

Abstract
(+)-Ryanodine is a natural product modulator of ryanodine receptors, important intracellular calcium ion channels that play a critical role in signal transduction leading to muscle movement and synaptic transmission. Chemical derivatization of (+)-ryanodine has demonstrated that certain peripheral structural modifications can alter its pharmacology, and that the pyrrole-2-carboxylate ester is critical for high affinity binding to ryanodine receptors. However, the structural variation of available ryanodine analogues has been limited by the challenge of site-specific functionalization of semisynthetic intermediates, such as (+)-ryanodol. Here we report a synthetic strategy that provides access to (+)-ryanodine and the related natural product (+)-20-deoxyspiganthine in 18 and 19 steps, respectively. A key feature of this strategy is the reductive cyclization of an epoxide intermediate that possesses the critical pyrrole-2-carboxylate ester. This approach allows for the direct introduction of this ester in the final stage of the synthesis and provides a framework for the synthesis of previously inaccessible synthetic ryanoids.
Short abstract
A strategy to synthesize (+)-ryanodine and (+)-20-deoxyspiganthine—natural products that modulate calcium release from ryanodine receptors—in 18 and 19 steps, respectively, is reported.
The ryanodine receptors (RyRs) are large, tetrameric protein assemblies that mediate rapid Ca2+ release from the sarco/endoplasmic reticulum to the cytoplasm, thereby triggering numerous physiological processes, including excitation–contraction coupling (ECC).1,2 There are three mammalian RyR isoforms (RyR1, RyR2, and RyR3) that are expressed primarily in muscle and brain tissue, although the expression levels of each depend on the tissue.1 Several diseases have been traced to aberrant RyR function:3 for example, malignant hypothermia and central core disease are disorders of skeletal muscle that result from mutations in RyR1,4 whereas mutations in RyR2 have been linked to inherited arrhythmogenic cardiac diseases.5 In addition, dysregulation of RyR2- and RyR3-mediated calcium release has been linked to the pathogenesis of Alzheimer’s disease.6,7
The ryanodine receptors derive their name from a natural product, (+)-ryanodine (1) (Figure 1), a tritiated version of which was used as a radiochemical tag for their isolation and purification.8,91 is a structurally complex diterpenoid that was first isolated in 1948 by Folkers and co-workers from Ryania speciosa Vahl, an insecticidal shrub native to Central and South America.10 Although a molecular formula was provided in the initial report, two decades of chemical degradation and derivatization studies were required before the structure was correctly assigned.11−13 Since the isolation of 1, several structurally related natural products that share a common carbocyclic core but vary in peripheral oxidation, such as (+)-spiganthine (2) and (+)-20-deoxyspiganthine (3),14,15 have also been identified as modulators of RyR function.
Figure 1.
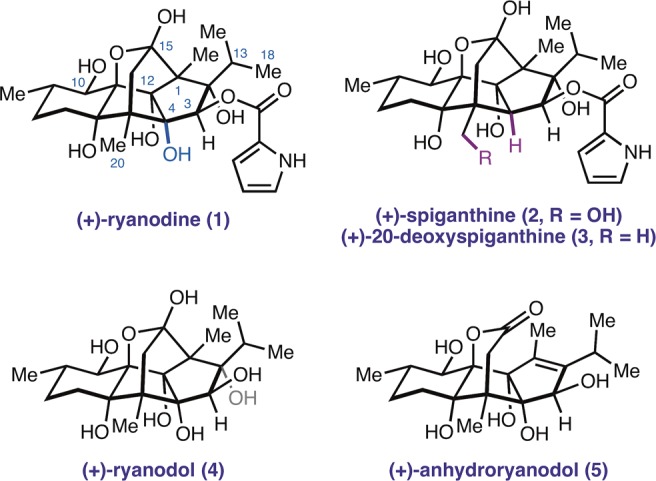
Ryanodine, ryanodol, and related natural products.
(+)-Ryanodine binds with high affinity to the conducting state of RyRs and is a valuable tool that has been used to interrogate the functional state of these ion channels.16 Notably, the hydrolysis product (+)-ryanodol (4) binds ∼1700 and ∼800 times more weakly than 1 to RyRs derived from rabbit skeletal and cardiac tissue, respectively.17 This structure–activity relationship (SAR) implicates the C3 pyrrole-2-carboxylate ester as a critical structural component for receptor binding. Unfortunately, the challenge of modifying (+)-ryanodol (or appropriately protected derivatives) at C3 has hampered the synthesis of ester analogues that would provide a more detailed SAR. In this report, we describe an 18-step chemical synthesis of ryanodine that overcomes this challenge by directly incorporating the C3 pyrrole-2-carboxylate ester at a late stage. We also demonstrate that the same approach can be used to prepare the related natural product (+)-20-deoxyspiganthine (3) in 19 steps. The strategy described here will serve as a platform for the synthesis and study of previously inaccessible ryanoids as tools to investigate and modulate RyR function.
Previous synthetic efforts toward the ryanoids have focused primarily on the preparation of the deacylated compound, (+)-ryanodol (4), with the first chemical synthesis reported by Deslongchamps and co-workers in 1979 (37 linear steps).18−22 Deslongchamps’s synthesis was guided by the discovery that (+)-anhydroryanodol (5) could be converted to 4 in two steps.18 Deslongchamps, Ruest, and co-workers,23 as well as an independent team lead by Casida,24 subsequently went on to prepare a number of semisynthetic derivatives of 1 and 4 for use in pharmacological studies. These investigations revealed a key challenge: the difficulty of converting (+)-ryanodol to (+)-ryanodine. This challenge is one of chemoselectivity, in that a site-specific acylation of a molecule containing seven hydroxyl groups (including the C15 hemiacetal) is required; this is further exacerbated by the fact that the sterically encumbered C3 alcohol is much less intrinsically reactive than the surrounding O–H groups.
The question of whether 4 can be converted to 1 was finally answered in recent studies by Inoue and co-workers.25 Having completed a 35 step synthesis of (±)-ryanodol in 2014,26 they subsequently determined that it can be advanced in 4 steps to an intermediate in which all but the C3 alcohol is protected. Although this compound could not be directly coupled with pyrrole-2-carboxylic acid (or its derivatives), it could be esterified with N,N-di(tert-butoxycarbonyl)glycine, and elaborated in an additional 5 steps to 1.25 In sum, the conversion of 4 to 1 required ten synthetic steps. Alternatively, Inoue and co-workers demonstrated that a synthetic precursor to 4 could be advanced directly to 1, allowing access to ryanodine in 42 linear steps.27,28
We recently described a 15-step synthesis of (+)-ryanodol from the terpene (S)-pulegone.29 The synthesis was enabled by the expedient, gram scale preparation of tetracycle 6, which can be oxidized using SeO2, then triflated, to give either of two oxidation products, 7 or 8 (Figure 2A). In considering how to translate our synthesis of 4 to a synthesis of 1, our guiding strategic objective was to incorporate the pyrrole-2-carboxylate ester directly—rather than building it from a glycinate ester25,28—by an esterification reaction in the final stage of the synthesis. Given the challenges encountered by others when trying to acylate 4, or appropriately protected derivatives of 4, we decided to target the acylation of a protected derivative of (+)-anhydroryanodol (5) (Figure 2B).
Figure 2.
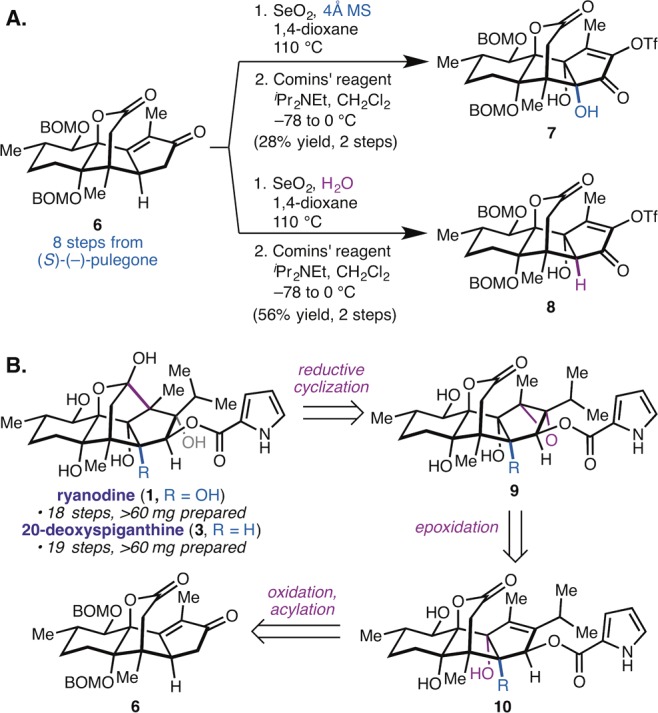
Synthetic considerations. (A) Oxidation products 7 and 8, each accessible from 6 by modification of the SeO2 reaction conditions, can be elaborated to either (+)-ryanodine (1) or (+)-20-deoxyspiganthine (3), respectively. (B) Retrosynthetic analysis for 1 and 3. Key strategic innovation is to directly incorporate pyrrole-2-carboxylate ester (as in 10) prior to reductive cyclization.
This decision was driven by the hypothesis that the C3 hydroxyl group of 5 is less sterically hindered, and by the fact that 5 presents two fewer hydroxyl groups than 4, thereby simplifying the chemoselectivity challenge. Although this approach was considered advantageous from the perspective of the C3-acylation, it presents its own challenge in that the pyrrole-2-carboxylate ester would need to survive the required epoxidation of the C1–C2 alkene (10 → 9), as well as the reductive ring closure event (9 → 1). The reductive cyclization of anhydroryanodol epoxide intermediates bearing C3 ester functionality (e.g., 9) had not been reported previously. Finally, we hoped to adapt the chemistry developed for the synthesis of (+)-ryanodine to the synthesis of (+)-20-deoxyspiganthine (3), beginning from 8 (Figure 2A), with minimal amounts of reaction reoptimization. Although this study would focus on the synthesis of 1 and 3, we anticipated that, if successful, it would ultimately enable the incorporation of previously inaccessible pyrrole esters at C3 through simple acylation chemistry, thus allowing access to new synthetic ryanoids.
The synthesis of ryanodine (1) commenced with tetracycle 12, accessible in 11 steps from (S)-pulegone (11) and prepared in gram quantities on a single pass through the sequence (Scheme 1).29 Prior studies have shown that the C4 and C12 alcohols, although tertiary, react faster with standard acylating reagents than the C3 alcohol.17,18,28 Building off Inoue’s findings that cyclic boronates can be used to strategically protect the alcohols of ryanodol,25 the ring fusion diol of 12 was converted to the dioxaborinane by treatment with methylboronic acid. Subsequent LiBH4-mediated 1,2-reduction of 13 furnished the C3 alcohol with high diastereoselectivity. We initially explored a route involving hydrogenation of the isopropenyl group prior to acylation; however, poor reactivity was observed under a variety of conditions using pyrrole-2-carboxylic acid derivatives.
Scheme 1. Chemical Synthesis of (+)-Ryanodine.

Instead, we turned to the acylation of 14, under the hypothesis that the smaller steric profile of the adjacent C(sp2) isopropenyl group (relative to isopropyl) would render the C3 alcohol more accessible. Whereas several standard acylating reagents derived from pyrrole-2-carboxylic acid failed to deliver 16 (see the Supporting Information), we were pleased to find that deprotonation of alcohol 14 with sodium hydride (NaH) at 0 °C, followed by addition of 2,2,2-trichloro-1-(1H-pyrrol-2-yl)ethan-1-one (15), afforded the desired pyrrole ester 16 in 57% isolated yield (structure verified by X-ray crystallography).30
Having incorporated the key pyrrole-2-carboxylate ester, 16 was elaborated to anhydroryanodine (18) in two steps. First, the methyl boronate of 16 was cleaved with aqueous KHF2 in methanol to afford diol 17, which was then subjected to H2 and Pd(OH)2/C to simultaneously reduce the terminal olefin and remove the benzyloxymethyl ether (BOM) groups, providing 18 in 90% yield. The structural assignment of 18 was confirmed by single crystal X-ray diffraction.
With access to 18, completion of the synthesis required epoxidation and reductive ring closure. Unfortunately, mild epoxidation reagents (e.g., 3-chloroperoxybenzoic acid (mCPBA)) returned starting material, whereas more reactive reagents (e.g., trifluoroperacetic acid) resulted in decomposition, likely due to undesired reactivity at the pyrrole. In an attempt to harness the directing ability of the proximal C4 and C12 alcohols, we turned to vanadyl-mediated epoxidation conditions; however, no reaction was observed. We hypothesized that competing, nonproductive coordination of vanadium by the C6, C4, and C12 alcohols was precluding formation of the active epoxidation species, and therefore investigated the reaction of a partially protected intermediate. Chemoselective hydrogenation of the terminal alkene of 17 was achieved using H2 with PtO2, and, to our delight, exposure of 19 to VO(acac)2/TBHP in benzene at room temperature delivered epoxide 20 in 60% yield. Subsequent hydrogenolytic deprotection of the BOM protecting groups under previously developed conditions provided anhydroryanodine epoxide 21.
The final step of the synthesis required formation of the C1–C15 bond through a reductive cyclization step, in analogy to the chemistry developed for the synthesis of (+)-ryanodol (4). Although submission of 21 to Li/NH3 indeed affected the desired bond construction, 4 was the only product recovered under these conditions, highlighting the additional challenge presented by the lability of the ester in 21 (Table 1, entry 1).
Table 1. Evaluation of Reductive Cyclization Conditions.
| entry | conditions | 21:4:1a | yield of 1 (%)b |
|---|---|---|---|
| 1 | Li/NH3, THF, −78 °C | 0:1:0 | |
| 2 | LDA (5.9 equiv), THF, −78 °C; thenLi/NH3, −78 °C | 0:2:1 | 20 |
| 3 | LDA (10.0 equiv), THF, −78 °C; then Li/NH3, −78 °C | 1:0:0 | |
| 4 | LiNap (4.5 equiv), THF, −78 °C | 2:0:1 | |
| 5 | LiDBB (3.5 equiv), THF, −78 °C | 1:0:4 | 64 |
Determined by 1H NMR spectroscopy.
Isolated yield of 1 after purification by silica gel preparative TLC.
Hypothesizing that deprotonation of the pyrrole-2-carboxylate ester would render it less susceptible to cleavage by reduction or aminolysis processes, 21 was first treated with LDA (5.9 equiv), and the resulting solution was added to a mixture of Li0 in NH3, which provided 1 (20% yield) in a 1:2 ratio with 4 (Table 1, entry 2).
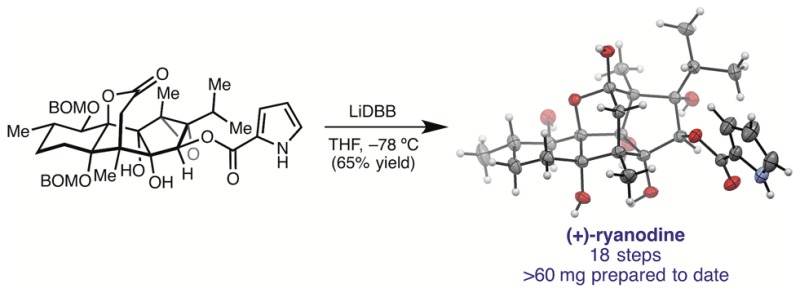
Reactions conducted with 10 equiv of LDA under otherwise identical conditions resulted in recovery of starting material, presumably due to deprotonation of the lactone, which precludes ketyl formation (entry 3). Concerned that formation of 4 under the Li/NH3 conditions results in part from aminolysis of the ester, we investigated soluble single electron reductants that could be used in solvents other than liquid ammonia. Gratifyingly, when lithium naphthalenide (LiNap) in THF was used, a 2:1 ratio of 21 to 1 was produced, without detectable amounts of 4 (entry 4). The reaction could be further improved by employing the more reducing lithium di-tert-butylbiphenylide (LiDBB), which afforded 1 in 64% isolated yield (entry 5). Moreover, the step count of the synthesis could be reduced by performing the reductive ring closure on BOM protected epoxide 20, which smoothly undergoes C–C bond formation and BOM deprotection to provide (+)-1 in 65% yield. Using this 18-step route, we have prepared >60 mg of synthetic (+)-ryanodine to date and were able to use this material to solve the first X-ray crystal structure of the molecule.
Having established a concise route to (+)-1, we turned to the synthesis of (+)-20-deoxyspiganthine (3). Preliminary biological studies reported by the isolation team determined that this compound also exhibits potent activity in cardiac tissue assays of RyR function. Given the improved yields for the formation of 8 in the SeO2-mediated oxidation/triflation of tetracycle 6 (Figure 2A), 3 could be an appealing starting point for the future development of RyR probe molecules. Starting with tetracycle 22 (which is prepared from 8 by Stille cross-coupling), the C12 alcohol was protected as a TMS ether and the ketone was reduced with LiBH4 to give alcohol 23 (Scheme 2). Unfortunately, efforts to acylate the C3 alcohol under the previously described conditions resulted in rapid TMS deprotection and very low yields of the desired ester (10–15%). Ultimately we found that treatment of 23 with 2 equiv of potassium bis(trimethylsilyl)amide (KHMDS) followed by addition of a solution of TBS-protected pyrrole 24 provided ester 25, which had also undergone chlorination at C14 (5.8:1 dr). The undesired chlorination reaction could, in principle, be mitigated by using less KHMDS; however, under these conditions translactonization by the C3 alcohol precedes esterification. Thus, enolization of the lactone prevents translactonization, but also results in α-chlorination at temperatures required to observe good levels of reactivity.
Scheme 2. Chemical Synthesis of (+)-20-Deoxyspiganthine.

Undeterred by this result, dechlorination of 25 was accomplished without complication using tris(trimethylsilyl)silane ((TMS)3SiH) and the low-temperature initiator triethylborane (Et3B) to give 26. Deprotection of both silyl groups, hydrogenation of the terminal alkene, and hydroxyl-directed epoxidation of the remaining tetrasubstituted alkene furnished 27. Treatment of 27 with 10 equiv of freshly prepared LiDBB resulted in cyclization and deprotection of both BOM groups, affording (+)-20-deoxyspiganthine (3) in 58% yield. The structure of 3 was unambiguously confirmed by single crystal X-ray diffraction. At 19 steps, this is the first total synthesis of a spiganthine-type natural product. It is notable that, with the exception of the C3-acylation reaction, no reoptimization was required to translate the synthesis of 1 to 3, demonstrating the versatility of this strategy.
In summary, the chemical syntheses of (+)-ryanodine (1) and (+)-20-deoxyspiganthine (3) were achieved in 18 and 19 steps, respectively. A key discovery is that anhydroryanodol derivatives (and related C4-deoxy analogues) that possess C13–C18 unsaturation can be acylated directly to incorporate the crucial pyrrole-2-carboxylate ester. This represents a practical solution to one of the key challenges encountered in previous efforts to prepare ryanodine,25 which should allow for the incorporation of an array of ester substituents at C3 for future SAR studies. In addition, we determined that a LiDBB-mediated reductive cyclization can be used to forge the final C1–C15 bond with the pyrrole ester intact. Taken together, these discoveries provide a synthetic platform that will enable the synthesis of a variety of natural and designed ryanoids. Ongoing studies in our laboratory seek to leverage this chemistry for the development of synthetic, molecular probes that can aid the study of RyR dysfunction and Ca2+ ion channel-based diseases.
Acknowledgments
The Caltech Center for Catalysis and Chemical Synthesis is gratefully acknowledged for access to analytical equipment. We thank Dr. Michael Takase and Larry Henling for acquiring the X-ray diffraction data and Julie Hofstra for solving the structures of compounds 1, 3, 16, and 18. Fellowship support was provided by the Shenzhen UV-ChemTech Inc. (postdoctoral fellowship to C.X.) and the NIH (training grant 5T32GM007616-37 to A.H.). S.E.R. is an American Cancer Society Research Scholar and Heritage Medical Research Institute investigator. Financial support from the NIH (NIGMS RGM097582-01, R35GM118191-01), Eli Lilly, and Novartis is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00361.
The authors declare no competing financial interest.
Supplementary Material
References
- Lanner J. T.; Georgiou D. K.; Joshi A. D.; Hamilton S. L. Ryanodine Receptors: Structure, Expression, Molecular Details, and Function in Calcium Release. Cold Spring Harbor Perspect. Biol. 2010, 2, a003996. 10.1101/cshperspect.a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryanodine Receptors: Structure, Function and Dysfunction in Clinical Disease; Wehrens X. H. T., Marks A. R., Eds.; Springer US: Boston, 2005. [Google Scholar]
- Lanner J. T.Ryanodine Receptor Physiology and Its Role in Diseasec. In Calcium Signaling, Advances in Experimental Medicine and Biology; Islam S., Ed.; Springer: Dordrecht, 2012. [DOI] [PubMed] [Google Scholar]
- McCarthy T. V.; Quane K. A.; Lynch P. J. Ryanodine receptor mutations in malignant hyperthermia and central core disease. Hum. Mutat. 2000, 15, 410–417. 10.1002/(SICI)1098-1004(200005)15:5<410::AID-HUMU2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Laitinen P. J.; Brown K. M.; Piippo K.; Swan H.; Devaney J. M.; Brahmbhatt B.; Donarum E. A.; Marino M.; Tiso N.; Viitasalo M.; Toivonen L.; Stephan D. A.; Kontula K. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 2001, 103, 485–490. 10.1161/01.CIR.103.4.485. [DOI] [PubMed] [Google Scholar]
- Kelliher M.; Fastbom J.; Cowburn R. F.; Bonkale W.; Ohm T. G.; Ravid R.; Sorrentino V.; O’Neill C. Alterations in the ryanodine receptor calcium release channel correlate with Alzheimer’s disease neurofibrillary and beta-amyloid pathologies. Neuroscience 1999, 92, 499–513. 10.1016/S0306-4522(99)00042-1. [DOI] [PubMed] [Google Scholar]
- Bruno A. M.; Huang J. Y.; Bennett D. A.; Marr R. A.; Hastings M. L.; Stutzmann G. E. Altered ryanodine receptor expression in mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 2012, 33, 1001.e1–1001.e6. 10.1016/j.neurobiolaging.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessah I. N.; Waterhouse A. L.; Casida J. E. The calcium-ryanodine receptor complex of skeletal and cardiac muscle. Biochem. Biophys. Res. Commun. 1985, 128, 449–456. 10.1016/0006-291X(85)91699-7. [DOI] [PubMed] [Google Scholar]
- Inui M.; Saito A.; Fleischer S. Isolation of the ryanodine receptor from cardiac sarcoplasmic reticulum and identity with the feet structures. J. Biol. Chem. 1987, 262, 15637–15642. [PubMed] [Google Scholar]
- Rogers E. F.; Koniuszy F. R.; Shavel J.; Folkers K. Plant Insecticides. 1. Ryanodine, a new alkaloid from Ryania speciosa Vahl. J. Am. Chem. Soc. 1948, 70, 3086–3088. 10.1021/ja01189a074. [DOI] [PubMed] [Google Scholar]
- Wiesner K.; Valenta Z.; Findlay J. A. Structure of ryanodine. Tetrahedron Lett. 1967, 8, 221. 10.1016/S0040-4039(00)90521-5. [DOI] [Google Scholar]
- Srivastava S. N.; Przybylska M. The molecular structure of ryanodol-p-bromo benzyl ether. Can. J. Chem. 1968, 46, 795–797. 10.1139/v68-133. [DOI] [Google Scholar]
- Wiesner K. The structure of ryanodine. Adv. Org. Chem. 1972, 8, 295–316. [Google Scholar]
- Achenbach H.; Hübner H.; Vierling W.; Brandt W.; Reiter M. Spiganthine, the cardioactive principle of Spigelia anthelmia. J. Nat. Prod. 1995, 58, 1092–1096. 10.1021/np50121a019. [DOI] [PubMed] [Google Scholar]
- Hübner H.; Vierling W.; Brandt W.; Reiter M.; Achenbach H. Minor constituents of Spigelia anthelmia and their cardiac activities. Phytochemistry 2001, 57, 285–296. 10.1016/S0031-9422(01)00020-6. [DOI] [PubMed] [Google Scholar]
- Meissner G. Ryanodine activation and inhibition of the Ca(2+)-release channel of sarcoplasmic-reticulum. J. Biol. Chem. 1986, 261, 6300–6306. [PubMed] [Google Scholar]
- Sutko J. L.; Airey J. A.; Welch W.; Ruest L. The pharmacology of ryanodine and related compounds. Pharmacol. Rev. 1997, 49, 53–98. [PubMed] [Google Scholar]
- Bélanger A.; Berney D. J. F.; Borschberg H. J.; Brousseau R.; Doutheau A.; Durand R.; Katayama H.; Lapalme R.; Leturc D. M.; Liao C. C.; MacLachlan F. N.; Maffrand J. P.; Marazza F.; Martino R.; Moreau C.; Saint-Laurent L.; Saintonge R.; Soucy P.; Ruest L.; Deslongchamps P. Total Synthesis of Ryanodol. Can. J. Chem. 1979, 57, 3348–3354. 10.1139/v79-547. [DOI] [Google Scholar]
- Deslongchamps P.; Bélanger A.; Berney D. J. F.; Borschberg H. J.; Brousseau R.; Doutheau A.; Durand R.; Katayama H.; Lapalme R.; Leturc D. M.; Liao C. C.; Maclachlan F. N.; Maffrand J. P.; Marazza F.; Martino R.; Moreau C.; Ruest L.; Saint-Laurent L.; Saintonge R.; Soucy P. The Total Synthesis of (+)-Ryanodol. 1. General Strategy and Search for a Convenient Diene for the Construction of a Key Tricyclic Intermediate. Can. J. Chem. 1990, 68, 115–126. 10.1139/v90-021. [DOI] [Google Scholar]
- Deslongchamps P.; Bélanger A.; Berney D. J. F.; Borschberg H. J.; Brousseau R.; Doutheau A.; Durand R.; Katayama H.; Lapalme R.; Leturc D. M.; Liao C. C.; Maclachlan F. N.; Maffrand J. P.; Marazza F.; Martino R.; Moreau C.; Ruest L.; Saint-Laurent L.; Saintonge R.; Soucy P. The Total Synthesis of (+)-Ryanodol. 2. Model Studies for Ring B and Ring C of (+)-Anhydroryanodol. Preparation of a Key Pentacyclic Intermediate. Can. J. Chem. 1990, 68, 127–152. 10.1139/v90-022. [DOI] [Google Scholar]
- Deslongchamps P.; Bélanger A.; Berney D. J. F.; Borschberg H. J.; Brousseau R.; Doutheau A.; Durand R.; Katayama H.; Lapalme R.; Leturc D. M.; Liao C. C.; Maclachlan F. N.; Maffrand J. P.; Marazza F.; Martino R.; Moreau C.; Ruest L.; Saint-Laurent L.; Saintonge R.; Soucy P. The Total Synthesis of (+)-Ryanodol. 3. Preparation of (+)-Anhydroryanodol from a Key Pentacyclic Intermediate. Can. J. Chem. 1990, 68, 153–185. 10.1139/v90-023. [DOI] [Google Scholar]
- Deslongchamps P.; Bélanger A.; Berney D. J. F.; Borschberg H. J.; Brousseau R.; Doutheau A.; Durand R.; Katayama H.; Lapalme R.; Leturc D. M.; Liao C. C.; Maclachlan F. N.; Maffrand J. P.; Marazza F.; Martino R.; Moreau C.; Ruest L.; Saint-Laurent L.; Saintonge R.; Soucy P. The Total Synthesis of (+)-Ryanodol. 4. Preparation of (+)-Ryanodol from (+)-Anhydroryanodol. Can. J. Chem. 1990, 68, 186–192. 10.1139/v90-024. [DOI] [Google Scholar]
- Welch W.; Ahmad S.; Airey J. A.; Gerzon K.; Humerickhouse R. A.; Besch H. R.; Ruest L.; Deslongchamps P.; Sutko J. L. Structural determinants of high-affinity binding of ryanoids to the verterbrate skeletal-muscle ryanodine receptor–a comparative molecular-field analysis. Biochemistry 1994, 33, 6074–6085. 10.1021/bi00186a006. [DOI] [PubMed] [Google Scholar]
- Waterhouse A. L.; Pessah I. N.; Francini A. O.; Casida J. E. Structural aspects of ryanodine action and selectivity. J. Med. Chem. 1987, 30, 710–716. 10.1021/jm00387a022. [DOI] [PubMed] [Google Scholar]
- Masuda K.; Nagatomo M.; Inoue M. Chemical Conversion of Ryanodol to Ryanodine. Chem. Pharm. Bull. 2016, 64, 874–879. 10.1248/cpb.c16-00214. [DOI] [PubMed] [Google Scholar]
- Nagatomo M.; Koshimizu M.; Masuda K.; Tabuchi T.; Urabe D.; Inoue M. Total Synthesis of Ryanodol. J. Am. Chem. Soc. 2014, 136, 5916–5919. 10.1021/ja502770n. [DOI] [PubMed] [Google Scholar]
- Nagatomo M.; Hagiwara K.; Masuda K.; Koshimizu M.; Kawamata T.; Matsui Y.; Urabe D.; Inoue M. Symmetry-Driven Strategy for the Assembly of the Core Tetracycle of (+)-Ryanodine: Synthetic Utility of a Cobalt-Catalyzed Olefin Oxidation and alpha-Alkoxy Bridgehead Radical Reaction. Chem. - Eur. J. 2016, 22, 222–229. 10.1002/chem.201503640. [DOI] [PubMed] [Google Scholar]
- Masuda K.; Koshimizu M.; Nagatomo M.; Inoue M. Asymmetric Total Synthesis of (+)-Ryanodol and (+)-Ryanodine. Chem. - Eur. J. 2016, 22, 230–236. 10.1002/chem.201503641. [DOI] [PubMed] [Google Scholar]
- Chuang K. V.; Xu C.; Reisman S. E. A 15-step synthesis of (+)-ryanodol. Science 2016, 353, 912–915. 10.1126/science.aag1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The major side product of this reaction is an α-chloroester resulting from chlorination of the lactone. See the Supporting Information for structure and additional details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.