

Abstract
Aims: Macropinocytosis has been implicated in cardiovascular and other disorders, yet physiological factors that initiate fluid-phase internalization and the signaling mechanisms involved remain poorly identified. The present study was designed to examine whether matrix protein thrombospondin-1 (TSP1) stimulates macrophage macropinocytosis and, if so, to investigate the potential signaling mechanism involved.
Results: TSP1 treatment of human and murine macrophages stimulated membrane ruffle formation and pericellular solute internalization by macropinocytosis. Blockade of TSP1 cognate receptor CD47 and NADPH oxidase 1 (Nox1) signaling, inhibition of phosphoinositide 3-kinase, and transcriptional knockdown of myotubularin-related protein 6 abolished TSP1-induced macropinocytosis. Our results demonstrate that Nox1 signaling leads to dephosphorylation of actin-binding protein cofilin at Ser-3, actin remodeling, and macropinocytotic uptake of unmodified native low-density lipoprotein (nLDL), leading to foam cell formation. Finally, peritoneal chimera studies suggest the role of CD47 in macrophage lipid macropinocytosis in hypercholesterolemic ApoE−/− mice in vivo.
Innovation: Activation of a previously unidentified TSP1-CD47 signaling pathway in macrophages stimulates direct receptor-independent internalization of nLDL, leading to significant lipid accumulation and foam cell formation. These findings reveal a new paradigm in which delimited Nox1-mediated redox signaling, independent of classical lipid oxidation, contributes to early propagation of vascular inflammatory disease.
Conclusions: The findings of the present study demonstrate a new mechanism of solute uptake with implications for a wide array of cell types, including macrophages, dendritic cells, and cancer cells, and multiple pathological conditions in which matrix proteins are upregulated. Antioxid. Redox Signal. 26, 886–901.
Keywords: : macropinocytosis, NADPH oxidase, thrombospondin 1, CD47, macrophages
Introduction
Fluid-phase macropinocytosis is a highly conserved endocytic process by which extracellular fluid and pericellular solutes are internalized into cells via large heterogeneous vesicles known as macropinosomes. Macropinocytosis differs from other endocytic processes, such as phagocytosis or clathrin- and caveolin-mediated endocytosis, in that it is initiated by formation of F-actin-rich sheet-like membrane projections, also known as membrane ruffles (9). These dynamic membrane projections transition to curved ruffles, form macropinocytotic cups, and internalize as macropinosomes. Macrophages and dendritic cells undergo constitutive membrane ruffling and utilize macropinocytosis to sample their environment for antigens. The physiological ligands that stimulate macropinocytosis are limited to growth factors and cytokines (4, 9), and signaling pathways responsible for the initiation of membrane ruffle formation remain poorly understood. In addition, fundamental questions persist regarding the role of macropinocytosis in pathophysiological conditions, including cardiovascular diseases (CVD), cancer, and immune system disorders.
Innovation.
The findings of the current study demonstrate a new mechanism by which macrophage NADPH oxidase 1 (Nox1) signaling is stimulated by extracellular matrix protein thrombospondin-1 (TSP1), leading to alteration in actin-binding protein cofilin, membrane ruffle formation, and fluid-phase macropinocytosis. Activation of this novel signaling pathway stimulates direct receptor-independent internalization of native low-density lipoprotein (nLDL), lipid accumulation, and foam cell formation. These findings support a new paradigm, in which Nox1-mediated redox signaling independent of broader lipid oxidation propagates vascular inflammatory disease.
NADPH oxidase (Nox) is a major source of reactive oxygen species (ROS) in the brain, immune system, vasculature, and digestive tract (11, 14). The Nox family consists of seven members, namely Nox1, Nox2, Nox3, Nox4, Nox5, dual oxidase (DUOX) 1, and DUOX2. These Nox isoforms differ in tissue distribution, subcellular localization, activation, and pathophysiological function, but retain their ability to transfer electrons from NADPH to oxygen to form either superoxide anion (O2•−) or its dismuted metabolite hydrogen peroxide (H2O2) (8). Strong evidence reveals that excessive Nox-derived ROS production via protein oxidation, lipid peroxidation, and DNA damage contributes to pathological processes. In addition, Nox-derived ROS are involved in the regulation of tightly controlled signaling transduction pathways that are important in cellular physiology and pathophysiology (1, 15).
Atherosclerosis, the buildup of occlusive lipid-rich plaques in arterial walls, is a major trigger for fatal cardiovascular conditions, including heart attack and ischemic stroke (31). Over the past three decades, the oxidative modification of LDL hypothesis has emerged as the major paradigm explaining the initiation of early atherogenic processes (18, 22). This paradigm describes that the apoB-100 of native LDL (nLDL) is oxidized in the subendothelial matrix, in turn rendering the lipoprotein susceptible to uptake by macrophage scavenger receptors and contributing to arterial lipid accumulation (6, 19). Despite the dominance of this theory, not all previously published data are consistent with this paradigm. For instance, accumulation of nonoxidized lipids precedes appearance of oxidized lipoproteins in human atherosclerotic vessels (52). In addition, previous findings suggest that for LDL oxidation to occur, all alpha-tocopherol in the lipoprotein must be depleted (46). Relevant to these findings, lipids isolated from human atherosclerotic vessels from the earliest stages to advanced lesions are not depleted of alpha-tocopherol (52), suggesting an alternative internalization pathway, independent of LDL oxidation during lesion development. Furthermore, previous studies show that ApoE−/− mice lacking scavenger receptors A and CD36, major scavenger receptors responsible for >90% of ox-LDL uptake, are not protected from atherosclerosis (35). Taken together, these findings suggest a yet undefined pathway for direct cellular uptake of nLDL. The precise mechanism, however, for uptake and signaling and the pathophysiological role of nonoxidized lipids in vascular disease are woefully underappreciated.
Matricellular proteins are nonstructural proteins integrated in the extracellular matrix that regulate important cell signaling pathways via binding to specific cell surface receptors and modulate the activity of inflammatory cytokines and growth factors (10). Matricellular protein TSP1 is present at low levels in the wall of healthy blood vessels and is highly upregulated in the vessel wall in CVD, including atherosclerosis (20, 45). However, while increasing evidence supports the role of matricellular proteins in vascular disease (25, 36), their ability to regulate lipid homeostasis in macrophages and their pathological significance in CVD, in general, remain unknown. In this study, we postulated that TSP1 stimulates uptake of nLDL via macropinocytosis, leading to macrophage lipid accumulation: a key early step in the initiation of atherosclerosis.
The data presented herein demonstrate the involvement of a previously unidentified mechanism by which TSP1 via CD47 receptor-stimulated Nox1 signaling dephosphorylates cofilin in macrophages, leading to extensive membrane ruffle formation and fluid-phase macropinocytosis of nLDL. Importantly, activation of this highly regulated and delimited redox signaling pathway involving Nox1 stimulates oxidation-independent internalization of nLDL by macrophages. This, in turn, leads to extensive lipid accumulation and foam cell formation. These findings are expected to provide a shift in our understanding of the etiology of vascular inflammatory disorders and inform new therapeutic strategies targeting arterial lipid accumulation. The current data are also expected to stimulate new inquiries into matrix regulation of macropinocytosis in other cell types, including dendritic cells (30), neutrophils (16), and cancer cells (12). In addition, these findings are the first to describe a signaling role for Nox in macropinocytosis.
Results
TSP1 at physiologically relevant concentrations stimulates macrophage plasma membrane ruffling
Levels of matricellular protein TSP1 are significantly elevated in chronic vascular diseases (36), yet knowledge of its pathological role in the vessel wall remains scant. In this study, we first evaluated whether TSP1 stimulates macrophage membrane ruffling using scanning electron microscopy (SEM). The presence of numerous microvilli was observed on the otherwise smooth surface of vehicle-treated macrophages (Fig. 1A). In contrast, macrophages challenged with TSP1 at concentrations found in the plasma of patients with vascular disease (10 nM, 2 h) (34) exhibited marked cell surface ruffling (Fig. 1A). Emerging membrane extensions, fully formed dorsal membrane ruffles (projections on the apical cell surface), and formation of macropinocytotic cups (∼1 μm in diameter) were visualized following TSP1 treatment (Fig. 1A). Quantification of membrane ruffles revealed ∼10-fold increase in membrane ruffling following TSP1 treatment (0.2 ± 0.1 and 2.2 ± 0.2; *p < 0.05, for vehicle and TSP1 treatment, respectively; Fig. 1B). Next, high-resolution differential interference contrast (DIC) microscopy was employed to elucidate the dynamics of TSP1-induced membrane ruffling. Live cell DIC imaging showed initiation of membrane ruffling (red arrowheads) following 30 min of TSP1 stimulation (Fig. 1C and Supplementary Movie S1; Supplementary Data are available online at www.liebertpub.com/ars).
FIG. 1.
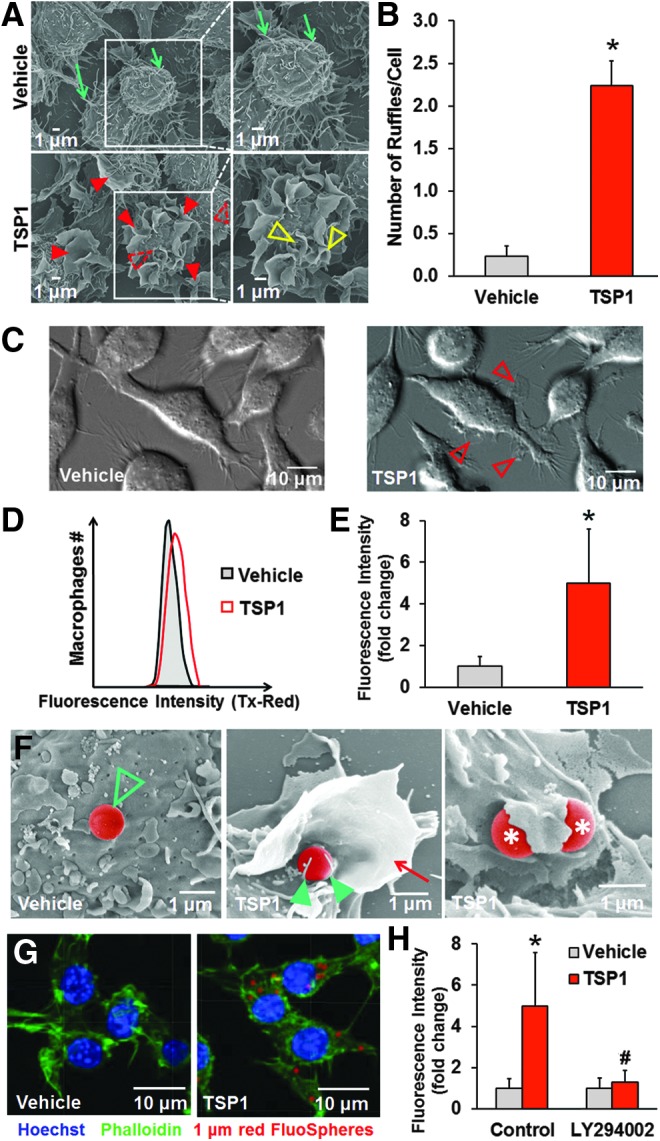
TSP1 stimulates fluid-phase macropinocytosis in macrophages. (A) RAW 264.7 macrophages were treated with vehicle (PBS) or challenged with exogenous TSP1 (10 nM) for 2 h, and then processed for SEM (scale bars, 1 μm). Note the presence of numerous microvilli (green arrows) and absence of membrane ruffles on the surface of vehicle-treated macrophages. Emerging membrane extensions (dotted line red arrowheads) and numerous fully formed ruffles (red arrowheads) can be observed across the dorsal surface of TSP1-treated cells. A higher magnification of the ruffling region shows formation of macropinocytotic cups in TSP1-treated cells that are ∼1 μm in diameter (open yellow arrowheads). Representative images of three independent experiments. (B) Number of membrane ruffles in vehicle- and TSP1-treated macrophages normalized to total cell number in the microscopic field evaluated. *p < 0.05 versus vehicle (n = 3). (C) Macrophages were treated with vehicle or TSP1 (10 nM), and dynamics of membrane ruffle formation visualized using differential interference contrast (DIC) microscopy. DIC images were taken 30 min after vehicle or TSP1 treatment. Open arrowheads indicate membrane ruffle formation on the surface of TSP1-treated cells (scale bars, 10 μm). Images are representative of three independent experiments (see Supplementary Movie S1) (D) Macrophages were incubated with 1 μm red (580/605) fluorescent FluoSpheres and treated with vehicle or TSP1 (10 nM) for 24 h. Cells were fixed in 2% paraformaldehyde (PFA) and subjected for fluorescence-activated cell sorting (FACS) analysis. Macrophages were gated based on forward scatter/side scatter, doublets were excluded, and then Zombie Yellow-negative (live) cells were selected for analysis. Representative FACS histogram plots are shown. (E) FACS results are expressed as fold change relative to vehicle treatment. *p < 0.05 (n = 4). (F) Macrophages were incubated with 1 μm FluoSpheres, treated with vehicle (left) or TSP1 (10 nM, 2 h; middle and right), and then processed for SEM (scale bars, 1 μm). Note the presence of a FluoSphere (pseudocolored) on the smooth cell surface following vehicle treatment (green open arrowhead). Higher magnification reveals a FluoSphere on the dorsal surface of a TSP1-treated cell (middle), stabilized by two microvilli (green arrowheads), and surrounded by a membrane ruffle (red arrow). Partially internalized FluoSpheres following TSP1 treatment are indicated by asterisks (right). (G) RAW 264.7 macrophages were treated as described in (1D) and fixed in 2% PFA. Nuclei were stained by Hoechst (blue) and F-actin was labeled by 488 phalloidin (green); red = fluorescent FluoSpheres (580/605). Images were taken with an Olympus FluoView™ FV1000 confocal microscope. (H) RAW 264.7 macrophages were preincubated with LY294002 (10 μM, 30 min), treated with 1 μm red (580/605) fluorescent FluoSpheres (1:20,000), and incubated with vehicle or TSP1 (10 nM) for 24 h and processed for FACS analysis. *p < 0.05 indicates significant difference between vehicle versus TSP1. #p < 0.05 indicates significant difference between TSP1 versus TSP1 + LY294002 (n = 4). Data represent the mean ± SD. PBS, phosphate-buffered saline; SD, standard deviation; SEM, scanning electron microscopy; TSP1, thrombospondin-1.
TSP1 promotes fluid-phase macropinocytosis in macrophages
Fluid-phase macropinocytosis is a complex process involving membrane ruffling, followed by formation and closure of macropinocytotic cups, scission of macropinosomes, and internalization (9). Thus, we next investigated whether TSP1-induced membrane ruffling is followed by macropinocytotic internalization of extracellular solute. Macrophages were incubated with 1 μm red (580/605) fluorescent FluoSpheres®, treated with vehicle or TSP1 (10 nM, 24 h), and uptake of fluorescent beads was quantified using fluorescence-activated cell sorting (FACS). As shown in Figure 1D, E, TSP1 treatment significantly stimulated uptake of 1 μm FluoSpheres in macrophages (the gating strategy is shown in Supplementary Fig. S1). Additional SEM images show 1 μm FluoSpheres (red pseudocolor) resting on the dorsal surface of vehicle-treated cells (Fig. 1F, left). In contrast, beads stabilized by microvilli and surrounded by a large ∼6 μm diameter membrane ruffle were visualized following TSP1 treatment (Fig. 1F, middle). Partially internalized FluoSpheres have been also observed following TSP1 treatment (Fig. 1F, right). Internalization of FluoSpheres into the cytosol was confirmed using confocal Z-stack imaging (Fig. 1G, Supplementary Movies S2A, B). Previous studies demonstrated that particles smaller than 0.2 μm and 0.5 μm in diameter are internalized by clathrin- and caveolin-mediated endocytosis, respectively (41). As the diameter of macropinosomes extends beyond this range and up to 5 μm (9), internalization of 1 μm FluoSpheres in TSP1-treated cells in and of itself suggests involvement of macropinocytosis. Consistent with this statement, we demonstrated that gene silencing clathrin heavy chain (CLTC) or caveolin (CAV1) does not attenuate TSP1-induced solute internalization by macrophages (Supplementary Fig. S2). As macropinocytosis is often characterized by its sensitivity to phosphatidylinositol-3-kinase (PI3K) inhibitors, we next investigated whether the PI3K inhibitor LY294002 inhibits TSP1-induced internalization of 1 μm FluoSpheres (4). Figure 1H demonstrates that preincubation of macrophages with LY294002 completely abolished TSP1-induced uptake of FluoSpheres. Taken together, these findings suggest a dorsal ruffle-constituted macropinocytotic pathway in the internalization of macromolecules that is activated by matrix protein TSP1.
TSP1 stimulates native LDL uptake into macrophages
The key initiating process in atherogenesis is the macrophage accumulation of apolipoprotein B-containing lipoproteins, such as nLDL, in the subendothelial matrix (31). It is generally accepted that LDL oxidation and its subsequent internalization by macrophage scavenger receptors cause foam cell formation and contribute to atherosclerosis (48). Meanwhile, extracellular matrix proteins are increased in the vessel wall in various vascular disorders (36, 45), yet their pathological significance in atherosclerosis or other CVD remains largely unknown. We postulated that matricellular protein TSP1 stimulates oxidation-independent uptake of nLDL in macrophages via fluid-phase macropinocytosis. To test this hypothesis, human THP-1 macrophages were incubated with nLDL (250 μg/ml), challenged with exogenous human TSP1 (10 nM, 24 h), and lipid accumulation quantified using the fluorescent lipophilic dye Nile Red. In this experiment, we used human macrophages to test the clinical relevance of our findings. It is also important to note that the reconstituted TSP1 solution contains 20 mM Tris-HCl, 600 mM NaCl, 2 mM CaCl2, and 20% sucrose (reconstitution buffer). We tested whether the reconstitution buffer alone without soluble TSP1 stimulates macropinocytosis. Our FACS results indicated that the reconstitution buffer without TSP1 did not stimulate solute uptake compared with untreated cells (data not shown). As shown in Figure 2A, however, TSP1 stimulated Nile Red fluorescence in nLDL-treated human macrophages by ∼4-fold compared with vehicle. TSP1-induced nLDL internalization was confirmed by confocal laser scanning microscopy (Fig. 2B), FACS analysis (Fig. 2C), and quantification of lipid accumulation using the Amplex® Red Cholesterol Quantification Assay (cholesterol [μg/ml/106 cells]: 9.8 ± 3.1 and 34.4 ± 12.1 for vehicle and TSP1 treatment, respectively). As shown in Supplementary Figure S3A, B, TSP1 treatment did not alter low-density lipoprotein receptor (LDLR) levels in macrophages and stimulated lipid accumulation in LDLR−/− macrophages. These data suggest that TSP1-stimulated nLDL internalization is independent of LDLR.
FIG. 2.
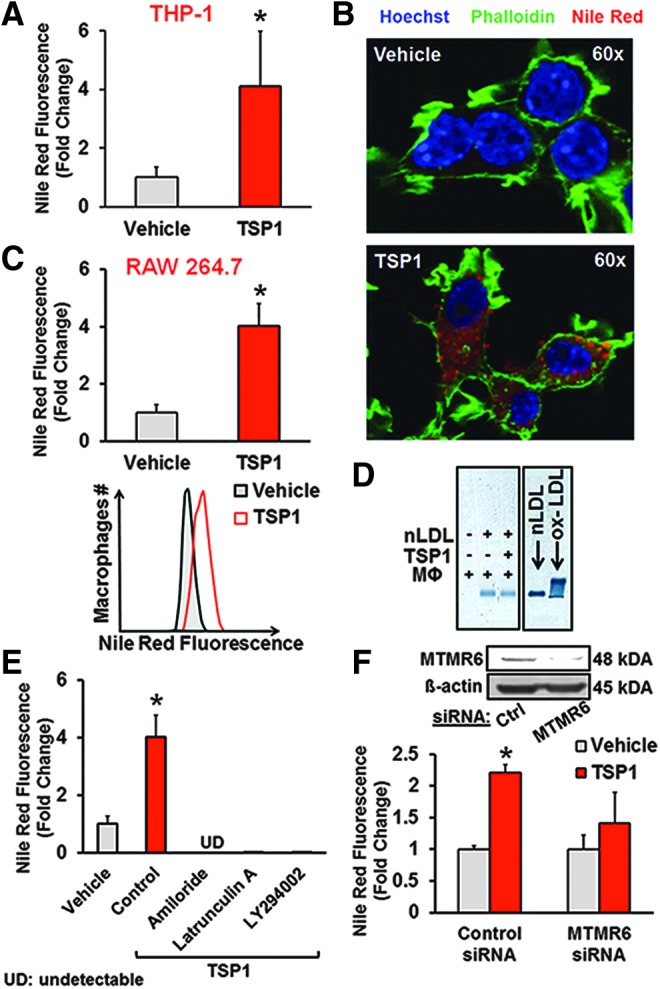
TSP1 promotes macropinocytosis of nLDL in macrophages. (A) Human THP-1-derived macrophages were incubated with nLDL (250 μg/ml), treated with vehicle or TSP1 (10 nM, 24 h), and fixed in 2% PFA. Lipid accumulation was quantified using Nile Red, a fluorescence lipophilic dye, and analyzed via FACS. Bar graphs show the mean percentages of Nile Red-positive cells. *p < 0.05 (n = 4). (B) RAW 264.7 macrophages were treated with nLDL (250 μg/ml) and incubated with vehicle or TSP1 (10 nM) for 24 h. Cells were fixed in 2% PFA, nuclei were stained by Hoechst (blue), F-actin was labeled by 488 phalloidin (green), and lipids were stained by Nile Red (red). Images were taken with an Olympus FluoView FV1000 confocal microscope (magnification: 60 × ). Similar results have been observed in three independent experiments. (C) Following treatment as described in (B), RAW 264.7 cells were processed for FACS analysis. Bar graphs show the mean percentages of Nile Red-positive cells. Representative FACS histogram plots are also shown. *p < 0.05 (n = 12). (D) RAW 264.7 macrophages (MΦ) were incubated in the absence or presence of nLDL (250 μg/ml) and treated with vehicle or TSP1 (10 nM). Twenty-four hours later, aliquots were collected and subjected to agarose gel electrophoresis (HYDRASYS® 2, Sebia). nLDL and ox-LDL (Kalen Biomedicals, Montgomery Village, MD) were used as negative and positive controls, respectively. (E) Macrophages were treated with nLDL (250 μg/ml) and vehicle, nLDL and TSP1, or pretreated with amiloride (300 μM, 30 min), latrunculin A (15 μM, 1 h), or LY294002 (10 μM, 30 min) and then treated with nLDL+TSP1. Cells were processed for FACS analysis. *p < 0.05 versus vehicle (n = 3–4). (F) RAW 264.7 macrophages were transfected with control or MTMR6 siRNA (48 h), treated with nLDL (250 μg/ml), and incubated with vehicle or TSP1 (10 nM) for 24 h. Cells were fixed in 2% PFA, stained with Nile Red, and processed for FACS analysis. *p < 0.05 versus vehicle (n = 3). Data represent the mean ± SD. MTMR6, myotubularin-related protein 6; nLDL, native low-density lipoprotein.
As esterification and hydrolysis of cholesterol are mediated by acyl-CoA:cholesterol O-acyltransferase (ACAT) and cholesteryl ester hydrolase (NCEH-1), respectively, we next tested whether TSP1 treatment alters ACAT and NCEH-1 expression in macrophages. Supplementary Figure S4 demonstrates that TSP1 does not change ACAT and NCEH-1 expression in macrophages. As oxidatively modified LDL binds to scavenger receptors leading to their internalization, we investigated whether TSP1 induces LDL oxidation in the media of cultured macrophages using agarose gel electrophoresis. Macrophages were incubated in the absence or presence of nLDL and treated with vehicle or TSP1 (10 nM) for 24 h. The electrophoretic mobility of LDL showed no difference between vehicle and TSP1 treatment, demonstrating that TSP1 does not induce oxidative modification of nLDL (Fig. 2D). Uptake of either nLDL or ox-LDL will result in increased total intracellular cholesterol. Thus, we compared macrophage cholesterol content following TSP1-induced nLDL uptake versus ox-LDL internalization; the latter known to be mediated by scavenger receptors. Importantly, ox-LDL uptake by macrophages was comparable with TSP1-induced nLDL internalization (cholesterol [μg/ml/106 cells]: 33.0 ± 13.1 and 34.4 ± 12.1 for ox-LDL alone and nLDL + TSP1 treatment, respectively).
Next, we interrogated whether TSP1 stimulates internalization of nLDL via macropinocytosis or other endocytic processes. Macropinosome formation is uniquely susceptible to inhibition by amiloride (26) (an inhibitor of Na+/H+ antiporter) by way of decreasing submembranous pH, which is critical for actin recruitment to the membrane and its polymerization. As shown in Figure 2E, pretreatment of macrophages with amiloride completely abolished TSP1-induced lipid uptake. Moreover, TSP1-induced lipid accumulation was dramatically attenuated by pretreatment with latrunculin A, an inhibitor of actin polymerization, and the PI3K inhibitor, LY294002, all consistent with macropinocytosis (Fig. 2E). In contrast, chlorpromazine [an inhibitor of clathrin-coated pit formation (47)] and nystatin [a sterol-binding agent that disassembles caveolae (54)] did not inhibit TSP1-induced lipid accumulation in macrophages (Nile Red+ (%): 3.5 ± 1.0, 14.3 ± 2.7*, 10.3 ± 2.7*, and 53.0 ± 15.7* for vehicle, TSP1, TSP1 + chlorpromazine, and TSP1 + nystatin treatment, respectively, *p < 0.05 vs. vehicle). Mindful of possible off-target effects of pharmacological inhibitors, we gene-silenced myotubularin-related protein 6 (MTMR6), a protein tyrosine phosphatase recently shown to be essential for macropinocytosis through dephosphorylation of phosphatidylinositol 3-phosphate (32). Importantly, MTMR6 is not required for clathrin-dependent endocytosis (32). TSP1 significantly increased lipid accumulation in scrambled siRNA-transfected macrophages. In contrast, siRNA knockdown of MTMR6 inhibited TSP1-stimulated lipid uptake (Fig. 2F). Effective gene silencing of MTMR6 is shown in Figure 2F and Supplementary Figure S5A, B. In aggregate, these data suggest that TSP1 stimulates uptake of nLDL in macrophages via fluid-phase macropinocytosis.
Nox1 activation is essential for TSP1-induced macropinocytosis
The next series of experiments were designed to investigate the mechanisms by which TSP1 stimulates macropinocytosis. Activation of the diacylglycerol (DAG)–protein kinase C (PKC) pathway stimulates macropinocytosis in various cell types (27, 43, 49); however, the precise signaling mechanism downstream of PKC activation remains unknown. We found that inhibition of classical and novel PKC isoforms by calphostin c completely abolished TSP1-induced lipid uptake (Fig. 3A). Given our previous findings that TSP1 via PKC increases Nox1-derived superoxide anion (O2•−) production in vascular smooth muscle cells (15), we speculated that (a) TSP1 stimulates O2•− generation in macrophages and (b) TSP1-induced macropinocytosis involves Nox signaling. To date, the ability of TSP1, or any other matricellular protein for that matter, to induce Nox activation and macropinocytosis in macrophages has never been demonstrated.
FIG. 3.

Nox1 is essential in TSP1-induced macropinocytosis. (A) RAW 264.7 macrophages were treated with vehicle and nLDL (250 μg/ml, 24 h) or TSP1 (10 nM) and nLDL in the absence or presence of calphostin c pretreatment (1 μM, 30 min). Cells were fixed in 2% PFA, stained by Nile Red, and processed for FACS analysis. *p < 0.05 indicates significant difference between vehicle versus TSP1. #p < 0.05 indicates significant difference between TSP1 versus TSP1 + calphostin c (n = 6). (B) Macrophages were treated with vehicle or TSP1 (10 nM) for 60 min. Superoxide generation was detected by the membrane-permeant EPR spin probe CMH. Representative CM• spectra are presented. Cumulative and averaged CM• radical formation in vehicle- and TSP1-treated macrophages is shown. *p < 0.05 versus vehicle (n = 3). (C) Macrophages were preincubated with DHE (10 μM, 30 min) and treated with vehicle or TSP1 (10 nM) for 60 min. 2-Hydroxyethidium (2-OH-E+), the O2•−-specific oxidation product of DHE, was quantified in vehicle- and TSP1-treated macrophages using HPLC. *p < 0.05 versus vehicle (n = 3). (D) Wild-type (WT) and Nox1y/− bone marrow-derived macrophages were incubated with nLDL (250 μg/ml) and treated with vehicle or TSP1 (10 nM) for 24 h. Cells were fixed in 2% PFA, stained by Nile Red, and processed for FACS analysis. *p < 0.05 versus WT vehicle. #p < 0.05 versus WT TSP1 (n = 4). (E) Representative FACS histogram plots (n = 4). (F) WT and Nox1y/− peritoneal macrophages were treated with vehicle or challenged with exogenous TSP1 (10 nM) for 2 h, and then processed for SEM (scale bars, 1 μm). Data represent the mean ± SD. EPR, electron paramagnetic resonance; DHE, dihydroethidium; Nox, NADPH oxidase. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Consequently, we first examined whether TSP1 stimulates O2•− production in macrophages using the membrane-permeant electron paramagnetic resonance (EPR) spin probe CMH. EPR spectra indicate that TSP1 stimulates intracellular O2•− generation in macrophages (Fig. 3B). Next, an HPLC-based approach was used to measure intracellular O2•− generation by quantifying 2-hydroxyethidium (2-OH-E+) (55), the O2•−-specific oxidation product of dihydroethidium, in macrophages. As shown in Figure 3C, TSP1 significantly increased 2-OH-E+ levels in macrophages compared with vehicle treatment. Extracellular O2•− production measured by the cell-impermeant EPR spin probe 1-hydroxy-4-phosphono-oxy-2,2,6,6-tetramethyl-piperidine (PPH) was not different between vehicle- and TSP1-treated cells (Supplementary Fig. S6). These results suggest that TSP1-stimulated intracellular O2•− in macrophages plays a role in nLDL uptake and concur with nLDL not being oxidized in the cell culture media of macrophages following TSP1 treatment (Fig. 2D). A time course and full concentration–response to TSP1 are shown in Supplementary Figure S7A, B. These data are consistent with TSP1 at pathophysiologically relevant concentrations stimulating intracellular O2•− and driving downstream signaling pathways in macrophages that in turn affect macropinocytosis.
Based on these results and our previous findings of TSP1-mediated Nox1 activation in smooth muscle cells, (15) we speculated that TSP1-induced macropinocytosis involves Nox1 activation and downstream oxidase-mediated signaling. Diphenyleneiodonium (DPI), a classical inhibitor of Nox and other oxidases, significantly suppressed TSP1-induced lipid uptake (Supplementary Fig. S8A, B). This finding supports a possible role of Nox in macropinocytosis. To explore the specific role of Nox1 in TSP1-induced lipid accumulation, bone marrow-derived monocytes were isolated from wild-type (WT) and Nox1y/− mice, differentiated into macrophages using macrophage colony-stimulating factor (M-CSF), treated with nLDL, and incubated with vehicle or TSP1. Lipid accumulation significantly increased in TSP1-treated macrophages from WT mice compared with vehicle treatment (Fig. 3D, E). Conversely, TSP1 did not stimulate lipid uptake in Nox1y/− macrophages (Fig. 3D, E). Finally, SEM data show that TSP1 stimulates membrane ruffling in WT, but not in Nox1y/− macrophages, corroborating a role for Nox1 in TSP1-induced membrane ruffle formation (Fig. 3F). Importantly, TSP1 stimulated macropinocytosis in primary murine macrophages, RAW cells, and human THP-1 macrophages, demonstrating the conservation of this pathway across different species and macrophage phenotypes.
CD47 is required for TSP1-induced macropinocytosis
TSP1 avidly binds CD47 (24) and stimulates Nox1 signaling in vascular cells (15). Therefore, we explored whether CD47 is required for TSP1-induced Nox activation and stimulation of macropinocytosis. In-cell Western analysis demonstrates that CD47 is expressed on macrophages (Supplementary Fig. S9). Figure 4A shows that pretreatment of macrophages with a CD47 blocking antibody (2 μg/ml, MIAP301) abolished TSP1-stimulated O2•− production. In addition, 7N3, a peptide activator of CD47 derived from the C terminus of TSP1 (Fig. 4B) (24), stimulated ROS signaling (Fig. 4C). We then asked whether CD47 plays a role in TSP1-induced membrane ruffling and fluid-phase macropinocytosis of nLDL. As demonstrated in Figure 4D–F, TSP1 stimulated membrane ruffling on the dorsal surface of WT macrophages and subsequent macropinocytosis of nLDL. In contrast, TSP1 did not stimulate membrane ruffling and lipid accumulation in CD47−/− macrophages (Fig. 4D–F).
FIG. 4.

CD47 is required for TSP1-induced macropinocytosis. (A) Macrophages were pretreated with a CD47 monoclonal antibody (2 μg/ml, CD47 [MIAP301], Santa Cruz Biotechnology) or isotype control IgG2α (2 μg/ml, Santa Cruz Biotechnology) for 30 min and treated with vehicle or TSP1 (10 nM, 60 min). O2•− production was measured using cytochrome c reduction (n = 4). (B) Multidomain structure of TSP1 and amino acid sequence of peptide 7N3. (C) Macrophages were treated with vehicle or peptide 7N3 (10 μM) for 60 min. Amplex Red fluorescence (RFU) was measured in macrophage lysates. Reaction was initiated by the addition of 36 μM NADPH and monitored at 24°C for 60 min (n = 3). (D) Wild-type and CD47−/− peritoneal macrophages were treated with vehicle or TSP1 (10 nM) for 2 h, and then processed for SEM (scale bars, 1 μm). (E) FACS analysis demonstrating Nile Red fluorescence in WT and CD47−/− macrophages. *p < 0.05 versus WT vehicle. #p < 0.05 versus WT TSP1 (n = 4). (F) Representative FACS histogram plots (n = 4). Data represent the mean ± SD. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Pivotal role of cofilin in TSP1-induced macropinocytosis
Previous studies demonstrated that dorsal ruffle formation in fibroblasts is stimulated by epidermal growth factor receptor (EGFR) activation (5). As intracellular Nox signaling is known to transactivate EGFR (51) and EGFR is upstream of PI3K signaling (38), we tested whether the EGFR tyrosine kinase inhibitor erlotinib inhibits TSP1-induced macropinocytosis. As shown in Figure 5A, erlotinib did not attenuate TSP1-induced solute uptake, suggesting that EGFR transactivation is not involved in TSP1-induced macropinocytosis. A previous study demonstrated that syndecan-mediated Rac1 and Cdc42 activation leads to stimulation of macropinocytosis (50). Therefore, we next tested whether TSP1 regulates syndecan expression in macrophages. Western blot results show that incubation of macrophages with TSP1 does not change syndecan-1 expression (Supplementary Fig. S10).
FIG. 5.

Role of cofilin in TSP1-induced macropinocytosis. (A) Macrophages were treated with nLDL plus vehicle, nLDL plus TSP1 (10 nM) or erlotinib (pretreated 1 μM, 1h) with nLDL plus TSP1. All treatments were for 24 hr. Cells were fixed in 2% PFA, stained by Nile Red, and processed for FACS analysis. *p < 0.05 versus vehicle (n = 3). (B) Interactome study was performed utilizing the STRING database (http://string-db.org/). (C) WT, Nox1y/−, and CD47−/− bone marrow-derived macrophages were incubated with vehicle or TSP1 and processed via Western blot for p-cofilin (Ser-3) and total cofilin expression. *p < 0.05 versus vehicle (n = 3). (D) A scheme summarizing the mechanism by which TSP1 stimulates membrane ruffling and macropinocytotic uptake of nLDL. Data represent the mean ± SD. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
We next utilized the STRING protein–protein interaction database (string-db.org/) to identify potential new targets linking TSP1/CD47/Nox1 signaling with dynamic actin cytoskeletal reorganization and membrane ruffling. Analysis of known and predicted protein–protein interactions revealed that cofilin (Cfl1) may be a downstream target of TSP1, thus linking PI3K, CD47, and Nox1 with actin reorganization (Fig. 5B). In recent years, cofilin has emerged as a key regulator of actin dynamics due to its ability to sever actin filaments and to create new actin barbed ends for Arp2/3 that in turn rapidly rebuilds the actin network in migrating cells (17). As the severing activity of cofilin is increased upon dephosphorylation of its Ser-3, we tested whether TSP1 dephosphorylates cofilin in macrophages. Western blot data indicated that TSP1 dephosphorylated cofilin on Ser-3 in WT macrophages (Fig. 5C). In contrast, TSP1 did not induce dephosphorylation of cofilin in Nox1y/− or CD47−/− macrophages. Gene silencing cofilin with siRNA inhibited TSP1-induced FITC-dextran uptake, indicative of a functional role of cofilin in TSP1-induced macropinocytosis (Supplementary Fig. S11). Together, our findings implicate that Nox1 internal cell signaling mediates TSP1-CD47-induced cofilin activation, leading to rapid remodeling of actin cytoskeleton, membrane ruffle formation and macropinocytosis (Fig. 5D).
Role of CD47 in macrophage lipid macropinocytosis in an animal model of atherosclerosis
To determine the role of CD47 in macropinocytosis in vivo, we evaluated the ability of macrophages derived from WT and CD47−/− mice (donors) to accumulate cholesterol following their adoptive transfer into the peritoneal cavities of high-fat diet-fed ApoE−/− mice (recipients). WT and CD47−/− macrophages were fluorescently labeled using the cell tracker dye carboxyfluorescein diacetate succinimidyl ester (CFDA), incubated with vehicle or treated with TSP1 (10 nM, 2 h) to stimulate membrane ruffling in vitro, and transferred into the peritoneal cavities of ApoE−/− mice. Twenty hours later, peritoneal macrophages were isolated and visualized under a fluorescence microscope (Fig. 6A). Cells were then fixed, stained with Nile Red, and lipids accumulated in gated CFDA+ (donor) macrophages analyzed by FACS (Fig. 6A). TSP1 significantly stimulated lipid accumulation in WT-CFDA+ (Nile Red fluorescence) macrophages compared with vehicle treatment (Fig. 6B). In contrast, TSP1-induced lipid accumulation was completely blocked in CD47−/−-CFDA+ macrophages (Fig. 6B). These data suggest that TSP1-CD47 signaling stimulates macropinocytosis of cholesterol in vivo leading to macrophage foam cell formation in hypercholesterolemic animals.
FIG. 6.

Role of CD47 in macropinocytotic uptake of lipids by macrophages in an atherosclerotic mouse model in vivo. (A) CFDA-labeled donor macrophages isolated from the peritoneal cavity of ApoE−/− mice assessed by fluorescent microscopy and FACS gated. (B) Representative Nile Red fluorescence histograms indicating lipid accumulation in vehicle- and TSP1-treated CFDA+-WT and CFDA+-CD47−/− macrophages. Bar graph indicates averaged Nile Red fluorescence in CFDA+-WT and CFDA+-CD47−/− macrophages following incubation with vehicle or TSP1 (n = 3). Data represent the mean ± SD. CFDA, carboxyfluorescein diacetate succinimidyl ester. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Discussion
In the present study, we show for the first time that matricellular protein TSP1 via Nox1 signaling, per se, sets in motion a dynamic and robust activation of dorsal membrane ruffle-mediated macropinocytosis. This unique signaling cascade includes CD47, PKC, Nox1, and cofilin dephosphorylation among other key intermediaries. Importantly, we also show here that TSP1 stimulates uptake of LDL independent of oxidative modification. Rather, a highly controlled intracellular redox signaling pathway involving Nox1 actuates macropinocytotic uptake of native LDL. The present study also supports the notion of pharmacologic Nox1 inhibition in vascular disease (40). These findings are expected to spur broader research into controlled, spatiotemporal Nox redox signaling. On a more translational level, they have broad implications for vascular inflammatory disorders and development of new interventions targeting macrophage lipid accumulation and atherogenesis.
The classical Nox is a multicomponent enzyme, which consists of the membrane-bound Nox subunit and p22phox and several cytosolic adaptor proteins. The canonical Nox1 system is constitutively active and requires the Nox organizer subunit 1 (NOXO1) and Nox activator subunit 1 (NOXA1) for O2•− generation. Our data suggest that the noncanonical Nox1 system, which supplants NOXO1 with p47phox, may be at play here as we demonstrate that PKC, which is the prototypical activator of p47phox, is involved in TSP1-induced macropinocytosis. However, we cannot rule out that transcriptional upregulation of Nox1 oxidase subunits is involved because we know that TSP1 via CD47 induces JAK/STAT signaling that may lead to Nox1 upregulation (33). The present study suggests that TSP1 stimulates Nox1 signaling in macrophages and initiates a highly regulated redox signaling pathway leading to dynamic actin cytoskeletal reorganization and solute internalization via macropinocytosis. Interestingly, a previous study demonstrated that Nox1 deficiency in hypercholesterolemic ApoE−/− mice reduces atherosclerotic lesion area (44); however, the mechanisms responsible for this remained unknown. The findings presented herein contend that redox signaling via Nox1 contributes to macropinocytosis, arterial lipid accumulation, and atherosclerosis.
To date, only one study investigated the functional role of TSP1 in atherosclerosis (36). This study revealed a protective effect of TSP1 deficiency at the early stages of plaque development. However, the underlying mechanism describing this effect is scant to none. Previous studies demonstrated that accumulation of nonoxidized lipids precedes that of oxidized lipoproteins in human atherosclerotic lesions, suggesting an important, but as yet undefined, mechanism for direct macrophage uptake of nLDL in the subendothelial matrix of atheroprone arteries (52). It is important to point out at this juncture, however, that it is widely held that cellular uptake, per se, of LDL requires its prior oxidation. Contrary to this theory, we postulated that pathophysiological concentrations of matrix protein TSP1 stimulate internalization of unmodified lipoproteins in macrophages and thereby bypass the need for oxidation and uptake by scavenger receptors. Indeed, our results demonstrated that treatment of human and rodent macrophages with TSP1 at concentrations found in patients with cardiovascular disease significantly increased nLDL uptake via a distinct signaling process (34). Importantly, TSP1 did not induce oxidative modification of nLDL in cell culture medium as indicated by the results of agarose gel electrophoresis.
To investigate the mechanism involved, we considered previous reports by Kruth et al. demonstrating that phorbol ester activation of macrophages stimulates macropinocytosis of nLDL and subsequent lipid accumulation (27, 28). Macropinocytosis begins with membrane ruffle formation. Membrane ruffles adhere to the nonextended plasma membrane or form circular macropinocytotic cups and following scission from the plasma membrane internalize as macropinosomes (3, 53). Based on these morphologically distinct plasma membrane activities, macropinocytosis can be divided into four consecutive steps: (a) membrane ruffling, (b) ruffle closure, (c) cup closure, and (d) internalization of extracellular medium and its contents via macropinosome formation (9). With further analysis utilizing SEM, we demonstrated initiation of membrane ruffle formation, fully formed C-shaped ruffles, and macropinocytotic cups on the dorsal surface of TSP1-treated cells. Time-lapse DIC indicated dynamic formation of membrane ruffles 30 min after TSP1 treatment. TSP1-induced membrane ruffling was still observed 120 min after stimulation. We went on to investigate whether TSP1 stimulates internalization of macromolecules, characteristics of macropinosome formation, and subsequent macropinocytotic uptake (49). Flow cytometry analysis and confocal imaging confirmed uptake of 1 μm FluoSpheres by macrophages in response to TSP1 treatment. Previous results demonstrate that the upper size limits for particles undergoing clathrin- and caveolin-mediated endocytosis are 200 and 500 nm in diameter, respectively (41). In contrast, the size range for particles entering macropinosomes extends beyond 500 nm and up to 5 μm in diameter (9), suggesting that the TSP1-stimulated macropinosome formation and internalization of 1 μm FluoSpheres we observed were a function of macropinocytosis. During the process of macropinosome formation, PI3K translocates to the ruffling region and its phosphorylation product PtdIns(3,4,5)P3 mediates spatiotemporal orchestration of actin polymerization and rearrangement through recruiting and activating a variety of actin-associated proteins, such as cofilin and Arp2/3 (37). It is logical then that previous reports went on to show that pharmacological inhibitors of PI3K, such as LY294002, inhibit cup closure and macropinosome formation in macrophages (4). In the current study, we found that preincubation of macrophages with LY294002 completely abolished TSP1-induced uptake of FluoSpheres. Collectively, these data are consistent with TSP1 stimulating membrane ruffling in macrophages, followed by cup closure, macropinosome formation, and subsequent internalization of macromolecules.
Next, we went on to challenge the notion that TSP1 stimulates internalization of nLDL via macropinocytosis by contrasting its potential contribution with that of other endocytic processes (39). Macropinocytosis is characterized by its sensitivity to PI3K inhibitors (4) or to actin perturbation by latrunculins (2). We observed that both LY294002 and latrunculin A completely inhibited TSP1-stimulated lipid accumulation in macrophages. An additional criterion was applied to identify macropinocytosis; namely, its susceptibility to inhibition by the Na+/H+ antiporter amiloride, which lowers submembranous pH and prevents Rac1 and Cdc42 signaling (26). Indeed, preincubation of macrophages with amiloride abolished lipid internalization following TSP1 treatment. In contrast, chlorpromazine, an inhibitor of clathrin-coated pit formation (47), and nystatin, a reagent that disassembles caveolae (54), did not inhibit TSP1-induced lipid accumulation. Considering possible off-target effects of pharmacological inhibitors, we silenced MTMR6, a protein tyrosine phosphatase, which is essential for macropinocytosis through dephosphorylation of PtdIns(3)P (32). Knockdown of MTMR6 inhibited TSP1-stimulated lipid internalization. To our knowledge, these data demonstrate for the first time that TSP1, or any matricellular protein for that matter, stimulates macropinocytosis.
Activation of the DAG-PKC pathway induces macropinocytosis in various cell types (27, 49). Incidentally, PKC activation stimulates Nox signaling through phosphorylation of the cytosolic organizer subunit p47phox (29). Indeed, recently published data from our laboratory indicated that TSP1 stimulates Nox1 activity via PKC and p47phox phosphorylation in vascular smooth muscle cells (15). In addition, the spike in actin polymerization during membrane ruffle formation involves the recruitment of the small GTPase Rac (42), which is an important positive regulator of Nox assembly in phagocytes (23). Those findings prompted us to investigate whether PKC could be an intermediary in Nox1 activation, ruffle formation, and macropinocytosis. Indeed, PKC inhibition by calphostin c and pretreatment with DPI, an inhibitor of flavin-containing oxidases, inhibited TSP1-induced lipid accumulation. With all this in mind, it was logical therefore to suggest a role for an active assembly of the Nox1 hybrid system. Intracellular O2•− production demonstrated by EPR and quantification of 2-OH-E+ support Nox1-mediated signal since Nox1 has been found in caveolae and endosomes and not on the plasma membrane. Focusing in on Nox1, we tested whether TSP1 stimulates membrane ruffling in Nox1y/− macrophages. SEM data indicated that TSP1 stimulates ruffle formation in WT, but not in Nox1y/−, macrophages. Consistent with these results, TSP1 did not stimulate nLDL uptake in Nox1y/− macrophages. Consequently, our results favor the view that Nox1 oxidase acts as a signaling intermediary in TSP1-mediated macropinocytosis. In line with this contention, a previous study demonstrated that Nox1 deficiency is associated with reduced lesion area in an atherosclerotic mouse model (44). At this juncture, it is important to point out that pharmacological PKC activation can also set in motion a similar sequence of events via Nox2 [see associated article in this issue (21)].
TSP1 via interaction with its high-affinity cell receptor, CD47, stimulates Nox1 activity in vascular cells (15) and inhibits nitric oxide-mediated signaling (7). TSP1 is also known to bind scavenger receptor CD36; however, this interaction is not required for Nox activation (15). The present study identifies a TSP1-CD47-Nox1 axis as a proximate promoter of macrophage macropinocytosis and thus provides multiple new targets to attenuate macrophage lipid internalization. Indeed, pharmacological blockade of CD47 completely abrogated TSP1-induced O2•− generation and CD47 deficiency abolished TSP1-induced membrane ruffling and macropinocytotic uptake of nLDL in macrophages. Although deficiency of TSP1 has been demonstrated to delay initial plaque formation in ApoE−/− mice (36), similar studies in CD47-deficient ApoE−/− mice had not been reported. With those findings in mind, we postulated that activation of CD47 by TSP1 stimulates macrophage macropinocytosis in vivo. To test this, we transplanted fluorescently labeled WT and CD47−/− macrophages into hypercholesterolemic ApoE−/− mice and evaluated their ability to internalize lipids via macropinocytosis. Before injection, cells were incubated with vehicle or TSP1 to stimulate membrane ruffling. Our data demonstrated that TSP1 pretreatment stimulated lipid accumulation in vivo in transplanted WT macrophages compared with vehicle treatment. In contrast, TSP1-induced lipid accumulation was attenuated in transplanted CD47−/− macrophages. These results led us to speculation that CD47 regulation of macrophage macropinocytosis contributes to the pathogenesis of atherosclerosis. Intriguingly, no prior studies investigated whether macrophage lipid macropinocytosis contributes to atherosclerotic plaque formation. We, therefore, anticipate a growing interest into the delineation of pinocytotic mechanisms controlling nLDL versus scavenger receptor-mediated ox-LDL in the vessel wall. These in vivo studies are particularly important as macrophages residing in the vessel wall may be functionally and phenotypically different from cell lines or primary cells kept in culture. In that respect, we expect future studies to investigate whether TSP1-CD47 signaling in the vessel wall regulates macrophage macropinocytosis.
In recent years, cofilin has emerged as a key regulator of actin filament dynamics, leading to lamellipodia formation in motile cells (37). Dephosphorylated cofilin at Ser-3 severs actin filaments and creates new actin barbed ends for Arp2/3 to rapidly rebuild actin network and simulate formation of membrane protrusions (17). Our data demonstrate for the first time that TSP1 activates cofilin (dephosphorylation at Ser-3) via CD47 and Nox1 in macrophages. We also showed that gene silencing cofilin inhibits TSP1-induced macropinocytosis. These data suggest that TSP1 via CD47-mediated Nox1 activation induces cofilin activation, leading to actin remodeling, membrane ruffle formation, and macropinocytosis. Interestingly, PI3K signaling stimulates the activity of slingshot phosphatase (SSH), leading to cofilin dephosphorylation and activation (37). As pharmacological blockade of PI3K completely inhibited TSP1-induced macropinocytosis, it is tempting to speculate that TSP1 via CD47-mediated Nox1 activation increases SSH activity, leading to dephosphorylation of cofilin and initiation of membrane ruffle formation. The role of SSH as another intermediary in this pathway will require further study, but is currently outside the scope of this article.
As growth factors and cytokines have been reported to stimulate macropinocytosis, we tested whether TSP1 induces secretion of cytokines and chemokines from macrophages. We found that TSP1 stimulates RANTES, interleukin-1 receptor antagonist (IL-1RA), and macrophage inflammatory protein-1α (MIP-1α) secretion from macrophages (Supplementary Fig. S12). It is therefore plausible that TSP1-induced cytokine release is involved in macropinocytosis stimulation.
In conclusion, our findings identify a previously uncharacterized mechanism of cellular solute uptake by a TSP1-CD47-Nox1-cofilin axis that is likely to have broad implications for vascular inflammatory disorders and multiple other cell types and pathologies in which macropinocytosis has been implicated. We have demonstrated that TSP1 at physiologically relevant concentration stimulates formation of circular dorsal ruffles in macrophages, leading to macropinocytotic uptake of nLDL and lipid accumulation. Importantly, TSP1 stimulates direct uptake of nLDL that does not require LDL oxidative modification or scavenger receptor-mediated internalization, but instead delimited intracellular Nox1 signaling and macropinocytosis. The results of the present study are expected to lead to broad new areas of exploration in the field of cardiovascular research and inform new pharmacological interventions to combat vascular disorders.
Materials and Methods
Reagents
Cytochrome c, superoxide dismutase (SOD), DPI chloride, phorbol 12-myristate 13-acetate (PMA), Nile Red, LY294002, calphostin C, amiloride, 5-(N-ethyl-N-isopropyl)amiloride (EIPA), latrunculin A, chlorpromazine, nystatin, and thioglycollate medium were purchased from Sigma-Aldrich (St. Louis, MO). Protease and phosphatase inhibitor cocktail tablets were purchased from Roche Diagnostics GmbH (Mannheim, Germany). Zombie Yellow™ was purchased from BioLegend (San Diego, CA). FluoSpheres®, negative control and MTMR6 siRNAs were purchased from Life Technologies (Grand Island, NY). Human TSP1 was purchased from Athens Research & Technology (Athens, GA). Human nLDL and ox-LDL were purchased from Kalen Biomedical, LLC (Montgomery Village, MD). CD47 monoclonal [MIAP301] and isotype control IgG2α antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX).
Cell culture
RAW 264.7 cells, a murine macrophage cell line (ATCC, Manassas, VA), and thioglycollate-elicited peritoneal macrophages were cultured in Dulbecco's modified Eagle medium (Mediatech, Inc., Manassas, VA) supplemented with 100 IU/ml of penicillin G, 100 μg/ml streptomycin, and 10% (vol/vol) fetal bovine serum (FBS). Bone marrow-derived monocytes were differentiated into macrophages using murine M-CSF (20 ng/ml, 6 days). THP-1 monocytes (ATCC, Manassas, VA) were cultured in RPMI-1640 medium supplemented with 2-mercaptoethanol (0.05 mM) and 10% FBS. THP-1 monocytes were differentiated into macrophages using PMA (8 nM, 48 h). Cells were incubated in humidified atmosphere of 5% CO2 in an incubator at 37°C. Cells were grown to 80% confluence and serum starved (1% FBS, 4 h) before experiments were run.
Animals
All animal studies were performed under a protocol approved by the IACUC of the University of Pittsburgh or Augusta University. Male C57BL/6 (wild-type), TSP1−/−, and LDLR−/− mice were purchased from The Jackson Laboratory (Bar Harbor, Maine). Nox1y/− mice on a C57BL/6 background were kindly provided by Dr. K. H. Krause (University of Geneva). CD47−/− mice were kindly provided by Dr. Jeffrey Isenberg (University of Pittsburgh).
Isolation of mouse macrophages
Collection of thioglycollate-elicited peritoneal macrophages
Three ml of 3% thioglycollate medium was injected into the peritoneum of 8-week-old male wild-type, Nox1y/−, and CD47−/− mice. Four days later, the mice were anesthetized (isoflurane inhalation, 3%) and sacrificed by cervical dislocation. Peritoneal cells were harvested by lavage with approximately 10 ml sterile ice-cold phosphate-buffered saline (PBS). Cells were sedimented by centrifugation at 300g for 5 min. Peritoneal macrophages were enriched by adherence to tissue culture dishes.
Generation of mouse bone marrow-derived macrophages
The femur and tibia of sacrificed mice were cleaned of adherent muscle and connective soft tissue. Bone marrow-containing cavities were exposed by removing the ends of each femur and tibia, and a 25-gauge needle containing Harvest Buffer (HBSS, containing 10 mM Hepes, 4 mM sodium bicarbonate, and 4% heat-inactivated FBS) was used to flush and collect the bone marrow. Bone marrow flushings were loaded onto Ficoll-Paque PLUS (GE Healthcare, Piscataway, NJ) for density gradient centrifugation (1.078 g/ml) for 30 min at 400g. The isolated interphase fraction containing bone marrow-derived mononuclear cells was washed using PBS and centrifuged for 7 min at 300g. Total viable cell numbers obtained from each isolation were assessed using Trypan Blue. In all isolations, cell viability was greater than 95%. Monocytes were cultured in RPMI-1640 medium and differentiated into macrophages using 20 ng/ml M-CSF (Miltenyi Biotec Inc., San Diego, CA) for 6 days.
Measurement of ROS in macrophages
Cytochrome c reduction assay
Macrophages were exposed to vehicle (PBS) or TSP1 (0.1–30 nM) for the indicated period of time. Cells were washed twice with ice-cold PBS, scraped in lysis buffer (8 mM potassium, sodium phosphate buffer pH 7.0, 131 mM NaCl, 340 mM sucrose, 2 mM NaN3, 5 mM MgCl2, 1 mM EGTA, 1 mM EDTA, and protease inhibitors). The cells were lysed by five freeze/thaw cycles and passed through a 30-gauge needle five times to disrupt cells. Cell disruption was confirmed by phase-contrast microscopy. The cell lysate was centrifuged at 400 g for 5 min at 4°C to remove unbroken cells, nuclei, and debris. Throughout all these procedures, extreme care was taken to maintain the lysate at a temperature close to 0°C. The cell lysate was centrifuged at 28,000 g for 15 min at 4°C. The supernatant was removed and the pellet was resuspended in lysis buffer. Protein concentration was measured using the Bradford microplate method. Superoxide production in particulate fractions (20 μg/ml) of vehicle- and TSP1-treated macrophages was measured in 0.1 ml of oxidase assay buffer (65 mM sodium phosphate buffer pH 7.0, 1 mM EGTA, 10 μM FAD, 1 mM MgCl2, 2 mM NaN3, and 0.2 mM cytochrome c). Superoxide production was initiated by the addition of 180 μM NADPH and was calculated from the initial linear rate of SOD (150 U/ml) inhibitable cytochrome c reduction quantified at 550 nm using an extinction coefficient of 21.1 mM−1 cm−1 (Biotek Synergy 4 Hybrid Multi-Mode Microplate Reader).
Electron paramagnetic resonance
Macrophages were preincubated for 30 min in EPR buffer (NaCl 130 mM, KCl 4.7 mM, KH2PO4 1.2 mM, MgSO4 1.2 mM, NaHCO3 14.9 mM, glucose 5.5 mM, CaCl2 1.6 mM, and EDTA 0.026 mM, pH 7.4) at 37°C and then treated with vehicle or 10 nM TSP1 for 60 min. The cell-permeant spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine hydrochloride (CMH; Alexis Corp., San Diego, CA) was added at a concentration of 500 μM and 15 min later O2•− was measured using the Bruker eScan tabletop EPR spectrometer (Bruker Biospin). EPR instrument settings were as follows: field sweep, 50 G; microwave frequency, 9.78 GHz; microwave power, 20 mW; modulation amplitude, 2 G; conversion time, 327 ms; time constant, 655 ms; and receiver gain, 1 × 105. Analyses of the upfield EPR spectra peak heights were used to quantify the amount of O2•− produced by the cells and were compared with buffer-only control spectra. To minimize the deleterious effects of contaminating metals, the buffers were treated with Chelex resin and contained 25 μM deferoxamine (Noxygen Science Transfer, Germany) and 5 μM diethyl-dithiocarbamate (Sigma-Aldrich, St. Louis, MO). In separate experiments, the cell-impermeant spin probe PPH was used (Noxygen, Germany).
Quantification of 2-OH-E ± using HPLC
Hydroethidine (HE), ethidium (E+), and 2-hydroxyethidium (2-OH-E+), the O2•−-specific oxidation product of HE, were separated on an HPLC system as described previously (55). Briefly, an ether-linked phenyl column (100 × 4.6 mm, Phenomenex, Torrance, CA) was employed using two mobile phases (Solution A: 50 mM phosphate buffer in 90% water and 10% acetonitrile and Solution B: 50 mM phosphate buffer in 40% water and 60% acetonitrile) and a gradient elution to increase the acetonitrile concentration from 25% to 60% over 10 min at a flow rate of 0.75 ml/min. Forty microliters of sample was injected into the HPLC system (ESA, Inc., with a CoulArray electrochemical detection array system, Sunnyvale, CA). The areas of the corresponding peaks were measured using the software provided by ESA, Inc. The limit of detection for this assay was determined from a standard curve and established at 20 fmoles. Concentration of 2-OH-E+ was calculated using a standard curve and normalized to total protein of the cell lysate.
Amplex Red fluorescence
Hydrogen peroxide (H2O2) production was quantified as described previously (13). Cells were suspended at a concentration of 5 × 107 cells/ml in ice-cold disruption buffer (PBS containing 0.1 mM EDTA, 10% glycerol, protease inhibitor cocktail, and 0.1 mM PMSF). Macrophages were lysed by five freeze/thaw cycles and passed through a 30-gauge needle five times. H2O2 production was measured in the lysate of vehicle- and TSP1-treated macrophages (10 μg/100 μL) in assay buffer (25 mM Hepes, pH 7.4, containing 120 mM NaCl, 3 mM KCl, 1 mM MgCl2, 25 μM FAD, 0.1 mM Amplex Red, and 0.32 U/ml of HRP). The reaction was initiated by the addition of 36 μM NADPH. Fluorescence measurements were made using a Biotek Synergy 4 Hybrid Multi-Mode Microplate Reader (excitation wavelength: 560 nm; emission wavelength: 590 nm). A standard curve of known H2O2 concentrations was generated to quantify H2O2. The reaction was monitored at room temperature for 60 min. The emission increase was linear during this interval.
Lipid accumulation
Confocal microscopy
Macrophages were grown on coverslips (Electron Microscopy Sciences, Hatfield, PA), serum starved, and incubated with nLDL (250 μg/ml) in the absence and presence of 10 nM TSP1 for 24 h. Cells were fixed in 2% paraformaldehyde, permeabilized with 0.1% Triton X-100, and then stained with Alexa Fluor 488® phalloidin, Hoechst (Life Technologies, Grand Island, NY), and Nile Red (50 ng/ml; 10 min). Images were captured with an Olympus FluoView™ FV1000 confocal microscope.
Cholesterol quantification using a fluorometric assay
The Amplex Red Cholesterol Assay Kit was used to quantify cholesterol content in macrophages according to the manufacturer's protocol (Thermo Fisher Scientific, MA).
Flow cytometry
RAW 264.7 and bone marrow-derived macrophages were cultured to ∼80% confluence, serum starved, incubated with nLDL (250 μg/ml), and treated with vehicle or TSP1 (10 nM, 24 h). Cells were stained with Nile Red (25 ng/ml), fixed in 2% paraformaldehyde, and analyzed using a BD LSRFortessa flow cytometer. The gating strategy for flow cytometry analysis is shown in Supplementary Figure S1.
Scanning electron microscopy
Coverslips of cells were fixed in 2.5% glutaraldehyde, secondarily fixed in 1% OsO4, dehydrated through a graded ethanol series (30%–100%), and washed with 100% ethanol before drying in hexamethyldisilazane solution. Samples were then mounted onto aluminum stubs, sputter coated with 3.5 nm of gold/palladium (Cressington Auto 108, Cressington, United Kingdom), and viewed with a JEOL JSM-6335F scanning electron microscope (Peabody, MA) at 3 kV with the SEI detector.
DIC microscopy
Cells were grown on 35-mm glass bottom culture dishes (Mattek Corp), mounted in an appropriately controlled environmental chamber (37°C, 5% CO2) on a Nikon TI-E inverted microscope. The microscope was set up appropriately for DIC imaging using a high-shear prism and Kohler illumination. Cells were imaged using a 60 × 1.49 Plan Apochromat objective and images were collected with a Zyla 5.5 sCMOS camera (Andor Corp, Belfast Ireland). Baseline images (vehicle treatment) were collected for 60 min and then TSP1 was added as described in the figure legend. Ten representative fields were chosen from each dish, experiments were performed in triplicate. Image collection, microscope control, and movie generation were performed using Nikon Elements software.
In-cell Western and Western blot experiments
In-cell Western assay was performed according to the manufacturer's instructions (Li-Cor Biotechnology, Lincoln, Nebraska). Cells were incubated with a rabbit polyclonal antibody against CD47 (Abcam, Cambridge, MA) overnight at 4°C. This was followed by incubation with secondary antibody IRDye 800CW (green) and CellTag™ 700 (red) for 1 h with protection from light. Plates were scanned at 680 and 800 nm using the Odyssey CLx Infrared Imaging System. In separate experiments, TSP1- and vehicle-treated macrophages were assessed via Western blot for p-cofilin (Ser-3), total cofilin (Cell Signaling, Danvers, MA), ACAT1 (Abcam, Cambridge, MA), and syndecan-1 expression (Santa Cruz Biotechnology, Dallas, TX). Representative unedited Western blot images are shown in Supplementary Figures. S5 and S13.
siRNA transfection
RAW 264.7 macrophages were grown to 50% confluence and transfected with siRNAs against MTMR6 (Silencer®, Invitrogen), Cfl1 (Cell Signaling), CLTC, or CAV1(Origene) using the transfection reagents Lipofectamine 2000 (Invitrogen, Carlsbad, CA) or TransIT-TKO (Mirus). To control for possible nonspecific effects of siRNA, silencer-negative control siRNA was used. Knockdown of MTMR6, Cfl1, CLTC, and CAV1 by siRNA was confirmed by Western blot or real-time PCR. Cells were used 48 h after transfection.
Real-time PCR analysis
Total RNA was extracted from macrophages using an RNA extraction kit (IBI Scientific, Peosta). cDNA was generated using the TaqMan® Reverse Transcriptase kit (Applied Biosystems, Carlsbad). Real-time PCR was carried out using the SYBR Green Supermix (Applied Biosystems, Inc., Warrington, United Kingdom). All amplifications were performed in triplicate, and GAPDH was used as the internal control. The primers used for real-time PCR are as follows:
Cofilin: F-TTCAACGACATGAAGGTGCGT, R-TCCTCCAGGATGATGTTCTTCT,
Clathrin heavy chain: F-TGATCGCCATTCTAGCCTTGC, R-CTCCCACCACACGATTTTGCT, and
GAPDH: F-CATGTTCGTCATGGGTGTGAACCA,
R-AGTGATGGCATGGACTGTGGTCAT.
Adoptive transfer of macrophages into ApoE−/− mice
Bone marrow-derived macrophages from WT and CD47−/− mice (donors) were resuspended in sterile PBS at a concentration of 2 × 106 cells per ml and labeled with 10 μM CFDA (Molecular Probes, OR) as per the manufacturer's instructions. Cells were pretreated with vehicle or TSP1 (10 nM, 2 h) and injected into recipient ApoE−/− mice (i.p., 2 × 106 cells/mouse). ApoE−/− mice were fed a high-fat diet for 4 weeks before experiments. Twenty hours later, peritoneal macrophages were isolated, stained with Nile Red, and processed for FACS analysis.
Statistical analysis
Significance of the differences was assessed by Student's t-test and one- or two-way ANOVA, followed by a Bonferroni post hoc test, as appropriate for the particular experiment and treatment groups. A value of p < 0.05 was considered to be statistically significant.
Supplementary Material
Abbreviations Used
- ACAT
acyl-CoA:cholesterol O-acyltransferase
- CFDA
carboxyfluorescein diacetate-succinimidyl ester
- CLTC
clathrin heavy chain
- CVD
cardiovascular diseases
- DAG
diacylglycerol
- DHE
dihydroethidium
- DIC
differential interference contrast
- DPI
diphenyleneiodonium
- DUOX
dual oxidase
- EGFR
epidermal growth factor receptor
- EPR
electron paramagnetic resonance
- FACS
fluorescence-activated cell sorting
- FBS
fetal bovine serum
- HE
hydroethidine
- LDLR
low-density lipoprotein receptor
- M-CSF
macrophage colony-stimulating factor
- MTMR6
myotubularin-related protein 6
- nLDL
native low-density lipoprotein
- Nox
NADPH oxidase
- NOXA1
Nox activator subunit 1
- PBS
phosphate-buffered saline
- PFA
paraformaldehyde
- PI3K
phosphatidylinositol-3-kinase
- PKC
protein kinase C
- PMA
phorbol 12-myristate 13-acetate
- PPH
1-hydroxy-4-phosphono-oxy-2,2,6,6-tetramethyl-piperidine
- ROS
reactive oxygen species
- RT-PCR
real-time, quantitative polymerase chain reaction
- SD
standard deviation
- SEM
scanning electron microscopy
- SOD
superoxide dismutase
- SSH
slingshot phosphatase
- TSP1
thrombospondin-1
- WT
wild-type
Acknowledgments
The authors wish to thank Iulia Popescu (University of Pittsburgh) for her help with the flow cytometry analysis. The authors also gratefully acknowledge the technical assistance of Benjamin Zhu and Asma Naqvi (University of Pittsburgh). This work was supported by the following: P.J.P. receives support from National Institutes of Health (R01HL079207, R01HL112914, and P01HL103455–01) and is an Established Investigator of the American Heart Association. G.C. receives support from National Institutes of Health (K99HL114648 and 4R00HL114648-03). S.N. receives support from National Institutes of Health (R01HL086674). All Vascular Medicine Institute investigators receive support from the Institute for Transfusion Medicine and the Hemophilia Center of Western Pennsylvania, PA.
Author Disclosure Statement
J.S.I. is chair of the Scientific Advisory Boards of Vasculox, Inc., (St. Louis, MO) and Radiation Control Technologies, Inc., (Jersey City, NJ). All other authors have no competing financial interests to disclose.
References
- 1.Al Ghouleh I, Frazziano G, Rodriguez AI, Csányi G, Maniar S, St Croix CM, Kelley EE, Egana LA, Song GJ, Bisello A, Lee YJ, and Pagano PJ. Aquaporin 1, Nox1, and Ask1 mediate oxidant-induced smooth muscle cell hypertrophy. Cardiovasc Res 97: 134–142, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aleksandrowicz P, Marzi A, Biedenkopf N, Beimforde N, Becker S, Hoenen T, Feldmann H, and Schnittler HJ. Ebola virus enters host cells by macropinocytosis and clathrin-mediated endocytosis. J Infect Dis 204 Suppl 3: S957–S967, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Araki N, Hatae T, Furukawa A, and Swanson JA. Phosphoinositide-3-kinase-independent contractile activities associated with Fcgamma-receptor-mediated phagocytosis and macropinocytosis in macrophages. J Cell Sci 116: 247–257, 2003 [DOI] [PubMed] [Google Scholar]
- 4.Araki N, Johnson MT, and Swanson JA. A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J Cell Biol 135: 1249–1260, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Azimifar SB, Bottcher RT, Zanivan S, Grashoff C, Kruger M, Legate KR, Mann M, and Fassler R. Induction of membrane circular dorsal ruffles requires co-signalling of integrin-ILK-complex and EGF receptor. J Cell Sci 125: 435–448, 2012 [DOI] [PubMed] [Google Scholar]
- 6.Babaev VR, Gleaves LA, Carter KJ, Suzuki H, Kodama T, Fazio S, and Linton MF. Reduced atherosclerotic lesions in mice deficient for total or macrophage-specific expression of scavenger receptor-A. Arterioscler Thromb Vasc Biol 20: 2593–2599, 2000 [DOI] [PubMed] [Google Scholar]
- 7.Bauer EM, Qin Y, Miller TW, Bandle RW, Csányi G, Pagano PJ, Bauer PM, Schnermann J, Roberts DD, and Isenberg JS. Thrombospondin-1 supports blood pressure by limiting eNOS activation and endothelial-dependent vasorelaxation. Cardiovasc Res 88: 471–481, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bedard K. and Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87: 245–313, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Bohdanowicz M. and Grinstein S. Role of phospholipids in endocytosis, phagocytosis, and macropinocytosis. Physiol Rev 93: 69–106, 2013 [DOI] [PubMed] [Google Scholar]
- 10.Bornstein P. and Sage EH. Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol 14: 608–616, 2002 [DOI] [PubMed] [Google Scholar]
- 11.Brown DI. and Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med 47: 1239–1253, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, Rabinowitz JD, Metallo CM, Vander Heiden MG, and Bar-Sagi D. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497: 633–637, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Csányi G. and Pagano PJ. Strategies aimed at Nox4 oxidase inhibition employing peptides from Nox4 B-Loop and C-Terminus and p22 (phox) N-Terminus: an elusive target. Int J Hypertens 2013: 842827, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Csányi G, Taylor WR, and Pagano PJ. NOX and inflammation in the vascular adventitia. Free Radic Biol Med 47: 1254–1266, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Csányi G, Yao M, Rodriguez AI, Al Ghouleh I, Sharifi-Sanjani M, Frazziano G, Huang X, Kelley EE, Isenberg JS, and Pagano PJ. Thrombospondin-1 regulates blood flow via CD47 receptor-mediated activation of NADPH oxidase 1. Arterioscler Thromb Vasc Biol 32: 2966–2973, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davtyan TK, Avetisyan SA, and Hakobyan GS. Neutrophil F-actin dynamics in familial mediterranean fever: the unequal effect of colchicine on activated neutrophils. Antiinflamm Antiallergy Agents Med Chem 12: 165–172, 2013 [DOI] [PubMed] [Google Scholar]
- 17.DesMarais V, Ghosh M, Eddy R, and Condeelis J. Cofilin takes the lead. J Cell Sci 118: 19–26, 2005 [DOI] [PubMed] [Google Scholar]
- 18.Esterbauer H, Gebicki J, Puhl H, and Jurgens G. The role of lipid peroxidation and antioxidants in oxidative modification of LDL. Free Radic Biol Med 13: 341–390, 1992 [DOI] [PubMed] [Google Scholar]
- 19.Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, Hoff HF, Sharma K, and Silverstein RL. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest 105: 1049–1056, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frolova EG, Pluskota E, Krukovets I, Burke T, Drumm C, Smith JD, Blech L, Febbraio M, Bornstein P, Plow EF, and Stenina OI. Thrombospondin-4 regulates vascular inflammation and atherogenesis. Circ Res 107: 1313–1325, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghoshal P, Singla B, Lin H, Feck DM, Cantu-Medellin N, Kelley EE, Haigh S, Fulton D, and Csányi G. Nox2-mediated PI3K and cofilin activation confers alternate redox control of macrophage pinocytosis. Antioxid Redox Signal 26: 902–916, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heinecke JW, Baker L, Rosen H, and Chait A. Superoxide-mediated modification of low density lipoprotein by arterial smooth muscle cells. J Clin Invest 77: 757–761, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heyworth PG, Knaus UG, Settleman J, Curnutte JT, and Bokoch GM. Regulation of NADPH oxidase activity by Rac GTPase activating protein(s). Mol Biol Cell 4: 1217–1223, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Isenberg JS, Annis DS, Pendrak ML, Ptaszynska M, Frazier WA, Mosher DF, and Roberts DD. Differential interactions of thrombospondin-1, -2, and -4 with CD47 and effects on cGMP signaling and ischemic injury responses. J Biol Chem 284: 1116–1125, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jandeleit-Dahm K, Rumble J, Cox AJ, Kelly DJ, Dziadek M, Cooper ME, and Gilbert RE. SPARC gene expression is increased in diabetes-related mesenteric vascular hypertrophy. Microvasc Res 59: 61–71, 2000 [DOI] [PubMed] [Google Scholar]
- 26.Koivusalo M, Welch C, Hayashi H, Scott CC, Kim M, Alexander T, Touret N, Hahn KM, and Grinstein S. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J Cell Biol 188: 547–563, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kruth HS, Huang W, Ishii I, and Zhang WY. Macrophage foam cell formation with native low density lipoprotein. J Biol Chem 277: 34573–34580, 2002 [DOI] [PubMed] [Google Scholar]
- 28.Kruth HS, Jones NL, Huang W, Zhao B, Ishii I, Chang J, Combs CA, Malide D, and Zhang WY. Macropinocytosis is the endocytic pathway that mediates macrophage foam cell formation with native low density lipoprotein. J Biol Chem 280: 2352–2360, 2005 [DOI] [PubMed] [Google Scholar]
- 29.Li JM, Mullen AM, Yun S, Wientjes F, Brouns GY, Thrasher AJ, and Shah AM. Essential role of the NADPH oxidase subunit p47(phox) in endothelial cell superoxide production in response to phorbol ester and tumor necrosis factor-alpha. Circ Res 90: 143–150, 2002 [DOI] [PubMed] [Google Scholar]
- 30.Liu Z. and Roche PA. Macropinocytosis in phagocytes: regulation of MHC class-II-restricted antigen presentation in dendritic cells. Front Physiol 6: 1, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lusis AJ. Atherosclerosis. Nature 407: 233–241, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maekawa M, Terasaka S, Mochizuki Y, Kawai K, Ikeda Y, Araki N, Skolnik EY, Taguchi T, and Arai H. Sequential breakdown of 3-phosphorylated phosphoinositides is essential for the completion of macropinocytosis. Proc Natl Acad Sci U S A 111: E978–E987, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manea A, Tanase LI, Raicu M, and Simionescu M. Jak/STAT signaling pathway regulates nox1 and nox4-based NADPH oxidase in human aortic smooth muscle cells. Arterioscler Thromb Vasc Biol 30: 105–112, 2010 [DOI] [PubMed] [Google Scholar]
- 34.McCrohan MB, Huang SW, Sleasman JW, Klein PA, and Kao KJ. Plasma thrombospondin as an indicator of intravascular platelet activation in patients with vasculitis. Thromb Haemost 58: 850–852, 1987 [PubMed] [Google Scholar]
- 35.Moore KJ, Kunjathoor VV, Koehn SL, Manning JJ, Tseng AA, Silver JM, McKee M, and Freeman MW. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J Clin Invest 115: 2192–2201, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moura R, Tjwa M, Vandervoort P, Van Kerckhoven S, Holvoet P, and Hoylaerts MF. Thrombospondin-1 deficiency accelerates atherosclerotic plaque maturation in ApoE−/− mice. Circ Res 103: 1181–1189, 2008 [DOI] [PubMed] [Google Scholar]
- 37.Nishita M, Wang Y, Tomizawa C, Suzuki A, Niwa R, Uemura T, and Mizuno K. Phosphoinositide 3-kinase-mediated activation of cofilin phosphatase Slingshot and its role for insulin-induced membrane protrusion. J Biol Chem 279: 7193–7198, 2004 [DOI] [PubMed] [Google Scholar]
- 38.Nyati MK, Morgan MA, Feng FY, and Lawrence TS. Integration of EGFR inhibitors with radiochemotherapy. Nat Rev Cancer 6: 876–885, 2006 [DOI] [PubMed] [Google Scholar]
- 39.Orth JD, Krueger EW, Weller SG, and McNiven MA. A novel endocytic mechanism of epidermal growth factor receptor sequestration and internalization. Cancer Res 66: 3603–3610, 2006 [DOI] [PubMed] [Google Scholar]
- 40.Ranayhossaini DJ, Rodriguez AI, Sahoo S, Chen BB, Mallampalli RK, Kelley EE, Csányi G, Gladwin MT, Romero G, and Pagano PJ. Selective recapitulation of conserved and nonconserved regions of putative NOXA1 protein activation domain confers isoform-specific inhibition of Nox1 oxidase and attenuation of endothelial cell migration. J Biol Chem 288: 36437–36450, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rejman J, Oberle V, Zuhorn IS, and Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J 377: 159–169, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ridley AJ, Paterson HF, Johnston CL, Diekmann D, and Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 70: 401–410, 1992 [DOI] [PubMed] [Google Scholar]
- 43.Sarkar K, Kruhlak MJ, Erlandsen SL, and Shaw S. Selective inhibition by rottlerin of macropinocytosis in monocyte-derived dendritic cells. Immunology 116: 513–524, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sheehan AL, Carrell S, Johnson B, Stanic B, Banfi B, and Miller FJ., Jr. Role for Nox1 NADPH oxidase in atherosclerosis. Atherosclerosis 216: 321–326, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stenina OI, Krukovets I, Wang K, Zhou Z, Forudi F, Penn MS, Topol EJ, and Plow EF. Increased expression of thrombospondin-1 in vessel wall of diabetic Zucker rat. Circulation 107: 3209–3215, 2003 [DOI] [PubMed] [Google Scholar]
- 46.Stocker R. and Keaney JF., Jr. Role of oxidative modifications in atherosclerosis. Physiol Rev 84: 1381–1478, 2004 [DOI] [PubMed] [Google Scholar]
- 47.Subtil A, Hemar A, and Dautry-Varsat A. Rapid endocytosis of interleukin 2 receptors when clathrin-coated pit endocytosis is inhibited. J Cell Sci 107 (Pt 12): 3461–3468, 1994 [DOI] [PubMed] [Google Scholar]
- 48.Suzuki H, Kurihara Y, Takeya M, Kamada N, Kataoka M, Jishage K, Ueda O, Sakaguchi H, Higashi T, Suzuki T, Takashima Y, Kawabe Y, Cynshi O, Wada Y, Honda M, Kurihara H, Aburatani H, Doi T, Matsumoto A, Azuma S, Noda T, Toyoda Y, Itakura H, Yazaki Y, Kodama T, et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature 386: 292–296, 1997 [DOI] [PubMed] [Google Scholar]
- 49.Swanson JA. Phorbol esters stimulate macropinocytosis and solute flow through macrophages. J Cell Sci 94 (Pt 1): 135–142, 1989 [DOI] [PubMed] [Google Scholar]
- 50.Tkachenko E, Lutgens E, Stan RV, and Simons M. Fibroblast growth factor 2 endocytosis in endothelial cells proceed via syndecan-4-dependent activation of Rac1 and a Cdc42-dependent macropinocytic pathway. J Cell Sci 117: 3189–3199, 2004 [DOI] [PubMed] [Google Scholar]
- 51.Tseng HY, Liu ZM, and Huang HS. NADPH oxidase-produced superoxide mediates EGFR transactivation by c-Src in arsenic trioxide-stimulated human keratinocytes. Arch Toxicol 86: 935–945, 2012 [DOI] [PubMed] [Google Scholar]
- 52.Upston JM, Niu X, Brown AJ, Mashima R, Wang H, Senthilmohan R, Kettle AJ, Dean RT, and Stocker R. Disease stage-dependent accumulation of lipid and protein oxidation products in human atherosclerosis. Am J Pathol 160: 701–710, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoshida S, Hoppe AD, Araki N, and Swanson JA. Sequential signaling in plasma-membrane domains during macropinosome formation in macrophages. J Cell Sci 122: 3250–3261, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu XD, Zhuang Y, Ben JJ, Qian LL, Huang HP, Bai H, Sha JH, He ZG, and Chen Q. Caveolae-dependent endocytosis is required for class A macrophage scavenger receptor-mediated apoptosis in macrophages. J Biol Chem 286: 8231–8239, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zielonka J, Vasquez-Vivar J, and Kalyanaraman B. Detection of 2-hydroxyethidium in cellular systems: a unique marker product of superoxide and hydroethidine. Nat Protoc 3: 8–21, 2008 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.