

ABSTRACT
The normal cellular prion protein (PrPC) resides in detergent-resistant outer membrane lipid rafts in which conversion to the pathogenic misfolded form is believed to occur. Once misfolding occurs, the pathogenic isoform polymerizes into highly stable amyloid fibrils. In vitro assays have demonstrated an intimate association between prion conversion and lipids, specifically phosphatidylethanolamine, which is a critical cofactor in the formation of synthetic infectious prions. In the current work, we demonstrate an alternative inhibitory function of lipids in the prion conversion process as assessed in vitro by real-time quaking-induced conversion (RT-QuIC). Using an alcohol-based extraction technique, we removed the lipid content from chronic wasting disease (CWD)-infected white-tailed deer brain homogenates and found that lipid extraction enabled RT-QuIC detection of CWD prions in a 2-log10-greater concentration of brain sample. Conversely, addition of brain-derived lipid extracts to CWD prion brain or lymph node samples inhibited amyloid formation in a dose-dependent manner. Subsequent lipid analysis demonstrated that this inhibitory function was restricted to the polar lipid fraction in brain. We further investigated three phospholipids commonly found in lipid membranes, phosphatidylethanolamine, phosphatidylcholine, and phosphatidylinositol, and found all three similarly inhibited RT-QuIC. These results demonstrating polar-lipid, and specifically phospholipid, inhibition of prion-seeded amyloid formation highlight the diverse roles lipid constituents may play in the prion conversion process.
IMPORTANCE Prion conversion is likely influenced by lipid interactions, given the location of normal prion protein (PrPC) in lipid rafts and lipid cofactors generating infectious prions in in vitro models. Here, we use real-time quaking-induced conversion (RT-QuIC) to demonstrate that endogenous brain polar lipids can inhibit prion-seeded amyloid formation, suggesting that prion conversion is guided by an environment of proconversion and anticonversion lipids. These experiments also highlight the applicability of RT-QuIC to identify potential therapeutic inhibitors of prion conversion.
KEYWORDS: prions, real-time quaking-induced conversion
INTRODUCTION
The normal cellular prion protein (PrPC) is intimately associated with the outer lipid bilayer of the plasma membrane. PrPC is located within detergent-resistant lipid rafts and anchored to the outer membrane through a C-terminal glycophosphatidylinositol (GPI) anchor (1–3). This cellular location is believed to be crucial for the conformational misfolding that creates the infectious pathogenic isoform of prion diseases (PrPRES) (4, 5). The PrPC pathogenic conformational change to PrPRES has been shown to occur at the cell surface, and disruption of lipid rafts, with the accompanying dispersal of PrPC, prevents prion infection in cultured neurons (6–8). Additionally, studies using recombinant PrPC to create de novo infectious prion have identified lipids as a cofactor facilitating the pathogenic conversion (9). An important finding in understanding in vitro prion conversion was the observation that phosphatidylethanolamine (PE) in particular has been isolated as an efficient cofactor of the conversion process for every animal species prion examined (10).
Real-time quaking-induced conversion (RT-QuIC) is an amyloid-seeding assay that uses prion amyloid oligomers or fibrils as the seed to initiate amyloid formation and elongation from recombinant PrPC (rPrPC) substrate, which is detected by thioflavin T (ThT) binding and fluorescence (11, 12). Similar to other assays using rPrPC, amyloid conversion in RT-QuIC is influenced by cofactors and inhibitors present in the test sample milieu. Previous reports (personal and multiple investigators' observations) have consistently demonstrated that higher concentrations of brain homogenate are unable to seed the RT-QuIC reaction, due to presumed inhibitors of the amyloid formation reaction (12–14). To analyze brain homogenates for PrPRES seeding activity, samples must be diluted to 10−3 or 10−4 for initial RT-QuIC detection to achieve a linear dilution range (e.g., from 10−4 to 10−7), consistent with the removal of reaction inhibitors. The identification of these endogenous amyloid formation inhibitors present in certain sample types may not only enhance PrPRES detection by RT-QuIC, but illuminate biologic features of the prion conversion process.
Through the development of methods to detect prion-seeding activity in fixed paraffin-embedded tissues using RT-QuIC (FPE RT-QuIC), we were able to detect amyloid-seeding activity at higher sample concentrations, 10−1 and 10−2 dilutions, than previously possible for brain homogenates (15). We then endeavored to identify the components of brain homogenate responsible for inhibiting the RT-QuIC amyloid formation reaction. Here, we demonstrate that endogenous lipids in brain homogenate inhibit prion-seeded RT-QuIC amyloid formation in vitro. Our data suggest prion conversion in vitro is guided by a balance of proconversion and anticonversion lipids.
RESULTS
Identification of lipids as inhibitors of prion-seeded amyloid formation.
Our experiments performed during development of the FPE RT-QuIC method demonstrated that RT-QuIC amyloid seeding was achieved at higher seed concentrations of fixed paraffin-embedded (FPE) samples than had been previously reported using conventional frozen brain to prepare homogenates (15). Amyloid seeding in FPE brain-derived samples had a linear detection range extending from 10−1 to 10−7, whereas non-FPE frozen brain homogenate samples displayed a linear range extending from 10−4 to 10−8, likely due to inhibitors at high concentrations and consistent with published literature (Fig. 1A) (12, 13). In contrast, comparable assay of retropharyngeal lymph node homogenates did not inhibit amyloid formation at high tissue concentrations and successfully seeded RT-QuIC amyloid formation (Fig. 1B). These observations indicated that a component of brain homogenates not present to the same degree in lymph node homogenates could inhibit prion-seeded amyloid formation and that the FPE sample preparation protocol was capable of removing this inhibitor.
FIG 1.
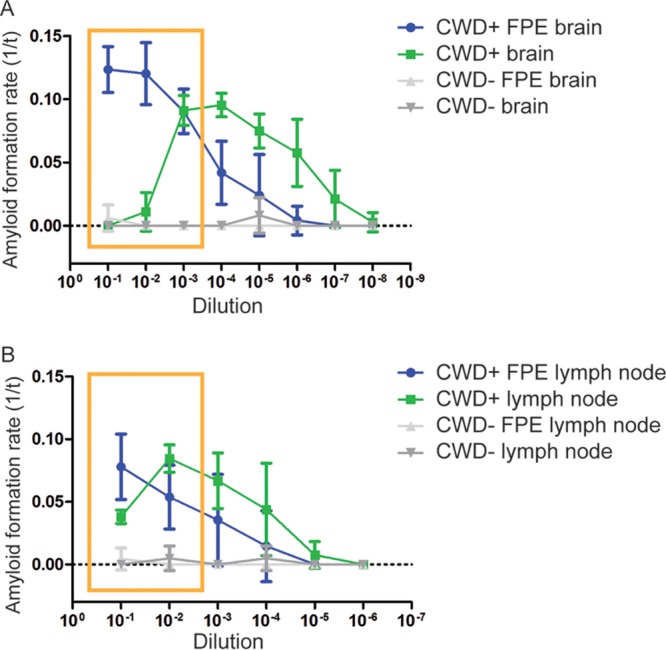
RT-QuIC dilutional series of FPE and frozen tissue homogenates. (A) RT-QuIC dilutional series of FPE and frozen brain homogenate samples. The FPE protocol allowed prion-templated RT-QuIC amyloid formation with higher sample seed concentrations, extending the linear range of detection from 10−1 to 10−7, while inhibitors in higher concentrations of brain homogenates prevented amyloid formation (yellow box). The data are displayed as means and standard errors of the mean (SEM) from 2 experiments with 8 replicates. (B) RT-QuIC dilutional series of FPE and frozen lymph node homogenate samples. In contrast to brain (A), higher concentrations of frozen lymph node homogenates were able to seed the RT-QuIC reaction, similar to FPE lymph node homogenates (yellow box), allowing detection at all dilutions examined, and displayed a linear range from 10−2 to 10−6. The data are displayed as means and SEM from 2 experiments with 8 replicates.
We then analyzed each component of the FPE RT-QuIC protocol to determine which was responsible for the removal of RT-QuIC reaction inhibitors. To assess the effects of tissue fixation on RT-QuIC amyloid-seeding ability, an unfixed, frozen sample of chronic wasting disease-positive (CWD+) obex was fixed in paraformaldehyde-lysine-periodate (PLP) fixative for 48 h prior to homogenization. The PLP CWD+ brain and standard unfixed frozen brain homogenates were further treated with either a combination of xylene and ethanol or ethanol alone prior to seeding the RT-QuIC (Fig. 2A). CWD+ PLP-fixed brain homogenate (Fig. 2A, green) displayed amyloid formation kinetics comparable to those of CWD+ unfixed, frozen brain homogenate (Fig. 2A, purple), characterized by the usual inhibition of amyloid formation at 10−1 dilution and lower rates of amyloid formation at 10−2 dilution, indicating PLP fixation did not remove RT-QuIC reaction inhibitors. However, treatment of both PLP-fixed and unfixed frozen brain homogenates with both xylene and ethanol steps created similar kinetic curves and enabled prion-seeded amyloid formation at 10−1 and 10−2 dilutions (Fig. 2A, black and red) that was indistinguishable from that of CWD+ FPE brain homogenate (Fig. 2A, blue). To determine which solvent, xylene or ethanol, was most responsible for removing inhibitors, unfixed CWD+ brain homogenate was treated with a series of ethanol dilutions. Ethanol treatment produced the highest rate of amyloid formation and displayed a linear dilution range from 10−1 to 10−3 (Fig. 2A, orange), and a dilutional series confirmed that the ethanol solvent did not alter amyloid formation kinetics (Fig. 2B). To evaluate the sample protein recovery following ethanol treatment, we measured the protein concentration of brain homogenate before and after precipitation by a bicinchoninic acid (BCA) assay. Approximately 20 to 40% of the protein was recovered following ethanol precipitation (an average of 2 ethanol precipitations and 6 samples).
FIG 2.
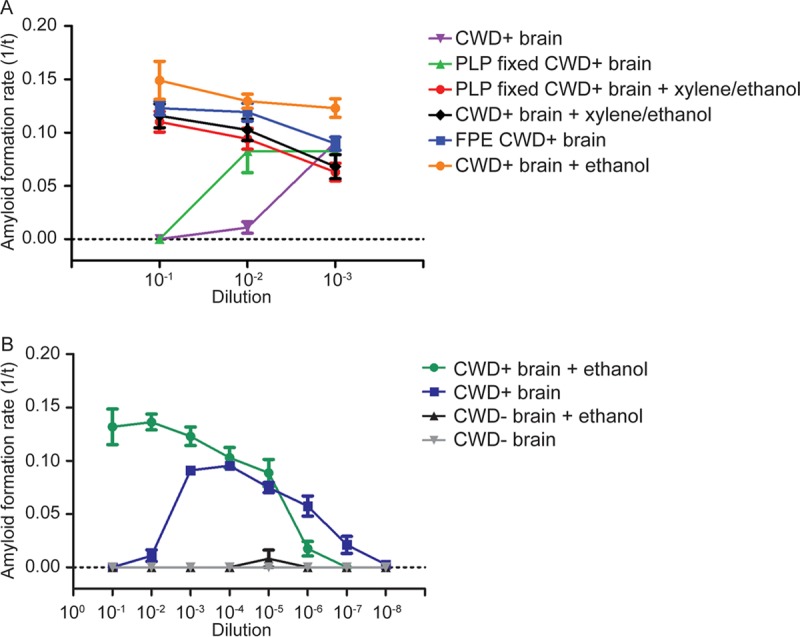
RT-QuIC detection of prion amyloid seeds following different solvent treatments. (A) RT-QuIC prion detection following FPE protocol solvent treatments. CWD+ brain homogenate prion seeding showed inhibition at 10−1 and 10−2 dilutions. PLP fixation of a CWD+ sample showed inhibition at 10-to-1 dilution. Treatment of CWD+ brain homogenate and PLP-fixed CWD+ brain homogenate with xylene and ethanol solvents allowed amyloid seeding at all dilutions, comparable to the FPE CWD+ sample. CWD+ brain homogenates treated only with ethanol allowed prion-seeded detection at all dilutions and had the highest rates of amyloid formation. The data are displayed as means and SEM from 2 experiments with 8 replicates. (B) RT-QuIC dilutional series following alcohol treatment. Ethanol treatment removed brain homogenate inhibitors at high concentrations and allowed prion-seeded amyloid formation from 10−1 to 10−7. The data are displayed as means and SEM from 2 experiments with 8 replicates.
Since alcohols are known classic lipid solvents, we hypothesized that brain-derived lipids were inhibiting the RT-QuIC amyloid formation reaction (16). To test this hypothesis, we added a commercial preparation of total brain lipid extract to CWD+ ethanol-precipitated brain samples approximately equal to physiologic lipid concentrations in brain (17). The addition of commercial lipids inhibited amyloid formation in a dose-dependent manner, with 6% commercial lipids in chloroform-methanol solvent completely inhibiting all sample dilutions tested and 3% commercial lipids inhibiting amyloid formation only at the 10−1 dilution (Fig. 3A), confirming that brain-derived lipids can inhibit the RT-QuIC amyloid formation reaction. The negative control (chloroform-methanol solvent) displayed minor inhibition at the 10−1 dilution but did not abolish prion-seeded amyloid formation.
FIG 3.
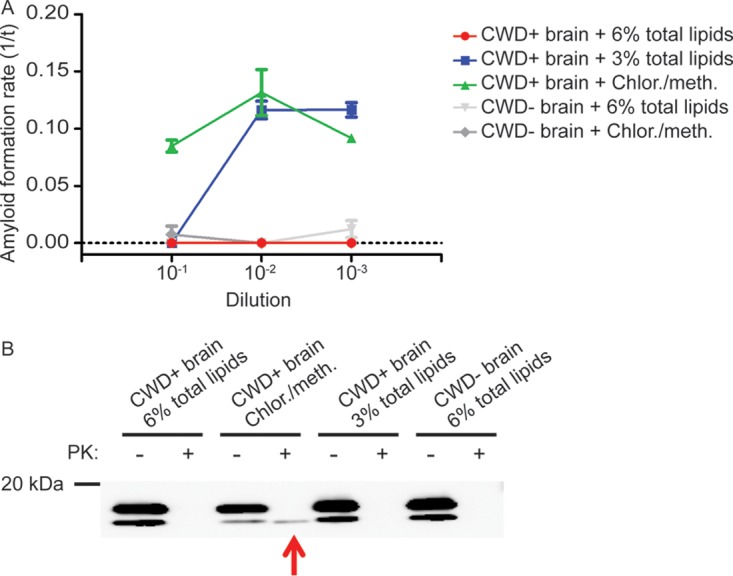
Total brain lipid extracts inhibit prion-seeded amyloid formation. (A) RT-QuIC of CWD+ ethanol-precipitated seeds with commercial total brain lipid extracts. The addition of total brain lipids to ethanol-precipitated CWD+ brain inhibited RT-QuIC in a dose-dependent manner. The lipid delivery solvent, chloroform-methanol, had a minimal effect on the RT-QuIC reaction. The data are displayed as means and SEM from 2 experiments with 8 replicates. (B) Western blot of RT-QuIC reaction products from 10−1 seed concentration. The Western blot of RT-QuIC products from the experiment shown in panel A detected only PK-resistant prion-seeded amyloid material from samples seeded with CWD+ brain–chloroform-methanol solvent (arrow). This result corresponded to the fluorescence observed at this seed dilution (green line, panel A).
The detection of amyloid formation in RT-QuIC relies on thioflavin T binding amyloid and subsequent fluorescence. To ensure that the lack of RT-QuIC fluorescence signal following the addition of lipids was due to inhibition of seeded amyloid formation and not inhibition of thioflavin T binding, we analyzed the RT-QuIC reaction products shown in Fig. 3A by Western blotting for prion-templated proteinase K (PK)-resistant amyloid material. Replicates from the 10−1 dilution-seeded reactions were pooled and treated with 10 μg/ml PK solution prior to Western blotting. PK-resistant material was observed only in the CWD+ chloroform-methanol solvent-seeded reaction (Fig. 3B), which corresponded to the positive fluorescence signal detected by RT-QuIC. The lack of PK-resistant RT-QuIC products confirmed that total brain lipids inhibited prion-seeded amyloid formation and not the detection of amyloid structures.
Next, we assessed the ability of extracted brain lipids to inhibit prion-seeded RT-QuIC amyloid formation in lymph node homogenates that did not contain the same inhibitors. First, we extracted brain lipids from CWD-negative white-tailed deer brain homogenate and added them to ethanol-precipitated CWD+ brain protein. This produced prion-seeded amyloid formation kinetics similar to those with frozen CWD+ brain homogenates (Fig. 4A). We then added extracted deer brain lipids to ethanol-precipitated CWD+ retropharyngeal lymph node samples. Prion-seeded amyloid formation in the lymph node treated with extracted deer brain lipids was inhibited at 10−1 dilution, whereas the ethanol-precipitated and frozen homogenate of the same lymph node displayed amyloid seeding at the same dilution (Fig. 4B). These results further demonstrated that brain-derived lipids could suppress prion-seeded amyloid formation in RT-QuIC.
FIG 4.

Deer brain lipids can be extracted and inhibit RT-QuIC. (A) Ethanol precipitation removes white-tailed deer brain lipids. The ethanol extraction protocol was used to remove lipids from CWD− brain homogenate. The removed lipids were added to ethanol-precipitated CWD+ brain homogenate (green) and inhibited the reaction at 10−1 and 10−2 dilutions with amyloid formation kinetics comparable to those with 10% brain homogenate (blue). The data are displayed as means and SEM from 2 experiments with 8 replicates. (B) Total brain lipids inhibit lymph node-seeded RT-QuIC. Brain lipids extracted from CWD− brain homogenate were added to ethanol-precipitated lymph node (LN) prion seeds (green). The addition of lipids inhibited RT-QuIC prion-seeded amyloid formation at Change text to 10−1 dilution where complete inhibition was not present in the initial lymph node homogenate (red). The data are displayed as means and SEM from 2 experiments with 8 replicates.
Polar lipids are responsible for inhibition of prion-seeded amyloid formation.
We used acetone precipitation to divide endogenous brain lipids into polar and neutral/nonpolar fractions and tested these classes for RT-QuIC inhibitor activity. Ethanol-precipitated CWD+ brain seeds treated with acetone-extracted polar lipids were inhibited at the 10−1 concentration (Fig. 5A) but displayed amyloid formation with additional dilution. In contrast, treating the same CWD+ brain seeds with the neutral/nonpolar lipid fraction resulted in amyloid formation at all dilutions, albeit with a lower formation rate at the 10−1 dilution. To confirm these results, CWD+ brain seeds were treated with commercially prepared polar lipids at physiologic brain concentrations. The addition of commercial polar lipids inhibited prion-seeded amyloid formation in a dose-dependent manner, with 5% polar lipids showing complete inhibition at all dilutions and with 0.5% inhibiting only the 10−1 concentration of prion seeds (Fig. 5B), indicating that polar lipids contain the majority of lipid amyloid-forming-inhibitory properties in brain lipid extracts.
FIG 5.
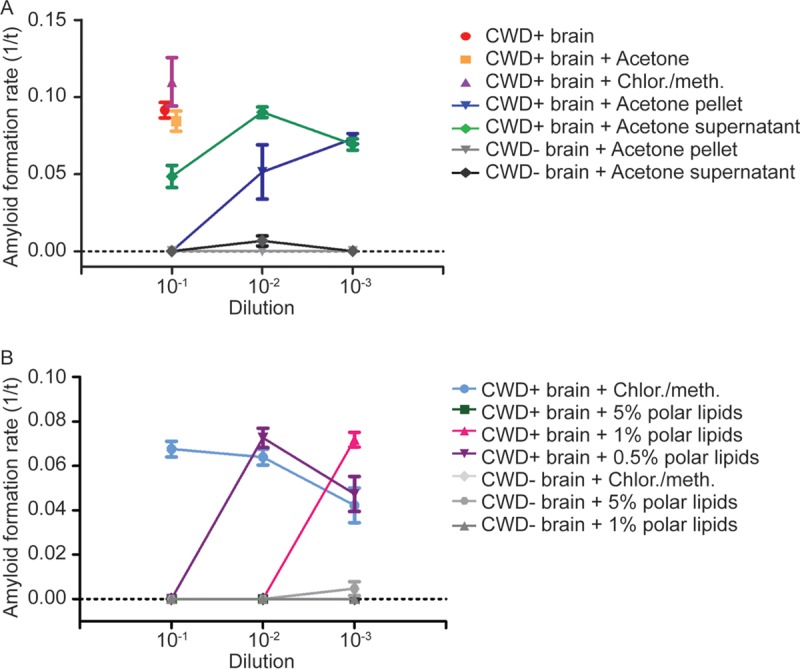
Polar lipids inhibit prion-seeded amyloid formation. (A) Acetone fractionation of total brain lipid extracts. Acetone was used to fractionate total brain lipids into polar lipids, contained in the pellet, and nonpolar lipids, contained in the supernatant. The addition of the pelleted lipid fraction to ethanol-precipitated CWD+ seeds inhibited RT-QuIC at 10−1 dilution, whereas addition of the acetone supernatant resulted in only minor inhibition of RT-QuIC at the same dilution. The data are displayed as means and SEM from 2 experiments with 8 replicates. (B) Commercial polar lipids inhibit RT-QuIC. Adding commercially prepared polar lipids to ethanol-precipitated CWD+ seeds caused inhibition of amyloid formation in a dose-dependent manner with complete absence of amyloid formation at a 5% lipid concentration and no inhibition at a 0.5% lipid concentration. The data are displayed as means and SEM from 2 experiments with 8 replicates.
Phospholipids inhibit CWD prion-seeded amyloid formation.
Deleault et al. identified the phospholipid phosphatidylethanolamine as a cofactor in the generation of synthetic infectious prions (10). To investigate the potential of phospholipids to induce or inhibit prion-seeded amyloid formation in RT-QuIC, we added 1% solutions of commercially prepared PE, phosphatidylcholine (PC), and phosphatidylinositol (PI) to CWD+ or CWD− ethanol-precipitated brain seeds. All three phospholipids inhibited prion-seeded amyloid formation at the 10−1 concentration, and dilution to 10−3 restored the conversion reaction (Fig. 6). Addition of PE, PC, or PI to recombinant Syrian hamster PrPC (SHrPrP) in unseeded RT-QuIC reactions did not induce amyloid formation (data not shown). These results suggest phospholipids may be the polar lipid component responsible for RT-QuIC inhibition.
FIG 6.
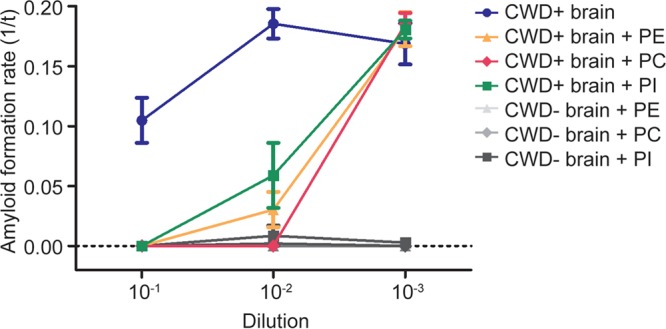
Phospholipids inhibit prion-seeded amyloid formation. The phospholipids PE, PC, and PI inhibit prion-seeded amyloid formation. The addition of 1% phospholipid solutions containing PE, PC, or PI to ethanol-precipitated CWD+ seeds caused complete inhibition at 10−1 dilution and allowed amyloid formation following dilution to 10−3. The data are displayed as means and SEM from 2 experiments with 8 replicates.
DISCUSSION
PrPC, like other GPI-anchored surface proteins, resides in lipid rafts within the outer leaflet of the cell membrane, a location that has been shown to be essential for the prion conversion process (2, 18, 19). Lipid raft microdomains are enriched in sphingolipids and cholesterol to maintain structural integrity and coalesce signaling molecules (20). Studies have shown that alterations in these components, such as disrupting cellular cholesterol homeostasis or treatment with the enzyme filipin, reduced surface PrPC and resulted in decreased formation of PrPRES (21–23). In contrast to cholesterol, sphingomyelin depletion in neuroblastoma cell membranes caused an increase in PrPRES through an unknown mechanism (24). The membrane lipid environment has been shown to directly promote prion propagation by destabilizing the C-terminal domain, facilitating PrPRES conversion and fibril formation (25, 26). These studies highlight the important role lipids play in prion propagation at a cellular level. Prion diseases also alter lipid homeostasis at the whole-organism level. A transgenic-mouse model of scrapie has shown alterations in lipid compositions in the brain in terminal disease (27).
The GPI anchor is equally important in the PrPC conversion process, as it facilitates PrPC interactions with lipids and supports PrPRES propagation. Cell culture and transgenic-mouse studies have established that the C-terminal GPI anchor is not required for PrPRES formation but is necessary for PrPRES propagation between cells and for cytopathology (28). Altering the location of PrPC from the cell surface to the transmembrane domain prevents PrPC conversion (5, 29, 30). In in vivo studies, GPI anchorless PrPRES formed large amyloid accumulations in mice without the formation of smaller oligomer and fibril forms (31). Thus, the GPI anchor appears to play an important role in the propagation of prion disease.
Interestingly, bacterially produced recombinant prion protein used as the substrate in RT-QuIC and some protein misfolding cyclic amplification (PMCA) experiments lacks posttranslational modifications, including the GPI anchor, and the conversion products displayed variable but generally lower infectivity (32–34). Currently, it is unclear whether RT-QuIC can generate infectious prions; further study is needed to investigate how the structure of the recombinant protein substrate may alter the prion-seeded amyloid fibrils produced in RT-QuIC (35).
Many of the lipid-protein interactions implicated in prion diseases mirror those described in the pathogenesis of amyloid-β (Aβ) aggregation in Alzheimer's disease. As with PrPC, the amyloid-β precursor protein (APP) is an integral plasma membrane protein, although APP contains a transmembrane domain instead of a GPI anchor (36, 37). The lipid environment surrounding APP and the secretase enzymes that perform its posttranslational processing impact the form of Aβ produced at the plasma membrane, and while APP is not a permanent resident, when located in lipid rafts it is more likely to be cleaved by γ-secretase and result in amyloidogenic forms (38–41). The influence of the lipid raft location on Aβ formation is also determined by cholesterol, and an increase in cholesterol shifts toward Aβ aggregations (42, 43). Similar to the influence on the prion conversion process, membrane lipids can also destabilize the Aβ conformational structure and induce the protein to acquire an additional β-sheet, which increases fibrillization and aggregation (44, 45). With the similarities in proamyloid protein-lipid interactions in prion and Alzheimer's diseases, it would be interesting to investigate whether the inhibitory lipid function we have identified in the RT-QuIC model can cross protein species.
Not only does the lipid bilayer influence protein misfolding, it may also contribute to prion pathology, as toxic nonfibrillar prion oligomers have been shown to directly interact with membrane lipids (46, 47). Proposed mechanisms of toxicity include prion oligomer membrane insertion as pores or channels, similar to Aβ in Alzheimer's disease pathology (47, 48); detergent-like activity, similar to islet amyloid precursor protein in diabetes pathology (49); and prion fibrillization on the surfaces of lipid rafts, leading to a functional loss of membrane domain organization (50). These toxic interactions appear to depend on the composition of the lipid bilayer. The ability of amyloid-β to form pores and fragment lipid bilayers depends on the presence of gangliosides (48). Studies altering the content of anionic cellular membrane lipids induced a switch in prion oligomer membrane interactions from micelle formation to increased formation of fibrils on lipid microdomains (50). Membrane lipids, specifically PE, have been shown to act as a cofactor in prion protein conversion in vitro by facilitating protein structural changes (10, 51). Taken together, these studies and the data presented illustrate a variety of roles for lipids in the prion conversion process and suggest that the composition of the lipid environment may be a crucial component of prion pathology.
In contrast to studies describing the stimulatory effect of PE on prion conversion, we found the addition of phospholipids, including PE, inhibited RT-QuIC amyloid formation (10). One potential explanation is the inherent differences in the PMCA and RT-QuIC assays used in the experiments. Investigations of the amyloid products produced by PMCA sonication have described a diverse population of conformers, including small oligomers, which may be more infectious than large aggregates (32, 52). Examinations of RT-QuIC products have demonstrated amyloid fibrils; however, full characterization of the sizes and types of fibrils remains to be done (35, 53, 54). A second difference between the present studies and those of Deleault et al. is the sequence of the recombinant protein used as the assay substrate (10, 12). The recombinant protein used in our RT-QuIC experiments lacks the N-terminal domain, in addition to posttranslational modifications, and previous studies have shown that the N-terminal domain can influence prion conversion (55). Further investigations into how lipids interact with PrPC in RT-QuIC would be helpful in understanding these different assay results.
Investigations of potential prion disease therapeutics have identified molecules that abolish or limit prion conversion, similar to our observations following the addition of lipids to RT-QuIC seeded reactions, and can provide insight into potential mechanisms of action. Glycosaminoglycans and the azo dye Congo red mechanistically decrease PrPRES formation by competitive inhibition. Both have been shown to bind PrPC and to decrease its availability for PrPRES interaction and prion conversion (56, 57). Congo red has also been shown to hyperstabilize prion amyloid fibrils, which prevents fragmentation and seeded amyloidogenesis (58, 59). Our Western blotting data (Fig. 4) show a lack of PK-resistant RT-QuIC products, which suggests that polar lipids interfere with the prion conversion process; however, the exact mechanism by which conversion is inhibited requires further investigation.
Our experiments highlight a potential exciting use of RT-QuIC to investigate molecules that can alter prion conversion kinetics. Currently, investigations into potential therapeutics are limited to cell culture assays and costly animal bioassays (60). A modified PMCA technique using recombinant protein, as well as other amyloid-seeding assays, have previously been used in a limited number of prion conversion cofactor investigations (10). We show that the different alcohol-based solvents used to deliver lipids did not themselves alter RT-QuIC amyloid formation kinetics. Additional study is needed to establish the robustness of real-time conversion methodology when used with a variety of delivery solvents or compounds. However, the ease and high-throughput nature of RT-QuIC can facilitate widespread screening for molecules that inhibit prion amyloid formation (61).
In summary, we have demonstrated that endogenous brain polar lipids can inhibit prion amyloid conversion in vitro. This appears to be the first identification of an inhibitory (or possibly regulatory) function of lipids and suggests prion conversion is a balance of proconversion and anticonversion lipid environments. Future experiments aim to identify which class of endogenous lipids or individual lipids contain the inhibiting function, how lipid moieties may influence which cells are permissive to prion infection, and whether the lipid environment can play a role in the generation of prion strains.
MATERIALS AND METHODS
Cervid tissue samples.
Brain and lymph node samples were harvested from white-tailed deer experimentally inoculated intranasally with CWD+ brain, or CWD− brain as a negative control (62). Deer were cared for and experimental studies were carried out in strict accordance with Colorado State University Institutional Animal Care and Use Committee-approved protocols. All CWD+ animals were allowed to progress to terminal disease prior to euthanasia and necropsy. At necropsy, each tissue sample was collected with individual, single-use, prion-free instruments to avoid cross-contamination and divided into sections for freezing at −80°C until use or fixation. Ten percent (wt/vol) tissue homogenates in 1× Dulbecco's phosphate-buffered saline (DPBS; Life Technologies) were prepared using zirconium oxide beads and a Blue Bullet Blender (Next Advance) tissue homogenizer.
Recombinant Syrian hamster PrPC protein substrate purification.
Purification of the recombinant, truncated (residues 90 to 231) SHrPrP used as the substrate in RT-QuIC was performed as previously described (15, 63). Escherichia coli BL21 Rosetta (Novagen) containing the truncated protein construct was cultured from a glycerol stock at 37°C in lysogeny broth (LB) medium with the selection antibiotics kanamycin and chloramphenicol to express SHrPrP until the culture optical density at 600 nm (OD600) reached at least 2.5. E. coli cell lysis was carried out using Bugbuster reagent supplemented with Lysonase (EMD Biosciences) according to the manufacturer's recommended protocol. Inclusion bodies were harvested by centrifugation at 15,000 rpm and dissolved in solubilization buffer (8 M guanidine hydrochloride, 100 mM Na2HPO4) prior to application to nickel-nitrilotriacetic acid (Ni-NTA) flow resin (Qiagen) that had been previously equilibrated with denaturation buffer (6 M guanidine hydrochloride, 100 mM Na2HPO4, 10 mM Tris, pH 8.0). The Ni-NTA resin–SHrPrP was loaded onto an XK16-60 column (GE Healthcare) and purified using Bio-Rad Duoflow fast protein liquid chromatography (FPLC). To induce protein refolding, a gradient from denaturation buffer to refolding buffer (100 mM Na2HPO4, 10 mM Tris, pH 5.5) was applied. Refolding was followed by a gradient from refolding to elution buffer (100 mM Na2HPO4, 10 mM Tris, 0.5 M imidazole), and fractions from the elution peak were pooled and dialyzed against two 4-liter changes of buffer (20 mM NaH2PO4, pH 5.5) overnight. The final protein concentration was calculated by measuring the A280 and using a coefficient of extinction of 25,900 in Beer's law (absorbance = coefficient × length × concentration). The purified SHrPrP was stored at 4°C until use.
RT-QuIC.
RT-QuIC was performed as previously described (63, 64). Tissue homogenates and ethanol-precipitated samples with lipids were diluted in 0.1% sodium dodecyl sulfate (SDS)-1× PBS to the desired concentration. The RT-QuIC reaction was performed by adding 2 μl of diluted sample to a buffer containing 20 mM NaH2PO4, 320 mM NaCl, 1.0 mM EDTA, 1 mM ThT, and 0.1 mg/ml SHrPrP in one well of a black, optical-bottom, 96-well plate (Nunc). RT-QuIC experiments were carried out in a BMG Labtech Polarstar fluorometer with cycles of 1 min of shaking (700 rpm; double orbital) followed by 1 min of rest, repeated for 15 min. ThT fluorescence was read (excitation, 450 nm; emission, 480 nm; gain, 1,700) at the conclusion of each 15-min shake/rest cycle, and each well was measured with 20 flashes per well, with an orbital average of 4. Each RT-QuIC experiment was performed for a minimum of 100 cycles. RT-QuIC amyloid formation was determined to be positive if the fluorescence exceeded a threshold determined to be 5 standard deviations above the average baseline fluorescence. RT-QuIC amyloid formation rates were calculated as the inverse of the lag time before fluorescence reached the threshold described above.
Lipid extraction from brain homogenates.
Lipids were extracted from 10% brain homogenates using a modification of the method of Folch et al. (65, 66). Briefly, 100 μl of a 10% brain homogenate was incubated with 900 μl of 100% ethanol at room temperature for 5 min. Following incubation, brain homogenate-alcohol solutions were centrifuged at 15,000 rpm for 5 min to precipitate proteins. The ethanol supernatant was saved, and the alcohol extraction process was repeated. The pooled ethanol supernatants were dried under a stream of nitrogen gas prior to resuspension in 100 μl of a chloroform-methanol solution (2:1) to maintain the original concentration (Fig. 7A). Extracted lipids in the chloroform-methanol solvent were added to the precipitated proteins from tissue samples (Fig. 7B). Commercially purchased total brain lipid extract (porcine), polar brain lipid extract (porcine), and the phospholipids phosphatidylethanolamine (brain; porcine), phosphatidylcholine (brain; porcine), and phosphatidylinositol (liver; bovine) (Avanti Polar Lipids) were dissolved in chloroform-methanol solvent (2:1) immediately prior to use to create the desired weight/volume concentration and added to ethanol-precipitated tissue samples.
FIG 7.
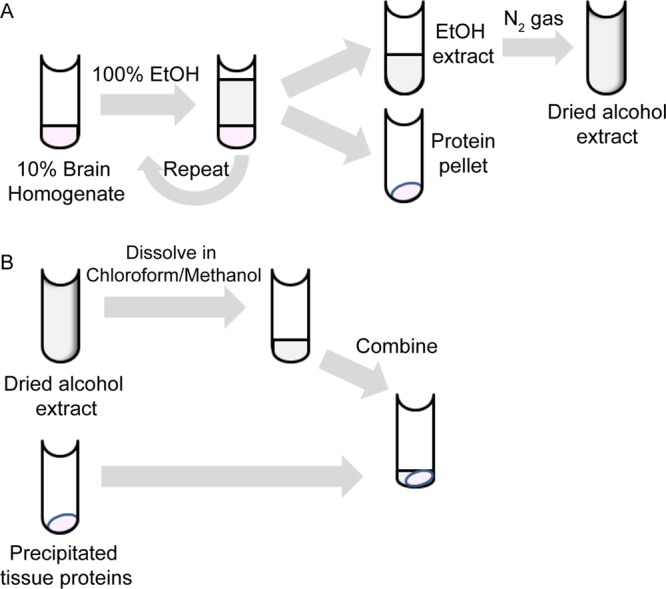
Overview of methods. (A) Lipid extraction from 10% brain homogenate. Brain homogenate was treated with 100% ethanol (EtOH), and the proteins were pelleted by centrifugation. The ethanol supernatant and its dissolved contents were removed and dried under a stream of nitrogen gas. (B) Sample preparation for RT-QuIC. Dried alcohol extracts were resuspended in chloroform-methanol solvent. The resuspended extracts were combined with alcohol-precipitated proteins (A) for use in the RT-QuIC assay.
The protein concentrations of brain and lymph node samples before and after ethanol precipitation were compared using BCA assays (Pierce BCA Protein Assay kit; Thermo Scientific). The BCA assays were performed according to the manufacturer's instructions, with each sample and standard tested in triplicate, and optical density was measured with an Opsys MR microplate reader (Dynex Technologies).
Isolation of polar lipids.
Total brain lipid extracts were separated into polar and neutral/nonpolar fractions by acetone precipitation. An approximately 20× to 30× volume of 100% acetone was used to suspend dried total lipid extracts and incubated on ice for 1 h. The acetone-lipid mixture was centrifuged at 15,000 rpm for 5 min to pellet polar lipids. The entire procedure was repeated, and the acetone supernatants were combined. The lipid pellets and acetone supernatants were dried under a stream of nitrogen gas prior to resuspension in chloroform-methanol solvent (2:1) in volumes equivalent to that of the starting material to maintain physiologic concentrations. Acetone -fractionated lipids were added to ethanol-precipitated proteins to seed the RT-QuIC reaction, as previously described.
FPE RT-QuIC.
FPE tissue blocks were processed, and proteins were extracted as previously described (15). Briefly, a microtome (Leica) was used to cut 8- to 10-μm-thick paraffin-embedded tissue-wax curls from paraffin blocks. The tissue-wax curls were treated sequentially with xylene to remove the paraffin, with a series of graded alcohol washes (100%, 95%, and 70%) to rehydrate the tissue sections, and with a final 1× PBS wash to remove any remaining solvents, with a 5-min centrifugation at 15,000 rpm to pellet tissue pieces between the wash steps. Rehydrated FPE tissue samples were homogenized in 1× PBS to create a standard 10% (wt/vol) concentration, using a bead homogenizer as described above. FPE homogenates were diluted in RT-QuIC dilution buffer (0.1% SDS-1× PBS) to the desired concentration prior to RT-QuIC analysis.
Western blotting of RT-QuIC products.
Protease-resistant amyloid products of the RT-QuIC reaction were detected by Western blotting. The RT-QuIC products were collected at the completion of the assay by washing each well with the RT-QuIC reaction buffer and combining replicates seeded by the same sample. Nine microliters of collected RT-QuIC products was digested with PK at a final concentration of 1 μg/ml and incubated at 37°C for 30 min with shaking, followed by 45°C for 10 min with shaking. Samples were mixed with reducing agent-LDS sample buffer (Invitrogen), heated at 95°C for 5 min, and then electrophoresed through a NuPAGE 10% Bis-Tris gel (Invitrogen) at 135 V for 1.5 h. The proteins were transferred to a polyvinylidene difluoride (PVDF) membrane using the Transblot Turbo system (Bio-Rad) following the manufacturer's instructions. The membrane was loaded into a prewetted SNAP i.d. blot holder (Millipore) and then sequentially blocked with blocking buffer (blocker casein in Tris-buffered saline [TBS] [Thermo-Scientific] and 0.1% Tween 20 [Sigma]) for 3 min and probed with the antibody BAR224 (Caymen Chemical) conjugated to horseradish peroxidase (HRP) and diluted to 0.2 μg/ml in blocking buffer for 10 min. The antibody was removed by vacuuming through the membrane using the SNAP i.d. system (Millipore), and the membrane was washed three times with 30 ml wash buffer (50% blocker casein in TBS, 50% 1× TBS, 0.1% Tween 20) with continuous vacuuming. The membrane was developed with ECL Plus Western blotting detection reagents (GE) and viewed on a luminescent image analyzer (LAS-3000; GE).
ACKNOWLEDGMENTS
We thank laboratory members Nathaniel Denkers for providing the white-tailed deer brain samples used in these experiments and Nikki Buhrdorf and Sarah Accardi for their assistance in sample preparation. We thank Byron Caughey for helping to seed the RT-QuIC assay in our laboratory.
This work was supported by NIH grant R01-NS061902 (E.A.H.). Clare Hoover was supported by NIH training grant T32-OD010437-14.
REFERENCES
- 1.Vey M, Pilkuhn S, Wille H, Nixon R, DeArmond SJ, Smart EJ, Anderson RG, Taraboulos A, Prusiner SB. 1996. Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc Natl Acad Sci U S A 93:14945–14949. doi: 10.1073/pnas.93.25.14945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naslavsky N, Stein R, Yanai A, Friedlander G, Taraboulos A. 1997. Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. J Biol Chem 272:6324–6331. doi: 10.1074/jbc.272.10.6324. [DOI] [PubMed] [Google Scholar]
- 3.Gorodinsky A, Harris DA. 1995. Glycolipid-anchored proteins in neuroblastoma cells form detergent-resistant complexes without caveolin. J Cell Biol 129:619–627. doi: 10.1083/jcb.129.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mange A, Nishida N, Milhavet O, McMahon HE, Casanova D, Lehmann S. 2000. Amphotericin B inhibits the generation of the scrapie isoform of the prion protein in infected cultures. J Virol 74:3135–3140. doi: 10.1128/JVI.74.7.3135-3140.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaneko K, Vey M, Scott M, Pilkuhn S, Cohen FE, Prusiner SB. 1997. COOH-terminal sequence of the cellular prion protein directs subcellular trafficking and controls conversion into the scrapie isoform. Proc Natl Acad Sci U S A 94:2333–2338. doi: 10.1073/pnas.94.6.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilch S, Kehler C, Schatzl HM. 2006. The prion protein requires cholesterol for cell surface localization. Mol Cell Neurosci 31:346–353. doi: 10.1016/j.mcn.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 7.Baron GS, Wehrly K, Dorward DW, Chesebro B, Caughey B. 2002. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrP(Sc)) into contiguous membranes. EMBO J 21:1031–1040. doi: 10.1093/emboj/21.5.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goold R, Rabbanian S, Sutton L, Andre R, Arora P, Moonga J, Clarke AR, Schiavo G, Jat P, Collinge J, Tabrizi SJ. 2011. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat Commun 2:281. doi: 10.1038/ncomms1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deleault NR, Kascsak R, Geoghegan JC, Supattapone S. 2010. Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry 49:3928–3934. doi: 10.1021/bi100370b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deleault NR, Piro JR, Walsh DJ, Wang F, Ma J, Geoghegan JC, Supattapone S. 2012. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc Natl Acad Sci U S A 109:8546–8551. doi: 10.1073/pnas.1204498109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atarashi R, Wilham JM, Christensen L, Hughson AG, Moore RA, Johnson LM, Onwubiko HA, Priola SA, Caughey B. 2008. Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat Methods 5:211–212. doi: 10.1038/nmeth0308-211. [DOI] [PubMed] [Google Scholar]
- 12.Wilham JM, Orru CD, Bessen RA, Atarashi R, Sano K, Race B, Meade-White KD, Taubner LM, Timmes A, Caughey B. 2010. Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog 6:e1001217. doi: 10.1371/journal.ppat.1001217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takatsuki H, Satoh K, Sano K, Fuse T, Nakagaki T, Mori T, Ishibashi D, Mihara B, Takao M, Iwasaki Y, Yoshida M, Atarashi R, Nishida N. 2015. Rapid and quantitative assay of amyloid-seeding activity in human brains affected with prion diseases. PLoS One 10:e0126930. doi: 10.1371/journal.pone.0126930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mori T, Atarashi R, Furukawa K, Takatsuki H, Satoh K, Sano K, Nakagaki T, Ishibashi D, Ichimiya K, Hamada M, Nakayama T, Nishida N. 2016. A direct assessment of human prion adhered to steel wire using real-time quaking-induced conversion. Sci Rep 6:24993. doi: 10.1038/srep24993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoover CE, Davenport KA, Henderson DM, Pulscher LA, Mathiason CK, Zabel MD, Hoover EA. 2016. Detection and quantification of CWD prions in fixed paraffin embedded tissues by real-time quaking-induced conversion. Sci Rep 6:25098. doi: 10.1038/srep25098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reis A, Rudnitskaya A, Blackburn GJ, Mohd Fauzi N, Pitt AR, Spickett CM. 2013. A comparison of five lipid extraction solvent systems for lipidomic studies of human LDL. J Lipid Res 54:1812–1824. doi: 10.1194/jlr.M034330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brady ST, Siegel GJ, Albers RW, Price DL, Benjamins J. 2012. Basic neurochemistry: principles of molecular, cellular, and medical neurobiology, 8th ed Elsevier, Amsterdam, the Netherlands. [Google Scholar]
- 18.Caughey B, Raymond GJ. 1991. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J Biol Chem 266:18217–18223. [PubMed] [Google Scholar]
- 19.Stahl N, Borchelt DR, Prusiner SB. 1990. Differential release of cellular and scrapie prion proteins from cellular membranes by phosphatidylinositol-specific phospholipase C. Biochemistry 29:5405–5412. doi: 10.1021/bi00474a028. [DOI] [PubMed] [Google Scholar]
- 20.Allen JA, Halverson-Tamboli RA, Rasenick MM. 2007. Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci 8:128–140. doi: 10.1038/nrn2059. [DOI] [PubMed] [Google Scholar]
- 21.Simons K, Toomre D. 2000. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 22.Gilch S, Bach C, Lutzny G, Vorberg I, Schatzl HM. 2009. Inhibition of cholesterol recycling impairs cellular PrP(Sc) propagation. Cell Mol Life Sci 66:3979–3991. doi: 10.1007/s00018-009-0158-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marella M, Lehmann S, Grassi J, Chabry J. 2002. Filipin prevents pathological prion protein accumulation by reducing endocytosis and inducing cellular PrP release. J Biol Chem 277:25457–25464. doi: 10.1074/jbc.M203248200. [DOI] [PubMed] [Google Scholar]
- 24.Naslavsky N, Shmeeda H, Friedlander G, Yanai A, Futerman AH, Barenholz Y, Taraboulos A. 1999. Sphingolipid depletion increases formation of the scrapie prion protein in neuroblastoma cells infected with prions. J Biol Chem 274:20763–20771. doi: 10.1074/jbc.274.30.20763. [DOI] [PubMed] [Google Scholar]
- 25.Morillas M, Swietnicki W, Gambetti P, Surewicz WK. 1999. Membrane environment alters the conformational structure of the recombinant human prion protein. J Biol Chem 274:36859–36865. doi: 10.1074/jbc.274.52.36859. [DOI] [PubMed] [Google Scholar]
- 26.Critchley P, Kazlauskaite J, Eason R, Pinheiro TJ. 2004. Binding of prion proteins to lipid membranes. Biochem Biophys Res Commun 313:559–567. doi: 10.1016/j.bbrc.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 27.Guan Z, Soderberg M, Sindelar P, Prusiner SB, Kristensson K, Dallner G. 1996. Lipid composition in scrapie-infected mouse brain: prion infection increases the levels of dolichyl phosphate and ubiquinone. J Neurochem 66:277–285. [DOI] [PubMed] [Google Scholar]
- 28.Priola SA, McNally KL. 2009. The role of the prion protein membrane anchor in prion infection. Prion 3:134–138. doi: 10.4161/pri.3.3.9771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marshall KE, Hughson A, Vascellari S, Priola SA, Sakudo A, Onodera T, Baron GS. 2017. PrP knockout cells expressing transmembrane PrP resist prion infection. J Virol 91:e01686-16. doi: 10.1128/JVI.01686-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taraboulos A, Scott M, Semenov A, Avrahami D, Laszlo L, Prusiner SB. 1995. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J Cell Biol 129:121–132. doi: 10.1083/jcb.129.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M. 2005. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 308:1435–1439. doi: 10.1126/science.1110837. [DOI] [PubMed] [Google Scholar]
- 32.Noble GP, Wang DW, Walsh DJ, Barone JR, Miller MB, Nishina KA, Li S, Supattapone S. 2015. A structural and functional comparison between infectious and non-infectious autocatalytic recombinant PrP conformers. PLoS Pathog 11:e1005017. doi: 10.1371/journal.ppat.1005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J, Rees JR, Supattapone S. 2012. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc Natl Acad Sci U S A 109:E1938–E1946. doi: 10.1073/pnas.1206999109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim JI, Cali I, Surewicz K, Kong Q, Raymond GJ, Atarashi R, Race B, Qing L, Gambetti P, Caughey B, Surewicz WK. 2010. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem 285:14083–14087. doi: 10.1074/jbc.C110.113464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sano K, Atarashi R, Ishibashi D, Nakagaki T, Satoh K, Nishida N. 2014. Conformational properties of prion strains can be transmitted to recombinant prion protein fibrils in real-time quaking-induced conversion. J Virol 88:11791–11801. doi: 10.1128/JVI.00585-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng H, Koo EH. 2006. The amyloid precursor protein: beyond amyloid. Mol Neurodegener 1:5. doi: 10.1186/1750-1326-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Selkoe DJ. 2001. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81:741–766. [DOI] [PubMed] [Google Scholar]
- 38.Zha Q, Ruan Y, Hartmann T, Beyreuther K, Zhang D. 2004. GM1 ganglioside regulates the proteolysis of amyloid precursor protein. Mol Psychiatry 9:946–952. doi: 10.1038/sj.mp.4001509. [DOI] [PubMed] [Google Scholar]
- 39.Lee SJ, Liyanage U, Bickel PE, Xia W, Lansbury PT Jr, Kosik KS. 1998. A detergent-insoluble membrane compartment contains A beta in vivo. Nat Med 4:730–734. doi: 10.1038/nm0698-730. [DOI] [PubMed] [Google Scholar]
- 40.Morishima-Kawashima M, Ihara Y. 1998. The presence of amyloid beta-protein in the detergent-insoluble membrane compartment of human neuroblastoma cells. Biochemistry 37:15247–15253. doi: 10.1021/bi981843u. [DOI] [PubMed] [Google Scholar]
- 41.Parkin ET, Hussain I, Karran EH, Turner AJ, Hooper NM. 1999. Characterization of detergent-insoluble complexes containing the familial Alzheimer's disease-associated presenilins. J Neurochem 72:1534–1543. [DOI] [PubMed] [Google Scholar]
- 42.Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K. 1998. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci U S A 95:6460–6464. doi: 10.1073/pnas.95.11.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, Sambamurti K, Duff K, Pappolla MA. 2000. Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiol Dis 7:321–331. doi: 10.1006/nbdi.2000.0304. [DOI] [PubMed] [Google Scholar]
- 44.Fezoui Y, Teplow DB. 2002. Kinetic studies of amyloid beta-protein fibril assembly. Differential effects of alpha-helix stabilization. J Biol Chem 277:36948–36954. [DOI] [PubMed] [Google Scholar]
- 45.McLaurin J, Franklin T, Chakrabartty A, Fraser PE. 1998. Phosphatidylinositol and inositol involvement in Alzheimer amyloid-beta fibril growth and arrest. J Mol Biol 278:183–194. doi: 10.1006/jmbi.1998.1677. [DOI] [PubMed] [Google Scholar]
- 46.Caughey B, Baron GS, Chesebro B, Jeffrey M. 2009. Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu Rev Biochem 78:177–204. doi: 10.1146/annurev.biochem.78.082907.145410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG. 2004. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem 279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- 48.Sciacca MF, Kotler SA, Brender JR, Chen J, Lee DK, Ramamoorthy A. 2012. Two-step mechanism of membrane disruption by Abeta through membrane fragmentation and pore formation. Biophys J 103:702–710. doi: 10.1016/j.bpj.2012.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brender JR, Salamekh S, Ramamoorthy A. 2012. Membrane disruption and early events in the aggregation of the diabetes related peptide IAPP from a molecular perspective. Acc Chem Res 45:454–462. doi: 10.1021/ar200189b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walsh P, Vanderlee G, Yau J, Campeau J, Sim VL, Yip CM, Sharpe S. 2014. The mechanism of membrane disruption by cytotoxic amyloid oligomers formed by prion protein(106-126) is dependent on bilayer composition. J Biol Chem 289:10419–10430. doi: 10.1074/jbc.M113.515866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller MB, Wang DW, Wang F, Noble GP, Ma J, Woods VL Jr, Li S, Supattapone S. 2013. Cofactor molecules induce structural transformation during infectious prion formation. Structure 21:2061–2068. doi: 10.1016/j.str.2013.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Noble GP, Supattapone S. 2015. Dissociation of recombinant prion autocatalysis from infectivity. Prion 9:405–411. doi: 10.1080/19336896.2015.1123843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ladner-Keay CL, Griffith BJ, Wishart DS. 2014. Shaking alone induces de novo conversion of recombinant prion proteins to beta-sheet rich oligomers and fibrils. PLoS One 9:e98753. doi: 10.1371/journal.pone.0098753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Groveman BR, Dolan MA, Taubner LM, Kraus A, Wickner RB, Caughey B. 2014. Parallel in-register intermolecular beta-sheet architectures for prion-seeded prion protein (PrP) amyloids. J Biol Chem 289:24129–24142. doi: 10.1074/jbc.M114.578344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davenport KA, Henderson DM, Mathiason CK, Hoover EA. 2016. Assessment of the PrPc amino-terminal domain in prion species barriers. J Virol 90:10752–10761. doi: 10.1128/JVI.01121-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caughey B, Brown K, Raymond GJ, Katzenstein GE, Thresher W. 1994. Binding of the protease-sensitive form of PrP (prion protein) to sulfated glycosaminoglycan and Congo red. J Virol 68:2135–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shyng SL, Lehmann S, Moulder KL, Harris DA. 1995. Sulfated glycans stimulate endocytosis of the cellular isoform of the prion protein, PrPC, in cultured cells. J Biol Chem 270:30221–30229. doi: 10.1074/jbc.270.50.30221. [DOI] [PubMed] [Google Scholar]
- 58.Caughey B, Race RE. 1992. Potent inhibition of scrapie-associated PrP accumulation by Congo red. J Neurochem 59:768–771. doi: 10.1111/j.1471-4159.1992.tb09437.x. [DOI] [PubMed] [Google Scholar]
- 59.Caughey B, Ernst D, Race RE. 1993. Congo red inhibition of scrapie agent replication. J Virol 67:6270–6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trevitt CR, Collinge J. 2006. A systematic review of prion therapeutics in experimental models. Brain 129:2241–2265. doi: 10.1093/brain/awl150. [DOI] [PubMed] [Google Scholar]
- 61.Ferreira NC, Marques IA, Conceicao WA, Macedo B, Machado CS, Mascarello A, Chiaradia-Delatorre LD, Yunes RA, Nunes RJ, Hughson AG, Raymond LD, Pascutti PG, Caughey B, Cordeiro Y. 2014. Anti-prion activity of a panel of aromatic chemical compounds: in vitro and in silico approaches. PLoS One 9:e84531. doi: 10.1371/journal.pone.0084531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Denkers ND, Hayes-Klug J, Anderson KR, Seelig DM, Haley NJ, Dahmes SJ, Osborn DA, Miller KV, Warren RJ, Mathiason CK, Hoover EA. 2013. Aerosol transmission of chronic wasting disease in white-tailed deer. J Virol 87:1890–1892. doi: 10.1128/JVI.02852-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Henderson DM, Davenport KA, Haley NJ, Denkers ND, Mathiason CK, Hoover EA. 2015. Quantitative assessment of prion infectivity in tissues and body fluids by real-time quaking-induced conversion. J Gen Virol 96:210–219. doi: 10.1099/vir.0.069906-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Henderson DM, Denkers ND, Hoover CE, Garbino N, Mathiason CK, Hoover EA. 2015. Longitudinal detection of prion shedding in saliva and urine by chronic wasting disease-infected deer by real-time quaking-induced conversion. J Virol 89:9338–9347. doi: 10.1128/JVI.01118-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Folch J, Lees M, Sloane Stanley GH. 1957. A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem 226:497–509. [PubMed] [Google Scholar]
- 66.Fajardo AR, Cerdán LE, Medina AR, Fernández FGA, Moreno PAG, Grima EM. 2007. Lipid extraction from the microalga Phaeodactylum tricornutum. Eur J Lipid Sci Technol 22:120–126. doi: 10.1002/ejlt.200600216. [DOI] [Google Scholar]