

ABSTRACT
Infection with Helicobacter pylori is a major risk factor for development of gastric disease, including gastric cancer. Patients infected with H. pylori strains that express CagA are at even greater risk of gastric carcinoma. Given the importance of CagA, this report describes a new molecular mechanism by which the cagA copy number dynamically expands and contracts in H. pylori. Analysis of strain PMSS1 revealed a heterogeneous population in terms of numbers of cagA copies; strains carried from zero to four copies of cagA that were arranged as direct repeats within the chromosome. Each of the multiple copies of cagA was expressed and encoded functional CagA; strains with more cagA repeats exhibited higher levels of CagA expression and increased levels of delivery and phosphorylation of CagA within host cells. This concomitantly resulted in more virulent phenotypes as measured by cell elongation and interleukin-8 (IL-8) induction. Sequence analysis of the repeat region revealed three cagA homologous areas (CHAs) within the cagA repeats. Of these, CHA-ud flanked each of the cagA copies and is likely important for the dynamic variation of cagA copy numbers. Analysis of a large panel of clinical isolates showed that 7.5% of H. pylori strains isolated in the United States harbored multiple cagA repeats, while none of the tested Korean isolates carried more than one copy of cagA. Finally, H. pylori strains carrying multiple cagA copies were differentially associated with gastric disease. Thus, the dynamic expansion and contraction of cagA copy numbers may serve as a novel mechanism by which H. pylori modulates gastric disease development.
IMPORTANCE
Severity of H. pylori-associated disease is directly associated with carriage of the CagA toxin. Though the sequences of the CagA protein can differ across strains, previous analyses showed that virtually all H. pylori strains carry one or no copies of cagA. This study showed that H. pylori can carry multiple tandem copies of cagA that can change dynamically. Isolates harboring more cagA copies produced more CagA, thus enhancing toxicity to host cells. Analysis of 314 H. pylori clinical strains isolated from patients in South Korea and the United States showed that 7.5% of clinical strains in the United States carried multiple cagA copies whereas none of the South Korean strains did. This study demonstrated a novel molecular mechanism by which H. pylori dynamically modulates cagA copy number, which affects CagA expression and activity and may impact downstream development of gastric disease.
INTRODUCTION
Helicobacter pylori is a Gram-negative microaerophilic bacterium that colonizes the human stomach and infects more than half of the world’s population (1–3). H. pylori infection is associated with various gastric diseases, ranging from gastritis to gastric adenocarcinoma and mucosa-associated lymphoid tissue (MALT) lymphoma (4, 5); the latter associations led the International Agency for Research on Cancer to classify H. pylori as a group I carcinogen (6). The high infection rates seen with this bacterium are believed to be responsible for making gastric cancer the third most common cause of cancer-related death worldwide (7).
H. pylori produces many virulence factors that contribute to pathogenesis (8, 9). Of these factors, cytotoxin-associated gene A (CagA) is one of the most widely studied proteins because of its association with increased risk of development of severe gastric diseases (10, 11). The cagA gene is carried on the cag pathogenicity island (PAI), which encodes a type IV secretion system (T4SS) that directly injects CagA into host cells (12). Once inside the host cell, CagA is phosphorylated by host cell kinases, forms a complex with SHP-2 (Src homology region 2-containing phosphatase 2) (13), and alters multiple host signaling pathways (13–17). Phosphorylation of CagA occurs in the carboxyl terminus on the conserved tyrosine residue within a repeated five-amino-acid sequence, Glu-Pro-Ile-Tyr-Ala, referred to as the EPIYA motif (13, 15). Both phosphorylated and nonphosphorylated forms of CagA modulate host cellular signaling pathways (18–20). The numbers of EPIYA motifs and the flanking regions surrounding these motifs differ dramatically across strains, making CagA highly polymorphic. On the basis of surrounding amino acid sequences, four distinct EPIYA motifs have been identified: EPIYA-A, EPIYA-B, EPIYA-C, and EPIYA-D. Interestingly, the distributions of EPIYA motif combinations differ geographically (15, 21); East Asian strains contain EPIYA-ABD, whereas Western strains contain EPIYA-ABC, and the EPIYA-C motif may be repeated up to five times (13, 15, 22). These different EPIYA combinations have been shown to have an impact on disease progression (21, 23). Indeed, our prior study showed a significant association between infection with H. pylori strains carrying the EPIYA-ABD cagA genotype and the development of gastric cancer (23). Similarly, other studies have shown that CagA variants containing an increased number of EPIYA-C motifs correlate to more virulent disease characteristics (15, 24, 25).
In addition to toxin amino acid variation, the cagA promoter region has also been shown to be genetically heterogeneous; an AATAAGATA motif located +59 bp upstream of the transcription start site is associated with higher levels of CagA expression in H. pylori isolates from Colombia (26, 27) and Portugal (28). These increased levels of CagA result in higher levels of interleukin-8 (IL-8) secretion by gastric cells in vitro. In populations at high risk for gastric carcinoma, strains containing the +59 motif are associated with more advanced precancerous lesions (27) and intestinal metaplasia (28). Thus, the amount of CagA expressed by H. pylori appears to be linked to downstream pathogenesis.
The host signaling pathways that are modified by CagA upon translocation into host cells stimulate cytoskeletal rearrangement, increased cellular mobility, and elongated cell shape, referred to as the “hummingbird phenotype” (14, 29). Additionally, H. pylori induces secretion of the proinflammatory cytokine IL-8 from gastric epithelial cells by a T4SS-dependent mechanism (30–33). The T4SS apparatus itself induces IL-8 secretion during the early phase of infection, and CagA augments IL-8 secretion in later phases (34).
As a bacterial species, H. pylori shows exceptionally high rates of genetic variability and intraspecies diversity (35–39). It is believed that these genetic differences likely influence the overall virulence potential of individual strains. Among the variable factors, the members of the outer membrane protein (OMP) families, such as adherence-associated lipoprotein A and B (AlpA and AlpB), blood group antigen binding adhesin (BabA), Helicobacter outer membrane B (HomB), Helicobacter outer membrane protein Z (HopZ), outer membrane inflammatory protein A (OipA), and sialic acid binding adhesin (SabA), show high genetic variability on the basis of the presence or absence of different closely related paralogs (40–46). For example, the bab family is made up of three paralogs (babA, babB, and babC) that can be located at three different chromosomal loci, referred to as locus A, locus B, and locus C (47–49). The notable exception is oipA, because duplicated oipA genes, not paralogs, may be located at two different loci (50). This DNA duplication event is thought to be mechanistically associated with DNA inversion (51).
Here, we describe a novel molecular mechanism by which H. pylori alters cagA copy number by dynamic expansion and contraction of cagA at the PAI locus. These changes in cagA copy number affect CagA expression; strains carrying multiple copies produce more toxin, which results in increased host cell elongation and IL-8 secretion. Thus, this mechanism of cagA variation promotes adaptation and pathogenesis of H. pylori.
RESULTS
H. pylori strain PMSS1 carries multiple cagA copies.
The PMSS1 strain of H. pylori is capable of persistently colonizing mice and is thus a useful strain for the study of H. pylori infection in this animal model (52). Our attempts to construct a PMSS1 derivative containing a clean deletion of cagA were repeatedly unsuccessful; PCR analysis of transformants yielded unexpected banding patterns that led us to postulate that PMSS1 may contain two tandem cagA genes, an observation that has not been made in any other strain of H. pylori. To test this theory, we designed two sets of PCR primers that would allow us to detect the presence of the cagA gene as well as the presence of multiple adjacent cagA genes (Fig. 1). First, primers F and R (Fig. 1A and B, panel a) were used to confirm the presence of the cagA gene in the common H. pylori strains PMSS1, G27 (53), 26695 (54), J99 (55), and 7.13 (56) (Fig. 1B). Next, the possibility of the presence of multiple cagA copies was examined. PCR performed using primer dF only (Fig. 1A and B, panel b) and primer dR only (Fig. 1A and B, panel c) would detect copies of cagA that were arranged as inverted repeats; this PCR was negative for all strains (Fig. 1B). Finally, PCR using primers dF and dR (Fig. 1A and B, panel d) would detect copies of cagA that were arranged as either inverted or tandem repeats; lack of a band in PCR using only primer dF or dR would imply that the cagA genes were carried in tandem. The PCR with primers dF and dR generated a strong amplicon from strain PMSS1 and a weaker amplicon from strain 7.13. These data suggest that H. pylori strains PMSS1 and 7.13 carry multiple copies of cagA that are arranged as tandem repeats.
FIG 1 .

Analysis of cagA copy number by PCR and real-time PCR. (A) A scheme for the PCR-based method designed to detect multiple cagA genes and to identify the gene orientation. Primer annealing sites are shown. (B) H. pylori strains PMSS1, G27, 26695, J99, and 7.13 were analyzed for the presence of multiple copies of cagA using the four PCR sets. (C) A 12,374-bp region of PMSS1 was mapped on the basis of the DNA sequence of the cagA genes and their flanking regions. Primer annealing sites used for PCR (filled triangles) and for DNA sequencing (empty triangles) are indicated. Three repeated cagA homologous areas (CHAs) were designated CHA-ud (red), CHA-u (yellow), and CHA-d (green). (D) The cagA copy numbers of PMSS1, G27, 26695, J99, and 7.13 were analyzed by real-time PCR using the 2−ΔΔCT method. ureA was used as a reference gene, and G27 was used as a calibrator. The bar graphs indicate the average cagA copy number of each strain, and error bars represent standard deviations, derived from results of 4 independent experiments.
To further investigate the presence of tandem cagA genes in PMSS1, a 12.4-kbp region encompassing cagA was sequenced; the strategy was designed with the assumption that two copies of cagA were present. As illustrated in Fig. 1C, the sequenced region began 2,162 bp upstream of the cagA initiation codon and ended 1,600 bp downstream of the cagA termination codon; the sequence ended in the middle of the glutamate racemase gene (glr) open reading frame (ORF). To encompass the entire area, three separate PCR amplicons were generated and sequenced (illustrated in Fig. 1C; primers are listed in Table S1 in the supplemental material, and the sequence is available in GenBank under accession KX673184). In order to confirm the sequence of the cagA ORF, the ORF was amplified with primers seqF2 and seqR10 and was sequenced as illustrated in Fig. 1C. The sequence of the cagA ORF was identical to the sequences obtained from the three overlapping PCR amplicons. Analysis confirmed that CagA from PMSS1 contained the EPIYA-ABC motif (52), as is characteristic of Western strains of H. pylori. Interestingly, sequencing also revealed three cagA homologous areas (CHAs): a CHA located both upstream and downstream of cagA (CHA-ud, red, 462 bp); a CHA located only upstream of cagA (CHA-u, yellow, 592 bp); and a CHA located only downstream of cagA (CHA-d, green, 478 bp). The sequences of CHA-ud, CHA-u, and CHA-d were identical to those of the same CHAs found at other positions within the tandem repeats. However, they did not share homology with the other CHAs. In total, PMSS1 contained two copies of cagA, CHA-u, and CHA-d and three copies of CHA-ud (Fig. 1C). We note, however, that the sequencing strategy was not able to distinguish between more than two copies of cagA; interior copies would not have been amplified during the initial PCR, as the primers anneal outside the repeat region. Further, amplification using primers that anneal within the cagA ORF would result in indistinguishable copies of cagA. Thus, this PCR and sequencing method would identify identical cagA genes carried as direct tandem repeats. For the remainder of this article, “cagA repeat” is used to refer to the following DNA sequence arrangement: CHA-ud, CHA u, cagA ORF, and CHA-d.
As mentioned, the designed PCR and sequencing strategies were unable to determine the absolute cagA copy number in PMSS1. Therefore, real-time PCR was utilized to tentatively quantify the copy number of cagA relative to that of the urease A gene (ureA). The same panel of H. pylori strains was analyzed using chromosomal DNA as a template (Fig. 1D). The amplification efficiencies of cagA and ureA were almost equal across strains (see Fig. S1 in the supplemental material). As expected on the basis of the earlier PCR results (Fig. 1B), the relative cagA copy numbers in G27, 26695, and J99 were close to 1 (means ± standard deviations [SD], 1.0 ± 0.0, 1.1 ± 0.1, and 1.3 ± 0.1, respectively) but were increased in strains 7.13 and PMSS1; approximately 1.4 (± 0.1) copies of cagA were detected in strain 7.13, and 3.7 (± 0.1) copies were detected in strain PMSS1. These data suggest that strain 7.13 may contain a mixed population of single-copy and multiple-copy cagA carriers and that PMSS1 might contain more than three cagA copies.
Primers used in this study. Download Table S1, XLSX file, 0.01 MB (13.5KB, xlsx) .
Copyright © 2017 Jang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Real-time PCR standard curves and CT deviation of cagA and ureA in H. pylori strains. The standard curves were calculated by conducting real-time PCR using serial 10-fold dilutions of chromosomal DNA isolated from PMSS1, G27, 26695, J99, and 7.13; concentrations ranged from 10 ng/μl to 0.01 ng/μl. CT deviations (ΔCT) of each strain were calculated and plotted. Download Figure S1, TIF file, 0.2 MB (244.4KB, tif) .
Copyright © 2017 Jang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Generation of PMSS1 H. pylori mutant strains containing different copy numbers of cagA.
Increasing the copy number of a gene may serve as a mechanism to increase protein expression. To determine whether carriage of multiple copies of cagA results in increased CagA expression, we generated PMSS1 isogenic mutant strains that contained no, single, or multiple copies of cagA. Three constructs were designed to replace the first, last, or all copies of cagA with a chloramphenicol resistance (cat) cassette (Fig. 2A). Homologous recombination with construct F, which contained CHA-d and a unique 3′ region, would replace all but the first copy of cagA with the cat cassette. Strains recovered from this transformation were designated PMSS1/cagA-SF. Homologous recombination with construct L, which contained a unique 5′ region and CHA-u, would replace all but the last cagA. Such strains were designated PMSS1/cagA-SL. Finally, homologous recombination with construct FL, which contained a unique 5′ region and a unique 3′ region, would replace all copies of cagA, generating PMSS1ΔcagAFL (Fig. 2B). Because a DNA region flanked by direct repeats can be duplicated or deleted by recombination (57), the three constructs were designed to remove any repeated CHAs flanking cagA or cat. Thus, the resulting PMSS1/cagA-SF, PMSS1/cagA-SL, and PMSS1ΔcagAFL strains cannot undergo further recombination at this region.
FIG 2 .

Generation and screening of PMSS1 isogenic mutant strains and determination of cagA copy number. (A) PMSS1 isogenic mutant strains were generated by transformation of PMSS1 with three different mutagenesis constructs: F, L, and FL. Putative homologous recombination events to generate cagA-SF, cagA-SL, and ΔcagAFL are illustrated. The three cagA homologous areas (CHAs) are indicated: CHA-ud (red), CHA-u (yellow), and CHA-d (green). A single cagA repeat is indicated with brackets. A subscript n indicates that the number of repeats is undetermined. (B) A PCR-based method was used to screen for the mutant strains cagA-SF, cagA-MF, cagA-SL, cagA-ML, and ΔcagAFL. Seven different PCRs (named a to g) were used for the screen; results from PCR f, which identified multiple cagA repeats, are denoted in panel A. The alignment sites of the primers are indicated with arrows, and the primers are listed in Table S1 in the supplemental material. (C) The cagA copy number was determined by real-time PCR using the 2−ΔΔCT method. ureA was used as a reference gene, and SF-1 was used as a calibrator. The bar graphs indicate the average cagA copy number of each strain, and error bars represent standard deviations, derived from results of 5 independent experiments.
Given that CHA-d and CHA-u are present at more than one location in PMSS1, the homologous recombination of constructs F and L may occur at any of the different CHA-d or CHA-u locations; the number of possible recombination sites is increased due to the carriage of multiple cagA repeats in PMSS1. Thus, chloramphenicol-resistant transformants may be generated by removing no cagA repeat or fewer cagA repeats than expected, depending on the site of recombination; such strains were recovered and were designated PMSS1/cagA-MF and PMSS1/cagA-ML, on the basis of the construct used for transformation. Notably, the resulting PMSS1/cagA-MF and PMSS1/cagA-ML strains can still undergo further recombination due to the remaining direct repeats of CHA-u, CHA-ud, CHA-d, and the cagA ORF. Transformants obtained with the three constructs were screened using seven PCRs, PCR a to PCR g (Fig. 2A and B; see also Fig. S2): PCR a was used to identify the first 5′ cagA; PCR b to identify a cat gene inserted at the first 5′ cagA location; PCR c to identify the cat gene inserted at the last 3′ cagA location; PCR d to identify the last 3′ cagA; PCR e to identify the deletion of all cagA genes; PCR f to identify multiple copies of cagA; and PCR g to detect the presence of any copies of cagA and thus to confirm total deletion of cagA. After screening, two transformants of PMSS1/cagA-SF (SF-1 and SF-2), four of PMSS1/cagA-MF (MF-1 to MF-4), four of PMSS1/cagA-SL (SL-1 to SL-4), two of PMSS1/cagA-ML (ML-1 and ML-2), and three of PMSS1ΔcagAFL (ΔcagAFL-1 to ΔcagAFL-3) were recovered (see Fig. S2). To confirm the results of the PCR screen, real-time PCR was performed to determine the cagA copy numbers (Fig. 2C). SF-1, SF-2, SL-1, SL-2, SL-3, and SL-4 contained approximately one copy of cagA (1.1 ± 0.1, 1.1 ± 0.1, 1.0 ± 0.1, 1.0 ± 0.2, 1.0 ± 0.1, and 1.0 ± 0.3, respectively). MF-1, MF-2, MF-3, and MF-4 contained 3.1 (± 0.3), 4.2 (± 0.2), 3.9 (± 0.1), and 1.9 (± 0.2) copies, and ML-1 and ML-2 contained 3.3 (± 0.1) and 3.4 (± 0.4) copies, respectively, indicating the presence of multiple copies of cagA. Strains SF-1, SL-2, MF-3, ML-1, and ΔcagAFL-2 were chosen for further characterization; the cag region was sequenced to confirm the expected recombination events.
PCR results of seven PCRs to screen mutant strains of PMSS1/cagA-SF, cagA-MF, cagA-SL, cagA-ML, and ΔcagAFL. (A) Results of PCRs a to f, which classified the strains into 5 groups: cagA-SF, cagA-MF, cagA-SL, cagA-ML, and ΔcagAFL, are shown. (B) Results of PCR g using primers F and R to confirm the absence of the cagA gene. Download Figure S2, TIF file, 1.2 MB (1.2MB, tif) .
Copyright © 2017 Jang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
More copies of cagA increase CagA expression and CagA phosphorylation and virulence phenotypes.
We hypothesized that increased numbers of copies of cagA would result in increased expression of CagA. Therefore, expression of CagA by PMSS1 and the SF-1, SL-2, MF-3, ML-1, and ΔcagAFL-2 transformants was measured, first within the bacterial cell and subsequently in a cell culture infection model. The levels of CagA and UreA were measured by Western blotting (Fig. 3), and strain SF-1 was used as a reference strain because the insertion of cat downstream of cagA resulted in a strain that carried a single copy of cagA (Fig. 2B). Thus, the CagA/UreA ratio of SF-1 was set as 1 and used for normalization of the other samples. Using this strategy, the relative expression level of CagA in strain SL-2 was 0.7 (± 0.1), which may indicate a slight polar effect due to the presence of the cat cassette upstream of cagA. In contrast, the relative expression levels of CagA in PMSS1, MF-3, and ML-1 were 3.1 (± 0.2), 2.2 (± 0.0), and 2.1 (± 0.1), respectively (Fig. 3A and B). These data suggest that strains of H. pylori that contain multiple copies of cagA express and produce more CagA than strains that contain only a single copy of the gene.
FIG 3 .

Relative levels of CagA protein and CagA phosphorylation. (A) The protein levels of CagA and UreA in lysates of H. pylori strains PMSS1, SF-1, SL-2, MF-3, and ML-1 were measured by Western blotting (upper panel). For each lysate, 0.5, 1, 1.5, and 2 μg of total protein were used to determine standard curves for CagA and UreA. The immunoblot images were analyzed using ImageJ software, and the values were plotted on a graph (lower panel). (B) Ratios of CagA to UreA were calculated, and each value was normalized to the value calculated for cagA-SF-1 to determine relative CagA protein levels. The bar graphs indicate average levels of CagA expression of each strain, and error bars represent standard deviations, derived from results of 2 independent experiments. (C) Lysates of AGS cells that were infected with H. pylori strains PMSS1, ΔcagAFL-2, SF-1, SL-2, MF-3, and ML-1 were immunoblotted for phosphorylated CagA (p-CagA), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), CagA, and UreA. GAPDH and UreA were used as controls.
To measure CagA expression in a cell culture infection model, the gastric adenocarcinoma cell line AGS was infected with strains PMSS1, SF-1, SL-2, MF-3, ML-1, and ΔcagAFL-2, and the levels of total and phosphorylated CagA were measured by Western blotting (Fig. 3C). As expected, and consistent with the deletion of all copies of cagA from this strain, no CagA or phosphorylated CagA was detected with the ΔcagAFL-2 strain. In contrast, for strains MF-3, ML-1, and PMSS1, which each contained multiple copies of cagA, more phosphorylated and more total CagA was detected than was seen with strains SF-1 and SL-2. Importantly, CagA is phosphorylated only by mammalian host cell kinases; thus, more CagA is produced and successfully translocated by H. pylori strains carrying multiple copies of cagA.
After translocation into mammalian cells, CagA causes host cell elongation and induces expression and secretion of IL-8. To assess the effect of cagA copy number on these virulence phenotypes, AGS cells were again infected with strains PMSS1, SF-1, SL-2, MF-3, ML-1, and ΔcagAFL-2 (Fig. 4). Cell elongation was measured by assessing the length/breadth ratio of 100 random AGS cells on micrographs. Significantly different levels of cell elongation were found among the groups (F[6, 693] = 38.44, P < 0.001, where we report degrees of freedom as F[between-groups, within-groups]). Strain PMSS1 significantly induced cell elongation compared with noninfected control cells (Mock, Fig. 4A). The average length/breadth ratio (± SD) of the mock control was 2.29 (± 0.70), while the ratio calculated for PMSS1 was 5.01 (± 2.88) (Fig. 4B). Similarly to the mock infection, strain PMSS1ΔcagAFL-2 exhibited a ratio of 2.40 (± 0.67). Thus, deletion of cagA resulted in loss of cell elongation. SF-1 and SL-2, which each contained a single copy of cagA, induced cell elongation ratios of 3.39 (± 2.04) and 3.26 (± 1.45), respectively. These ratios were statistically significantly different from those determined for the mock infection and strain ΔcagAFL-2 groups. Strains PMSS1, MF-3, and ML-1, which each contained multiple copies of cagA, induced significantly higher levels of cell elongation (5.01 [± 2.88], 4.92 [± 2.37], and 5.09 [± 2.52], respectively) than all other strains. Thus, H. pylori strains containing multiple copies of cagA are able to induce higher levels of cell elongation.
FIG 4 .

Cell elongation and IL-8 secretion in AGS cells infected by H. pylori. (A) AGS cells were infected with H. pylori strain PMSS1, ΔcagAFL-2, SF-1, SL-2, MF-3, or ML-1 at a MOI of 100 for 8 h. Micrographs were obtained under ×200 magnification. Cell elongation was calculated as the ratio of length to breadth of a cell. An example of these measurements is depicted in the figure (lower right image). (B) The cell elongation induced by each H. pylori strain was graphed using box plots. Thick center lines represent medians; box limits indicate the 25th and 75th percentiles; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles; and outliers are represented by open-circle dots. n = 100 for each group. *, P < 0.05 (compared to the results of both the mock-infected and ΔcagAFL-2 groups); #, P < 0.05 (compared to the results obtained for both the cagA-SF-1 and cagA-SL-2 groups). (C) AGS cells were infected with H. pylori strain PMSS1, ΔcagAFL-2, SF-1, SL-2, MF-3, or ML-1 at a MOI of 10 for 5 h or 30 h. Secretion of IL-8 was measured at 5 h and 30 h postinfection. The bar graphs indicate average levels of IL-8 secretion of AGS cells infected with each strain, and error bars represent standard deviations, derived from results of 3 independent experiments. *, P < 0.05 (compared to the results from the mock-infected group); #, P < 0.05 (compared to the results obtained with both cagA-SF-1 and cagA-SL-2 groups).
Next, the levels of secreted IL-8 were measured in AGS cells infected with strain PMSS1, SF-1, SL-2, MF-3, ML-1, or ΔcagAFL-2 (Fig. 4C). Infections were carried out for 5 h and 30 h. Due to the presence of the T4SS, all strains, including strain ΔcagAFL-2, significantly induced IL-8 expression compared to the mock-infected control. As expected, since CagA-dependent effects on IL-8 secretion occur at later time points (34), there were no significant differences among any of the six infected groups at 5 h (F[6, 14] = 127.86, P < 0.001). In contrast, at 30 h, CagA-dependent effects were more evident (F[6, 14] = 266.75, P < 0.001) (Fig. 4C). While strains cagA-SF-1 and cagA-SL-2 induced higher levels of IL-8 than strain ΔcagAFL-2, the difference did not reach statistical significance. However, each of the strains that contained multiple copies of cagA (strains PMSS1, MF-3, and ML-1) induced significantly larger amounts of IL-8 than strains ΔcagAFL-2, SF-1, and SL-2. Therefore, strains of H. pylori containing multiple copies of cagA are able to induce significantly higher levels of IL-8 expression. En masse, these data suggest that carriage of multiple copies of cagA leads to increased expression and translocation of the CagA toxin, which subsequently results in more pronounced virulence phenotypes.
Dynamic variation of cagA repeat numbers in PMSS1 H. pylori.
To begin to address the mechanism by which the PMSS1 strain came to carry multiple copies of cagA, we next sought to determine whether the PMSS1 population is homogeneous or heterogeneous in terms of cagA copy number. To this end, several hundred single colonies of PMSS1 were isolated and the cagA copy number for each was determined using colony PCR (Table 1; see also Fig. S3). This method was chosen to eliminate the additional culture time required for traditional genomic DNA isolation, as we anticipated that the number of cagA repeats could change temporally during culture. As shown in Fig. S3, five colonies that had a single cagA gene, as determined by real-time PCR, also generated a faint amplicon using PCR h; this suggests the presence of multiple cagA repeats (see Fig. S3). Furthermore, colonies that contained a single cagA gene or multiple cagA repeats, as determined by real-time PCR, also generated a strong amplicon using PCR i; this indicates the presence of cagA. However, these colonies also showed a faint amplicon using PCR j, which detected the ΔcagA deletion (Fig. S3). These results suggest that even during single-colony growth, the members of a minor population of H. pylori undergo a change in cagA copy number, forming a heterogeneous population. For downstream analysis of these colonies, the major population was used to assign the cagA gene number of the colony. Overall, among 389 first-passage colonies, 333 (85.6%) colonies carried multiple copies of cagA, 54 (13.8%) colonies carried a single cagA gene, and 2 (0.5%) colonies carried no cagA. Thus, the PMSS1 population was heterogeneous.
TABLE 1 .
Copy number of cagA in single colonies isolated from PMSS1, 1-107 (cagA-S), and 1-100 (cagA-M4)
| Original strain | No. (%) of isolates in indicated cagA copy number category |
||
|---|---|---|---|
| Multiple | Single | None | |
| PMSS1 | 333 (85.6) | 54 (13.8) | 2 (0.5) |
| 1-107 (cagA-S) | 1 (0.5) | 190 (99.5) | 0 (0.0) |
| 1-100 (cagA-M4) | 185 (98.4) | 3 (1.6) | 0 (0.0) |
Genotypic analysis of multiple copies of cagA in single-colony derivatives of PMSS1 using colony PCR and colony real-time PCR. (A) Three PCRs, named h to j, were used for this screen; the primers are listed in Table S1. PCR h was used to detect multiple cagA repeats (putative amplicon, 1,643 bp); PCR i was used to detect the cagA gene (putative amplicon, 540 bp); and PCR j was used to confirm the absence of cagA (putative amplicon, 851 bp). (B) Representative results from the colony PCR are shown. h, detection of multiple cagA repeats; i, detection of the cagA gene; j, confirmation of the absence of the cagA gene. (C) Relative cagA copy numbers were determined by colony real-time PCR. Download Figure S3, TIF file, 0.4 MB (404.6KB, tif) .
Copyright © 2017 Jang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
As mentioned previously (Fig. 1A), the PCR method used to type the strains was unable to differentiate between two copies of cagA and more copies. Thus, a subset of the colonies were also analyzed by real-time PCR and categorized into strains carrying no (strain ΔcagA), one (strain cagA-S), two (strain cagA-M2), three (strain cagA-M3), and four (strain cagA-M4) copies of cagA (data not shown). On the basis of this categorization, strains 1-89 (ΔcagA), 1-107 (cagA-S), 1-14 (cagA-M2), 1-77 (cagA-M3), and 1-100 (cagA-M4) were each selected for further analysis. First, the cagA region of each strain was sequenced. As expected, strains 1-89 (accession no. KX673186) and 1-107 (accession no. KX673185) were shown to carry no and one copy of cagA, respectively. The cagA repeats of strains 1-14, 1-77, 1-100, and PMSS1 were identical to each other and could not be distinguished by PCR and sequencing. Next, the PMSS1 strain and these other strains were used for Southern blot analysis performed with a probe that was specific for CHA-ud (Fig. 5A and C). As expected on the basis of predicted restriction patterns (Fig. 5C), bands were detected at 0.7 kbp for strain 1-89 (ΔcagA) and 5.8 kbp for strain 1-107 (cagA-S). Similarly, bands were detected at 10.8, 15.9, and 21.0 kbp for strains 1-14, 1-77, and 1-100, respectively. Of note, the PMSS1 strain showed detectable bands at 21.0 kbp, 15.9 kbp, and (faintly) 10.8 kbp, which correspond to the sizes expected for carriage of 4, 3, and 2 copies of cagA, respectively (indicated as cagA-M4, cagA-M3, and cagA-M2 in Fig. 5C). Thus, the Southern blot data further support the idea that the PMSS1 population is heterogeneous.
FIG 5 .

Identification of cagA copy number using Southern blot analysis. (A) Southern blot (upper panel) and real-time PCR (lower panel) analysis of PMSS1 and of single-colony derivatives 1-89, 1-107, 1-14, 1-77, and 1-100 at first passage from PMSS1. Control DNA used in the Southern blot was a mixture of a 3.5-kbp fragment that was linearized by SphI digestion of a pGEM clone of the hybridization probe and a 0.5-kbp fragment liberated by digestion of the same clone with EcoRI. Strain cagA-SF-1 was used for normalization of real-time PCR. The bar graphs indicate average cagA copy numbers, and error bars represent standard deviations, derived from results of 3 independent experiments. (B) Southern blot (upper panel) and real-time PCR (lower panel) analysis of the 1-107 and 1-100 colonies at first passage and of the single-colony derivatives 2-107-69 and 2-107-51 and single-colony derivatives 2-100-18 and 2-100-188 at second passage. (C) Schematic representation of the cagA repeats of PMSS1 derivatives cagA-M4, cagA-M3, cagA-M2, cagA-S, and ΔcagA. Three CHAs (CHA-ud, CHA-u, and CHA-d), SspI cleavage sites, and probe binding sites are indicated.
We next investigated whether the cagA copy number could dynamically change from a single copy to multiple copies and vice versa. To this end, strains 1-107 (cagA-S) and 1-100 (cagA-M4) were each passaged a second time, and the second-passage colonies were screened by PCR to detect any change in cagA copy number (Table 1; see also Fig. 5B). A total of 191 second-passage colonies from parental strain 1-107 (cagA-S) were screened. Of these colonies, one (0.5%, colony 2-107-51) changed from a single copy to multiple copies of cagA, while 190 (99.5%) colonies retained a single copy of cagA. On the other hand, of 188 second-passage colonies from parental strain 1-100 (cagA-M4), three colonies (1.6%) (for example, colony 2-100-188) changed to a single copy of cagA, while 185 colonies (98.4%) (for example, colony 2-100-18) retained multiple copies of cagA. Southern blot analysis revealed that strain 2-107-51 carried two copies of cagA, while strains 2-100-188 and 2-100-18 carried four copies and one copy of cagA, respectively. Thus, the cagA gene in PMSS1 is capable of dynamically changing copy number. We note that among the 379 second-passage colonies that we analyzed from both 1-107 (cagA-S) and 1-100 (cagA-M4) parental colonies, we did not identify any H. pylori isolates without any copies of the cagA gene. This may indicate that the recombination events required to generate this complete-loss genotype occur at a low frequency.
Clinical isolates possess multiple copies of cagA.
To validate that the novel finding of multiple copies of cagA in PMSS1 was relevant to other H. pylori strains, we next screened a large collection of clinical isolates that originated from South Korea and the United States (23, 41, 58–60). The presence of multiple copies of cagA was assessed by PCR as described for Fig. 1A, and the distribution of the results is shown in Table 2 (see also Table S2). Of 234 South Korean H. pylori isolates previously shown to contain cagA (23), 219 strains were positive for cagA using the F and R primers. All 219 isolates carried a single copy of the gene. In contrast, of the 80 cagA-containing United States H. pylori isolates, six (7.5%) strains harbored multiple copies of cagA. The differences in the associations between multiple copies of cagA and the geographical origin of the H. pylori isolates were significant (Fisher exact test, P < 0.001). The cagA repeats from these six H. pylori strains and from the 7.13 strain, which our previous data indicated might contain multiple copies of cagA (Fig. 1B and D), were sequenced next (GenBank accession no. KX673187 to KX673193); the results are schematized in Fig. 6A. For comparison, the previously published genome sequences of H. pylori strains G27, 26695, and J99 (GenBank accession no. CP001173, AE000511, and AE001439, respectively) were used for sequence analysis and are depicted in Fig. 6B. As expected, strain 7.13 contained multiple cagA repeats. Additionally, the number of examples and organization of CHA-u, CHA-ud, and CHA-d in five of the six H. pylori clinical strains (B128, B140, J166, B130A, and B125A) and in strain 7.13 were the same as in PMSS1; moreover, CHA-u, CHA-ud, and CHA-d were highly homologous to those found in PMSS1 (see Fig. S4). The finding of conservation suggests that this is a general pattern for the presence of multiple cagA repeats. Unfortunately, the sequence upstream of cagA could not be obtained for the sixth clinical isolate, B147. This may have been due to gene rearrangement of the 5′ cagA region. Of note, all three CHAs were present in G27, 26695, and J99 (Fig. 6B); however, while CHA-ud was highly homologous to the CHA-ud from PMSS1, it was not found in duplicate in these strains and existed only downstream of cagA. In addition, CHA-u and CHA-d showed a decreased level of homology to the CHAs found in PMSS1 (see Fig. S4). Thus, our overall analysis reveals that carriage of multiple copies of cagA is a phenomenon that is generalizable across a subset of laboratory strains and clinical isolates of H. pylori.
TABLE 2 .
Association between geographical origin of H. pylori strains and presence of multiple cagA repeats
| Geographical origin | No. (%) of isolates in indicated cagA repeat category (n = 299) |
P value | |
|---|---|---|---|
| Multiple | Single | ||
| South Korea | 0 (0) | 219 (100) | <0.001 |
| United States | 6 (7.5) | 74 (92.5) | |
FIG 6 .
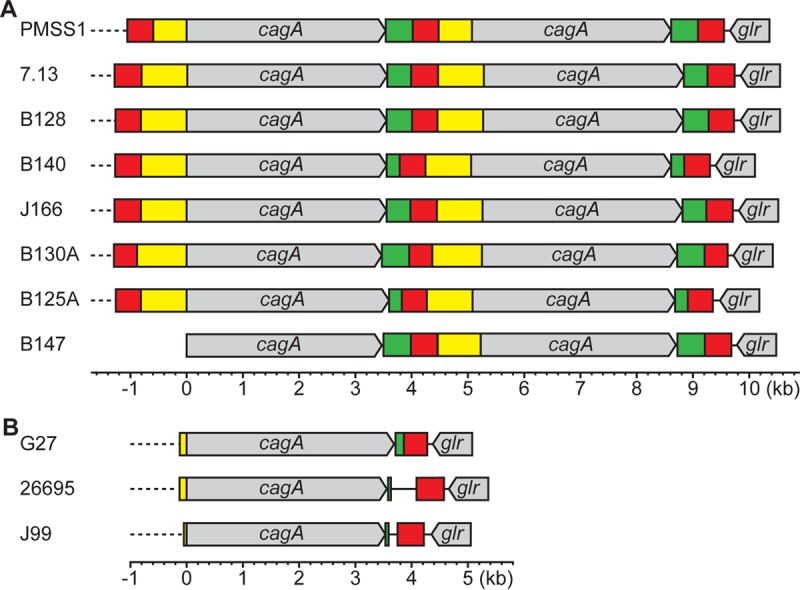
Comparison of multiple copies of cagA in clinical isolates. (A) Schematic representation of the cagA repeats in PMSS1 and 7.13 and in six clinical isolates containing multiple cagA repeats. In strain B147, the region upstream of cagA could not be sequenced. Color categorization was made according to sequence similarity to PMSS1. The three repeating cagA homologous areas (CHAs), CHA-ud (red), CHA-u (yellow), and CHA-d (green), are shown. (B) Schematic representation of the cagA repeats in G27, 26695, and J99.
cagA EPIYA polymorphism, vacA s/i/m polymorphism, homA/B genotype, babA/B/C genotype, and cagA repeat genotype of 80 American H. pylori strains. Download Table S2, XLSX file, 0.02 MB (22KB, xlsx) .
Copyright © 2017 Jang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Comparison of CHA sequences among cagA repeats. The DNA sequences were aligned and compared using the AlignX program (Invitrogen). (A) The full-length CHA-u sequences from PMSS1, 7.13, B128, B140, J166, B130A, and B125A were aligned (upper panel). The same regions from G27, 26695, and J99 were aligned to CHA-u from PMSS1 (lower panel). (B) The full-length CHA-ud sequences from PMSS1, 7.13, B128, B140, J166, B130A, B125A, G27, 26695, and J99 were aligned. (C) The full-length CHA-d sequences from PMSS1, 7.13, B128, B140, J166, B130A, and B125A were aligned (upper panel). The same regions from G27, 26695, and J99 were aligned to CHA-d from PMSS1 (lower panel). Download Figure S4, TIF file, 2.8 MB (2.9MB, tif) .
Copyright © 2017 Jang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Association between cagA copy number, genotype, and gastric disease.
The collection of H. pylori clinical isolates from the United States was previously characterized for several genotypic variations: cagA EPIYA polymorphism, vacA s/i/m polymorphism, homA/B genotype, and babA/B/C genotype (see Table S2 in the supplemental material) (59). Given our data that suggested that increased cagA copy numbers lead to increased pathogenesis (Fig. 4), we next assessed whether there were any epidemiological associations between the multiple cagA copies and the available strain and patient data (Table 3; see also Table S3). Despite the fact that all six isolates harboring multiple copies of cagA were from white patients, there was no significant association between copies of cagA and the patient’s ethnic group. However, there was a significant association of multiple cagA repeats with occupancy of bab locus C; this was true both when the locus was measured as simply occupied or empty and when it was measured as occupied by babA, babB, or babC or was empty. Of the six multicopy cagA isolates, three (50%) strains carried a bab paralog at locus C. In comparison, locus C was occupied in only 5 (7%) of the 74 single-copy isolates. This may suggest that there is a functional association between the presence of a bab paralog at locus C and the presence of multiple cagA repeats.
TABLE 3 .
Significant associations of multiple cagA copies with genotypic variations and disease state
| Parameter compared with cagA repeat genotypea (n = 80) | P value |
|---|---|
| EPIYA-AB or EPIYA-ABC or EPIYA-ABCC or other | 0.013 |
| Empty or babA or babB or babC at bab locus C | 0.005 |
| Empty or occupied at bab locus C | 0.012 |
| Cancer or Barrett’s esophagus or gastric ulcer or duodenal ulcer or gastritis or esophagitis | 0.036 |
| Cancer, Barrett’s esophagus, and gastric ulcer or duodenal ulcer and gastritis or esophagitis | 0.024 |
| Cancer and gastric ulcer or duodenal ulcer and gastritis or Barrett’s esophagus and esophagitis | 0.016 |
The cagA repeat genotype was categorized as single or multiple cagA repeats.
Associations of multiple cagA copies with ethnic groups, genotypic variations, and disease state. Download Table S3, XLSX file, 0.01 MB (10.8KB, xlsx) .
Copyright © 2017 Jang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Particular cagA polymorphisms are known to be associated with more severe disease outcomes (23, 60, 61). Furthermore, we found a significant association between multiple copies of cagA and EPIYA motif polymorphism (Table 3). Among the six isolates that contained multiple copies of cagA, two isolates carried EPIYA-AB and four isolates carried EPIYA-ABC. Notably, while two of the three isolates containing EPIYA-AB had multiple copies of cagA, none of the 27 strains carrying EPIYA-ABCC carried more than one copy of cagA.
Finally, the association between multiple copies of cagA and disease state was assessed (Table 3). The six isolates containing multiple cagA repeats were from patients diagnosed with gastric ulcer (n = 3), duodenal ulcer (n = 1), and esophagitis (n = 2). Despite the small number of strains carrying multiple copies of cagA, the distribution of single or multiple copies analyzed on the basis of individual disease state was significant (Table 3). Significant associations were also seen when various disease states were grouped on the basis of combinations of diseases that reflect differences in severity and/or anatomical location (Table 3). Three of the 6 multiple-copy cagA strains were from patients with gastric ulcers, of which there were only 10 in total within the entire collection. Furthermore, the fact that no strains harboring multiple copies of cagA were found among the large number of gastritis patients likely influences the observed statistical associations. En masse, these data suggest that the presence of multiple copies of cagA may impact the development of gastric disease.
DISCUSSION
Despite the presence of 84 completely sequenced and assembled H. pylori genomes in the NCBI database, no strains of H. pylori have been shown to carry identical copies of cagA, which encodes what is arguably the most-studied virulence factor of this pathogen. However, here we describe the novel finding that some strains of H. pylori harbor multiple tandem copies of cagA. Among the strains that carry multiple cagA copies, PMSS1 was originally isolated from a patient with a duodenal ulcer and has recently been widely studied since it can persistently colonize mice; this provides an invaluable tool to study gastric disease development in an animal model (52). Our data indicate that the PMSS1 strain represents a heterogeneous population where individual bacterial cells carry between zero and four copies of cagA (Fig. 5A). At the population level, real-time PCR analysis indicated that PMSS1 contained an average of 3.7 copies of cagA (Fig. 1D). Characterization of individual isolates that were engineered to contain various numbers of cagA repeats (PMSS1/cagA-SF and PMSS1/cagA-SL) showed that both the 5′ and 3′ cagA genes functionally expressed CagA (Fig. 3A). Furthermore, DNA sequence analysis showed that in strains containing multiple copies of cagA, these genes existed as identical tandem direct repeats that also contained three CHA types (CHA-u, CHA-ud, and CHA-d) (Fig. 1C). Remarkably, the cagA repeats in PMSS1 were able to expand and contract during culture in rich media (Fig. 5B); thus, even in vitro, cagA copy number can undergo dynamic change.
The existence of multiple copies of cagA led us to hypothesize that these strains would express more CagA, deliver more CagA to host cells, and, therefore, show increased levels of CagA-dependent virulence. Indeed, when the amount of CagA produced by PMSS1/cagA-SF-1, which contained a single copy of cagA, was set to 1, the relative CagA expression levels of strains harboring increasing copies of cagA were proportional, though not one to one. Specifically, the level of CagA expressed by strain PMSS1, with 3.7 copies of cagA, was 3.1; that expressed by strain PMSS1/cagA-MF-3, with 3.9 copies, was 2.2; and that expressed by strain PMSS1/cagA-ML-1, with 3.3 copies, was 2.1 (Fig. 3A). Furthermore, the cagA copy number was proportional to the levels of the CagA-dependent virulence phenotypes that were induced: cell elongation and late induction of IL-8. In sum, increased copies of cagA led to more CagA expression and, subsequently, to increased virulence.
Though the exact mechanism for expansion and contraction of the cagA repeats is not yet clear, an analysis of the DNA sequences of strains PMSS1ΔcagA and PMSS1/cagA-S allows us to hypothesize a potential mechanism by which this could occur. Previous studies showed that when a gene is flanked by direct repeats, the copy number of the gene can increase or decrease due to recombination between the repeats (57, 62–65). Therefore, it is likely that CHA-ud plays a critical and unique role in the generation of multiple cagA repeats from a homogeneous population containing only a single cagA repeat. This assertion is based on the fact that only CHA-ud exists as two copies in strain PMSS1/cagA-S; this type of repetition is necessary for standard homologous recombination to duplicate a gene. Additionally, CHA-ud is the only CHA that remained in the PMSS1ΔcagA strain. It is worth noting that a change from the PMSS1ΔcagA population to a PMSS1/cagA-S or PMSS1/cagA-M population could not happen if the PMSS1ΔcagA population were homogeneous. Given that CHA-ud is highly conserved among H. pylori strains (see Fig. S4) and that coinfection with multiple strains of H. pylori occurs clinically (66–69), this raises the intriguing issue of whether interstrain recombination at the cagA region occurs; this issue remains to be addressed.
Moreover, expansion to multiple repeats was not detected in the PMSS1/cagA-SF-1 and -SL-2 mutant strains. This is likely because generation of these strains left only a single copy each of CHA-ud, CHA-u, cagA, and CHA-d. We do note that, despite our hypothesis that CHA-ud is the central player in cagA change, in H. pylori cagA-M, all of the CHAs (CHA-u, CHA-d, and CHA-ud) and cagA are present at least in duplicate; thus, any of these could formally be involved in duplication or deletion of the cagA repeats. The frequency of recombination between repeats is proportional to the number (70, 71) and length (72) of the repeats. Our data indicated that approximately 1.6% of the PMSS1/cagA-M4 population recombined into the PMSS1/cagA-S population during one passage. However, only 0.5% of the PMSS1/cagA-S population recombined to form the PMSS1/cagA-M population, suggesting that this event is rarer. In PMSS1/cagA-S, only CHA-ud is present as a tandem repeat in a manner that could facilitate the recombination process. In contrast, PMSS1/cagA-M4 possesses four copies of cagA, four of CHA-u, four of CHA-d, and five of CHA-ud, all of which could possibly be involved in the recombination event. If this were the case, this would explain why the recombination rate in the PMSS1/cagA-M4 population was higher than that that seen in the PMSS1/cagA-S population. It is worth noting that the recombination frequency was measured in rich media and thus that the frequency may be different in in vitro cell culture or under in vivo conditions. Clearly, the exact mechanism of change for cagA copy numbers remains to be elucidated.
We analyzed the 84 complete H. pylori genomes but were not able to identify any strains that were shown to carry tandem repeats of cagA. We did note that two genome sequences, Shi470 and v225d (GenBank accession numbers CP001072 and CP001582, respectively), did show carriage of two cagA genes at two distant loci. However, the CagA copies in Shi470 shared only 85% homology and one of the cagA sequences in v225d was predicted to be a truncated pseudogene (73). Thus, our work is the first to suggest the presence of multiple functional copies of CagA in H. pylori. We do note that since we were able to readily identify numerous strains containing multiple repeats of cagA, it is highly likely that their occurrence had been previously missed in standard genome sequencing projects; this is due to the average length obtained by most high-throughput sequencing technologies combined with assembly programs that are designed to obtain a single “best fit” contig. This issue could be overcome either by technological advancements that allow greater read lengths or by significantly increasing the sequencing depth of the genome. The latter option would then require analysis of the data to identify areas showing increased sequencing coverage, which could indicate gene duplication. Indeed, this sequencing based strategy has concurrently been used to identify multiple copies of cagA in recently sequenced H. pylori strains (86). It will be of interest to see if new genome sequences generated by future DNA sequencing applications will be able to identify strains carrying multiple cagA repeats.
Among the relatively small number (n = 80) of United States clinical isolates that we analyzed, 7.5% were shown to contain multiple copies of cagA. In contrast, none of the 219 South Korean H. pylori isolates that we analyzed carried more than one copy of cagA. Numerous previous studies have shown that there are distinct genetic differences between H. pylori strains isolated in Western countries, including the United States, and those isolated in East Asian countries, including South Korea (23, 41, 58–60, 74, 75). Thus, this difference in cagA copy numbers between the South Korean and United States H. pylori isolates may not be surprising. The strains containing multiple cagA copies within this collection possessed various virulence factor polymorphisms and genotypes: cagA EPIYA polymorphism, vacA s/i/m polymorphism, and homA/B and bab genotypes (see Table S2). This indicates that these strains are not closely related to each other. Thus, to identify the origin of the multiple cagA copies, further future genomic analyses will be necessary. Perhaps harboring multiple copies of cagA may be a strategy by which H. pylori adapts to more diverse hosts within the population. Within this realm, it is worth mentioning that the two laboratory strains of H. pylori (PMSS1 and 7.13) that we showed to carry multiple copies of cagA are both animal-colonizing strains (52, 56). Thus, the ability to change the number of cagA copies may also be important for adaptation to new host environments. Clearly, the ability of H. pylori strains to affect genetic diversity plays a critical role in this adaptation process.
Since the cagA EPIYA polymorphism, vacA s/i/m polymorphism, homA/B genotype, and babA/B/C genotype of the 80 H. pylori clinical isolates from the United States were previously identified (see Table S2) (59), we were able to easily investigate whether the presence of multiple copies of cagA had any association with other virulence factors. To this end, we found a positive association with occupancy of bab locus C. Interestingly, Hennig et al. (48) previously reported an association of babA carried at any bab locus with the presence of cagA in United States H. pylori isolates. Furthermore, Kim et al. (59) described an association between the genotype of the presence of the bab gene at locus A and the cagA EPIYA-ABD genotype in H. pylori isolates from Korean and American populations. En masse, these studies suggest that there is a close relationship between the bab and cagA genotypes. The molecular basis for this relationship remains to be elucidated.
The other genotype which was shown to be associated with the presence of multiple cagA copies was the CagA EPIYA type. Multiple copies of cagA appeared only in strains carrying no or single EPIYA-C motifs. Since CagA variants containing an increased number of EPIYA-C motifs are known to correlate with more virulent disease characteristics (15, 24, 25), carriage of multiple copies of cagA in strains containing the AB or ABC motifs might provide a mechanism to compensate for the decreased virulence abilities of those CagA variants. Thus, the dynamic expansion and contraction of cagA copy number may serve as a novel mechanism by which H. pylori modulates gastric disease development.
MATERIALS AND METHODS
Bacterial strains and cultures.
H. pylori strains PMSS1, G27, 26695, J99, and 7.13 and the 15 PMSS1 isogenic mutant strains containing different numbers of cagA repeats were cultured and stored as previously described (76). Briefly, all H. pylori strains were grown on horse blood agar plates supplemented with antibiotics and stored at −80°C until use. Chloramphenicol was added to the horse blood agar plates or liquid culture medium at a concentration of 8 μg/ml for cultures of PMSS1 derivatives that contained the cat cassette. For infection of mammalian cells to measure cell elongation and IL-8 induction and for immunoblot assays, H. pylori strains were prepared as previously described (23), with minor modifications. H. pylori strains were initially cultured in brucella broth (BD, Franklin Lakes, NJ) containing 10% fetal bovine serum (FBS) (Gibco, Grand Island, NY, USA) and 10 μg/ml vancomycin (Duchefa, Haarlem, Netherlands) for 24 h and were then inoculated into new media to obtain an optical density of 0.05 at 600 nm. These cultures were grown for 18 h with shaking at 110 rpm. All H. pylori strains were cultured at 37°C under microaerophilic conditions generated by an Anaeropack-Microaero gas-generating system (Mitsubishi Gas Chemical, Tokyo, Japan).
AGS cell culture.
AGS (ATCC CRL-1739), a human gastric adenocarcinoma epithelial cell line, was maintained in RPMI 1640 (Gibco) media supplemented with 10% FBS, 100 U/ml penicillin, and 100 µg/ml streptomycin (Gibco). Cells were cultured at 37°C in a water-saturated 5% CO2 air atmosphere.
Clinical H. pylori isolates.
A total of 314 clinical H. pylori isolates obtained from South Korea and the United States were used in this study. The 234 South Korean H. pylori clinical isolates were a subset of a collection of South Korean strains used in previous studies (23, 41, 58, 60), and the 80 isolates from the United States were the population described in a previous study (59).
cagA repeat genotyping.
A PCR-based method was designed to identify the presence of multiple cagA repeats and the orientation of multiple cagA genes in H. pylori (Fig. 1A). The primers used for the genotyping of the cagA repeats are listed in Table S1 in the supplemental material. Chromosomal DNA was extracted from H. pylori strains as previously described (77) and was used as a template for PCR. Amplification with primer set F and R was used to determine the presence of the cagA gene (Fig. 1A, panel a). Primers F and R were designed to match a conserved cagA region in H. pylori strains PMSS1, G27, 26695, J99, and 7.13 on the basis of genome sequences available in GenBank. The dF and dR primers are complementary to the R and F primers, respectively. Amplification with just the dF or dR primer was used to determine tail-to-tail or head-to-head orientation of multiple cagA inverted repeats (Fig. 1A, panels b and c), respectively. Amplification with the primer set of dF and dR was used to identify adjacent multiple cagA repeats (Fig. 1A, panel d).
Real-time PCR.
Real-time PCR was conducted to determine numbers of cagA copies in the H. pylori strains. ureA was used as a reference gene to quantify the relative number of repeats of cagA. Specific primers for cagA (RTcagAF and RTcagAR) and ureA (RTureAF and RTureAR) were designed to yield 145-bp and 142-bp amplicons, respectively. Primers used for the real-time PCR are listed in Table S1. The relative numbers of cagA copies were quantified using the 2−ΔΔCT method (78, 79), where −ΔΔCT = ΔCT of target − ΔCT of calibrator; ΔCT = CT of cagA − CT of ureA; and CT = threshold cycle. G27 was used as the calibrator for cagA copy number determinations for the other wild-type strains, and strain cagA-SF-1 was used as the calibrator for the copy number determinations for PMSS1-derived mutant strains and single-colony isolates. The real-time PCR analysis was performed using SYBR Premix Ex Taq (TaKaRa, Kusatsu, Japan) on a 7300 real-time PCR system (Applied Biosystems, Foster City, CA) according to the instructions of the manufacturers. Briefly, samples were initially denatured for 30 s at 95°C and then amplified for 40 cycles of 5 s at 95°C and 31 s at 60°C. The data for the fluorescence signal were collected at the end of the 60°C step in each cycle. Melting curves of amplicons from cagA and ureA were analyzed to exclude amplification of nonspecific products. The amplification efficiencies of cagA and ureA for each strain were determined with a standard curve. The standard curve was generated by applying a 10-fold serial dilution of chromosomal DNA to real-time PCR quantification. The data were analyzed using 7300 System SDS software version 1.4 (Applied Biosystems) and are presented as means ± SD of the results from the indicated replicate.
Generation of PMSS1 cagA isogenic mutant strains.
Three constructs targeting the PMSS1 cagA gene at different positions were generated (Fig. 2). Construct F, which replaces the cagA gene at the last 3′ position with a cat cassette, was made as follows. Amplicons generated from PCRs using a primer set of cagAF-5′F and cagAF-5′RXS and a primer set of cagAF-3′FXS and cagAdownR were fused by splicing by overlap extension (SOE) PCR (80, 81). The fused amplicon contained XhoI and SmaI sites in the overlap-joining region. The fused amplicon was inserted into a pGEM-T Easy vector system (Promega, Madison, WI) by TA cloning. A cat cassette, liberated from plasmid pKJMSH (82) by double digestion with XhoI and SmaI, was ligated with the XhoI-SmaI doubly digested plasmid that contained the fused amplicon. The other two constructs were made with the same process except they used different primers for the SOE PCR. Construct L, which replaces the cagA gene at the first 5′ position with a cat cassette, was made using the primer set of cagAupF and cagAL-5′RXS and the primer set of cagAL-3′FXS and cagAL-3′R. Construct FL, which replaces all cagA genes with a cat cassette, was made using the primer set of cagAupF and cagAL-5′RXS and the primer set of cagAF-3′FXS and cagAdownR. The three constructs were then introduced into PMSS1 by natural transformation (83). Transformants were selected on horse blood agar plates supplemented with 8 µg/ml chloramphenicol, and the double homologous recombination of the construct into single-colony isolates of chloramphenicol-resistant H. pylori was verified by seven PCRs (Fig. 2A and B; see also Fig. S2 in the supplemental material) and DNA sequencing. The primers used to generate and verify the constructs are listed in Table S1. PCR a with primers seqF1 and dR was used to identify the first 5′ cagA, PCR b with primers seqF1 and catR was used to identify the cat gene inserted at the first 5′ cagA location, PCR c with primers catF and seqR12 was used to identify the cat gene inserted at the last 3′ cagA location, PCR d with primers dF and seqR12 was used to identify the last 3′ cagA, PCR e with primers seqF1 and seqR12 was used to identify the deletion of all cagA genes, and PCR f with primers dF and dR was used to identify multiple cagA repeats. Finally, PCR g with primers F and R was used to identify the presence of the cagA gene. The number of cagA genes in each colony was determined by real-time PCR.
Cell elongation assay.
AGS cells were seeded onto 6-well cell culture plates at a density of 4 × 105 cells per well and were then incubated for 1 day at 37°C. At 2 h prior to infection, cells were washed with phosphate-buffered saline (PBS) and the medium was changed to 2 ml of RPMI 1640 containing 2% FBS and no antibiotics. Liquid cultures of H. pylori were resuspended in RPMI 1640 containing 2% FBS, and AGS cells were infected at a multiplicity of infection (MOI) of 100. At 8 h postinfection, cells were fixed with 4% paraformaldehyde. Images of the cells were taken using a CKX41 inverted microscope and a DP20 microscope camera (Olympus, Tokyo, Japan) under ×200 magnification. One hundred cells were randomly selected from each well, and cell elongation was calculated by dividing the length of the longest protrusion of a cell by the breadth of the cell, as previously described (84). The length of the longest protrusion was defined as the length of the line that connects the end of the protrusion and the farthest border of the nucleus of a cell, and the breadth was defined as the diameter of the nucleus perpendicular to the line used to measure the length of the cell (Fig. 4A). The length and the breadth were measured using ImageJ software version 1.47 (National Institutes of Health, Bethesda, MD), and then averaged ratios in each group were statistically analyzed. Values corresponding to the elongation of cells induced by each H. pylori mutant strain are presented as box plots generated using the BoxPlotR website (http://boxplot.tyerslab.com), which utilizes R software to analyze data and generate plots.
IL-8 secretion assay.
AGS cells were seeded onto 6-well cell culture plates at a density of 4 × 105 cells per well and were then incubated for 2 days at 37°C. At 2 h prior to infection, cells were washed with PBS and the medium was changed to 2 ml of RPMI 1640 containing 2% FBS and no antibiotics. Liquid cultures of H. pylori were resuspended in RPMI 1640 containing 2% FBS, and AGS cells were infected at a MOI of 10; the lower MOI, compared to that used for the cell elongation assays, was used to prevent significant levels of cell death during the longer incubation required in the IL-8 assay. The infection was allowed to proceed for 5 h as a means to measure cag PAI-dependent immediate IL-8 secretion and for 30 h as a means to measure CagA-dependent late IL-8 secretion (34). At 5 and 30 h postinfection, 1 ml of cell culture medium was taken and centrifuged at 12,000 × g for 10 min at 4°C, and the supernatant was used for IL-8 enzyme-linked immunosorbent assay (ELISA). Assays were performed using human IL-8 ELISA Max Deluxe (BioLegend, San Diego, CA) following the manufacturer’s instructions, and absorbance was measured using an Epoch microplate spectrophotometer (BioTek, Winooski, VT). Data were presented as means ± SD of results from 3 independent replicates.
Immunoblot assay.
To prepare H. pylori bacterial lysates, 1.5 ml of overnight liquid culture of the various H. pylori strains was pelleted and then lysed with 100 µl of cell lysis buffer (Cell Signaling, Inc., Danvers, MA) supplemented with protease inhibitor cocktail (Roche, Basel, Switzerland). To prepare lysates of infected cells, AGS cells were seeded onto 6-well cell culture plates at a density of 4 × 105 cells per well and were then incubated for 2 days at 37°C. At 2 h prior to infection, cells were washed with PBS and the medium was changed to 2 ml of RPMI 1640 containing 2% FBS and no antibiotics. Liquid cultures of H. pylori were resuspended in RPMI 1640 containing 2% FBS, and AGS cells were infected at a MOI of 100. At 5 h postinfection, cells were washed with PBS and then lysed with 100 µl of cell lysis buffer supplemented with protease inhibitor cocktail. Protein concentrations of bacterial lysates and infected cell lysates were measured using Pierce bicinchoninic acid (BCA) protein assay reagent (Thermo Fisher Scientific, Waltham, MA). The indicated amount of each sample was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to a polyvinylidene fluoride membrane (Merck Millipore, Darmstadt, Germany). To detect CagA, phosphorylated CagA, UreA, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase), membranes were probed by the use of rabbit polyclonal anti-CagA antibody b-300 (Santa Cruz Biotechnology, Dallas, TX), mouse monoclonal anti-phosphotyrosine antibody pY99 (Santa Cruz Biotechnology), rabbit polyclonal anti-UreA antibody b-234 (Santa Cruz Biotechnology), and rabbit polyclonal anti-GAPDH antibody (Koma Biotech, Seoul, South Korea), respectively. These membranes were further probed with goat anti-mouse IgG-horseradish peroxidase (IgG-HRP) (Santa Cruz Biotechnology) or goat anti-rabbit IgG-HRP (Santa Cruz Biotechnology). Antibodies against CagA, phosphorylated CagA, UreA, and GAPDH were diluted to 1:10,000 in 3% bovine serum albumin dissolved in Tris-buffered saline with 0.1% Tween 20 (TBST), and HRP-conjugated antibodies were diluted to 1:10,000 in 3% skim milk dissolved in TBST. Probed membranes were then developed using WesternBright enhanced chemiluminescence-HRP (ECL-HRP) substrate (Advansta, Menlo Park, CA, USA) on X-ray film (Agfa, Mortsel, Belgium). Relative expression levels of CagA were measured as previously described (85). Briefly, developed films were scanned to image files, intensities of blots on the images were measured using ImageJ software version 1.47, and the values were plotted on a graph. A ratio of CagA to UreA was calculated, and each value was normalized to the value of cagA-SF-1 to determine relative CagA protein levels.
DNA sequencing.
Sanger dideoxy DNA sequencing was performed at Cosmo Genetech Co., Ltd. (Seoul, South Korea). The DNA sequencing primers are listed in Table S1. The resulting DNA sequences were analyzed using Vector NTI version 9.1 (Invitrogen, Carlsbad, CA) and Sequencher 5.1 (Gene Codes, Ann Arbor, MI).
Genotyping of H. pylori single colonies by colony PCR.
A culture of PMSS1 and its derivatives was diluted in brucella broth and was plated onto horse blood agar plates. Plates were incubated for 4 days under microaerobic conditions until single colonies appeared, which was considered to represent one passage. Single colonies were picked up and streaked onto a new blood agar plate for further culture. The cultures were used for isolation of genomic DNA and frozen stocks.
Colonies were transferred into 20 μl of Tris-EDTA (TE) buffer (pH 8.0) and then were heated at 99°C for 3 min. The supernatant of the TE buffer after centrifugation was used as the template for both PCR and real-time PCR to analyze cagA copy numbers in single colonies of PMSS1 and its derivatives.
A colony PCR was designed to identify the absence of or the presence of single or multiple cagA genes in colonies. Primers specific to PMSS1 were designed for more efficient and more accurate PCR (Table S1). The primer alignment sites are indicated in Fig. S3A. Three sets of colony PCRs were performed in parallel: PCR h with primers dF2 and dR2 was used to detect the presence of multiple cagA genes; PCR i with primers F2 and R2 was used to detect the presence or absence of cagA; and PCR j with primers cagAupF2 and cagAdownR2 was used to confirm the absence of cagA. PCR h was carried out as follows: a cycle at 94°C for 3 min; 35 cycles of 94°C for 15 s, 54°C for 15 s, and 72°C for 90 s; a final elongation step at 72°C for 10 min. For PCRs i and j, the same PCR parameters were used except a 60-s extension time was used instead of 90 s; this change decreased amplification of the larger (>5.5-kbp) PCR product generated from genomes containing cagA with PCR j. Thus, PCRs i and j generated a 540-bp amplicon to detect cagA and an 851-bp amplicon from a cagA deletion, respectively. A colony real-time PCR was performed to determine cagA copy numbers in the single colonies. The colony-boiled TE buffer supernatant was used as a template. The colony real-time PCR was conducted as described for the real-time PCR.
Southern blot assay.
A total of 0.5 μg of chromosomal DNA from each sample was digested with restriction enzyme SspI (Thermo Fisher Scientific) and resolved on a 0.8% agarose gel for 12 h at 25 V. SspI was used because the SspI site is not found within the cagA repeat region (Fig. 5C). The DNA fragments were transferred from the gel to a positively charged nylon membrane (Roche) via downward capillary action using alkaline transfer. A probe was generated using a PCR digoxigenin (DIG) Probe synthesis kit (Roche) according to the manufacturer’s instructions. Briefly, chromosomal DNA of PMSS1 was used as a template to amplify CHA-ud by PCR using the primer set of probe-F and probe-R (Table S1) and the 462-bp amplicon was cloned into a pGEM-T Easy vector system, resulting in the pGEM probe. The pGEM probe containing the amplicon was purified and used as a DNA template to synthesize the DIG-labeled probe. The probe was designed to target the CHA-ud sequence (Fig. 5C). Prehybridization and hybridization were performed at 45°C for 30 min and overnight in DIG Easy Hyb solution (Roche), respectively. Washing and blocking steps were done using a Wash buffer set and a Block buffer set (Roche), respectively, and the detection step was accomplished using anti-digoxigenin-alkaline phosphatase (AP)/Fab fragments and CSPD chemiluminescence substrate (Roche).
Statistical analysis.
Statistical analyses were performed using the IBM SPSS statistics 23 program (IBM, Armonk, NY). The results of the cell elongation assay and IL-8 induction assay were analyzed by one-way analysis of variance followed by Tukey’s post hoc test. P values were presented with F values and degrees of freedom (for between-group and within-group comparisons). The Fisher exact test was used to analyze the association between the geographical origin of H. pylori clinical isolates and the presence of multiple cagA repeats and between cagA copy number, genotype, and gastric disease. A P value of less than 0.05 was considered to be statistically significant.
Accession number(s).
The sequences for the cagA gene(s) and their flanking regions from 10 strains have been deposited in GenBank under accession no. KX673184 to KX673193.
ACKNOWLEDGMENTS
We thank Richard M. Peek, Jr., and Judith Romero-Gallo (Vanderbilt University) for providing the United States H. pylori clinical isolates, which were obtained from patients at Vanderbilt University Medical Center. We also thank Stephanie Servetas for assistance with statistical analysis.
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (NRF-2014R1A2A1A11051054) (to Jeong-Heon Cha). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
For a companion article on this topic, see https://doi.org/10.1128/mBio.02321-16.
Citation Jang S, Su H, Blum FC, Bae S, Choi YH, Kim A, Hong YA, Kim J, Kim J-H, Gunawardhana N, Jeon Y-E, Yoo Y-J, Merrell DS, Ge L, Cha J-H. 2017. Dynamic expansion and contraction of cagA copy number in Helicobacter pylori impact development of gastric disease. mBio 8:e01779-16. https://doi.org/10.1128/mBio.01779-16.
REFERENCES
- 1.The EUROGAST Study Group 1993. Epidemiology of, and risk factors for, Helicobacter pylori infection among 3194 asymptomatic subjects in 17 populations. Gut 34:1672–1676. doi: 10.1136/gut.34.12.1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marshall BJ, Warren JR. 1984. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet i:1311–1315. doi: 10.1016/S0140-6736(84)91816-6. [DOI] [PubMed] [Google Scholar]
- 3.Matysiak-Budnik T, Mégraud F. 1997. Epidemiology of Helicobacter pylori infection with special reference to professional risk. J Physiol Pharmacol 48(Suppl 4):3–17. [PubMed] [Google Scholar]
- 4.Blaser MJ. 1998. Helicobacter pylori and gastric diseases. BMJ 316:1507–1510. doi: 10.1136/bmj.316.7143.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suerbaum S, Michetti P. 2002. Helicobacter pylori infection. N Engl J Med 347:1175–1186. doi: 10.1056/NEJMra020542. [DOI] [PubMed] [Google Scholar]
- 6.IARC Working Group on the Evaluation of Carcinogenic Risks to Humans 1994. Schistosomes, liver flukes and Helicobacter pylori. IARC Monogr Eval Carcinog Risks Hum 61:1–241. [PMC free article] [PubMed] [Google Scholar]
- 7.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. 2015. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 8.Roesler BM, Rabelo-Gonçalves EM, Zeitune JM. 2014. Virulence factors of Helicobacter pylori: a review. Clin Med Insights Gastroenterol 7:9–17. doi: 10.4137/CGast.S13760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamaoka Y. 2010. Mechanisms of disease: Helicobacter pylori virulence factors. Nat Rev Gastroenterol Hepatol 7:629–641. doi: 10.1038/nrgastro.2010.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nomura AM, Lee J, Stemmermann GN, Nomura RY, Perez-Perez GI, Blaser MJ. 2002. Helicobacter pylori CagA seropositivity and gastric carcinoma risk in a Japanese American population. J Infect Dis 186:1138–1144. doi: 10.1086/343808. [DOI] [PubMed] [Google Scholar]
- 11.Parsonnet J, Friedman GD, Orentreich N, Vogelman H. 1997. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut 40:297–301. doi: 10.1136/gut.40.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Odenbreit S, Püls J, Sedlmaier B, Gerland E, Fischer W, Haas R. 2000. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287:1497–1500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- 13.Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, Hatakeyama M. 2002. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 295:683–686. doi: 10.1126/science.1067147. [DOI] [PubMed] [Google Scholar]
- 14.Higashi H, Nakaya A, Tsutsumi R, Yokoyama K, Fujii Y, Ishikawa S, Higuchi M, Takahashi A, Kurashima Y, Teishikata Y, Tanaka S, Azuma T, Hatakeyama M. 2004. Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J Biol Chem 279:17205–17216. doi: 10.1074/jbc.M309964200. [DOI] [PubMed] [Google Scholar]
- 15.Higashi H, Tsutsumi R, Fujita A, Yamazaki S, Asaka M, Azuma T, Hatakeyama M. 2002. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc Natl Acad Sci U S A 99:14428–14433. doi: 10.1073/pnas.222375399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neel BG, Gu H, Pao L. 2003. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci 28:284–293. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 17.Tsutsumi R, Takahashi A, Azuma T, Higashi H, Hatakeyama M. 2006. Focal adhesion kinase is a substrate and downstream effector of SHP-2 complexed with Helicobacter pylori CagA. Mol Cell Biol 26:261–276. doi: 10.1128/MCB.26.1.261-276.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones KR, Whitmire JM, Merrell DS. 2010. A tale of two toxins: Helicobacter pylori CagA and VacA modulate host pathways that impact disease. Front Microbiol 1:115. doi: 10.3389/fmicb.2010.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee IO, Kim JH, Choi YJ, Pillinger MH, Kim SY, Blaser MJ, Lee YC. 2010. Helicobacter pylori CagA phosphorylation status determines the gp130-activated SHP2/ERK and JAK/STAT signal transduction pathways in gastric epithelial cells. J Biol Chem 285:16042–16050. doi: 10.1074/jbc.M110.111054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suzuki M, Mimuro H, Kiga K, Fukumatsu M, Ishijima N, Morikawa H, Nagai S, Koyasu S, Gilman RH, Kersulyte D, Berg DE, Sasakawa C. 2009. Helicobacter pylori CagA phosphorylation-independent function in epithelial proliferation and inflammation. Cell Host Microbe 5:23–34. doi: 10.1016/j.chom.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 21.Yamaoka Y, Kodama T, Kashima K, Graham DY, Sepulveda AR. 1998. Variants of the 3' region of the cagA gene in Helicobacter pylori isolates from patients with different H. pylori-associated diseases. J Clin Microbiol 36:2258–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen LT, Uchida T, Murakami K, Fujioka T, Moriyama M. 2008. Helicobacter pylori virulence and the diversity of gastric cancer in Asia. J Med Microbiol 57:1445–1453. doi: 10.1099/jmm.0.2008/003160-0. [DOI] [PubMed] [Google Scholar]
- 23.Jones KR, Joo YM, Jang S, Yoo YJ, Lee HS, Chung IS, Olsen CH, Whitmire JM, Merrell DS, Cha JH. 2009. Polymorphism in the CagA EPIYA motif impacts development of gastric cancer. J Clin Microbiol 47:959–968. doi: 10.1128/JCM.02330-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Batista SA, Rocha GA, Rocha AM, Saraiva IE, Cabral MM, Oliveira RC, Queiroz DM. 2011. Higher number of Helicobacter pylori CagA EPIYA C phosphorylation sites increases the risk of gastric cancer, but not duodenal ulcer. BMC Microbiol 11:61. doi: 10.1186/1471-2180-11-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Honarmand-Jahromy S, Siavoshi F, Malekzadeh R, Sattari TN, Latifi-Navid S. 2015. Multiple repeats of Helicobacter pylori CagA EPIYA-C phosphorylation sites predict risk of gastric ulcer in Iran. Microb Pathog 89:87–92. doi: 10.1016/j.micpath.2015.09.005. [DOI] [PubMed] [Google Scholar]
- 26.Loh JT, Friedman DB, Piazuelo MB, Bravo LE, Wilson KT, Peek RM Jr, Correa P, Cover TL. 2012. Analysis of Helicobacter pylori cagA promoter elements required for salt-induced upregulation of CagA expression. Infect Immun 80:3094–3106. doi: 10.1128/IAI.00232-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loh JT, Shaffer CL, Piazuelo MB, Bravo LE, McClain MS, Correa P, Cover TL. 2011. Analysis of cagA in Helicobacter pylori strains from Colombian populations with contrasting gastric cancer risk reveals a biomarker for disease severity. Cancer Epidemiol Biomarkers Prev 20:2237–2249. doi: 10.1158/1055-9965.EPI-11-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferreira RM, Pinto-Ribeiro I, Wen X, Marcos-Pinto R, Dinis-Ribeiro M, Carneiro F, Figueiredo C. 2016. Helicobacter pylori cagA promoter region sequences influence CagA expression and interleukin 8 secretion. J Infect Dis 213:669–673. doi: 10.1093/infdis/jiv467. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki M, Mimuro H, Suzuki T, Park M, Yamamoto T, Sasakawa C. 2005. Interaction of CagA with Crk plays an important role in Helicobacter pylori-induced loss of gastric epithelial cell adhesion. J Exp Med 202:1235–1247. doi: 10.1084/jem.20051027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, Rappuoli R, Covacci A. 1996. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci U S A 93:14648–14653. doi: 10.1073/pnas.93.25.14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crabtree JE, Covacci A, Farmery SM, Xiang Z, Tompkins DS, Perry S, Lindley IJ, Rappuoli R. 1995. Helicobacter pylori induced interleukin-8 expression in gastric epithelial cells is associated with CagA positive phenotype. J Clin Pathol 48:41–45. doi: 10.1136/jcp.48.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fischer W, Püls J, Buhrdorf R, Gebert B, Odenbreit S, Haas R. 2001. Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol Microbiol 42:1337–1348. doi: 10.1046/j.1365-2958.2001.02714.x. [DOI] [PubMed] [Google Scholar]
- 33.Sharma SA, Tummuru MK, Blaser MJ, Kerr LD. 1998. Activation of IL-8 gene expression by Helicobacter pylori is regulated by transcription factor nuclear factor-κB in gastric epithelial cells. J Immunol 160:2401–2407. [PubMed] [Google Scholar]
- 34.Brandt S, Kwok T, Hartig R, König W, Backert S. 2005. NF-κB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci U S A 102:9300–9305. doi: 10.1073/pnas.0409873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Achtman M, Azuma T, Berg DE, Ito Y, Morelli G, Pan ZJ, Suerbaum S, Thompson SA, van der Ende A, van Doorn LJ. 1999. Recombination and clonal groupings within Helicobacter pylori from different geographical regions. Mol Microbiol 32:459–470. doi: 10.1046/j.1365-2958.1999.01382.x. [DOI] [PubMed] [Google Scholar]
- 36.Björkholm B, Sjölund M, Falk PG, Berg OG, Engstrand L, Andersson DI. 2001. Mutation frequency and biological cost of antibiotic resistance in Helicobacter pylori. Proc Natl Acad Sci U S A 98:14607–14612. doi: 10.1073/pnas.241517298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Falush D, Kraft C, Taylor NS, Correa P, Fox JG, Achtman M, Suerbaum S. 2001. Recombination and mutation during long-term gastric colonization by Helicobacter pylori: estimates of clock rates, recombination size, and minimal age. Proc Natl Acad Sci U S A 98:15056–15061. doi: 10.1073/pnas.251396098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kraft C, Suerbaum S. 2005. Mutation and recombination in Helicobacter pylori: mechanisms and role in generating strain diversity. Int J Med Microbiol 295:299–305. doi: 10.1016/j.ijmm.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 39.Suerbaum S. 2000. Genetic variability within Helicobacter pylori. Int J Med Microbiol 290:175–181. doi: 10.1016/S1438-4221(00)80087-9. [DOI] [PubMed] [Google Scholar]
- 40.de Jonge R, Durrani Z, Rijpkema SG, Kuipers EJ, van Vliet AH, Kusters JG. 2004. Role of the Helicobacter pylori outer-membrane proteins AlpA and AlpB in colonization of the guinea pig stomach. J Med Microbiol 53:375–379. doi: 10.1099/jmm.0.45551-0. [DOI] [PubMed] [Google Scholar]
- 41.Kang J, Jones KR, Jang S, Olsen CH, Yoo YJ, Merrell DS, Cha JH. 2012. The geographic origin of Helicobacter pylori influences the association of the homB gene with gastric cancer. J Clin Microbiol 50:1082–1085. doi: 10.1128/JCM.06293-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mahdavi J, Sondén B, Hurtig M, Olfat FO, Forsberg L, Roche N, Ångström J, Larsson T, Teneberg S, Karlsson KA, Altraja S, Wadström T, Kersulyte D, Berg DE, Dubois A, Petersson C, Magnusson KE, Norberg T, Lindh F, Lundskog BB, Arnqvist A, Hammarström L, Borén T. 2002. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 297:573–578. doi: 10.1126/science.1069076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oleastro M, Ménard A. 2013. The role of Helicobacter pylori outer membrane proteins in adherence and pathogenesis. Biology (Basel) 2:1110–1134. doi: 10.3390/biology2031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Talebi Bezmin Abadi A, Taghvaei T, Mohabbati Mobarez A, Vaira G, Vaira D. 2013. High correlation of babA2-positive strains of Helicobacter pylori with the presence of gastric cancer. Intern Emerg Med 8:497–501. doi: 10.1007/s11739-011-0631-6. [DOI] [PubMed] [Google Scholar]
- 45.Yamaoka Y, Kita M, Kodama T, Imamura S, Ohno T, Sawai N, Ishimaru A, Imanishi J, Graham DY. 2002. Helicobacter pylori infection in mice: role of outer membrane proteins in colonization and inflammation. Gastroenterology 123:1992–2004. doi: 10.1053/gast.2002.37074. [DOI] [PubMed] [Google Scholar]
- 46.Yamaoka Y, Ojo O, Fujimoto S, Odenbreit S, Haas R, Gutierrez O, El-Zimaity HM, Reddy R, Arnqvist A, Graham DY. 2006. Helicobacter pylori outer membrane proteins and gastroduodenal disease. Gut 55:775–781. doi: 10.1136/gut.2005.083014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Armitano RI, Matteo MJ, Goldman C, Wonaga A, Viola LA, De Palma GZ, Catalano M. 2013. Helicobacter pylori heterogeneity in patients with gastritis and peptic ulcer disease. Infect Genet Evol 16:377–385. doi: 10.1016/j.meegid.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 48.Hennig EE, Allen JM, Cover TL. 2006. Multiple chromosomal loci for the babA gene in Helicobacter pylori. Infect Immun 74:3046–3051. doi: 10.1128/IAI.74.5.3046-3051.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pride DT, Meinersmann RJ, Blaser MJ. 2001. Allelic variation within Helicobacter pylori babA and babB. Infect Immun 69:1160–1171. doi: 10.1128/IAI.69.2.1160-1171.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawai M, Furuta Y, Yahara K, Tsuru T, Oshima K, Handa N, Takahashi N, Yoshida M, Azuma T, Hattori M, Uchiyama I, Kobayashi I. 2011. Evolution in an oncogenic bacterial species with extreme genome plasticity: Helicobacter pylori East Asian genomes. BMC Microbiol 11:104. doi: 10.1186/1471-2180-11-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Furuta Y, Kawai M, Yahara K, Takahashi N, Handa N, Tsuru T, Oshima K, Yoshida M, Azuma T, Hattori M, Uchiyama I, Kobayashi I. 2011. Birth and death of genes linked to chromosomal inversion. Proc Natl Acad Sci U S A 108:1501–1506. doi: 10.1073/pnas.1012579108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arnold IC, Lee JY, Amieva MR, Roers A, Flavell RA, Sparwasser T, Müller A. 2011. Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterology 140:199–209. doi: 10.1053/j.gastro.2010.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Covacci A, Censini S, Bugnoli M, Petracca R, Burroni D, Macchia G, Massone A, Papini E, Xiang Z, Figura N, Rappuoli R. 1993. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc Natl Acad Sci U S A 90:5791–5795. doi: 10.1073/pnas.90.12.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 55.Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, Smith DR, Noonan B, Guild BC, deJonge BL, Carmel G, Tummino PJ, Caruso A, Uria-Nickelsen M, Mills DM, Ives C, Gibson R, Merberg D, Mills SD, Jiang Q, Taylor DE, Vovis GF, Trust TJ. 1999. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397:176–180. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- 56.Franco AT, Israel DA, Washington MK, Krishna U, Fox JG, Rogers AB, Neish AS, Collier-Hyams L, Perez-Perez GI, Hatakeyama M, Whitehead R, Gaus K, O’Brien DP, Romero-Gallo J, Peek RM Jr.. 2005. Activation of β-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci U S A 102:10646–10651. doi: 10.1073/pnas.0504927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sandegren L, Andersson DI. 2009. Bacterial gene amplification: implications for the evolution of antibiotic resistance. Nat Rev Microbiol 7:578–588. doi: 10.1038/nrmicro2174. [DOI] [PubMed] [Google Scholar]
- 58.Jang S, Jones KR, Olsen CH, Joo YM, Yoo YJ, Chung IS, Cha JH, Merrell DS. 2010. Epidemiological link between gastric disease and polymorphisms in VacA and CagA. J Clin Microbiol 48:559–567. doi: 10.1128/JCM.01501-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim A, Servetas SL, Kang J, Kim J, Jang S, Cha HJ, Lee WJ, Kim J, Romero-Gallo J, Peek RM Jr, Merrell DS, Cha JH. 2015. Helicobacter pylori bab paralog distribution and association with cagA, vacA, and homA/B genotypes in American and South Korean clinical isolates. PLoS One 10:e0137078. doi: 10.1371/journal.pone.0137078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jones KR, Jang S, Chang JY, Kim J, Chung IS, Olsen CH, Merrell DS, Cha JH. 2011. Polymorphisms in the intermediate region of VacA impact Helicobacter pylori-induced disease development. J Clin Microbiol 49:101–110. doi: 10.1128/JCM.01782-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bridge DR, Merrell DS. 2013. Polymorphism in the Helicobacter pylori CagA and VacA toxins and disease. Gut Microbes 4:101–117. doi: 10.4161/gmic.23797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bzymek M, Lovett ST. 2001. Instability of repetitive DNA sequences: the role of replication in multiple mechanisms. Proc Natl Acad Sci U S A 98:8319–8325. doi: 10.1073/pnas.111008398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chédin F, Dervyn E, Dervyn R, Ehrlich SD, Noirot P. 1994. Frequency of deletion formation decreases exponentially with distance between short direct repeats. Mol Microbiol 12:561–569. doi: 10.1111/j.1365-2958.1994.tb01042.x. [DOI] [PubMed] [Google Scholar]
- 64.Corn PG, Anders J, Takala AK, Käyhty H, Hoiseth SK. 1993. Genes involved in Haemophilus influenzae type b capsule expression are frequently amplified. J Infect Dis 167:356–364. doi: 10.1093/infdis/167.2.356. [DOI] [PubMed] [Google Scholar]
- 65.Mekalanos JJ. 1983. Duplication and amplification of toxin genes in Vibrio cholerae. Cell 35:253–263. doi: 10.1016/0092-8674(83)90228-3. [DOI] [PubMed] [Google Scholar]
- 66.Cao Q, Didelot X, Wu Z, Li Z, He L, Li Y, Ni M, You Y, Lin X, Li Z, Gong Y, Zheng M, Zhang M, Liu J, Wang W, Bo X, Falush D, Wang S, Zhang J. 2015. Progressive genomic convergence of two Helicobacter pylori strains during mixed infection of a patient with chronic gastritis. Gut 64:554–561. doi: 10.1136/gutjnl-2014-307345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morales-Espinosa R, Castillo-Rojas G, Gonzalez-Valencia G, Ponce de León S, Cravioto A, Atherton JC, López-Vidal Y. 1999. Colonization of Mexican patients by multiple Helicobacter pylori strains with different vacA and cagA genotypes. J Clin Microbiol 37:3001–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Patra R, Chattopadhyay S, De R, Ghosh P, Ganguly M, Chowdhury A, Ramamurthy T, Nair GB, Mukhopadhyay AK. 2012. Multiple infection and microdiversity among Helicobacter pylori isolates in a single host in India. PLoS One 7:e43370. doi: 10.1371/journal.pone.0043370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Secka O, Antonio M, Berg DE, Tapgun M, Bottomley C, Thomas V, Walton R, Corrah T, Thomas JE, Adegbola RA. 2011. Mixed infection with cagA positive and cagA negative strains of Helicobacter pylori lowers disease burden in The Gambia. PLoS One 6:e27954. doi: 10.1371/journal.pone.0027954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anderson RP, Roth JR. 1977. Tandem genetic duplications in phage and bacteria. Annu Rev Microbiol 31:473–505. doi: 10.1146/annurev.mi.31.100177.002353. [DOI] [PubMed] [Google Scholar]
- 71.Peterson BC, Rownd RH. 1985. Drug resistance gene amplification of plasmid NR1 derivatives with various amounts of resistance determinant DNA. J Bacteriol 161:1042–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aras RA, Kang J, Tschumi AI, Harasaki Y, Blaser MJ. 2003. Extensive repetitive DNA facilitates prokaryotic genome plasticity. Proc Natl Acad Sci U S A 100:13579–13584. doi: 10.1073/pnas.1735481100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mane SP, Dominguez-Bello MG, Blaser MJ, Sobral BW, Hontecillas R, Skoneczka J, Mohapatra SK, Crasta OR, Evans C, Modise T, Shallom S, Shukla M, Varon C, Mégraud F, Maldonado-Contreras AL, Williams KP, Bassaganya-Riera J. 2010. Host-interactive genes in Amerindian Helicobacter pylori diverge from their Old World homologs and mediate inflammatory responses. J Bacteriol 192:3078–3092. doi: 10.1128/JB.00063-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Argent RH, Hale JL, El-Omar EM, Atherton JC. 2008. Differences in Helicobacter pylori CagA tyrosine phosphorylation motif patterns between western and East Asian strains, and influences on interleukin-8 secretion. J Med Microbiol 57:1062–1067. doi: 10.1099/jmm.0.2008/001818-0. [DOI] [PubMed] [Google Scholar]
- 75.Yamaoka Y, Kodama T, Gutierrez O, Kim JG, Kashima K, Graham DY. 1999. Relationship between Helicobacter pylori iceA, cagA, and vacA status and clinical outcome: studies in four different countries. J Clin Microbiol 37:2274–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Carpenter BM, McDaniel TK, Whitmire JM, Gancz H, Guidotti S, Censini S, Merrell DS. 2007. Expanding the Helicobacter pylori genetic toolbox: modification of an endogenous plasmid for use as a transcriptional reporter and complementation vector. Appl Environ Microbiol 73:7506–7514. doi: 10.1128/AEM.01084-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Owen RJ, Bickley J. 1997. Isolation of H. pylori genomic DNA and restriction analysis. Methods Mol Med 8:81–88. doi: 10.1385/0-89603-381-3:81. [DOI] [PubMed] [Google Scholar]
- 78.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 79.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 80.Horton RM, Cai ZL, Ho SN, Pease LR. 1990. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 8:528–535. doi: 10.2144/000114017. [DOI] [PubMed] [Google Scholar]
- 81.Horton RM, Ho SN, Pullen JK, Hunt HD, Cai Z, Pease LR. 1993. Gene splicing by overlap extension. Methods Enzymol 217:270–279. [DOI] [PubMed] [Google Scholar]
- 82.Kim J, Kim SW, Jang S, Merrell DS, Cha JH. 2011. Complementation system for Helicobacter pylori. J Microbiol 49:481–486. doi: 10.1007/s12275-011-1196-9. [DOI] [PubMed] [Google Scholar]
- 83.Israel DA, Lou AS, Blaser MJ. 2000. Characteristics of Helicobacter pylori natural transformation. FEMS Microbiol Lett 186:275–280. doi: 10.1111/j.1574-6968.2000.tb09117.x. [DOI] [PubMed] [Google Scholar]
- 84.Bourzac KM, Satkamp LA, Guillemin K. 2006. The Helicobacter pylori cag pathogenicity island protein CagN is a bacterial membrane-associated protein that is processed at its C terminus. Infect Immun 74:2537–2543. doi: 10.1128/IAI.74.5.2537-2543.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Taylor SC, Berkelman T, Yadav G, Hammond M. 2013. A defined methodology for reliable quantification of Western blot data. Mol Biotechnol 55:217–226. doi: 10.1007/s12033-013-9672-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Draper JL, Hansen LM, Bernick DL, Abedrabbo S, Underwood JG, Kong N, Huang BC, Weis AM, Weimer BC, van Vliet AHM, Pourmand N, Solnick JV, Karplus K, Ottemann KM. 2017. Fallacy of the unique genome: sequence diversity within single Helicobacter pylori strains. mBio 8:e02321–16. doi: 10.1128/mBio.02321-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primers used in this study. Download Table S1, XLSX file, 0.01 MB (13.5KB, xlsx) .
Copyright © 2017 Jang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Real-time PCR standard curves and CT deviation of cagA and ureA in H. pylori strains. The standard curves were calculated by conducting real-time PCR using serial 10-fold dilutions of chromosomal DNA isolated from PMSS1, G27, 26695, J99, and 7.13; concentrations ranged from 10 ng/μl to 0.01 ng/μl. CT deviations (ΔCT) of each strain were calculated and plotted. Download Figure S1, TIF file, 0.2 MB (244.4KB, tif) .
Copyright © 2017 Jang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
PCR results of seven PCRs to screen mutant strains of PMSS1/cagA-SF, cagA-MF, cagA-SL, cagA-ML, and ΔcagAFL. (A) Results of PCRs a to f, which classified the strains into 5 groups: cagA-SF, cagA-MF, cagA-SL, cagA-ML, and ΔcagAFL, are shown. (B) Results of PCR g using primers F and R to confirm the absence of the cagA gene. Download Figure S2, TIF file, 1.2 MB (1.2MB, tif) .
Copyright © 2017 Jang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Genotypic analysis of multiple copies of cagA in single-colony derivatives of PMSS1 using colony PCR and colony real-time PCR. (A) Three PCRs, named h to j, were used for this screen; the primers are listed in Table S1. PCR h was used to detect multiple cagA repeats (putative amplicon, 1,643 bp); PCR i was used to detect the cagA gene (putative amplicon, 540 bp); and PCR j was used to confirm the absence of cagA (putative amplicon, 851 bp). (B) Representative results from the colony PCR are shown. h, detection of multiple cagA repeats; i, detection of the cagA gene; j, confirmation of the absence of the cagA gene. (C) Relative cagA copy numbers were determined by colony real-time PCR. Download Figure S3, TIF file, 0.4 MB (404.6KB, tif) .
Copyright © 2017 Jang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
cagA EPIYA polymorphism, vacA s/i/m polymorphism, homA/B genotype, babA/B/C genotype, and cagA repeat genotype of 80 American H. pylori strains. Download Table S2, XLSX file, 0.02 MB (22KB, xlsx) .
Copyright © 2017 Jang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Comparison of CHA sequences among cagA repeats. The DNA sequences were aligned and compared using the AlignX program (Invitrogen). (A) The full-length CHA-u sequences from PMSS1, 7.13, B128, B140, J166, B130A, and B125A were aligned (upper panel). The same regions from G27, 26695, and J99 were aligned to CHA-u from PMSS1 (lower panel). (B) The full-length CHA-ud sequences from PMSS1, 7.13, B128, B140, J166, B130A, B125A, G27, 26695, and J99 were aligned. (C) The full-length CHA-d sequences from PMSS1, 7.13, B128, B140, J166, B130A, and B125A were aligned (upper panel). The same regions from G27, 26695, and J99 were aligned to CHA-d from PMSS1 (lower panel). Download Figure S4, TIF file, 2.8 MB (2.9MB, tif) .
Copyright © 2017 Jang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Associations of multiple cagA copies with ethnic groups, genotypic variations, and disease state. Download Table S3, XLSX file, 0.01 MB (10.8KB, xlsx) .
Copyright © 2017 Jang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.