

Abstract
The DNA damage response (DDR) is activated when DNA is altered by intrinsic or extrinsic agents. This pathway is a complex signaling network and plays important roles in genome stability, tumor transformation, and cell cycle regulation. Human papillomaviruses (HPVs) are the main etiological agents of cervical cancer. Cervical cancer ranks as the fourth most common cancer among women and the second most frequent cause of cancer-related death worldwide. Over 200 types of HPVs have been identified and about one third of these infect the genital tract. The HPV life cycle is associated with epithelial differentiation. Recent studies have shown that HPVs deregulate the DDR to achieve a productive life cycle. In this review, I summarize current findings about how HPVs mediate the ataxia-telangiectasia mutated kinase (ATM) and the ATM-and RAD3-related kinase (ATR) DDRs, and focus on the roles that ATM and ATR signalings play in HPV viral replication. In addition, I demonstrate that the signal transducer and activator of transcription-5 (STAT)-5, an important immune regulator, can promote ATM and ATR activations through different mechanisms. These findings may provide novel opportunities for development of new therapeutic targets for HPV-related cancers.
Keywords: Papillomavirus, DNA damage; Amplification, Differentiation; ATM/CHK2, ATR/CHK1, STAT-5
1. Introduction
DNA damage occurs naturally in cells and is associated with intrinsic reagents such as reactive oxygen species (ROS) or extrinsic agents such as ultraviolet (UV), ionizing radiation (IR), and chemotherapeutic drugs. The process begins with an alteration in the structure of DNA through formation of single and double DNA breaks, as well as other changes. In response to DNA damage, cells signal through a complex network. This response plays important roles in processes such as genome stability, tumor transformation, and cell cycle regulation (Matt and Hofmann, 2016). To maintain genome stability, cells must repair damage-induced DNA lesions. If the damage cannot be fixed, cells activate cell cycle check points to trigger cell death. Previous studies have shown that deregulation of the DNA damage response (DDR) can contribute to the development of various cancers such as hereditary non-polyposis colorectal cancer, non-small-cell lung cancer, breast cancer, and ovarian cancer, as well as other cancer-causing conditions such as Fanconi anaemia (FA), xeroderma pigmentosum, and ataxia telangiectasia (Lord and Ashworth, 2012; Sperka et al., 2012; O'Connor, 2015; Seebode et al., 2016). Many small molecule modulators targeting the DDR have been proposed as potential therapeutic treatments against cancers ranging from acute myeloid leukaemia to lung squamous cell carcinoma, as well as cervical squamous cell carcinoma (Pearl et al., 2015).
Worldwide, cervical cancer is the fourth most common cancer in women and the second most frequent cause of cancer-related death. Human papillomaviruses (HPVs) are the causative agents of cervical cancer as well as other anogenital cancers (Zur Hausen, 2002). There are more than 200 types of HPVs. Around 40 of them infect the genital tracts and are sexually transmitted. HPVs can be classified as high-risk or low-risk according to their association with cancers. Infection by low-risk HPVs can lead to cutaneous lesions or benign mucosal lesions while high-risk HPV infection may develop into anogenital or oropharyngeal cancers (Zur Hausen, 2009; Doorbar et al., 2012). Evidence suggests that persistent infection of high-risk HPVs causes genetic instability (Ho et al., 1995; Herrero et al., 2000) and DNA damage repair machinery is utilized by HPVs for productive viral replication (Hong and Laimins, 2013b; Galloway and Laimins, 2015; McKinney et al., 2015). The purpose of this review is to understand better how HPVs employ the DDR, especially ataxia-telangiectasia mutated kinase (ATM)/ATM-and RAD3-related kinase (ATR) signaling, in their life cycle. Before describing what is known about the pathway, I will summarize the cellular pathways involved in the DDR and give a general description of mechanisms that have been reported to be involved in regulating the productive life cycle of HPVs. Then I will discuss the relationship between the DDR and the HPV viral life cycle in detail, including the roles of the signal transducer and activator of transcription-5 (STAT-5).
2. DNA damage response
2.1. Significance and classification of DNA damage response
It has been estimated that up to 200 000 DNA damage lesions occur naturally in each cell every day (Atamna et al., 2000). The causes of DNA damage can be either endogenous or environmental. Endogenous causes of DNA damage include replication errors, unrepaired single strand lesions, and base deamination or loss (Curtin, 2012). Environmental causes result mainly from exposure to damaging agents, including UV, IR, ROS, S-adenosyl methionine, dietary nitrosamines, and tobacco. In response to DNA damage, cells mount collective cellular events such as cell-cycle arrest, regulation of gene expression and DNA replication, activation of DNA damage repair, and cell fate decisions (Matt and Hofmann, 2016). Proper DNA damage repair is necessary for genome stability, prevention of transformation, and tumor suppression. Deregulation of the DDR leads to many clinically relevant diseases such as premature aging, neurodegenerative disorders, and cancer formation (Jackson and Bartek, 2009).
DNA damage often results in DNA mutations, crosslinking, and single-strand breaks (SSBs) as well as double-strand breaks (DSBs). Damaged cells activate various pathways to repair these lesions, including base excision repair (BER), nucleotide excision repair (NER), mismatch repair, non-homologous end joining (NHEJ), homologous recombination repair (HRR), and interstrand crosslink repair (Curtin, 2012). Among these, NHEJ and HRR are the two major repair mechanisms for DSBs in eukaryotes, BER enzymes are major players for SSB repair (Dianov and Hubscher, 2013), and NER works for other bulk single-strand lesions. During these processes, various complexes of cellular proteins are recruited to DNA damage loci to activate DNA damage-responsive phosphatidylinositide 3 kinase (PI3K)-like Ser/Thr kinases, which consist of ATM, ATR, and DNA-dependent protein kinase catalytic subunit kinase (DNA-PKcs). For example, the heterotrimeric meiotic recombination 11 (Mre11)/Rad50/Nijmegen breakage syndrome protein 1 (NBS1) (MRN) complex (Carney et al., 1998) and the Ku70/Ku80 complex (Cary et al., 1997) are responsible for repairing DSBs. When cells experience SSBs, the Rad9-Hus1-Rad1 (9-1-1) complex is activated to facilitate recruitment of the replication protein A (RPA) and other factors. The functions of ATM and ATR (Fig. 1) will be addressed in detail below.
Fig. 1.

Signaling pathways of ATM and ATR
The DNA damage response is activated by ATM, ATR, and DNA-PKcs. All three play central roles in DDR. The ATM-CHK2 and ATR-CHK1 signaling pathways activate the HRR. The ATM and ATR pathways can be activated respectively by DSBs and SSBs. ATM activation can be regulated by Tip60, MRN complex, ATR, and PP2A as well as Wip1. The activated ATM can further trigger the activation of CHK2, SMC-1, FANCD2, BRCA1, as well as H2AX. In addition, the activated CHK2 can phosphorylate Cdc25A and p53. The ATR pathway can be activated by ATRIP, TopBP1, claspin, and 9-1-1 complex. The activation of ATR leads to phosphorylation of various downstream targets such as CHK1, SMC-1, ATM, and p21. Furthermore, CHK1 can facilitate phosphorylation of Wee1, RAD51, Cdc25A, p65, and Rb
2.2. Function and regulation of ATM
ATM function and activation in the DDR depend on its phosphorylation. When cells encounter DNA damage signals, the MRN complex provides the platform for inactivated ATM to be phosphorylated (Lee and Paull, 2004). Several other cellular factors are also necessary to activate ATM directly or indirectly, such as Tat interactive protein 60 (Tip60) (Sun et al., 2005), poly adenosine diphosphate (ADP)-ribose polymerase (PARP) (Aguilar-Quesada et al., 2007), and phosphatase and tensin homolog (PTEN) (Zhang R. et al., 2016). A number of factors can attenuate ATM phosphorylation including the serine/threonine protein phosphatases such as PP2A, Wip1, and PP5 (Ali et al., 2004; Goodarzi et al., 2004; Shreeram et al., 2006). The activated ATM kinase phosphorylates Kruppel-associated box (KRAB)-associated protein-1 (White et al., 2006), releases this substrate from the DNA lesion, and facilitates the recruitment of MRE11 and C-terminal binding protein (CtBP)-interacting protein (Ziv et al., 2006; Goodarzi et al., 2008). In addition, ATM activation leads to the formation of RAD51 nucleofilaments and the formation of RAD51/γ-H2AX foci (Bakr et al., 2015). The histone variant H2AX, which spreads to sites and DNA breaks, can be also phosphorylated by ATM (Burma et al., 2001).
ATM activation is important for the regulation of cell cycle checkpoints and downstream pathways. For the G1/S checkpoint, ATM triggers a p53-independent checkpoint kinase 2 (CHK2)/Cdc25A arm (Falck et al., 2001) and an MDM2/p53 arm (Maya et al., 2001). S-phase checkpoint control depends on the activation of NBS1, breast cancer type 1 susceptibility protein (BRCA1) and Fanconi anemia group D2 protein (FANCD2) (Gatei et al., 2001; D'Amours and Jackson, 2002; Taniguchi et al., 2002). ATM activation inhibits Cdc25 phosphatase resulting in S/G2 arrest (Matsuoka et al., 1998). G2/M arrest can be mediated by BRCA1 or p53 in a Cdc2-or Cdc25C-dependent manner. The downstream effector kinase CHK2 activation triggers Cdc25C phosphorylation causing a G2/M arrest (Zhou et al., 2000). In addition, ATM mediates p53 phosphorylation and stabilization to regulate cyclin-dependent kinase 2 (CDK2)/cyclin E-dependent G1 cell cycle arrest (Canman et al., 1994; Banin et al., 1998). Many substrates, including γ-H2AX (Fernandez-Capetillo et al., 2004), the cohesin factor structural maintenance chromosome-1 (SMC-1) (Kitagawa et al., 2004), and CHK2 (Bouwman and Jonkers, 2012), are recruited to the sites of damage. As a consequence, CHK2 is activated, which then phosphorylates additional substrates such as BRCA1 tumor suppressor (Cortez et al., 1999), p53 tumor suppressor (Chen et al., 2005), and the Cdc25 family of phosphatases (Blasina et al., 1999).
2.3. Function and regulation of ATR
In parallel to ATM signaling, ATR signaling is critical to HRR in response to SSBs. ATR is known to be important to cell survival and its inactivation in mice by disruption of the kinase domain results in early embryonic lethality (de Klein et al., 2000). Furthermore, a splicing mutation of ATR leads to Seckel syndrome (O'Driscoll et al., 2003). In response to DNA damage stimuli, long stretches of single strand DNA (ssDNA) are generated, which are coated with RPA (Coverley et al., 1992). ATR is recruited to RPA-ssDNA complex and interacts with its canonical partners: ATR-interacting protein (ATRIP) (Zou and Elledge, 2003), claspin (Kumagai and Dunphy, 2003) and topoisomerase IIβ-binding protein 1 (TopBP1) (Kumagai et al., 2006). Among these, TopBP1 interacts with the 9-1-1 complex, which is recruited to ssDNA lesions. ATR kinase activities can be regulated by several factors such as ATRIP (Zou and Elledge, 2003) and TopBP1 (Kumagai et al., 2006). Once activated, ATR phosphorylates various downstream substrates including BRCA1 (Chen, 2000), MCM proteins (Cortez et al., 2004), and checkpoint kinase 1 (CHK1) (Liu et al., 2000). In turn, CHK1 phosphorylates Cdc25A (Falck et al., 2002), Wee1 kinase (Lee et al., 2001), and RAD51 (Sorensen et al., 2005).
The ATR pathway can be activated not only by SSBs but also by DSBs. Recent studies have provided evidence that the ATR and ATM pathways are interlinked (Smith J. et al., 2010). For example, ATM phosphorylation is necessary for a rapid activation of ATR when cells are exposed to DSBs (Byun et al., 2005; Myers and Cortez, 2006). ATR activation by radiation-stimulated DSBs shares a common pattern of induction of an S/G2 phase arrest with ATM signaling (Jazayeri et al., 2006; Walker et al., 2009). In addition, activation of ATR signaling depends on the MRN complex that is also necessary for ATM signaling (Yarden et al., 2002; Myers and Cortez, 2006). Both the ATM and ATR pathways can be manipulated by various viruses, such as Epstein-Barr virus (EBV), Kaposi’s sarcoma-associated herpesvirus (KSHV), and HPV (McFadden and Luftig, 2013; Hollingworth and Grand, 2015). The regulation of the ATM/ATR pathways by HPV will be discussed later.
3. Life cycle of HPVs
3.1. HPV life cycle
HPVs are the major etiological agents responsible for cervical cancer (Zur Hausen, 2002). In recent years HPVs have been shown also to be involved in many other genital cancers such as those of the vulva, vagina, anus, penis, and oral cavity. About nine types of HPVs are considered as high-risk types, including HPV16, HPV18, HPV31, and HPV45, that are the causative agents of most anogenital cancers. The low-risk types, including HPV6 and HPV11, can cause genital warts. The life cycle of HPVs is dependent on epithelial differentiation (Fig. 2). During infection, HPVs escape immune surveillance and can remain latent for decades. HPVs infect stratified squamous epithelia by entering the cells in the basal layer that becomes exposed following trauma or wounding (Hebner and Laimins, 2006; Doorbar et al., 2012). The infected basal cell divides into a new basal cell and a daughter cell that migrates to the upper layers of the epithelium as it undergoes terminal differentiation. HPV episomes are replicated and equally distributed to the new basal cell and the daughter cell.
Fig. 2.

Life cycle of human papillomaviruses
HPVs infect basal layer keratinocytes when the basal layer of stratified epithelia is exposed to the virus. On the left, a normal uninfected epithelium is shown for the regular differentiation. On the right, the HPV-infected epithelium is shown with the progress of HPV viral proteins expression. After persisting infection, HPVs replicate with cellular chromosomes in basal cells. Upon the differentiation, more viral genes are observed in differentiated cells. The late gene expression and viral replication are activated, followed by virion assembly and release of newly synthesized virions from the top layers of epithelium
HPVs maintain low copy numbers of viral genomes (around 100 copies per cell) in undifferentiated cells. Upon differentiation, the viral copy number increases rapidly to levels of around 1000 copies per cell in a process referred to as amplification. This is followed by virion assembly and viral release from the upper layers of the epithelium (Longworth and Laimins, 2004).
3.2. HPV viral protein function
The genomes of HPVs are about 8 kb in size and encode multiple viral gene products, including E1, E2, E4, E5, E6, E7, as well as L1 and L2 (Hebner and Laimins, 2006). E1 and E2 are two genes that regulate initiation of HPV genome replication (Sedman and Stenlund, 1995; McBride, 2013), whereas E4 and E5 regulate late viral functions (Dimaio and Petti, 2013). E6 and E7 are necessary for HPV genome maintenance and amplification, and act as two oncogenes to alter the host environment to be advantageous for viral replication (Thomas et al., 1999; Munger and Howley, 2002; Wise-Draper and Wells, 2008; Wallace and Galloway, 2015). L1 and L2 are the two capsid proteins synthesized following productive replication (Hebner and Laimins, 2006; Thomas et al., 2008).
HPV viral proteins function cooperatively to regulate the HPV life cycle. The E1 protein acts as a DNA helicase/ATPase to facilitate DNA unwinding and recruits host DNA polymerases to viral origins (Hughes and Romanos, 1993; Conger et al., 1999). E2 has DNA-binding activities and is important for DNA segregation in mitotic cells (Oliveira et al., 2006; Poddar et al., 2009). Bromodomain-containing protein 4 (Brd4) has been implicated in this E2 function, and the interaction between Brd4 and E2 is also required for E2-dependent transcriptional regulation (McPhillips et al., 2006; Wu et al., 2006). In addition, E2 regulates expressions of E6, E7, E1, as well as E2 itself via controlling transcription of the early viral promoter (Steger and Corbach, 1997). E8^E2C is a truncated form of E2 that inhibits early gene expression and viral replication (Stubenrauch et al., 2000). E1 and E2 work cooperatively in the initiation of viral replication (Frattini and Laimins, 1994; Sedman and Stenlund, 1995). E6 and E7 promote genome maintenance and viral replication (Cheng et al., 1995; Thomas et al., 1999) in addition to their roles in cell transformation and immortalization. An HPV31 genome containing an E6 or E7 mutation is not able to be maintained stably as an episome (Thomas et al., 1999). The high-risk E6 binds to the cellular E3 ubiquitin ligase E6-associated protein (E6AP) to degrade p53 (Scheffner et al., 1990; 1993; Huibregtse et al., 1991) and to inhibit p53 function by blocking acetylation (Hebner et al., 2007) in a p300-dependent manner (Patel et al., 1999). The E7 protein binds to pRb to facilitate the regulation of cell cycle events by E2F family members (Dyson et al., 1989; Munger et al., 1989; Longworth et al., 2005). E5 has been shown to be involved in cell motility, adhesion, and proliferation (Kivi et al., 2008; Liao et al., 2013). L1 and L2 capsid proteins are responsible for viral chromatin packaging and virion assembly (Nelson et al., 2002; Darshan et al., 2004). The lack of viral DNA polymerases and other necessary factors leads HPVs to rely on host factors to accomplish genome amplification.
4. Regulation of HPV viral replication
4.1. Classification of regulators for HPV viral replication
HPV viral replication is dependent on epithelial differentiation, and is regulated by viral factors, such as E1 and E2 (Kadaja et al., 2009), and host factors. Because HPVs do not encode their own DNA polymerases and other factors, their replication is largely dependent on host factors such as transcriptional factors, microRNAs (miRs), kinases, apoptotic caspases, epigenetic enzymes, and DNA damage signaling (Moody and Laimins, 2010; Kajitani et al., 2012; Hong and Laimins, 2013b). Before describing the interaction between the HPV life cycle and the DDR, I will provide a general description of what is known about the cellular factors involved in HPV viral replication, though the actual process of amplification is still not clear. The regulation of HPV viral replication by the DDR will be then discussed in detail in the following section.
4.2. Cellular enzymes
Several of the host factors that are important for HPV viral replication have been identified as cellular enzymes associated with the DDR. For example, the ubiquitin-specific protease 1 (USP1)-associated factor 1 (UAF1)-USP deubiquitinase complex, which is suggested to be associated with the DDR (Chen et al., 2011), can be recruited by E1 to facilitate HPV DNA replication (Lehoux et al., 2014). The activated CDK2 phosphorylates NBS1 (Wohlbold et al., 2012) and mediates HPV genome maintenance (Fradet-Turcotte et al., 2010). The activity of caspase-3 is required for HPV genome replication (Moody et al., 2007), and caspase-3-dependent apoptosis can be inhibited by ATR/CHK1 (Myers et al., 2009). Activation of the nuclear factor-(B (NF-(B) pathway has been implicated in HPV genome amplification (Nakahara et al., 2015; Satsuka et al., 2015) and can be induced by DNA damage (Janssens and Tschopp, 2006). The deregulation of these factors suggests that HPV induces the DDR while suppressing cellular apoptotic events. Furthermore, binding of histone deacetylases (HDAC) to E7 contributes to E7-facilitated HPV genome amplification (Longworth et al., 2005) and HDACs have been reported to function in the DDR (Miller et al., 2010; Thurn et al., 2013). The deacetylase sirtuin 1 facilitates HPV DNA replication, partly by modulating histone acetylation (Langsfeld et al., 2015). In addition, repression of Brd4, which insulates chromatin from DNA damage signaling (Floyd et al., 2013), deregulates E2-dependent HPV oncogene expression (Smith J.A. et al., 2010). The association of these factors with HPV genomes further indicates that the DDR is involved in HPV viral replication.
4.3. Transcription factors
Cellular transcription factors also play a role in HPV viral replication. Some, including YY-1 (Ai et al., 2000), TATA-binding protein (TBP) (Hartley and Alexander, 2002), activator protein 1 (AP-1) (Offord and Beard, 1990), Oct-1 (O'Connor and Bernard, 1995), and Sp1 (Stunkel and Bernard, 1999), act on the HPV early promoters located upstream of the E6 open reading frame (ORF). Others, such as CCAAT/enhancer binding protein β (C/EBPβ) isoforms, liver-enriched inhibitory protein (LIP), and liver-enriched transcriptional activator protein (LAP) (Gunasekharan et al., 2012), act on the late promoter. Several of these transcription factors have been shown to be related to the DDR. For example, YY-1 is essential in homologous recombination-based DNA repair (Wu et al., 2007). Downregulation of the subunit of TBP is responsible for DNA damage-induced repression of RNA polymerase III transcription (Ghavidel and Schultz, 2001). Activation of Oct-1 can be induced by DNA damage (Zhao et al., 2000). The phosphorylated Sp1 colocalizes with (-H2AX and depletion of Sp1 inhibits the repair of DSBs (Beishline et al., 2012). In addition, members of the STAT family can be differently regulated by HPVs to facilitate viral replication. Inhibition of STAT-1 by E6 and E7 is necessary for HPV genome amplification (Hong et al., 2011). In contrast, STAT-5 is activated by E7 and this activation is important for HPV genome amplification (Hong and Laimins, 2013a). Another transcription factor, Kruppel-like factor 13, can regulate STAT-5 expression, and is important for the differentiation-dependent HPV life cycle (Zhang W. et al., 2016). The relationship between STAT-5 and the DDR will be discussed later.
4.4. MicroRNAs
miRs are among other cellular factors involved in HPV viral replication. The miRs are noncoding regulatory RNAs, 18–25 nucleotides in size. They post-transcriptionally regulate mRNA stability and translation and have been reported to be associated with the DDR (Wan et al., 2011; Wang and Taniguchi, 2013). miRs are also associated with cervical cancer (Pedroza-Torres et al., 2014) and can be considered as biomarkers for high-risk HPV infection (Wang et al., 2014). HPV E7 down-regulates miR-203 and this suppression is required for HPV genome amplification through regulation of p63 (Melar-New and Laimins, 2010) and ATM DDR (Mighty and Laimins, 2011). miR-145, which is also suppressed by E7 and targets E1 ORFs, is important for HPV genome amplification (Gunasekharan and Laimins, 2013). Ectopic expression of miR-125b suppresses HPV gene expression (Nuovo et al., 2010) possibly due to the sequence homology between HPV16 L2 and miR-125b. The role of these HPV-related miRs in the DDR is still not clear.
5. HPV and DDR
5.1. HPV regulates the DDR
Upon DNA damage, many host repair factors are recruited to the damage loci to repair single or double strand breaks. HPVs hijack this repair machinery, by both inhibiting and activating the DDR, to replicate viral genes (Hong and Laimins, 2013b; Wallace and Galloway, 2014; 2015; McKinney et al., 2015). For example, HPV oncogenes have been suggested to abrogate radiation-induced DDRs (Song et al., 1998), and recent studies support the idea that HPVs regulate the DDR to mediate their life cycle. HPV E7 activates the FA repair pathway, enhances FANCD2 foci, and recruits FANCD2 and BRCA2 to chromatin (Spardy et al., 2007). Both E6 and E7 interact with BRCA1 to inhibit its transcriptional regulation (Zhang et al., 2005). HPV E6 disrupts p53 signaling (Thomas and Chiang, 2005) and interferes SSBs through interactions with XRCC1 and O6-methylguanine-DNA methyltransferase (Iftner et al., 2002; Srivenugopal and Ali-Osman, 2002).
Besides regulation of the DDR proteins mentioned above, HPVs are capable of manipulating host genomic destabilization. E6 and E7 can induce DNA damage independently by causing host chromosome instability (Duensing and Munger, 2002), and collaboratively by uncoupling centrosome duplication from the cell division cycle (Duensing et al., 2000). Delocalization of the microtubule motor dynein by E7 results in failed chromosome alignment (Nguyen et al., 2008). The expression of E7 results in polyploidy by inducing HPV rereplication in response to DNA damage (Fan et al., 2013). In addition, E1 and E2 act as recruiters to facilitate colocalization of the DDR proteins with the HPV replication complex, which will be addressed below.
5.2. HPV regulates ATM and ATR pathways
The two major arms of the DDR are the ATM/CHK2 and ATR/CHK1 pathways. Little is known about how HPVs employ these two arms for the viral life cycle. ATM activation can be induced by high-risk HPV E1 and E7 proteins. For example, HPV18 E1 could cause DSBs and lead to the induction of an ATM-dependent signaling cascade (Reinson et al., 2013). The HPV31 E7 oncoprotein binds to ATM and induces phosphorylation of ATM and CHK2 (Moody and Laimins, 2009). Similarly, HPV18 E7 induces elevated expression of phosphorylated ATM, as well as CHK2 and c-Jun N-terminal kinases (JNKs) (Banerjee et al., 2011). The activated ATM and CHK2 are recruited to the DNA repair centers that are colocalized with the HPV integrated replication sites (Kadaja et al., 2009), suggesting a critical role of ATM signaling in recruitment of HPV DNA to the DNA repair centers to start viral DNA synthesis.
Less is known about the effect of the viral proteins on ATR activity. Several contradictory results have been reported by different groups. An elevation of ATR protein levels was indicated in HPV16 E7-expressing cells (Spardy et al., 2008), whereas other studies showed that HPV16 E7 attenuates CHK1 phosphorylation by increasing claspin turnover (Spardy et al., 2009). Activation of the ATR pathway is induced by HPV18 E1 and E2, but not E6 or E7 (Reinson et al., 2013). However, recent studies have shown that HPV31 E7 increases ATR phosphorylation as well as CHK1 phosphorylation (Hong et al., 2015b). The levels of phosphorylated CHK1 are also enhanced and sustained in HPV16 E6-expressing fibroblasts (Chen et al., 2009). The complexity of ATR regulation by high-risk HPV proteins indicates that differences between HPV species might lead to different modulation of ATR activation and that HPV proteins employ different mechanisms to regulate ATR and ATM.
Unlike high-risk α HPV types, β HPV E6 reduces ATM protein levels (Wallace et al., 2013) and abrogates ATR activities (Wallace et al., 2012). The reduction of ATR protein levels can be explained by the degradation of p300 (Wallace et al., 2012), which is regulated by protein kinase B (PKB/AKT) (Howie et al., 2011). Furthermore, β HPV E6 inhibits the stability of p53 (Wallace et al., 2014) and attenuates BRCA1 and BRCA2 expression as well as foci formation (Wallace et al., 2015). The suppressive effect of β E6 on ATM/ATR signaling may reflect an increased cellular tolerance of DNA lesions and a reduced induction of apoptosis by the DDR. It is not known how this regulation contributes to β HPV genome amplification.
E1 and E2, instead of modulating the phosphorylation of factors of the ATM/ATR pathway, are parts of the viral replication complex that colocalize with these factors (Kadaja et al., 2009). Several cellular replication factors colocalize with HPV18 E1 in HeLa cells, including RPA, proliferating cell nuclear antigen (PCNA), and death-associated protein (DAXX). HPV18 E1 also recruits the MRN and Ku70/Ku80 complexes to the viral replication complex, and induces ATM/CHK2 activation. The same study provided some evidence that E1 is colocalized with ATRIP and CHK1. Another set of DDR proteins, including (-H2AX and 53BP1, colocalizes with the HPV DNA foci (Gillespie et al., 2012). It has been suggested that the function of E2 is to facilitate the translocation of the complex of E1 and the associated DDR proteins to the nuclear foci (Sakakibara et al., 2011). In addition, Boner et al. (2002) showed that HPV16 E2 interacts with TopBP1, and that this interaction enhances the ability of E2 to activate transcription and replication. All these findings suggest that interactions of the DDR proteins and E1/E2 play an important role in the aggregation of the HPV viral replication complex and DNA damage repair factors in the nuclear foci.
5.3. ATM and ATR DDR are necessary for HPV viral replication
The fact that many DDR proteins are deregulated by HPVs suggests that the DDR plays a significant role in HPV genome amplification. This is supported by the finding that activation of the ATM pathway is necessary for HPV genome amplification (Moody and Laimins, 2009). The same study also showed that inhibition of ATM activities has no effect on HPV genome maintenance. NBS1, as one component of the MRN complex that facilitates ATM activation, is also required for HPV genome amplification (Anacker et al., 2014). Consistently, BRCA1, as a downstream target of ATM, is important for HPV genome amplification (Chappell et al., 2016). Another downstream target, SMC-1, is activated by HPV and is required for HPV genome amplification (Mehta et al., 2015), whereas loss of FANCD2 stimulates HPV replication (Hoskins et al., 2012). In addition, RAD51 plays essential roles in the DDR and binds to HPV31 genomes. Depletion of RAD51 or inhibition of RAD51’s recombinase activity abolishes HPV viral replication upon differentiation (Chappell et al., 2016). HPV’s association with Brd4 leads to an increase in the rate of asynchronous DNA replication (Jang et al., 2014).
Compared to the ATM pathway, the role of ATR signaling for HPV replication is less well known. HPV18 transient replication induces an accumulation of ATR signaling, indicating a role of ATR in the initiation of HPV18 replication (Reinson et al., 2013). Recent studies have shown that inhibition of ATR activities reduces HPV genome amplification, and that inhibition of CHK1 activities suppresses HPV genome amplification (Hong et al., 2015b). Edwards et al. (2013) also showed that inhibition of CHK1 significantly reduces HPV episome levels in undifferentiated cells. In addition, knockdown of TopBP1, which is upstream of the ATR activation, suppresses HPV viral replication (Hong et al., 2015b). The interaction between TopBP1 and E2 also contributes to HPV genome amplification (Donaldson et al., 2012). Taken together, both ATM signaling and the ATR pathway are important for HPV viral replication.
5.4. STAT-5 activation promotes both ATM and ATR signaling for HPV viral replication
Although there is mounting evidence that the DDR is important for HPV genome amplification, less is known about how HPV regulates it to accomplish its life cycle. Recent studies investigated the relationships between HPVs and the immune response, and found that HPVs might utilize immune factors to promote the DDR by deregulation of the STAT family that contains important regulators of the innate immune response (Beglin et al., 2009; Stanley, 2012; Hong and Laimins, 2013b).
STAT signaling is part of the interferon (IFN) pathway. Canonically, IFN-α or IFN-β binds to a heterodimeric transmembrane receptor termed IFN ax-receptor (IFNAR) (Abbas et al., 2014) and activates the receptor-associated Janus kinase 1 (JAK1) and tyrosine kinase 2 (TYK2), resulting in the phosphorylation of inactive STAT proteins translocating from the cytoplasm to the nucleus and the induction of hundreds of genes that act to block viral propagation. The STAT family consists of STAT-1, -2, -3, -4, -5, and -6. Upon phosphorylation, STAT-1 forms a complex referred to as IFN-stimulated gene factor (ISGF3) along with STAT-2 and IFN regulatory factor (IRF)-9. The ISGF3 complex binds to the IFN stimulated elements (ISRE) located in promoter regions of many IFN-stimulated genes (ISGs) to induce their expressions. Similarly, unphosphorylated STAT-5 becomes activated following binding of cytokines to the cytokine receptors resulting in the formation of either homo-or heterodimers between its two isoforms STAT-5α and STAT-5β, and translocation from the cytoplasm to the nucleus (Ferbeyre and Moriggl, 2011). STAT-1 can induce many ISGs such as IFN-inducible double-stranded RNA-dependent protein kinase (PKR) and IFN-induced protein with tetratricopeptide repeats (IFIT) (Diamond and Farzan, 2013), while STAT-5 activates a different set of downstream genes (Doan et al., 2008). STAT-5 activities are important for the development and survival of lymphocytes (Heltemes-Harris and Farrar, 2012), as well as epithelial cells (Groner, 2002).
Evidence suggests that the STAT proteins are associated with the DDR. For example, STAT-1 is suggested to confer resistance to DNA damage (Cheon et al., 2013). STAT-3 has been shown to disrupt ATR/CHK1 signaling (Koganti et al., 2014) and phosphorylation of STAT-3 requires ATM activation (Zhang et al., 2003). STAT-5 shares 50% homology with STAT-1. Knockout mice lacking STAT-5 exhibit a perinatal lethal phenotype (Cui et al., 2004) and severely impaired lymphoid development (Yao et al., 2006). Over-expression of STAT-5 promotes the DDR and is associated with CHK2 activity (Eilon and Barash, 2011). This suggests that HPV might regulate the STAT proteins to promote the DDR.
Before discussing the relationship between the STAT proteins and the DDR, I will discuss the expression of the STAT family in HPV-positive cells. The Laimins group has shown that HPV specifically suppresses the expression of STAT-1, but not STAT-2 (Hong et al., 2011) or STAT-3 (unpublished). In contrast, HPV induces the constitutive activation of STAT-5 while only minimally affecting total levels (Hong and Laimins, 2013a). HPVs can also deregulate STAT expression by controlling the IRF transcription factors and synthesis of IFNs. For example, IRF-1 expression can be downregulated by HPV16 E7 (Park et al., 2000) and HPV38 E6E7 (Cordano et al., 2008). HPV16 E6 binds to IRF-3 to inhibit its transcriptional activity (Ronco et al., 1998). Consequently, IRF deregulation by HPV proteins leads to a reduced expression of IFN-α (Chang and Laimins, 2000), IFN-β (Ronco et al., 1998; Park et al., 2000), and IFN-κ(Rincon-Orozco et al., 2009). Among these, IFN-β has been shown to be associated with the DDR (Moiseeva et al., 2006; Cheon et al., 2013).
Hong and Laimins (2013a) have shown that inhibition of STAT-5 phosphorylation suppresses HPV genome amplification and late gene expression upon epithelial differentiation. This STAT-5-dependent regulation can be explained by two DDR mechanisms: the ATM (Hong and Laimins, 2013a) and the ATR pathways (Hong et al., 2015b) (Fig. 3). Inhibition of STAT-5 suppresses the phosphorylation of ATM, CHK2, ATR, CHK1, and BRCA1, as well as the level of RAD51, but not BRCA2 or SMC-1. These findings suggest that the DDR crosslinks with the immune response and both responses could be important for HPV viral replication.
Fig. 3.
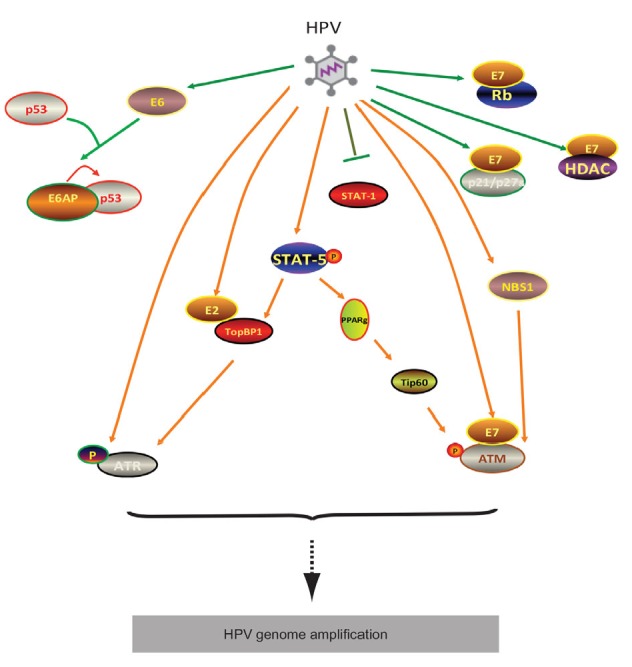
STAT-5-dependent activation of ATM and ATR pathways, required for HPV genome amplification
High-risk HPV activates STAT-5 to mediate TopBP1 transcriptionally to promote ATR pathway. Meanwhile, STAT-5 does not directly regulate Tip60; instead, it partially work through PPARg to manipulate Tip60 activation which consequently facilitates ATM activation. HPV E7 oncogene is responsible for STAT-5 activation as well as interaction with other factors such as Rb, ATM, NBS1, HDAC, and p21. E6 gene may collaborate with other viral proteins to act on Tip60 in addition to its role in p53 degradation. E2 is capable of interacting with TopBP1 to mediate HPV initial replication
As a transcription factor, STAT-5 does not directly mediate the level of ATM or ATR. Instead, it deregulates the acetyltransferase Tip60 (Hong et al., 2015a) or TopBP1 (Hong et al., 2015b) to promote the activation of ATM or ATR signaling. Tip60 knockdown blocks ATM activation and HPV genome amplification upon differentiation. Both p53 and histone H2AX, the downstream targets of Tip60, may also play a role in the DDR in HPV-positive cells. Inhibition of STAT-5 activation blocks Tip60 activation and this regulation might be mediated by the kinase glycogen synthase kinase 3β (GSK3β). Consistently, GSK3β inhibition also leads to reduced levels of viral episomes and impaired genome amplification.
STAT-5 regulates transcription of the TopBP1 gene to mediate ATR activation in HPV-positive cells and knockdown of TopBP1 blocks HPV genome amplification (Hong et al., 2015b). Knockdown of TopBP1 moderately increases short-term replication of transiently transfected HPV31 plasmids and has a modest effect on stable maintenance of HPV31 episomes (Kanginakudru et al., 2015). Hong et al. (2015b) identified a critical role for TopBP1 in the differentiation-dependent late phase of the viral life cycle. TopBP1 is also recruited to viral genomes by forming complexes with the HPV E2 protein. The failure of E2 to bind TopBP1 results in a reduced replication ability, indicating that it is a positive regulator of viral replication (Donaldson et al., 2012). TopBP1 also helps to load replication factors onto origins (Gauson et al., 2015) and acts as a transcriptional regulator (Liu et al., 2003), but it is unclear if these functions are important for HPV genome amplification.
6. Perspectives
The complexity of the role of the DDR in the viral life cycle has been a recent focus of research. In the HPV field, although there have been some studies demonstrating the necessary roles of ATM or ATR signaling in HPV viral replication, more effort should be placed on the roles of the DDR factors in the HPV life cycle. What are the substrates for ATM or ATR that contribute to the HPV life cycle What other DNA damage factors are important in HPV viral replication Are DNA damage repair foci the same as HPV replication centers An additional question raised here is whether the DDR is important for the development of cervical cancer. Moreover, I presented evidence that the immune regulator STAT-5 participates in activation of the ATM or ATR pathway to facilitate HPV genome amplification, suggesting that the interaction between the innate immune response and the DNA damage pathway needs to be dissected in detail. The role of other members of STATs, such as STAT-3, in DNA damage regulation and HPV genome amplification is still not clear. Further, is the DDR able to mediate the innate immune response as a feedback loop for HPV persistent infection Resolving these questions will improve our understanding of the replication mechanisms of HPVs and provide insight for developing new therapeutic targets against HPV-related diseases.
Footnotes
Compliance with ethics guidelines: Shi-yuan HONG declares that he has no conflict of interest.
This article does not contain any studies with human or animal subjects performed by the author.
References
- 1.Abbas AK, Lichtman AH, Pillai S. Cellular and Molecular Immunology. Philadelphia, PA: Elsevier Saunders; 2014. [Google Scholar]
- 2.Aguilar-Quesada R, Munoz-Gamez JA, Martin-Oliva D, et al. Interaction between ATM and PARP-1 in response to DNA damage and sensitization of ATM deficient cells through PARP inhibition. BMC Mol Biol. 2007;8:29. doi: 10.1186/1471-2199-8-29. (Available from: http://dx.doi.org/10.1186/1471-2199-8-29) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ai W, Narahari J, Roman A. Yin yang 1 negatively regulates the differentiation-specific E1 promoter of human papillomavirus type 6. J Virol. 2000;74(11):5198–5205. doi: 10.1128/jvi.74.11.5198-5205.2000. (Available from: http://dx.doi.org/10.1128/JVI.74.11.5198-5205.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ali A, Zhang J, Bao S, et al. Requirement of protein phosphatase 5 in DNA-damage-induced ATM activation. Genes Dev. 2004;18(3):249–254. doi: 10.1101/gad.1176004. (Available from: http://dx.doi.org/10.1101/gad.1176004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anacker DC, Gautam D, Gillespie KA, et al. Productive replication of human papillomavirus 31 requires DNA repair factor NBS1. J Virol. 2014;88(15):8528–8544. doi: 10.1128/JVI.00517-14. (Available from: http://dx.doi.org/10.1128/JVI.00517-14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Atamna H, Cheung I, Ames BN. A method for detecting abasic sites in living cells: age-dependent changes in base excision repair. PNAS. 2000;97(2):686–691. doi: 10.1073/pnas.97.2.686. (Available from: http://dx.doi.org/10.1073/pnas.97.2.686) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bakr A, Oing C, Kocher S, et al. Involvement of ATM in homologous recombination after end resection and RAD51 nucleofilament formation. Nucleic Acids Res. 2015;43(6):3154–3166. doi: 10.1093/nar/gkv160. (Available from: http://dx.doi.org/10.1093/nar/gkv160) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banerjee NS, Wang HK, Broker TR, et al. Human papillomavirus (HPV) E7 induces prolonged G2 following S phase reentry in differentiated human keratinocytes. J Biol Chem. 2011;286(17):15473–15482. doi: 10.1074/jbc.M110.197574. (Available from: http://dx.doi.org/10.1074/jbc.M110.197574) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banin S, Moyal L, Shieh S, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281(5383):1674–1677. doi: 10.1126/science.281.5383.1674. (Available from: http://dx.doi.org/10.1126/science.281.5383.1674) [DOI] [PubMed] [Google Scholar]
- 10.Beglin M, Melar-New M, Laimins L. Human papillomaviruses and the interferon response. J Interferon Cytokine Res. 2009;29(9):629–635. doi: 10.1089/jir.2009.0075. (Available from: http://dx.doi.org/10.1089/jir.2009.0075) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beishline K, Kelly CM, Olofsson BA, et al. Sp1 facilitates DNA double-strand break repair through a nontranscriptional mechanism. Mol Cell Biol. 2012;32(18):3790–3799. doi: 10.1128/MCB.00049-12. (Available from: http://dx.doi.org/10.1128/MCB.00049-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blasina A, de Weyer IV, Laus MC, et al. A human homologue of the checkpoint kinase Cds1 directly inhibits Cdc25 phosphatase. Curr Biol. 1999;9(1):1–10. doi: 10.1016/s0960-9822(99)80041-4. (Available from: http://dx.doi.org/10.1016/S0960-9822(99)80041-4) [DOI] [PubMed] [Google Scholar]
- 13.Boner W, Taylor ER, Tsirimonaki E, et al. A functional interaction between the human papillomavirus 16 transcription/replication factor E2 and the DNA damage response protein TopBP1. J Biol Chem. 2002;277(25):22297–22303. doi: 10.1074/jbc.M202163200. (Available from: http://dx.doi.org/10.1074/jbc.M202163200) [DOI] [PubMed] [Google Scholar]
- 14.Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer. 2012;12(9):587–598. doi: 10.1038/nrc3342. (Available from: http://dx.doi.org/10.1038/nrc3342) [DOI] [PubMed] [Google Scholar]
- 15.Burma S, Chen BP, Murphy M, et al. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276(45):42462–42467. doi: 10.1074/jbc.C100466200. (Available from: http://dx.doi.org/10.1074/jbc.C100466200) [DOI] [PubMed] [Google Scholar]
- 16.Byun TS, Pacek M, Yee MC, et al. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19(9):1040–1052. doi: 10.1101/gad.1301205. (Available from: http://dx.doi.org/10.1101/gad.1301205) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Canman CE, Wolff AC, Chen CY, et al. The p53-dependent G1 cell cycle checkpoint pathway and ataxia-telangiectasia. Cancer Res. 1994;54(19):5054–5058. [PubMed] [Google Scholar]
- 18.Carney JP, Maser RS, Olivares H, et al. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93(3):477–486. doi: 10.1016/s0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- 19.Cary RB, Peterson SR, Wang J, et al. DNA looping by Ku and the DNA-dependent protein kinase. PNAS. 1997;94(9):4267–4272. doi: 10.1073/pnas.94.9.4267. (Available from: http://dx.doi.org/10.1073/pnas.94.9.4267) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang YE, Laimins LA. Microarray analysis identifies interferon-inducible genes and STAT-1 as major transcriptional targets of human papillomavirus type 31. J Virol. 2000;74(9):4174–4182. doi: 10.1128/jvi.74.9.4174-4182.2000. (Available from: http://dx.doi.org/10.1128/JVI.74.9.4174-4182.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chappell WH, Gautam D, Ok ST, et al. Homologous recombination repair factors RAD51 and BRCA1 are necessary for productive replication of human papillomavirus 31. J Virol. 2016;90(5):2639–2652. doi: 10.1128/JVI.02495-15. (Available from: http://dx.doi.org/10.1128/JVI.02495-15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen B, Simpson DA, Zhou Y, et al. Human papilloma virus type16 E6 deregulates CHK1 and sensitizes human fibroblasts to environmental carcinogens independently of its effect on p53. Cell Cycle. 2009;8(11):1775–1787. doi: 10.4161/cc.8.11.8724. (Available from: http://dx.doi.org/10.4161/cc.8.11.8724) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J. Ataxia telangiectasia-related protein is involved in the phosphorylation of BRCA1 following deoxyribonucleic acid damage. Cancer Res. 2000;60(18):5037–5039. [PubMed] [Google Scholar]
- 24.Chen J, Dexheimer TS, Ai Y, et al. Selective and cell-active inhibitors of the USP1/UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem Biol. 2011;18(11):1390–1400. doi: 10.1016/j.chembiol.2011.08.014. (Available from: http://dx.doi.org/10.1016/j.chembiol.2011.08.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L, Gilkes DM, Pan Y, et al. ATM and Chk2-dependent phosphorylation of mdmx contribute to p53 activation after DNA damage. EMBO J. 2005;24(19):3411–3422. doi: 10.1038/sj.emboj.7600812. (Available from: http://dx.doi.org/10.1038/sj.emboj.7600812) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng S, Schmidt-Grimminger DC, Murant T, et al. Differentiation-dependent up-regulation of the human papillomavirus E7 gene reactivates cellular DNA replication in suprabasal differentiated keratinocytes. Genes Dev. 1995;9(19):2335–2349. doi: 10.1101/gad.9.19.2335. (Available from: http://dx.doi.org/10.1101/gad.9.19.2335) [DOI] [PubMed] [Google Scholar]
- 27.Cheon H, Holvey-Bates EG, Schoggins JW, et al. IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 2013;32(20):2751–2763. doi: 10.1038/emboj.2013.203. (Available from: http://dx.doi.org/10.1038/emboj.2013.203) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Conger KL, Liu JS, Kuo SR, et al. Human papillomavirus DNA replication. Interactions between the viral E1 protein and two subunits of human DNA polymerase α/PRIMASE. J Biol Chem. 1999;274(5):2696–2705. doi: 10.1074/jbc.274.5.2696. (Available from: http://dx.doi.org/10.1074/jbc.274.5.2696) [DOI] [PubMed] [Google Scholar]
- 29.Cordano P, Gillan V, Bratlie S, et al. The E6E7 oncoproteins of cutaneous human papillomavirus type 38 interfere with the interferon pathway. Virology. 2008;377(2):408–418. doi: 10.1016/j.virol.2008.04.036. (Available from: http://dx.doi.org/10.1016/j.virol.2008.04.036) [DOI] [PubMed] [Google Scholar]
- 30.Cortez D, Wang Y, Qin J, et al. Requirement of ATM-dependent phosphorylation of BRCA1 in the DNA damage response to double-strand breaks. Science. 1999;286(5442):1162–1166. doi: 10.1126/science.286.5442.1162. (Available from: http://dx.doi.org/10.1126/science.286.5442.1162) [DOI] [PubMed] [Google Scholar]
- 31.Cortez D, Glick G, Elledge SJ. Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. PNAS. 2004;101(27):10078–10083. doi: 10.1073/pnas.0403410101. (Available from: http://dx.doi.org/10.1073/pnas.0403410101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coverley D, Kenny MK, Lane DP, et al. A role for the human single-stranded DNA binding protein HSSB/RPA in an early stage of nucleotide excision repair. Nucleic Acids Res. 1992;20(15):3873–3880. doi: 10.1093/nar/20.15.3873. (Available from: http://dx.doi.org/10.1093/nar/20.15.3873) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cui Y, Riedlinger G, Miyoshi K, et al. Inactivation of STAT5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004;24(18):8037–8047. doi: 10.1128/MCB.24.18.8037-8047.2004. (Available from: http://dx.doi.org/10.1128/MCB.24.18.8037-8047.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12(12):801–817. doi: 10.1038/nrc3399. (Available from: http://dx.doi.org/10.1038/nrc3399) [DOI] [PubMed] [Google Scholar]
- 35.D'Amours D, Jackson SP. The MRE11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat Rev Mol Cell Biol. 2002;3(5):317–327. doi: 10.1038/nrm805. (Available from: http://dx.doi.org/10.1038/nrm805) [DOI] [PubMed] [Google Scholar]
- 36.Darshan MS, Lucchi J, Harding E, et al. The L2 minor capsid protein of human papillomavirus type 16 interacts with a network of nuclear import receptors. J Virol. 2004;78(22):12179–12188. doi: 10.1128/JVI.78.22.12179-12188.2004. (Available from: http://dx.doi.org/10.1128/JVI.78.22.12179-12188.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Klein A, Muijtjens M, van Os R, et al. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol. 2000;10(8):479–482. doi: 10.1016/s0960-9822(00)00447-4. (Available from: http://dx.doi.org/10.1016/S0960-9822(00)00447-4) [DOI] [PubMed] [Google Scholar]
- 38.Diamond MS, Farzan M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat Rev Immunol. 2013;13(1):46–57. doi: 10.1038/nri3344. (Available from: http://dx.doi.org/10.1038/nri3344) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dianov GL, Hubscher U. Mammalian base excision repair: the forgotten archangel. Nucleic Acids Res. 2013;41(6):3483–3490. doi: 10.1093/nar/gkt076. (Available from: http://dx.doi.org/10.1093/nar/gkt076) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dimaio D, Petti LM. The E5 proteins. Virology. 2013;445(1-2):99–114. doi: 10.1016/j.virol.2013.05.006. (Available from: http://dx.doi.org/10.1016/j.virol.2013.05.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doan T, Melvold R, Viselli S, et al. Lippincott’s Illustrated Reviews: Immunology. Wolters Kluwer Health/Lippincott Williams & Wilkins; 2008. [Google Scholar]
- 42.Donaldson MM, Mackintosh LJ, Bodily JM, et al. An interaction between human papillomavirus 16 E2 and TopBP1 is required for optimum viral DNA replication and episomal genome establishment. J Virol. 2012;86(23):12806–12815. doi: 10.1128/JVI.01002-12. (Available from: http://dx.doi.org/10.1128/JVI.01002-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doorbar J, Quint W, Banks L, et al. The biology and life-cycle of human papillomaviruses. Vaccine. 2012;30(Suppl. 5):F55–F70. doi: 10.1016/j.vaccine.2012.06.083. (Available from: http://dx.doi.org/10.1016/j.vaccine.2012.06.083) [DOI] [PubMed] [Google Scholar]
- 44.Duensing S, Munger K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002;62(23):7075–7082. [PubMed] [Google Scholar]
- 45.Duensing S, Lee LY, Duensing A, et al. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. PNAS. 2000;97(18):10002–10007. doi: 10.1073/pnas.170093297. (Available from: http://dx.doi.org/10.1073/pnas.170093297) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dyson N, Howley PM, Munger K, et al. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243(4893):934–937. doi: 10.1126/science.2537532. (Available from: http://dx.doi.org/10.1126/science.2537532) [DOI] [PubMed] [Google Scholar]
- 47.Edwards TG, Helmus MJ, Koeller K, et al. Human papillomavirus episome stability is reduced by aphidicolin and controlled by DNA damage response pathways. J Virol. 2013;87(7):3979–3989. doi: 10.1128/JVI.03473-12. (Available from: http://dx.doi.org/10.1128/JVI.03473-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eilon T, Barash I. Forced activation of Stat5 subjects mammary epithelial cells to DNA damage and preferential induction of the cellular response mechanism during proliferation. J Cell Physiol. 2011;226(3):616–626. doi: 10.1002/jcp.22381. (Available from: http://dx.doi.org/10.1002/jcp.22381) [DOI] [PubMed] [Google Scholar]
- 49.Falck J, Mailand N, Syljuasen RG, et al. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001;410(6830):842–847. doi: 10.1038/35071124. (Available from: http://dx.doi.org/10.1038/35071124) [DOI] [PubMed] [Google Scholar]
- 50.Falck J, Petrini JH, Williams BR, et al. The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat Genet. 2002;30(3):290–294. doi: 10.1038/ng845. (Available from: http://dx.doi.org/10.1038/ng845) [DOI] [PubMed] [Google Scholar]
- 51.Fan X, Liu Y, Heilman SA, et al. Human papillomavirus E7 induces rereplication in response to DNA damage. J Virol. 2013;87(2):1200–1210. doi: 10.1128/JVI.02038-12. (Available from: http://dx.doi.org/10.1128/JVI.02038-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ferbeyre G, Moriggl R. The role of STAT5 transcription factors as tumor suppressors or oncogenes. Biochim Biophys Acta. 2011;1815(1):104–114. doi: 10.1016/j.bbcan.2010.10.004. (Available from: http://dx.doi.org/10.1016/j.bbcan.2010.10.004) [DOI] [PubMed] [Google Scholar]
- 53.Fernandez-Capetillo O, Lee A, Nussenzweig M, et al. H2AX: the histone guardian of the genome. DNA Repair (Amst) 2004;3(8-9):959–967. doi: 10.1016/j.dnarep.2004.03.024. (Available from: http://dx.doi.org/10.1016/j.dnarep.2004.03.024) [DOI] [PubMed] [Google Scholar]
- 54.Floyd SR, Pacold ME, Huang Q, et al. The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature. 2013;498(7453):246–250. doi: 10.1038/nature12147. (Available from: http://dx.doi.org/10.1038/nature12147) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fradet-Turcotte A, Moody C, Laimins LA, et al. Nuclear export of human papillomavirus type 31 E1 is regulated by Cdk2 phosphorylation and required for viral genome maintenance. J Virol. 2010;84(22):11747–11760. doi: 10.1128/JVI.01445-10. (Available from: http://dx.doi.org/10.1128/JVI.01445-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frattini MG, Laimins LA. Binding of the human papillomavirus E1 origin-recognition protein is regulated through complex formation with the E2 enhancer-binding protein. PNAS. 1994;91(26):12398–12402. doi: 10.1073/pnas.91.26.12398. (Available from: http://dx.doi.org/10.1073/pnas.91.26.12398) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Galloway DA, Laimins LA. Human papillomaviruses: shared and distinct pathways for pathogenesis. Curr Opin Virol. 2015;14:87–92. doi: 10.1016/j.coviro.2015.09.001. (Available from: http://dx.doi.org/10.1016/j.coviro.2015.09.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gatei M, Zhou BB, Hobson K, et al. Ataxia telangiectasia mutated (ATM) kinase and ATM and Rad3 related kinase mediate phosphorylation of Brca1 at distinct and overlapping sites. In vivo assessment using phospho-specific antibodies. J Biol Chem. 2001;276(20):17276–17280. doi: 10.1074/jbc.M011681200. (Available from: http://dx.doi.org/10.1074/jbc.M011681200) [DOI] [PubMed] [Google Scholar]
- 59.Gauson EJ, Donaldson MM, Dornan ES, et al. Evidence supporting a role for TopBP1 and Brd4 in the initiation but not continuation of human papillomavirus 16 E1/E2-mediated DNA replication. J Virol. 2015;89(9):4980–4991. doi: 10.1128/JVI.00335-15. (Available from: http://dx.doi.org/10.1128/JVI.00335-15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ghavidel A, Schultz MC. TATA binding protein-associated CK2 transduces DNA damage signals to the RNA polymerase III transcriptional machinery. Cell. 2001;106(5):575–584. doi: 10.1016/s0092-8674(01)00473-1. (Available from: http://dx.doi.org/10.1016/S0092-8674(01)00473-1) [DOI] [PubMed] [Google Scholar]
- 61.Gillespie KA, Mehta KP, Laimins LA, et al. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J Virol. 2012;86(17):9520–9526. doi: 10.1128/JVI.00247-12. (Available from: http://dx.doi.org/10.1128/JVI.00247-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goodarzi AA, Jonnalagadda JC, Douglas P, et al. Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J. 2004;23(22):4451–4461. doi: 10.1038/sj.emboj.7600455. (Available from: http://dx.doi.org/10.1038/sj.emboj.7600455) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goodarzi AA, Noon AT, Deckbar D, et al. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell. 2008;31(2):167–177. doi: 10.1016/j.molcel.2008.05.017. (Available from: http://dx.doi.org/10.1016/j.molcel.2008.05.017) [DOI] [PubMed] [Google Scholar]
- 64.Groner B. Transcription factor regulation in mammary epithelial cells. Domest Anim Endocrinol. 2002;23(1-2):25–32. doi: 10.1016/s0739-7240(02)00142-x. (Available from: http://dx.doi.org/10.1016/S0739-7240(02)00142-X) [DOI] [PubMed] [Google Scholar]
- 65.Gunasekharan V, Laimins LA. Human papillomaviruses modulate microRNA 145 expression to directly control genome amplification. J Virol. 2013;87(10):6037–6043. doi: 10.1128/JVI.00153-13. (Available from: http://dx.doi.org/10.1128/JVI.00153-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gunasekharan V, Hache G, Laimins L. Differentiation-dependent changes in levels of C/EBPβ repressors and activators regulate human papillomavirus type 31 late gene expression. J Virol. 2012;86(9):5393–5398. doi: 10.1128/JVI.07239-11. (Available from: http://dx.doi.org/10.1128/JVI.07239-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hartley KA, Alexander KA. Human TATA binding protein inhibits human papillomavirus type 11 DNA replication by antagonizing E1-E2 protein complex formation on the viral origin of replication. J Virol. 2002;76(10):5014–5023. doi: 10.1128/JVI.76.10.5014-5023.2002. (Available from: http://dx.doi.org/10.1128/JVI.76.10.5014-5023.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hebner C, Beglin M, Laimins LA. Human papillomavirus E6 proteins mediate resistance to interferon-induced growth arrest through inhibition of p53 acetylation. J Virol. 2007;81(23):12740–12747. doi: 10.1128/JVI.00987-07. (Available from: http://dx.doi.org/10.1128/JVI.00987-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hebner CM, Laimins LA. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity. Rev Med Virol. 2006;16(2):83–97. doi: 10.1002/rmv.488. (Available from: http://dx.doi.org/10.1002/rmv.488) [DOI] [PubMed] [Google Scholar]
- 70.Heltemes-Harris LM, Farrar MA. The role of STAT5 in lymphocyte development and transformation. Curr Opin Immunol. 2012;24(2):146–152. doi: 10.1016/j.coi.2012.01.015. (Available from: http://dx.doi.org/10.1016/j.coi.2012.01.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Herrero R, Hildesheim A, Bratti C, et al. Population-based study of human papillomavirus infection and cervical neoplasia in rural Costa Rica. J Natl Cancer Inst. 2000;92(6):464–474. doi: 10.1093/jnci/92.6.464. (Available from: http://dx.doi.org/10.1093/jnci/92.6.464) [DOI] [PubMed] [Google Scholar]
- 72.Ho GY, Burk RD, Klein S, et al. Persistent genital human papillomavirus infection as a risk factor for persistent cervical dysplasia. J Natl Cancer Inst. 1995;87(18):1365–1371. doi: 10.1093/jnci/87.18.1365. (Available from: http://dx.doi.org/10.1093/jnci/87.18.1365) [DOI] [PubMed] [Google Scholar]
- 73.Hollingworth R, Grand RJ. Modulation of DNA damage and repair pathways by human tumour viruses. Viruses. 2015;7(5):2542–2591. doi: 10.3390/v7052542. (Available from: http://dx.doi.org/10.3390/v7052542) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hong S, Laimins LA. The JAK-STAT transcriptional regulator, STAT-5, activates the ATM DNA damage pathway to induce HPV 31 genome amplification upon epithelial differentiation. PLoS Pathog. 2013;9(4):e1003295. doi: 10.1371/journal.ppat.1003295. (Available from: http://dx.doi.org/10.1371/journal.ppat.1003295) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hong S, Laimins LA. Regulation of the life cycle of HPVs by differentiation and the DNA damage response. Future Microbiol. 2013;8(12):1547–1557. doi: 10.2217/fmb.13.127. (Available from: http://dx.doi.org/10.2217/fmb.13.127) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hong S, Mehta KP, Laimins LA. Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J Virol. 2011;85(18):9486–9494. doi: 10.1128/JVI.05007-11. (Available from: http://dx.doi.org/10.1128/JVI.05007-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hong S, Dutta A, Laimins LA. The acetyltransferase Tip60 is a critical regulator of the differentiation-dependent amplification of human papillomaviruses. J Virol. 2015;89(8):4668–4675. doi: 10.1128/JVI.03455-14. (Available from: http://dx.doi.org/10.1128/JVI.03455-14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hong S, Cheng S, Iovane A, et al. STAT-5 regulates transcription of the topoisomerase IIβ-binding protein 1 (TopBP1) gene to activate the ATR pathway and promote human papillomavirus replication. MBio. 2015;6(6):e02006–e02015. doi: 10.1128/mBio.02006-15. (Available from: http://dx.doi.org/10.1128/mBio.02006-15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hoskins EE, Morreale RJ, Werner SP, et al. The fanconi anemia pathway limits human papillomavirus replication. J Virol. 2012;86(15):8131–8138. doi: 10.1128/JVI.00408-12. (Available from: http://dx.doi.org/10.1128/JVI.00408-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Howie HL, Koop JI, Weese J, et al. Beta-HPV 5 and 8 E6 promote p300 degradation by blocking AKT/p300 association. PLoS Pathog. 2011;7(8):e1002211. doi: 10.1371/journal.ppat.1002211. (Available from: http://dx.doi.org/10.1371/journal.ppat.1002211) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hughes FJ, Romanos MA. E1 protein of human papillomavirus is a DNA helicase/ATPase. Nucleic Acids Res. 1993;21(25):5817–5823. doi: 10.1093/nar/21.25.5817. (Available from: http://dx.doi.org/10.1093/nar/21.25.5817) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huibregtse JM, Scheffner M, Howley PM. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991;10(13):4129–4135. doi: 10.1002/j.1460-2075.1991.tb04990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Iftner T, Elbel M, Schopp B, et al. Interference of papillomavirus E6 protein with single-strand break repair by interaction with XRCC1. EMBO J. 2002;21(17):4741–4748. doi: 10.1093/emboj/cdf443. (Available from: http://dx.doi.org/10.1093/emboj/cdf443) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–1078. doi: 10.1038/nature08467. (Available from: http://dx.doi.org/10.1038/nature08467) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jang MK, Shen K, McBride AA. Papillomavirus genomes associate with Brd4 to replicate at fragile sites in the host genome. PLoS Pathog. 2014;10(5):e1004117. doi: 10.1371/journal.ppat.1004117. (Available from: http://dx.doi.org/10.1371/journal.ppat.1004117) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Janssens S, Tschopp J. Signals from within: the DNA-damage-induced NF-κB response. Cell Death Differ. 2006;13(5):773–784. doi: 10.1038/sj.cdd.4401843. (Available from: http://dx.doi.org/10.1038/sj.cdd.4401843) [DOI] [PubMed] [Google Scholar]
- 87.Jazayeri A, Falck J, Lukas C, et al. ATM-and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8(1):37–45. doi: 10.1038/ncb1337. (Available from: http://dx.doi.org/10.1038/ncb1337) [DOI] [PubMed] [Google Scholar]
- 88.Kadaja M, Isok-Paas H, Laos T, et al. Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog. 2009;5(4):e1000397. doi: 10.1371/journal.ppat.1000397. (Available from: http://dx.doi.org/10.1371/journal.ppat.1000397) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kajitani N, Satsuka A, Kawate A, et al. Productive lifecycle of human papillomaviruses that depends upon squamous epithelial differentiation. Front Microbiol. 2012;3:152. doi: 10.3389/fmicb.2012.00152. (Available from: http://dx.doi.org/10.3389/fmicb.2012.00152) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kanginakudru S, Desmet M, Thomas Y, et al. Levels of the E2 interacting protein TopBP1 modulate papillomavirus maintenance stage replication. Virology. 2015;478:135–142. doi: 10.1016/j.virol.2015.01.011. (Available from: http://dx.doi.org/10.1016/j.virol.2015.01.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kitagawa R, Bakkenist CJ, McKinnon PJ, et al. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev. 2004;18(12):1423–1438. doi: 10.1101/gad.1200304. (Available from: http://dx.doi.org/10.1101/gad.1200304) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kivi N, Greco D, Auvinen P, et al. Genes involved in cell adhesion, cell motility and mitogenic signaling are altered due to HPV 16 E5 protein expression. Oncogene. 2008;27(18):2532–2541. doi: 10.1038/sj.onc.1210916. (Available from: http://dx.doi.org/10.1038/sj.onc.1210916) [DOI] [PubMed] [Google Scholar]
- 93.Koganti S, Hui-Yuen J, McAllister S, et al. STAT3 interrupts ATR-Chk1 signaling to allow oncovirus-mediated cell proliferation. PNAS. 2014;111(13):4946–4951. doi: 10.1073/pnas.1400683111. (Available from: http://dx.doi.org/10.1073/pnas.1400683111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kumagai A, Dunphy WG. Repeated phosphopeptide motifs in Claspin mediate the regulated binding of Chk1. Nat Cell Biol. 2003;5(2):161–165. doi: 10.1038/ncb921. (Available from: http://dx.doi.org/10.1038/ncb921) [DOI] [PubMed] [Google Scholar]
- 95.Kumagai A, Lee J, Yoo HY, et al. TopBP1 activates the ATR-ATRIP complex. Cell. 2006;124(5):943–955. doi: 10.1016/j.cell.2005.12.041. (Available from: http://dx.doi.org/10.1016/j.cell.2005.12.041) [DOI] [PubMed] [Google Scholar]
- 96.Langsfeld ES, Bodily JM, Laimins LA. The deacetylase sirtuin 1 regulates human papillomavirus replication by modulating histone acetylation and recruitment of DNA damage factors NBS1 and RAD51 to viral genomes. PLoS Pathog. 2015;11(9):e1005181. doi: 10.1371/journal.ppat.1005181. (Available from: http://dx.doi.org/10.1371/journal.ppat.1005181) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee J, Kumagai A, Dunphy WG. Positive regulation of Wee1 by Chk1 and 14-3-3 proteins. Mol Biol Cell. 2001;12(3):551–563. doi: 10.1091/mbc.12.3.551. (Available from: http://dx.doi.org/10.1091/mbc.12.3.551) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lee JH, Paull TT. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 2004;304(5667):93–96. doi: 10.1126/science.1091496. (Available from: http://dx.doi.org/10.1126/science.1091496) [DOI] [PubMed] [Google Scholar]
- 99.Lehoux M, Gagnon D, Archambault J. E1-mediated recruitment of a UAF1-USP deubiquitinase complex facilitates human papillomavirus DNA replication. J Virol. 2014;88(15):8545–8555. doi: 10.1128/JVI.00379-14. (Available from: http://dx.doi.org/10.1128/JVI.00379-14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liao S, Deng D, Zhang W, et al. Human papillomavirus 16/18 E5 promotes cervical cancer cell proliferation, migration and invasion in vitro and accelerates tumor growth in vivo. Oncol Rep. 2013;29(1):95–102. doi: 10.3892/or.2012.2106. (Available from: http://dx.doi.org/10.3892/or.2012.2106) [DOI] [PubMed] [Google Scholar]
- 101.Liu K, Lin FT, Ruppert JM, et al. Regulation of E2F1 by BRCT domain-containing protein TopBP1. Mol Cell Biol. 2003;23(9):3287–3304. doi: 10.1128/MCB.23.9.3287-3304.2003. (Available from: http://dx.doi.org/10.1128/MCB.23.9.3287-3304.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu Q, Guntuku S, Cui XS, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint. Genes Dev. 2000;14(12):1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 103.Longworth MS, Laimins LA. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol Mol Biol Rev. 2004;68(2):362–372. doi: 10.1128/MMBR.68.2.362-372.2004. (Available from: http://dx.doi.org/10.1128/MMBR.68.2.362-372.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Longworth MS, Wilson R, Laimins LA. HPV31 E7 facilitates replication by activating E2F2 transcription through its interaction with HDACs. EMBO J. 2005;24(10):1821–1830. doi: 10.1038/sj.emboj.7600651. (Available from: http://dx.doi.org/10.1038/sj.emboj.7600651) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481(7381):287–294. doi: 10.1038/nature10760. (Available from: http://dx.doi.org/10.1038/nature10760) [DOI] [PubMed] [Google Scholar]
- 106.Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282(5395):1893–1897. doi: 10.1126/science.282.5395.1893. (Available from: http://dx.doi.org/10.1126/science.282.5395.1893) [DOI] [PubMed] [Google Scholar]
- 107.Matt S, Hofmann TG. The DNA damage-induced cell death response: a roadmap to kill cancer cells. Cell Mol Life Sci. 2016;73(15):2829–2850. doi: 10.1007/s00018-016-2130-4. (Available from: http://dx.doi.org/10.1007/s00018-016-2130-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Maya R, Balass M, Kim ST, et al. ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev. 2001;15(9):1067–1077. doi: 10.1101/gad.886901. (Available from: http://dx.doi.org/10.1101/gad.886901) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McBride AA. The papillomavirus E2 proteins. Virology. 2013;445(1-2):57–79. doi: 10.1016/j.virol.2013.06.006. (Available from: http://dx.doi.org/10.1016/j.virol.2013.06.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McFadden K, Luftig MA. Interplay between DNA tumor viruses and the host DNA damage response. Curr Top Microbiol Immunol. 2013;371:229–257. doi: 10.1007/978-3-642-37765-5_9. (Available from: http://dx.doi.org/10.1007/978-3-642-37765-5_9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McKinney CC, Hussmann KL, McBride AA. The role of the DNA damage response throughout the papillomavirus life cycle. Viruses. 2015;7(5):2450–2469. doi: 10.3390/v7052450. (Available from: http://dx.doi.org/10.3390/v7052450) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.McPhillips MG, Oliveira JG, Spindler JE, et al. Brd4 is required for E2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J Virol. 2006;80(19):9530–9543. doi: 10.1128/JVI.01105-06. (Available from: http://dx.doi.org/10.1128/JVI.01105-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mehta K, Gunasekharan V, Satsuka A, et al. Human papillomaviruses activate and recruit SMC1 cohesin proteins for the differentiation-dependent life cycle through association with ctcf insulators. PLoS Pathog. 2015;11(4):e1004763. doi: 10.1371/journal.ppat.1004763. (Available from: http://dx.doi.org/10.1371/journal.ppat.1004763) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Melar-New M, Laimins LA. Human papillomaviruses modulate expression of microRNA 203 upon epithelial differentiation to control levels of p63 proteins. J Virol. 2010;84(10):5212–5221. doi: 10.1128/JVI.00078-10. (Available from: http://dx.doi.org/10.1128/JVI.00078-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mighty KK, Laimins LA. p63 is necessary for the activation of human papillomavirus late viral functions upon epithelial differentiation. J Virol. 2011;85(17):8863–8869. doi: 10.1128/JVI.00750-11. (Available from: http://dx.doi.org/10.1128/JVI.00750-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Miller KM, Tjeertes JV, Coates J, et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010;17(99):1144–1151. doi: 10.1038/nsmb.1899. (Available from: http://dx.doi.org/10.1038/nsmb.1899) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Moiseeva O, Mallette FA, Mukhopadhyay UK, et al. DNA damage signaling and p53-dependent senescence after prolonged β-interferon stimulation. Mol Biol Cell. 2006;17(4):1583–1592. doi: 10.1091/mbc.E05-09-0858. (Available from: http://dx.doi.org/10.1091/mbc.E05-09-0858) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Moody CA, Laimins LA. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009;5(10):e1000605. doi: 10.1371/journal.ppat.1000605. (Available from: http://dx.doi.org/10.1371/journal.ppat.1000605) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10(8):550–560. doi: 10.1038/nrc2886. (Available from: http://dx.doi.org/10.1038/nrc2886) [DOI] [PubMed] [Google Scholar]
- 120.Moody CA, Fradet-Turcotte A, Archambault J, et al. Human papillomaviruses activate caspases upon epithelial differentiation to induce viral genome amplification. PNAS. 2007;104(49):19541–19546. doi: 10.1073/pnas.0707947104. (Available from: http://dx.doi.org/10.1073/pnas.0707947104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Munger K, Howley PM. Human papillomavirus immortalization and transformation functions. Virus Res. 2002;89(2):213–228. doi: 10.1016/s0168-1702(02)00190-9. (Available from: http://dx.doi.org/10.1016/S0168-1702(02)00190-9) [DOI] [PubMed] [Google Scholar]
- 122.Munger K, Werness BA, Dyson N, et al. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989;8(13):4099–4105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Myers JS, Cortez D. Rapid activation of ATR by ionizing radiation requires ATM and MRE11. J Biol Chem. 2006;281(14):9346–9350. doi: 10.1074/jbc.M513265200. (Available from: http://dx.doi.org/10.1074/jbc.M513265200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Myers K, Gagou ME, Zuazua-Villar P, et al. ATR and Chk1 suppress a caspase-3-dependent apoptotic response following DNA replication stress. PLoS Genet. 2009;5(1):e1000324. doi: 10.1371/journal.pgen.1000324. (Available from: http://dx.doi.org/10.1371/journal.pgen.1000324) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nakahara T, Tanaka K, Ohno S, et al. Activation of NF-κB by human papillomavirus 16 E1 limits E1-dependent viral replication through degradation of E1. J Virol. 2015;89(9):5040–5059. doi: 10.1128/JVI.00389-15. (Available from: http://dx.doi.org/10.1128/JVI.00389-15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nelson LM, Rose RC, Moroianu J. Nuclear import strategies of high risk HPV16 L1 major capsid protein. J Biol Chem. 2002;277(26):23958–23964. doi: 10.1074/jbc.M200724200. (Available from: http://dx.doi.org/10.1074/jbc.M200724200) [DOI] [PubMed] [Google Scholar]
- 127.Nguyen CL, McLaughlin-Drubin ME, Munger K. Delocalization of the microtubule motor dynein from mitotic spindles by the human papillomavirus E7 oncoprotein is not sufficient for induction of multipolar mitoses. Cancer Res. 2008;68(21):8715–8722. doi: 10.1158/0008-5472.CAN-08-1303. (Available from: http://dx.doi.org/10.1158/0008-5472.CAN-08-1303) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nuovo GJ, Wu X, Volinia S, et al. Strong inverse correlation between microRNA-125b and human papillomavirus DNA in productive infection. Diagn Mol Pathol. 2010;19(3):135–143. doi: 10.1097/PDM.0b013e3181c4daaa. (Available from: http://dx.doi.org/10.1097/PDM.0b013e3181c4daaa) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.O'Connor M, Bernard HU. Oct-1 activates the epithelial-specific enhancer of human papillomavirus type 16 via a synergistic interaction with NFI at a conserved composite regulatory element. Virology. 1995;207(1):77–88. doi: 10.1006/viro.1995.1053. (Available from: http://dx.doi.org/10.1006/viro.1995.1053) [DOI] [PubMed] [Google Scholar]
- 130.O'Connor MJ. Targeting the DNA damage response in cancer. Mol Cell. 2015;60(4):547–560. doi: 10.1016/j.molcel.2015.10.040. (Available from: http://dx.doi.org/10.1016/j.molcel.2015.10.040) [DOI] [PubMed] [Google Scholar]
- 131.O'Driscoll M, Ruiz-Perez VL, Woods CG, et al. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in seckel syndrome. Nat Genet. 2003;33(4):497–501. doi: 10.1038/ng1129. (Available from: http://dx.doi.org/10.1038/ng1129) [DOI] [PubMed] [Google Scholar]
- 132.Offord EA, Beard P. A member of the activator protein 1 family found in keratinocytes but not in fibroblasts required for transcription from a human papillomavirus type 18 promoter. J Virol. 1990;64(10):4792–4798. doi: 10.1128/jvi.64.10.4792-4798.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Oliveira JG, Colf LA, McBride AA. Variations in the association of papillomavirus E2 proteins with mitotic chromosomes. PNAS. 2006;103(4):1047–1052. doi: 10.1073/pnas.0507624103. (Available from: http://dx.doi.org/10.1073/pnas.0507624103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Park JS, Kim EJ, Kwon HJ, et al. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J Biol Chem. 2000;275(10):6764–6769. doi: 10.1074/jbc.275.10.6764. (Available from: http://dx.doi.org/10.1074/jbc.275.10.6764) [DOI] [PubMed] [Google Scholar]
- 135.Patel D, Huang SM, Baglia LA, et al. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J. 1999;18(18):5061–5072. doi: 10.1093/emboj/18.18.5061. (Available from: http://dx.doi.org/10.1093/emboj/18.18.5061) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pearl LH, Schierz AC, Ward SE, et al. Therapeutic opportunities within the DNA damage response. Nat Rev Cancer. 2015;15(3):166–180. doi: 10.1038/nrc3891. (Available from: http://dx.doi.org/10.1038/nrc3891) [DOI] [PubMed] [Google Scholar]
- 137.Pedroza-Torres A, Lopez-Urrutia E, Garcia-Castillo V, et al. MicroRNAs in cervical cancer: evidences for a miRNA profile deregulated by hpv and its impact on radio-resistance. Molecules. 2014;19(5):6263–6281. doi: 10.3390/molecules19056263. (Available from: http://dx.doi.org/10.3390/molecules19056263) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Poddar A, Reed SC, McPhillips MG, et al. The human papillomavirus type 8 E2 tethering protein targets the ribosomal DNA loci of host mitotic chromosomes. J Virol. 2009;83(2):640–650. doi: 10.1128/JVI.01936-08. (Available from: http://dx.doi.org/10.1128/JVI.01936-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Reinson T, Toots M, Kadaja M, et al. Engagement of the ATR-dependent DNA damage response at the human papillomavirus 18 replication centers during the initial amplification. J Virol. 2013;87(2):951–964. doi: 10.1128/JVI.01943-12. (Available from: http://dx.doi.org/10.1128/JVI.01943-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rincon-Orozco B, Halec G, Rosenberger S, et al. Epigenetic silencing of interferon-κ in human papillomavirus type 16-positive cells. Cancer Res. 2009;69(22):8718–8725. doi: 10.1158/0008-5472.CAN-09-0550. (Available from: http://dx.doi.org/10.1158/0008-5472.CAN-09-0550) [DOI] [PubMed] [Google Scholar]
- 141.Ronco LV, Karpova AY, Vidal M, et al. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998;12(13):2061–2072. doi: 10.1101/gad.12.13.2061. (Available from: http://dx.doi.org/10.1101/gad.12.13.2061) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sakakibara N, Mitra R, McBride AA. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J Virol. 2011;85(17):8981–8995. doi: 10.1128/JVI.00541-11. (Available from: http://dx.doi.org/10.1128/JVI.00541-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Satsuka A, Mehta K, Laimins L. p38MAPK and MK2 pathways are important for the differentiation-dependent human papillomavirus life cycle. J Virol. 2015;89(3):1919–1924. doi: 10.1128/JVI.02712-14. (Available from: http://dx.doi.org/10.1128/JVI.02712-14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Scheffner M, Werness BA, Huibregtse JM, et al. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63(6):1129–1136. doi: 10.1016/0092-8674(90)90409-8. (Available from: http://dx.doi.org/10.1016/0092-8674(90)90409-8) [DOI] [PubMed] [Google Scholar]
- 145.Scheffner M, Huibregtse JM, Vierstra RD, et al. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75(3):495–505. doi: 10.1016/0092-8674(93)90384-3. (Available from: http://dx.doi.org/10.1016/0092-8674(93)90384-3) [DOI] [PubMed] [Google Scholar]
- 146.Sedman J, Stenlund A. Co-operative interaction between the initiator E1 and the transcriptional activator E2 is required for replicator specific DNA replication of bovine papillomavirus in vivo and in vitro. EMBO J. 1995;14(24):6218–6228. doi: 10.1002/j.1460-2075.1995.tb00312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Seebode C, Lehmann J, Emmert S. Photocarcinogenesis and skin cancer prevention strategies. Anticancer Res. 2016;36(3):1371–1378. [PubMed] [Google Scholar]
- 148.Shreeram S, Demidov ON, Hee WK, et al. Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol Cell. 2006;23(5):757–764. doi: 10.1016/j.molcel.2006.07.010. (Available from: http://dx.doi.org/10.1016/j.molcel.2006.07.010) [DOI] [PubMed] [Google Scholar]
- 149.Smith J, Tho LM, Xu N, et al. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. (Available from: http://dx.doi.org/10.1016/B978-0-12-380888-2.00003-0) [DOI] [PubMed] [Google Scholar]
- 150.Smith JA, White EA, Sowa ME, et al. Genome-wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. PNAS. 2010;107(8):3752–3757. doi: 10.1073/pnas.0914818107. (Available from: http://dx.doi.org/10.1073/pnas.0914818107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Song S, Gulliver GA, Lambert PF. Human papillomavirus type 16 E6 and E7 oncogenes abrogate radiation-induced DNA damage responses in vivo through p53-dependent and p53-independent pathways. PNAS. 1998;95(5):2290–2295. doi: 10.1073/pnas.95.5.2290. (Available from: http://dx.doi.org/10.1073/pnas.95.5.2290) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sorensen CS, Hansen LT, Dziegielewski J, et al. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005;7(2):195–201. doi: 10.1038/ncb1212. (Available from: http://dx.doi.org/10.1038/ncb1212) [DOI] [PubMed] [Google Scholar]
- 153.Spardy N, Duensing A, Charles D, et al. The human papillomavirus type 16 E7 oncoprotein activates the Fanconi anemia (FA) pathway and causes accelerated chromosomal instability in FA cells. J Virol. 2007;81(23):13265–13270. doi: 10.1128/JVI.01121-07. (Available from: http://dx.doi.org/10.1128/JVI.01121-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Spardy N, Duensing A, Hoskins EE, et al. HPV-16 E7 reveals a link between DNA replication stress, fanconi anemia D2 protein, and alternative lengthening of telomere-associated promyelocytic leukemia bodies. Cancer Res. 2008;68(23):9954–9963. doi: 10.1158/0008-5472.CAN-08-0224. (Available from: http://dx.doi.org/10.1158/0008-5472.CAN-08-0224) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Spardy N, Covella K, Cha E, et al. Human papillomavirus 16 E7 oncoprotein attenuates DNA damage checkpoint control by increasing the proteolytic turnover of claspin. Cancer Res. 2009;69(17):7022–7029. doi: 10.1158/0008-5472.CAN-09-0925. (Available from: http://dx.doi.org/10.1158/0008-5472.CAN-09-0925) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sperka T, Wang J, Rudolph KL. DNA damage checkpoints in stem cells, ageing and cancer. Nat Rev Mol Cell Biol. 2012;13(9):579–590. doi: 10.1038/nrm3420. (Available from: http://dx.doi.org/10.1038/nrm3420) [DOI] [PubMed] [Google Scholar]
- 157.Srivenugopal KS, Ali-Osman F. The DNA repair protein, O6-methylguanine-DNA methyltransferase is a proteolytic target for the E6 human papillomavirus oncoprotein. Oncogene. 2002;21(38):5940–5945. doi: 10.1038/sj.onc.1205762. (Available from: http://dx.doi.org/10.1038/sj.onc.1205762) [DOI] [PubMed] [Google Scholar]
- 158.Stanley MA. Epithelial cell responses to infection with human papillomavirus. Clin Microbiol Rev. 2012;25(2):215–222. doi: 10.1128/CMR.05028-11. (Available from: http://dx.doi.org/10.1128/CMR.05028-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Steger G, Corbach S. Dose-dependent regulation of the early promoter of human papillomavirus type 18 by the viral E2 protein. J Virol. 1997;71(1):50–58. doi: 10.1128/jvi.71.1.50-58.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Stubenrauch F, Hummel M, Iftner T, et al. The E8^E2C protein, a negative regulator of viral transcription and replication, is required for extrachromosomal maintenance of human papillomavirus type 31 in keratinocytes. J Virol. 2000;74(3):1178–1186. doi: 10.1128/jvi.74.3.1178-1186.2000. (Available from: http://dx.doi.org/10.1128/JVI.74.3.1178-1186.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Stunkel W, Bernard HU. The chromatin structure of the long control region of human papillomavirus type 16 represses viral oncoprotein expression. J Virol. 1999;73(3):1918–1930. doi: 10.1128/jvi.73.3.1918-1930.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sun Y, Jiang X, Chen S, et al. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. PNAS. 2005;102(37):13182–13187. doi: 10.1073/pnas.0504211102. (Available from: http://dx.doi.org/10.1073/pnas.0504211102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Taniguchi T, Garcia-Higuera I, Xu B, et al. Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell. 2002;109(4):459–472. doi: 10.1016/s0092-8674(02)00747-x. (Available from: http://dx.doi.org/10.1016/S0092-8674(02)00747-X) [DOI] [PubMed] [Google Scholar]
- 164.Thomas JT, Hubert WG, Ruesch MN, et al. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes. PNAS. 1999;96(15):8449–8454. doi: 10.1073/pnas.96.15.8449. (Available from: http://dx.doi.org/10.1073/pnas.96.15.8449) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Thomas M, Narayan N, Pim D, et al. Human papillomaviruses, cervical cancer and cell polarity. Oncogene. 2008;27(55):7018–7030. doi: 10.1038/onc.2008.351. (Available from: http://dx.doi.org/10.1038/onc.2008.351) [DOI] [PubMed] [Google Scholar]
- 166.Thomas MC, Chiang CM. E6 oncoprotein represses p53-dependent gene activation via inhibition of protein acetylation independently of inducing p53 degradation. Mol Cell. 2005;17(2):251–264. doi: 10.1016/j.molcel.2004.12.016. (Available from: http://dx.doi.org/10.1016/j.molcel.2004.12.016) [DOI] [PubMed] [Google Scholar]
- 167.Thurn KT, Thomas S, Raha P, et al. Histone deacetylase regulation of ATM-mediated DNA damage signaling. Mol Cancer Ther. 2013;12(10):2078–2087. doi: 10.1158/1535-7163.MCT-12-1242. (Available from: http://dx.doi.org/10.1158/1535-7163.MCT-12-1242) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Walker M, Black EJ, Oehler V, et al. Chk1 C-terminal regulatory phosphorylation mediates checkpoint activation by de-repression of Chk1 catalytic activity. Oncogene. 2009;28(24):2314–2323. doi: 10.1038/onc.2009.102. (Available from: http://dx.doi.org/10.1038/onc.2009.102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wallace NA, Galloway DA. Manipulation of cellular DNA damage repair machinery facilitates propagation of human papillomaviruses. Semin Cancer Biol. 2014;26:30–42. doi: 10.1016/j.semcancer.2013.12.003. (Available from: http://dx.doi.org/10.1016/j.semcancer.2013.12.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wallace NA, Galloway DA. Novel functions of the human papillomavirus E6 oncoproteins. Annu Rev Virol. 2015;2(1):403–423. doi: 10.1146/annurev-virology-100114-055021. (Available from: http://dx.doi.org/10.1146/annurev-virology-100114-055021) [DOI] [PubMed] [Google Scholar]
- 171.Wallace NA, Robinson K, Howie HL, et al. HPV 5 and 8 E6 abrogate ATR activity resulting in increased persistence of UVB induced DNA damage. PLoS Pathog. 2012;8(7):e1002807. doi: 10.1371/journal.ppat.1002807. (Available from: http://dx.doi.org/10.1371/journal.ppat.1002807) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wallace NA, Gasior SL, Faber ZJ, et al. HPV 5 and 8 E6 expression reduces ATM protein levels and attenuates LINE-1 retrotransposition. Virology. 2013;443(1):69–79. doi: 10.1016/j.virol.2013.04.022. (Available from: http://dx.doi.org/10.1016/j.virol.2013.04.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Wallace NA, Robinson K, Galloway DA. Beta human papillomavirus E6 expression inhibits stabilization of p53 and increases tolerance of genomic instability. J Virol. 2014;88(11):6112–6127. doi: 10.1128/JVI.03808-13. (Available from: http://dx.doi.org/10.1128/JVI.03808-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wallace NA, Robinson K, Howie HL, et al. β-HPV 5 and 8 E6 disrupt homology dependent double strand break repair by attenuating BRCA1 and BRCA2 expression and foci formation. PLoS Pathog. 2015;11(3):e1004687. doi: 10.1371/journal.ppat.1004687. (Available from: http://dx.doi.org/10.1371/journal.ppat.1004687) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wan G, Mathur R, Hu X, et al. miRNA response to DNA damage. Trends Biochem Sci. 2011;36(9):478–484. doi: 10.1016/j.tibs.2011.06.002. (Available from: http://dx.doi.org/10.1016/j.tibs.2011.06.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wang X, Wang HK, Li Y, et al. MicroRNAs are biomarkers of oncogenic human papillomavirus infections. PNAS. 2014;111(11):4262–4267. doi: 10.1073/pnas.1401430111. (Available from: http://dx.doi.org/10.1073/pnas.1401430111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wang Y, Taniguchi T. MicroRNAs and DNA damage response: implications for cancer therapy. Cell Cycle. 2013;12(1):32–42. doi: 10.4161/cc.23051. (Available from: http://dx.doi.org/10.4161/cc.23051) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.White DE, Negorev D, Peng H, et al. KAP1, a novel substrate for PIKK family members, colocalizes with numerous damage response factors at DNA lesions. Cancer Res. 2006;66(24):11594–11599. doi: 10.1158/0008-5472.CAN-06-4138. (Available from: http://dx.doi.org/10.1158/0008-5472.CAN-06-4138) [DOI] [PubMed] [Google Scholar]
- 179.Wise-Draper TM, Wells SI. Papillomavirus E6 and E7 proteins and their cellular targets. Front Biosci. 2008;13:1003–1017. doi: 10.2741/2739. (Available from: http://dx.doi.org/10.2741/2739) [DOI] [PubMed] [Google Scholar]
- 180.Wohlbold L, Merrick KA, De S, et al. Chemical genetics reveals a specific requirement for Cdk2 activity in the DNA damage response and identifies Nbs1 as a Cdk2 substrate in human cells. PLoS Genet. 2012;8(8):e1002935. doi: 10.1371/journal.pgen.1002935. (Available from: http://dx.doi.org/10.1371/journal.pgen.1002935) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wu S, Shi Y, Mulligan P, et al. A YY1-INO80 complex regulates genomic stability through homologous recombination-based repair. Nat Struct Mol Biol. 2007;14(12):1165–1172. doi: 10.1038/nsmb1332. (Available from: http://dx.doi.org/10.1038/nsmb1332) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wu SY, Lee AY, Hou SY, et al. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 2006;20(17):2383–2396. doi: 10.1101/gad.1448206. (Available from: http://dx.doi.org/10.1101/gad.1448206) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yao Z, Cui Y, Watford WT, et al. STAT5a/b are essential for normal lymphoid development and differentiation. PNAS. 2006;103(4):1000–1005. doi: 10.1073/pnas.0507350103. (Available from: http://dx.doi.org/10.1073/pnas.0507350103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Yarden RI, Pardo-Reoyo S, Sgagias M, et al. BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat Genet. 2002;30(3):285–289. doi: 10.1038/ng837. (Available from: http://dx.doi.org/10.1038/ng837) [DOI] [PubMed] [Google Scholar]
- 185.Zhang R, Zhu L, Zhang L, et al. PTEN enhances G2/M arrest in etoposide-treated MCF7 cells through activation of the ATM pathway. Oncol Rep. 2016;35(5):2707–2714. doi: 10.3892/or.2016.4674. (Available from: http://dx.doi.org/10.3892/or.2016.4674) [DOI] [PubMed] [Google Scholar]
- 186.Zhang W, Hong S, Maniar KP, et al. KLF13 regulates the differentiation-dependent human papillomavirus life cycle in keratinocytes through STAT5 and IL-8. Oncogene. 2016;35:5565–5575. doi: 10.1038/onc.2016.97. (Available from: http://dx.doi.org/10.1038/onc.2016.97) [DOI] [PubMed] [Google Scholar]
- 187.Zhang Y, Cho YY, Petersen BL, et al. Ataxia telangiectasia mutated proteins, mapks, and RSK2 are involved in the phosphorylation of STAT3. J Biol Chem. 2003;278(15):12650–12659. doi: 10.1074/jbc.M210368200. (Available from: http://dx.doi.org/10.1074/jbc.M210368200) [DOI] [PubMed] [Google Scholar]
- 188.Zhang Y, Fan S, Meng Q, et al. BRCA1 interaction with human papillomavirus oncoproteins. J Biol Chem. 2005;280(39):33165–33177. doi: 10.1074/jbc.M505124200. (Available from: http://dx.doi.org/10.1074/jbc.M505124200) [DOI] [PubMed] [Google Scholar]
- 189.Zhao H, Jin S, Fan F, et al. Activation of the transcription factor Oct-1 in response to DNA damage. Cancer Res. 2000;60(22):6276–6280. [PubMed] [Google Scholar]
- 190.Zhou BB, Chaturvedi P, Spring K, et al. Caffeine abolishes the mammalian G2/M DNA damage checkpoint by inhibiting ataxia-telangiectasia-mutated kinase activity. J Biol Chem. 2000;275(14):10342–10348. doi: 10.1074/jbc.275.14.10342. (Available from: http://dx.doi.org/10.1074/jbc.275.14.10342) [DOI] [PubMed] [Google Scholar]
- 191.Ziv Y, Bielopolski D, Galanty Y, et al. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM-and KAP-1 dependent pathway. Nat Cell Biol. 2006;8(8):870–876. doi: 10.1038/ncb1446. (Available from: http://dx.doi.org/10.1038/ncb1446) [DOI] [PubMed] [Google Scholar]
- 192.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300(5625):1542–1548. doi: 10.1126/science.1083430. (Available from: http://dx.doi.org/10.1126/science.1083430) [DOI] [PubMed] [Google Scholar]
- 193.Zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2(5):342–350. doi: 10.1038/nrc798. (Available from: http://dx.doi.org/10.1038/nrc798) [DOI] [PubMed] [Google Scholar]
- 194.Zur Hausen H. Papillomaviruses in the causation of human cancers–a brief historical account. Virology. 2009;384(2):260–265. doi: 10.1016/j.virol.2008.11.046. (Available from: http://dx.doi.org/10.1016/j.virol.2008.11.046) [DOI] [PubMed] [Google Scholar]