce CM, Huebner K. Proc Natl Acad Sci U S A. 2000;97:4742–4747. doi: 10.1073/pnas.080063497. 10.1073/pnas.080063497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zanesi N, Fidanza V, Fong LY, Mancini R, Druck T, Valtieri M, Rüdiger T, McCue PA, Croce CM, Huebner K. Proc Natl Acad Sci U S A. 2001;98:10250–10255. doi: 10.1073/pnas.191345898. 10.1073/pnas.191345898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dumon KR, Ishii H, Fong LY, Zanesi N, Fidanza V, Mancini R, Vecchione A, Baffa R, Trapasso F, During MJ, Huebner K, Croce CM. Proc Natl Acad Sci U S A. 2001;98:3346–3351. doi: 10.1073/pnas.061020098. 10.1073/pnas.061020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishii H, Zanesi N, Vecchione A, Trapasso F, Yendamuri S, Sarti M, Baffa R, During MJ, Huebner K, Fong LY, Croce CM. FASEB J. 2003;17:1768–1770. doi: 10.1096/fj.03-0241fje. 10.1096/fj.03-0241fje. [DOI] [PubMed] [Google Scholar]
- 14.Ji L, Fang B, Yen N, Fong K, Minna JD, Roth JA. Cancer Res. 1999;59:3333–3339. [PubMed] [Google Scholar]
- 15.Ramesh R, Saeki T, Templeton NS, Ji L, Stephens LC, Ito I, Wilson DR, Wu Z, Branch CD, Minna JD, Roth JA. Mol Ther. 2001;3:337–350. doi: 10.1006/mthe.2001.0266. 10.1006/mthe.2001.0266. [DOI] [PubMed] [Google Scholar]
- 16.Pichiorri F, Okumura H, Nakamura T, Garrison PN, Gasparini P, Suh SS, Druck T, McCorkell KA, Barnes LD, Croce CM, Huebner K. J Biol Chem. 2009;284:1040–1049. doi: 10.1074/jbc.M806638200. 10.1074/jbc.M806638200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trapasso F, Pichiorri F, Gaspari M, Palumbo T, Aqeilan RI, Gaudio E, Okumura H, Iuliano R, Di Leva G, Fabbri M, Birk DE, Raso C, Green-Church K, Spagnoli LG, Venuta S, Huebner K, Croce CM. J Biol Chem. 2008;283:13736–13744. doi: 10.1074/jbc.M709062200. 10.1074/jbc.M709062200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Siprashvili Z, Sozzi G, Barnes LD, McCue P, Robinson AK, Eryomin V, Sard L, Tagliabue E, Greco A, Fusetti L, Schwartz G, Pierotti MA, Croce CM, Huebner K. Proc Natl Acad Sci U S A. 1997;94:13771–13776. doi: 10.1073/pnas.94.25.13771. 10.1073/pnas.94.25.13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pace HC, Garrison PN, Robinson AK, Barnes LD, Draganescu A, Rosler A, Blackburn GM, Siprashvili Z, Croce CM, Huebner K, Brenner C. Proc. Natl. Acad. Sci. USA. 1998;95:5484–5489. doi: 10.1073/pnas.95.10.5484. 10.1073/pnas.95.10.5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joannes A, Grelet S, Duca L, Gilles C, Kileztky C, Dalstein V, Birembaut P, Polette M, Nawrocki-Raby B. Mol Cancer Res. 2014;12:775–783. doi: 10.1158/1541-7786.MCR-13-0386-T. 10.1158/1541-7786.MCR-13-0386-T. [DOI] [PubMed] [Google Scholar]
- 21.Suh SS, Yoo JY, Cui R, Kaur B, Huebner K, Lee TK, Aqeilan RI, Croce CM. PLoS Genet. 2014;10:e1004652. doi: 10.1371/journal.pgen.1004652. 10.1371/journal.pgen.1004652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saldivar JC, Miuma S, Bene J, Hosseini SA, Shibata H, Sun J, Wheeler LJ, Mathews CK, Huebner K. PLoS Genet. 2012;8:e1003077. doi: 10.1371/journal.pgen.1003077. 10.1371/journal.pgen.1003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miuma S, Saldivar JC, Karras JR, Waters CE, Paisie CA, Wang Y, Jin V, Sun J, Druck T, Zhang J, Huebner K. PLoS One. 2013;8:e80730. doi: 10.1371/journal.pone.0080730. 10.1371/journal.pone.0080730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Waters CE, Saldivar JC, Amin ZA, Schrock MS, Huebner K. Oncotarget. 2015;6:3409–3419. doi: 10.18632/oncotarget.2636. 10.18632/oncotarget.2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cantor JP, Iliopoulos D, Rao AS, Druck T, Semba S, Han SY, McCorkell KA, Lakshman TV, Collins JE, Wachsberger P, Friedberg JS, Huebner K. Int J Cancer. 2007;120:24–31. doi: 10.1002/ijc.22073. 10.1002/ijc.22073. [DOI] [PubMed] [Google Scholar]
- 26.Druck T, Hadaczek P, Fu TB, Ohta M, Siprashvili Z, Baffa R, Negrini M, Kastury K, Veronese ML, Rosen D, Rothstein J, McCue P, Cotticelli MG, Inoue H, Croce CM, Huebner K. Cancer Res. 1997;57:504–512. [PubMed] [Google Scholar]
- 27.Arcot SS, Flemington EK, Deininger PL. J Biol Chem. 1989;264:2343–2349. [PubMed] [Google Scholar]
- 28.Peng J, Murray EL, Schoenberg DR. Methods Mol. Biol. 2008;419:215–230. doi: 10.1007/978-1-59745-033-1_15. 10.1007/978-1-59745-033-1_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu CM, Chang ZF. J Biomed Sci. 2007;14:491–497. doi: 10.1007/s11373-007-9175-1. 10.1007/s11373-007-9175-1. [DOI] [PubMed] [Google Scholar]
- 30.Hinnebusch AG, Ivanov IP, Sonenberg N. Science. 2016;352:1413–1416. doi: 10.1126/science.aad9868. 10.1126/science.aad9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Semba S, Trapasso F, Fabbri M, McCorkell KA, Volinia S, Druck T, Iliopoulos D, Pekarsky Y, Ishii H, Garrison PN, Barnes LD, Croce CM, Huebner K. Oncogene. 2006;25:2860–2872. doi: 10.1038/sj.onc.1209323. 10.1038/sj.onc.1209323. [DOI] [PubMed] [Google Scholar]
- 32.Hosseini SA, Horton S, Saldivar JC, Miuma S, Stampfer MR, Heerema NA, Huebner K. Genes Chromosomes Cancer. 2013;52:1017–1029. doi: 10.1002/gcc.22097. 10.1002/gcc.22097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kruse R, Lamberti C, Wang Y, Ruelfs C, Bruns A, Esche C, Lehmann P, Ruzicka T, Rütten A, Friedl W, Propping P. Hum Genet. 1996;98:747–750. doi: 10.1007/s004390050298. 10.1007/s004390050298. [DOI] [PubMed] [Google Scholar]
- 34.Goldberg M, Rummelt C, Foja S, Holbach LM, Ballhausen WG. Hum Mutat. 2006;27:155–162. doi: 10.1002/humu.20281. 10.1002/humu.20281. [DOI] [PubMed] [Google Scholar]
- 35.Becker K, Goldberg M, Helmbold P, Holbach LM, Loeffler KU, Ballhausen WG. Br J Dermatol. 2008;159:1282–1289. doi: 10.1111/j.1365-2133.2008.08783.x. 10.1111/j.1365-2133.2008.08783.x. [DOI] [PubMed] [Google Scholar]
- 36.Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, Krijgsveld J, Hentze MW. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 37.Heyer EE, Moore MJ. Cell. 2016;164:757–769. doi: 10.1016/j.cell.2016.01.003. 10.1016/j.cell.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 38.Rakotondrafara AM, Hentze MW. Nat Protoc. 2011;6:563–571. doi: 10.1038/nprot.2011.314. 10.1038/nprot.2011.314. [DOI] [PubMed] [Google Scholar]
- 39.Pelletier J, Graff J, Ruggero D, Sonenberg N. Cancer Res. 2015;75:250–263. doi: 10.1158/0008-5472.CAN-14-2789. 10.1158/0008-5472.CAN-14-2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Truitt ML, Conn CS, Shi Z, Pang X, Tokuyasu T, Coady AM, Seo Y, Barna M, Ruggero D. Cell. 2015;162:59–71. doi: 10.1016/j.cell.2015.05.049. 10.1016/j.cell.2015.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.