

Abstract
Achieving complete, accurate, and cost-effective assembly of human genomes is of great importance for realizing the promise of precision medicine. The abundance of repeats and genetic variations in human genomes and the limitations of existing sequencing technologies call for the development of novel assembly methods that can leverage the complementary strengths of multiple technologies. We propose a Hybrid Structural variant Assembly (HySA) approach that integrates sequencing reads from next-generation sequencing and single-molecule sequencing technologies to accurately assemble and detect structural variants (SVs) in human genomes. By identifying homologous SV-containing reads from different technologies through a bipartite-graph-based clustering algorithm, our approach turns a whole genome assembly problem into a set of independent SV assembly problems, each of which can be effectively solved to enhance the assembly of structurally altered regions in human genomes. We used data generated from a haploid hydatidiform mole genome (CHM1) and a diploid human genome (NA12878) to test our approach. The result showed that, compared with existing methods, our approach had a low false discovery rate and substantially improved the detection of many types of SVs, particularly novel large insertions, small indels (10–50 bp), and short tandem repeat expansions and contractions. Our work highlights the strengths and limitations of current approaches and provides an effective solution for extending the power of existing sequencing technologies for SV discovery.
The complete, accurate, and cost-effective assembly of human genomes is a prerequisite for genomic medicine. Advances in translational genomics are hampered by technical challenges in assembling structurally altered regions in human genomes, which are shown to be essential for generating genetic diversities and in human diseases (Feuk et al. 2006; Sharp et al. 2006; Lupski 2007). Advances in next-generation sequencing (NGS) technologies have greatly facilitated the assembly and detection of structural variations (SVs) in human genomes (Alkan et al. 2011a). Many computational methods have been developed to identify SVs through examining the alignment of paired-end reads to the human reference genome, scanning for abnormally aligned reads (such as unmapped reads, discordant read pairs, clipped reads, and reads with large gaps) and variation of read depths, and inferring SV positions and orientations (Chen et al. 2009; Wang et al. 2011; Rausch et al. 2012; Sindi et al. 2012; Layer et al. 2014). Other methods perform whole-genome assembly (WGA) or targeted assembly of sequencing reads and identify SVs from pairwise alignment of assembled contigs against the reference (Iqbal et al. 2012; Chen et al. 2014; Xie et al. 2014). Although NGS reads have low base-calling error rates (Ross et al. 2013), their read lengths are often limited (e.g., 100–200 bp for Illumina HiSeq instruments). The short read length leads to a bias against the assembly and detection of SVs, which often occur near segmental duplications or large repeats in the genome (Alkan et al. 2011b). Moreover, complex sequence alterations around SV breakpoints (e.g., microhomology, micro-indels [Hackl et al. 2014], and kataegis [Alexandrov et al. 2013]) substantially hamper the sensitivity and specificity of SV detection methods that depend on aligning individual reads against the reference (The 1000 Genomes Project Consortium 2010).
The advent of single-molecule sequencing (SMS) technologies greatly changed the landscape of genome assembly approaches by providing much longer reads (e.g., 12 kb on average for Pacific Biosciences (PacBio) reads in P6-C4 chemistry). As a result, many SVs missed by NGS could be detected (Chaisson et al. 2015b) by SMS. Unfortunately, SMS technologies are error-prone due to the use of one molecule for real-time sequencing. For example, PacBio reads typically have an error rate of 15%, and the majority (14%) of the errors are indels (Ross et al. 2013). This high error rate has posed new challenges to bioinformatics tools that perform alignment or assembly. To tackle these challenges, BLASR (Chaisson and Tesler 2012) was developed to align noisy PacBio reads in a computationally efficient way. The widely used BWA algorithm (Li and Durbin 2009) was also extended to align PacBio reads (Li 2013). It is plausible to infer SVs based on the analysis of the alignment of long reads to the reference by searching for indel signals (gaps inside a read alignment) and stop signals (clipped ends) (English et al. 2014; Chaisson et al. 2015b). However, it is often difficult for the aligners to assign gaps or stops accurately due to high error rates, long read lengths, and the prevalence of repeats. Thus, it is challenging to parse PacBio read alignment to accurately determine SVs and breakpoints, particularly those resulting from novel (nonreference) insertions.
On the other hand, de novo genome assembly approaches have rapidly advanced to achieve high quality and computational efficiency (Chaisson et al. 2015a). Overall, three different paradigms have been developed: (1) overlap-layout-consensus (OLC) (e.g., Mira [Chevreux et al. 2004], Newbler [Margulies et al. 2005], and Celera Assembler [Myers et al. 2000]); (2) de Bruijn graph (e.g., Velvet [Zerbino and Birney 2008], SOAPdenovo [Xie et al. 2014], ABySS [Simpson et al. 2009], and ALLPATHS [Butler et al. 2008]); and (3) string graph (Myers 2005). Approaches based on the de Bruijn graph require high quality reads and are only applicable to NGS data, whereas approaches based on the OLC and string graphs are applicable to both NGS and SMS data. For example, FALCON (Chin et al. 2016), a recently developed assembly algorithm, utilizes string graphs to assemble a diploid genome from PacBio reads. Although constructing string graphs using the Ferragina-Manzini (FM)-index only takes time linear to the number of reads, FALCON is still computationally intensive due to an error correction step that requires pairwise alignment of all PacBio reads. Chin et al. (2013) also utilized relatively short PacBio reads to correct long PacBio reads before performing assembly. Berlin et al. (2015) made use of a statistical hashing technique for identifying pairwise overlaps, which greatly reduces the computation time. For SV detection, however, de novo whole-genome assembly is not optimal since the majority of the genome does not contain SVs. WGA often requires computational resources not widely available when assembling large mammalian genomes (∼3 Gbp) (Pendleton et al. 2015), and such approaches are often not optimized to assemble diploid genomes containing heterogeneous SVs. In comparison, targeted SV assembly approaches (Chen et al. 2014; Chong et al. 2016) that aim to assemble sequences spanning SVs are often more effective in terms of computational efficiency and SV detection power, as they dissect a WGA problem into a set of independent local assembly problems that can be more effectively solved. However, in addition to performing powerful local assembly, targeted approaches need to (1) achieve comprehensive unbiased selection of targets, and (2) ensure that the results obtained from local solutions are also globally optimal.
Considering the advantages and disadvantages of the technologies, i.e., NGS reads are short but accurate, whereas SMS reads are long but inaccurate, a hybrid assembly approach that combines data from the two or more technologies can potentially achieve more powerful assembly and SV detection. Ideally, the accuracy of NGS reads can be used to correct errors in SMS reads, whereas the length of SMS reads can be used to anchor assemblies confidently to the reference. A few efforts aiming to achieve such combination, although not specifically for SV detection, have been proposed. A toolbox has been developed to simulate the integration of multiple technologies for optimal personal genome assembly (Du et al. 2009). PacBioToCA (Koren et al. 2012) performs hybrid de novo WGA by aligning all NGS short reads to all PacBio reads for error correction. LSC (Au et al. 2012) applies a similar strategy but aims to reduce the error rate in homopolymer runs. While these methods utilize the high fidelity of NGS reads and the long length of PacBio reads, they tend to be computationally intensive and are not designed for SV detection. MultiBreak-SV (Ritz et al. 2014), on the other hand, uses a probabilistic approach that combines the alignment of individual SMS and NGS reads for detecting SVs, particularly in regions involving multiple SVs. However, it is not suitable for detecting novel insertions since it relies on accurate alignment of individual reads to the reference. Moreover, it only examines discordantly aligned NGS read pairs (but not unmapped or clipped NGS reads).
In short, there lacks a computationally efficient hybrid assembly approach for accurate SV detection despite a pressing demand in applying the technologies. The main obstacle is a lack of computationally efficient algorithms that can effectively synergize heterogeneous data sources of highly discrepant properties (e.g., read length and sequencing error) with highly structured contents (e.g., sequence homology in human genomes).
Results
Method overview
We propose a novel Hybrid Structural variant Assembly (HySA) method that identifies and performs genome-wide SV assembly from both NGS and SMS data (Fig. 1A). The complementary properties of NGS and SMS data allow ascertainment of SVs in genomic regions that cannot be confidently mapped by short reads and improve accuracy of gap or novel sequence assignment in noisy long reads. HySA requires two sets of input data: Set A, the reference alignment of paired-end short reads generated by low-error-rate NGS (such as Illumina HiSeq); and set B, long reads generated by high-error-rate SMS (such as PacBio SMRT-seq). HySA first identifies and extracts unmapped, discordantly paired and end-clipped short reads in set A and then aligns them to the set of long reads in set B (Methods; Supplemental Results). The set of aligned short and long reads form a bipartite graph in which one set of nodes represents short reads, the other represents long reads, and the edges between them represent confident pairwise alignments. The extracted short reads are often from disjointed regions of unique sequence context, due to the sparseness of SVs and the short fragment size of set A. Consequently, the bipartite graph is often sparsely connected and can be computationally efficiently (near linear complexity with regard to the number of nodes) decomposed into connected components (CC) using the Union-Find algorithm. Each CC often corresponds to one SV-containing sequence with at least one breakpoint and its size (number of nodes and edges) is proportional to the average physical coverage in both sets A and B. False alignments between short and long reads can lead to inaccurate nodes and edges in the graph and result in erroneously large connected components (ELCC). When that occurs, we further decompose the ELCCs into small communities via a network flow-based graph algorithm (Fig. 1B; Rosvall and Bergstrom 2008). This algorithm iteratively merges and splits small communities in a random order until they reach expected sizes and no better partitioning can be found. Each resulting CC or community contains a cluster of long and short reads that are expected to come from a single genomic origin. Assembling long reads in each cluster into contigs and aligning them to the reference facilitates the discovery of SVs. To reduce the false discovery rate, short reads in the same clusters as the long reads are aligned to the assembled contigs to confirm the identified SVs (Fig. 1A).
Figure 1.

Diagram of HySA for SV assembly and detection. (A). Abnormally aligned Illumina reads are extracted from a BAM file and aligned to a set of PacBio reads (light blue) generated from the same DNA sample. The cluster of reads associated with an SV is identified using a set of bipartite-graph partitioning algorithms. Contigs are assembled from PacBio reads in each cluster and are aligned to the reference, from which SVs and breakpoints are identified and further confirmed by Illumina reads in the same cluster. An insertion (yellow segment in the reference and yellow segments in PacBio reads) is used for illustration. The Illumina reads are in red and green, corresponding to the forward and the reverse strands, respectively. The subsequence or whole read that cannot be mapped is in gray. (B) Clustering strategy. A bipartite graph is built from the pairwise alignment of Illumina reads to PacBio reads. One set of the nodes corresponds to PacBio reads (top row, black open circles), and the other set corresponds to Illumina read pairs (second row, red solid circles). An edge is added when there is a reliable alignment between an Illumina read pair and a PacBio read. The bipartite graph is decomposed into connected components (green and red boxes) using the Union-Find algorithm. Large components, e.g., the one in the red box, are further decomposed into communities of expected sizes using a graph decomposition algorithm called Infomap. (C) False alignments between Illumina and PacBio reads are illustrated in dashed red box: (1) single-end alignments; (2) paired ends with abnormal insert size; and (3) paired ends with abnormal orientation. (D) False alignments due to repetitive regions. Illumina read alignments against a PacBio read (dotted red box) are filtered out when the depth of Illumina reads significantly exceeds the expected coverage. (E) A competitive alignment strategy that eliminates false alignments between PacBio and Illumina reads. For each Illumina read pair, a pseudo (ref) read pair is synthesized from the reference sequence in identical positions and orientations. An alignment between an Illumina read pair and a PacBio read is false when (1) the Illumina pair has a shorter aligned sequence against the PacBio read than does its pseudo pair (left), or (2) the alignment of the Illumina pair has a split whereas the pseudo pair does not (right).
Important features of our algorithm include:
The read-clustering approach via the partitioning of a bipartite graph is reference-agnostic, allowing reads containing novel nonreference sequences to be clustered and assembled together and thus facilitates the assembly of nonreference insertions;
Only the subset of reads that potentially contain variants is analyzed, which leads to substantial savings in computational cost, as compared with the WGA approaches; and
No direct alignment of individual long reads to the reference is needed. This not only reduces computation but also alleviates challenges in assigning gaps or stops in aligning noisy long reads.
Obtaining accurate clustering is important for subsequent analysis. Thus, we used simulation data to examine how clustering accuracy changes with respect to Illumina coverage, PacBio coverage, and Illumina read length (Supplemental Results). At 25× PacBio coverage, the clustering quality metric JI90 (Supplemental Results) reached 0.8, indicating that Illumina and PacBio reads were effectively hybridized and generated a reasonably accurate representation of the targeted regions in the genome (Supplemental Results; Supplemental Fig. 1).
SV detection in a haploid genome CHM1
The performance of our algorithm can be measured by comparing the sensitivity and specificity of our algorithm with those of other algorithms using the same data sets. We ran our algorithm on 50× Illumina and 46× PacBio whole-genome sequencing data generated from a hydatidiform mole haploid genome (CHM1) (Chaisson et al. 2015b). SVs in this genome have been well characterized in previous studies using approaches that analyze BLASR alignment of PacBio reads to the reference (Chaisson et al. 2015b). Moreover, a high quality de novo WGA constructed from PacBio reads (Berlin et al. 2015) and further confirmed by an independent high coverage (200×) Illumina WGA (Steinberg et al. 2014) was available as a reference to validate our results.
Our algorithm extracted 0.28% of the 50× Illumina reads and 6.8% of 46× PacBio reads. In all, 130,058 (72,354 from Union-Find, 57,704 from Infomap decomposed from one ELCC) clusters were formed and 114,230 (71,092 from Union-Find, 43,138 from Infomap) were successfully assembled into at least one contig, which led to the detection of 32,121 SVs, including 3007 large deletions (size > 50 bp), 4587 large insertions (size > 50 bp), 12,401 small deletions (size ≤ 50 bp), and 12,126 small insertions (size ≤ 50 bp) (Supplemental Table S1). The two main steps of HySA—alignment, and assembly—took a total of around 36,000 CPU hours on a high performance BL465c G7 blade with AMD 6174 processors and <12 GB memory per node. Both the CPU and memory cost were at least an order of magnitude lower than what were required to perform a de novo WGA (Chin et al. 2016).
Large deletions in CHM1
Among the 3007 large deletions we called (referred to below as HALD), 2557 (85%) were directly validated by aligning the variant sequences to the WGA of Berlin et al. (2015) (Supplemental Results). A detailed look at the calls that could not be validated by the WGA of Berlin et al. provided an estimated false discovery rate (FDR) of 7.5% (Supplemental Results). For comparison, we generated a merged call-set (MGLD) containing 2645 deletions discovered, respectively, by DELLY (Rausch et al. 2012) from the 50× Illumina data and by Chaisson et al. (2015b) from the 46× PacBio data (Supplemental Results). Thus, the differences between HALD and MGLD can reveal the uniqueness of our approach relative to a naive approach that merges call-sets independently derived from a single technology without performing hybrid assembly. In total, 1961 deletions (74.1% of MGLD, 65.2% of HALD) were shared between these two sets (requiring 50% reciprocal overlap). Importantly, 659 (21.9% of HALD) deletions were uniquely discovered by HySA and were validated by the Berlin et al. assembly, indicating a shear gain in discovery power attributable to our hybrid methodology.
We further found that these 659 deletions uniquely discovered by our approach were associated with significantly fewer variant-supporting Illumina reads (7.28 versus 29.28, P-value = 1.382 × 10−14, Student's t-test) than were the deletions in the MGLD (Table 1). No significant differences were found in the numbers of variant-supporting PacBio reads (Supplemental Results). However, gaps in the PacBio reads appeared at significantly different locations in the HALD than in the MGLD (P-value < 2.2 × 10−16, two-sample Kolmogorov-Smirnov test). Gap opening positions are much closer to the breakpoints (mean: 18.06 bp versus 43.77 bp), more tightly clustered (standard deviation: 19.56 bp versus 42.24 bp), and thus easier to detect in the MGLD than in the HALD. These observations confirmed the challenges in accurately aligning PacBio reads to the reference. It is difficult to obtain consistent gap positions from BLASR alignment, due likely to not only the high error rates of PacBio reads but also the repetitive sequence context, as indicated by the significant difference between the proportion of calls overlapping short tandem repeats (STRs) in the HALD and that in the MGLD (0.86 versus 0.67, P-value < 2.2 × 10−16, χ2 test for two independent proportions). Overall, these statistics confirmed that the novel deletions discovered by our approach were indeed associated with weak signals in either Illumina or PacBio data and thus were difficult to identify using DELLY or approaches described by Chaisson et al. (2015b)
Table 1.
Comparison of CHM1 deletions detected by single technologies with those detected by HySA
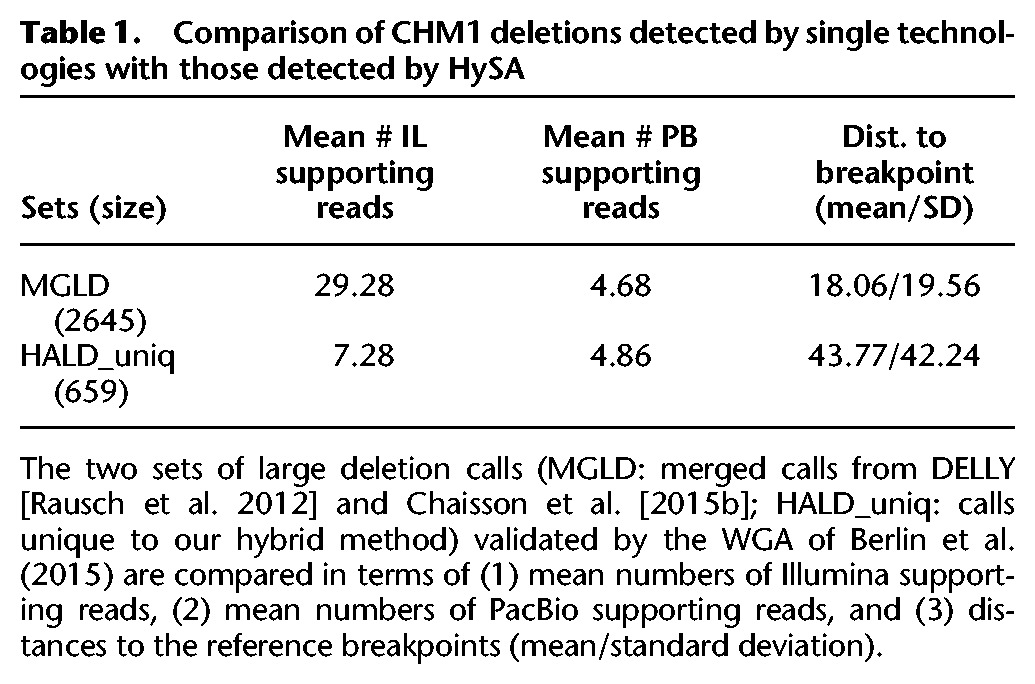
Our approach missed 684 deletions in MGLD, potentially because of the challenge to extract and cluster the variant supporting reads. Nonetheless, the results indicate that our approach can complement existing approaches and improve the overall discovery power.
Large insertions in CHM1
Among the insertions (size > 50 bp) detected by HySA, 1165 contigs contained inserted sequences longer than 500 bp. Among them, 778 could not be aligned to the Genome Reference Consortium GRCh37 reference assembly and were novel (nonreference) insertions (Supplemental Results). Two hundred and eleven (211) of them could be aligned to the GRCh38 assembly, including nine uniquely identified by our approach that were not reported by Chaisson et al. (2015b). Among the 567 insertions that could not be aligned to the GRCh38, 522 could be aligned to the Berlin et al. (2015) assembly, including 20 uniquely identified by our approach that were not reported by Chaisson et al. Thirty-five of the remaining 45 insertions that were neither aligned to the Berlin et al. assembly nor to the GRCh38 were also reported by Chaisson et al., indicating their potential validity and the possibility of further improving the Berlin et al. assembly as well as the GRCh38 assembly using our results. Only 10 (1.3%) of the novel insertions discovered had no evidence of support from the available data.
In summary, our approach discovered 29 validated large novel insertions that were missed by Chaisson et al. This can be largely credited to our approach, which does not rely on having accurate alignment of PacBio reads to the reference. It is easier to target segments of novel sequences from the alignment of Illumina reads to the reference than from the alignment of long PacBio reads due to the accuracy of Illumina reads. Apparently, hybridizing Illumina and PacBio reads together through the HySA algorithm has improved the assembly of insertions over approaches that involve error-prone reference alignments. As shown by an example of a 3-kbp novel insertion validated by GRCh38 (Supplemental Fig. 2), PacBio reads containing novel insertions could not be accurately aligned to the reference. However, they could be correctly clustered together through short Illumina read pairs (Supplemental Fig. 3), a large portion of which were both-end-unmapped (Supplemental Fig. 2b), and those with one-end unmapped could be used to anchor the insertion onto the reference genome.
Small indels in CHM1
We compared the small indels (≤50 bp) detected by HySA with those detected by Pindel (Ye et al. 2009) and GATK (Supplemental Results; Supplemental Figs. 4, 5a,b; DePristo et al. 2011). The minimum size of the indels we detected was 11 bp. A majority (95.1% for deletions and 84.5% for insertions) of HySA calls overlapped with those of either Pindel or GATK, and a large portion (85.9% for deletions and 68.4% for insertions) overlapped with the calls detected by both methods, showing the specificity of the HySA calls. In addition, HySA was able to identify 2538 novel indels that were missed by Pindel or GATK.
SV detection in a diploid genome NA12878
We further examined HySA using data from a well-studied diploid genome, NA12878, that contains two alleles of each chromosome. We downloaded raw Illumina (300×) and PacBio reads (31×) from the Genome in a Bottle (GIAB) Consortium (Zook et al. 2014) and assessed results based on the GRCh38 assembly (Supplemental Results). In total, HySA identified 59,640 SVs, including 5801 large deletions (>50 bp), 18,418 small deletions (≤50 bp), 9299 large insertions (>50 bp), and 26,122 small insertions (≤50 bp) (Supplemental Table S1). The two main steps of HySA—alignment and assembly—took a total of around 165,000 CPU hours on a high performance BL465c G7 blade with AMD 6174 processors and <12 GB memory per node. The CPU hours were much higher than that required for CHM1 because the majority of time was spent on performing BLASR alignment between the 300× Illumina reads and the 31× PacBio reads. For a diploid genome with 50× Illumina reads and 30× PacBio reads, the total CPU hours is around 37,000 (∼4.2 yr), roughly an order of magnitude less than what was required (∼35 yr) to perform WGA of 30× PacBio reads (Pendleton et al. 2015).
The de novo WGA of NA12878 (Pendleton et al. 2015), which used Celera Assembler (Myers et al. 2000) and FALCON for error-corrected PacBio reads, followed by scaffolding with genome maps produced by BioNano technology and phasing with Illumina and PacBio reads, can be used as a reference to assess the accuracy of SV assemblies.
Large deletions in NA12878
For large deletions, we created a curated deletion set (referred to below as GD) by merging five deletion call-sets produced, respectively, by (1) HySA, (2) DELLY (Rausch et al. 2012), (3) PBHoney (English et al. 2014), (4) a customized pipeline (CP) from Pendleton et al. (2015), and (5) svclassify (Parikh et al. 2016) and validating each deletion at sequence resolution using the assembly of Pendleton et al. (2015) (Supplemental Results). The svclassify call-set was a high-confidence set obtained from Personalis and the 1000 Genomes Project (The 1000 Genomes Project Consortium 2010; Mills et al. 2011), which was the result of a machine learning method that integrated signals in Illumina, PacBio, and Moleculo reads and thus also resulted from a hybrid approach.
We plotted the receiver operating characteristic (ROC) curves based on the comparison of each of these five sets with the GD set (Fig. 2). For the HySA, DELLY, and svclassify sets, a series of cutoffs were applied, respectively, to the number of supporting Illumina reads, the number of supporting split reads, and a score that combines various features from all the technologies (Parikh et al. 2016). By a fairly large margin, HySA outperformed DELLY, PBHoney, CP, and svclassify. The svclassify call-set was slightly inferior to that obtained from HySA (specificity difference <0.1 at a sensitivity of 0.33) even though it incorporated data from additional Moleculo long reads. In summary, the HySA approach achieved evidently better accuracy than approaches based on single technologies such as DELLY, PBHoney, and CP and was favorable over another hybrid approach.
Figure 2.
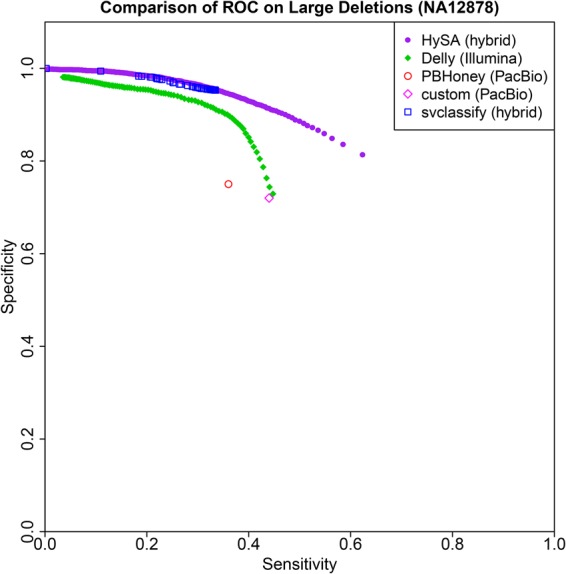
A comparison of the sensitivity and specificity for detecting large deletions in NA12878 among five competing approaches (1) HySA, (2) DELLY, (3) PBHoney, (4) a custom pipeline based on PacBio data alone, and (5) svclassify.
Large insertions in NA12878
Among the 1672 large (>500 bp) insertions we detected, 783 could not be properly aligned to the GRCh38 assembly (Supplemental Results). Among them, 642 were aligned to the assembly of Pendleton et al. (2015) or the fosmid data of NA12878 (Kidd et al. 2008). Only 141 (8.4%) had no supporting evidence from the available data.
Small indels in NA12878
We compared our small indel set (≤50 bp) with the Platinum set (Eberle et al. 2016) and the GIAB set. Interestingly, for the small insertions, we observed more overlapping calls between our set and the GIAB set than between the Platinum set and the GIAB set, allowing a 30-bp or more offset in the indel positions (Fig. 3; Supplemental Figs. 5c,d). In addition, we discovered 10,881 novel insertions and 9930 novel deletions that were neither in the Platinum nor in the GIAB sets. Manual inspection of our novel calls indicated that they were likely missed due to insufficient coverage or lack of alignment accuracy in a single source.
Figure 3.
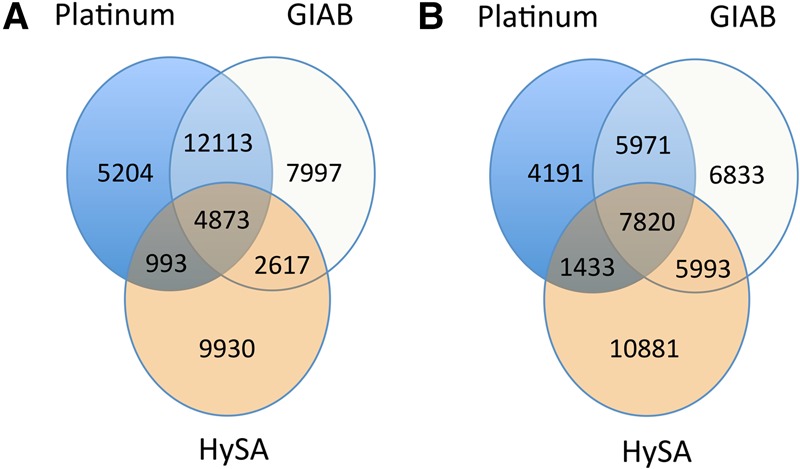
Comparison of small indels detected in NA12878 by HySA with those in the Platinum and GIAB sets. (A) Deletions with 1-bp overlap criterion. (B) Insertions with 1-bp overlap criterion allowing 50-bp offset on the left and right of the insertion breakpoints.
SV validation using fosmid data
We further validated these SV calls using the fosmid end sequencing data available in the 1000 Genomes Project (Supplemental Results). We found that a larger proportion of large deletions in the HySA set can be validated by the fosmid data, compared with the proportions in the other sets (Supplemental Table S2; Supplemental Results), and the low overall proportions are due largely to the low coverage of the fosmid data. On the other hand, more small indels in the HySA set can be validated by the fosmid data with proportions similar to those in the other call-sets (Supplemental Tables S3, S4; Supplemental Results), which were already shown to have low false discovery rates (Zook et al. 2014).
Complex structural variation
The long contigs assembled by HySA can be used to discover complex SVs. We focused on analyzing complex deletions with insertions (Supplemental Fig. 6) at the breakpoints. Overall, we detected 962 (Supplemental Table S5; Methods), including 22 with spacers (10 with inverted spacers), three duplications (≤70 bp), and one with both (see an example of an inverted spacer in Supplemental Fig. 7). Of these, 11 overlapped fosmid data and five were validated at nucleotide resolution (Supplemental Results).
Coverage analysis
We further analyzed the sensitivity of HySA at different Illumina and PacBio coverage. We found a likely optimal combination of coverage would be 60× Illumina and 25× PacBio reads in order to obtain the most cost-effective hybrid SV assembly in a diploid genome using HySA (Supplemental Results). As a reference, we also computed the sensitivity of DELLY from the Illumina data. We found DELLY had higher sensitivity than HySA when the PacBio coverage was lower than 5× regardless of the Illumina coverage. However, once PacBio coverage reached 10× or above, HySA achieved higher sensitivity (Fig. 4). This was expected because HySA requires at least 10× PacBio coverage to successfully assemble heterozygous deletions in a diploid genome. Notably, with 10× PacBio coverage and 30× Illumina coverage, HySA achieved sensitivity comparable to that of DELLY at 150× Illumina coverage. Compared to PBHoney at 30× PacBio coverage, HySA achieved higher sensitivity at 10× PacBio and 30× Illumina coverage. Given the current lower throughput and higher cost of PacBio data, our approach clearly provides a more cost-effective solution than approaches that utilize only Illumina or PacBio data but not both.
Figure 4.
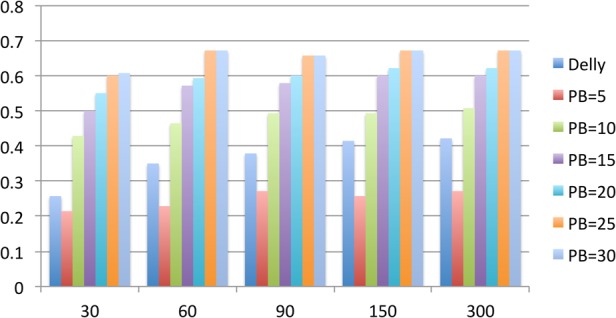
Coverage analysis. Sensitivities of HySA are estimated at combinations of 5, 10, 15, 20, 25, and 30× PacBio coverage and 30, 60, 90, 150, and 300× Illumina coverage, respectively. Sensitivity of DELLY is also shown on the leftmost bar of each Illumina coverage.
Discussion
In this work, we developed HySA that performs targeted hybrid SV assembly from NGS and SMS reads for SV detection. HySA combines the advantages of two technologies, the accuracy of the Illumina reads and the length of the PacBio reads, and was able to discover novel SVs missed by algorithms that detect SVs from a single technology, or by naively merging technology-specific call-sets. It complements existing approaches and can be applied to substantially enhance the discovery power of ongoing personal genomic projects. In particular, we found HySA to be advantageous in detecting SVs that have weak evidence in data generated by one technology. Those SVs tend to occur in repeats (e.g., STRs) or contain novel insertions (i.e., sequences absent from the reference). The FDRs of HySA appeared to be less than 10%, owing partly to the combined use of orthogonal technologies. Although HySA was developed and assessed using data produced by Illumina and PacBio technologies, the general framework is potentially applicable to data produced by other NGS and SMS technologies such as Ion Proton and Oxford Nanopore.
Dramatically different error profiles and lengths between sequencing reads generated by different technologies made it difficult to perform hybrid assembly using standard approaches such as OLC and de Bruijn and string graphs. The graph-theoretic approach that we developed and assessed in this study appears to be effective for constructing accurate hybrid SV assembly. Focusing on SVs that are difficult to assemble by the WGA approaches highlights the computational efficiency of HySA and its applicability in translational research.
We quantified the advantage of having both Illumina and PacBio coverage in an assembly project and found that a combination of 25× PacBio coverage and 60× Illumina coverage is likely optimal for comprehensively assembling a diploid genome, as many of the SVs uniquely identified by HySA lie in highly repetitive regions that cannot be mapped confidently by Illumina alone. On the other hand, we found that at least 10× PacBio coverage is required for HySA to perform well. This requirement appeared to result from a limitation of the Celera Assembler that we used to perform the local assembly. New tools under development may alleviate such limitations. For example, Canu (Koren et al. 2017) as a fork of the Celera Assembler has been developed to assemble high-noise single-molecule sequencing and is more capable of assembling lower coverage (<10×) PacBio data. Employing these new assemblers in HySA may further enhance the discovery of heterogeneous SVs, particularly those represented in low coverage (e.g., subclonal SVs in bulk tumor tissue sequencing). Although the relationship we revealed between discovery power and coverage is specific to our algorithm, it is potentially generalizable to any discovery approach that utilizes both Illumina and PacBio data.
Our work further highlights the complexity of human genomes and the limitations of current technologies and approaches. To obtain a perfect genome assembly and detect all the SVs, multiple technologies and computational algorithms that are advantageous in complementary ways should be employed. Despite the combined use of PacBio and Illumina technologies, our approach may have missed a substantial portion of SVs, particularly in highly repetitive areas of the genomes. Overcoming such limitation will require further development of sequencing technologies, as well as hybrid approaches that leverage the unique strengths of each technology.
Methods
Let I be a set of Illumina reads and P be a set of PacBio reads. The overall objective is to identify for each unknown SV location x, the subsets I ⊂ I and P ⊂ P of Illumina and PacBio reads that contain x, assemble the genomic region around x using I and P, and recover x. To achieve this objective, we propose a two-step solution in which the first step (Algorithm 1 in Supplemental Results) clusters the Illumina and PacBio reads by SV sequences, and the second step (Algorithm 2 in Supplemental Results) conducts the assembly and SV calling from the clusters. A cluster is the pair (I,P) that corresponds to one potential SV, as discussed above.
Detailed description of key steps in Algorithms 1 and 2
In Algorithm 1, we extract Illumina reads that are not well aligned to the reference, including those that are discordant, unmapped, or have at least one read clipped or containing a large gap (Fig. 1A; Supplemental Results). We then align these reads to all PacBio reads by BLASR (Fig. 1A; Supplemental Results). Due to the high error rate of PacBio reads, false alignment may occur between an Illumina read and a PacBio read. To reduce the number of false alignments, we require at least 70 bp of the Illumina read sequence to be aligned to the PacBio reads with at least 70% identity. Moreover, Illumina reads’ paired signal is used for selecting concordant and thus reliable alignments with the same criteria described in Step 2 of Algorithm 1 (Fig. 1C; Supplemental Results).
On each PacBio read, we expect a set of Illumina reads to be aligned and piled at a certain location, where the breakpoint lies. The piling of an excessive number of Illumina reads indicates a potential repetitive region. On the other hand, a small number of Illumina reads piling at one location indicates a potential false alignment. We remove the alignments (Fig. 1D) involved in these two situations by setting up a range (3,λK1), in which K1 is the mean coverage of Illumina reads, and λ is a threshold set by the user (heuristically, a reasonable value for λ is within [1, 1.2]).
In a diploid genome, PacBio reads corresponding to the reference genome can be falsely extracted by clipped Illumina reads. To avoid extracting these PacBio reads, we synthesize pseudo (a.k.a., “ref”) read pairs from the reference sequence, with positions, lengths, and orientations identical to those clipped Illumina reads. The purpose of constructing these “ref” reads is to discern the allele to which the PacBio read belongs. Both clipped and pseudo (ref) reads are aligned to all PacBio reads. A pseudo (ref) read is considered to align better if (1) it matches more (>10 bp) bases than its clipped counterpart, or (2) its alignment is continuous, whereas the clipped counterpart is aligned with a large (>30 bp) gap (Fig. 1E). When that happens, the alignment between the Illumina clipped read and the PacBio read is regarded as false.
We use all reliable alignments between Illumina read pairs and PacBio reads as the edges to build the bipartite graph and apply the Union-Find algorithm (Sedgewick and Wayne 2011) to partition the graph into connected components and further partition the large connected components into communities by Infomap (Fig. 1B; Supplemental Results; Rosvall and Bergstrom 2008).
In Algorithm 2, we assemble the PacBio reads in each connected component or community into contigs using Celera Assembler (Myers et al. 2000), and align the contigs to the reference using BLASR (Fig. 1A; Chaisson and Tesler 2012). The alignment of the contigs to the reference is ignored if either clipped end is >500 bp. The rest of the alignments are analyzed to search for large indel gaps (>10 bp). We require a matching flanking region >10 bp. For large gaps, BLASR tends to chop them into small ones separated by short matching subsequences. To accurately infer breakpoints for these gaps, we implement a local realignment algorithm, pair-HMM (Durbin et al. 1998), which realigns the assembled contig to the reference. The HMM has three states (‘M’ for matching, ‘I’ for insertion, and ‘D’ for deletion). Transitions are encouraged from ‘M’ to ‘M’, ‘D’ to ‘D’, and ‘I’ to ‘I’, with transition probabilities 0.99. Other transitions are discouraged with small transition probabilities (<0.01). Through this process, the small indels segmented by BLASR could be concatenated in a big one. We notice a similar procedure in MultiBreak-SV (Ritz et al. 2014) and Pendleton et al. (2015). To confirm the inferred SV, we align the Illumina reads in the same cluster to the contig using the customized BLASR and examine the alignment (Supplemental Results).
Complex deletion detection
To identify complex deletions, we examine the set of contigs whose BLASR alignment against the reference indicates a large deletion with ≥20-bp near-perfect flanking alignments and a series of D and I (cumulatively ≥10 bp) but no M in the CIGAR string between the deletion breakpoints. These D's and I's (indels) may result from additional rearrangements (e.g., insertions) and thus cannot be well aligned against the reference by BLASR.
For each of these deletions, we extract the portion of contig sequence that is not well aligned (including 50-bp flanking sequences) and the corresponding local reference sequence (including 70-bp flanking sequences) from the breakpoints. We realign them using BWA-MEM (0.7.5a-r405, default parameter). If the resulting alignments indicate a deletion (with at least 10 bp in length with an inserted/unaligned sequence of at least 10 bp at the breakpoint), we call this SV a complex deletion (i.e., deletion with insertion).
We then further examine the origin of the inserted/unaligned sequences from additional/secondary alignments returned by BWA-MEM. If the inserted/unaligned sequence (plus maximally 10-bp flanking sequence on either end) can be mapped onto the flanking reference sequence, it is called “duplication.” If an inserted/unaligned sequence can be aligned onto the deleted sequence on the reference, it is called a “spacer” (i.e., it separates a deletion into two or more constituent deletions). In all cases, the alignment of the inserted/unaligned sequences could be in either the same or the “inverted” orientations, compared with the alignment of their flanking sequences (Supplemental Fig. 6).
Software availability
The developed pipeline and the scripts used in this manuscript are available in the Supplemental Material (Supplemental Software) and also online at https://bitbucket.org/xianfan/hybridassemblysv/overview (with commit ID number eee31f6).
Data access
The large (≥50 bp) SVs identified in this study have been submitted to NCBI's dbVar (https://www.ncbi.nlm.nih.gov/dbvar) under accession number nstd140, and the small indels (<50 bp) have been submitted to dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/) under batch numbers 1062737 and 1062738 (release number B151).
Supplementary Material
Acknowledgments
This work was supported in part by the Division of Cancer Prevention, National Cancer Institute (NCI) grant R01-CA172652 to K.C., National Human Genome Research Institute (NHGRI) grant U41-HG007497-01, and the National Cancer Institute Cancer Center Support Grant P30-CA016672. We thank E. Eichler, J. Korbel, C. Lee, and other members of the Human Genome Structural Variation Consortium for ideas and discussions. We also thank W. Zhou for suggesting community decomposition algorithms and Z. Chong for discussion of ideas.
Author contributions: X.F. and K.C. conceived the study. X.F. and M.C. designed and implemented the code. X.F. performed the analysis. X.F., L.N., and K.C. wrote the manuscript. K.C. and L.N. provided oversight and coordinated the project. All authors read, revised, and approved the final manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.214767.116.
References
- The 1000 Genomes Project Consortium. 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. 2013. Signatures of mutational processes in human cancer. Nature 500: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C, Coe B, Eichler E. 2011a. Genome structural variation discovery and genotyping. Nat Rev Genet 12: 363–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C, Sajjadian S, Eichler EE. 2011b. Limitations of next-generation genome sequence assembly. Nat Methods 8: 61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au KF, Underwood JG, Lee L, Wong WH. 2012. Improving PacBio long read accuracy by short read alignment. PLoS One 7: e46679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin K, Koren S, Chin CS, Drake JP, Landolin JM, Phillippy AM. 2015. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat Biotechnol 33: 623–630. [DOI] [PubMed] [Google Scholar]
- Butler J, MacCallum I, Kleber M, Shlyakhter IA, Belmonte MK, Lander ES, Nusbaum C, Jaffe DB. 2008. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res 18: 810–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, Tesler G. 2012. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, Wilson RK, Eichler EE. 2015a. Genetic variation and the de novo assembly of human genomes. Nat Rev Genet 16: 627–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJP, Huddleston J, Dennis MY, Sudmant PH, Malig M, Hormozdiari F, Antonacci F, Surti U, Sandstrom R, Boitano M, et al. 2015b. Resolving the complexity of the human genome using single-molecule sequencing. Nature 517: 608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, et al. 2009. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods 6: 677–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Chen L, Fan X, Wallis J, Ding L, Weinstock G. 2014. TIGRA: a targeted iterative graph routing assembler for breakpoint assembly. Genome Res 24: 310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Muller WEG, Wetter T, Suhai S. 2004. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res 14: 1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, et al. 2013. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10: 563–569. [DOI] [PubMed] [Google Scholar]
- Chin C-S, Peluso P, Sedlazeck FJ, Nattestad M, Concepcion GT, Clum A, Dunn C, O'Malley R, Figueroa-Balderas R, Morales-Cruz A, et al. 2016. Phased diploid genome assembly with single molecule real-time sequencing. Nat Methods 13: 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong Z, Ruan J, Gao M, Zhou W, Chen T, Fan X, Ding L, Lee AY, Boutros P, Chen J, et al. 2016. novoBreak: local assembly for breakpoint detection in cancer genomes. Nat Methods 14: 65–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Bjornson RD, Zhang ZD, Kong Y, Snyder M, Gerstein MB. 2009. Integrating sequencing technologies in personal genomics: optimal low cost reconstruction of structural variants. PLoS Comput Biol 5: e1000432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin R, Eddy SR, Krogh A, Mitchison G. 1998. Biological sequence analysis: probabilistic models of proteins and nucleic acids, 1st ed Cambridge University Press, New York. [Google Scholar]
- Eberle MA, Fritzilas E, Krusche P, Kallberg M, Moore BL, Bekritsky MA, Iqbal Z, Chuang HY, Humphray SJ, Halpern AL, et al. 2016. A reference data set of 5.4 million phased human variants validated by genetic inheritance from sequencing a three-generation 17-member pedigree. Genome Res 27: 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English AC, Salerno WJ, Reid JG. 2014. PBHoney: identifying genomic variants via long-read discordance and interrupted mapping. BMC Bioinformatics 15: 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuk L, Carson AR, Scherer SW. 2006. Structural variation in the human genome. Nat Rev Genet 7: 85–97. [DOI] [PubMed] [Google Scholar]
- Hackl T, Hedrich R, Schultz J, Forster F. 2014. proovread: large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 30: 3004–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal Z, Caccamo M, Turner I, Flicek P, McVean G. 2012. De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat Genet 44: 226–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, et al. 2008. Mapping and sequencing of structural variation from eight human genomes. Nature 453: 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Schatz MC, Walenz BP, Martin J, Howard JT, Ganapathy G, Wang Z, Rasko DA, McCombie WR, Jarvis ED, et al. 2012. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol 30: 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res (this issue). 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layer RM, Chiang C, Quinlan AR, Hall IM. 2014. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 15: R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arxiv:1303.3997 [q-bio.GN]. [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR. 2007. Structural variation in the human genome. N Engl J Med 356: 1169–1171. [DOI] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, et al. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437: 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills R, Walter K, Stewart C, Handsaker R, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham K, et al. 2011. Mapping copy number variation by population-scale genome sequencing. Nature 470: 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers EW. 2005. The fragment assembly string graph. Bioinformatics 21: i79–ii85. [DOI] [PubMed] [Google Scholar]
- Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, Kravitz SA, Mobarry CM, Reinert KH, Remington KA, et al. 2000. A whole-genome assembly of Drosophila. Science 287: 2196–2204. [DOI] [PubMed] [Google Scholar]
- Parikh H, Mohiyuddin M, Lam HY, Iyer H, Chen D, Pratt M, Bartha G, Spies N, Losert W, Zook JM, et al. 2016. svclassify: a method to establish benchmark structural variant calls. BMC Genomics 17: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton M, Sebra R, Pang AW, Ummat A, Franzen O, Rausch T, Stutz AM, Stedman W, Anantharaman T, Hastie A, et al. 2015. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat Methods 12: 780–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T, Zichner T, Schlattl A, Stütz A, Benes V, Korbel J. 2012. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28: i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz A, Bashir A, Sindi S, Hsu D, Hajirasouliha I, Raphael BJ. 2014. Characterization of structural variants with single molecule and hybrid sequencing approaches. Bioinformatics 30: 3458–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross MG, Russ C, Costello M, Hollinger A, Lennon NJ, Hegarty R, Nusbaum C, Jaffe DB. 2013. Characterizing and measuring bias in sequence data. Genome Biol 14: R51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosvall M, Bergstrom CT. 2008. Maps of random walks on complex networks reveal community structure. Proc Natl Acad Sci 105: 1118–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedgewick R, Wayne K. 2011. Algorithms, 4th ed Addison-Wesley Professional, Boston. [Google Scholar]
- Sharp AJ, Cheng Z, Eichler EE. 2006. Structural variation of the human genome. Annu Rev Genomics Hum Genet 7: 407–442. [DOI] [PubMed] [Google Scholar]
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. 2009. ABySS: a parallel assembler for short read sequence data. Genome Res 19: 1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindi S, Onal S, Peng L, Wu H, Raphael B. 2012. An integrative probabilistic model for identification of structural variation in sequencing data. Genome Biol 13: R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg KM, Schneider VA, Graves-Lindsay TA, Fulton RS, Agarwala R, Huddleston J, Shiryev SA, Morgulis A, Surti U, Warren WC, et al. 2014. Single haplotype assembly of the human genome from a hydatidiform mole. Genome Res 24: 2066–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Mullighan C, Easton J, Roberts S, Heatley S, Ma J, Rusch M, Chen K, Harris C, Ding L, et al. 2011. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods 8: 652–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Wu G, Tang J, Luo R, Patterson J, Liu S, Huang W, He G, Gu S, Li S, et al. 2014. SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 30: 1660–1666. [DOI] [PubMed] [Google Scholar]
- Ye K, Schulz M, Long Q, Apweiler R, Ning Z. 2009. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25: 2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zook JM, Chapman B, Wang J, Mittelman D, Hofmann O, Hide W, Salit M. 2014. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat Biotechnol 32: 246–251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.