

Abstract
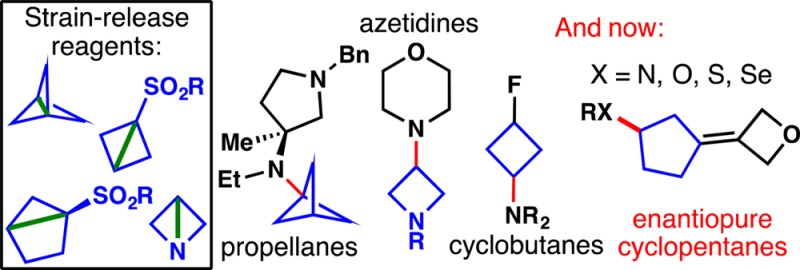
Driven by the ever-increasing pace of drug discovery and the need to push the boundaries of unexplored chemical space, medicinal chemists are routinely turning to unusual strained bioisosteres such as bicyclo[1.1.1]pentane, azetidine, and cyclobutane to modify their lead compounds. Too often, however, the difficulty of installing these fragments surpasses the challenges posed even by the construction of the parent drug scaffold. This full account describes the development and application of a general strategy where spring-loaded, strained C–C and C–N bonds react with amines to allow for the “any-stage” installation of small, strained ring systems. In addition to the functionalization of small building blocks and late-stage intermediates, the methodology has been applied to bioconjugation and peptide labeling. For the first time, the stereospecific strain-release “cyclopentylation” of amines, alcohols, thiols, carboxylic acids, and other heteroatoms is introduced. This report describes the development, synthesis, scope of reaction, bioconjugation, and synthetic comparisons of four new chiral “cyclopentylation” reagents.
Introduction
The unique opportunities afforded by strained bonds in organic synthesis have been appreciated for decades.1 The potential energy stored in such constructs can have applications in total synthesis, diversity generation, materials science, and even bioorthogonal chemistry. Some notable examples are depicted in Figure 1A. Some of the best illustrations of strain-enabled reactivity stem from the groups of Sharpless and Finn with their studies of thiiranium and quadricyclane ring openings.2,3 These high energy systems could be harnessed to provide simple and rapid, click-like4 access to new connections. A multitude of cycloadditions have also benefited from the release of strain, as exemplified by the work of Kerr,5 Wipf,6 Shi,7 and others. The area of C–C bond functionalization has also largely relied on the release of strain such as in Mitsudo’s studies of cyclobutanone ring opening.8 Numerous examples of strain-assisted generation of complexity in total synthesis are also apparent as illustrated in Wender’s classic cedrene synthesis9 and Danishefsky’s aplykuroinone synthesis10 as well as some examples from our laboratory.11 The advantages of using strained systems extend beyond the aforementioned cases where bonds are broken. For example, Myers and Denmark’s use of strained silacyclobutanes as a method to enhance Lewis-acidity in a Mukaiyama aldol reaction demonstrated that strained bonds can influence reactivity at distal sites.12 Bertozzi’s use of strained cyclooctynes enabled rapid metal-free 3+2 cycloadditions with applications in materials chemistry and bioconjugation.13 Strain even manifests itself in commonly used reagents such as dimethyldioxirane (DMDO) and trifluoromethyldioxirane (TFDO) for epoxidations and C–H oxidation.14 Also worth mentioning here is the large body of literature on exotic and strained hydrocarbons.15 This science has largely been buried in the realm of physical organic chemistry literature but forms much of the foundation of the work cited above as well as the present report. Taken together, this historical backdrop served as a robust inspiration for the work described in this Article.
Figure 1.
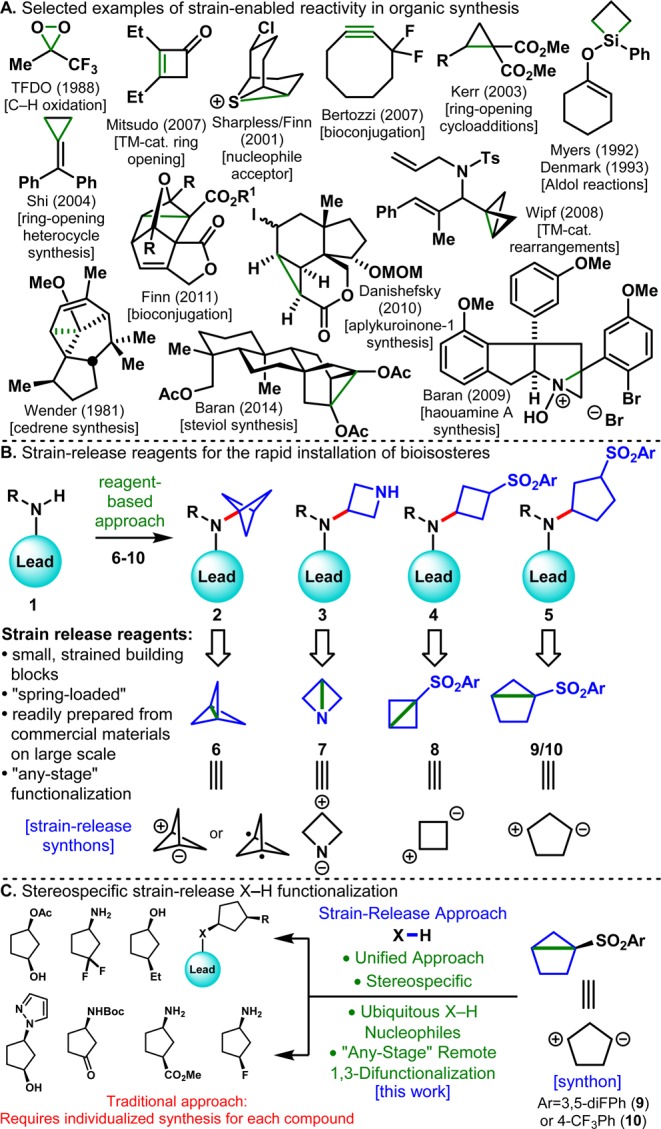
(A) Examples of the utility of strained bonds in organic synthesis. (B) Suite of strain-release reagents for the installation of bioisosteres. (C) Installation of chiral 1,3-disubstituted cyclopentanes via stereospecific strain-release X–H functionalization.
Outlined herein is the development of a series of “spring-loaded”4 reagents (6–10) that enable the direct installation of small ring bioisosteres (Figure 1B) onto heteroatoms. Borne out of pragmatic considerations, these reagents can be used at any stage of a synthesis. More importantly, they free practitioners from a retrosynthetic design wedded to small fragments of a target and instead allow for a “scaffold-first” analysis. This full account follows up on our recent publication16 and includes a number of new substrates and details on the optimization and invention of each reagent.
A major addition to this area is also unveiled for the first time, as depicted in Figure 1C: stereospecific strain-release. Thus, the invention of unique chiral strained “housane” derivatives is described enabling a wide array of direct functionalizations of amines, heterocycles, alcohols, thiols, selenols, amides, and even carboxylic acids. From a basic reactivity standpoint, these studies represent the first asymmetric syntheses of such strained hydrocarbons. The applications of these systems in the context of medicinal chemistry is also demonstrated.
Direct Bicyclo[1.1.1]pentylation (“Propellerization”) via Strain Release
Our entry into this area was borne directly from an academic–industrial partnership between our laboratory and Pfizer.17 Specifically, scientists at Pfizer had settled upon a lead candidate incorporating an intriguing bicyclo[1.1.1]pentane bioisosteric motif. Over the past few decades, interest in exotic bioisosteres has blossomed, and discovery chemists are constantly seeking routes to high-value bioisosteres to improve the properties of compounds and to create new chemical space in the extremely competitive intellectual property landscape.18 Although interest in bioisosteres has grown, methods and routes to install these groups are not well developed. From a strategic perspective, bioisosteric replacements often shift the focus of retrosynthetic analysis away from the primary scaffold. Thus, there is a need to develop simple methods for the rapid incorporation of such motifs onto pre-existing structures.
Bicyclo[1.1.1]pentanes (11–15, Figure 2A) in particular have emerged as a bioisostere for phenyl rings and tert-butyl groups due to their unique chemical and physical properties. Specifically, bicyclo[1.1.1]pentanes have been shown to impart favorable properties onto drug-like molecules over their phenyl counterparts with respect to passive permeability, aqueous solubility, and metabolic stability.19 Carreira evaluated bicyclo[1.1.1]pentane, along with pentafluorosulfanyl and cyclopropyl-trifluoromethyl groups as bioisosteric replacements for tert-butyl groups in bosentan and vercimon.20 Additionally, bicyclo[1.1.1]pentanes have been shown to increase three-dimensionality of a molecule and have also been shown to act as a rigid spacer compared to their phenyl congeners (16 and 17, Figure 2B).21 Reports have emerged from Pfizer, Bristol-Myers Squibb, Cephalon, and others in which the bicyclo[1.1.1]pentane (BCP) motif has been incorporated among their series of lead compounds.22
Figure 2.

(A) Lead compounds containing the BCP bioisostere. (B) BCP as a phenyl bioisostere and rigidifying linker. (C) Previous syntheses of bicyclo[1.1.1]pentan-1-amine.
As alluded to above, the story of strain-release functionalization began in our lab when Pfizer approached us to invent a new method for the synthesis of bicyclo[1.1.1]pentan-1-amine (44, Figure 2C). Since the pioneering synthesis of bicyclo[1.1.1]pentan-1-amine by Wiberg in 1970, chemists have sought out new and scalable routes to this elusive compound.
The first synthesis by Wiberg commenced with an electrochemical Wurtz reaction to access bicyclopentane, which was further elaborated to bicyclo[1.1.1]pentan-1-amine in three steps via carboxylic acid 18.23 All subsequent routes take advantage of Szeimies’ synthesis of [1.1.1]propellane24 from tetrahalide 40 (Figure 3E).25 Timberlake converted Wiberg’s intermediate, carboxylic acid 18, to 44 via the Schmidt reaction.26 During the course of their NMR studies on bridgehead-substituted polycycloalkanes, Della converted 19 to 44 via a Hofmann rearrangement with iodosobenzene.27 Barbachyn showed that organotin species 20 could be transmetalated with n-BuLi and quenched with LiNHOMe to afford 44.28 Bunker and Adsool independently reported the reduction of azide 21 to 44 with H2/Pd(OH)2/C (16% yield) and tris(trimethylsilyl)silane/AIBN/2-mercaptoethanol (82%), respectively.29,30 The same research groups also reported the reduction of azide 23 to 44.30,31 Of all of the syntheses presented in Figure 2C, perhaps the most scalable route was developed by Bunker at Pfizer where a hydrohydrazination reaction was used to append an amine equivalent onto the bicyclopentane moiety through intermediate 22.29 However, this route was ultimately deemed unscalable by Pfizer’s process group for a number of reasons: (1) 6 had to be isolated via distillation resulting in clogging of the distillation head; (2) flash chromatography, low boiling solvents (e.g., CH2Cl2), and large exotherms complicated the hydrohydrazination step; and (3) high-energy functional groups were incorporated in several of the later intermediates (e.g., hydrazine derivatives). During this time, Pfizer was concerned that the scale-up of bicyclo[1.1.1]pentan-1-amine would be a limiting factor if compounds containing the bicyclo[1.1.1]pentane structure were to be considered for clinical trials. Thus, there was an urgent need for a practical large scale synthesis of 44 that did not involve the use of toxic or dangerous reagents or even the discrete isolation of 6.
Figure 3.
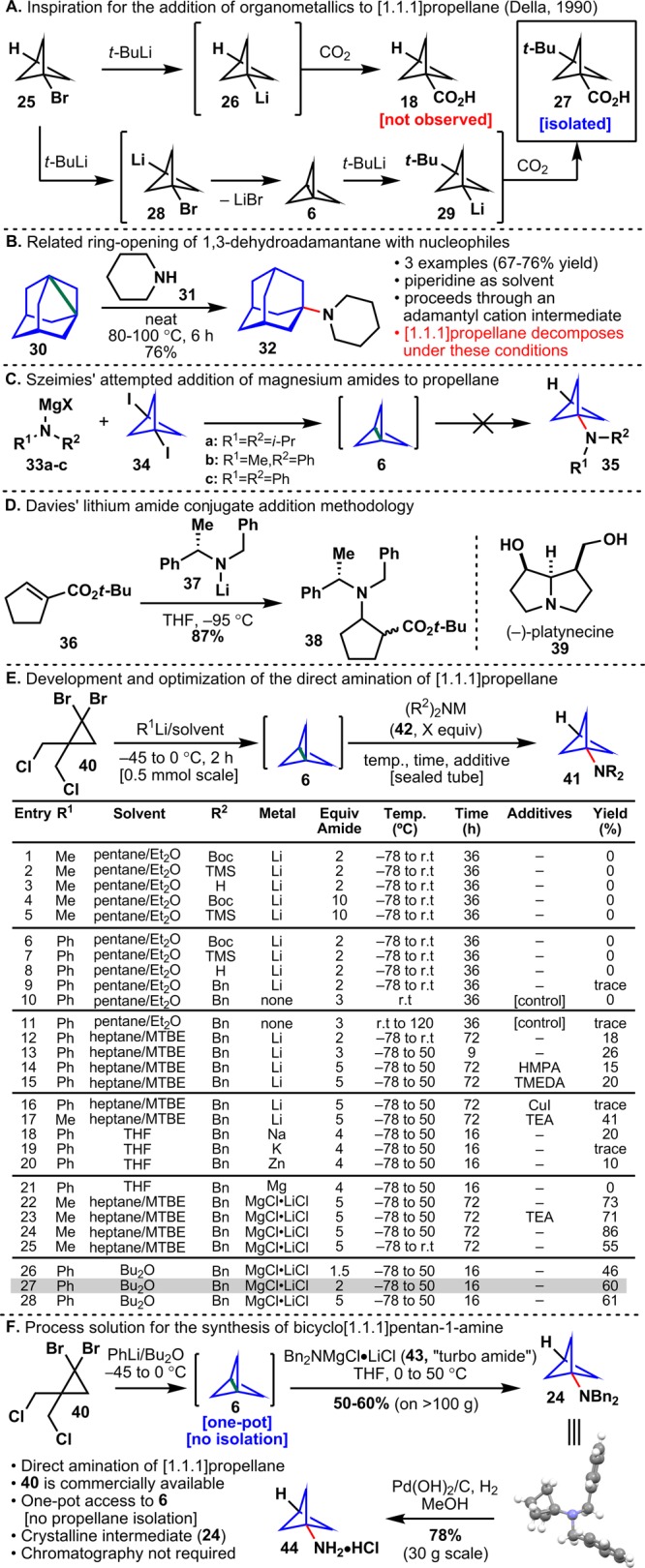
(A) Della’s addition of alkyl lithium reagents into [1.1.1]propellane. (B) Addition of amines to 1,3-dehydroadamantane. (C) Attempted addition of Hauser bases to [1.1.1]propellane. (D) Davies’ addition of chiral lithium amides to enones. (E) Development and optimization of the direct amination of [1.1.1]propellane. (F) Process-scale synthesis of bicyclo[1.1.1]pentan-1-amine.
Inspiration for a potential route to 44 stemmed from the work of Della (Figure 3A) where an attempted halogen–metal exchange of bromide 25 led to the tert-butylated adduct 27 instead of the expected carboxylic acid 18.32 Della reasoned that lithiation of the bridgehead C–H bond in 25 led to 6 via formation of 28 followed by addition of t-BuLi across the strained C–C bond. Subsequent trapping of 29 with CO2 afforded adduct 27. Another precedent stems from Kogay’s work on the direct amination of strained systems such as 1,3-dehydroadamantane (DHA, Figure 3B) wherein strained bonds were directly functionalized with amines and amine surrogates.33 In those initial studies it was shown that DHA could be directly functionalized with sulfonamides in refluxing toluene. Since then, Butov and co-workers have shown that DHA could be directly substituted with heterocycles, amides, hydrazones, azides and amines (e.g., 30 → 32).34 It is instructive to note that an acidic N–H bond is required for these reactions to proceed as it is proposed that an adamantyl cation is the key intermediate (although a radical pathway cannot be excluded completely). However, the feasibility of a direct amination strategy toward 44 was questioned by a student in Szeimies’ laboratory where attempted amination of 6 with magnesiated amines 33a–33c failed (Figure 3C).35
With two promising precedents in hand, (Figure 3A,B) a search for conditions that would allow for direct (without isolation of 6) amination of 6 was pursued (Figure 3E). Initial screens of lithium amides (entries 1–8) were chosen with two factors foremost in mind: convenient access to the amide reagents and a facile deprotection that would ultimately furnish 44. Unfortunately, despite extensive screening, these lithium amides were not sufficiently reactive to aminate 6, even in large excess (entries 4, 5). In addition, it was immediately discovered that the use of MeLi was problematic. For each equivalent of 6 made from tetrahalide 40, 2 equiv of MeBr were generated and swiftly methylated the lithium amides (entries 1–5). Since a one-pot reaction was required (and MeBr and 6 have similar boiling points) PhLi was instead used to generate 6 from 40. It was reasoned that bromobenzene, the side product under the new reactions conditions, would be unreactive toward the amides. In considering lithium amides with higher nucleophilicity, the work of Davies stood out (Figure 3D).36 The finding that lithium- and magnesium amides 37 derived from dibenzylamine readily undergo 1,4-addition to enone 36 at low temperatures has been extensively applied in both academic and industrial venues, such as the total synthesis of (−)-platynecine (39).37 Gratifyingly, treatment of 6 with lithium dibenzylamide gave traces of aminated bicycle 24 (entry 9). This enthusiasm was tempered somewhat by the observation of large amounts of an unexpected byproduct in the reaction mixture: N-phenyl-dibenzylamine. Presumably, this adduct is formed upon addition of lithium dibenzylamide to benzyne (formed in situ from bromobenzene). Separation of this byproduct from 24 proved incredibly challenging and no efficient methods (recrystallization, chromatography, selective precipitation) were identified. After extensive optimization of temperature (entries 12, 13), additives (e.g., HMPA, TMEDA, Et3N, CuI, entries 14–17), metal (e.g., Na, K, Zn, Mg, entries 18–21), and solvent, yields of 24 were boosted to 41%, but unfortunately all reactions contained the inseparable N-phenyl-dibenzylamine byproduct. Control reactions with the free amine (no metal, Butov’s dehydroadamantane conditions) gave only traces of product by LC/MS (entries 10, 11). The key breakthrough was achieved when dibenzylamine was treated with Knochel’s turbo Grignard (i-PrMgCl·LiCl)38 to give the corresponding “turbo amide” (43, Bn2N-MgCl·LiCl); this reagent reacted smoothly with 6 to deliver 24 in 73% yield (entry 22). Perhaps most importantly, the “turbo amide” was completely unreactive with bromobenzene, giving a very clean reaction profile and greatly simplifying purification. Optimization of this reaction on small scale gave yields as high as 86% (entries 23–28). However, with the constraints of process chemistry in mind, reductions in reaction time (16 h), “turbo amide” equivalents (from 5 to 2), the temperature of PhLi addition (kept at −45 °C instead of −78 °C), and the number of solvents in the reaction mixture (Bu2O and THF only) were achieved, all while maintaining a yield of 60% (entry 27) from 40 to 24 on over 100 g scale (Figure 3F). Deprotection of 24 was achieved by hydrogenolysis with Pd(OH)2/H2 in MeOH to afford 44 in 78% yield (30 g scale). This scalable reaction sequence represents the first direct amination of 6, obviates the need for the isolation of 6, gives stable, crystalline intermediate 24 without the need for chromatography, and is the shortest synthesis of 44 to date.
This result met Pfizer’s immediate process chemistry need in order to advance a specific clinical candidate; however, it did very little to aid medicinal chemists that needed to incorporate this motif in numerous different settings. If the direct amination of 6 with “turbo amides” could be generalized for the “propellerization” of a variety of amine scaffolds (Figure 4A), it would have an immediate impact on drug discovery. Traditionally, when seeking to install the bicyclo[1.1.1]pentane bioisostere onto an existing scaffold, one is restricted to a building block approach based on both the existing methodology and commercially available materials. In other words, the smallest fragment (the bioisostere) of the target counterintuitively becomes the focus of the retrosynthetic plan. For example, when designing a synthesis of tetrahydroisoquinoline derivative 48, the BCP unit would derive from amine 44, and the rest of the scaffold would ultimately trace back to diacid 45 resulting in a three-step synthesis. Alternatively, a direct “propellerization” of tetrahydroisoquinoline (49) would give 48 in one step from a commercial material. Although the difference in step count in this example (3 vs 1) is not large, a building block approach to sertraline derivative 75 or paroxetine derivative 76 would require an enormous amount of time and effort in redesigning and implementing the synthetic route around the availability of 44 rather than sertraline or paroxetine themselves! Such an effort is unlikely to occur during the normal course of analog design and that chemical space would likely remain unexplored. In order to adapt the “propellerization” for general use in medicinal chemistry, stock solutions of 6 were prepared, and found to be stable for weeks to months at −20 and −78 °C, respectively. With the stock solution of 6 in hand, the scope of the direct “propellerization” was explored (Figure 4B). A total of 31 different tertiary, BCP-containing amines were synthesized. These structures are unprecedented since all previous routes to related compounds were forced to rely on 44 as the building block. The structural diversity among the scaffolds include both cyclic and acyclic amines as well as functional groups like acetals (56), ethers (57, 69, 70), olefins (58), aromatic heterocycles (61, 63, 71, 72, 78), ketals (65), aryl halides (76, 77, 79), and Lewis-basic groups (62–64, 68, 69, 71–73, 78, 79). Many examples incorporate N- or O-Bn groups which, after deprotection, afford primary or secondary amines and alcohols, which increase the accessible structural diversity of this method. Perhaps the most intriguing compounds in Figure 4B are 74–79, six structurally distinct commercial drugs to which BCP units have been appended. These compounds would have required laborious multi-step syntheses to access otherwise, emphasizing the powerful nature of “any-stage” functionalization. Field-testing of this chemistry at Pfizer was rapid: compounds 54, 55, 57, 60, 62, and 71 were prepared at Pfizer as part of their ongoing discovery programs. The enthusiastic uptake of this chemistry at Pfizer inspired further research into the direct incorporation of other bioisosteres using a strain-release approach.
Figure 4.

(A) Rationale for the development of a medicinal chemistry version of the “propellerization”. (B) Scope of the direct “propellerization” of amines. aConditions: amine substrate (1 equiv). bThe HCl salt of the amine starting material was used. cConditions: amine substrate (2 equiv). dThe extra equivalent of the amine starting material was recovered in ca. 90% yield (see SI for details).
Direct Azetidinylation via Strain Release
Azetidines were a natural choice for the next structure to be explored due to their extensive use in both the rigidification of amine backbones and as phenyl bioisosteres (80–82, Figure 5A).39 Like propellane systems, access to amino-azetidines is largely limited to a building-block approach that relies on the multistep synthesis of protected azetidinones. Azabicyclobutane (ABB, 7) was first prepared by Funke in 1969 and has been used sporadically ever since as a method for preparing functionalized azetidines.40 In contrast to [1.1.1]propellane, which decomposes in the presence of electrophiles, 7 is highly nucleophilic (at the nitrogen atom) and readily attacks electrophiles like tosyl chloride or ethyl chloroformate to give synthetically useful azetidine derivatives.41 During the course of synthetic studies toward new quinolone antibiotics, Nagao demonstrated that aniline attacks ABB at C3 to afford azetidine 83 (Figure 5B).42 The reaction requires superstoichiometric amounts of Lewis acids (e.g., Mg(ClO4)2) and also works with some thiols and dibenzylamine. Unfortunately, this approach failed to give 85 when ABB was instead treated with other alkyl amines such as piperidine.
Figure 5.
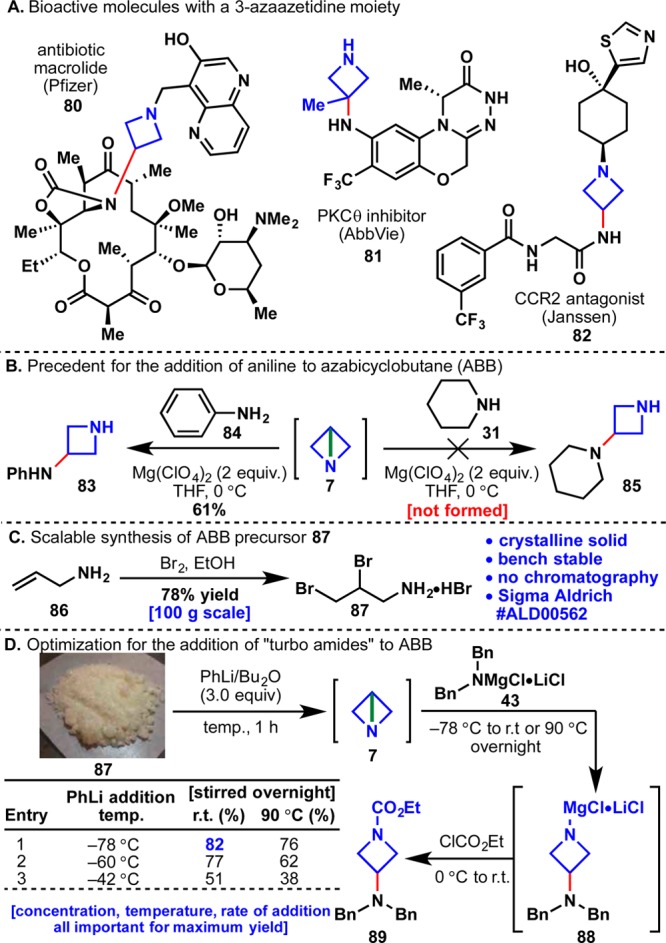
(A) Azetidines in lead compounds. (B) Nagao’s addition of anilines to ABB. (C) Scalable preparation of ABB precursor 87. (D) Development of the reaction of “turbo amides” with ABB.
Strain-release amination of ABB was therefore evaluated as a means to simplify the preparation of functionalized azetidines. As shown in Figure 5C, ABB precursor 87 was readily prepared in one step by adding allylamine (86) to a solution of Br2/EtOH (100 g scale, 78% yield).42,43 Dibromide 87 is a bench-stable, crystalline solid that is isolated and purified by recrystallization (no chromatography). Upon treatment with PhLi, 87 undergoes sequential ring-closing reactions (analogous to forming propellane: 40 → 6) to give ABB in situ. To test the feasibility of using “turbo amides” with ABB, a solution of Bn2N-MgCl·LiCl (43) was added to 7 and stirred at room temperature overnight. After quenching the reaction with ethyl chloroformate, 89 was isolated in 82% yield. Further optimization revealed the importance of temperature, concentration, and rate of addition in maximizing the yield of the reaction (Figure 5D).
Before exploring the scope of the reaction further, a series of electrophilic quenching agents was examined. Although the free azetidines could be obtained directly from the reaction mixture if desired (90, 53%), in situ treatment of 88 with ethyl chloroformate (89, 82%), Boc anhydride (91, 93%), or tosyl chloride (92, 78%) simplified purification and made the product easier to handle (Figure 6A). The substrate scope mirrors that of the “propellerization”: the reaction tolerates cyclic and acyclic amines along with functional groups like olefins (93), ethers (97), heterocycles (98, 107), aryl halides (108), and Lewis-basic groups (102, 103, 107). Eighteen different azetidines were synthesized in total, including three late-stage pharmaceuticals (Figure 6B). As before, N- and O-Bn groups were used to mask primary or secondary amines and alcohols.
Figure 6.

(A) Screen of the trapping agents for the “azetidinylation” of amines. (B) Scope of the “azetidinylation” of amines.
Direct Cyclobutylation via Strain Release
With the direct installation of azetidines enabled, cyclobutane was identified as the next target for a stain-release approach. Cyclobutanes are found in a variety of natural products44 and medicinal agents45 (Figure 7A), and yet methods for the direct installation and functionalization remain limited despite their useful lipophilic and rigidifying properties.18,46 In considering a strain-release “cyclobutylation” the goal was two-fold: develop a bench-stable source of bicyclobutane and have a built-in handle for further functionalization. Bicyclobutane chemistry has a rich history, dating back to their first preparations in 1959.47 Many of the studies since have either been focused on using bicyclobutane as a nucleophile or in using a transition-metal mediated process to fragment the strained ring system.48 Pursuit of the parent ring system, which has a boiling point of 8 °C,49 appeared impractical, so attention was instead turned to arylsulfone-substituted bicyclobutanes that are reported to be solids at room temperature.
Figure 7.
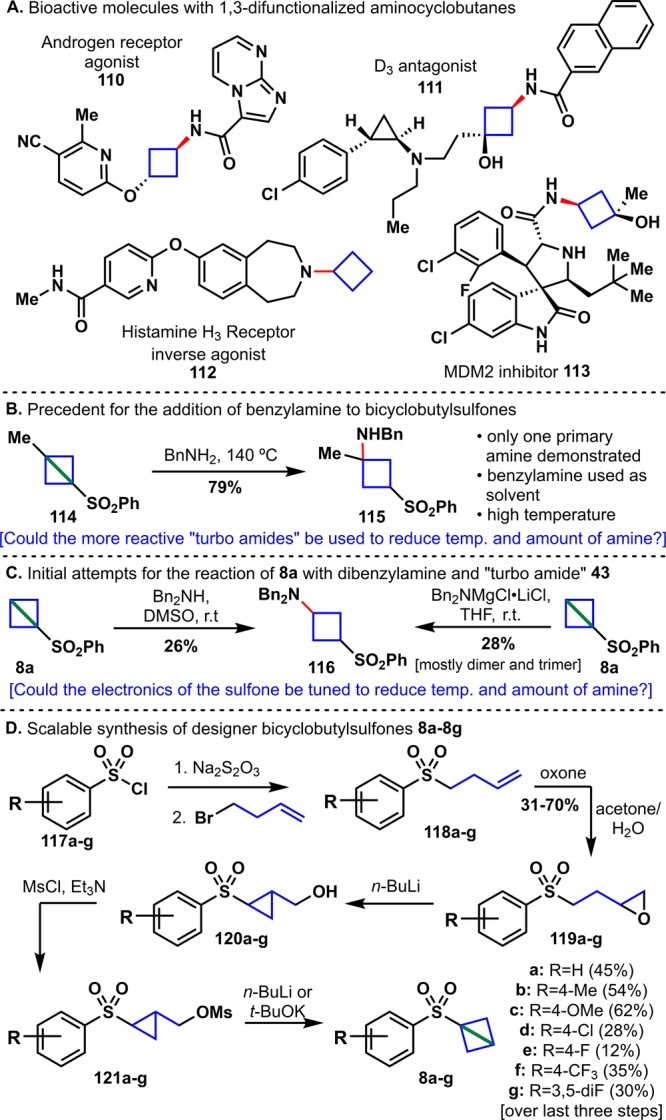
(A) Cyclobutanes in lead compounds. (B) Gaoni’s addition of benzylamine to sulfone 114. (C) Initial studies for the addition of dibenzylamine and “turbo amide” 43 to sulfone 8a. (D) Scalable synthesis of 8a and designer sulfones 8b–8g.
A series of studies by Gaoni on the addition of benzylamine to bicyclobutane 114 served to encourage this approach (Figure 7B).50 While this provided a proof-of-concept, several drawbacks were immediately apparent: the amine was used as solvent, the reaction was run at high temperatures (140 °C), and few other examples were reported.
With the goal of reducing the temperature and amount of amine needed for the reaction, bicyclobutane 8a was treated with a solution of Bn2N-MgCl·LiCl (Figure 7C). Although 28% of the desired product 116 was isolated, most of the reaction mixture contained dimers and trimers where the intermediate addition product reacted with more equivalents of 8a. When the reaction was run with dibenzylamine instead, the desired product 116 was isolated in 26% yield with no side reactions. Instead of trying to optimize the reaction based on the “turbo amide”, a series of electronically tuned bicyclobutanes 8b–8g was prepared (Figure 7D) in order to adjust the reactivity of the strained bond. Commercially available sulfonyl chlorides 117b–117g were converted to the corresponding sulfinate salts and added to 4-bromobut-1-ene to furnish olefins 118b–118g. Epoxidation with Oxone gave 119b–119g in 31–70% yield over three steps. The penultimate intermediate was prepared by ring-opening via α-deprotonation of the sulfone followed by protection of the alcohol as the mesylate. The final strained bond was installed by addition of n-BuLi or t-BuOK to the mesylates to afford designer bicyclobutane sulfones 8b–8g in 12–62% yield over three steps.
When sulfones 8b–8g were treated with dibenzylamine in DMSO at room temperature, strain-release products 122b–122g were obtained in 16–87% yield (by NMR). Perhaps unsurprisingly, sulfones appended with electron-donating groups (e.g., Me, OMe) were significantly less reactive, while those with electron-withdrawing groups (e.g., 4-F, 4-Cl, 4-CF3, 3,5-diF) were much more reactive toward the amine (Figure 8A). A screen of additives identified LiCl as a helpful accelerant for the strain-release reaction. In order to obtain the unsubstituted cyclobutanes, intermediates 122a–122g were treated in the same pot with Mg/MeOH to afford cyclobutanes 123a–123g.51 Using this protocol, cyclobutanes were installed onto 15 structurally diverse cyclic and acylic, primary and secondary amines or anilines, including four late-stage pharmaceuticals (Figure 8B). Compared to the “propellerization” and “azetidinylation”, the “cyclobutylation” benefits from an increased functional group tolerance due to the use of the free amines. In addition to heterocycles (131), olefins (134), ethers (136), and aryl halides (136, 137), carbamates (128) and silyl ethers (133) are also compatible under the reaction conditions. Impressively, the nucleophilic addition of amines to 8g is chemoselective in the presence of free hydroxyl groups. The piperidine derivative 133 was also prepared from 4-hydroxypiperidine in 43% yield over three steps (see SI for details). Aside from imparting bench-stability to reagent 8g, the arylsulfone handle provides an excellent opportunity for diversification of the cyclobutane moiety. Intermediate 122g was readily converted to valuable 3-substituted building blocks containing deuterium (139), fluorine (141), allyl (143), and olefin (145) groups (Figure 8C).
Figure 8.

(A) Design and optimization of a one-pot “cyclobutylation” of amines and anilines. (B) Scope of the “cyclobutylation”. (C) Diversification of strain-release product 122. aGeneral procedure A with 8g: one-pot, no purification of intermediates. bGeneral procedure B with 8g: intermediates isolated by column chromatography (first yield for strain-release step, second yield for desulfonylation). cGeneral procedure C with 8g: one-pot, no purification of intermediates, reduction initiated by sonication. dThis compound was also prepared from 4-hydroxy-piperidine (43% over three steps, see SI for details).
The conceptual appeal of a practical, strain-release approach to peptide functionalization prompted an initial evaluation of the reactivity of bicyclobutylsulfones with various nucleophilic proteinogenic amino acid side chains. Remarkably, in a mixed aqueous/organic solvent system, phenylsulfonyl bicyclobutane 8a reacted exclusively at the thiol side chain of cysteine (Figure 9A). This chemoselectivity translated directly into a complex peptide system (146), which, in the absence of cysteine, exhibited no off-target reactivity with 3,5-difluorophenylsulfone 8g (Figure 9B). In contrast, N-ethylmaleimide, an oft-employed electrophile for bioconjugation,52 reacted unselectively with peptide 146 to yield multiple labeled products. With chemoselectivity established, strain-release reagents 8a and 8g were next employed under optimized conditions for the high-yielding cyclobutylation of various cysteine residues, including cysteine methyl ester (148), glutathione (149 and 150), and a functionalized polypeptide variant bearing an internal cysteine residue (151 and 152) (Figure 9C). Peptide labeling with 3,5-difluorophenylsulfone reagent 8g was complete in less than 1 h with no detectable byproducts. The protocol was also compatible with aqueous phosphate buffer, and reagent 8g was shown to be stable to the common protein disulfide reductant tris(2-carboxyethyl)phosphine (TCEP) (see SI for details), emphasizing the suitability of the reagent for rapid and chemoselective “click” reactions with cysteine-containing peptides.53
Figure 9.

(A) Chemoselectivity of bicyclobutylsulfones for Cys side chains over other proteinogenic amino acids. (B) Superior selectivity of reagent 8g over N-ethylmaleimide 147. (C) Optimized conditions and substrate scope of Cys “cyclobutylation.” (D) Temporal control of Cys labeling using electronically distinct bicyclobutylsulfones.
Modulation of the electronic properties of the aryl sulfone unit also enabled careful control of the kinetic parameters of peptide labeling (Figure 9D). The rates of reaction with polypeptide 153 were highly sensitive to substitution on the aryl ring of the sulfone, with electron-withdrawing substituents (8e, 8f, and 8g) enhancing the rate of labeling relative to the parent phenylsulfone (8a) and an electron-donating substituent (8c) attenuating the reactivity of the strain-release reagent. Such precise temporal control afforded by the tunable electrophilicity of arylsulfonyl bicyclobutane reagents has promising implications for the use of custom strain-release electrophiles for drug design and activity-based protein profiling.54
Stereospecific Strain Release
Up to this point the amination of three different classes of strained electrophiles enabled the rapid incorporation of useful small-ring bioisosteres. Applying this approach to five-membered ring systems was then questioned from a strategic perspective. After all, reductive amination using cyclopentanone is commonplace and thus the simple amination of a reagent such as 9 (Figure 10A) might have very little strategic value.55 Furthermore, such reagents would be chiral, and it was not clear if they could be easily synthesized in enantiopure form and if that chirality would be transferred upon addition of a nucleophile. However, numerous applications could be envisaged in medicinal chemistry if exotic reagents such as 9 or 10, known colloquially as “housanes”, could be fashioned and if they would engage multiple nucleophile classes in stereospecific strain-release events. To be sure, the rapid addition of a chiral 1,3-disubstituted cyclopentane spacer unit could reduce the time and effort required to explore these popular fragments.
Figure 10.
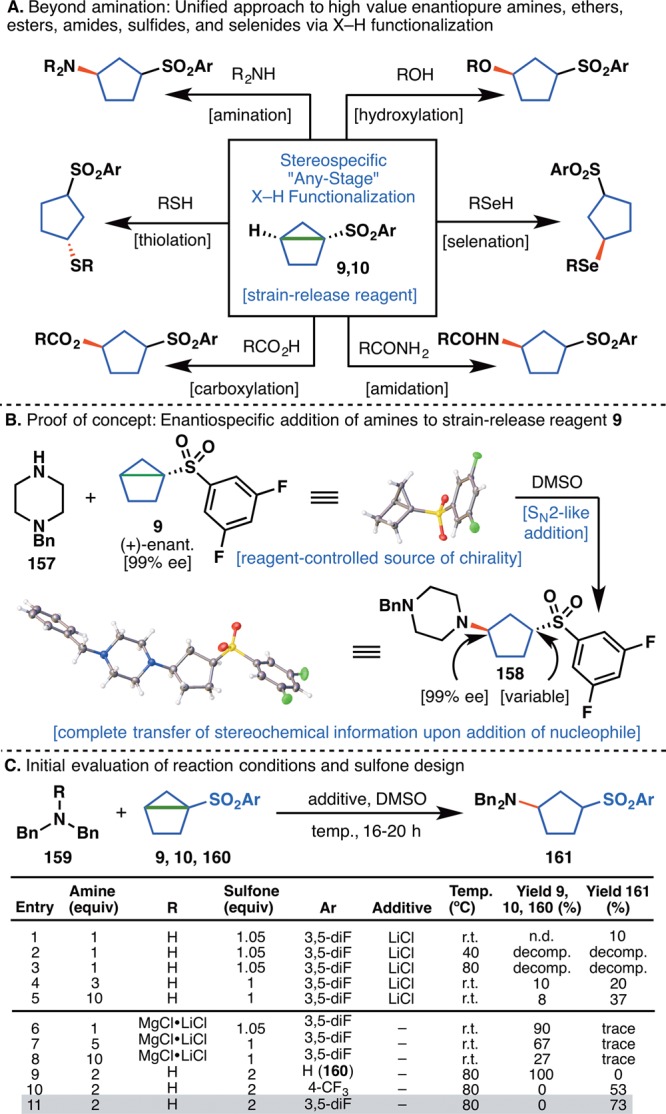
(A) Unified approach to chiral 1,3-disubstituted cyclopentanes. (B) Proof of concept for the stereospecific ring-opening of housane reagent 9 with amine 157. (C) Initial optimization of the stereospecific “cyclopentylation”.
In a striking proof of concept (Figure 10B), the chiral sulfonyl housane (+)-9 was prepared in 99% ee (initially through separation of racemic material by chiral supercritical fluid chromatography (SFC)), assigned by X-ray crystallography, and reacted with amine 157 to deliver adduct 158 in 99% ee with complete transfer of stereochemical information at the C–N bond as a ∼3.5:1 mixture of diastereoisomers (structure of both diastereomers verified by X-ray crystallography, see SI for full details). To the best of our knowledge, this represents the first preparation of an enantiopure housane derivative whose sole chirality originates at the bridgehead along with nucleophilic opening and confirmation that such an opening proceeds stereospecifically.56 This result gave confidence in the overall approach and propelled all additional studies in this area. Subsequent optimization of this reaction led to a number of critical observations (Figure 10C). The addition of LiCl, a key additive with related bicyclobutane 8g, decomposed sulfone 9 (an effect amplified with an increase in temperature, entries 1–5). An attempt to increase reactivity with “turbo amides” gave only traces of the desired product 161. Ultimately, the removal of LiCl entirely, and an increase in temperature led to the best yield and final conditions (entry 11). Notably, both new designer housanes (9 and 10) that were synthesized gave good yields of 161 (entries 10, 11), while the previously known “parent” housane 160(57) (Ar = Ph, entry 9) failed to react. With an optimized set of conditions for the addition of amine nucleophiles in hand, attention turned to the identification of practical means to prepare enantiopure sulfonylated housane derivatives, because chiral chromatography was not a sustainable option. With a high-yielding, scalable racemic synthesis already in hand (Figure 11A), the first approach focused on performing a kinetic resolution of intermediate alcohols 167 and 168 (Figure 11B).58 After extensive screening on alcohol 167 (see SI for details), porcine pancreas lipase (enzymatic catalyst), vinyl acetate (acetyl donor), and MTBE were identified as the optimum protocol.59 After 48–60 h at room temperature, enantioenriched alcohol 167a was isolated in 54% yield and 81% ee.60 To obtain the other enantiomer of alcohol 167, acetate 169 was cleaved by brief reaction with Na/MeOH to afford enantioenriched alcohol 167b in 96% yield and 84% ee. Both alcohols were converted to mesylates 165a and 165b in 94 and 98% yield, respectively. At this stage, recrystallization from EtOAc at −20 °C upgraded the ee’s of both mesylates to 91–92%. Treatment of 165a and 165b with n-BuLi formed the housane framework and completed the synthesis of enantioenriched (+)-9 and (−)-9. The same set of conditions was used to generate trifluoromethyl derivatives (+)-10 and (−)-10. All structures were confirmed by X-ray crystallography (Figure 11C).
Figure 11.
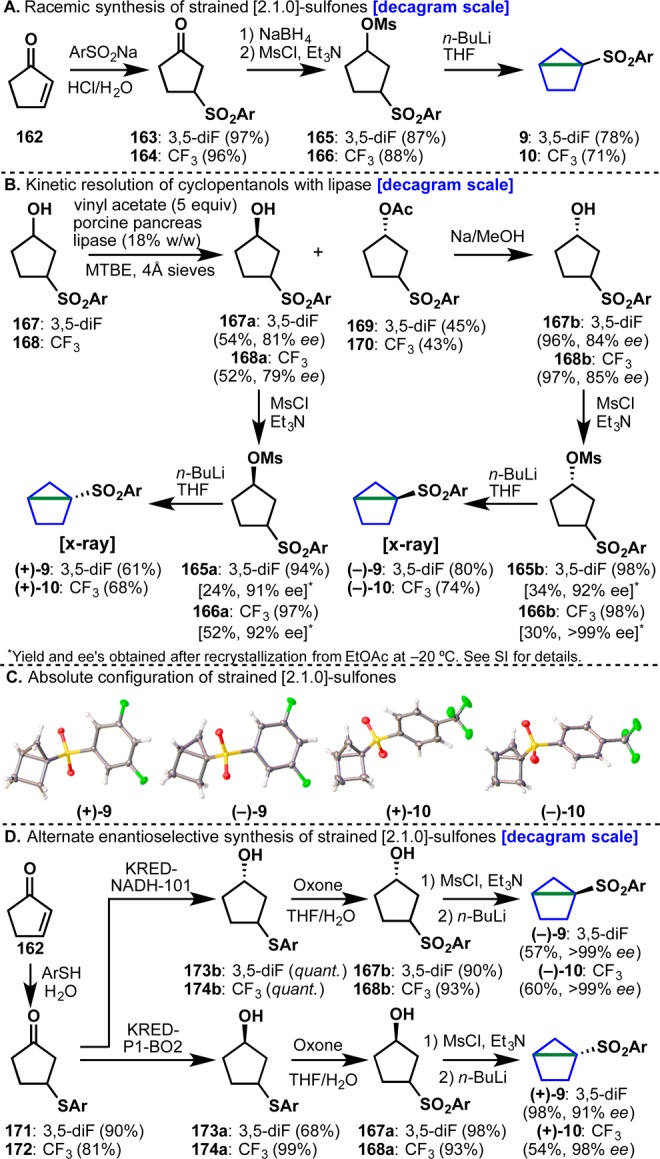
(A) Racemic synthesis of sulfones 9 and 10. (B) Lipase-based synthesis of chiral sulfones 9 and 10. (C) X-ray structures of reagents (+)-9, (−)-9, (+)-10, and (−)-10. (D) Ketoreductase-based asymmetric synthesis of chiral sulfones 9 and 10.
An alternative synthesis using a ketoreductase was explored in parallel and also proved successful (Figure 11D).61 Thus, enzymatic enantioselective reduction of ketones 171 and 172, derived from the Michael addition of the appropriate arylthiol to 162 led to either enantiomer of 173/174.62 Subsequent oxidation to the sulfones using Oxone intercepted the prior route to enantiopure housanes 9/10. It is worth noting that attempted enzymatic reduction of 163/164 proceeded in low conversion due to instability of 163 under the ketoreductase conditions. With two robust routes to enantiopure housanes (decagram-scale) the scope of stereospecific strain release could be explored. The initial substrate scope comprised 28 different amines and anilines with diverse structural features (Table 1).63 In accord with prior strain-release functionalizations, the chemoselectivity is high with broad functional group tolerance including olefins (176), alcohols (183, 195, 201), heterocycles (179, 193, 199, 201), carbamates (187, 192), amides (190), and aryl halides (197, 200). Ammonia was also found to be an excellent nucleophile for the “cyclopentylation” reaction and delivered 188 in 88% yield. Several unprotected amino acids were monoalkylated with reagent (+)-9 (193–196).64 Finally, five late-stage pharmaceutical agents were “cyclopentylated” including sertraline 197, fluoxetine 198, varenicline 199, amoxapine 200, and mefloquine 201. Notably, complete stereotransfer at the site of nucleophilic addition was observed in all substrates.
Table 1. Stereospecific Strain-Release “Cyclopentylation” of Amines, N-Heterocycles, Carboxylic Acids, Thiols, and Selenolsa.

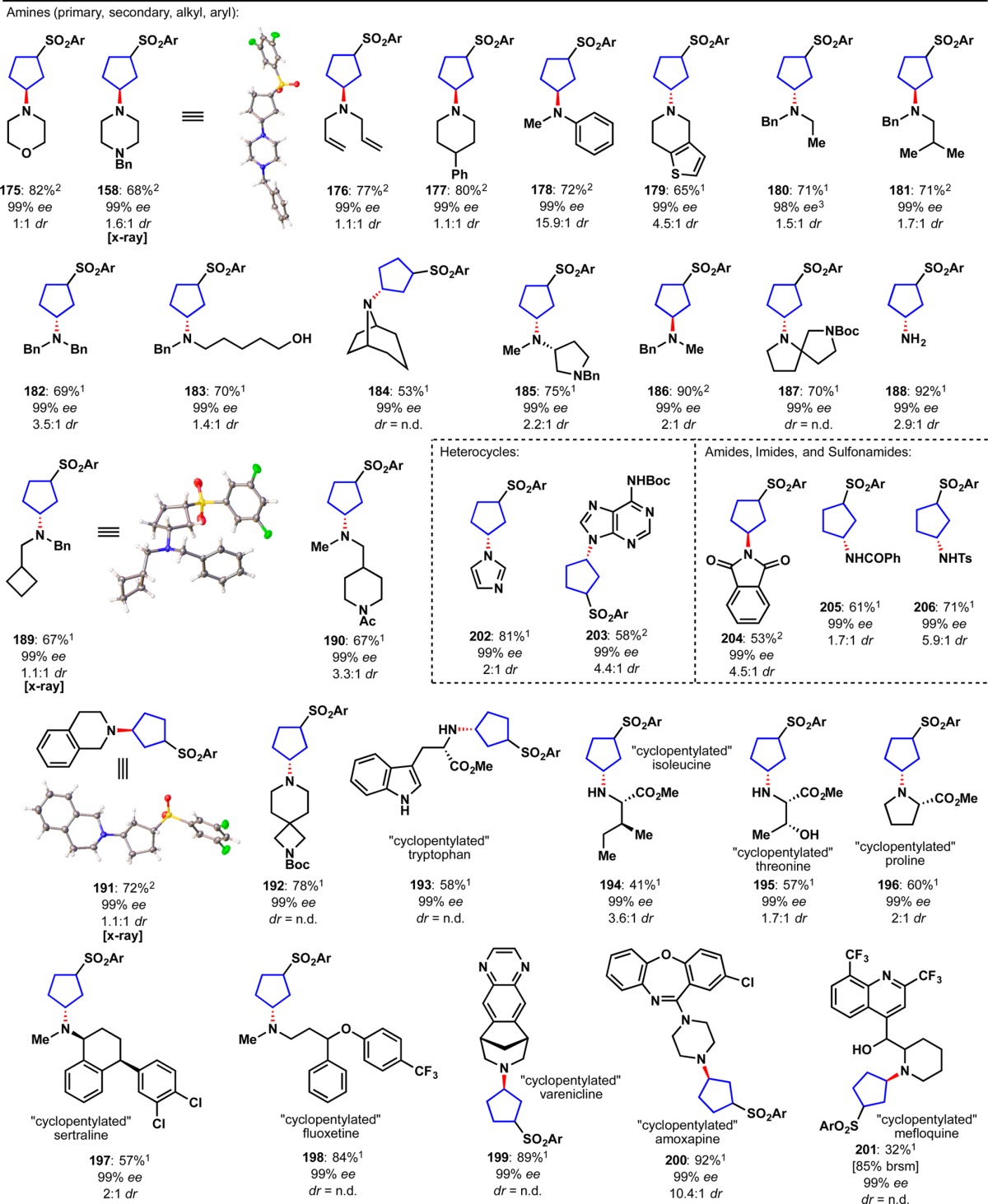
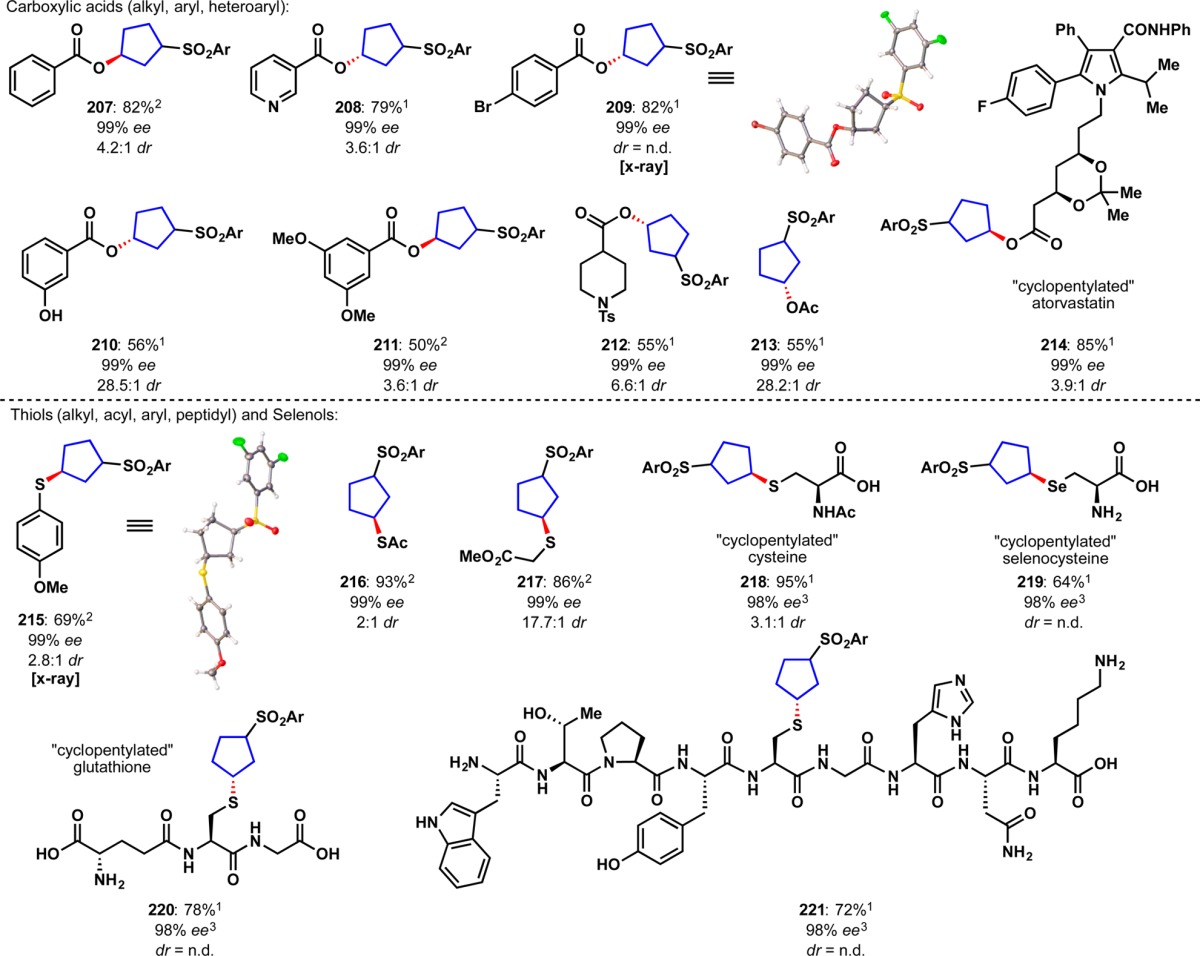
Notes: 1Ar = 3,5-diF, reaction run with reagent (+)-9; 2Ar = 3,5-diF, reaction run with reagent (−)-9; 3(+)-9 at 98% ee was used in this reaction (complete stereotransfer was observed).
The versatility of chiral housanes to engage not only amines but also a host of other nucleophiles was next explored. To our delight, N-heterocycles, amides, sulfonamides, imides, thiols, selenols, and even carboxylic acids were also found to engage these species thus allowing rapid access to enantiopure disubstituted cyclopentane derivatives that would be otherwise difficult to access. Thus, as illustrated in Table 1, nitrogen nucleophiles such as imidazole 202, adenine derivative 203, phthalimide 204, benzamide 205, and sulfonamide 206 were all competent in the “cyclopentylation”. Both aromatic and alkyl carboxylic acids were suitable substrates; functional groups tolerated included free hydroxyls (210), ethers (211), aryl halides (209, 214), heterocycles (208), sulfonamides (212), and acetals (214). Notably, an atorvastatin derivative underwent smooth “cyclopentylation” to furnish 214 in 85% yield. As was observed with the strain-release “cyclobutylation”, thiols proved to be excellent substrates. Structural diversity in this class includes thiophenol 215, thioacetic acid 216, alkyl thiol 217, cysteine 218, glutathione 220 and complex polypeptide 221. Even selenols were suitable with selenocysteine 219 obtained in 64% yield.
The next class of nucleophiles explored were alcohols, since the rapid installation of chiral ethers would have immense value in medicinal chemistry. Previous results had shown that free hydroxyl groups were generally unreactive in strain-release reactions with amines and thiols both reacting chemoselectively in their presence (183, 195, 201, 210, 221). Indeed, when 222 and 9 were heated to 150 °C in either DMSO or THF, both starting materials were returned unreacted (Figure 12A). However, when the corresponding alkoxides of 222 were generated with NaHMDS, KHMDS, or LiHMDS, clean conversion to SNAr product 224 was observed.65 Despite extensive screening of various solvents and bases, 224 predominated in all cases. It was reasoned that a switch from the SNAr-prone fluorine substituents to a 4-CF3 group would minimize the chances of SNAr and instead direct the innate reactivity back to the strained housane bond. Gratifyingly, when alcohol 222 was treated with LiHMDS and heated to 90 °C in the presence of 10, desired ether 225 was obtained in 50% yield with complete stereotransfer at the site of alkoxide addition (Figure 12A). While either NaHMDS or LiHMDS worked well as the base in these reactions, both KHMDS and t-BuOK still tended to promote SNAr, but this time giving ipso substitution at the sulfone (resulting in 4-(trifluoromethyl)phenylated ethers). With the method optimized, the scope of alcohols and phenols was explored (Figure 12B). Functional group tolerance of strain-release etherification included carbamates (225), ethers (226), olefins (227), and ketones (229). Although the yield was low (228, 12%), the reaction of menthol with reagent 10 demonstrated the ability for the “cyclopentylation” to occur, even with sterically hindered hydroxyl groups. The reaction with estrone occurred in 72% yield, and the product 229 was confirmed by X-ray crystallography. In addition to the alcohols, several substrates are shown in Figure 12C that were prone to SNAr when using 3,5-diF reagents (+)-9 and (−)-9: these include pyrazole 230, indole 231, piperidine 232, and azetidine 233. It should be noted that 3,5-diF 9 is more reactive and generally results in higher yields compared to 4-CF310 for most substrates: for example, amines 182, 234, and 235 were obtained in 69% yield with 9, 53% yield with 10, and 0% with the “parent” housane 160 where Ar = Ph (Figure 12D).
Figure 12.

(A) Development of reagent 10 to avoid SNAr side reactions. (B) Substrate scope of alcohols. (C) Substrate scope of other heteroatoms. (D) Comparison of the reaction of dibenzylamine with 9, 10, and the “parent” housane 160 (Ar = Ph). Notes: 1Ar = 4-CF3, reaction run with reagent (+)-10. 2Ar = 4-CF3, reaction run with reagent (−)-10. 3(−)-10 at ∼97% ee was used in this reaction (complete stereotransfer was observed). 4(−)-10 at 98% ee was used in this reaction (complete stereotransfer was observed).
Although it is not atom economic, the sulfone motif of the housane reagents serves to stabilize the system, activate it for opening, render the reagents chiral, and, perhaps most importantly, set the stage for a variety of downstream functionalizations (Figure 13A). The resulting structures are useful chiral building blocks that would be difficult to rapidly access in other ways (particularly in a diversity-oriented pathway). Using the adduct of dibenzylamine and 9 as an example, 182 was submitted to six different diversification steps initiated by deprotonation with n-BuLi followed by quenching with an electrophile and reductive desulfonylation. Allylated (236), carboxylated (237), alkylated (238), olefinated (239, 240), and fluorinated (241) chiral 1,3-disubstituted cyclopentane building blocks were easily prepared using this strategy. In the case of 236, 237, 238, and 241, no diastereoselectivity was observed in the desulfonylative steps (attempted optimization for substrate 236 is included in the SI).
Figure 13.

(A) Diversification of strain-release intermediate 182. (B) Strain-release “cyclopentylation” of polypeptide 244 on solid phase.
The successful functionalization of proteinogenic α-amines (e.g., 193–196, Table 1) inspired the pursuit of a convenient approach to peptide N-terminal derivatization. Given the ability of diverse nucleophiles to productively engage housanes, it was reasoned that the orthogonal protection strategy employed in standard Fmoc-solid-phase peptide synthesis (Fmoc-SPPS) could be strategically exploited to facilitate selective functionalization of the N-terminus over possible side-chain modifications on a resin-bound peptide substrate (Figure 13B). To test this hypothesis, a model peptide was prepared using Fmoc-SPPS beginning with 2-chlorotrityl chloride resin. Following resin loading with Fmoc-Gly-OH, the peptide was elongated to afford the N-α-Fmoc-protected hexapeptide 243. Standard orthogonal deprotection of the N-terminal Fmoc-group using 20% piperidine/DMF unmasked the target α-amine 244. Treatment of the resin-bound amine with strain-release reagent 9 at 95 °C afforded the N-terminally functionalized peptide 245 in good overall yields following acidic resin-cleavage and global side-chain deprotection. Notably, elevated reaction temperatures proved crucial on the solid-phase, with no reactivity observed at or below 70 °C. The need for more forcing conditions might be a consequence of reduced accessibility of the terminal amine within the resin-bound substrate. Nevertheless, this on-resin extension of the strain-release manifold serves as a powerful proof-of-concept for the rapid and efficient generation of high-value peptide targets bearing non-native structural motifs. All four chiral housanes as well as racemic versions are now commercially available from Sigma-Aldrich.
Strategic Application of Stereospecific Strain Release
It is worth reflecting on the power of this methodology in certain contexts, specifically when rapid access to enantiopure 1,3-substituted cyclopentane adducts is needed or when mild heteroatom functionalization is desired to probe structure–activity relationships (SAR). Figure 14 (panels A–H) summarizes eight different case studies where strain-release with chiral housanes can simplify access to a specific or similar target. For instance, access to enantiopure cyclopentane scaffolds linked through a sulfur atom can be facilitated (panel A) using reagent (−)-9 in high ee and yield as compared to designer catalytic systems.66 In a similar vein, amine-based nucleophiles of all types can be appended (panel B).67 Panel C shows how a low-yielding SN2 reaction can be avoided to access chiral indole-linked systems.68 Purine systems can also be rapidly functionalized to produce nucleoside analogs in high ee and good dr as opposed to arduous sequences (panel D).69 The alkylation of estrone with cyclopentyl electrophiles is known to be problematic70,71 and a four-step alternative route was developed to access such systems (panel E). Using housane (−)-10, the same intermediate (after desulfonylation) or chiral analogs can be accessed in short order. Chiral aminocyclopentanes such as 239 are also directly accessed in high ee using strain-release followed by Julia olefination as opposed to a counter-intuitive approach commencing from an alkenyl-aziridine (panel F).72 Even access to simple β-amino ester derivatives such as 237 and 255 can be simplified and allow for a diverse array of amino-substituents to be incorporated (panel G).73 Finally, the fluorinated chiral cyclopentanes 241/256, previously accessed in eight steps from a costly starting material, could be easily procured in three steps with complete enantiopurity albeit with 1:1 dr (panel H).74
Figure 14.

Synthetic comparisons of stereospecific strain-release “cyclopentylation” vs current state of the art.
Use of Strain-Release in Covalent Reactive Groups
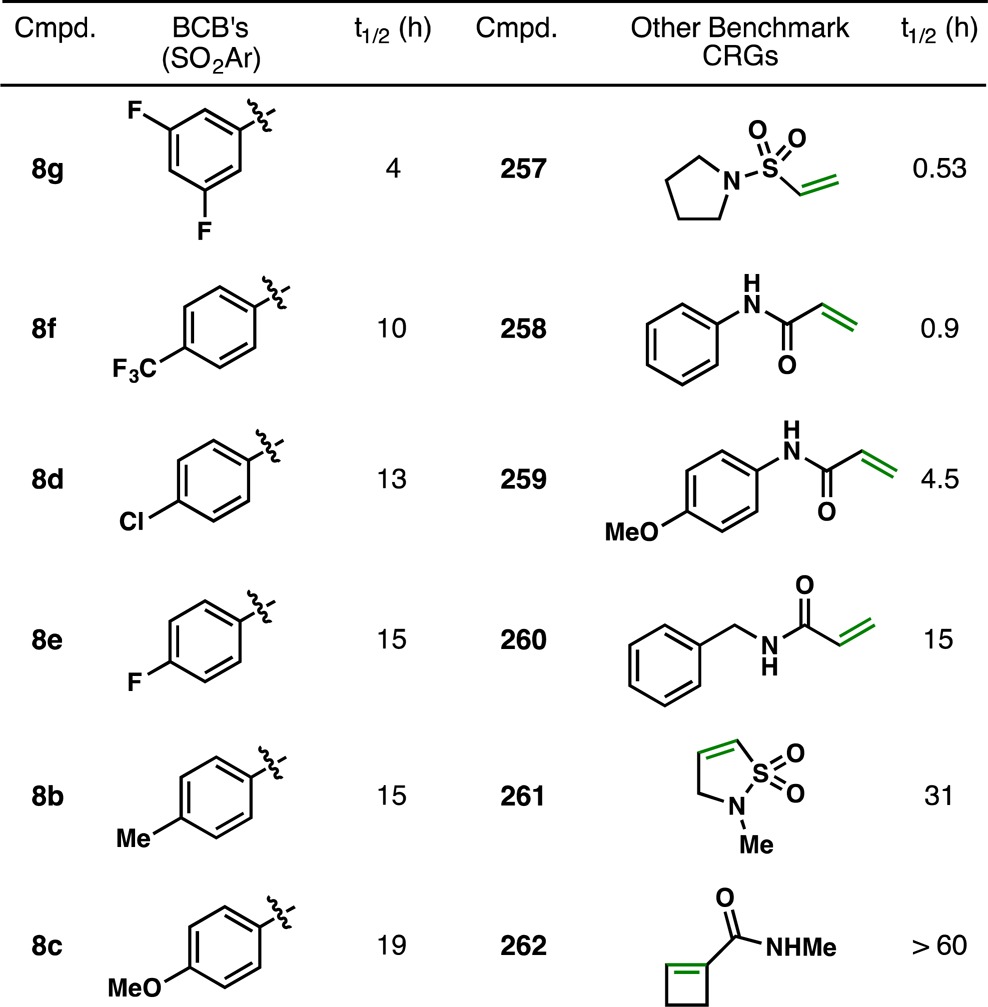
Given the remarkable chemoselectivity observed for bicyclobutane 8g with biogenic thiols (Figure 9), it was thought that these strained fragments might be suitable as “covalent reactive groups” (CRGs) that could be incorporated into irreversible inhibitors in drug design. Furthermore, recent accounts have shown the utility of modulating the strength of electrophilic sites when designing both reversible and irreversible inhibitors.75 Recently, Pfizer reported a glutathione (GSH)-based assay in which a variety of electrophilic acceptors (Table 2, 257–262) were treated with GSH under physiological conditions (phosphate buffer, pH 7.4, 37 °C).76 Half-lives (t1/2) were measured with mass spectrometry or NMR and based on the consumption of GSH. Some of the most reactive groups tested were vinyl sulfonamide 257 (t1/2 = 0.53 h) and phenyl acrylamide 258 (t1/2 = 0.9 h). Notably, the enrichment of electron density in 259 increased t1/2 to 4.5 h, while a one carbon homologation in 260 (Ph to Bn) resulted in a half-life of 15 h. The strained bonds in vinyl sulfonamide 261 and acrylamide 262 were much less reactive, with half-lives of 31 h and >60 h, respectively. Strain-release fragments 8b–8g demonstrated half-lives between 4 and 19 h, placing them roughly in the range of 259 to 260. Encouragingly, changes in the electronics of the aryl rings had the desired effect: substituents such as 3,5-diF and 4-CF3 were substantially more reactive (t1/2 = 4 and 10 h, respectively) compared to groups like 4-Me or 4-OMe (t1/2 = 15 and 19 h, respectively). The comparatively large range of half-lives for 8b–8g (15 h between 8c and 8g) suggests that fine control over the reactivity of the strained bicyclobutane bond may be possible. In addition, as has been demonstrated previously in the synthesis of polypeptides 151, 152, and 154–156, bicyclobutanes 8c–8g are not promiscuous, so reaction with nucleophiles other than thiols should be minimal. The results for 8b–8g are encouraging, and the evaluation of 9, 10, and other strained systems as CRGs is ongoing.
Table 2. Evaluation of Reagents 8b–8g as Covalent Reactive Groups.
Outlook and Conclusion
The construction of strained C–C bonds for the sole purpose of heteroatom diversification is a powerful maneuver for both the early and late stages of a synthesis. As shown in Figure 15A, the use of [1.1.1]propellane to access amino propellanes is representative of this type of C–C activation.77,78 This Article has traced the development of this strategy as a means to incorporate not only propellanes but also azetidines, cyclobutanes, and, for the first time, chiral cyclopentanes (Figure 15B). Nearly every type of heteroatom can be functionalized with this method, including those embedded into complex peptide architectures. Enantiopure housanes facilitate the rapid introduction of valuable cyclopentane scaffolds that can be diversified in a multitude of ways. In medicinal chemistry where the exact target structure is unknown, this rapid method for exploring “vectors” of chemical space is likely to find widespread use. Further diversification of the adducts formed (in the case of azetidines, cyclobutanes, and cyclopentanes) allows for a limitless array of possibilities. The user-friendly nature of strain-release reagents, chemoselective reactivity, and experimental ease will further lower the barrier to adoption in numerous areas of chemical science. It is thus anticipated that strain-release functionalization will find use in many other contexts such as bioconjugation, covalent drug design, polymer science, and materials chemistry.
Figure 15.
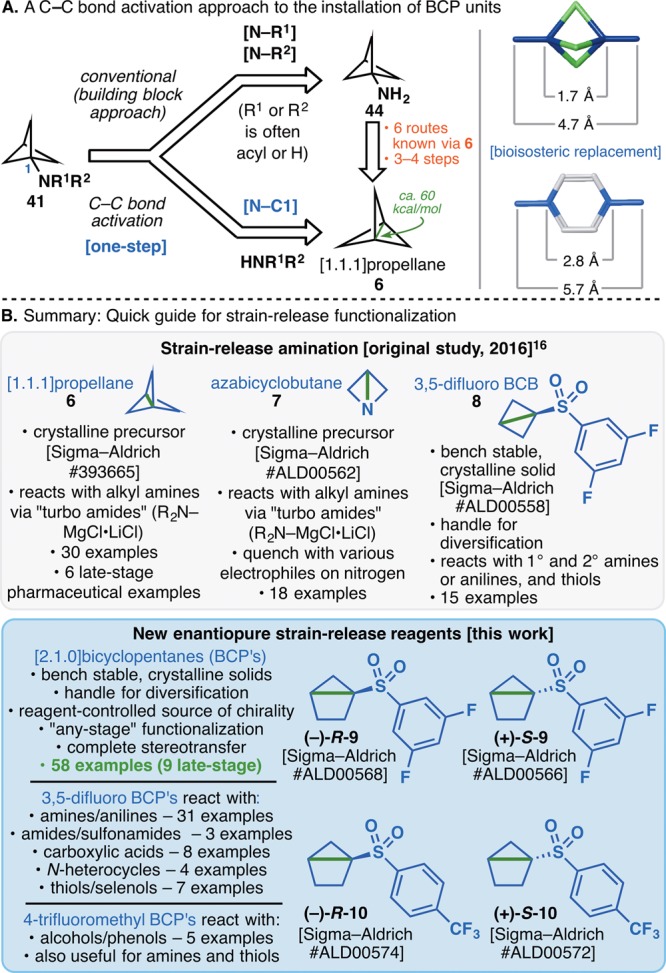
(A) C–C bond activation provides a new disconnection for the installation of BCP units. (B) A reference guide for the use of 6–10 in strain-release functionalization.
Acknowledgments
Financial support for this work was provided by Pfizer, Inc., and the National Institutes of Health (GM-118176). The National Institutes of Health provided a postdoctoral fellowship to J.M.L. and L.R.M.; the Danish Agency for Science, Technology and Innovation and H. Lundbeck A/S provided funding for K.F.; the Shanghai Institute of Organic Chemistry, Zhejiang Medicine Co., and Pharmaron provided a postdoctoral fellowship to J.W.; and the University of the Basque Country provided a predoctoral fellowship to L.P. We are grateful to D.-H. Huang and L. Pasternack (The Scripps Research Institute) for assistance with NMR spectroscopy and A. L. Rheingold, C. E. Moore, and M. Gembicky (University of California, San Diego) for X-ray crystallographic analyses.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b13229.
Experimental procedures and analytical data (1H, 13C, 19F NMR, MS) for all new compounds (PDF). Graphical procedures for reagent synthesis and representative reactions (PDF)
X-ray crystallographic data for compounds 8a, (±)-9, (−)-9, (+)-9, (−)-10, (+)-10, 24, 52, 103, 158, cis-158, ent-158, 165, 189, 191, ent-191, 209, 215, 229, 230, and additional compound S7 found in the SI PDF (CIF)
Author Contributions
⊥ K.F. and Y.K. contributed equally.
The authors declare no competing financial interest.
Author Status
# R.O.: deceased, November 9, 2016.
Supplementary Material
References
- Wiberg K. B. Angew. Chem., Int. Ed. Engl. 1986, 25, 312–322. 10.1002/anie.198603121. [DOI] [Google Scholar]
- a Converso A.; Saaidi P.-L.; Sharpless K. B.; Finn M. G. J. Org. Chem. 2004, 69, 7336–7339. 10.1021/jo0489869. [DOI] [PubMed] [Google Scholar]; b Accurso A. A.; Cho S.-H.; Amin A.; Potapov V. A.; Amosova S. V.; Finn M. G. J. Org. Chem. 2011, 76, 4392–4395. 10.1021/jo102440k. [DOI] [PubMed] [Google Scholar]
- Kislukhin A. A.; Higginson C. J.; Finn M. G. Org. Lett. 2011, 13, 1832–1835. 10.1021/ol103153f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. . [DOI] [PubMed] [Google Scholar]
- a Young I. S.; Kerr M. A. Angew. Chem., Int. Ed. 2003, 42, 3023–3026. 10.1002/anie.200351573. [DOI] [PubMed] [Google Scholar]; b Carson C. A.; Kerr M. A. J. Org. Chem. 2005, 70, 8242–8244. 10.1021/jo0512251. [DOI] [PubMed] [Google Scholar]; c Dias D. A.; Kerr M. A. Org. Lett. 2009, 11, 3694–3697. 10.1021/ol901454y. [DOI] [PubMed] [Google Scholar]; d Humenny W. J.; Kyriacou P.; Sapeta K.; Karadeolian A.; Kerr M. A. Angew. Chem., Int. Ed. 2012, 51, 11088–11091. 10.1002/anie.201206177. [DOI] [PubMed] [Google Scholar]; e Grover H. K.; Emmett M. R.; Kerr M. A. Org. Biomol. Chem. 2015, 13, 655–671. 10.1039/C4OB02117G. [DOI] [PubMed] [Google Scholar]
- a Wipf P.; Stephenson C. R. J.; Okumura K. J. Am. Chem. Soc. 2003, 125, 14694–14695. 10.1021/ja038623a. [DOI] [PubMed] [Google Scholar]; b Wipf P.; Walczak M. A. A. Angew. Chem., Int. Ed. 2006, 45, 4172–4175. 10.1002/anie.200600723. [DOI] [PubMed] [Google Scholar]; c Walczak M. A. A.; Wipf P. J. Am. Chem. Soc. 2008, 130, 6924–6925. 10.1021/ja802906k. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ueda M.; Walczak M. A. A.; Wipf P. Tetrahedron Lett. 2008, 49, 5986–5989. 10.1016/j.tetlet.2008.07.179. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Walczak M.; Krainz T.; Wipf P. Acc. Chem. Res. 2015, 48, 1149–1158. 10.1021/ar500437h. [DOI] [PubMed] [Google Scholar]; f Milligan J. A.; Busacca C. A.; Senanayake C. H.; Wipf P. Org. Lett. 2016, 18, 4300–4303. 10.1021/acs.orglett.6b02051. [DOI] [PubMed] [Google Scholar]; g Milligan J. A.; Wipf P. Nat. Chem. 2016, 8, 296–297. 10.1038/nchem.2485. [DOI] [PubMed] [Google Scholar]
- a Xu G.-C.; Liu L.-P.; Lu J.-M.; Shi M. J. Am. Chem. Soc. 2005, 127, 14552–14553. 10.1021/ja054988e. [DOI] [PubMed] [Google Scholar]; b Huang J.-W.; Shi M. Synlett 2004, 2343–2346. 10.1055/s-2004-832838. [DOI] [Google Scholar]; c Shao L.-X.; Shi M. Curr. Org. Chem. 2007, 11, 1135–1153. 10.2174/138527207781662483. [DOI] [Google Scholar]; d Shi M.; Shao L.-X.; Lu J.-M.; Wei Y.; Mizuno K.; Maeda H. Chem. Rev. 2010, 110, 5883–5913. and references therein 10.1021/cr900381k. [DOI] [PubMed] [Google Scholar]
- a Kondo T.; Nakamura A.; Okada T.; Suzuki N.; Wada K.; Mitsudo T. J. Am. Chem. Soc. 2000, 122, 6319–6320. 10.1021/ja000238n. [DOI] [Google Scholar]; b Kondo T.; Taguchi Y.; Kaneko Y.; Niimi M.; Mitsudo T. Angew. Chem., Int. Ed. 2004, 43, 5369–5372. 10.1002/anie.200461002. [DOI] [PubMed] [Google Scholar]
- Wender P. A.; Howbert J. J. J. Am. Chem. Soc. 1981, 103, 688–690. 10.1021/ja00393a041. [DOI] [Google Scholar]
- Zhang Y.; Danishefsky S. J. J. Am. Chem. Soc. 2010, 132, 9567–9569. 10.1021/ja1035495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Baran P. S.; Burns N. Z. J. Am. Chem. Soc. 2006, 128, 3908–3909. 10.1021/ja0602997. [DOI] [PubMed] [Google Scholar]; b Cherney E. C.; Green J. C.; Baran P. Angew. Chem., Int. Ed. 2013, 52, 9019–9022. 10.1002/anie.201304609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Myers A. G.; Kephart S. E.; Chen H. J. Am. Chem. Soc. 1992, 114, 7922–7923. 10.1021/ja00046a054. [DOI] [Google Scholar]; b Denmark S. E.; Griedel B. D.; Coe D. M. J. Org. Chem. 1993, 58, 988–990. 10.1021/jo00057a002. [DOI] [Google Scholar]; c Denmark S. E.; Griedel B. D.; Coe D. M.; Schnute M. E. J. Am. Chem. Soc. 1994, 116, 7026–7043. 10.1021/ja00095a004. [DOI] [Google Scholar]
- a Agard N. J.; Prescher J. A.; Bertozzi C. R. J. Am. Chem. Soc. 2004, 126, 15046–15047. 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]; b Baskin J. M.; Prescher J. A.; Laughlin S. T.; Agard N. J.; Chang P. V.; Miller I. A.; Lo A.; Codelli J. A.; Bertozzi C. R. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 16793–16797. 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Codelli J. A.; Baskin J. M.; Agard N. J.; Bertozzi C. R. J. Am. Chem. Soc. 2008, 130, 11486–11493. 10.1021/ja803086r. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Jewett J. C.; Bertozzi C. R. Org. Lett. 2011, 13, 5937–5939. 10.1021/ol2025026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Murray R. W.; Jeyaraman R. J. Org. Chem. 1985, 50, 2847–2853. 10.1021/jo00216a007. [DOI] [Google Scholar]; b Mello R.; Fiorentino M.; Sciacovelli O.; Curci R. J. Org. Chem. 1988, 53, 3890–3891. 10.1021/jo00251a053. [DOI] [Google Scholar]; c Mello R.; Fiorentino M.; Fusco C.; Curci R. J. Am. Chem. Soc. 1989, 111, 6749–6757. 10.1021/ja00199a039. [DOI] [Google Scholar]
- Dodziuk H.Strained Hydrocarbons: Beyond the van’t Hoff and Le Bell Hypothesis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2009. [Google Scholar]
- Gianatassio R.; Lopchuk J. M.; Wang J.; Pan C.-M.; Malins L. R.; Prieto L.; Brandt T. A.; Collins M. R.; Gallego G. M.; Sach N. W.; Spangler J. E.; Zhu H.; Zhu J.; Baran P. S. Science 2016, 351, 241–246. 10.1126/science.aad6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaudel Q.; Ishihara Y.; Baran P. S. Acc. Chem. Res. 2015, 48, 712–721. 10.1021/ar500424a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Meanwell N. A. J. Med. Chem. 2011, 54, 2529–2591. 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]; b Meanwell N. A. Chem. Res. Toxicol. 2016, 29, 564–616. 10.1021/acs.chemrestox.6b00043. [DOI] [PubMed] [Google Scholar]
- Stepan A. F.; Subramanyam C.; Efremov I. V.; Dutra J. K.; O’Sullivan T. J.; DiRico K. J.; McDonald W. S.; Won A.; Dorff P. H.; Nolan C. E.; Becker S. L.; Pustilnik L. R.; Riddell D. R.; Kauffman G. W.; Kormos B. L.; Zhang L.; Lu Y.; Capetta S. H.; Green M. E.; Karki K.; Sibley E.; Atchison K. P.; Hallgren A. J.; Oborski C. E.; Robshaw A. E.; Sneed B.; O’Donnell C. J. J. Med. Chem. 2012, 55, 3414–3424. 10.1021/jm300094u. [DOI] [PubMed] [Google Scholar]
- Westphal M. V.; Wolfstädter B. T.; Plancher J.-M.; Gatfield J.; Carreira E. M. ChemMedChem 2015, 10, 461–469. 10.1002/cmdc.201402502. [DOI] [PubMed] [Google Scholar]
- a Pellicciari R.; Raimondo M.; Marinozzi M.; Natalini B.; Costantino G.; Thomsen C. J. Med. Chem. 1996, 39, 2874–2876. 10.1021/jm960254o. [DOI] [PubMed] [Google Scholar]; b Pätzel M.; Sanktjohanser M.; Doss A.; Henklein P.; Szeimies G. Eur. J. Org. Chem. 2004, 2004, 493–498. 10.1002/ejoc.200300554. [DOI] [Google Scholar]; c de Meijere A.; Zhao L.; Belov V. N.; Bossi M.; Noltemeyer M.; Hell S. W. Chem. - Eur. J. 2007, 13, 2503–2516. 10.1002/chem.200601316. [DOI] [PubMed] [Google Scholar]
- a Andrews M. D.; Bagal S. K.; Gibson K. R.; Omoto K.; Ryckmans T.; Skerratt S. E.; Stupple P. A. U.S. Patent Appl. US2014364415A1, 2014.; b Gammill R. B.; Bisaha S. N.; Timko J. M.; Judge T. M.; Barbachyn M. R.; Kim K. S. U.S. Patent Appl. US5262417A, 1993.; c Breslin H. J.; Dorsey B. D.; Dugan B. J.; Fowler K. M.; Hudkins R. L.; Mesaros E. F.; Monck N. J. T. Int. Patent Appl. WO2013US62085A1, 2013.; d Hayashi K.; Watanabe T.; Toyama K.; Kamon J.; Minami M., Uni M. Int. Patent WO2012JP70876A1, 2012.; e Bennett M. J.; Zehnder L. R.; Ninkovic S.; Kung P.; Meng J. J.; Huang B. U.S. Patent Appl. US2010093696A1, 2010.; f Johnson T. W.; Richardson P. F.; Collins M. R.; Richter D. T.; Burke B. J.; Gajiwala K.; Ninkovic S.; Linton M. A.; Le P. T. Q.; Hoffman J. E. Can. Patent Appl. CA2915356A1, 2016.
- Wiberg K. B.; Williams V. Z. J. Org. Chem. 1970, 35, 369–373. 10.1021/jo00827a018. [DOI] [Google Scholar]
- a Wiberg K. B. Chem. Rev. 1989, 89, 975–983. 10.1021/cr00095a001. [DOI] [Google Scholar]; b Delia E. W.; Lochert I. J. Org. Prep. Proced. Int. 1996, 28, 411–441. 10.1080/00304949609356550. [DOI] [Google Scholar]; c Levin M. D.; Kaszynski P.; Michl J. Chem. Rev. 2000, 100, 169–234. 10.1021/cr990094z. [DOI] [PubMed] [Google Scholar]; d Kaszynski P.; Michl J. In The Chemistry of the Cyclopropyl Group; Rappoport Z., Ed.; John Wiley & Sons: New York, 2004; Vol. 2, Chapter 13, pp 773–812. [Google Scholar]
- Semmler K.; Szeimies G.; Belzner J. J. Am. Chem. Soc. 1985, 107, 6410–6411. 10.1021/ja00308a053. [DOI] [Google Scholar]
- Hossain M. T.; Timberlake J. W. J. Org. Chem. 2001, 66, 6282–6285. 10.1021/jo010212u. [DOI] [PubMed] [Google Scholar]
- Della E. W.; Kasum B.; Kirkbride K. P. J. Am. Chem. Soc. 1987, 109, 2746–2749. 10.1021/ja00243a030. [DOI] [Google Scholar]
- Toops D. S.; Barbachyn M. R. J. Org. Chem. 1993, 58, 6505–6508. 10.1021/jo00075a063. [DOI] [Google Scholar]
- Bunker K. D.; Sach N. W.; Huang Q. H.; Richardson P. F. Org. Lett. 2011, 13, 4746–4748. 10.1021/ol201883z. [DOI] [PubMed] [Google Scholar]
- Goh Y. L.; Tam E. K. W.; Bernardo P. H.; Cheong C. B.; Johannes C. W.; William A. D.; Adsool V. A. Org. Lett. 2014, 16, 1884–1887. 10.1021/ol500635p. [DOI] [PubMed] [Google Scholar]
- Bunker K. D. PCT Int. Appl. WO201589170, 2015.
- Della E. W.; Taylor D. K.; Tsanaktsidis J. Tetrahedron Lett. 1990, 31, 5219–5220. 10.1016/S0040-4039(00)97847-X. [DOI] [Google Scholar]
- Kogay B. E.; Sokolenko W. A. Tetrahedron Lett. 1983, 24, 613–616. 10.1016/S0040-4039(00)81478-1. [DOI] [Google Scholar]
- a Butov G. M.; Mokhov V. M.; Parshin G. Y.; Panyushkina O. A. Russ. J. Org. Chem. 2009, 45, 1732–1733. 10.1134/S107042800911030X. [DOI] [Google Scholar]; b Butov G. M.; Mokhov V. M.; Parshin G. Y.; Lysykh B. A.; Konyushkin L. D.; Firgang S. I. Russ. J. Org. Chem. 2011, 47, 150–151. 10.1134/S1070428011010210. [DOI] [Google Scholar]; c Butov G. M.; Mokhov V. M. Russ. J. Org. Chem. 2013, 49, 1403–1404. 10.1134/S1070428013090303. [DOI] [Google Scholar]; d Butov G. M.; Mokhov V. M. Russ. J. Org. Chem. 2014, 50, 447–448. 10.1134/S1070428014030270. [DOI] [Google Scholar]
- Guffler S. Ph.D. Dissertation, Humboldt-Universität zu Berlin, 1998. [Google Scholar]
- Davies S. G.; Fletcher A. M.; Roberts P. M.; Thomson J. E. Tetrahedron: Asymmetry 2012, 23, 1111–1153. 10.1016/j.tetasy.2012.08.009. [DOI] [Google Scholar]
- Brambilla M.; Davies S. G.; Fletcher A. M.; Roberts P. M.; Thomson J. E. Tetrahedron 2016, 72, 4523–4535. 10.1016/j.tet.2016.06.011. [DOI] [Google Scholar]
- Krasovskiy A.; Knochel P. Angew. Chem., Int. Ed. 2004, 43, 3333–3336. 10.1002/anie.200454084. [DOI] [PubMed] [Google Scholar]; b Bao R. L.-Y.; Zhao R.; Shi L. Chem. Commun. 2015, 51, 6884–6900. 10.1039/C4CC10194D. [DOI] [PubMed] [Google Scholar]
- a Brown A.; Ellis D.; Wallace O.; Ralph M. Bioorg. Med. Chem. Lett. 2010, 20, 1851–1853. 10.1016/j.bmcl.2010.01.143. [DOI] [PubMed] [Google Scholar]; b Han Y.; Han M.; Shin D.; Song C.; Hahn H.-G. J. Med. Chem. 2012, 55, 8188–8192. 10.1021/jm3008294. [DOI] [PubMed] [Google Scholar]; c Han M.; Song C.; Jeong N.; Hahn H.-G. ACS Med. Chem. Lett. 2014, 5, 999–1004. 10.1021/ml500187a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funke W. Angew. Chem., Int. Ed. Engl. 1969, 8, 70–71. 10.1002/anie.196900701. [DOI] [Google Scholar]
- Bartnik R.; Marchand A. P. Synlett 1997, 1997, 1029–1039. 10.1055/s-1997-1520. [DOI] [Google Scholar]
- a Hayashi K.; Sato C.; Hiki S.; Kumagai T.; Tamai S.; Abe T.; Nagao Y. Tetrahedron Lett. 1999, 40, 3761–3764. 10.1016/S0040-4039(99)00603-6. [DOI] [Google Scholar]; b Hayashi K.; Hiki S.; Kumagai T.; Nagao Y. Heterocycles 2002, 56, 433–442. 10.3987/COM-01-S(K)65. [DOI] [Google Scholar]; c Hayashi K.; Ikee Y.; Goto S.; Shiro M.; Nagao Y. Chem. Pharm. Bull. 2004, 52, 89–94. 10.1248/cpb.52.89. [DOI] [PubMed] [Google Scholar]; d Ikee Y.; Hashimoto K.; Nakashima M.; Hayashi K.; Sano S.; Shiro M.; Nagao Y. Bioorg. Med. Chem. Lett. 2007, 17, 942–945. 10.1016/j.bmcl.2006.11.048. [DOI] [PubMed] [Google Scholar]; e Ikee Y.; Hashimoto K.; Kamino M.; Nakashima M.; Hayashi K.; Sano S.; Shiro M.; Nagao Y. Chem. Pharm. Bull. 2008, 56, 346–356. 10.1248/cpb.56.346. [DOI] [PubMed] [Google Scholar]
- For other preparations of ABB, see:; a Dave P. J. J. Org. Chem. 1996, 61, 5453–5455. 10.1021/jo9602579. [DOI] [Google Scholar]; b Bartnik R.; Cal D.; Marchand A. P.; Alihodzic S.; Devasagayaraj A. Synth. Commun. 1998, 28, 3949–3954. 10.1080/00397919808004953. [DOI] [Google Scholar]
- Dembitsky V. M. J. Nat. Med. 2008, 62, 1–33. 10.1007/s11418-007-0166-3. [DOI] [PubMed] [Google Scholar]
- Wheate N. J.; Walker S.; Craig G. E.; Oun R. Dalton Trans. 2010, 39, 8113–8127. 10.1039/c0dt00292e. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; Tice C. M.; Singh S. B. Bioorg. Med. Chem. Lett. 2014, 24, 3673–3682. 10.1016/j.bmcl.2014.06.081. [DOI] [PubMed] [Google Scholar]
- Wiberg K. B.; Ciula R. P. J. Am. Chem. Soc. 1959, 81, 5261–5262. 10.1021/ja01528a060. [DOI] [Google Scholar]
- Wiberg K. B. Adv. Alicyclic Chem. 1968, 2, 185–254. 10.1016/B978-1-4831-9918-4.50009-0. [DOI] [Google Scholar]
- Lampman G. M.; Aumiller J. C. Org. Synth. 1971, 51, 55–59. 10.1002/0471264180.os051.13. [DOI] [Google Scholar]
- a Gaoni Y. Tetrahedron Lett. 1988, 29, 1591–1594. 10.1016/S0040-4039(00)80361-5. [DOI] [Google Scholar]; b Gaoni Y. Tetrahedron 1989, 45, 2819–2840. 10.1016/S0040-4020(01)80112-5. [DOI] [Google Scholar]; c Gaoni Y. Org. Prep. Proced. Int. 1995, 27, 185–212. 10.1080/00304949509458453. [DOI] [Google Scholar]
- Nájera C.; Yus M. Tetrahedron 1999, 55, 10547–10658. 10.1016/S0040-4020(99)00600-6. [DOI] [Google Scholar]
- a Kalia J.; Raines R. T. Curr. Org. Chem. 2010, 14, 138–147. 10.2174/138527210790069839. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Stephanopoulos N.; Francis M. B. Nat. Chem. Biol. 2011, 7, 876–884. 10.1038/nchembio.720. [DOI] [PubMed] [Google Scholar]; c Spicer C. D.; Davis B. G. Nat. Commun. 2014, 5, 4740. 10.1038/ncomms5740. [DOI] [PubMed] [Google Scholar]; d Boutureira O.; Bernardes G. J. Chem. Rev. 2015, 115, 2174–2195. 10.1021/cr500399p. [DOI] [PubMed] [Google Scholar]
- a Chalker J. M.; Bernardes G. J. L.; Lin Y. A.; Davis B. G. Chem. - Asian J. 2009, 4, 630–640. 10.1002/asia.200800427. [DOI] [PubMed] [Google Scholar]; b Hoyle C. E.; Bowman C. N. Angew. Chem., Int. Ed. 2010, 49, 1540–1573. 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]; c Hoyle C. E.; Lowe A. B.; Bowman C. N. Chem. Soc. Rev. 2010, 39, 1355–1387. 10.1039/b901979k. [DOI] [PubMed] [Google Scholar]; d Nair D. P.; Podgórski M.; Chatani S.; Gong T.; Xi W.; Fenoli C. R.; Bowman C. N. Chem. Mater. 2014, 26, 724–744. 10.1021/cm402180t. [DOI] [Google Scholar]; e McKay C. S.; Finn M. G. Chem. Biol. 2014, 21, 1075–1101. 10.1016/j.chembiol.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cravatt B. F.; Wright A. T.; Kozarich J. W. Annu. Rev. Biochem. 2008, 77, 383–414. 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]; b Willems L. I.; Overkleeft H. S.; van Kasteren S. I. Bioconjugate Chem. 2014, 25, 1181–1191. 10.1021/bc500208y. [DOI] [PubMed] [Google Scholar]
- a Nugent T. C.; El-Shazly M. Adv. Synth. Catal. 2010, 352, 753–891. 10.1002/adsc.200900719. [DOI] [Google Scholar]; b Guizzetti S.; Benaglia M. Eur. J. Org. Chem. 2010, 2010, 5529–5541. 10.1002/ejoc.201000728. [DOI] [Google Scholar]; c Wang C.; Xiao J. Top. Curr. Chem. 2013, 343, 261–282. 10.1007/128_2013_484. [DOI] [PubMed] [Google Scholar]; d Alinezhad H.; Yavari H.; Salehian F. Curr. Org. Chem. 2015, 19, 1021–1049. 10.2174/1385272819666150311233021. [DOI] [Google Scholar]
- For examples of housanes with chirality on other positions, see:; a Steliou K.; Poupart M.-A. J. Org. Chem. 1989, 54, 5128–5131. 10.1021/jo00282a032. [DOI] [Google Scholar]; b Magar S. S.; Fuchs P. L. Tetrahedron Lett. 1992, 33, 745–748. 10.1016/S0040-4039(00)77705-7. [DOI] [Google Scholar]
- Jeffery S. M.; Stirling C. J. M. J. Chem. Soc., Perkin Trans. 2 1993, 2, 1617–1624. 10.1039/p29930001617. [DOI] [Google Scholar]
- Ghanem A.; Aboul-Enein H. Y. Chirality 2005, 17, 1–15. 10.1002/chir.20089. [DOI] [PubMed] [Google Scholar]
- Ghanem A.; Schurig V. Chirality 2001, 13, 118–123. . [DOI] [PubMed] [Google Scholar]
- Hoye T. R.; Jeffrey C. S.; Shao F. Nat. Protoc. 2007, 2, 2451–2458. (see SI for specific details) 10.1038/nprot.2007.354. [DOI] [PubMed] [Google Scholar]
- Huisman G. W.; Liang J.; Krebber A. Curr. Opin. Chem. Biol. 2010, 14, 122–129. 10.1016/j.cbpa.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Liang J.; Mundorff E.; Voladri R.; Jenne S.; Gilson L.; Conway A.; Krebber A.; Wong J.; Huisman G.; Truesdell S.; Lalonde J. Org. Process Res. Dev. 2010, 14, 188–192. 10.1021/op9002714. [DOI] [Google Scholar]
- An attempted SN2 reaction with amine dibenzylamine and mesylate 165 (DMSO or DMF, rt up to 150 °C) gave only a trace of product 182 by LC/MS (elimination of the sulfone predominates).
- The amino acids were used as their free acids in the reactions and converted to their methyl esters during workup to simplify purification.
- Rodriguez J. R.; Agejas J.; Bueno A. B. Tetrahedron Lett. 2006, 47, 5661–5663. 10.1016/j.tetlet.2006.06.029. [DOI] [Google Scholar]
- a Saito M.; Nakajima M.; Hashimoto S. Chem. Commun. 2000, 1851–1852. 10.1039/b005994n. [DOI] [Google Scholar]; b Sundararajan G.; Prabagaran N. Org. Lett. 2001, 3, 389–392. 10.1021/ol006898e. [DOI] [PubMed] [Google Scholar]; c Rana N. K.; Unhale R.; Singh V. K. Tetrahedron Lett. 2012, 53, 2121–2124. 10.1016/j.tetlet.2012.02.052. [DOI] [Google Scholar]
- Perdicchia D.; Jorgensen K. A. J. Org. Chem. 2007, 72, 3565–3568. 10.1021/jo0626717. [DOI] [PubMed] [Google Scholar]
- Cusack K. P.; Breinlinger E. C.; Fix-Stenzel S. R.; Stoffel R. H.; Woller K. R. PCT Int. Appl. WO2011071570A1, 2011.
- Marcé P.; Díaz Y.; Matheu M. I.; Castillón S. Org. Lett. 2008, 10, 4735–4738. 10.1021/ol801791g. [DOI] [PubMed] [Google Scholar]
- Zheng D.-Q.; Jing Y.; Zheng B.-Y.; Ye Y.-F.; Xu S.; Tian W.-S.; Ma H.-Y.; Ding K. Tetrahedron 2016, 72, 2164–2169. 10.1016/j.tet.2016.03.002. [DOI] [Google Scholar]
- The cyclopentylation of estrones with cyclopentyl bromide took 7 days to complete.Peters R. H.; Crowe D. F.; Tanabe M.; Avery M. A.; Chong W. K. M. J. Med. Chem. 1987, 30, 646–652. 10.1021/jm00387a011. [DOI] [PubMed] [Google Scholar]
- Cerai G. P.; Morandi B. Chem. Commun. 2016, 52, 9769–9772. 10.1039/C6CC04410G. [DOI] [PubMed] [Google Scholar]
- Yang L.; Mills S. G.; Jiao R. PCT Int. Patent WO2005120505A2, 2005.
- Thorarensen A.; Brown M. F.; Casimiro-Garcia A.; Che Y.; Coe J. W.; Flanagan M. E.; Gilbert A. M.; Hayward M. M.; Langille J. D.; Montgomery J. I.; Telliez J.-B.; Unwalla R. J. U.S. Patent Appl. US20150158864A1, 2015.
- a Krishnan S.; Miller R. M.; Tian B.; Mullins R. D.; Jacobson M. P.; Taunton J. J. Am. Chem. Soc. 2014, 136, 12624–12630. 10.1021/ja505194w. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jost C.; Nitsche C.; Scholz T.; Roux L.; Klein C. D. J. Med. Chem. 2014, 57, 7590–7599. 10.1021/jm5006918. [DOI] [PubMed] [Google Scholar]
- Flanagan M. E.; Abramite J. A.; Anderson D. P.; Aulabaugh A.; Dahal U. P.; Gilbert A. M.; Li C.; Montgomery J.; Oppenheimer S. R.; Ryder T.; Schuff B. P.; Uccello D. P.; Walker G. S.; Wu G. S.; Brown M. F.; Chen J. M.; Hayward M. M.; Noe M. C.; Obach R. S.; Philippe L.; Shanmugasundaram V.; Shapiro M. J.; Starr J.; Stroh J.; Che Y. J. Med. Chem. 2014, 57, 10072–10079. 10.1021/jm501412a. [DOI] [PubMed] [Google Scholar]
- For some reports regarding the bond energy of the central C–C bond of [1.1.1]propellane, see:; a Wiberg K. B.; Walker F. H. J. Am. Chem. Soc. 1982, 104, 5239–5240. 10.1021/ja00383a046. [DOI] [Google Scholar]; b Wiberg K. B. J. Am. Chem. Soc. 1983, 105, 1227–1233. 10.1021/ja00343a025. [DOI] [Google Scholar]; c Jackson J. E.; Allen L. C. J. Am. Chem. Soc. 1984, 106, 591–599. 10.1021/ja00315a022. [DOI] [Google Scholar]; d Feller D.; Davidson E. R. J. Am. Chem. Soc. 1987, 109, 4133–4139. 10.1021/ja00248a001. [DOI] [Google Scholar]
- For a recent report describing the central C–C bond of [1.1.1]propellane as a charge-shift bond, see:Wu W.; Gu J.; Song J.; Shaik S.; Hiberty P. C. Angew. Chem., Int. Ed. 2009, 48, 1407–1410. 10.1002/anie.200804965. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.