

Abstract
Single-molecule tracking can extract quantitative kinetic information and identify possible state transitions of diffusing molecules (such as switching between binding and unbinding) in the in vivo environment of living cells. Confined diffusion, caused by the encompassing membrane boundary of the cell, results in increased uncertainties in identifying state-associated diffusion coefficients and transition probabilities. This problem is particularly acute in bacterial cells because of their small sizes. A standard approach to eliminating confinement errors in bacterial cells is to analyze molecule displacements only along the long axis of the cell, where molecules experience the least confinement, and hence turn three-dimensional tracking into a one-dimensional problem. However, this approach dramatically decreases the amount of data usable for statistical analysis and leads to increased uncertainties in identifying different states. Here, we present a simple algorithm, termed single-particle tracking improvement with confinement error reduction (SPICER), which significantly decreases confinement errors by selectively incorporating data not only from the long axis but also from the short axes of the cell. We validate SPICER using both reaction-diffusion simulations and experimental single-molecule tracking (SMT) data of RNA polymerase in live Escherichia coli cells. SPICER is easy to implement with existing SMT analysis routines and should find broad applications in SMT analysis.
Introduction
Single-molecule tracking (SMT) is a powerful technique for probing possible functional states of biomolecules in living cells (1, 2, 3). In a typical SMT experiment, a molecule’s cellular positions are recorded by acquiring its fluorescent images consecutively at defined time intervals. From these images, an SMT trajectory, a time series of corresponding spatial coordinates of the molecule in reference to the cell, is extracted. From the statistical analysis of these SMT trajectories, different diffusive states of the molecule, each characterized by a different diffusion coefficient, D, can be obtained. These diffusive states and the associated population percentages can provide valuable information regarding possible functional states of the molecule. Recent SMT in bacterial cells have indeed shed light on the working mechanisms of transcription factors (4, 5), RNA polymerase (6, 7, 8), DNA polymerase (9), ribosomes (10), cytoskeletal proteins (11, 12), and more (13, 14).
In addition to measuring a molecule’s diffusion coefficients, SMT experiments offer another significant advantage, which is to obtain transition probabilities of molecules between different diffusive states. These transition probabilities provide crucial information regarding the kinetics of state switching, such as the binding and unbinding rates of a protein molecule to its target site and the lifetime of a particular functional state of the molecule (15, 16). Such information is often difficult to obtain in live cells by other means.
Various algorithms based on the statistical analyses of a large number of SMT trajectories have been developed to obtain the transition probabilities and associated diffusive states from SMT experiments. Among them, the vbSPT algorithm developed by Persson et al. (16) has proven robust. vbSPT assumes a hidden Markov model (HMM) in which diffusing molecules make a memoryless jump in states defined by different diffusion coefficients, and uses a variational Bayesian approach to identify individual states and their associated kinetics (16).
Successful application of analysis methods like vbSPT requires the correct identification of different diffusive states, which are characterized by unique diffusion coefficients. However, in bacterial cells, the small cell size (1–2 μm) spatially confines a molecule’s diffusion such that the measured apparent diffusion coefficient, , of a molecule appears smaller than the actual value, leading to difficulties in identifying the correct diffusive state. A common practice for minimizing confinement effects is to use displacements measured only along the long axis of the cell, where molecules would experience the least confinement; we refer to this method as “1d” analysis throughout the work (7, 16, 17). However, using information only along one dimension rather than using all available dimensions leads to a less accurate determination of diffusion parameters. The reduced amount of data limits the available number of well-defined trajectories, which is particularly important for calculating transition probabilities (18, 19).
Here, we present, to our knowledge, a new, simple algorithm, termed single-particle tracking improvement with confinement error reduction (SPICER). SPICER maintains the full length of an SMT trajectory and maximizes the amount of data used by calculating displacements in all dimensions available (2d or 3d) and only selectively switches to 1d (along the cell’s long axis) when a molecule is likely to experience confinement. As such, the accuracy in determining both the diffusion states and transition probabilities is dramatically improved. We demonstrate the use of SPICER on both simulated and experimental SMT data of Escherichia coli RNA polymerase (RNAP). The simple implementation of the SPICER algorithm in SMT analysis should allow its wide application in probing in vivo dynamics of molecular events in small bacterial cells.
Results
Operational principle of SPICER
To illustrate the operational principle of SPICER, we show in Fig. 1 a schematic 3d SMT trajectory of a diffusing molecule in a typical rod-shaped bacterial cell with two cross sections along the long, x (top), and short, y (bottom) axes of the cell. The parameter R defines the confinement zone (red) and is the distance from the membrane boundary of the cell to the edge of the midcell region where the molecule diffuses freely and does not experience confinement (green).
Figure 1.

An example 3d SMT trajectory of a molecule in a rod-shaped bacterial cell. The purple solid circles are localizations of the molecule inside the confinement-free region (green). Displacements using these localizations as initial positions, i.e., displacements 2, 4, and 5, are calculated using their full 3d coordinates. Yellow solid circles are localizations of the molecule inside the R-region, where the molecule experiences confinement (red). Displacements using these localizations as initial positions, e.g., displacements 1, 3, and 6, are calculated using only 1d coordinates along the x (long) axis of the cell.
In previous studies, to avoid confinement, only displacements along the cell’s long axis were used for HMM analysis. We refer to this method as “1d” analysis in this work (7, 16, 17). Consequently, in the 1d analysis, the available data in the trajectory ω shown in Fig. 1, represented by a series of single-step displacements, ω = and containing information in all three dimensions (3d), , is reduced to a third of the original amount of data, . SPICER increases the amount of data available and reduces the proportion of data that experiences confinement by analyzing a modified trajectory, , where displacements are calculated in 1d along the long axis of the cell to avoid confinement in the R region, whereas , are calculated in 3d, as the initial positions of these displacements are in the midcell and outside of the R region. As such, the full length of the trajectory is maintained, there is an increase of data being utilized with coordinates of all available dimensions, and there is a decrease in the proportion of data experiencing confinement. Note here that the same principle can be applied to 2d tracking experiments because of the symmetry of rod-shaped bacterial cells along the short axis. An example and associated discussion are provided in the Supporting Material.
Next, we demonstrate that switching dimensions within an SMT trajectory as described above does not modify the ability of SPICER to identify a set of most suitable parameters (diffusion coefficients, D, and transition probabilities, P) describing the trajectory using the maximum likelihood method (Supporting Material) (15).
The likelihood of having a diffusion coefficient, D, given a single displacement in d dimensions, , is proportional to the probability of having that displacement given the diffusion coefficient, , and is defined by the equation
| (1) |
where for d = 1, for d = 2, and for d = 3; D is the corresponding diffusion coefficient and τ is the time interval for each displacement. Equation 2 is the direct result of solving the diffusion equation with no barriers. If a molecule stays in one state, as defined by a single diffusion coefficient, D, the likelihood of having a particular trajectory specified by a series of experimentally measured displacements, w, will be
| (2) |
Maximizing the likelihood, L, with respect to D results in the well-known relation for d = 1, for d = 2, and so on (15). In previous studies, the value d is set constant for all displacements in a trajectory. However, note here that the true diffusion coefficient, D, is independent of the d value used, that the probability of each displacement is independent of the other displacements at each time point, and that the likelihood, L, is an arbitrary multiplicative constant with no significance in its absolute value in isolation. Therefore, varying d values along a trajectory does not prevent maximizing the likelihood to find the best-fit parameter D. In the Supporting Material, we provide a further validation of this concept.
Selection of an optimal R value
Before one can analyze SMT data with SPICER, an optimal R-value for a given experimental system must be identified. The R-value defines the size of the confinement zone, within which the displacements of a trajectory are calculated using only 1d coordinates along the long axis. Displacements outside of the R region, toward the center of the cell, are computed using the full coordinates available in 2d or 3d, depending on the experimental setup.
Intuitively, the size of the confinement zone, or the R-value, is primarily dependent on how fast the molecule diffuses. Molecules diffusing quickly require a large R-value to avoid confinement, whereas molecules diffusing slowly do not. Therefore, for a mixed population of molecules, the optimal R-value, should be set for molecules that diffuse the fastest. Consequently, it follows that at a given imaging speed, one can construct a lookup table so that each estimated value of a system corresponds to an optimal R-value.
To create the lookup table, we simulated five sets of 3d SMT experiments in a rod-shaped cell with radius r = 500 nm and length l = 2 μm; each set contains 10,000 single-state SMT trajectories with a fixed ranging from 0.4 to 4 μm2/s, tracked with an imaging speed of 200 f/s. For each data set, we varied the R-value systematically from 50 to 500 nm at 50-nm intervals and used SPICER to identify the corresponding apparent D value at each R-value. We then plotted the approximation percentage of the data set at different R values (Fig. 2 A). The optimal R-value was then identified as the one at which the approximation percentage reaches a maximum. The reasoning is that if an R-value is correctly selected, should contain the least confinement error in SPICER, hence approaching maximally the value.
Figure 2.
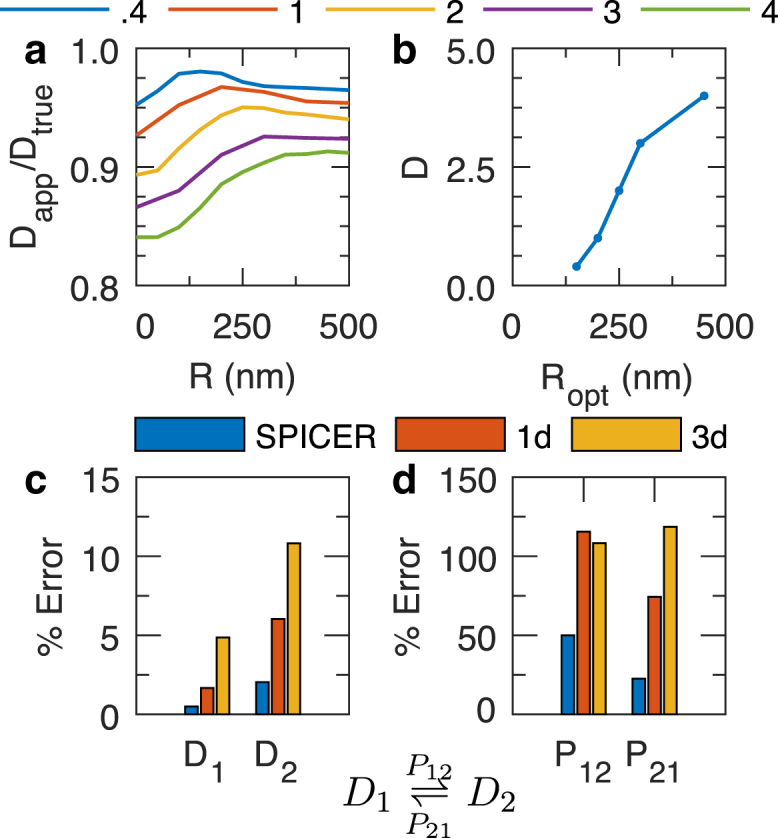
(a and b) Construction of a lookup table for finding the optimal R-value in a 3d tracking system. (a) Approximation percentage of five simulated systems at different R-values, with varying from 0.4 to 4 μm2/s. (b) Optimal R-values at different diffusion coefficients are identified from (a) as the R-value at which the maximal is reached. (c and d) Comparison of the performance of SPICER and conventional 1d and 3d analyses in identifying the diffusion coefficients (c) and transition probabilities (d) in a two-state system with μm2/s, μm2/s, and The percentage error is defined as
As shown in Fig. 2 A, data sets with small diffusion coefficients reach their maximal apparent diffusion coefficients, , at small R values, consistent with the notion that slowly diffusing molecules experience less confinement and hence the R-region would be small. However, for the data set that has a D = 4 μm2/s, = 450 nm, indicating that for molecules diffusing faster than 4 μm2/s, the small size of a bacterial cell itself confines diffusion, regardless of how far away from the membrane the molecule is. In Fig. 2 B, we plotted the optimal R-value for each simulated value. It can be seen that monotonically increases with D. Note here that although this lookup table is coarse-grained, with the R-value changing in 50-nm increments and the D-value changing in ∼1-μm2/s increments, finer grains on the order of 5 nm and 0.1 μm2/s are not necessary. The typical spatial resolution in an SMT experiment in live bacterial cells is in the range 30–50 nm, and a change of 0.1 μm2/s in D does not lead to a significant change in the corresponding R-value within the 50-nm increment. Therefore, to use this lookup table, one can first estimate the largest diffusion coefficient of a given system using the 1d analysis, which approximates the true D value by eliminating confinement error along the cell long axis, and then use Fig. 2 B to estimate the value. A similar simulation and lookup table using 2d SMT data are shown in Fig. S3. A particular note here is that the lookup table is also related to the imaging speed, so that a fast-diffusing molecule imaged at a slow speed (long time intervals between subsequent acquisitions) will naturally require a larger R-value to accommodate the longer distance it travels during the time. Therefore, it is important to construct the lookup table based on the actual imaging condition, as we described above.
Next, we verified whether the utilization of an optimal R-value in SPICER indeed improves SMT analysis when molecules exist in two different diffusive states. We simulated 25,000 trajectories of a two-state system. The two diffusion coefficients are μm2/s and μm2/s, and the transition probabilities which corresponds to a transition rate of ∼5 s−1. We then analyzed this data set using 1d (only displacements along the cell long axis), 3d (using all displacements in three dimensions), or SPICER, in which the was chosen to be 200 nm (the larger at 1 μm2/s) according to Fig. 2 B.
In Fig. 2, C and D, we plotted the percent error in each of the four parameters analyzed using the three methods. Clearly, 3d analysis led to the highest amount of error for all the four parameters, consistent with the presence of significant confinement errors when displacements in all three dimensions were used under this condition. The 1d analysis showed improvement compared to the 3d analysis, especially in the identification of D, but it was significantly outperformed by SPICER, in which the percentage errors in all four parameters were the smallest. Note that localizations in the R-region of cell poles are still confined in SPICER, even though only their displacements along the cell’s long axis are used, just as in 1d analysis. Nevertheless, SPICER outperforms the 1d analysis, because in SPICER, the full coordinates of localizations outside of the R-region are given more weight than their corresponding 1d coordinates in the search for optimal parameters. We further verified that the same trend holds for a variety of systems with different diffusive parameters (Fig. S4 Table S1). These results demonstrate that by increasing the proportion of data containing full coordinates, SPICER, with an optimal R-value identified from the lookup table (Fig. 2 B), indeed improves the accuracy in determining both diffusion coefficients and transition probabilities of a diffusive system.
SPICER improves accuracy in identifying states with close diffusion coefficients
One important criterion used by the HMM to identify different diffusive states is the difference between diffusion coefficients associated with each state. If the diffusion coefficients of the two states are close to each other, the considerable overlap of the displacement distributions will lead to difficulties in determining the associated state of a displacement, and consequently to large errors in identifying corresponding transition probabilities. SPICER should be especially useful in improving data analysis under this circumstance, as it can effectively eliminate confinement error without significantly reducing the available data.
To compare the performance of SPICER with traditional 1d and 3d analyses under these scenarios, we simulated eight different systems with 50,000 trajectories each, with and set to 0.0224 (k = 5 s−1), to 1 μm2/s, and varied between 0.8 and 0.2 μm2/s. We analyzed these systems as described in the Supporting Material and plotted the average percentage errors in D and P for the three methods (Fig. 3).
Figure 3.

Comparison of the averaged percent errors in identifying diffusion coefficients (a) and transition probabilities (b) of systems with varying separations between the diffusion coefficients of the two states using SPICER, 1d, or 3d analysis. The larger D is fixed at 1 μm2/s, with the smaller D varying between 0.8 and 0.2 μm2/s. The average percent error is calculated as or The shaded region indicates the uncertainty in the parameter and is defined as the standard deviation of the parameter during the Markov chain Monte Carlo approach.
Consistent with what we expected, when decreases, the percentage errors in D and P increase for all three methods, but SPICER consistently outperforms the 1d and 3d analyses, in particular with smaller values. Only at larger values, 0.6 μm2/s, is the improvement less dramatic. These results thus demonstrate SPICER’s unique advantage in systems where the diffusion coefficients of two states are closely spaced relative to each other.
SPICER requires fewer trajectories compared to 1d or 3d analysis to achieve the same level of error reduction
SMT analysis usually requires a large number of trajectories (on the order of if the average length of trajectories is short (16)), so that diffusion coefficients and state transitions can be determined with statistical significance. However, experimentally, it is time-consuming to collect tens of thousands of SMT trajectories. To investigate whether SPICER helps in lowering this requirement, we used the same two-state system analyzed in Fig. 2 and varied the number of trajectories used in the analysis. In Fig. 4, we show that for all three methods (1d, 3d, and SPICER) the averaged percent error in D plateaus when the number of trajectories is >5000; the averaged percent error in P plateaus when the number is greater than 10,000, as accurate determination of P requires a higher number of trajectories. However, even at a low number of trajectories (∼3000), averaged percent errors of D and P in SPICER are substantially lower than those in 1d and 3d analyses, approaching the level that would be achieved by 10,000 trajectories with the 1d analyses. Note here that the decreases in the total error are mainly brought about by the minimization of confinement error in SPICER, which compensates for errors caused by an insufficient number of trajectories, as that occurs in 1d and 3d analyses. These results demonstrate that increasing the number of trajectories used will not improve the error in the calculated parameters when confinement error is present in the commonly used 1d and 3d analyses. The application of SPICER raises the proportion of data without confinement and leads to the least amount of error in determining the diffusion coefficients and transition probabilities in these systems.
Figure 4.

Comparison of the averaged percent errors in identifying diffusion coefficients (a) and transition probabilities (b) of the two-state system with a varying number of trajectories, shown in Fig. 2, c and d, using SPICER, 1d, and 3d analyses. The averaged percent error and shaded region are calculated the same way as for Fig. 3. The solid lines are the 10-point moving averages of raw data (scattered dots), and the shaded areas are the moving averages of the standard deviations of the parameters during the Markov chain Monte Carlo approach.
Validating SPICER using experimental RNAP tracking data
To further validate SPICER with experimentally obtained data, we performed 2d SMT on RNA polymerase (RNAP) in live E. coli cells. RNAP is primarily found within the nucleoid; because of its frequent interactions with chromosomal DNA, it has relatively small diffusion coefficients (6). Thus, RNAP would experience less confinement from the membrane than would other freely diffusing protein molecules in cells, and it can serve as a control system with negligible confinement to validate the SPICER algorithm.
We used a functional RNAP-PAmCherry fusion (gift from Dr. D. J. Jin of the National Cancer Institute) that is integrated into the E. coli chromosome, replacing the endogenous rpoC gene, which encodes the β′ subunit of RNAP (Supporting Material). Under our imaging conditions, we collected a total of ∼25,000 trajectories, with the average trajectory length at ∼3, in RNAP-PAmCherry-expressing cells grown in minimal M9 medium with a 5-ms exposure time. We first used conventional 1d and 2d analyses to determine that under this condition, the best model describing the diffusive behaviors of RNAP is a two-state model. The two D values from 1d and 2d analyses are similar to each other ( = 0.38 μm2/s and = 0.1 μm2/s; Fig. 5 A) and are consistent with values from previous SMT studies of RNAP (6). However, transition probabilities obtained from the 1d analysis are significantly lower than those obtained from the 2d analysis (Fig. 5 B). The lower transition probabilities of the 1d analysis are most likely due to short trajectory lengths (with around three displacements) combined with the reduced amount of data in the 1d analysis, which makes it difficult to observe rare transitions between states. Using simulations, we further verified that indeed at this slow-diffusion condition, 2d analysis describes the system more accurately than 1d analysis (Fig. 5, C and D).
Figure 5.

Validation of SPICER using experimentally acquired 2d SMT data of RNAP in live E. coli cells. (a and b) Comparison of the identified D1 and D2 (a) and and (b) values using SPICER, 1d, and 2d analyses. (c and d) Simulation of a similar system showing the same trend, that 2d and SPICER analyses are significantly more accurate than the 1d analysis, with SPICER reflecting the true values most closely. The true values for the simulation are shown as horizontal black lines.
Next, we applied SPICER using an R-value of 200 nm, identified as optimal using the procedure described in the previous section (Fig. S3), and obtained a new set of , , , and . As shown in Fig. 5 A, diffusion coefficients obtained using the three methods are similar to each other, suggesting that at this slow diffusing rate the confinement error is low, and that all the methods are capable of identifying D sufficiently well with the acquired number of trajectories. However, transition probabilities from SPICER and the 2d analysis are similar to each other and are both significantly higher than those obtained from the 1d analysis (Fig. 5 B). These results further demonstrate that SPICER can be used to analyze experimental SMT data with high accuracy.
Conclusions
In this work, we present a simple algorithm, SPICER, to reduce the confinement error in SMT analysis in small bacterial cells. SPICER calculates displacements in all dimensions available (2d or 3d) and only selectively switches to 1d (along the cell’s long axis) when a molecule is within a pre-defined R-region where it likely experiences confinement. We provided lookup tables and experimental guidelines for finding an optimal R-value. The complete package of SPICER is available for download at https://github.com/XiaoLabJHU/SPICER.git. Using simulations, we compared SPICER with commonly used SMT analyses and show that SPICER consistently improves the accuracy in determining diffusion coefficients and state-transition probabilities in SMT analyses. Even when compared to the 1d analysis, the traditional method used to relieve confinement in multistate systems, the confinement in the poles of the cells allows SPICER to outperform the 1d analysis. This improvement is achieved by increasing the overall proportion of molecules experiencing free diffusion during the maximization of the likelihood. Furthermore, SPICER performs significantly better than previous methods when the separation between diffusion coefficients of two different states is small, and when the acquired number of SMT trajectories is low ( 3000). We further validated SPICER using experimentally obtained SMT trajectories of RNAP in live E. coli cells. SPICER should be particularly useful for comparing SMT results in bacterial cell size mutants, as the influence of confinement in cells of different sizes can be easily accounted for with SPICER. Furthermore, the central concept of SPICER can be generalized to other cell geometries as long as localizations in the R-region can be used along a particular dimension in which the molecule experiences the least confinement.
Author Contributions
C.H.B. designed research, performed research, developed ideas, analyzed data, and wrote the article. K.B. performed experimental research and analyzed data. J.X. designed research and wrote the article.
Editor: Thomas Perkins.
Footnotes
Supporting Materials and Methods, six figures, and one table are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(17)30043-7.
Supporting Material
References
- 1.Sauer M. Localization microscopy coming of age: from concepts to biological impact. J. Cell Sci. 2013;126:3505–3513. doi: 10.1242/jcs.123612. [DOI] [PubMed] [Google Scholar]
- 2.Gahlmann A., Moerner W.E. Exploring bacterial cell biology with single-molecule tracking and super-resolution imaging. Nat. Rev. Microbiol. 2014;12:9–22. doi: 10.1038/nrmicro3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vrljic M., Nishimura S.Y., Moerner W.E. Single-molecule tracking. Methods Mol. Biol. 2007;398:193–219. doi: 10.1007/978-1-59745-513-8_14. [DOI] [PubMed] [Google Scholar]
- 4.Izeddin I., Récamier V., Darzacq X. Single-molecule tracking in live cells reveals distinct target-search strategies of transcription factors in the nucleus. eLife. 2014;3:e02230. doi: 10.7554/eLife.02230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elf J., Li G.-W., Xie X.S. Probing transcription factor dynamics at the single-molecule level in a living cell. Science. 2007;316:1191–1194. doi: 10.1126/science.1141967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stracy M., Lesterlin C., Kapanidis A.N. Live-cell superresolution microscopy reveals the organization of RNA polymerase in the bacterial nucleoid. Proc. Natl. Acad. Sci. USA. 2015;112:E4390–E4399. doi: 10.1073/pnas.1507592112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bakshi S., Dalrymple R.M., Weisshaar J.C. Partitioning of RNA polymerase activity in live Escherichia coli from analysis of single-molecule diffusive trajectories. Biophys. J. 2013;105:2676–2686. doi: 10.1016/j.bpj.2013.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bakshi S., Siryaporn A., Weisshaar J.C. Superresolution imaging of ribosomes and RNA polymerase in live Escherichia coli cells. Mol. Microbiol. 2012;85:21–38. doi: 10.1111/j.1365-2958.2012.08081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uphoff S., Reyes-Lamothe R., Kapanidis A.N. Single-molecule DNA repair in live bacteria. Proc. Natl. Acad. Sci. USA. 2013;110:8063–8068. doi: 10.1073/pnas.1301804110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanamrad A., Persson F., Elf J. Single-particle tracking reveals that free ribosomal subunits are not excluded from the Escherichia coli nucleoid. Proc. Natl. Acad. Sci. USA. 2014;111:11413–11418. doi: 10.1073/pnas.1411558111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niu L., Yu J. Investigating intracellular dynamics of FtsZ cytoskeleton with photoactivation single-molecule tracking. Biophys. J. 2008;95:2009–2016. doi: 10.1529/biophysj.108.128751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim S.Y., Gitai Z., Moerner W.E. Single molecules of the bacterial actin MreB undergo directed treadmilling motion in Caulobacter crescentus. Proc. Natl. Acad. Sci. USA. 2006;103:10929–10934. doi: 10.1073/pnas.0604503103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plochowietz A., Farrell I., Kapanidis A.N. In vivo single-RNA tracking shows that most tRNA diffuses freely in live bacteria. Nucleic Acids Res. 2016 doi: 10.1093/nar/gkw787. Published online September 12, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liao Y., Schroeder J.W., Biteen J.S. Single-molecule motions and interactions in live cells reveal target search dynamics in mismatch repair. Proc. Natl. Acad. Sci. USA. 2015;112:E6898–E6906. doi: 10.1073/pnas.1507386112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Das R., Cairo C.W., Coombs D. A hidden Markov model for single particle tracks quantifies dynamic interactions between LFA-1 and the actin cytoskeleton. PLoS Comput. Biol. 2009;5:e1000556. doi: 10.1371/journal.pcbi.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Persson F., Lindén M., Elf J. Extracting intracellular diffusive states and transition rates from single-molecule tracking data. Nat. Methods. 2013;10:265–269. doi: 10.1038/nmeth.2367. [DOI] [PubMed] [Google Scholar]
- 17.Bakshi S., Bratton B.P., Weisshaar J.C. Subdiffraction-limit study of Kaede diffusion and spatial distribution in live Escherichia coli. Biophys. J. 2011;101:2535–2544. doi: 10.1016/j.bpj.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chung I., Akita R., Mellman I. Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature. 2010;464:783–787. doi: 10.1038/nature08827. [DOI] [PubMed] [Google Scholar]
- 19.Beausang J.F., Zurla C., Nelson P.C. DNA looping kinetics analyzed using diffusive hidden Markov model. Biophys. J. 2007;92:L64–L66. doi: 10.1529/biophysj.107.104828. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.