

Abstract
CTCF is an architectural protein with a critical role in connecting higher-order chromatin folding in pluripotent stem cells. Recent reports have suggested that CTCF binding is more dynamic during development than previously appreciated. Here, we set out to understand the extent to which shifts in genome-wide CTCF occupancy contribute to the 3D reconfiguration of fine-scale chromatin folding during early neural lineage commitment. Unexpectedly, we observe a sharp decrease in CTCF occupancy during the transition from naïve/primed pluripotency to multipotent primary neural progenitor cells (NPCs). Many pluripotency gene-enhancer interactions are anchored by CTCF, and its occupancy is lost in parallel with loop decommissioning during differentiation. Conversely, CTCF binding sites in NPCs are largely preexisting in pluripotent stem cells. Only a small number of CTCF sites arise de novo in NPCs. We identify another zinc finger protein, Yin Yang 1 (YY1), at the base of looping interactions between NPC-specific genes and enhancers. Putative NPC-specific enhancers exhibit strong YY1 signal when engaged in 3D contacts and negligible YY1 signal when not in loops. Moreover, siRNA knockdown of Yy1 specifically disrupts interactions between key NPC enhancers and their target genes. YY1-mediated interactions between NPC regulatory elements are often nested within constitutive loops anchored by CTCF. Together, our results support a model in which YY1 acts as an architectural protein to connect developmentally regulated looping interactions; the location of YY1-mediated interactions may be demarcated in development by a preexisting topological framework created by constitutive CTCF-mediated interactions.
The spatial organization of the genome within the three-dimensional nucleus is dynamic during development and linked to spatiotemporal regulation of gene expression. Recent advances in proximity-ligation and deep-sequencing technologies have enabled the interrogation of genome organization at a genome-wide scale and nucleosome resolution (de Laat and Dekker 2012; Denker and de Laat 2016). Within individual chromosomes, open chromatin and active genes tend to spatially cluster into “A” compartments, whereas closed, inactive chromatin spatially segregates into “B” compartments (Lieberman-Aiden et al. 2009; Rao et al. 2014). Although compartments undergo marked reorganization during cell fate transitions, the restructuring only modestly correlates with changes in gene expression, suggesting that transcription is not deterministically regulated at the compartment level (Dixon et al. 2015). Within compartments, the mammalian genome is partitioned into megabase-sized topologically associating domains (TADs) that are largely invariant across cell types (Dixon et al. 2012; Nora et al. 2012). TAD structural integrity is critical for proper gene expression; perturbation of TAD boundaries leads to ectopic enhancer looping and aberrant activation of nontarget genes (Dowen et al. 2014; Lupiáñez et al. 2015; Narendra et al. 2015; Flavahan et al. 2016; Hnisz et al. 2016). Finally, at the sub-megabase scale within TADs, two classes of highly dynamic architectural features exist: (1) small-scale contact domains termed sub-TADs (Phillips-Cremins et al. 2013; Dowen et al. 2014; Rao et al. 2014), and (2) loops (Rao et al. 2014). Looping interactions and sub-TADs often link genes to developmentally regulated enhancers and are markedly reorganized between cellular states (Tolhuis et al. 2002; Sanyal et al. 2012; Phillips-Cremins et al. 2013; Dowen et al. 2014; Smith et al. 2016). Thus, the emerging model is that mammalian genomes are arranged into a nested hierarchy of unique structural features, of which the finer, sub-megabase scale configurations within TADs are critical for the proper activation and inactivation of genes during development.
CCCTC-binding factor (CTCF) is a ubiquitously expressed zinc finger protein implicated in the regulation of a wide range of genome functions including transcription, insulation, splicing, replication, recombination, and repair (Ong and Corces 2014). A leading hypothesis is that CTCF's diverse regulatory roles can be explained by a unifying mechanism in which it functions as an architectural protein to connect higher-order chromatin configurations (Phillips and Corces 2009). CTCF is found at the base of looping interactions, and knockdown of the protein abrogates chromatin connections (Kurukuti et al. 2006; Hadjur et al. 2009; Handoko et al. 2011; Splinter et al. 2012). In a recent high-resolution, genome-wide proximity ligation study, approximately 10,000 looping interactions were reported in human cells. Importantly, of the subset of loops bound by CTCF with clear consensus sequences, 92% were anchored by consensus sequences pointed toward each other in a convergent orientation (Rao et al. 2014; Vietri Rudan et al. 2015). CTCF-mediated interactions can be disrupted by mutation, inversion, and/or deletion of the CTCF motif, indicating that consensus orientation is a critical contributing factor in loop establishment and/or maintenance (de Wit et al. 2015; Guo et al. 2015; Sanborn et al. 2015). CTCF is also enriched at the boundaries of TADs (Dixon et al. 2012; Nora et al. 2012), and deletion or inversion of these motifs can perturb domain boundaries and disrupt nearby gene expression (Dowen et al. 2014; de Wit et al. 2015; Guo et al. 2015; Lupiáñez et al. 2015; Narendra et al. 2015; Sanborn et al. 2015; Flavahan et al. 2016). Together, these data indicate the CTCF is an architectural protein that functions in an orientation-dependent manner to organize mammalian genomes across several length scales.
Genome-wide CTCF occupancy patterns have been mapped across more than 100 mammalian cell types (Kim et al. 2007; Chen et al. 2008; Cuddapah et al. 2009; Wang et al. 2012; Maurano et al. 2015). Early studies comparing ChIP-seq signal between two or three cell types reported that CTCF binding was largely invariant, with 65%–90% of approximately 35,000 binding sites detected in all cellular states queried (Kim et al. 2007; Cuddapah et al. 2009). More recent studies comparing CTCF occupancy across 40 cell lines showed a range of 35,000–75,000 binding sites per cellular state, with a total of about 110,000 possible unique genomic locations (Maurano et al. 2015). Notably, at most 20% of possible unique sites were classified as constitutive when comparing 40 cellular states, indicating that CTCF binding is more dynamic during development than previously reported (Wang et al. 2012; Maurano et al. 2015). Thus, it is critically important to understand the dynamic patterns of CTCF binding and whether/how they are causally linked to chromatin architecture and gene expression during cellular state transitions in development.
Recent genetic studies have confirmed that CTCF is essential for proper spatiotemporal gene expression in the developing mammalian brain. Conditional knockdown of Ctcf at early, embryonic stages of mouse development triggered marked apoptosis of primary neural progenitor cells (NPCs), premature neurogenesis, and disruption of tissue architecture (Watson et al. 2014). Moreover, Ctcf knockout in post-mitotic cortical and hippocampal neurons (Hirayama et al. 2012), or the hippocampus more broadly (Sams et al. 2016), resulted in defects in gene expression, synaptic connectivity, and learning and memory behavior. Finally, CTCF binding is also required for the differential expression of protocadherin (Pcdh) isoforms that enable branching neurites to self-recognize (Guo et al. 2012). Together, these studies indicate that CTCF plays an essential role in early neural development and highlight the importance of unraveling the currently unknown mechanisms linking occupancy with genome architecture and expression in the brain.
Here, we set out to understand the dynamic CTCF occupancy landscape and how it is linked to the restructuring of fine-scale chromatin architecture at the earliest stages of the establishment of neuronal gene expression programs. We used well-established cellular models of early neural lineage commitment: (1) mouse embryonic stem (ES) cells cultured in GSK3/MEK inhibitors (“2i” conditions) representing a state of naïve pluripotency from the earliest stages of a preimplantation embryo (Ying et al. 2008); (2) ES cells cultured in serum/LIF representing a slightly more mature state of pluripotency with increased poising of developmentally regulated genes (Galonska et al. 2015); and (3) primary multipotent neural progenitor cells (NPCs) representing the earliest departure from pluripotency and commitment to lineages in the mammalian brain (Sharma et al. 2016). By integrating ChIP-seq data, 3D genome folding maps, and functional knock down studies, we aimed to uncover several new organizing principles governing higher-order genome folding during neural lineage commitment.
Results
CTCF engagement with the genome decreases during neural development
To investigate CTCF dynamics during the earliest stages of neural development, we performed ChIP-seq in NPCs derived from neonatal mouse brains as well as embryonic stem (ES) cells cultured under both 2i/LIF (2i) and serum/LIF (serum) conditions. The three cellular states were chosen to capture the initial establishment of neural gene expression programs and to benchmark the changes against a presumably less dramatic transition between naïve and primed/mature pluripotency. Equivalent genetic backgrounds were achieved by utilizing v6.5 ES cells (C57Bl6×129SvJae) and NPCs from mice maintained on a mixed C57Bl6/129SvJae background (Stadtfeld et al. 2008). We first noticed that the number of CTCF binding sites decreased in a stepwise manner during the transition from naïve pluripotency to multipotency, with the sharpest drop in binding sites between ES serum and NPC conditions (Fig. 1A). To further explore dynamic CTCF during neural development, we utilized available ENCODE CTCF ChIP-seq data sets from the mouse (Shen et al. 2012). Consistent with trends in our cellular models, published ENCODE CTCF ChIP-seq peaks also showed a global decrease in adult brain regions (cortex, cerebellum, olfactory) compared to the E14.5 brain tissue (Fig. 1B). Notably, when we investigated other developmental lineages, we found that CTCF binding can display the opposite trend, in some cases increasing between the embryonic and adult stages (Supplemental Fig. 1A,B). Thus, although CTCF occupancy appears to decrease during the transition from pluripotency to early neuronal lineage commitment, it is not a pervasive trend across all developmental lineages.
Figure 1.
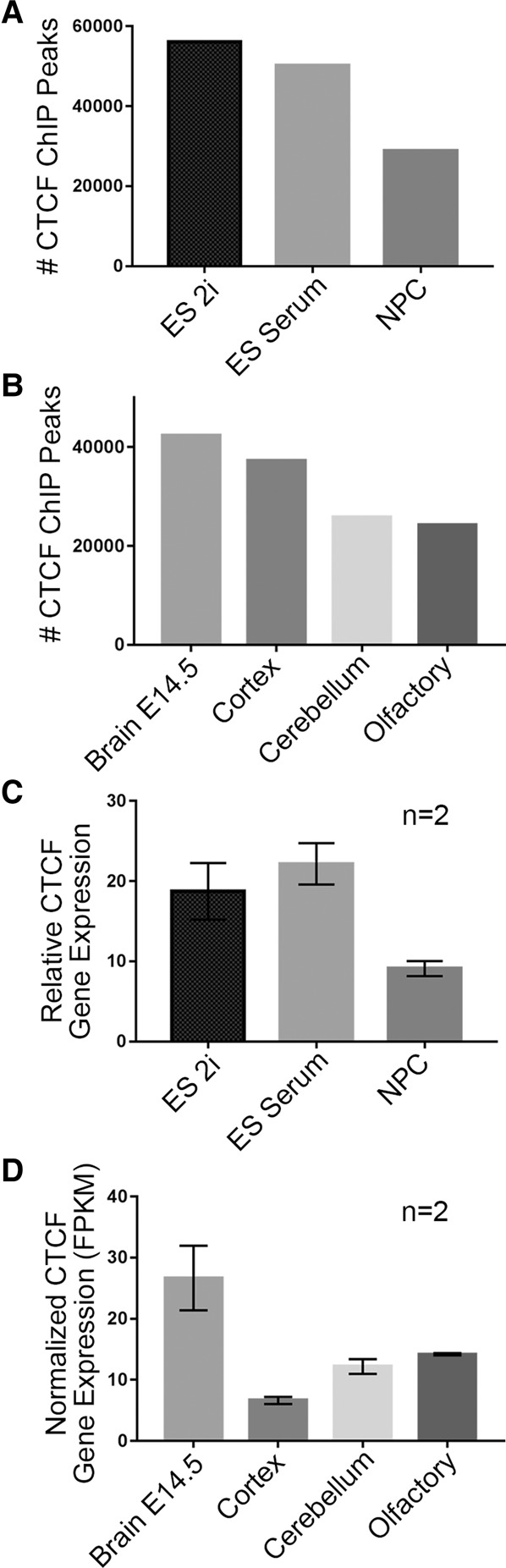
CTCF binding and expression decrease during neural development. (A) Number of CTCF ChIP-seq peaks called across the ES 2i, ES serum, and NPC cellular states. (B) Number of CTCF ChIP-seq peaks across several mouse ENCODE brain tissues (Shen et al. 2012). (C) Relative CTCF gene expression across three developmental cell types (error bars represent 1 SD from mean across two replicates). (D) Normalized CTCF gene expression (FPKM) across mouse ENCODE brain tissues (error bars represent 1 SD from mean across two replicates) (Shen et al. 2012).
To gain insight into why NPCs have a unique pattern of decreased CTCF occupancy during early neuronal lineage commitment, we next conducted an analysis of CTCF gene expression and protein levels. We observed a general accordance between the ChIP-seq and RNA-seq results in our cellular models: CTCF gene expression decreased between the pluripotent stem cell states and multipotent NPCs (Fig. 1C) and also between the embryonic mouse brain and mature adult brain regions (Fig. 1D). Moreover, Western blot analysis of CTCF protein levels showed a similar decrease in NPCs compared to pluripotent ES cells (Supplemental Fig. 1C). Corroborating our results, while this manuscript was under review, an independent study also reported a decrease in CTCF protein levels in whole mouse brains during the transition from E15 to post-natal week 1 (Sams et al. 2016). Sams et al. also identified differential CTCF levels across neurons, astrocytes, and oligodendrocytes from the hippocampus, highlighting that we cannot rule out the possibility that heterogeneity in cells derived from ENCODE tissues may contribute to the aggregate decrease in CTCF levels. Our NPC cultures exhibited a highly consistent morphology throughout the population, and >90% were SOX2 positive (Beagan et al. 2016), suggesting that our NPC preparations were substantially less heterogeneous than brain tissue lysates. Our data indicate that CTCF gene and protein expression levels decrease in the transition from pluripotency to multipotent neural progenitor cells in parallel with a global decrease in the number of genome-wide CTCF binding sites.
CTCF occupancy in NPCs is largely preestablished in the pluripotent state
To better understand the CTCF sites that are dynamic among our cellular states, we parsed CTCF peaks present in the ES 2i, ES serum, and NPC conditions into classes based on their cell-type–specific occupancy (Fig. 2A; Supplemental Methods). We identified 56,138, 50,185, and 28,860 binding sites in ES 2i, ES serum, and NPCs, respectively, with a total of 60,688 unique, nonredundant sites across all three cell types. We found that approximately 44% of CTCF sites (n = 26,435) displayed constant occupancy across our three cell types of interest and were thus classified as “constitutive.” We also explored several classes of dynamically occupied CTCF sites, including: (1) the “2i only” class present in naïve pluripotency conditions and lost in the transition to a more primed/mature pluripotent cellular state; (2) the “2i+serum” class present across pluripotency conditions and lost in NPCs; and (3) the “NPC-only” class arising only upon the departure from pluripotency. We confirmed the validity of our parsing scheme for our four CTCF classes of interest by plotting the composite ChIP-seq signal for all three cell types centered on the midpoint genomic location of a given class (Fig. 2B; Supplemental Fig. 2). The ChIP-seq pileup plots indicate that constitutive CTCF binding sites display markedly higher occupancy signal than sites that are dynamically altered upon changes in cellular state. These results confirm and extend recent reports suggesting that there is a larger class of dynamically occupied CTCF sites than previously appreciated (Shen et al. 2012; Maurano et al. 2015).
Figure 2.

Sites bound by CTCF in NPCs are predominantly preexisting from earlier stages of development. (A) Classification of CTCF binding sites parsed between three developmental cell states. (B) Composite CTCF ChIP-seq signal in NPCs (green), ES serum (blue), and ES 2i (red) centered around the peaks of Constitutive, 2i+Serum, NPC-only, and 2i only CTCF classes. (C) Stacked bar plot representing the distribution of CTCF binding classes across ES cells in 2i, ES cells in serum, and NPCs. (D) Theorized landscape plot depiction of constitutive and dynamic CTCF during the early time points of development. Colors represent same CTCF classes as presented in C. (E) Library read depth is comparable across conditions. After redundant read removal and down-sampling, 11 million reads were utilized for the CTCF ChIP-seq analysis of each cell type.
We next sought to understand dynamic CTCF occupancy patterns in the naïve to mature pluripotency transition and the mature pluripotency to multipotency transition. At the outset of our analysis, we hypothesized that CTCF binding may decrease severely between 2i and serum conditions due to the known hypomethylated state of naïve pluripotent stem cells (Ficz et al. 2013; Habibi et al. 2013; Leitch et al. 2013), but we observed only a relatively minor reduction in CTCF occupancy between ES 2i and ES serum (“2i only” class, n = 8832). In contrast, we noticed that the number of CTCF sites lost between ES serum and NPCs nearly matched that of the constitutive class (“2i+Serum” class, n = 20,068), suggesting that the transition from pluripotency to multipotent progenitor cells represents a critical developmental window in establishing the neural CTCF landscape. Importantly, the number of “NPC-only” CTCF sites that arose during differentiation was relatively small (n = 1119), indicating that the vast majority of CTCF peaks called in NPCs were already present in the pluripotent cell types (Fig. 2C). Our results suggest that the CTCF occupancy landscape in NPCs does not result from a marked reshuffling and/or extensive de novo acquisition of new CTCF binding sites. Rather, a large proportion of CTCF sites are preestablished at least as early in development as naïve pluripotency and selectively lost in early neural lineage commitment (Fig. 2D, orange and purple classes). ChIP-seq experiments were conducted in the same batch, sequenced on the same flow cell, and down-sampled to the same read depth to attenuate technical artifacts that might influence our observed results (Fig. 2E).
The 3D genome is reconfigured during early neural development
CTCF has a well-established role in connecting long-range looping interactions (de Wit et al. 2015; Guo et al. 2015). Given the large number of CTCF peaks that are dynamic across development, we sought to investigate how chromatin folding is altered as a function of occupancy during each cell fate transition. We generated fine-scale chromatin architecture maps (∼4- to12-kb matrix resolution) across >7 Mb of the mouse genome surrounding key developmentally regulated genes with Chromosome-Conformation-Capture-Carbon-Copy (5C) and high-throughput sequencing. In a previous study focused on chromatin folding during somatic cell reprogramming, we generated 5C libraries (n = 2 biological replicates) in ES serum, ES 2i, and NPC conditions (Beagan et al. 2016). Here, we elected to begin our 3D analyses and mechanistic exploration with the raw reads from our published 5C libraries because they were genetically and culture condition matched to the pellets used to generate our RNA-seq and CTCF ChIP-seq libraries.
Building on the foundation of our previously published 5C analysis pipeline (Beagan et al. 2016), we further developed and applied a new set of computational methods to better resolve punctate looping interactions present within each cell type. We normalized the intrinsic biases in 5C data, corrected for library complexity and sequencing depth differences, and attenuated spatial noise via a 16-kb blocked smoothing window. The resultant “Relative Interaction Frequency” data binned at 4-kb matrix resolution exhibited high reproducibility between biological replicates (Fig. 3A; Supplemental Table 1; Supplemental Fig. 3A). Additionally, our 5C data showed strong biological concordance with published Hi-C data from the murine cortex (Dixon et al. 2012) across a 1-Mb region surrounding the Sox2 gene in NPCs (Supplemental Fig. 3B).
Figure 3.

Dynamic classes of 3D interactions arise during neural lineage commitment. (A) Heatmaps displaying the relative chromatin contact frequency in a 1-Mb region surrounding the Sox2 gene in ES 2i, ES serum, and NPCs. Color bars range from low (gray) to high (red/black). (B) Schematic depiction of donut (blue) and lower left (green) expected background models. (C–E) Expected background heatmaps for the region surrounding the Sox2 gene. (C) Donut filter, (D) lower left filter, and (E) maximum value of donut and lower left filters. (F) Interaction score heatmaps at the Sox2 locus. Color bar ranges from low (blue) to high (red/black). (G) Schematic of looping classes parsed by their dynamic behavior across three cellular states. (H) Scatter plot of 5C interaction scores for each pixel classified as part of a looping interaction across the ES 2i, ES serum, and NPC states (Supplemental Methods). (I) Number of significant looping clusters in each dynamic 3D interaction class. (J) Box plots representing interaction scores across each cell type for the pixels classified into each looping class. (K) Visualization of a Sox2-pluripotency enhancer interaction in relative interaction frequency heatmaps (top left), interaction score heatmaps (bottom left), and classified loop clusters (right).
Looping interactions can be detected in 5C heatmaps as concentrated points of high interaction frequency compared to the surrounding local background (Rao et al. 2014). Although one universal distance-dependence expected model could be computed on 5C data, we have found that application of a global expected model often leads to over- or underestimation of looping strength. To compute a local expected interaction frequency, we applied the “donut” and “lower-left” background filters (Fig. 3B, blue and green outlines, respectively) recently proposed by Lieberman-Aiden and colleagues (Rao et al. 2014). Local background filters capture the more nuanced aspects of the distance-dependence expected interaction frequency and the TAD/subTAD domain structure (Fig. 3C,D; Supplemental Figs. 4, 5). To take a conservative approach for loop detection, we corrected our 5C counts with the maximum of the two filters (Fig. 3E; Supplemental Fig. S6). We next modeled the “expected-corrected interaction frequency” data as a continuous random variable with a logistic distribution (Supplemental Fig. S7). The resultant P-values for each pixel were converted to an “Interaction Score” (IS = −10*log2[P-value]), allowing for systematic comparison of looping signal within and between 5C regions and experiments. Punctate looping structures were readily apparent in the uncorrected and interaction score heatmaps (Fig. 3A,F).
As a critical first step toward understanding the relationship between CTCF occupancy and 3D chromatin architecture changes, we computationally parsed looping interactions into subclasses based on their interaction score in each cell type (Fig. 3G). Pixels in which both replicates of each biological condition similarly passed or failed each threshold (Supplemental Fig. S8A) were classified into one of seven looping classes (Fig. 3G,H). Thresholds were chosen so that our top five largest dynamic looping classes achieved an empirical false discovery rate less than 15% (Supplemental Methods; Supplemental Fig. S8B–D). Consistent with previous reports (Rao et al. 2014), we noticed that pixels of the same looping class were often adjacent to each other and therefore could be clustered together into a contiguous architectural feature. Altogether, we identified several classes of 3D interactions (Fig. 3I) and elected to focus our analysis on three main groups: (1) 141 loops present in all three cell types (“Constitutive,” gray class); (2) 46 loops present in both ES 2i and ES serum but lost in NPCs (“2i+Serum,” purple class); and (3) 75 loops specific to the NPC state (“NPC-only,” green class). We confirmed that our looping class interaction scores trended across the three cellular states in a manner that was commensurate with their intended classification (Fig. 3J). Visual inspection of the data confirmed that gold-standard looping interactions, such as the “NPC-only” interaction between the Sox2 gene and an upstream regulatory element were accurately detected, clustered, and classified (Fig. 3K, colored green). Finally, we assessed the orientation of the CTCF motifs at loop anchors and confirmed that loops in all three cell types were highly enriched for convergently oriented motifs compared with divergent or tandem orientations (Supplemental Fig. 9A,B). Altogether, our analysis pipeline allowed us to accurately identify and visualize looping interactions critical to each cellular state within our 5C regions.
CTCF binding correlates with loss of 3D interactions during the departure from pluripotency
We next investigated the relationship between the significant loss of CTCF binding between the pluripotent and multipotent states and coincident architectural rearrangements. Sox2 forms a pluripotency-specific loop with a putative ES-specific enhancer ∼120 kb downstream that is essential for proper expression of the gene in ES cells (Fig. 4A,B, magenta arrowhead; Li et al. 2014). We identified several “2i+Serum” CTCF sites at the putative ES-specific enhancer (Fig. 4B, green boxes on the x-axis). During the departure from pluripotency, CTCF binding is lost, and the looping interaction connecting the Sox2 gene to the putative ES-specific enhancer concurrently breaks apart (Fig. 4B,C; Supplemental Fig. 10A,B, red arrow). In concordance with these locus-specific examples, “2i+Serum” looping interactions across our 5C regions were enriched with “2i+Serum” CTCF sites (Fig. 4D). Together, these data suggest that the loss of CTCF occupancy at key looping interactions during the departure from pluripotency is accompanied by a decrease in looping strength.
Figure 4.

Pluripotency interactions that disengage in multipotent NPCs display reduced CTCF occupancy. (A) Global view of relative interaction frequency heatmaps of 1 Mb surrounding the Sox2 gene. (B) Zoom in highlighting a strong pluripotency-specific looping interaction between Sox2 and an ES-specific enhancer. CTCF binds at both loop anchors (note green boxes). Heatmaps include relative interaction frequency (top) and background corrected interaction score (bottom). The Sox2 gene is colored green. (C) Classified interaction clusters are plotted above relevant ChIP-seq tracks. (D) Fold enrichment/depletion of chromatin features in 2i+serum and NPC-only looping interaction classes compared to presence in background interactions. P-values included in each entry are calculated using Fisher's exact test. (E) Stacked bar plot contrasting the proportion of loops connected by CTCF in one or both anchoring fragments versus not anchored by CTCF.
We questioned some conflicting observations: although the loss of CTCF often coincides with the loss of a looping interaction (Fig. 4D) and NPCs have substantially fewer CTCF peaks than the pluripotent states (Fig. 2C), NPCs have roughly the same number of looping interactions as ES serum/ES 2i in the genomic regions covered by our 5C primers (Fig. 3J). Notably, when we explored the percentage of key looping classes anchored by CTCF, we found that almost 40% of “NPC-only” interactions were not anchored by CTCF binding, whereas only <5% and <10% of “constitutive” and “2i+serum” interactions lacked CTCF binding, respectively (Fig. 4E, black bars). Moreover, “NPC-only” looping interactions across our 5C regions were not enriched for “NPC-only” CTCF binding (Fig. 4D). These results suggested that CTCF might not be the critical architectural protein connecting developmentally regulated looping interactions that arise de novo in differentiated NPCs.
YY1 binding is enriched at looping interactions connecting NPC-specific genes and distal regulatory elements
We posited that an additional class of looping proteins might connect developmentally regulated “NPC-only” interactions. We searched for the presence of candidate architectural proteins at the base of 3D interactions between genes critical to the NPC phenotype and their putative target enhancers. Since its discovery, the Nes gene has been a widely referenced marker of proliferating NPCs (Lendahl et al. 1990; Park et al. 2010). Therefore, we began our search by investigating published NPC ChIP-seq libraries for their signal at the “NPC-only” long-range interactions between Nes and Bcan and a putative NPC-specific enhancer ∼200 kb downstream from the genes (Fig. 5A,B, magenta arrowhead; Supplemental Fig. 11A,B, green box). As expected, Nes and Bcan expression markedly increased in NPCs in concert with the increase in 3D contact with the putative enhancer element (Fig. 5C). Interestingly, we observed strong occupancy of the zinc-finger protein Yin Yang 1 (YY1) at the putative NPC-specific enhancer (Fig. 5B,D, green box, x-axis; Supplemental Fig. 11B). Moreover, globally across all our 5C loops, we observed that YY1 was strongly enriched in “NPC-only” 3D interactions compared to background nonloops (Fig. 5E). These data demonstrate that YY1 binding is enriched at “NPC-only” looping interactions.
Figure 5.

YY1 is enriched at NPC-specific enhancers that form developmentally regulated loops. (A) Relative interaction frequency heatmaps of the global view of 1 Mb surrounding Nes (top), and zoom in of 400 kb surrounding nestin with putative NPC enhancer annotations (bottom). Nes (upstream) and Bcan (downstream) genes are colored green. (B) Zoom-in interaction score heatmaps of the Nes/Bcan genes interacting with a downstream putative NPC enhancer. Heatmaps are overlaid with ChIP-seq tracks of CTCF in NPCs and YY1 in ES serum and NPCs. The Nes (upstream) and Bcan (downstream) genes are colored green. (C) Relative gene expression of Nes and Bcan across ES 2i, ES serum, and NPC cellular states. (D) Interaction cluster outlines of the loop boxed in magenta in B. Plot is overlaid with ChIP-seq tracks of H3K27ac, YY1, and CTCF in the ES 2i, ES serum, and NPC conditions. Cluster outline classifications include NPC-only (green), serum+NPC (yellow), and constitutive (gray). (E) Fold enrichment/depletion of the presence of chromatin features in NPC-only interaction class compared to the presence in background. P-values are computed with Fisher's exact test and listed in each entry. (F,G) YY1 ChIP-seq signal in NPCs (green), ES serum (blue), and ProB cells (red), centered at (F) putative NPC enhancers at the base of NPC-only loops, and (G) NPC enhancers that do not fall at the base of any looping interactions. (H) YY1 binding sites parsed by their occupancy across ES cells, NPCs, and ProB cells. (I) Fold enrichment/depletion of YY1 peak classes and NPC enhancers parsed based on the presence/absence of CTCF/YY1 in NPC-only loops compared to their presence in background interactions. (J) Stacked bar plot of the breakdown of ES and NPC enhancers that are bound with confidence by a combination of CTCF and/or YY1.
To better understand the role for YY1 in NPC looping, we parsed our putative genome-wide NPC-specific enhancers (Supplemental Methods) into those that engage in “NPC-only” loops (Fig. 5F) and those that do not participate in long-range interactions (Fig. 5G). We found strong YY1 signal at NPC-specific enhancers engaged in “NPC-only” looping interactions and negligible YY1 binding at NPC-specific enhancers that do not loop. Similarly, YY1 signal was also enriched at NPC-specific and constitutively expressed genes in looping interactions compared to nonloops (Supplemental Fig. 12A–C). These data suggest that YY1 is present at NPC regulatory elements engaged in 3D interactions and support our working hypothesis that YY1 might serve as an architectural protein to connect NPC-specific genes and enhancers.
We next set out to understand YY1 occupancy across cellular states and its cobinding with respect to CTCF. In the case of CTCF, 47%, 53%, and 92% of classified binding sites were constitutive (n = 26,435) in the ES 2i, ES serum, and NPC cellular conditions, respectively (Fig. 2A). In contrast, a markedly lower proportion of classified YY1 sites were constitutive among ES serum, NPCs, and primary pro-B cells (36%, 39%, and 25%, respectively; n = 3474), indicating that YY1 might exhibit more cell-type–specific binding than CTCF (Fig. 5H). To understand if YY1 colocalizes with CTCF, we explored pileup plots of average ChIP signal over the different classes of dynamic YY1 binding sites (Supplemental Fig. 12F–H). CTCF signal was negligible at all classes of YY1 binding, suggesting that YY1 and CTCF do not, for the most part, directly colocalize (Supplemental Fig. 12F,G). We also observed a striking overlap of YY1 with H3K27ac signal (Supplemental Fig. 12H), which prompted us to query the overlap of cell-type–specific genes and regulatory elements with CTCF and YY1 and how this impacts looping. Importantly, NPC-specific enhancers were strongly enriched in “NPC-only” looping interactions when bound by YY1 without CTCF, but not when bound by CTCF without YY1 (Fig. 5I). Similarly, constitutive and NPC-specific genes were also significantly enriched in “constitutive” and “NPC-only” looping interactions, respectively, when bound by YY1 without CTCF (Supplemental Fig. 12K,M). Together, these data indicate that NPC-specific enhancers, constitutive genes, and NPC-specific genes can engage in strong 3D interactions in NPCs when bound by YY1 in the absence of CTCF.
In contrast to the role for YY1 at NPC regulatory elements, the role for YY1 at ES-specific genes and enhancers was less clear. YY1 occupancy signal was low and diffuse across putative ES-specific regulatory elements and did not show a clear preference between those engaged in loops versus nonloops (Supplemental Fig. 12D,E). By focusing on distal cell-type–specific regulatory elements that overlap binding sites of CTCF, YY1, or both (and not considering those bound by neither), we observed that the majority of architectural protein-bound NPC-specific enhancers were bound by YY1 without CTCF, whereas the majority of ES-specific enhancers were bound by CTCF without YY1 (Fig. 5J). Additionally, “2i+serum” looping interactions were enriched for ES-specific enhancers regardless of CTCF and YY1 occupancy, whereas ES-specific genes were only enriched in “2i+serum” looping interactions when bound by CTCF without YY1 (Supplemental Fig. 12J,L). It is not clear to what extent our observed differences between ES and NPC YY1 are due to (1) an increased reliance by ES cells on CTCF as the primary architectural protein; (2) different ChIP methods between the ES and NPC YY1 data sets (Supplemental Methods); or (3) a different regulatory role for YY1 in the two cell types. Thus, although our data indicate that YY1 might be important for developmentally regulated looping in somatic cells, we cannot conclusively define or rule out any role for YY1 in mediating loops in ES cells.
YY1-mediated developmentally regulated looping interactions are often nested within a larger framework mediated by constitutive CTCF
While exploring the gene-enhancer interaction formed by the Nes and Bcan genes, we noticed a “constitutive” interaction at the outer corner of the larger “NPC-only” and “Serum+NPC” looping interaction cluster (Fig. 5D). At the base of this “constitutive” interaction, we identified convergently oriented constitutive CTCF sites (Fig. 5D, lower red boxes on both axes, consensus orientation not shown). We hypothesized that a subset of constitutive CTCF sites might form loops that create a preexisting topological framework ,within which critical developmentally dynamic chromatin interactions form (Symmons et al. 2014; Tang et al. 2015; Smith et al. 2016). Constitutive topological frameworks may be critical for proper gene expression because they create insulated neighborhoods around coregulated genes and enhancers that will interact during subsequent differentiation steps (Dowen et al. 2014). Consistent with this idea, constitutive CTCF was the most significantly enriched chromatin mark underlying “constitutive” interactions (Fig. 6A). Importantly, NPC genes were also slightly enriched in “constitutive” interactions (Fig. 6A), corroborating our observation that NPC gene-enhancer loops connected by YY1 often appear adjacent to and nested within constitutive looping events.
Figure 6.

YY1 connects neural regulatory elements nested within and adjacent to a framework of constitutive CTCF-mediated interactions. (A) Fold enrichment/depletion of chromatin regulatory elements in the constitutive looping class compared to background interactions. P-values are computed with Fisher's exact test and listed in each entry. (B,C) Relative interaction frequency heatmaps of ∼1 Mb region (B) and ∼200 kb region (C) surrounding the Olig1 and Olig2 genes in ES 2i, ES serum, and NPCs. Heatmaps in C are overlaid with ChIP-seq tracks of H3K27ac in ES serum cells and NPCs. (D) Relative gene expression of Olig1 and Olig2 genes across the ES 2i, ES serum, and NPC cellular states. (E) Zoom-in interaction score heatmaps of looping interactions between the Olig1 and Olig2 genes and surrounding putative NPC enhancers (green boxes). (F) Zoom-in cluster map of classified looping interactions at Olig2 and Olig1 with NPC only (green), serum+NPC (blue), and constitutive class looping interactions (gray). (G–I) Heatmaps and cluster map at different length scales around the Sox2 gene in ES 2i, ES serum, and NPCs. Zoom-in heatmaps of relative interaction frequencies (G) and background corrected interaction scores (H) across ∼500 kb downstream from Sox2. Relative interaction frequency heatmaps are overlaid H3K27ac tracks. Interaction score heatmaps are overlaid with ChIP-seq tracks of YY1 and CTCF across cell types. The Sox2 gene is colored green. (I) Zoom-in classified cluster map of a ∼100-kb window around a Sox2-enhancer interaction with NPC only (green), serum+NPC (yellow), and constitutive classified looping interactions (gray), overlaid on ChIP-seq tracks.
We sought additional examples of punctate “constitutive” interactions adjacent to “NPC-only” interactions. The Olig1 and Olig2 genes encode bHLH transcription factors involved in differentiation along the oligodendrocyte lineage (Zhou et al. 2000). In NPCs, putative enhancer(s) marked by NPC-specific H3K27ac connect to both Olig1 and Olig2 in a rosette-like structure, and these genes show markedly increased expression in NPCs compared to ES 2i/ES serum (Fig. 6B–D, magenta arrows). We observed significant NPC YY1 signal at all NPC-specific genes and enhancers at the Olig1/2 locus (Fig. 6E; Supplemental Fig. 13A–D). Similar to the Nes locus, we observed two “constitutive” interactions anchored by constitutive CTCF sites in a convergent orientation adjacent to the NPC specific interactions formed by the Olig1 and Olig2 genes (Fig. 6F, red boxes/green arrows). Similarly, the Sox2 gene also forms a long-range “NPC-only” interaction with a putative NPC-specific enhancer marked by H3K27ac (Fig. 6G, magenta arrow; Supplemental Fig. 10A,B, second green box). YY1 is detected at both Sox2 and the putative NPC-specific enhancer (Fig. 6H,I, upper green boxes). Again, the “NPC-only” interaction exists adjacent to and nested within a punctate “constitutive” interaction anchored by convergent CTCF (Fig. 6I, lower red boxes, consensus orientation not shown). Together, these results support a working model of 3D genome folding in which developmentally regulated genes such as Nes, Olig1, Olig2, and Sox2 form de novo connections to their target enhancers via YY1 within a larger topological framework preexisting from naïve pluripotency and connected by constitutive CTCF.
Yy1 knockdown results in the loss of key NPC enhancer to gene looping interactions
Finally, to better understand the role for YY1 in fine-scale chromatin architecture, we knocked down Yy1 in NPCs and assessed changes in looping. We performed Yy1 knockdown using an siRNA pool purchased from Dharmacon (Supplemental Methods) to target multiple sites along the Yy1 transcript. Transfection of the Yy1-targeting siRNA pool produced a >50% decrease in Yy1 expression and protein levels compared to a control nontargeting pool condition (Fig. 7A,B). Reduction in YY1 levels resulted in a striking loss of interaction frequency between the upstream putative NPC-specific enhancer and the Sox2 gene (Figs. 3K, 7C; Supplemental Fig. 10A,B, second green box) and a decrease in Sox2 expression (Fig. 7D). We also observed loop ablation upon Yy1 knockout at interactions between the Klf4 gene and a downstream putative NPC-enhancer (Supplemental Figs. 14A,B, 15A,B, second green box) and at the Zfp462 gene (Supplemental Fig. 14C,D). Due to technical issues related to poor library complexity across all conditions in this batch of experiments, we were unable to obtain high complexity 5C maps at Olig1, Olig2, and Nes regions. However, upon Yy1 knockdown we observed a reduction in the expression of these genes, suggesting that the enhancer-promoter loops that Nes, Olig1, and Olig2 engage in might be disrupted by Yy1 knockdown (Supplemental Fig. 14E,F). Together, these results support our working hypothesis that YY1 is critical for the formation developmentally regulated looping interactions in NPCs.
Figure 7.

YY1-mediated developmentally regulated looping interactions form within a constitutive framework demarcated by CTCF. (A) Western blot analysis querying YY1 and GAPDH protein levels in NPCs exposed to nontargeting control and Yy1-targeting siRNA. (B) Gene-expression quantified by qPCR of the Yy1 gene in NPCs exposed to control and Yy1-targeting siRNA. (C) Zoom-in interaction score heatmaps of a loop between the Sox2 gene and an upstream enhancer (originally presented in Fig. 3K) in NPCs exposed to nontargeting control siRNA (left) and an siRNA targeting Yy1 (right). (D) Gene-expression quantified by qPCR of the Sox2 gene in NPCs exposed to control and Yy1-targeting siRNA. (E) Schematic depicting a CTCF-mediated constitutive interaction, present across all early stages of neural lineage commitment, and a YY1-mediated gene-enhancer interaction, present only in NPCs.
Discussion
CTCF is ubiquitously expressed across cell types and developmental stages and has a well-established role in connecting higher-order genome architecture. Here, we seek to shed light on the dynamic CTCF binding landscape and how it is linked to the reconfiguration of chromatin architecture during the earliest stages of the establishment of neuronal expression programs. We present evidence for several organizing principles governing 3D genome folding during early brain development. First, we find that CTCF occupancy is predominantly lost in the transition from ES cells to multipotent NPCs, suggesting that the CTCF occupancy landscape might be saturated in naïve pluripotency and regulated primarily through selective pruning of CTCF binding sites. Second, reduced CTCF occupancy is correlated with the loss of chromatin interactions between ES-specific genes and enhancers, indicating that loss of CTCF binding is a critical step during the decommissioning of pluripotency gene expression programs. Third, we did not observe a strong correlation between CTCF occupancy and NPC-specific interactions. Rather, we detected high levels of occupancy of the zinc finger protein YY1 at NPC-specific genes and enhancers when engaged in NPC-specific 3D interactions and negligible YY1 levels when these regulatory elements did not interact. Upon knockdown of Yy1 in NPCs, many 3D interactions break apart, suggesting that YY1 may serve as an architectural protein connecting developmentally regulated genes and enhancers in NPCs. Finally, we found that key YY1-mediated NPC-specific looping interactions occur adjacent to and nested within punctate constitutive looping interactions anchored by convergently oriented, constitutively bound CTCF. Our data support a model in which YY1-anchored looping interactions arise de novo in NPCs within a larger topological framework established prior to or during naïve pluripotency and connected by constitutively bound CTCF (Fig. 7E).
Seminal genome-wide CTCF occupancy studies based on two to three cell types initially suggested that the CTCF binding landscape remains largely unchanged across mammalian lineages (Kim et al. 2007; Cuddapah et al. 2009). A more recent comparison of CTCF occupancy across 40 cell types revealed that at least 80% of CTCF sites are dynamic across cellular states (Maurano et al. 2015). Here, we find that CTCF occupancy is highest in the naïve pluripotent stem cell state and globally decreases in parallel with its expression during the commitment to multipotent NPCs. A large cohort of roughly 8000 and roughly 20,000 CTCF sites are lost during the transition from ES 2i to ES serum and ES serum to NPCs, respectively. In contrast, we only observe a small group of about 1200 CTCF sites that are acquired de novo in NPCs, suggesting that the vast majority of the CTCF sites occupied in NPCs were preexisting from earlier stages in development. We speculate that one hallmark of the initial establishment of the neuronal lineage is a wave of CTCF occupancy loss to remove residual topological configurations required for pluripotency-specific gene expression and off-target lineages that will not be expressed in neural cell types. In the future, additional studies across non-neuronal lineages will also be important to determine how widely our model of CTCF pruning in neural development applies, as our initial analyses of ENCODE data indicated that CTCF occupancy does not always decrease during development across all lineages.
DNA methylation is a critical regulator of neural lineage commitment (Meissner et al. 2008) and CTCF binding (Renda et al. 2007). Recent reports suggest that the largest rearrangement of DNA methylation during neural development occurs upon the departure from pluripotency (Ziller et al. 2015; Sharma et al. 2016). The transition from ES cells to early NPCs is associated with a large increase in DNA methylation (Sharma et al. 2016). Importantly, a large proportion of genomic loci that are methylated in NPCs maintain the mark through the duration of neural development (Ziller et al. 2015; Sharma et al. 2016). Because CTCF binding is anti-correlated with DNA methylation, we posit that a notable proportion of the large class (n = ∼20,000) of “2i+serum” CTCF sites might be methylated during the initial establishment of the neural lineage and subsequently remain methylated and unbound through terminal differentiation in the developing/maturing brain. In support of this hypothesis, we observe the highest levels of CTCF occupancy in our naïve pluripotency cellular state (ES 2i) in which cells have consistently been found to exhibit an extreme state of hypomethylation across the genome (Ficz et al. 2013; Habibi et al. 2013; Leitch et al. 2013). The large-scale shifts in DNA methylation and CTCF binding during the transition from ES cells to NPCs suggest that elucidating the CTCF landscape in the progenitor state of development is critically important for understanding the CTCF sites and 3D topological configurations available for binding across terminally differentiated lineages in the brain.
Although CTCF is the best understood protein-mediated mechanism for connecting 3D chromatin interactions, we hypothesized that additional architectural proteins might exist to connect the 3D genome. Here, unexpectedly, we found that NPC-specific CTCF was not significantly enriched in NPC-specific loops in our 5C regions. Rather, we observed high levels of the zinc finger protein YY1 at NPC-specific genes and enhancers when engaged in 3D looping interactions and negligible/low YY1 occupancy when these regulatory elements were not connected. Several key NPC-specific enhancer-gene looping interactions were ablated upon Yy1 knockdown. YY1 is an intriguing architectural protein candidate: (1) it is strongly enriched in genome-wide looping interactions in human cell lines (Rao et al. 2014); (2) it is necessary for the formation of specific 3D interactions in B cells (Medvedovic et al. 2013); (3) it can connect a long-range interaction in B cells in the absence of its transcriptional activation domain (Mehra et al. 2016); and (4) it is required for proper neural development (He et al. 2007). Future studies should aim to elucidate YY1's function as an architectural protein in non-neural lineages and determine the extent to which YY1's critical role in looping is due to indirect effects on chromatin activity.
Our results open the question of the mechanism by which YY1 connects distal regulatory regions. Biochemical studies have indicated that the zinc fingers of YY1 may interact with the N terminus of CTCF (Donohoe et al. 2007), suggesting that YY1 could anchor loops via homodimerization (Supplemental Fig. 16A) or heterodimerization (Supplemental Fig. 16B) mechanisms. It is also important to consider YY1's mechanistic functions in the context of the recently proposed “loop extrusion” model. Marko, Mirny, Lieberman-Aiden and colleagues have posited that a chromatin molecular motor, such as cohesin/condensin, might extrude chromatin until blocked by architectural proteins, resulting in looped out chromatin (Alipour and Marko 2012; Sanborn et al. 2015; Fudenberg et al. 2016). Previous studies have shown that the REPO domain of YY1 physically interacts with cohesin (Pan et al. 2013). In the context of the loop extrusion model, it is possible that YY1 works as an architectural protein to physically impede loop extrusion (Supplemental Fig. 16C) within larger loops where extrusion is blocked by CTCF (Sanborn et al. 2015; Fudenberg et al. 2016). Future studies should aim to shed light on the plausibility of the heterodimerization, homodimerization, and extrusion models for YY1-mediated loop formation.
A key finding of this manuscript is that YY1-mediated looping interactions in NPCs are nested within larger constitutive interactions anchored by constitutively occupied CTCF sites. A leading hypothesis is that sub-TAD/TAD boundaries, anchored by constitutive CTCF, might constrain developmentally regulated enhancers from aberrantly looping to off-target genes (Dowen et al. 2014; Symmons et al. 2014; Lupiáñez et al. 2015; Flavahan et al. 2016; Hnisz et al. 2016). We and others have previously reported that pluripotency genes connect to enhancers in smaller looping interactions nested within larger constitutive structures (Phillips-Cremins et al. 2013; Dowen et al. 2014). Here, our results confirm and extend this model to suggest that CTCF-mediated constitutive interactions might also might serve to pre-mark genomic locations of connections between somatic developmentally regulated gene-enhancer interactions through punctate, constitutive “seed” interactions. In agreement with this idea, Ruan and colleagues reported evidence that CTCF-mediated looping interactions might function to coordinate nearby interactions involving RNA Pol II (Tang et al. 2015). Furthermore, a recent genomics analysis showed that up to 30% of YY1 sites bind at locations directly adjacent to CTCF and might work together to cooperatively influence occupancy (Schwalie et al. 2013). Thus we posit that architectural proteins such as YY1 might cooperatively build upon a constitutive CTCF architectural “seed” scaffold to connect nearby developmentally regulated genes and enhancers. Future work teasing out the causal interplay between architectural seeds, CTCF and additional architectural proteins will shed light on the fundamental mechanisms governing proper spatiotemporal regulation of gene expression during development.
Methods
Cell culture
V6.5 ES cells from Novus Biologicals were grown under standard pluripotent (serum/LIF) conditions on Mitomycin-C inactivated MEFs. To generate the 2i/LIF condition, ES cells were transitioned to serum-free media with 3 µM CHIR99021 and 1 µM PD0325901 and propagated for two passages on feeder cells. Before fixation, both ES cell conditions were passaged onto 0.1% gelatin-coated plates to remove the feeder layer and fixed at ∼60% confluency. NPCs (previously isolated from whole brain of newborn pups) were cultured as neurospheres for two passages to purify the population of nonadherent NPCs. Neurospheres were dissociated and passaged onto poly-D-lysine hydrobromide (100 µg/mL) and laminin (15 µg/mL)-coated plates to be fixed next day (for further details, see Supplemental Methods).
CTCF ChIP-seq
Chromatin immunoprecipitation was performed as previously described (Beagan et al. 2016). Sequencing libraries were prepared with the NEBNext Ultra Library Prep Kit following the manufacturer's protocol for ChIP-seq library preparation, with amplification over 18 PCR cycles using NEBNext Multiplex Oligos for Illumina. The library was confirmed to contain DNA fragments ranging roughly from 250 to 1200 bp by running a High-Sensitivity DNA assay on an Agilent Bioanalyzer and sequenced with 75 cycles per paired-end on the Illumina NextSeq500.
ChIP-seq peak calling
Published ChIP-seq data was downloaded from GEO (http://www.ncbi.nlm.nih.gov/geo/) and reanalyzed according to Supplemental Table S4. Reads were aligned to mouse genome build mm9 using Bowtie with default mismatch parameters (Langmead et al. 2009). Reads were considered if they had two or fewer reportable alignments. To facilitate the comparison of ChIP-seq libraries across cell types, the mapped reads were filtered to remove optical and PCR duplicates and then down-sampled to equivalent read numbers across cellular states. The CTCF ChIP-seq libraries for ES 2i, ES serum, and NPC were down-sampled to 11 million reads, and the whole-cell extract libraries were down-sampled to 15 million reads. The YY1 ChIP-seq libraries for ES serum, NPC and ProB cells were down-sampled to 7.2 million reads, and the whole-cell extract libraries were down-sampled to 7.7 million reads. The H3K27ac ChIP-seq libraries for ES serum and NPC were down-sampled to 7 million reads, and the whole-cell extract libraries were down-sampled to 7 million reads. Peaks were identified using Model-based Analysis for ChIP Sequencing v2.0 (MACS2 [Zhang et al. 2008]). For CTCF ChIP-seq, default parameters were used with a P-value cutoff of P < 1 × 10−8. For YY1 and H3K27ac ChIP-seq, we modified the parameters to facilitate accurate detection of broad peaks (--broad --broad-cutoff 1 × 10−4 -p 1 × 10−8).
5C analyses
Custom scripts were developed to characterize and analyze 5C data as reported elsewhere with some modifications (Beagan et al. 2016; Supplemental Materials). Briefly, 5C primer biases were corrected by adapting and modifying the Express matrix balancing algorithm (Sauria et al. 2015). 5C counts were binned at 4-kb matrix resolution, and spatial noise was reduced by applying a 16-kb-sized smoothing window. Binned 5C counts were normalized for the expected background interaction frequency with local “donut” and “lower left” filters (Rao et al. 2014) as detailed in the Supplemental Methods. Background-corrected interaction frequencies for each independent replicate and region were fitted with a logistic distribution, and P-values were assigned to each bin–bin pair. P-values were transformed to an Interaction Score (−10*log2[P-value]) for downstream analyses (for details, see Supplemental Methods). Seven classes of constitutive and dynamic looping interactions across three cellular states were classified and correlated with linear epigenetic modifications as described in detail in the Supplemental Methods.
Yy1 knockdown
siRNA pools were purchased from Dharmacon for the knockdown of Yy1 (# L-050273-00-0005) along with a nontargeting pool (# D-001810-10-05). NPCs were transfected with RNAimax (Lifetech #13778-075) for 78 h at a final siRNA concentration of 20 nM (replaced every 24 h). Cells were then harvested for Western blot, qPCR, and 3C/5C analysis. For these Yy1 knockdown samples, we utilized a modification of in situ 3C (Rao et al. 2014). Further details can be found in Supplemental Methods.
Data access
ChIP-seq and 5C data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE85185. Code for full reproducibility of all analyses is provided in the Supplemental Materials.
Supplementary Material
Acknowledgments
We thank members of the Cremins laboratory for helpful discussions, in particular, Jesi Kim and Thomas Gilgenast for computational assistance. J.E.P.-C. is a New York Stem Cell Foundation – Robertson Investigator and an Alfred P. Sloan Foundation Fellow. This research was supported by The New York Stem Cell Foundation (J.E.P.-C.), the Alfred P. Sloan Foundation (J.E.P.-C.), the NIH Director's New Innovator Award from the National Institute of Mental Health (1DP2MH11024701; J.E.P.-C.), a 4D Nucleome (NIH) Common Fund grant (1U01HL12999801; J.E.P.-C.), and a joint NSF-NIGMS grant to support research at the interface of the biological and mathematical sciences (1562665; J.E.P.-C.). This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1321851 (J.A.B.).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.215160.116.
Freely available online through the Genome Research Open Access option.
References
- Alipour E, Marko JF. 2012. Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Res 40: 11202–11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beagan JA, Gilgenast TG, Kim J, Plona Z, Norton HK, Hu G, Hsu SC, Shields EJ, Lyu X, Apostolou E, et al. 2016. Local genome topology can exhibit an incompletely rewired 3D-folding state during somatic cell reprogramming. Cell Stem Cell 18: 611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinka S, Reimer MH Jr, Pulakanti K, Rao S. 2016. Super-enhancers at the Nanog locus differentially regulate neighboring pluripotency-associated genes. Cell Rep 17: 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. 2008. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133: 1106–1117. [DOI] [PubMed] [Google Scholar]
- Cuddapah S, Jothi R, Schones DE, Roh TY, Cui K, Zhao K. 2009. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res 19: 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Laat W, Dekker J. 2012. 3C-based technologies to study the shape of the genome. Methods 58: 189–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denker A, de Laat W. 2016. The second decade of 3C technologies: detailed insights into nuclear organization. Genes Dev 30: 1357–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit E, Vos ES, Holwerda SJ, Valdes-Quezada C, Verstegen MJ, Teunissen H, Splinter E, Wijchers PJ, Krijger PH, de Laat W. 2015. CTCF binding polarity determines chromatin looping. Mol Cell 60: 676–684. [DOI] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. 2012. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. 2015. Chromatin architecture reorganization during stem cell differentiation. Nature 518: 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohoe ME, Zhang LF, Xu N, Shi Y, Lee JT. 2007. Identification of a Ctcf cofactor, Yy1, for the X chromosome binary switch. Mol Cell 25: 43–56. [DOI] [PubMed] [Google Scholar]
- Dowen JM, Fan ZP, Hnisz D, Ren G, Abraham BJ, Zhang LN, Weintraub AS, Schuijers J, Lee TI, Zhao K, et al. 2014. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 159: 374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficz G, Hore TA, Santos F, Lee HJ, Dean W, Arand J, Krueger F, Oxley D, Paul YL, Walter J, et al. 2013. FGF signaling inhibition in ESCs drives rapid genome-wide demethylation to the epigenetic ground state of pluripotency. Cell Stem Cell 13: 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suva ML, Bernstein BE. 2016. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529: 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, Mirny LA. 2016. Formation of chromosomal domains by loop extrusion. Cell Rep 15: 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galonska C, Ziller MJ, Karnik R, Meissner A. 2015. Ground state conditions induce rapid reorganization of core pluripotency factor binding before global epigenetic reprogramming. Cell Stem Cell 17: 462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Monahan K, Wu H, Gertz J, Varley KE, Li W, Myers RM, Maniatis T, Wu Q. 2012. CTCF/cohesin-mediated DNA looping is required for protocadherin α promoter choice. Proc Natl Acad Sci 109: 21081–21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU, Jung I, Wu H, Zhai Y, Tang Y, et al. 2015. CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell 162: 900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habibi E, Brinkman AB, Arand J, Kroeze LI, Kerstens HH, Matarese F, Lepikhov K, Gut M, Brun-Heath I, Hubner NC, et al. 2013. Whole-genome bisulfite sequencing of two distinct interconvertible DNA methylomes of mouse embryonic stem cells. Cell Stem Cell 13: 360–369. [DOI] [PubMed] [Google Scholar]
- Hadjur S, Williams LM, Ryan NK, Cobb BS, Sexton T, Fraser P, Fisher AG, Merkenschlager M. 2009. Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature 460: 410–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, Lee CW, Ye C, Ping JL, Mulawadi F, et al. 2011. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet 43: 630–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Dupree J, Wang J, Sandoval J, Li J, Liu H, Shi Y, Nave KA, Casaccia-Bonnefil P. 2007. The transcription factor Yin Yang 1 is essential for oligodendrocyte progenitor differentiation. Neuron 55: 217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayama T, Tarusawa E, Yoshimura Y, Galjart N, Yagi T. 2012. CTCF is required for neural development and stochastic expression of clustered Pcdh genes in neurons. Cell Rep 2: 345–357. [DOI] [PubMed] [Google Scholar]
- Hnisz D, Weintraub AS, Day DS, Valton AL, Bak RO, Li CH, Goldmann J, Lajoie BR, Fan ZP, Sigova AA, et al. 2016. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 351: 1454–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B. 2007. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell 128: 1231–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurukuti S, Tiwari VK, Tavoosidana G, Pugacheva E, Murrell A, Zhao Z, Lobanenkov V, Reik W, Ohlsson R. 2006. CTCF binding at the H19 imprinting control region mediates maternally inherited higher-order chromatin conformation to restrict enhancer access to Igf2. Proc Natl Acad Sci 103: 10684–10689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch HG, McEwen KR, Turp A, Encheva V, Carroll T, Grabole N, Mansfield W, Nashun B, Knezovich JG, Smith A, et al. 2013. Naive pluripotency is associated with global DNA hypomethylation. Nat Struct Mol Biol 20: 311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendahl U, Zimmerman LB, McKay RD. 1990. CNS stem cells express a new class of intermediate filament protein. Cell 60: 585–595. [DOI] [PubMed] [Google Scholar]
- Li Y, Rivera CM, Ishii H, Jin F, Selvaraj S, Lee AY, Dixon JR, Ren B. 2014. CRISPR reveals a distal super-enhancer required for Sox2 expression in mouse embryonic stem cells. PLoS One 9: e114485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326: 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, et al. 2015. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161: 1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Wang H, John S, Shafer A, Canfield T, Lee K, Stamatoyannopoulos JA. 2015. Role of DNA methylation in modulating transcription factor occupancy. Cell Rep 12: 1184–1195. [DOI] [PubMed] [Google Scholar]
- Medvedovic J, Ebert A, Tagoh H, Tamir IM, Schwickert TA, Novatchkova M, Sun Q, Huis In ’t Veld PJ, Guo C, Yoon HS, et al. 2013. Flexible long-range loops in the VH gene region of the Igh locus facilitate the generation of a diverse antibody repertoire. Immunity 39: 229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehra P, Gerasimova T, Basu A, Jha V, Banerjee A, Sindhava V, Gray F, Berry CT, Sen R, Atchison ML. 2016. YY1 controls Eμ–3′RR DNA loop formation and immunoglobulin heavy chain class switch recombination. Blood Adv 2016: 15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. 2008. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454: 766–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra V, Rocha PP, An D, Raviram R, Skok JA, Mazzoni EO, Reinberg D. 2015. Transcription. CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347: 1017–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. 2012. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485: 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong CT, Corces VG. 2014. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet 15: 234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Papasani M, Hao Y, Calamito M, Wei F, Quinn WJ Iii, Basu A, Wang J, Hodawadekar S, Zaprazna K, et al. 2013. YY1 controls Igκ repertoire and B-cell development, and localizes with condensin on the Igκ locus. EMBO J 32: 1168–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Xiang AP, Mao FF, Zhang L, Di CG, Liu XM, Shao Y, Ma BF, Lee JH, Ha KS, et al. 2010. Nestin is required for the proper self-renewal of neural stem cells. Stem Cells 28: 2162–2171. [DOI] [PubMed] [Google Scholar]
- Phillips JE, Corces VG. 2009. CTCF: master weaver of the genome. Cell 137: 1194–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips-Cremins JE, Sauria ME, Sanyal A, Gerasimova TI, Lajoie BR, Bell JS, Ong CT, Hookway TA, Guo C, Sun Y, et al. 2013. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153: 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. 2014. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159: 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renda M, Baglivo I, Burgess-Beusse B, Esposito S, Fattorusso R, Felsenfeld G, Pedone PV. 2007. Critical DNA binding interactions of the insulator protein CTCF: a small number of zinc fingers mediate strong binding, and a single finger-DNA interaction controls binding at imprinted loci. J Biol Chem 282: 33336–33345. [DOI] [PubMed] [Google Scholar]
- Sams DS, Nardone S, Getselter D, Raz D, Tal M, Rayi PR, Kaphzan H, Hakim O, Elliott E. 2016. Neuronal CTCF is necessary for basal and experience-dependent gene regulation, memory formation, and genomic structure of BDNF and arc. Cell Rep 17: 2418–2430. [DOI] [PubMed] [Google Scholar]
- Sanborn AL, Rao SS, Huang SC, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, et al. 2015. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci 112: E6456–E6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal A, Lajoie BR, Jain G, Dekker J. 2012. The long-range interaction landscape of gene promoters. Nature 489: 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauria ME, Phillips-Cremins JE, Corces VG, Taylor J. 2015. HiFive: a tool suite for easy and efficient HiC and 5C data analysis. Genome Biol 16: 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalie PC, Ward MC, Cain CE, Faure AJ, Gilad Y, Odom DT, Flicek P. 2013. Co-binding by YY1 identifies the transcriptionally active, highly conserved set of CTCF-bound regions in primate genomes. Genome Biol 14: R148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Klein SS, Barboza L, Lohdi N, Toth M. 2016. Principles governing DNA methylation during neuronal lineage and subtype specification. J Neurosci 36: 1711–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. 2012. A map of the cis-regulatory sequences in the mouse genome. Nature 488: 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EM, Lajoie BR, Jain G, Dekker J. 2016. Invariant TAD boundaries constrain cell-type-specific looping interactions between promoters and distal elements around the CFTR locus. Am J Hum Genet 98: 185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splinter E, de Wit E, van de Werken HJ, Klous P, de Laat W. 2012. Determining long-range chromatin interactions for selected genomic sites using 4C-seq technology: from fixation to computation. Methods 58: 221–230. [DOI] [PubMed] [Google Scholar]
- Stadtfeld M, Maherali N, Breault DT, Hochedlinger K. 2008. Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell Stem Cell 2: 230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmons O, Uslu VV, Tsujimura T, Ruf S, Nassari S, Schwarzer W, Ettwiller L, Spitz F. 2014. Functional and topological characteristics of mammalian regulatory domains. Genome Res 24: 390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Luo OJ, Li X, Zheng M, Zhu JJ, Szalaj P, Trzaskoma P, Magalska A, Wlodarczyk J, Ruszczycki B, et al. 2015. CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell 163: 1611–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolhuis B, Palstra RJ, Splinter E, Grosveld F, de Laat W. 2002. Looping and interaction between hypersensitive sites in the active β-globin locus. Mol Cell 10: 1453–1465. [DOI] [PubMed] [Google Scholar]
- Vietri Rudan M, Barrington C, Henderson S, Ernst C, Odom DT, Tanay A, Hadjur S. 2015. Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep 10: 1297–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Maurano MT, Qu H, Varley KE, Gertz J, Pauli F, Lee K, Canfield T, Weaver M, Sandstrom R, et al. 2012. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res 22: 1680–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson LA, Wang X, Elbert A, Kernohan KD, Galjart N, Bérubé NG. 2014. Dual effect of CTCF loss on neuroprogenitor differentiation and survival. J Neurosci 34: 2860–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, Smith A. 2008. The ground state of embryonic stem cell self-renewal. Nature 453: 519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Wang S, Anderson DJ. 2000. Identification of a novel family of oligodendrocyte lineage-specific basic helix–loop–helix transcription factors. Neuron 25: 331–343. [DOI] [PubMed] [Google Scholar]
- Ziller MJ, Edri R, Yaffe Y, Donaghey J, Pop R, Mallard W, Issner R, Gifford CA, Goren A, Xing J, et al. 2015. Dissecting neural differentiation regulatory networks through epigenetic footprinting. Nature 518: 355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.