

Supplemental Digital Content is available in the text.
Keywords: AKT, ARQ 087, ARQ 092, combination, FGFR, inhibitor, kinase, small molecule
Abstract
The PI3K/AKT pathway plays an important role in the initiation and progression of cancer, and the drug development efforts targeting this pathway with therapeutic interventions have been advanced by academic and industrial groups. However, the clinical outcome is moderate. Combination of inhibition of PI3K/AKT and other targeted agents became a feasible approach. In this study we assessed the combined effect of ARQ 092, a pan-AKT inhibitor, and ARQ 087, a pan-FGFR inhibitor, in vitro and in vivo. In a panel of 45 cancer cell lines, on 24% (11 out of 45) the compounds showed synergistic effect, on 62% (28 out of 45) additive, and on 13% (6 out of 45) antagonistic. The highest percentage of synergism was found on endometrial and ovarian cancer cell lines. Mutational analysis revealed that PIK3CA/PIK3R1 mutations and aberrant activation of FGFR2 predicted synergism, whereas Ras mutations showed a reverse correlation. Pathway analysis revealed that a combination of ARQ 092 and ARQ 087 enhanced the inhibition of both the AKT and FGFR pathways in cell lines in which synergistic effects were found (AN3CA and IGROV-1). Cell cycle arrest and apoptotic response occurred only in AN3CA cell, and was not seen in IGROV-1 cells. Furthermore, enhanced antitumor activity was observed in mouse models with endometrial cancer cell line and patient-derived tumors when ARQ 092 and ARQ 087 were combined. These results from in-vitro and in-vivo studies provide a strong rationale in treating endometrial and other cancers with the activated PI3K/AKT and FGFR pathways.
Introduction
PI3K/AKT is a major signaling pathway involved in various biological functions including cell cycle, proliferation/growth, survival, metabolism, and metastasis, and it is one of the most frequently dysregulated pathways in cancer 1,2. It is evident that dysregulation, including hyperactivity, amplification, or activating mutations of genes encoding PI3K and AKT, or PTEN deficiency, can cause aberrant signaling of this pathway. Targeting the PI3K/AKT pathway is a rational approach to fight cancer, and different PI3K/AKT inhibitors are in various stages of development 3–6.
In preclinical studies, PI3K and AKT inhibitors have shown significant single-agent efficacy both in vitro and in vivo 6–11. However, in the clinic, blockage of these targets has not produced the responses that the preclinical data set suggested 12,13. Unlike the resistance caused by acquired mutations on genes such as BCR-ABL and EGFR 14,15, compensatory signaling pathway changes appear to be the major event for the diminished efficacy of PI3K/AKT inhibitors. Inhibition of the PI3K/AKT pathway increased phosphorylation of MAPK through an S6-mediated negative feedback loop 16. Chandarlapaty et al. 17 have shown that inhibition of AKT activity upregulated the expression and activity of many receptor kinases such as HER2 and HER3 through the feedback action of FOXO proteins 17. Combination of PI3K/AKT/mTOR inhibitors with other targeted agents became an emerging approach to overcome such resistance 6,10,18–20.
The efficacy of PI3K/AKT inhibitors is attenuated if alternative pathway(s) activated in parallel to the PI3K/AKT pathway 21–23. For instance, inhibition of the PI3K pathway did not induce significant apoptotic response in EGFR-addicted NSCLC cell lines, but combined inhibition of the PI3K and MAPK pathways achieved superior efficacy in vitro and in vivo 23. Furthermore, in the cancer cell lines with genetically altered FGFR2, concomitant inhibition of FGFR and mTORC1 performed better in terms of antiproliferation and antitumor activity compared with single agents 24,25. Clinical studies have been undertaken to evaluate the effect of PI3K/AKT pathway inhibitors in combination with estrogen antagonists, aromatase inhibitors, and HER2 inhibitors 26,27.
In this study, we assessed the combined effect of ARQ 092 and ARQ 087 in a panel of 45 cancer cell lines and showed that the highest synergism was found in endometrial and ovarian cancer cells. ARQ 092 is an oral, selective pan-AKT inhibitor that is potent against the AKTE17K activating mutation. ARQ 092 also demonstrates anticancer effect on cells bearing PIK3CA mutations 6 and is currently in phase I single-agent and combination clinical trials. ARQ 087 is a multikinase inhibitor with potent activity against FGFR1/2/3 28, being tested in a phase I/II trial for patients with intrahepatic cholangiocarcinoma. Inhibition of both AKT and FGFR resulted in cell cycle arrest and apoptosis in AN3CA cells but not in IGROV-1 cells. Combination of ARQ 092 and ARQ 087 exerted enhanced antitumor activity in in-vivo models with endometrial cancer cell line and one of the patient-derived tumors compared with the single agents. Both compounds are orally delivered agents with good bioavailability and good tissue/tumor exposure. These results provide us a rationale in treating endometrial and other cancers with activated AKT and FGFR signaling using combined therapy of ARQ 092 and ARQ 087.
Materials and methods
Reagents
ARQ 092 and ARQ 087 were synthesized at ArQule, Incorperated. Cycletest plus DNA reagent kit was purchased from BD Biosciences (Franklin Lakes, New Jersey, USA). Primary antibodies for western blot analysis were purchased from Cell Signaling Technology (Danvers, Massachusetts, USA): phospho-AKT1 (Ser473) (Cat# 9271), pan-AKT (Cat# 2920), phospho-ERK (Thr202/Tyr204) (Cat# 9101), total ERK (Cat# 9107), phospho-S6 ribosomal protein (Ser235/236) (Cat# 4858), phospho-S6 ribosomal protein (S240/244) (Cat# 2215), phospho-GSK-3β(S9) (Cat#9336), phospho-FoxO1 (T24)/3a(T32) (Cat# 9464), phospho-Bad (S136) (Cat# 4366), and total FGFR2 (Cat# 11835). Phospho-FGFR1–4 (Y653/Y654) was purchased from R&D systems (Minneapolis, Minnesota, USA) and β-actin (Cat# A2228) from Sigma-Aldrich (St Louis, Missouri, USA). Precast Tris Glycine gels and PVDF were purchased from Life Technologies (Carlsbad, California, USA).
Cell culture
Cancer cells were grown in cell culture media recommended by the vendors, and supplemented with 10% FBS maintained at 37°C in a humidified atmosphere at 5% CO2 (Supplementary Table 1, Supplemental digital content 1, http://links.lww.com/ACD/A194). IGROV-1 cells were donated by Daiichi Sankyo (Chuo-ku, Tokyo, Japan).
Cell proliferation MTS assays
For single-agent assays, cells were seeded at an optimal number per well in 130 μl of full growth media in 96-well tissue culture plates, incubated overnight, and then treated with three-fold serial dilutions of ARQ 087 or ARQ 092 at a starting concentration of 33.3 or 100 μmol/l. Treated cells were incubated at 37°C for 72 h in 5% CO2.
For the combination study, cells were seeded in 96-well tissue-culture plates at optimal number of cells per well overnight and subsequently treated with serial dilutions of ARQ 087 at a starting concentration of 33.3 μmol/l in combination with serial dilutions of ARQ 092 at a starting concentration of 100 μmol/l. Treated cells were incubated at 37°C for 72 h in 5% CO2.
Thirty microliters of the mixture of MTS reagent (18.4 mg/ml) and PMS (0.92 mg/ml) at a ratio of 20 : 1 was added to each well, and the plates were incubated at 37°C for 4 h in 5% CO2. The absorbance was measured at 490 nmol/l using the Victor microplate reader. For single-agent assay, IC50 was determined using Microsoft ExcelFit software (Microsoft Corporation, Redmond, Washington, USA) in ArQule Activity Base. For the combination studies, a Combination Index (CI) of 50% effect was determined in Activity Base using the Chou–Talalay method 29 (synergistic: CI≤0.85; additive: CI≥0.85 and ≤1.2; and antagonistic: CI≥1.2).
Mutation analysis
Mutational status of cell lines was obtained from the COSMIC database (http://cancer.sanger.ac.uk/cosmic). Fisher’s exact test was applied, with P value less than 0.05 considered as statistically significant.
Reverse phase protein analysis
AN3CA, IGROV-1, and Caov-3 cells were plated in a six-well plate and then treated with various concentrations of ARQ 092 or ARQ 087 as single agents or combination for 2 h. The cell pellet was collected, snap frozen, and shipped to Theranostics Health (Gaithersburg, Maryland, USA) (http://www.theranosticshealth.com). pAKT (S473), pPRAS40 (T246), pS6 (S235/S236), and p4EBP1 (T37/T46) levels were assessed using reverse phase protein array.
Cell cycle analysis using flow cytometry
AN3CA and IGROV-1 cells were treated with ARQ 092 at 1 μmol/l or ARQ 087 at 1 μmol/l as single agents or combination for 72 h, trypsinized, washed in cold PBS, and stained using Cyletest Plus reagent kit (BD biosciences) according to the manufacturer’s instruction. Cell cycle profile was analyzed using a FACS Calibur flow cytometer (BD Biosciences).
Western blot analysis
Cells treated under designated conditions were lysed using cell lysis buffer supplemented with Phosphatase and Protease Inhibitor cocktail (Sigma-Aldrich). For tumor tissue process, tumor samples were kept on dry ice at all times and a tissue pulverizer was dipped in liquid nitrogen before use. Tumor tissues were first pulverized and then lysed in cell lysis buffer supplemented with Halt Protease Phosphatase inhibitor (ThermoFisher, Waltham, Massachusetts, USA). Proteins were resolved in SDS-PAGE and transferred to PVDF membranes. After blockage with 5% BSA in TBX (1×) blocking buffer, the membranes were incubated with primary antibodies with 1% BSA in TBST overnight at 4°C and then blotted with HRP-conjugated secondary antibodies (Cell Signaling Technology). Immunoblotting detection was performed with GE ECL Prime reagent (GE Healthcare, Piscataway, New Jersey, USA) and images were captured using FuJi LAS 3000 system (FUJIFILM Corporation, Tokyo, Japan).
In-vivo studies
All experimental procedures were performed according to the Institutional Animal Care and Use Committee of Crown Biosciences Inc. (Santa Clara, California, USA) or START (San Antonio, Texas, USA).
AN3CA cells (5×106) were inoculated into female BALB/c nude mice. ARQ 092 was formulated in vehicle of 0.01 mol/l phosphoric acid pH22.25, and ARQ 087 was formulated in a vehicle of dimethylacetamide : cremophor EL : propylene glycol : 0.2 mol/l acetate buffer pH 5 (10 : 10 : 30 : 50). Tumor-bearing mice were dosed with ARQ 092 at 50, 75, or 100 mg/kg, or with ARQ 087 at 50 and 75 mg/kg or their combination orally once daily for 12 days. The efficacy of ARQ 092 and ARQ 087 in human endometrial PDX models was determined in START. Tumor-bearing mice were dosed with ARQ 092 at 100 mg/kg or ARQ 087 at 75 g/kg alone or in combination for 4 days followed by a 3-day break, for four cycles.
The tumor length and width (mm) were measured using a digital caliper. Estimated tumor volume (mm3) was calculated according to the formula 1/2×(tumor length)×(tumor width)2, shown as mean±SEM. Percent inhibition or tumor growth inhibition (%TGI) was calculated using the formula: 1−(mean tumor value of treated/mean tumor value of control)×100.
Statistical analysis
All statistical analyses were performed using an unpaired t-test. Data were presented as mean±SEM. A P value of less than 0.05 was considered to be statistically significant.
Pharmacokinetic analysis
Plasma and tumor samples were collected at 24 h after the last dose of ARQ 092, and plasma and tumor levels of ARQ 092 and ARQ 087 were determined by means of LC-MS/MS.
Results
Synergistic effect of ARQ 092 and ARQ 087 in endometrial and ovarian cancer cells in vitro
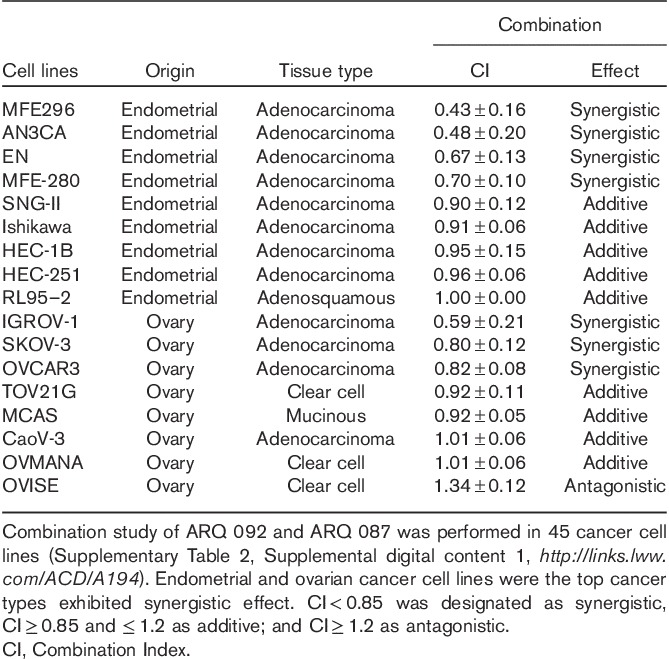
Inhibition of AKT by ARQ 092 has shown antiproliferative and antitumor effect 6. Previous studies have shown that concurrent inhibition of the PI3K/AKT and FGFR pathways enhanced the antiproliferative effect in FGFR-addicted cells compared with single pathway inhibition 11,24,25,30. We tested the combination of ARQ 092 and ARQ 087 on a panel of 45 cancer cell lines originating from ovarian, endometrial, colorectal, bladder, breast, lung, central nervous system, head and neck, cervix, vulvar cancer, lymphoma, and leukemia. CI at 50% effectiveness was determined on the basis of the median-effect principle by Chou–Talalay 29. Combined treatment of ARQ 092 and ARQ 087 resulted in a synergistic effect in 24% (11 out of 45), additive in 62% (28 out 0 f 45), and antagonistic in 13% (6 out of 45) of cancer cell lines tested (Supplementary Table 2, Supplemental digital content 1, http://links.lww.com/ACD/A194). Interestingly, endometrial (44%) and ovarian (38%) cancer cells were the two top cancer types showing most synergism (Table 1). These data indicate that ARQ 092 and ARQ 087 are combinable and exerted synergism in certain cancer cells lines, particularly endometrial and ovarian cancers.
Table 1.
Combination of ARQ 092 and ARQ 087 showed more synergism in endometrial and ovarian cancer cell lines
It has been shown that the PI3K/AKT and FGFR pathways are frequently aberrant in endometrial and ovarian cancers 31–34. To determine whether synergism of ARQ 087 and ARQ 092 can be predicted by genetic characteristics of treated cells, we analyzed the somatic mutational status of selected genes in the entire panel of cancer cell lines treated and assessed any potential correlation. As shown in Fig. 1a, 38% (11 out of 29) of cell lines with PIK3CA/PIK3R1 mutations showed synergistic effect when ARQ 092 and ARQ 087 were combined, whereas cell lines with wild-type PIK3CA/R1 showed no synergism (P=0.004). It has been shown that the mutational status of Ras (KRas, HRas, and NRas) could be a negative factor for various targeted therapies 35. We analyzed the Ras mutation status in all tested cell lines to determine whether it has any effect on the combinatorial treatment of ARQ 092 and ARQ 087. As shown in Fig. 1b and Supplementary Table 2, Supplemental digital content 1, http://links.lww.com/ACD/A194, in cell lines with mutant PIK3CA/PIK3R1, 59% (10 out of 17) of cell lines with wild-type Ras showed synergism; however, only 8% (1 out of 12) of cell lines with mutant Ras displayed synergism following ARQ 087 and ARQ 092 treatment. The only cell line with Ras mutation showing synergism was AN3CA cells, which bears a substitution of phenylalanine by leucine at position 82 of HRas, and may not have any impact on HRas activity. Accumulated evidence has shown that activating mutations of Ras commonly occurs at codons 12, 13, and 61 36. Interestingly, FGFR2 mutations were only observed in endometrial cancer cell lines showing synergism but not in ovarian cell lines. Mutations of FGFR in ovarian cancer are rare but the FGFR pathway plays a very important role in ovarian cancer initiation and progression through paracrine or autocrine effect 32,37–39. Taken together, PIK3CA/PIK3R1 mutations and aberrant activation of FGFR2 may predict synergism of combination treatment with ARQ 092 and ARQ 087, whereas Ras mutation is a negative indicator for synergism.
Fig. 1.
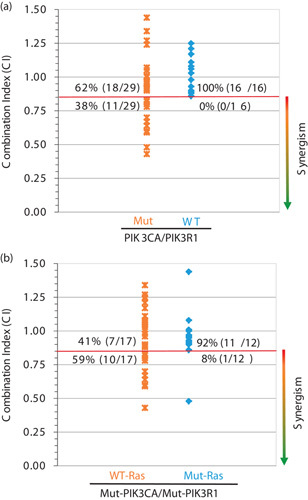
PI3K3CA mutation may predict synergism of combination of ARQ 092 and ARQ 087. Somatic mutation analysis was performed for 45 cancer cell lines tested. Synergistic effect of ARQ 092 and ARQ 087 combination was defined as CI<0.85. (a) Scatter plot shows the correlation between synergism and PIK3CA and PIK3R1 mutations. (b) Scatter plot shows the correlation between synergism and Ras mutations. CI, Combination Index; WT, wild type.
Combination of ARQ 092 and ARQ 087 regulates the AKT and FGFR pathways
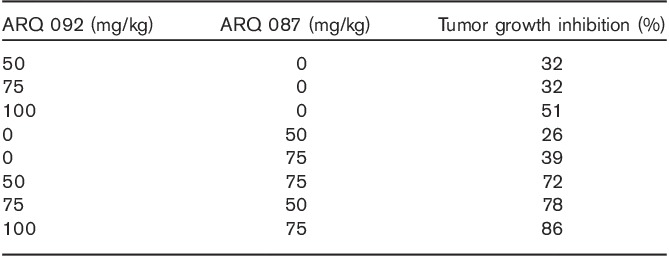
To determine how the combined treatment of ARQ 092 and ARQ 087 has effect on the AKT and FGFR pathways, we selected two synergistic cell lines, AN3CA (CI=0.48) and IGROV-1 (CI=0.59), and one additive cell line, Caov3 (CI=0.98). AN3CA cells have several major mutations, including PIK3R1 (R557 K561Q), PTEN (R130fs*), and FGFR2 (N549K & K310R). IGROV-1 cells contain mutations of PIK3CA (R38C, *1069 W) and PTEN (T319fs*1, Y155C) and express FGFRs, and Caov3 cells have no mutations in PIK3CA, PIK3R1, PTEN, and FGFRs and express FGFRs 39. Synergistic effect was also observed in AN3CA and IGROV-1 cells when ARQ 092 and AZD4547, a FGFR inhibitor, were combined (data not shown). Cells were treated with ARQ 092 or ARQ 087 at various concentrations as single agents or combination, and phospho proteins pAKT (S473), pPRAS40 (T246), pS6 (S235/S236), and p4EBP1 (T37/T46) were assessed using reverse phase protein array. As shown in Fig. 2a, ARQ 092 but not ARQ 087 inhibited phosphorylation of AKT, PRAS40, S6, and 4EBP1 in both AN3CA and IGROV-1 cells. In CaoV3 cells, only inhibition of phospho-S6 was observed upon treatment with ARQ 087. Combined treatment with ARQ 092 and ARQ 087 resulted in the enhanced inhibition of pAKT (S473), pS6 (S235/236), p4EBP1 (T37/T46), and pPRAS40 (T246) in both AN3CA and IGROV-1 cells (synergistic) but not in Caov3 cells (additive). Furthermore, to assess changes in pathway in AN3CA and IGROV-1 cells after treatment with ARQ 092 or ARQ 087 as single agents or combination, we performed western blot analysis. As shown in Fig. 2b, ARQ 092 inhibited AKT and its downstream targets, whereas ARQ 087 suppressed phosphorylation of FGFR and ERK but had no effect on the direct AKT targets pFOXO, pGSK-3β, and pBAD. IGROV-1 cells exhibited no detectable pBAD. Combination of ARQ 092 and ARQ 087 markedly inhibited phosphorylation for S6 phosphorylation in both AN3CA and IGROV-1 cells compared with the single agents. However, ARQ 087 did not significantly potentiate ARQ 092-induced decrease in pFOXO, pGSK-3β, and pBAD. Interestingly, ARQ 092 had no effect on pFGFR in AN3CA cells but increased phosphorylation of FGFR in IGROV-1 cells. Concurrent treatment of ARQ 092 and ARQ 087 at a high concentration (1 μmol/l) enhanced dephosphorylation of FGFR induced in IGROV-1 cells but not AN3CA cells. The exact mechanism underlining ARQ 092 that induced an increase in phosphorylation of FGFR in IGROV-1 cells still needs to be further investigated. These data suggest that the enhanced inhibition of phosphorylation of S6 with combined treatment of ARQ 092 and ARQ 087 may drive synergism for two agents.
Fig. 2.
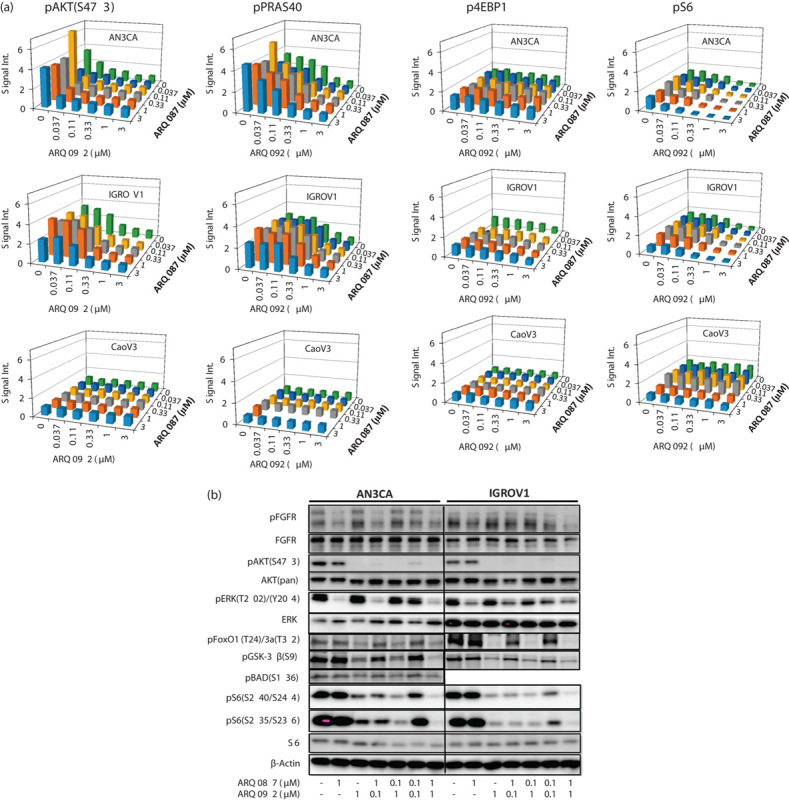
Combined effect of ARQ 092 and ARQ 087 on signaling pathway. (a) AN3CA, IGROV-1, and Caov3 cells were treated with various concentrations of ARQ 092 or ARQ 087 as single agents or combination for 2 h. pAKT (S473), pPRAS40 (T246), pS6 (S235/S236), and p4EBP1 (T37/T46) were assessed using RPMA. (b) AN3CA and IGROV-1 cells were treated with ARQ 092 or ARQ 087 at 0.1 or 1 μmol/l as single agents or combination for 2 h. pFGFR, pAKT (S473), pERK1/2 (T202/Y204), pFoxO1 (T24)/3a(T32), pGSK-3β (S9), pBAD (S136), pS6 (S235/S236), pS6 (S240/S244), and their total proteins were assessed using western blot analysis.
Combination of ARQ 092 and ARQ 087 induced cell cycle arrest and apoptosis
Next, we assessed the effect of combination of ARQ 092 and ARQ 087 on cell cycle and apoptotic response. AN3CA and IGROV-1 cells were treated with ARQ 092 and ARQ 087 at 1 μmol/l as single agents or combination for 72 h. As shown in Fig. 3, and Supplementary Fig. 1 (Supplemental digital content 1, http://links.lww.com/ACD/A194) in AN3CA cells, combined treatment led to cell cycle arrest in G1 phase, concomitant with a decrease in the percentage of cells in S phase. Significant apoptotic response was observed in AN3CA cells as an increase in sub-G1 phase. Interestingly, no change in cell cycle and apoptosis were observed in IGROV-1 cells.
Fig. 3.
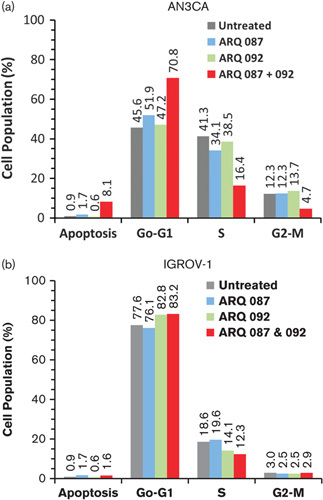
Combination of ARQ 092 and ARQ 087 induced G1 phase arrest. AN3CA (a) and IGROV-1 (b) cells were treated with ARQ 092 or ARQ 087 at 1 μmol/l or combination for 72 h. Cells were harvested, stained, and analyzed using flow cytometry. The percentage of cells in G1, S, or G2/M phase was calculated.
Combination of ARQ 092 and ARQ 087 exhibited enhanced antitumor activity in vivo
To determine whether the combination of ARQ 092 with ARQ 087 exerted enhanced antitumor activity, we assessed the combined effect of ARQ 092 and ARQ 087 in the AN3CA xenograft model. In previous studies, we have demonstrated that ARQ 092 and ARQ 087 as single agents suppressed tumor growth in the AN3CA xenograft model 6,28. Mice bearing AN3CA tumors were treated with various doses of ARQ 092 and ARQ 087. As single agents, ARQ 092 at 50, 75, and 100 mg/kg showed %TGI of 32, 32, and 51%, whereas ARQ 087 at 50 and 75 mg/kg showed a %TGI of 26 and 39%, respectively. Combination of ARQ 092 at 100 mg/kg and ARQ 087 at 75 mg/kg exerted an enhanced %TGI of 86% compared with single agents (P<0.05) (Fig. 4a and Table 2). Furthermore, either combination of ARQ 092 at 75 mg/kg with ARQ 087 at 50 mg/kg or combination of ARQ 092 at 50 mg/kg with ARQ 087 at 75 mg/kg exhibited a TGI of 78 and 72% (P<0.05), respectively. However, significant toxicity was observed in the group receiving ARQ 092 at 100 mg/kg and ARQ 087 at 75 mg/kg with a body weight loss of about 21.4% after 12 days (Fig. 4b). Moderate body weight loss (10.9 and 11.5% on day 12) was observed for the groups receiving ARQ 092 at 75 mg/kg with ARQ 087 at 50 mg/kg and ARQ 092 at 50 mg/kg with ARQ 087 at 75 mg/kg. Furthermore, we assessed the combined effect of ARQ 092 and ARQ 087 in two human endometrial PDX models (ST413 and ST259). ST413 model contains PIK3CAH1047R and FGFR1E486Q&L417R mutations; and ST259 model has PIK3CAR93W&D350G and FGFR4V10I&P136L mutations. In a dosing schedule of 3 days on and 4 days off for 4 cycles, a combination of ARQ 092 at 100 mg/kg and ARQ 087 at 75 mg/kg exhibited enhanced antitumor activity (67%) in ST413 model compared with single agents (43% for ARQ 092 and 0% for ARQ 087). However, there is no statistical difference from ARQ 092 as single agent. In ST259 model, combination of two agents did not perform better than ARQ 092 as single agent. These results indicate that combination of ARQ 092 with ARQ 087 improved each single agent in terms of antitumor activity in vivo, but the toxicity of such combination needs further investigation.
Fig. 4.
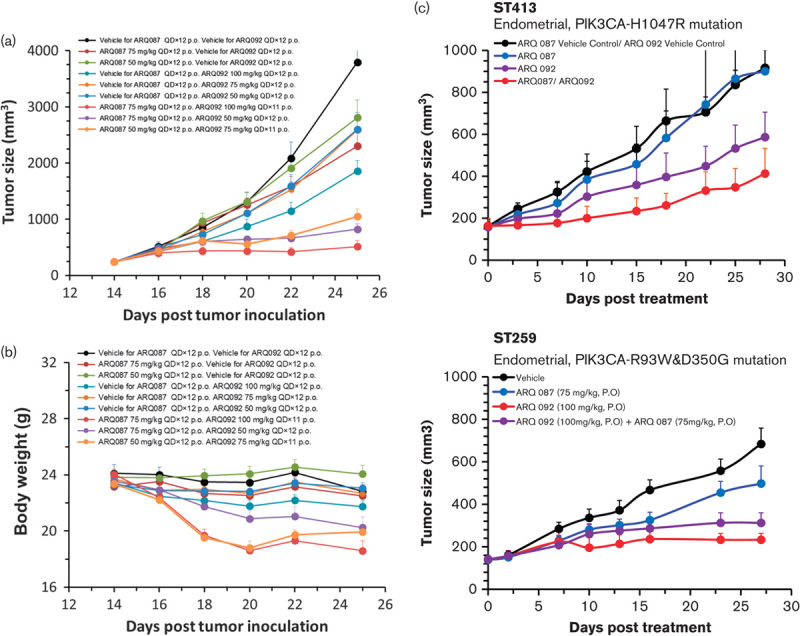
Combination of ARQ 092 and ARQ 087 exerted enhanced antitumor activity in AN3CA cells. (a) Mice bearing AN3CA cells were dosed with ARQ 092 at 50, 75, or 100 mg/kg or ARQ 087 at 50 and 75 mg/kg as single agents or combination orally once daily for 12 days (n=10). (b) Body weight loss of mice bearing AN3CA cells was recorded during the dosing period. (c) Mice bearing human endometrial PDX models ST413 (PIK3CAH1047R and FGFR1E486Q&L417R) and ST259 (PIK3CAR93W&D350G and FGFR4V10I&P136L) were dosed with ARQ 092 at 100 mg/kg (n=3) or ARQ 087 at 75 g/kg (n=3) or combination (n=5) in a schedule of 4 days on and 3 days of for four cycles. The statistical significance as shown in comparison with the vehicle group (*P<0.05; **P<0.01).
Table 2.
Summary of in-vivo efficacy of combination of ARQ 092 and ARQ 087 in AN3CA xenograft model
In-vivo pharmacodynamics and pharmacokinetic studies of combination of ARQ 092 and ARQ 087
To further assess target inhibition, we performed western blot analysis and evaluated the changes in the AKT pathway and FGFR phosphorylation. pFGFR, pAKT, pERK, pPRAS40, and pS6 were analyzed in the tumor samples from the control group and the group treated with ARQ 092 and ARQ 087. As shown in Fig. 5a and Supplementary Fig. 2 (Supplemental digital content 1, http://links.lww.com/ACD/A194), ARQ 092 inhibited AKT phosphorylation and its downstream targets PRAS40 and S6 but elevated phopho ERK, whereas ARQ 087 suppressed FGFR phosphorylation. Combination of ARQ 092 and ARQ 087 enhanced the inhibition of AKT, ERK, PRAS40, and S6. These data suggest that combination of ARQ 092 and ARQ 087 exerted enhanced AKT and FGFR pathway inhibition in vivo.
Fig. 5.
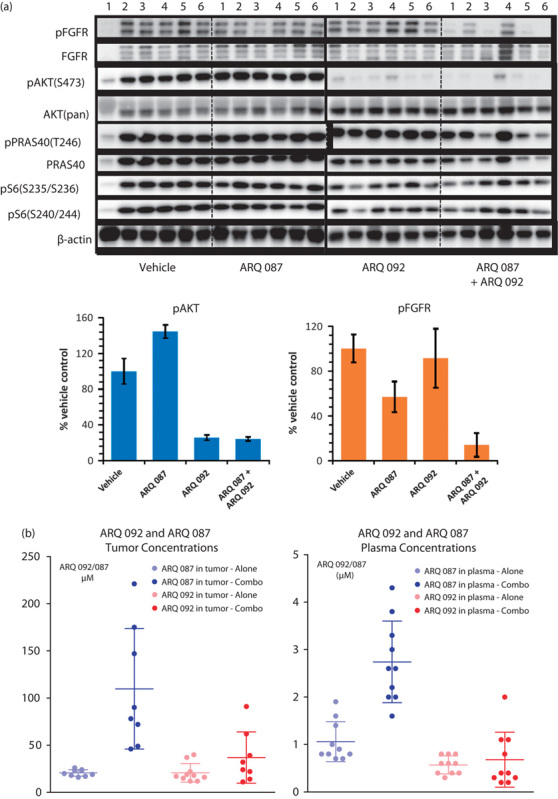
Combination of ARQ 092 and ARQ 087 enhanced the inhibition of the AKT and FGFR pathways in vivo. (a) Tumor samples from AN3CA bearing mice treated with ARQ 092 at 100 mg/kg or ARQ 087 at 75 mg/kg or combined treatment were assessed for pFGFR (Y653/Y654), FGFR2, pAKT (S473), AKT, pPRAS40 (T246), pS6 (S235/S236), and pS6 (S240/S244) using western blot analysis. Six samples from each group were analyzed. One of the samples from vehicle was not analyzed due to poor quality. (b) Plasma and tumor levels for both compounds in single agent and combination dose groups.
ARQ 092 and ARQ 087 concentrations were determined for three treatment groups: the ARQ 087 75 mg/kg, 092 100 mg/kg, and ARQ 087/092 75/100 mg/kg combination groups. In each group both tumor and plasma were assessed by means of LC-MS/MS analysis (5b). In the animals treated with ARQ 087, the mean plasma concentration was 1.1±0.4 µmol/l, and in the combined dosing group the mean ARQ 087 concentration was 2.7±0.8 µmol/l. For ARQ 092, plasma concentrations were 0.6±0.2 µmol/l in the single agent group and 0.7±0.6 µmol/l in the combination group. In tumors, the mean ARQ 087 levels were 20±0.4 µmol/l (0.4 µmol/l SD) in the single-agent treatment group and 110±64 µmol/l in the combination group, whereas the mean ARQ 092 tumor concentrations were 25±10 and 37±27 µmol/l, respectively.
Discussion
AKT acts as a critical node connecting the PI3K and mTOR pathways and many AKT inhibitors are in various development stages in addition to the development of PI3K and mTOR inhibitors. To improve the efficacy or overcome the resistance, the combination of AKT inhibitors and other agents appears a valid strategy. In combination with MEK inhibitors, inhibition of AKT enhanced antiproliferation in vitro and antitumor activity in vivo 6,10,19,40. Concurrent inhibition of the AKT pathway with other pathway inhibitors (e.g. estrogen receptor, androgen receptor, and HER2) also showed more effectiveness in preclinical studies 41–43, and clinical studies for such combination have been undertaken 12,27. ARQ 092 is a selective and potent allosteric AKT inhibitor currently under phase I/Ib clinical investigation 6. As a multikinase inhibitor, ARQ 087 preferably suppresses FGFR family members and is in phase I/II trial in patients with FGFR genetic alterations 28. In this study, we report that the combination therapy of ARQ 092 and ARQ 087 exhibited more effectiveness in antitumor activity in-vitro and in-vivo compared with the same experimental drugs administered as single agents. We assessed the combined effect of ARQ 092 and ARQ 087 in 45 cancer cell lines representing 13 different tumor types. Synergism was observed in 24% (11 out of 45), and endometrial and ovarian cancers showed the highest percentage of synergism of 44 and 38%, respectively. Mutational analysis showed that PIK3CA and PIK3R1 significantly increased chance of synergism (38%) for ARQ 092 and ARQ 087 compared with the cell lines with wild type (0%). Our findings also demonstrated that activating mutations of Ras (including KRas, HRas, and NRas) are a negative predictor for synergism in combination of ARQ 092 and ARQ 087.
Furthermore, in addition to mutations of PIK3CA/PIK3R1, the activated FGFR pathway through mutations or ligand engagement may be required for such synergism to occur. It has been shown that endometrial cancers contain a high frequency of FGFR mutations, which were accompanied with PIK3CA mutations in many cases 31,34. FGFR mutations are rare in ovarian cancers but the majority of ovarian cancers express FGFR 32. However, activation of the FGFR pathway by its ligands promotes tumor progression and associates with resistance to therapeutic agents 37–39. Knockdown of FGFR1 and 2 improved the response of ovarian cancer cells to cisplatin treatment in vitro and in vivo 39. It is evident that concurrent inhibition of the FGFR and AKT/mTOR pathways resulted in more significant antitumor activity in models with activated AKT/mTOR and FGFR signaling 11,24,25. Thus, our data support the notion that combined therapy with ARQ 092 and ARQ 087 could be a feasible approach in endometrial and ovarian cancers with PIK3CA/PIK3R1 mutation and activated FGFR pathway. However, activating mutations of Ras may counteract such synergistic effect 35.
To understand the molecular mechanism for the changes in pathways, we analyzed the changes in AKT and FGFR pathways in vitro and in vivo. Enhanced inhibition of AKT and FGFR pathways only occurred in the cell lines (AN3CA and IGROV-1 cells) that exhibited synergistic sensitivity to combined treatment with ARQ 092 and ARQ 087 but not with Caov3 cells that exhibited only additive sensitivity. AN3CA cells bear mutations of PIK3R1, PTEN, and FGFR2, whereas IGROV-1 contains PIK3CA mutations and express wild-type FGFRs. Interestingly, suppression of phosphorylation of ribosomal protein S6 was particularly more significant in combination groups compared with single-agent groups in in-vitro cell models. The observations were further confirmed by our in-vivo study. However, the combination of ARQ 092 and ARQ 087 did not result in enhanced reduction of phosphorylation of direct AKT downstream targets (FOXO, GSK, and BAD), suggesting that suppression of FGFR and AKT affects distinct pathways. Thus, it is reasonable to believe that S6 could be a converging point for the PI3K/AKT and FGFR pathways. Further inhibition of S6 phosphorylation may be required for the synergistic effect of ARQ 092 and ARQ 087 combination. It is evident that MAPK-associated RSK activity contributed to S6 phosphorylation through two distinct mechanisms: (a) inhibition of TSC1/TSC2 complex, which subsequently elevated mTORC activity toward S6 kinase/S6, or (b) direct phosphorylation of S6 at serine 235/236 sites 44–46. Other studies have also shown that, in FGFR-addicted cell lines, concomitant inhibition of mTOR and FGFR exerted more reduction in S6 phosphorylation compared with the single agents 24,25. In-vivo efficacy study in xenograft model with endometrial AN3CA cells showed that the combination of ARQ 092 at 50, 75, or 100 mg/kg with ARQ 087 at 50 and 75 mg/kg exerted significant TGI. At maximum tolerated doses of ARQ 092 (100 mg/kg) and ARQ 087 (75 mg/kg), the highest TGI (86%) resulted, but was associated with severe toxicity with a body weight loss of 21.4% in the end of study. The toxicity was minimized when mice were dosed with ARQ 092 at 50 mg/kg+ARQ 087 (TGI=72%) at 75 mg/kg, and ARQ 092 at 75 mg/kg+ARQ 087 at 50 mg/kg (TGI=78%). Only 11.5 and 10.9% body weight loss was observed, respectively, in each group. Interestingly, plasma and tumor concentrations from this study revealed that, when dosed in combination with ARQ 092, ARQ 087 plasma and tissue levels are elevated by 2–5-fold. The precise reason for the higher ARQ 087 levels is incompletely understood, and may be due to alterations in the absorption and metabolism of the combined agents. In addition, this may be the cause for the increased toxicity observed in the animals dosed with both compounds; however it is notable that ARQ 087 concentrations as high as those observed in the combo group have been seen in previous xenograft studies without any increased toxicity 28. The toxicity data suggests that continuous dosing of both agents may be suboptimal. Subsequently, in a modified dosing regimen of ARQ 092 at 100 mg/kg and ARQ 087 at 75 mg/kg 4 days on and 3 days off per week, enhanced antitumor activity was observed in one of the two endometrial PDX with PIK3CA and FGFR mutations, with no toxicity observed. Therefore, our in-vivo efficacy data provide a basis for the combination of these two agents only if the ideal dosing regimen can be achieved. Finally, in 7-day toxicity studies in rats, ARQ 092 (15 mg/kg) and ARQ 087 (50 mg/kg) combinations were well tolerated, suggesting that the toxicity observed in the xenograft models may be driven by species-specific metabolism (Supplementary Fig. 3, Supplemental digital content 1, http://links.lww.com/ACD/A194).
ARQ 092 and ARQ 087 are being tested in clinical trials as single agents (NCT01473095, NCT01752920), with encouraging signs of efficacy in different cancer types. An increasingly common strategy in cancer treatment involves the combination of agents targeting different pathways or targeting the same pathway at multiple points 26,27, and as recently shown by Davies et al. 30 in endometrial and bladder tumors, there is a preclinical and clinical rationale for combining AKT and FGFR inhibitors.
In summary, we have demonstrated in this study that the combination of ARQ 092 and ARQ 087 exerted enhanced antiproliferative activity in vitro and antitumor activity in vivo. Mutations of PIK3CA/PIK3R1 and FGFR may predict patients who are most likely to respond to the therapy, whereas activating mutations of Ras may prevent responses to these agents. Thus, the results provide an avenue for combined therapy of ARQ 092 and ARQ 087 in endometrial and other cancer patients with defined molecular signatures.
Supplementary Material
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website (www.anti-cancerdrugs.com).
Acknowledgements
The authors thank Enkeleda Nakuci, Chang-Rung Chen, and Daniel T. Dransfield for their contribution to this project.
The authors acknowledge the financial support from ArQule, Inc. and critical review by ArQule colleagues.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer 2002; 2:489–501. [DOI] [PubMed] [Google Scholar]
- 2.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 2006; 7:606–619. [DOI] [PubMed] [Google Scholar]
- 3.Mayer IA, Arteaga CL. The PI3K/AKT pathway as a target for cancer treatment. Annu Rev Med 2016; 67:11–28. [DOI] [PubMed] [Google Scholar]
- 4.Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 2015; 15:7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nitulescu GM, Margina D, Juzenas P, Peng Q, Olaru OT, Saloustros E, et al. Akt inhibitors in cancer treatment: the long journey from drug discovery to clinical use (review). Int J Oncol 2016; 48:869–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu Y, Savage RE, Eathiraj S, Meade J, Wick MJ, Hall T, et al. Targeting AKT1-E17K and the PI3K/AKT pathway with an allosteric AKT inhibitor, ARQ 092. PLoS One 2015; 10:e0140479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burger MT, Pecchi S, Wagman A, Ni ZJ, Knapp M, Hendrickson T, et al. Identification of NVP-BKM120 as a potent, selective, orally bioavailable class I PI3 kinase inhibitor for treating cancer. ACS Med Chem Lett 2011; 2:774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glauer J, Pletz N, Schon M, Schneider P, Liu N, Ziegelbauer K, et al. A novel selective small-molecule PI3K inhibitor is effective against human multiple myeloma in vitro and in vivo. Blood Cancer J 2013; 3:e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao YY, Tian Y, Zhang J, Xu F, Yang YP, Huang Y, et al. Effects of an oral allosteric AKT inhibitor (MK-2206) on human nasopharyngeal cancer in vitro and in vivo. Drug Des Devel Ther 2014; 8:1827–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dumble M, Crouthamel MC, Zhang SY, Schaber M, Levy D, Robell K, et al. Discovery of novel AKT inhibitors with enhanced anti-tumor effects in combination with the MEK inhibitor. PLoS One 2014; 9:e100880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies BR, Greenwood H, Dudley P, Crafter C, Yu DH, Zhang J, et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol Cancer Ther 2012; 11:873–887. [DOI] [PubMed] [Google Scholar]
- 12.Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol 2013; 10:143–153. [DOI] [PubMed] [Google Scholar]
- 13.Khan KH, Yap TA, Yan L, Cunningham D. Targeting the PI3K-AKT-mTOR signaling network in cancer. Chin J Cancer 2013; 32:253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Branford S, Rudzki Z, Walsh S, Grigg A, Arthur C, Taylor K, et al. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood 2002; 99:3472–3475. [DOI] [PubMed] [Google Scholar]
- 15.Huang L, Fu L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm Sin B 2015; 5:390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest 2008; 118:3065–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer cell 2011; 19:58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carracedo A, Baselga J, Pandolfi PP. Deconstructing feedback-signaling networks to improve anticancer therapy with mTORC1 inhibitors. Cell Cycle 2008; 7:3805–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meng J, Dai B, Fang B, Bekele BN, Bornmann WG, Sun D, et al. Combination treatment with MEK and AKT inhibitors is more effective than each drug alone in human non-small cell lung cancer in vitro and in vivo. PLoS One 2010; 5:e14124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ren H, Chen M, Yue P, Tao H, Owonikoko TK, Ramalingam SS, et al. The combination of RAD001 and NVP-BKM120 synergistically inhibits the growth of lung cancer in vitro and in vivo. Cancer Lett 2012; 325:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faber AC, Li D, Song Y, Liang MC, Yeap BY, Bronson RT, et al. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Natl Acad Sci USA 2009; 106:19503–19508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Serra V, Scaltriti M, Prudkin L, Eichhorn PJ, Ibrahim YH, Chandarlapaty S, et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene 2011; 30:2547–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Temraz S, Mukherji D, Shamseddine A. Dual inhibition of MEK and PI3K pathway in KRAS and BRAF mutated colorectal cancers. Int J Mol Sci 2015; 16:22976–22988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu Y, Lu H, Zhang J, Chen J, Chai Z, Zhang J. Essential role of AKT in tumor cells addicted to FGFR. Anticancer Drugs 2014; 25:183–188. [DOI] [PubMed] [Google Scholar]
- 25.Gozgit JM, Squillace RM, Wongchenko MJ, Miller D, Wardwell S, Mohemmad Q, et al. Combined targeting of FGFR2 and mTOR by ponatinib and ridaforolimus results in synergistic antitumor activity in FGFR2 mutant endometrial cancer models. Cancer Chemother Pharmacol 2013; 71:1315–1323. [DOI] [PubMed] [Google Scholar]
- 26.Castel P, Toska E, Zumsteg ZS, Carmona FJ, Elkabets M, Bosch A, et al. Rationale-based therapeutic combinations with PI3K inhibitors in cancer treatment. Mol Cell Oncol 2014; 1:e963447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JJ, Loh K, Yap YS. PI3K/Akt/mTOR inhibitors in breast cancer. Cancer Biol Med 2015; 12:342–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hall T, Yu Y, Savage RE, Eathiraj S, Wang Y, Lapierre JM, et al. Preclinical activity of ARQ 087, a novel inhibitor targeting FGFR dysregulation. PLoS One 2016; 11:e0162594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chou TC. Drug combination studies and their synergy quantification using the Chou–Talalay method. Cancer Res 2010; 70:440–446. [DOI] [PubMed] [Google Scholar]
- 30.Davies B, Guan N, Logie A, Crafter C, Hanson L, Jacobs V, et al. Tumors with AKT1E17K mutations are rational targets for single agent or combination therapy with AKT inhibitors. Molecular cancer therapeutics 2015; 14:2441–2451. [DOI] [PubMed] [Google Scholar]
- 31.Byron SA, Gartside M, Powell MA, Wellens CL, Gao F, Mutch DG, et al. FGFR2 point mutations in 466 endometrioid endometrial tumors: relationship with MSI, KRAS, PIK3CA, CTNNB1 mutations and clinicopathological features. PLoS One 2012; 7:e30801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Byron SA, Gartside MG, Wellens CL, Goodfellow PJ, Birrer MJ, Campbell IG, et al. FGFR2 mutations are rare across histologic subtypes of ovarian cancer. Gynecol Oncol 2010; 117:125–129. [DOI] [PubMed] [Google Scholar]
- 33.Cheaib B, Auguste A, Leary A. The PI3K/Akt/mTOR pathway in ovarian cancer: therapeutic opportunities and challenges. Chin J Cancer 2015; 34:4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cancer Genome Atlas Research Network, Kandoth C, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013; 497:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 2008; 14:1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takashima A, Faller DV. Targeting the RAS oncogene. Expert Opin Ther Targets 2013; 17:507–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei W, Mok SC, Oliva E, Kim SH, Mohapatra G, Birrer MJ. FGF18 as a prognostic and therapeutic biomarker in ovarian cancer. J Clin Invest 2013; 123:4435–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steele IA, Edmondson RJ, Bulmer JN, Bolger BS, Leung HY, Davies BR. Induction of FGF receptor 2-IIIb expression and response to its ligands in epithelial ovarian cancer. Oncogene 2001; 20:5878–5887. [DOI] [PubMed] [Google Scholar]
- 39.Cole C, Lau S, Backen A, Clamp A, Rushton G, Dive C, et al. Inhibition of FGFR2 and FGFR1 increases cisplatin sensitivity in ovarian cancer. Cancer Biol Ther 2010; 10:495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu R, Liu D, Xing M. The Akt inhibitor MK2206 synergizes, but perifosine antagonizes, the BRAF(V600E) inhibitor PLX4032 and the MEK1/2 inhibitor AZD6244 in the inhibition of thyroid cancer cells. J Clin Endocrinol Metab 2012; 97:E173–E182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vilquin P, Villedieu M, Grisard E, Ben Larbi S, Ghayad SE, Heudel PE, et al. Molecular characterization of anastrozole resistance in breast cancer: pivotal role of the Akt/mTOR pathway in the emergence of de novo or acquired resistance and importance of combining the allosteric Akt inhibitor MK-2206 with an aromatase inhibitor. Int J Cancer 2013; 133:1589–1602. [DOI] [PubMed] [Google Scholar]
- 42.Toren P, Kim S, Cordonnier T, Crafter C, Davies BR, Fazli L, et al. Combination AZD5363 with enzalutamide significantly delays enzalutamide-resistant prostate cancer in preclinical models. Eur Urol 2015; 67:986–990. [DOI] [PubMed] [Google Scholar]
- 43.Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 2010; 9:1956–1967. [DOI] [PubMed] [Google Scholar]
- 44.Steelman LS, Chappell WH, Abrams SL, Kempf RC, Long J, Laidler P, et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging (Albany NY) 2011; 3:192–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, et al. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem 2007; 282:14056–14064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci USA 2004; 101:13489–13494. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website (www.anti-cancerdrugs.com).