

Abstract
Mesothelin overexpression in lung adenocarcinomas correlates with the presence of activating KRAS mutations and poor prognosis. Hence SS1P, a mesothelin‐targeted immunotoxin, could offer valuable treatment options for these patients, but its use in solid tumor therapy is hampered by high immunogenicity and non‐specific toxicity. To overcome both obstacles we developed RG7787, a de‐immunized cytotoxic fusion protein comprising a humanized SS1 Fab fragment and a truncated, B‐cell epitope silenced, 24 kD fragment of Pseudomonas exotoxin A (PE24). Reactivity of RG7787 with sera from immunotoxin‐treated patients was >1000 fold reduced. In vitro RG7787 inhibited cell viability of lung cancer cell lines with picomolar potency. The pharmacokinetic properties of RG7787 in rodents were comparable to SS1P, yet it was tolerated up to 10 fold better without causing severe vascular leak syndrome or hepatotoxicity. A pharmacokinetic/pharmacodynamic model developed based on NCI‐H596 xenograft studies showed that for RG7787 and SS1P, their in vitro and in vivo potencies closely correlate. At optimal doses of 2–3 mg/kg RG7787 is more efficacious than SS1P. Even large, well established tumors (600 mm3) underwent remission during three treatment cycles with RG7787. Also in two patient‐derived lung cancer xenograft models, Lu7336 and Lu7187, RG7787 showed anti‐tumor efficacy. In monotherapy two treatment cycles were moderately efficacious in the Lu7336 model but showed good anti‐tumor activity in the KRAS mutant Lu7187 model (26% and 80% tumor growth inhibition, respectively). Combination of RG7787 with standard chemotherapies further enhanced efficacy in both models achieving near complete eradication of Lu7187 tumors.
Keywords: Immunotoxin, De-immunization, Pharmacokinetics, Lung cancer, Patient-derived xenografts, Targeted therapy
Highlights
Immunogenicity and off‐target toxicity hamper the therapeutic use of immunotoxins.
Re‐engineering the immunotoxin format reduces antigenicity and toxicity.
A re‐engineered immunotoxin against mesothelin potently kills lung cancer cells.
Combining it with chemotherapy shrinks patient‐derived KRAS mutant lung tumors.
Abbreviations
- ADAs
anti-drug antibodies
- CI
confidence interval
- Fab
antibody fragment comprising the variable and the first constant domains
- Fv
antibody fragment comprising only the variable domains
- h
hour(s)
- PDX
patient-derived xenograft
- PE
Pseudomonas exotoxin A
- RIT
recombinant immunotoxin
- TGI
tumor growth inhibition
- TV
tumor volume
- VLS
vascular leak syndrome
1. Introduction
Recombinant immunotoxins (RITs) consist of a bacterial toxin linked to an antibody fragment that is directed against a tumor‐specific cell‐surface antigen. While different RITs have shown promising preclinical efficacy, their clinical use has been severely hampered by immunogenicity and vascular leak syndrome (VLS) as dose‐limiting toxicity (Weidle et al., 2014). The SS1P molecule developed at the National Cancer Institute (NCI) is a typical RIT consisting of a disulfide‐stabilized single chain Fv fragment (dsFv) for tumor targeting, in this case derived from the SS1 mouse anti‐mesothelin antibody, and a 38 kDa Pseudomonas Exotoxin A fragment (PE38) comprising domains II and III as the cytotoxic payload. In phase 1 trials, SS1P monotherapy given either as bolus intravenous infusion (Hassan et al., 2007) or by continuous infusion (Kreitman et al., 2009) showed only minor antitumor activity after a single treatment cycle. About 90% of treated patients developed neutralizing antibodies to SS1P after only one therapy cycle, which prohibited successful re‐treatment and sustained clinical benefit (Pastan and Hassan, 2014). Hence the primary goal for generating a clinically more useful, re‐engineered version of SS1P was to reduce its immunogenicity thereby allowing for multiple treatment cycles. To achieve this, the bacterial toxin portion was reduced to 24 kD consisting essentially only of domain III and the furin cleavage motif of PE. In addition seven amino acid exchanges were introduced into domain III to “silence” previously mapped B‐cell epitopes (Weldon et al., 2013). The murine antibody fragment of SS1P was also humanized and in addition converted to a Fab fragment to compensate for the molecule size reduction by truncation of the toxin and prevent enhanced renal clearance. A further goal of the re‐engineering effort was to reduce the adverse effect potential of the molecule, particularly with regards to causing VLS, which can result in a clinically difficult to manage combination of edema, weight gain, and hypotension (Kelly et al., 2012). An improved off‐target toxicity profile should enable to clinically exploit more aggressively combination regimen with standard of care chemotherapies. We describe here the generation of RG7787, the first mesothelin‐targeted RIT with substantially reduced antigenicity and improved tolerability in animals that retains cytotoxic potency and pharmacokinetic properties comparable to SS1P.
Evaluation of SS1P has primarily focused on solid tumor indications reported to show close to 100% prevalence of mesothelin positivity, like epithelial mesotheliomas (Ordonez, 2003) and pancreatic adenocarcinomas (Argani et al., 2001). Literature reports of mesothelin expression in lung adenocarcinomas suggest much lower prevalence ranging from 30% to 50%. However, since lung cancer causes globally more than 1.5 million deaths per year (Torre et al., 2015), it is an important indication for clinical development of RG7787. Moreover, for almost half of all advanced non‐small cell lung cancer cases there is no targeted therapy available, particularly for the ∼25% of lung adenocarcinomas, that carry a mutated KRAS allele (Kris et al., 2014). A recent immunohistochemical analysis of samples from 93 lung adenocarcinoma patients performed at the NCI found that mesothelin expression, as defined by positive staining in >25% of the tumor cells, was an independent predictor for poor survival and was strongly associated (p < 0.0001) with KRAS mutation status (Thomas et al., 2015). In the present study we characterized the in vitro and in vivo potency of RG7787 in lung cancer models. RG7787 showed high potency on a series of lung cancer cell lines that were preselected for mesothelin expression based on Affymetrix chip data. In efficacy studies with standard as well as patient‐derived xenografts, it dose‐dependently induced tumor regressions at an optimal dose of ∼2.5 mg/kg. Combination with chemotherapy led to almost complete tumor eradication in a KRAS mutated lung cancer model suggesting this could be a promising therapeutic approach for a subpopulation of lung adenocarcinoma patients that currently lack targeted therapy options.
2. Material and methods
2.1. Cell lines and culture conditions
Human lung carcinoma cells NCI‐H596, obtained from the NCI at the National Institute of Health (Bethesda, Maryland, USA), and AsPC‐1‐luc cells, a stably luciferase transfected pancreatic adenocarcinoma cell line purchased from ProQinase (Freiburg, Germany), were cultured at 37 °C with 5% CO2 in RPMI 1640 high glucose (4.5 g/L) medium supplemented with 10% FCS, 2 mM l‐glutamine, 1 mM sodium pyruvate, and 10 mM Hepes. All other lung cancer cell lines were purchased by Horizon Discovery from commercial cell banks and cultivated exactly as described in the Cancer Cell Line Encyclopedia (CCLE) by the Broad Institute (Barretina et al., 2012). All cultured cells retained the characteristic phenotype described by the commercial providers and the CCLE. Cells were immediately expanded, multiple aliquots cryopreserved and used for no more than 20 passages after resuscitation.
2.2. Humanization of the targeting moiety of RG7787
Humanization was done by generating specific combinations of certain framework regions of the mouse anti‐mesothelin antibody SS1. The resulting clones were screened for retaining high binding affinity for human and cynomolgus mesothelin. Fab‐PE24 fusion proteins of the best binders were then screened for cellular potency on As‐PC‐1‐luc cells, and for thermal stability, and technical developability. The protein sequence of the candidate selected based on these criteria is given in the Supplement (SI 2).
2.3. In vitro potency assays
1 × 104 cells in 50 μl medium were seeded in 96 well plates and incubated overnight. On the following day, 1:3 serial dilutions of RG7787 in medium were prepared starting at 3 μg/ml. An untargeted PE24 was used as control for non‐specific cytotoxicity. 50 μl of each RIT dilution was added to the wells and incubated for 72 h. Reduction in cell viability was determined using the CellTiter‐Glo® reagent (Promega) except for the AsPC‐1‐luc cells, for which the cytotoxic effect of RITs was determined by measuring luciferase activity with Steady‐Glo® assay (Promega). In both cases results were plotted as percent inhibition versus untreated controls. Assessment of cytotoxicity for the lung cancer cell lines from the CCLE panel were performed by Horizon Discovery Inc. (Cambridge, MA, U.S.A.) as described in the Supplement (SI 1).
2.4. Study medications
SS1P was provided by Dr. I. Pastan (NIH). SS1(dsFv)‐PE24, SS1(Fab)‐PE24, and RG7787 were obtained by E. coli inclusion body expression of heavy and light chains and pulse refolding (Buchner et al., 1992; for details see Supplement SI 2 and Figure S1).
2.5. In vivo efficacy studies
Female 4–6 weeks‐old SCID beige mice from Charles River (Sulzfeld, Germany) were subcutaneously inoculated in the presence of matrigel with 5 × 106 NCI‐H596 cells. For the patient‐derived lung cancer models (Lu7187, Lu7336) performed at EPO Berlin‐Buch GmbH, tumor fragments were subcutaneously passaged on male NMRI nu/nu mice (Charles River) as described in detail in Fichtner et al., 2008. Xenograft size was determined by caliper and tumor volume (TV) was calculated according to the formula TV = (length × width2)/2. Depending on the study purpose and tumor model intravenous tail vain injection of study medications or solvent was initiated when TV reached a median value of approximately 150, 250 or 600 mm3. The treatment schedules applied for the different in‐vivo studies are provided as Supplementary Table 5. All animal studies were conducted in accordance with the German animal welfare law and approved by the local governmental animal ethics committee.
2.6. Pharmacokinetic studies and determination of drug serum concentrations
For determination of pharmacokinetic properties, mice were given intravenously the indicated doses of SS1P or RG7787. Serum concentrations of study medications were assessed by a sequential target capture sandwich ELISA using human mesothelin as capturing reagent and a rabbit derived anti‐PE antibody preparation as detection reagent. Further details on measurement of total‐intact versus free‐intact drug plasma concentrations are provided in the Supplement (SI 3).
2.7. Modeling and simulation
The PK/PD model was built in Simbiology and fitted to the experimental data in MATLAB (R2013b; Mathworks). Detailed equations and resulting parameters are provided in the Supplement (SI 4). The pharmacokinetic parameters for SS1P and RG7787 were calculated with non‐compartmental analysis approach in MATLAB.
2.8. Vascular leak syndrome and hepatotoxicity in rats
To investigate the relative risk for induction of VLS by RG7787 and SS1P, a previously described rat model was used (Siegall et al., 1994) and extended by an assessment for hepatotoxicity. Three female 6–8 week old Wistar Furth rats per treatment group were given SS1P (2 mg/kg), RG7787 (10 mg/kg) or vehicle as single i.v. bolus. Dose selection was based on the described off‐target toxicity of SS1P in this strain. Hepatotoxicity and vascular leakage, was assessed by clinical biochemistry of liver parameters, presence of serum protein changes, and thoracic fluid accumulation at 24 h post dose as well as by macroscopic examination of lung and liver. Body weight, food intake and clinical signs were also recorded.
2.9. Statistics
Xenograft data were processed in the statistics software SAS‐JMP version 8.0.2.2 (SAS Inc., Cary, NC, 2007). Data regarding primary tumor growth were statistically analyzed using non‐parametric methods, when the data showed an asymmetrical distribution (Hothorn, 2006), or parametric methods, when tumor volume values were normally distributed (Fieller, 1954).
3. Results
3.1. Generation of RG7787 – a RIT with reduced antigenicity
Anti‐drug antibodies (ADAs) from immunotoxin‐treated patients are always directed against the PE38 moiety (Alewine et al., 2015). However, once the immune‐dominant toxin portion is successfully de‐immunized, an ADA response could conceivably also be mounted against the murine targeting moiety of SS1P. Therefore, we humanized the Fab fragment used in RG7787 as described in patent WO2015051199. To de‐immunize the toxin moiety of RG7787 most of domain II (residues 251–273 and 284–394 of PE) was replaced by a peptide linker with an extended furin cleavage site (DKTHKASGGRHRQPR↓GWEQLGGGGGS). In addition all known human B‐cell epitopes of domain III (Liu et al., 2012) were removed by exchanging key charged surface residues: R505A, R427A, R490A, R467A, D463A, R456A, and R538A (Figure 1A). Using an in‐solution competition ELISA we could demonstrate that reactivity of RG7787 with sera from patients previously treated with SS1P was substantially reduced (Figure 1B). For 10 of 20 patient sera the antigenicity of RG7787 was >1000 fold reduced when benchmarked against SS1P as ratio of the IC50 values of competition dose response curves (Figure 1C). PE24‐based RITs have reduced circulation half‐life due to smaller size as previously shown (Weldon et al., 2013). In order to retain a molecular size of around 70 kD a Fab rather than a disulfide stabilized single chain Fv fragment was used in RG7787 (Figure 1A).
Figure 1.
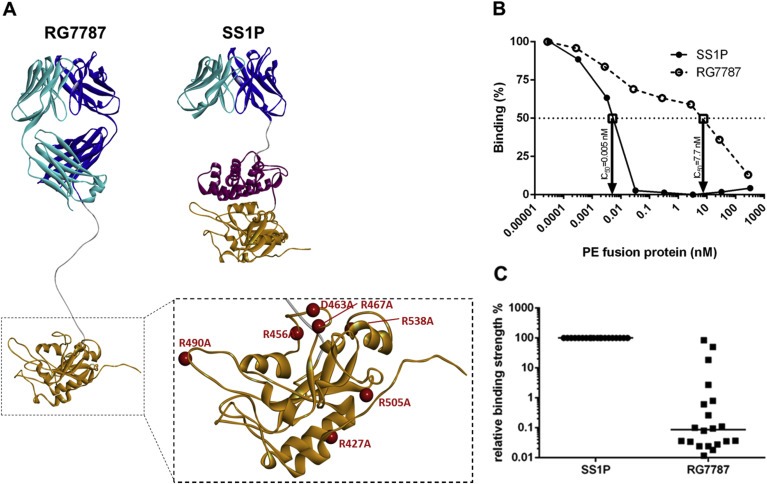
Structures of RG7787 and SS1P and assessment of their relative antigenicity (A) Ribbon diagrams showing the different domain structures of SS1P and RG7787 as well as the 7 amino acid substitutions (red balls) within the catalytic domain III of PE that were introduced in RG7787 to silence all 6 known human B‐cell epitopes. The mutated PE24 was modeled onto the crystal structure of PE (structure 1IKQ in the Research Collaboratory for Structural Bioinformatics protein data base), the linker with the furin cleavage site was modeled as linear peptide chain and energy minimized, and the Fab fragment was modeled onto fragments of different Fv structures and refined as described in Bujotzek et al., 2015. (B) The graph shows typical competition curves obtained using serum from an SS1P‐treated patient with a neutralizing anti‐drug antibody response. In this case the concentrations of RG7787 and SS1P at which binding to PE38 was inhibited by 50% (IC50) were 7.7 nM and 0.005 nM, respectively. Based on these IC50 values the binding ratio of SS1P/RG7787 was 0.065%. (C) Binding ratios for sera from 20 patients were plotted and the median binding ratio is indicated by a horizontal line.
3.2. In vitro potency of RG7787
To exclude that humanization compromised functionality of RG7787 we compared the cytotoxic potency of variants containing either the murine or the humanized SS1 Fab fragment on NCI‐H596 cells. As control for cytotoxicity by unspecific uptake we also included treatment with an untargeted PE24 (free PE24). To assess, whether the targeting moiety per se had any effect on cell viability, we also tested the humanized SS1 Fab fragment without PE fusion (huSS1Fab). RG7787 variants with the murine and humanized targeting moiety gave overlapping dose‐response curves (Figure 2A), while the Fab fragment alone had no effect and the free PE24 was ∼200 fold less potent in this 72 h assay format. A shorter incubation time with the RIT of ≤2 h reflects more closely the pharmacokinetic situation in vivo and limits unspecific uptake. Under such conditions the potency difference between RG7787 and free PE24 or a misdirected RIT increased to >7000 fold (Supplement Table S1). Next we tested in vitro a panel of 13 lung cancer cell lines, pre‐selected for mesothelin positivity based on Affymetrix gene expression data, for sensitivity to RG7787 treatment. In 9 out of 13 lines RG7787 induced tumor cell death as indicated by max GI values >100%. Two other lines, T3M‐10 and NCI‐H292, were completely growth inhibited, while proliferation of NCI‐H2052 and DV90 cells was only partially inhibited by 61% and 19%, respectively. For all 11 responsive cell lines GI50 values were below 160 pM (Figure 2B).
Figure 2.
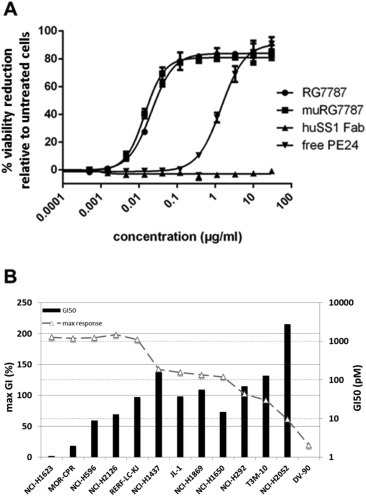
Cytotoxic potency of RG7787 (A) Humanization of the targeting Fab fragment did not negatively affect the cytotoxic potency of RG7787. Dose‐response curves comparing reduction of cell viability after 72 h incubation with just the humanized Fab fragment of SS1 (▴), free PE24 toxin (▾), RG7787 (●) or RG7787 with the murine SS1 Fab fragment (■) are shown. Cell viability was determined by luciferase‐based ATP measurement (CellTiter‐Glo®). (B) RG7787 has high cytotoxic potency on 11 of 13 lung cancer cell lines that were preselected based on Affymetrix data for mesothelin expression. GI50 values represent the concentrations necessary to reduce the ATP signal of vehicle‐treated control cells after 72 h by 50% and are shown as bars logarithmically scaled according to the y‐axis on the right. No bar is shown for cell line DV‐90 since a 50% reduction was not achieved. The maximum responses in % reduction versus viability signal before treatment start are plotted as line according to the linear scale of the y axis on the left. A max GI of 100% signifies that the ATP signal after 72 h was the same as at the starting time point t0 indicating complete inhibition of cell growth. A max GI of 200% indicates that all cells were dead after 72 h.
In the initial potency screening on AsPC‐1‐luc cells SS1P (IC50: 0.02 nM ± 0.004, n = 3) was about twofold more potent than RG7787 (0.05 nM ± 0.01, n = 3). In‐vitro on NCI‐H596 cells, a lung adenocarcinoma cell line that we selected for subsequent in‐vivo studies, SS1P (IC50: 0.07 nM ± 0.02, n = 3) was also somewhat more potent than RG7787 (IC50: 0.28 nM ± 0.02, n = 3), however, when comparing their potency on a wider range of cancer cell lines, we often also observed a potency difference in favor of RG7787, e.g., on 3 of 6 pancreatic cancer cell lines tested (Hollevoet et al., 2014) or on 2 of 2 triple negative breast cancer cell lines (Alewine et al., 2014). Overall the subnanomolar in‐vitro potency of RG7787 on multiple lung cancer cell lines suggests that mesothelin‐positive lung cancer might be a promising indication for clinical development of RG7787.
3.3. Tumor growth inhibition by RG7787 in cell line based standard xenografts
Because blocking protein synthesis affects not only the proliferative capacity of tumor cells, but also all other hallmarks of cancer, RITs have the potential to also affect slow growing tumors and perhaps to eliminate non‐proliferating cancer stem cells. We chose the NCI‐H596 adenosquamous lung carcinoma as a lung cancer xenograft model that more closely resembles the slow growth rate of human tumors. NCI‐H596 tumors took 62 days post inoculation to reach a median volume of 250 mm3 for randomization. A single therapy cycle with SS1P at the maximally tolerated dose of 0.4 mg/kg was more efficacious than RG7787 at an equimolar dose of 0.46 mg/kg (non‐parametric tumor‐to‐control ratio: 0.28 CI: 0.16–0.44 versus 0.48 CI: 0.36–0.67, respectively; p‐value calculated by non‐parametric Wilcoxon test: 0.0043). However, RG7787 was much better tolerated and could be dosed 5 to 10 fold higher. Between 0.46 mg/kg and 2 mg/kg the antitumor effect of RG7787 increased dose‐dependently from tumor stasis to tumor regression (Figure 3A). We next tested whether repeated treatment cycles at a dose of 2 mg/kg are able to shrink very large, established tumors. For this study, therapy was initiated when the median tumor volume reached 600 mm3 and animals received three treatment cycles with a one‐week break between consecutive cycles. This dose and regimen shrank 600 mm3 tumors to a nadir volume of ∼120 mm3 (Figure 3B). Histological examination of residual lesions showed many necrotic/pre‐necrotic tumor cells but also some that appeared still viable. The membranes of ∼50% of the residual tumor cells stained weakly positive for mesothelin. In summary our data show that at higher doses RG7787 achieves better in vivo efficacy than SS1P and repeated treatment cycles can control even large tumors.
Figure 3.
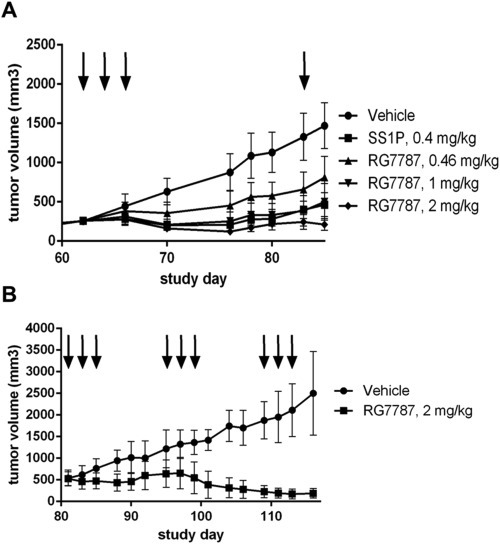
In vivo efficacy of RG7787 in NCI‐H596 xenografts (A) RG7787 dose‐dependently inhibits growth of NCI‐H596 xenografts in female SCID beige mice. Groups of mice (n = 10–12) were treated at the time points indicated by vertical arrows either with vehicle (●) or the maximally tolerated dose of SS1P (0.4 mg/kg: ■) or different doses of RG7787 (0.46 mg/kg: ▲; 1 mg/kg: ▼; 2 mg/kg: ♦). Data points indicate median tumor volumes and error bars represent interquartile ranges. (B) Three cycles of RG7787 therapy with intermittent breaks of 1 week shrank even large tumors in this model. Treatment of groups of mice (n = 12) with either vehicle or 2 mg/kg RG7787 started when subcutaneous NCI‐H596 tumors reached an average volume of 600 mm3. The three treatment cycles are indicated by the vertical arrows.
3.4. Pharmacokinetics of RG7787
To relate drug exposure to the measured antitumor effect, we determined in parallel efficacy and pharmacokinetic parameters of RG7787 and SS1P. In female SCID beige mice bearing H596 xenografts free drug levels of RG7787 increased dose proportionally. For all doses during the 1st and 2nd treatment cycle plasma levels declined over time with similar kinetics (Figure 4A). Because of the short circulation half‐life of RG7787 drug plasma levels were not different between the 1st (day 1) and 3rd (day 5) dose of the same treatment cycle. Figure 4B shows that dose normalized concentration‐time curves of RG7787 and SS1P were superimposable. Plasma levels of free and total drug were highly correlated over a wide concentration range (Figure 4C). Over all doses tested, RG7787 showed a mean half‐life of 53 min (±5 min) for free drug and 58 min (±9.9 min) for total drug and similar results were obtained for SS1P. Detailed pharmacokinetic parameters for both agents calculated with non‐compartmental analysis are tabulated in the supplement (Tables S2 and S3). In summary, SS1P and RG7787 showed superimposable linear PK profiles in mice.
Figure 4.
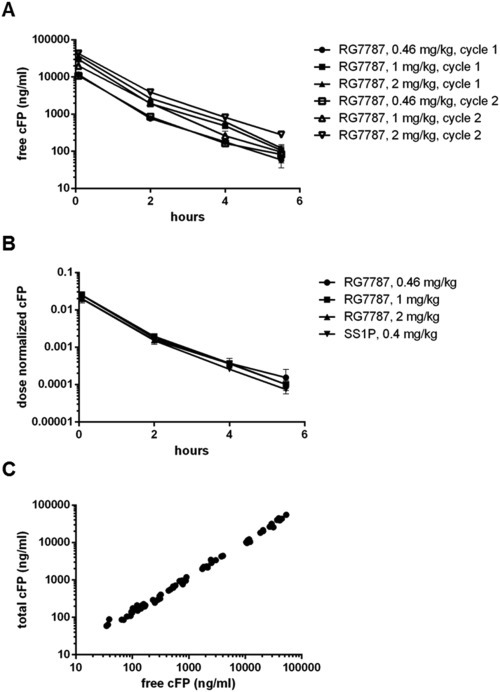
Pharmacokinetics of RG7787 and SS1P (A) Average free drug plasma levels of RG7787 are dose proportional during the 1st (filled symbols) and 2nd (open symbols) treatment cycle in female SCID beige mice bearing NCI‐H596 xenografts. RG7787 was given at 0.46 mg/kg (●, □), 1 mg/kg (■, ▵), and 2 mg/kg (▴,▿). (B) SS1P and RG7787 have similar pharmacokinetics. Dose‐normalized free drug plasma levels are plotted for the maximally tolerated dose of SS1P (0.4 mg/kg▼) and three different doses of RG7787 (0.46 mg/kg ●, 1 mg/kg ■, 2 mg/kg ▲). (A, B) All data points are mean values with standard deviations (n = 3/time point). (C) RG7787 shows a tight linear correlation between free and total drug plasma levels over a large concentration range. For all samples the free drug levels are plotted against the total drug levels.
3.5. Pharmacokinetic/pharmacodynamic (PK/PD) modeling of RG7787
Figure 5A shows a graphical representation of key components of a PK/PD model that was developed and fitted to the in vivo efficacy and drug exposure data. The underlying equations and fitted parameters are provided in the Supplement (SI 4). To determine the antitumor effect of a single cycle of RIT therapy the model was fitted to tumor volumes measured between day 49 (when tumor growth became detectable) and day 94 (when the controls needed to be sacrificed) while at the same time incorporating the measured drug concentrations from different doses. Figure 5B illustrates that tumor growth curves predicted by the model (solid lines) are in good agreement with actual measurements (symbols) for both SS1P and RG7787. Based on the model, RG7787 eliminates NCI‐H‐596 xenografts with half maximal rate at 6.8 μg/ml (±36%) plasma concentration. Doses of RG7787 that resulted in plasma concentrations above that level caused potent tumor regression. In the Supplement (SI 4) other calculated model parameters (e.g. the tumor recovery rate) are provided. Doses of 1.0 mg/kg RG7787 and 0.4 mg/kg SS1P achieved similar tumor growth inhibition. Using the model and the PK data, the in‐vivo potency of SS1P was found to be 2.9 fold greater than RG7787 when accounting for the slightly different molecular weights. In vitro SS1P was about twofold more potent than RG7787 on NCI‐H596 cells suggesting a close correlation between the observed in vitro and in vivo potencies.
Figure 5.
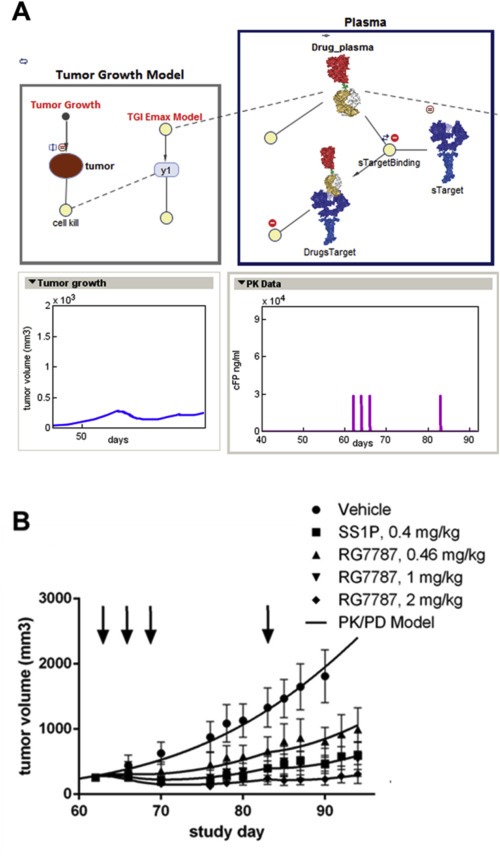
Modeling the relationship between pharmacokinetics and pharmacodynamics (A) The scheme illustrates the different model parameters and their relationships to each other. (B) Model predictions correlate well with the observed experimental data. Tumor growth curves calculated by the model are represented by the solid lines, while the data symbols show actual measured tumor volumes. Vehicle data were used for model fitting until day 90. Up to this time point no animals had to be sacrificed based on predefined termination criteria.
3.6. Vascular leak syndrome and hepatotoxicity in rats
In rodents the main off‐target effects of immunotoxins are hepatotoxicity and VLS. Deletion of domain II of PE has recently been shown to improve tolerability of an immunotoxin in mice as well as VLS in rats (Weldon et al., 2013) but this could have been, at least in part, attributed to its much shorter plasma half‐life. The fact that RG7787 retains similar PK properties as SS1P enabled us to assessed the true effect of domain II deletion on off‐target toxicities in a rat VLS model. As expected a single i.v. dose of 2 mg/kg of SS1P resulted in overall poor physical condition as evidenced by clinical signs like hunched posture, rolling gait, and lung rales as well as slight body weight loss and markedly increased blood urea nitrogen levels. At necropsy 24 h post dose, clear indications for vascular leakage were detected in all SS1P‐treated animals: marked decreases in serum protein and albumin levels and tan‐stained fluid in the lung of 2 out of 3 rats. SS1P‐mediated hepatotoxicity was evident not only from brown, clay‐like discoloration of the livers, but also from up to 40‐fold increases in several liver enzymes (alanine and aspartate aminotransferases, gamma‐glutamyltransferase, glutamate dehydrogenase, sorbitol dehydrogenase). In contrast RG7787 given at a 5 fold higher dose of 10 mg/kg did not cause any of these symptoms. Only a marginal increase in sorbitol dehydrogenase, a marker of acute hepatocellular injury (Ozer et al., 2008), was observed. The dose‐normalized AUC values determined from the study animals confirmed comparable PK properties of both agents in rats. Taken together these results strongly suggest that domain II of PE contributes significantly to the off‐target toxicity of PE fusion proteins, at least in rodents.
3.7. In vivo efficacy of RG7787 in patient‐derived lung cancer models as monotherapy and in combination with chemotherapy
Patient‐derived xenograft (PDX) models more closely resemble the parental human tumors with regards to histology, and gene expression and may have better predictive power for translational medicine (Siolas and Hannon, 2013). Based on the results of prescreening 24 PDX lung cancer models from EPO (Berlin‐Buch, Germany) for mesothelin expression by Western blot and immunohistochemistry (Supplement Figures S2 and S3), we chose two mesothelin‐positive PDX lung cancer models to evaluate the antitumor effects of RG7787 as monotherapy and in combination with standard of care chemotherapies. For both models all tumor cells showed moderate to strong apically confined membrane mesothelin staining, but they differed in the intensity of cytoplasmic staining, which was weak for Lu7187, but high for Lu7336. Despite similar surface mesothelin levels the two models showed different sensitivity to monotherapy with RG7787 (Figure 6).
Figure 6.
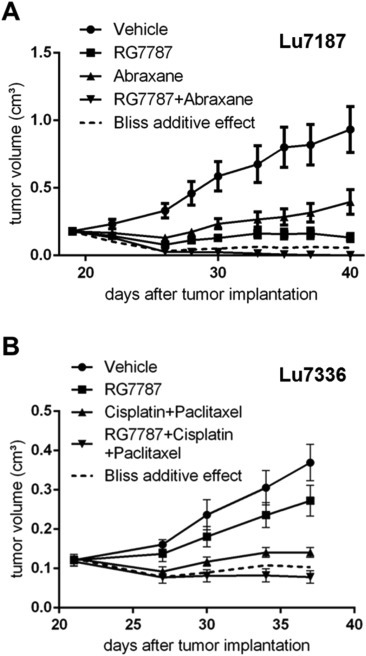
Efficacy of RG7787 in PDX lung cancer xenografts (A) In the KRASmut lung cancer model Lu7187 monotherapy with RG7787 was more efficacious than Abraxane® monotherapy and 2 cycles of combination therapy with both agents achieved near complete remissions. Animals were treated with either vehicle (●) or 2 mg/kg RG7787 (■, i.v. 2 cycles q2dx3, 2 days off then a final 7th treatment) or with 12.5 mg/kg Abraxane® (▴, i.v. q1dx5) or with a combination of both agents (▾). (B) In the KRASwt lung cancer model Lu7336 RG7787 had modest antitumor activity as monotherapy, but in combination with the chemotherapy doublet cisplatin plus paclitaxel also achieved tumor regressions. Animals were treated with either vehicle (●) or 2 mg/kg RG7787 (■, i.v. 2 cycles of q2dx3, 2 days off then a final 7th treatment) or with 5 mg/kg cisplatin (i.p. q7dx2) plus 12.5 mg/kg paclitaxel (i.v. q7dx2) (▴) or with the triple combination (▾). (A, B) Animals were randomized into treatment groups (n = 10) and therapy was started, when tumor volumes had reached a size of ∼100 mm3. Median tumor volumes are plotted with standard deviations as error bars. The dashed line indicates the calculated tumor growth assuming bliss additivity of the respective combination regimen.
In the Lu7187 model, RG7787 monotherapy (2 mg/kg i.v. q2dx3) was more efficacious than Abraxane® (12.5 mg/kg i.v. daily ×3) resulting in 80% and 64% TGI, respectively. The chosen Abraxane® dose regimen was borderline tolerable (7% body weight loss, 1 animal dead). Combining Abraxane® with RG7787 increased toxicity further (10% maximum body weight loss, 3 animals dead), but achieved almost complete tumor eradication in the surviving 7 animals. The antitumor effect of the combination therapy exceeded the calculated bliss additivity curve (dashed line in Figure 6A). In a repeat study using the same dose of Abraxane® but a reduced q3dx5 regimen, Abraxane® monotherapy as well as its combination with RG7787 were well tolerated without visible signs of toxicity and ≤3% body weight loss. Between day 17, when treatment was started, and day 34, when the study was terminated, the relative median tumor volumes increased 5.1 fold for the vehicle group, 2 fold for the RG7787 group, 4.6 fold for the Abraxane group and not at all for the combination group confirming the beneficial effect of the combination treatment.
In the Lu7336 model, treatment with 5 mg/kg cisplatin (i.p. q7d x2) plus 12.5 mg/kg paclitaxel (i.v. q7d x2) was well tolerated (maximum body weight loss 4%) and resulted in 62% TGI, whereas RG7787 monotherapy achieved only 26% TGI. However, while the chemotherapy doublet alone only slowed tumor growth (the mean tumor volume increased under treatment from 125 mm3 ± 25 to 141 mm3 ± 40), the triple combination with RG7787 was highly efficacious and actually shrunk the tumors (from a mean volume of 118 mm3 ± 37 to 78 mm3 ± 50). In addition the triple combination was reasonably well tolerated (maximum body weight loss 8%, no deaths).
4. Discussion
The re‐engineered immunotoxin RG7787 has potential to overcome the two main hurdles for broad clinical use of immunotoxins in solid tumor therapy: immunogenicity and off‐target toxicity of PE. Liu et al. have previously described a CD22‐targeted immunotoxin, HA22‐LR‐LO10, with B‐cell epitopes silenced and domain II deleted from the PE moiety (Liu et al., 2012). The PE24 variant used in RG7787 differs from the LR‐LO10 variant by using an R456A instead of an R458A exchange to silence the human epitope H3, since this resulted in better in vitro potency on some lung cancer cell lines. The concept underlying de‐immunization by removal of B‐cell epitopes assumes that there are only a limited number of antigenic hot spots on the surface of the toxin that can lead to production of mature high affinity antibodies from the human germ line repertoire (Nagata and Pastan, 2009). This concept has been proven in mice, since a PE variant with mutations that remove all known murine B‐cell epitopes was less immunogenic in three different mouse strains (Onda et al., 2008). While the affinities of neutralizing anti‐drug antibodies can vary >10 000 fold, antibodies with high affinities predominate in neutralization of pharmacological activity and drug elimination by formation of drug–antibody complexes (Marzocchi‐Machado et al., 1999). Based on the substantially reduced reactivity of RG7787 with sera from many immunotoxin‐treated patients and by analogy to the proof‐of‐concept in mouse, RG7787 can be expected to be less immunogenic in patients. However, while the data presented here show reduced antigenicity of RG7787, successful de‐immunization of the PE moiety by human B‐cell epitope removal needs to be ultimately proven by monitoring ADA as well as free drug titers in a clinical phase I trial. Alternative strategies for de‐immunization of PE24 by removal of T‐cell epitopes or different combinations of B‐ and T‐cell epitopes are currently also being pursued preclinically (Mazor et al., 2015 Dec, 2016 May 4). Although the PE portion dominates the antibody response in clinical trials with immunotoxins, we humanized the Fab fragment in RG7787, because for bouganin, a plant toxin used in RITs, an ADA response against a non‐human germline sequence of the targeting moiety emerged once the toxin itself was successfully de‐immunized (Entwistle et al., 2013).
A PE24 variant lacking domain II has previously been found to cause less toxicity in rodents than wild type PE38 fusion proteins (Weldon et al., 2013). However, whether removing domain II per se reduced off‐target toxicity or whether increased renal clearance largely accounted for the observed improved tolerability remained unclear in that previous study. Answering this question by assessing the toxicity profile of RG7787 and SS1P that have comparable PK properties in animals was important, since the relatively short plasma half‐life of immunotoxins limits drug uptake into the tumor and improving tolerability at the expense of further reducing tumor uptake would have been undesirable. In rodents RG7787 was significantly better tolerated than SS1P at comparable dose‐normalized systemic exposure suggesting that indeed domain II of PE contributes significantly to off‐target adverse effects. On target toxicity of RG7787 and SS1P can only be studied in cynomolgus monkeys, since the SS1 Fab is not rodent cross‐reactive and only binds to cynomolgus and human mesothelin with similar affinity. In a 5‐day repeat dose toxicity study of RG7787 in cynomolgus monkey intravenous doses of 0.1 and 0.3 mg/kg/day showed only minimal histopathological findings (e.g., hypertrophy of mesothelium in lung and spleen) considered as evidence of mesothelin‐related pharmacology (unpublished data). Since the maximally tolerated dose of SS1P as bolus infusion in humans is 45 μg/kg (Hassan et al., 2007), one can reasonably expect that RG7787 can potentially also be dosed higher than SS1P in humans, although this remains to be proven in a phase I clinical trial. In general, clinical experience with on‐target toxicity of SS1P suggests that it does not represent a major hurdle for clinical development: While pleuritis was the dose‐limiting toxicity in a phase I study with bolus i.v. injection of SS1P it appeared to be self‐limited, since pain and hypoxia resolved within a few days and pleural effusion also improved over time without drainage (Hassan et al., 2007). Moreover, pleuritis was successfully prevented in 3 patients that received concurrent anti‐inflammatory treatment with prednisone (Hassan et al., 2007). No significant pericardial toxicity was observed, despite the fact that normal pericardial cells also express mesothelin suggesting that pericardial cells might not take up enough SS1P to be affected by it. Whether the better tolerability of RG7787 in different animal species shown in this study ultimately translates into a higher maximally tolerated dose than for SS1P in the clinic is being determined in a phase I trial.
In rodents the dose‐limiting off‐target toxicities of SS1P are VLS, weight loss, and hepatotoxicity (Onda et al., 2000) while in humans VLS is critical. The etiology of these adverse effects is incompletely understood, but they require the catalytic activity of PE and are associated with an inflammatory component (Siegall et al., 1997). A surface exposed three amino acid motif (x)D(y) present in PE and other VLS causing agents, like IL2 and ricin, has been suggested to cause binding to and damaging of endothelial cells (Baluna et al., 1999; Wang et al., 2007). PE domain II contains one such exposed GDL motif at position 348–350, but its relevance for VLS is hitherto unproven. RG7787 retains two other such motifs within domain III (GDV 430–432 and GDL 605–607). Our results suggest that the two surface exposed motifs within domain III do not contribute significantly to VLS. This is consistent with the finding that a PE fragment comprising only domain III (residues 400–613) did not induce VLS in rats when given i.v. as single bolus of 12 mg/kg (Siegall et al., 1997). Binding of domain II to yet unidentified immune modulatory cells might contribute substantially to off‐target toxicity of immunotoxins. The catalytic activity of PE in these cells then probably leads to local release of pro‐inflammatory cytokines, like TNFα, that can ultimately cause killing of endothelial cells and hepatocytes by natural killer cells and T‐lymphocytes (Schumann et al., 1998; Muhlen et al., 2004). Alveolar macrophages and Kupffer cells are potential candidates for domain II mediated effects (Schumann et al., 2000). While eliminating domain II greatly improved the tolerability of PE, it had no or little negative impact on the cytotoxic potency of RG7787 compared to SS1P for most cancer cell lines and in case of primary mesothelioma cells and some established cancer cell lines even improved potency. Potency differences between RG7787 and SS1P are likely related to differences in intracellular trafficking and processing that can vary between different cell types. We did not see any major differences in the enzymatic activities of RG7787 and SS1P (data not shown), but several molecular differences between the two molecules can form the mechanistic basis for differences in cytosolic escape efficiency. Not only is the furin cleavage site presented very differently in RG7787, but also an intramolecular disulfide bond within domain II of SS1P that is absent from RG7787 requires in addition to furin cleavage a reducing intracellular milieu for separating the toxin from its targeting moiety. Moreover, removing domain II eliminates the majority of cleavage sites for lysosomal proteases from the PE moiety (Weldon et al., 2009). On NCI‐H596 cells SS1P was more potent than RG7787 and extensive PK/PD modeling showed that this translated into a similar in vivo efficacy difference suggesting that other properties of SS1P and RG7787, like e.g., tumor penetration, are comparable. RG7787 has a greater therapeutic window than SS1P and achieves better antitumor efficacy at doses ≥2 mg/kg. Based on our PK/PD model we found that the effect of RG7787 on tumor growth lasted much longer than the drug could be measured in plasma. We described this behavior using an indirect response model. The calculated half‐life of this long lasting antitumor effect is 16 days, which explains why, despite a short circulation half‐life of RG7787, even very large tumors regressed with three consecutive therapy cycles spaced by one week drug holiday in between. Dose response curves obtained in NCI‐H596 and other xenograft models showed that the maximally efficacious dose of RG7787 in mice was reached between 2 and 3 mg/kg. Based on PK/PD modeling, ∼7 μg/ml RG7787 plasma concentration was necessary to trigger the onset of a long lasting antitumor effect and a 2 mg/kg dose achieved exposure above this value for ∼1.5 h. Sufficient receptor mediated uptake of RG7787 by cells can occur within 1 h as suggested by the fact that in vitro prolonging incubation with RG7787 from 1 h to 24 h only slightly improved the IC50 value (Table S1). This might be why in mice a maximally efficacious dose is reached between 2 and 3 mg/kg. However, tumor penetration rather than cellular uptake of RG7787 can be a main limiting factor in vivo as previously shown in KLM‐1 pancreatic cancer xenografts (Mason‐Osann et al., 2015). At a dose of 2.5 mg/kg only a fraction of the KLM‐1 tumor cells was loaded with a lethal amount of RG7787 at the achievable peak level. Poor tolerability prohibits increasing peak levels by dosing RG7787 higher: dependent on the mouse strain doses ≥5 mg/kg can be given once weekly, but cause intolerable body weight loss when given q2dx3. Also according to our PK/PD model there is a limit to enhancing the antitumor effect in NCI‐H596 xenografts by dosing higher, because exposures achieved with doses between 2 and 3 mg/kg already trigger a maximal long lasting effect. Tumor uptake and antitumor efficacy might be further enhanced without causing intolerability by prolonging the circulation half‐life of RG7787 in mice by continuous infusion (Benhar et al., 1995). Alternatively, instead of enhancing tumor uptake the threshold level for tumor elimination by RG7787 could be lowered by combination treatment with chemotherapeutics that sensitize tumor cells to protein synthesis inhibition (Mason‐Osann et al., 2015). In humans SS1P had an unexpectedly long serum half‐life of ∼8 h (Hassan et al., 2007). The reason for this is unclear hence one cannot predict whether the same will be the case for RG7787 or not. Either way, better tolerability should allow dosing RG7787 in the clinic higher than the maximally tolerated dose of SS1P (45 μg/kg).
For further efficacy testing of RG7787 in lung tumors, we focused on patient‐derived models because they retain histologic and genetic characteristics of the respective donor tumor and are assumed to have more translational relevance for the clinic. Apical localization of mesothelin is a feature that is frequently observed in human lung cancers (Kushitani et al., 2007; Thomas et al., 2015) and both PDX models (Lu7187, Lu7336) tested showed predominantly apical surface mesothelin staining (see Supplement Figure S3). The fact that RG7787 monotherapy was highly efficacious in the Lu7187 model indicates that apical confinement of mesothelin positivity does not limit its uptake into tumor cells. By amplicon panel sequencing covering 212 target regions in 48 cancer‐related genes Lu7187 tumors have been found to carry mutations in the genes KRAS, STK11, TP53. Lung tumors with activating KRAS mutations and LKB1 (STK11) deficiency are known to depend on YAP‐mediated induction of the anti‐apoptotic onco‐protein surviving (Zhang et al., 2015). The survivin protein is rapidly degraded by the ubiquitin‐proteasome pathway and therefore has only a very short protein half‐life of 30 min (Zhao et al., 2000). KRAS mutant cancer cells are also highly sensitive to apoptosis induction by disturbance of Bcl‐2 homeostasis (Peng et al., 2015). Thus the KRAS mutation might make lung tumors very sensitive to apoptosis induction by RG7787 mediated inhibition of protein synthesis. Lu7336 tumors were much less sensitive to RG7787 monotherapy despite similar mesothelin membrane staining as in Lu7187 tumors. Since Lu7336 tumor cells also showed strong cytoplasmic staining for mesothelin, differences in internalization kinetics, intracellular trafficking, and/or processing of RG7787 could affect its potency. Alternatively, the different mutational status of Lu7336 tumor cells might make them less susceptible to apoptosis induction by RG7787. Interestingly, recent characterization of Lu7336 tumor samples with a next generation sequencing panel did not confirm the presence of a previously reported activating G12D KRAS mutation (Hoffmann, 2014), but instead only found mutations in KDR and TP53 (personal communication, Hoffmann and http://www.epo‐berlin.com/epo‐tumor‐models‐xenografts.html).
Current standard of care first line treatments for NSCLC without a targeted therapy option are platinum‐based doublet chemotherapy (e.g., carboplatin plus Abraxane®) for patients with favorable performance status and mono‐chemotherapy (taxanes, gemcitabine, or vinorelbine) for frail patients. In second line docetaxel is approved for all NSCLC histological subtypes. Immunotoxins are attractive combination agents for taxane therapy, because the two agents have been shown to synergize at several levels: i) by increased tumor uptake of the immunotoxin (Zhang et al., 2010), ii) by reduced shedding of mesothelin (Zhang et al., 2007), or iii) by synergistic cell death induction (Hollevoet et al., 2014).
In conclusion the reduced antigenicity and improved tolerability of RG7787 together with high in‐vitro potency on different lung cancer cell lines and achievement of tumor remissions in two PDX lung cancer models by combining RG7787 with a taxane‐based chemotherapy regimen suggests that this re‐engineered RIT offers a promising new therapeutic option for NSCLC patients, particularly those with activating KRAS mutations.
Financial information
This research was supported by the Center for Cancer Research of the National Cancer Institute and by a Cooperative Research and Development Agreement with Roche Pharmaceuticals for the development of Pseudomonas exotoxin‐derived entities in targeted cancer therapy.
Conflict of interest declaration
The authors disclose the following potential conflicts of interest:
IP, MO, UB, WS, GN and SIJ are inventors on immunotoxin patents; all rights have been assigned to NIH. Except for MO and IP, who are employees of the NIH, all other authors are currently employed by Roche.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Acknowledgements
The authors thank P. Falkner and D. Geiss for providing excellent technical assistance during the animal studies, Gregor Jordan, Uwe Dahl and Miriam Moheysen‐Zadeh for dedicated bioanalytical support and H. Herrmuth and J. Brandl for great technical assistance for in vitro work and tumor sample analyses. Dr. N. Rieder is thanked for her great help with immunohistochemical evaluations and Dr. G. Georges and Dr. S. Dengl for their computer modeling support. The authors thank also L. Kifle from Horizon Discovery for excellent project management.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2016.07.003.
Bauss Frieder, Lechmann Martin, Krippendorff Ben-Fillippo, Staack Roland, Herting Frank, Festag Matthias, Imhof-Jung Sabine, Hesse Friederike, Pompiati Marc, Kollmorgen Gwendlyn, da Silva Mateus Seidl Rita, Bossenmaier Birgit, Lau Wilma, Schantz Christian, Stracke Jan O., Brinkmann Ulrich, Onda Masanori, Pastan Ira, Bosslet Klaus, Niederfellner Gerhard, (2016), Characterization of a re-engineered, mesothelin-targeted Pseudomonas exotoxin fusion protein for lung cancer therapy, Molecular Oncology, 10, doi: 10.1016/j.molonc.2016.07.003.
References
- Alewine, C. , Xiang, L. , Yamori, T. , Niederfellner, G. , Bosslet, K. , Pastan, I. , 2014 Nov. Efficacy of RG7787, a next-generation mesothelin-targeted immunotoxin, against triple-negative breast and gastric cancers. Mol. Cancer Ther. 13, (11) 2653–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alewine, C. , Hassan, R. , Pastan, I. , 2015 Feb. Advances in anticancer immunotoxin therapy. Oncologist 20, (2) 176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argani, P. , Iacobuzio-Donahue, C. , Ryu, B. , Rosty, C. , Goggins, M. , Wilentz, R.E. , 2001 Dec. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin. Cancer Res. 7, (12) 3862–3868. [PubMed] [Google Scholar]
- Baluna, R. , Rizo, J. , Gordon, B.E. , Ghetie, V. , Vitetta, E.S. , 1999 Mar 30. Evidence for a structural motif in toxins and interleukin-2 that may be responsible for binding to endothelial cells and initiating vascular leak syndrome. Proc. Natl. Acad. Sci. USA 96, (7) 3957–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina, J. , Caponigro, G. , Stransky, N. , Venkatesan, K. , Margolin, A.A. , Kim, S. , 2012 Mar 29. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, (7391) 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhar, I. , Reiter, Y. , Pai, L.H. , Pastan, I. , 1995 Jul 28. Administration of disulfide-stabilized Fv-immunotoxins B1(dsFv)-PE38 and B3(dsFv)-PE38 by continuous infusion increases their efficacy in curing large tumor xenografts in nude mice. Int. J. Cancer 62, (3) 351–355. [DOI] [PubMed] [Google Scholar]
- Buchner, J. , Pastan, I. , Brinkmann, U. , 1992 Sep. A method for increasing the yield of properly folded recombinant fusion proteins: single-chain immunotoxins from renaturation of bacterial inclusion bodies. Anal Biochem. 205, (2) 263–270. [DOI] [PubMed] [Google Scholar]
- Bujotzek, A. , Fuchs, A. , Qu, C. , Benz, J. , Klostermann, S. , Antes, I. , George, G. , 2015. MoFvAb: modeling the Fv region of antibodies. MAbs 7, (5) 838–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entwistle, J. , Kowalski, M. , Brown, J. , Cizeau, J. , MacDonald, G.C. , 2013. The Preclinical and Clinical Evaluation of VB6-845: an Immunotoxin with a De-immunized Payload for the Systemic Treatment of Solid Tumors. Antibody-drug Conjugates and Immunotoxins Springer; 349–367. [Google Scholar]
- Fichtner, I. , Rolff, J. , Soong, R. , Hoffmann, J. , Hammer, S. , Sommer, A. , Becker, M. , Merk, J. , 2008 Oct 15. Establishment of patient-derived non-small cell lung cancer xenografts as models for the identification of predictive biomarkers. Clin. Cancer Res. 14, (20) 6456–6468. [DOI] [PubMed] [Google Scholar]
- Fieller, E.C. , 1954. Some problems in interval estimation. J. R. Stat. Soc. Ser. B 16, (2) 175–185. [Google Scholar]
- Hassan, R. , Bullock, S. , Premkumar, A. , Kreitman, R.J. , Kindler, H. , Willingham, M.C. , 2007 Sep 1. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin. Cancer Res. 13, (17) 5144–5149. [DOI] [PubMed] [Google Scholar]
- Hoffmann, J. , 2014 Jun. Integrative oncology drug discovery accompanied by preclinical translational research as prerequisite for clinical development. Chin Clin. Oncol. 3, (2) 15 [DOI] [PubMed] [Google Scholar]
- Hollevoet, K. , Mason-Osann, E. , Liu, X.F. , Imhof-Jung, S. , Niederfellner, G. , Pastan, I. , 2014 Aug. In vitro and in vivo activity of the low-immunogenic antimesothelin immunotoxin RG7787 in pancreatic cancer. Mol. Cancer Ther. 13, (8) 2040–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hothorn, L.A. , 2006. Multiple comparisons and multiple contrasts in randomized dose-response trials - confidence interval oriented approaches. J. Biopharm. Stat. 16, (5) 711–731. [DOI] [PubMed] [Google Scholar]
- Kelly, R.J. , Sharon, E. , Pastan, I. , Hassan, R. , 2012 Mar. Mesothelin-targeted agents in clinical trials and in preclinical development. Mol. Cancer Ther. 11, (3) 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitman, R.J. , Hassan, R. , Fitzgerald, D.J. , Pastan, I. , 2009 Aug 15. Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin. Cancer Res. 15, (16) 5274–5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kris, M.G. , Johnson, B.E. , Berry, L.D. , Kwiatkowski, D.J. , Iafrate, A.J. , Wistuba, I.I. , 2014 May 21. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 311, (19) 1998–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushitani, K. , Takeshima, Y. , Amatya, V.J. , Furonaka, O. , Sakatani, A. , Inai, K. , 2007 Apr. Immunohistochemical marker panels for distinguishing between epithelioid mesothelioma and lung adenocarcinoma. Pathol. Int. 57, (4) 190–199. [DOI] [PubMed] [Google Scholar]
- Liu, W. , Onda, M. , Lee, B. , Kreitman, R.J. , Hassan, R. , Xiang, L. , 2012 Jul 17. Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human B-cell epitopes. Proc. Natl. Acad. Sci. USA 109, (29) 11782–11787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzocchi-Machado, C.M. , Polizello, A.C. , Azzolini, A.E. , Lucisano-Valim, Y.M. , 1999 Mar. The influence of antibody functional affinity on the effector functions involved in the clearance of circulating immune complexes anti-BSA IgG/BSA. Immunol. Invest 28, (2–3) 89–101. [DOI] [PubMed] [Google Scholar]
- Mason-Osann, E. , Hollevoet, K. , Niederfellner, G. , Pastan, I. , 2015. Quantification of recombinant immunotoxin delivery to solid tumors allows for direct comparison of in vivo and in vitro results. Sci. Rep. 5, 10832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazor, R. , Zhang, J. , Xiang, L. , Addissie, S. , Awuah, P. , Beers, R. , Hassan, R. , Pastan, I. , 2015 Dec. Recombinant immunotoxin with T-cell epitope mutations that greatly reduce immunogenicity for treatment of mesothelin-expressing tumors. Mol. Cancer Ther. 14, (12) 2789–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazor, R. , Onda, M. , Park, D. , Addissie, S. , Xiang, L. , Zhang, J. , Hassan, R. , Pastan, I. , 2016 May 4. Dual B- and T-cell de-immunization of recombinant immunotoxin targeting mesothelin with high cytotoxic activity. Oncotarget 7, (21) 29916–29926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlen, K.A. , Schumann, J. , Wittke, F. , Stenger, S. , Van, R.N. , Van, K.L. , 2004 Mar 1. NK cells, but not NKT cells, are involved in Pseudomonas aeruginosa exotoxin A-induced hepatotoxicity in mice. J. Immunol. 172, (5) 3034–3041. [DOI] [PubMed] [Google Scholar]
- Nagata, S. , Pastan, I. , 2009 Sep 30. Removal of B cell epitopes as a practical approach for reducing the immunogenicity of foreign protein-based therapeutics. Adv. Drug Deliv. Rev. 61, (11) 977–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onda, M. , Beers, R. , Xiang, L. , Nagata, S. , Wang, Q.C. , Pastan, I. , 2008 Aug 12. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc. Natl. Acad. Sci. USA 105, (32) 11311–11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onda, M. , Willingham, M. , Wang, Q.C. , Kreitman, R.J. , Tsutsumi, Y. , Nagata, S. , 2000 Dec 15. Inhibition of TNF-alpha produced by Kupffer cells protects against the nonspecific liver toxicity of immunotoxin anti-Tac(Fv)-PE38, LMB-2. J. Immunol. 165, (12) 7150–7156. [DOI] [PubMed] [Google Scholar]
- Ordonez, N.G. , 2003 Mar. Value of mesothelin immunostaining in the diagnosis of mesothelioma. Mod. Pathol. 16, (3) 192–197. [DOI] [PubMed] [Google Scholar]
- Ozer, J. , Ratner, M. , Shaw, M. , Bailey, W. , Schomaker, S. , 2008 Mar 20. The current state of serum biomarkers of hepatotoxicity. Toxicology 245, (3) 194–205. [DOI] [PubMed] [Google Scholar]
- Pastan, I. , Hassan, R. , 2014 Jun 1. Discovery of mesothelin and exploiting it as a target for immunotherapy. Cancer Res. 74, (11) 2907–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, B. , Ganapathy, S. , Shen, L. , Huang, J. , Yi, B. , Zhou, X. , 2015 Sep 8. Targeting Bcl-2 stability to sensitize cells harboring oncogenic ras. Oncotarget 6, (26) 22328–22337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann, J. , Wolf, D. , Pahl, A. , Brune, K. , Papadopoulos, T. , Van, R.N. , 2000 Nov. Importance of Kupffer cells for T-cell-dependent liver injury in mice. Am. J. Pathol. 157, (5) 1671–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann, J. , Angermuller, S. , Bang, R. , Lohoff, M. , Tiegs, G. , 1998 Nov 15. Acute hepatotoxicity of Pseudomonas aeruginosa exotoxin A in mice depends on T cells and TNF. J. Immunol. 161, (10) 5745–5754. [PubMed] [Google Scholar]
- Siegall, C.B. , Liggitt, D. , Chace, D. , Mixan, B. , Sugai, J. , Davidson, T. , 1997 Mar. Characterization of vascular leak syndrome induced by the toxin component of Pseudomonas exotoxin-based immunotoxins and its potential inhibition with nonsteroidal anti-inflammatory drugs. Clin. Cancer Res. 3, (3) 339–345. [PubMed] [Google Scholar]
- Siegall, C.B. , Liggitt, D. , Chace, D. , Tepper, M.A. , Fell, H.P. , 1994 Sep 27. Prevention of immunotoxin-mediated vascular leak syndrome in rats with retention of antitumor activity. Proc. Natl. Acad. Sci. USA 91, (20) 9514–9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siolas, D. , Hannon, G.J. , 2013 Sep 1. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res. 73, (17) 5315–5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, A. , Chen, Y. , Steinberg, S.M. , Luo, J. , Pack, S. , Raffeld, M. , 2015 May 10. High mesothelin expression in advanced lung adenocarcinoma is associated with KRAS mutations and a poor prognosis. Oncotarget 6, (13) 11694–11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torre, L.A. , Bray, F. , Siegel, R.L. , Ferlay, J. , Lortet-Tieulent, J. , Jemal, A. , 2015 Mar. Global cancer statistics, 2012. CA Cancer J. Clin. 65, (2) 87–108. [DOI] [PubMed] [Google Scholar]
- Wang, H. , Song, S. , Kou, G. , Li, B. , Zhang, D. , Hou, S. , 2007 Nov. Treatment of hepatocellular carcinoma in a mouse xenograft model with an immunotoxin which is engineered to eliminate vascular leak syndrome. Cancer Immunol. Immunother. 56, (11) 1775–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidle, U.H. , Tiefenthaler, G. , Schiller, C. , Weiss, E.H. , Georges, G. , Brinkmann, U. , 2014 Jan. Prospects of bacterial and plant protein-based immunotoxins for treatment of cancer. Cancer Genomics Proteomics 11, (1) 25–38. [PubMed] [Google Scholar]
- Weldon, J.E. , Xiang, L. , Chertov, O. , Margulies, I. , Kreitman, R.J. , FitzGerald, D.J. , 2009 Apr 16. A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood 113, (16) 3792–3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weldon, J.E. , Xiang, L. , Zhang, J. , Beers, R. , Walker, D.A. , Onda, M. , 2013 Jan. A recombinant immunotoxin against the tumor-associated antigen mesothelin reengineered for high activity, low off-target toxicity, and reduced antigenicity. Mol. Cancer Ther. 12, (1) 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W. , Gao, Y. , Li, F. , Tong, X. , Ren, Y. , Han, X. , 2015 Nov 1. YAP Promotes malignant progression of Lkb1-deficient lung adenocarcinoma through downstream regulation of survivin. Cancer Res. 75, (21) 4450–4457. [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Hansen, J.K. , Xiang, L. , Kawa, S. , Onda, M. , Ho, M. , 2010 Feb 1. A flow cytometry method to quantitate internalized immunotoxins shows that taxol synergistically increases cellular immunotoxins uptake. Cancer Res. 70, (3) 1082–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Xiang, L. , Hassan, R. , Pastan, I. , 2007 Oct 23. Immunotoxin and taxol synergy results from a decrease in shed mesothelin levels in the extracellular space of tumors. Proc. Natl. Acad. Sci. USA 104, (43) 17099–17104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, J. , Tenev, T. , Martins, L.M. , Downward, J. , Lemoine, N.R. , 2000 Dec. The ubiquitin-proteasome pathway regulates survivin degradation in a cell cycle-dependent manner. J. Cell Sci. 113, (Pt 23) 4363–4371. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data