

Abstract
Risk factors indicate the importance of oxidative stress during ovarian carcinogenesis. To tolerate oxidative stress, cells activate the transcription factor Nrf2 (Nfe2l2), the master regulator of antioxidant and cytoprotective genes. Indeed, for most cancers, hyperactivity of Nrf2 is observed, and siRNA studies assigned Nrf2 as therapeutic target. However, the cancer‐protective role of Nrf2 in healthy cells highlights the requirement for an adequate therapeutic window. We engineered artificial transcription factors to assess the role of Nrf2 in healthy (OSE‐C2) and malignant ovarian cells (A2780). Successful NRF2 up‐ and downregulation correlated with decreased, respectively increased, sensitivity toward oxidative stress. Inhibition of NRF2 reduced the colony forming potential to the same extent in wild‐type and BRCA1 knockdown A2780 cells. Only in BRCA1 knockdown A2780 cells, the effect of Nrf2 inhibition could be enhanced when combined with PARP inhibitors. Therefore, we propose that this combination therapy of PARP inhibitors and Nrf2 inhibition can further improve treatment efficacy specifically in BRCA1 mutant cancer cells without acquiring the side‐effects associated with previously studied Nrf2 inhibition combinations with either chemotherapy or radiation. Our findings stress the dual role of Nrf2 in carcinogenesis, while offering approaches to exploit Nrf2 as a potent therapeutic target in ovarian cancer.
Keywords: Ovarian carcinogenesis, Oxidative stress, Nrf2 (NFE2L2), BRCA1, PARP inhibitor
Highlights
Nrf2 plays a dual role in ovarian carcinogenesis.
Inhibition of Nrf2 increases ovarian cancer cell death.
The combination with PARP inhibitors enhances the effectivity of monotherapy.
Abbreviations
- ARE
antioxidant response element
- ATF
artificial transcription factor
- BER
base excision repair
- CRISPR/Cas9
clustered regularly interspaced short palindromic repeats associated 9 protein
- EOC
epithelial ovarian cancer
- ER UPR
endoplasmatic reticulum unfolded protein response
- HR
homologous recombination
- KEAP1
Kelch like-ECH-associated protein 1
- N4Py
ligand 1 of the iron(II) complex of the pentadentate ligand N,N-bis(2-pyridylmethyl)-N-bis(2-pyridyl)-methylamine
- NER
nuclear protein extract
- NRF2, NFE2L2
nuclear factor erythroid 2-like 2
- PARP
poly ADP ribose polymerase
- ROS
reactive oxygen species
- SKD
super KRAB domain
- TALE
transcription activator-like effector
- VP64
four copies of the Herpes Simplex Viral Protein 16
- ZFP
zinc finger protein
1. Introduction
Oxidative stress is a well‐established risk factor for cancer (Sosa et al., 2013). It reflects an imbalance between production and detoxification of reactive oxygen species (ROS) and causes oxidative damage to biomolecules, including DNA. The cell has several defense mechanisms to protect itself against oxidative stress, including antioxidants, DNA‐repair enzymes and the endoplasmatic reticulum unfolded protein response (ER UPR). The latter response is activated when cells accumulate an overload of (ROS‐induced) unfolded or misfolded proteins, causing ER stress (Walter and Ron, 2011). Activation of the ER UPR results either in an adaptive response, in which protein synthesis is inhibited and protein folding capacity is increased, or, when not sufficient to overcome the ER stress, a pro‐apototic response (Rutkowski et al., 2006; Walter and Ron, 2011). Oxidative stress and the ER UPR can activate cytoprotective responses mediated by the master regulator Nrf2 (NFE2L2, nuclear factor erythroid 2‐like 2) (Cullinan et al., 2003; Kansanen et al., 2012).
Normally, Nrf2 is present in the cytoplasm where it is bound by Kelch like‐ECH‐associated protein 1 (Keap1). There, Keap1 mediates the ubiquitination of Nrf2, whereupon Nrf2 is targeted to the proteasomes for degradation. When cells are exposed to oxidative stress, the cysteine residues of Keap1 become oxidized, resulting in disruption of the binding between Keap1 and Nrf2 (van der Wijst et al., 2014). Nrf2 then translocates to the nucleus and binds to antioxidant response elements (AREs). Binding of Nrf2 to AREs results in the transcription of its target genes (Malhotra et al., 2010).
Not only healthy cells, but also various tumor cells, including ovarian tumors (Konstantinopoulos et al., 2011; Liao et al., 2012; Martinez et al., 2014), can acquire protection against oxidative stress by constitutively activating Nrf2 (Bauer et al., 2013; Jiang et al., 2010; Stacy et al., 2006; Wang et al., 2010). Thus the role of Nrf2 in carcinogenesis is bivalent as Nrf2 has been assigned a protective, but also a cancer promoting role (Lau et al., 2008). Therefore, activation of Nrf2 can be exploited for cancer prevention (Cornblatt et al., 2007; Kou et al., 2013), whereas inhibition of Nrf2 can be of therapeutic value (Homma et al., 2009; Lister et al., 2011; Ma et al., 2012; Singh et al., 2008). However, the cancer‐protective role of Nrf2 in healthy cells highlights the requirement for an adequate therapeutic window. Therefore, insights into differential effects of Nrf2 modulation in normal and malignant cells are essential to allow optimal exploitation of Nrf2 as therapeutic target.
Combination therapies provide an opportunity to enhance the therapeutic window of Nrf2 inhibition in cancer. Previously, the cytotoxic effects of Nrf2 inhibition could be further enhanced by chemotherapeutics (Cho et al., 2008; Lister et al., 2011; Wang et al., 2008), including cisplatin, or gamma‐radiation (Lister et al., 2011). However, inhibition of Nrf2 has also been shown to exacerbate chemotherapy‐induced myelosuppression (Cao et al., 2012). This side‐effect severely limits the use of such combination therapies. The combination with PARP inhibitors may be an alternative strategy to enhance the efficacy of Nrf2 inhibition preferentially in cancer cells without increasing adverse effects in healthy cells. PARP inhibitors are small molecule inhibitors particularly effective in tumors harboring a defect in double strand DNA (dsDNA) break repair as they act by blocking single strand DNA (ssDNA) repair by base excision repair (BER) (Banerjee and Kaye, 2011; Fong et al., 2009). Interestingly, clinical trials have already shown efficacy of PARP inhibitors as single agent in BRCA1 mutant ovarian tumors (Audeh et al., 2010; Fong et al., 2010). Furthermore, the DNA damaging effect of PARP inhibitors can be potentiated when combined with chemotherapy (as reviewed in (Chen et al., 2013; Sessa, 2011)). In line with this, we expect that Nrf2 inhibition combined with PARP inhibitor treatment can enhance the DNA damaging effect of PARP inhibitors while diminishing the serious side‐effects of chemotherapeutics.
A unique approach to validate and obtain better insights into the therapeutic potential of Nrf2 is the bivalent modulation of NRF2 by artificial transcription factors (ATFs) targeted to the NRF2 promoter region. ATFs contain a DNA‐binding domain, for example an engineered zinc finger protein (ZFP), a transcription activator‐like effector (TALE) protein or a clustered regularly interspaced short palindromic repeats (CRISPR) associated 9 (Cas9) protein (Gaj et al., 2013; Gersbach and Perez‐Pinera, 2014), fused to a transcriptional activation or repression domain such as VP64 (four copies of the viral protein VP16) or super KRAB domain (SKD), respectively. ATFs have been successfully used to modulate gene expression and study gene function in many disease models both in vitro and in vivo (as reviewed in (Gaj et al., 2013; Gersbach and Perez‐Pinera, 2014; Sera, 2009)).
In the present study, we engineered ZFP‐ATFs to modulate NRF2 expression allowing investigation into the cancer‐preventive and therapeutic potential of Nrf2 in healthy (Nrf2 activation) and malignant ovarian epithelial cells (Nrf2 inhibition), respectively. We combined Nrf2 inhibition with PARP inhibitor treatment to further improve on the therapeutic window of Nrf2 inhibition in cancer without acquiring the side‐effects associated with other previously studied combinations such as chemotherapy and radiation.
2. Materials and methods
2.1. Reagents
Ligand 1 of the iron(II) complex of the pentadentate ligand N,N‐bis(2‐pyridylmethyl)‐N‐bis(2‐pyridyl)‐methylamine (Fe(II)‐1‐N4Py), from now on abbreviated as N4Py, was synthesized according to literature procedures and all characterization data are in agreement with those reported (Li et al., 2010). N4Py is a synthetic mimic of the metal‐binding domain of bleomycin. It acts as a catalyst that converts less‐reactive primary ROS in highly reactive secondary ROS. The main advantage of using N4Py over directly adding ROS, such as H2O2, to the culture medium, is the more constant and stable production of ROS (Bjorkman and Ekholm, 1995; Li et al., 2010). l‐Buthionine sulphoximine (BSO), l‐glutathione reduced (GSH), GSH reductase, 5,5'dithiobis‐(2‐nitrobenzoic acid) (DTNB) and NADPH were bought from Sigma–Aldrich.
2.2. Cell culture
The human ovarian carcinoma cell lines A2780 and SKOV3, and the human embryonic kidney cell line HEK293T were obtained from the ATCC. The temperature‐sensitive, conditionally immortalized human ovarian surface epithelial (OSE‐C2) cells were kindly provided by Dr. Richard Edmondson (Newcastle University, UK) (Davies et al., 2003). All cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) (Lonza) supplemented with 10% FCS (Perbio Hyclone), 50 μg/mL gentamycine sulfate (Invitrogen), 2 mM l‐glutamine (BioWhittaker). A2780, SKOV3 and HEK293T cells were cultured at 37 °C, whereas OSE‐C2 cells were cultured at 33 °C, both under humidified conditions and 5% CO2.
2.3. Engineering and delivery of artificial transcription factors (ATFs)
The promoter region of NRF2 transcript variant 1 (NCBI‐id: NM_006164_4) surrounding its transcription start site (TSS) was screened with the online tool www.zincfingertools.org for potential target sites for engineering zinc finger proteins (ZFPs) consisting of six fingers fused together to target a 18 bp DNA region (Mandell and Barbas, 2006). Six ZFPs, named OX1‐OX6 (Table 1), were selected based on predicted high affinity for its designed target region and uniqueness in the human genome, as confirmed by an NCBI BLAST search. ZFPs were synthesized at Bio Basic Canada and were flanked by SfiI restriction sites. Each ZFP was subcloned with SfiI restriction enzymes into the pMX‐IRES‐GFP retroviral vector (Royer et al., 2004), which contains an HA‐tag, nuclear localization signal (NLS) and either the transcriptional repressor (SKD), the transcriptional activator (VP64) or no effector domain (NoED). NRF2 transcript variant 1 cDNA was subcloned into pMX‐IRES‐GFP with BamHI and NotI digestion enzymes. The pMX‐IRES‐GFP (empty vector) and NRF2 cDNA constructs functioned as controls. The OX2‐SKD ATF, including its HA‐tag and NLS, was further subcloned into the lentiviral vector pCDH‐CMV‐MCS‐EF1‐copGFP (System Biosciences) using BamHI and NotI digestion.
Table 1.
Zinc finger protein (ZFP) target sequences.
| ZFP | DNA strand | Target region (5′–3′) |
|---|---|---|
| OX1 | Sense | TGA GTA CGT GAA AAA GAA |
| OX2 | Antisense | GCA AAC GGA GAA GCC CCT |
| OX3 | Antisense | GTG GGC CCT GCC TAG GGG |
| OX4 | Antisense | GCC GGG GTG GGG GGG GCT |
| OX5 | Sense | GCG GTA AAG TGA GAT AAA |
| OX6 | Sense | GCA ACT CCA AAT CAG GGA |
In order to produce retroviral or lentiviral particles containing the ATFs, HEK293T packaging cells were co‐transfected using the calcium phosphate method with plasmids containing the ATF, and viral packaging plasmids containing gag/pol and the vesicular stomatitis virus G protein in a 3:2:1 ratio, as described before (Huisman et al., 2013). Viral supernatant was collected 48 h and 72 h post transfection and was used in combination with 6 μg/ml polybrene (Sigma–Aldrich) to infect ovarian host cells. Three days post transduction, efficient viral delivery was evaluated in host cells by the percentage of GFP‐positive cells using flow cytometry (FACSCalibur, BD Bioschiences) or by fluorescent microscopy (Leica DC300). Only transductions resulting in ≥90% GFP positive cells were included for analysis (Suppl. Figure 1A). In order to further improve the delivery of lentiviral particles in A2780, cells were superinfected and the top 25% GFP positive cells was sorted with the MoFlo XDP (Beckman Coulter). By minimizing the variation in GFP positivity, variation in the multiplicity of infection (MOI) was reduced and any toxicity associated with GFP expression would be equal in all conditions analyzed.
2.4. Quantitative real‐time PCR (qRT‐PCR)
Total RNA was extracted using the GeneJET RNA purification kit (Thermo Scientific) according to the manufacturer's protocol, including a 15 min DNaseI (Roche) treatment to avoid gDNA contamination. 1 μg of total RNA was reverse transcribed to cDNA using the RevertAid First Strand cDNA synthesis kit (Thermo Scientific). qRT‐PCR reactions were performed in triplicate, containing 10 ng of cDNA, ABsolute qPCR SYBR Green ROX Mix (Thermo Scientific) and gene‐specific primers (Table 2). qRT‐PCR reactions were conducted on the ViiA7 Real time PCR (Applied Biosystems) for 15 min at 95 °C, followed by 40 cycles of 15 s at 95 °C, 30 s at 60 °C and 30 s at 72 °C. GAPDH was used as housekeeping gene. Data was analyzed using ViiA7 RUO software and relative expression compared to controls was calculated using the ΔΔCt method (Livak and Schmittgen, 2001).
Table 2.
qRT‐PCR Primer sequences.
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| NRF2 | ACACACGGTCCACAGCTCAT | CCGTCGCTGACTGAAGTCAAAT |
| NRF2 cDNA | GGTTGCCCACATTCCCAAATC | TGACTGAAACGTAGCCGAAGA |
| NQO1 | GTGGAGTCGGACCTCTATGC | AATATCACAAGGTCTGCGGCT |
| GCLC | GCTGTTGCAGGAAGGCATTG | AACAGTGTCAGTGGGTCTCT |
| HMOX1 | GGGTGATAGAAGAGGCCAAGA | AGCTCCTGCAACTCCTCAAA |
| OGG1 | CCGAGCCATCCTGGAAGAAC | GTCTAGGGCCATCAGGCAGA |
| BRCA1 | TGCTCTTCGCGTTGAAGAAGT | TGGTCACACTTTGTGGAGACA |
| GRP78 | GTTCTTGCCGTTCAAGGTGG | TGGTACAGTAACAACTGCATG |
| HERP | CCAAAGCAGGAAAAACGGCAT | TGTCCCCGATTAGAACCAGC |
| p58IPK | CGTTTGCGTTCACAAGCACT | CCCGTAGTTCTGCATCCCAA |
| ERp72 | ACCGCAAGGTGTCAAACGAT | CTCTAGGACTTTGCTCCGCC |
| GAPDH | CCACATCGCTCAGACACCAT | GCGCCCAATACGACCAAAT |
| OX2‐ChIP6 | GGAGACACGTGGGAGTTCAG | TGCCTAGGGGAGATGTGGAC |
| OX5‐ChIP5 | AGGGCAAGGTTCTGCAACTC | TGGAGTTCGGACGCTTTGAA |
2.5. Chromatin immunoprecipitation (ChIP)
Three days after transduction, binding of the ZFP to its designed target region and histone modifications associated with ZFP binding in this region were determined by ChIP. Cells were fixed with 1% formaldehyde for 8 min at RT. After two PBS washes, cells were lysed and subsequently sonicated for 8 min using a Bioruptor (Diagenode; 4 cycles of 30″ on, 30″ off). Sheared chromatin was collected by centrifugation for 10 min at 13.000 rpm at 4 °C. 40 μl magnetic beads (Life technologies) were coated for 10 min at RT with 4 μg antibody (Rabbit IgG, Ab46540; HA‐tag, Ab9110; H3K9me3, Ab8899 (Abcam); H3K4me3, 07‐473 (Millipore)). After washing the beads with 0.02% PBS‐Tween, sheared chromatin of 25 × 104 cells was added to the precoated magnetic beads and incubated O/N at 4 °C. The following day, unbound chromatin was collected from the IgG control IP. All beads were washed 3 times with PBS and DNA was eluted with elution buffer (1% SDS, 100 mM NaHCO3). Eluted DNA was RNase (Roche) treated under high salt conditions O/N at 62 °C. The next day, after a 1 h incubation at 62 °C with proteinase K (Thermo Scientific), DNA was purified with the Qiagen Qiaquick PCR Purification kit (Qiagen). qRT‐PCR was performed using primers specific for the OX2 and OX5 ZFP target region (Table 2) to determine relative enrichment of the HA‐tag and histone marks in these regions with the formula: percentage input = 2(Ctinput – CtChIP) * dilution factor * 100.
2.6. Western blotting
Total protein extracts were collected in RIPA buffer (Thermo Scientific). Nuclear (NER) protein extracts were obtained with NE‐PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific). Protein quantification was performed with the DC BioRad Protein Assay (BioRad). 70 μg protein was loaded on a 7% SDS‐PAGE gel for the detection of BRCA1, whereas 20 μg of NER protein was loaded on a 10% SDS‐PAGE gel for the detection of Nrf2. Blots were blocked for 1 h with 5% skimmed milk in TBS‐Tween. For detection, the following antibodies were used: 1:200 rabbit anti‐BRCA1 (C‐20: sc‐642, Santa Cruz Biotechnology) 1:1000 rabbit anti‐Nrf2 (Ab31163, Abcam), 1:1000 mouse anti‐lamin B1 (clone L‐5, Invitrogen), 1:5000 mouse anti‐Actin (clone C4 MAB15O1, Millipore), and 1:2500 horseradish peroxidase‐conjugated rabbit anti‐mouse (P0260, Dako) and swine anti‐rabbit (P0217, Dako). Western blot signal was generated with Pierce ECL Plus Western blot substrate (Thermo Scientific) and detected with the Biorad ChemiDoc MP imaging system (Biorad). Western blots were analyzed with the Image Lab 5.0 software (Biorad).
2.7. Luciferase reporter assay
The promoter region of NRF2 transcript variant 1 (−288 to +555 bp), containing both OX2 and OX5 target sites, was cloned into pCpG‐luc basic (promoterless) with BglII and HindIII restriction sites. Two days after viral delivery of ATFs, host cells were transiently transfected with lipofectamine LTX Plus (Life Technologies) in a 1:1:2 ratio of DNA (pCpG‐luc NRF2 promoter): plus reagent: lipofectamine LTX according to company's instructions. Luciferase activity relative to empty vector was determined 48 h post transfection with the Luciferase assay system (Promega) on a Luminoskan ascent (Thermo Scientific). For each condition, the total luciferase signal of 75,000 cells was measured. Each experiment was performed in triplicate.
2.8. siRNA transfection
A2780 cells were reverse transfected with either BRCA1 esiRNA (EHU096311, Sigma–Aldrich) or RLUC esiRNA (EHURLUC, Sigma–Aldrich) using Lipofectamine RNAiMAX reagent (Life Technologies) in a 10:3 ratio of DNA: Lipofectamine RNAiMAX according to the manufacturer's protocol. For every experiment, BRCA1 knockdown was confirmed by qRT‐PCR. FACS analyses (ROS production, cell death analysis, dsDNA breaks) were performed 4 days after transfection. Cells that were used for the MTS assay and colony forming assay (CFA), were reverse transfected a second time 2 days after the first transfection. The MTS assay was performed 2 days after the second transfection, the CFA was seeded 1 day after the second transfection.
2.9. Metabolic activity
0.35 × 104 OSE‐C2 cells (empty, OX2‐SKD, OX5‐SKD, NRF2 cDNA) were seeded and 2 × 104 A2780 cells were (for the second time) siRNA reverse transfected (empty RLUC/BRCA1 siRNA, OX2‐SKD RLUC/BRCA1 siRNA) in 96‐well plates. The following day, OSE‐C2 cells were treated for 48 h with 200 μM H2O2 (Sigma–Aldrich), 5 μM N4Py or 0.1% DMSO as a control. A2780 cells were treated for 24 h with 1 μM or 5 μM of the PARP inhibitor olaparib (AZD‐2281, KU‐0059436, Cayman Chemical) or 0.1% DMSO. After treatment, metabolic activity was measured by incubating the cells with CellTiter 96 Aqueous One Solution (Promega) for 3 h at 33 °C (OSE‐C2) or 37 °C (A2780). The absorbance at 490 nm was measured using a Biorad iMark microplate reader (Biorad) and subtracted with the absorbance of cell‐free medium. For every experiment, triplicates were measured of each condition.
2.10. Total glutathione assay
A2780 cells were treated with 0, 0.5, 1, 2 and 10 mM BSO for 48h. After treatment, cells were lysed in 5% trichloroacetic acid. After a 5 min centrifugation at 10,000 g, the supernatant was collected and diluted 1:15 in GSH buffer (0.125 M phosphate buffer, 5 mM EDTA, pH 7.5). Total glutathione levels were determined in the resulting supernatant by the Tietze enzymatic recycling assay (Tietze, 1969). Standards of known GSH content were serial‐diluted in order to make a standard curve. The GSH content of all samples was determined by reference to this standard curve. Each reaction mixture contained 150 μl sample, 1.5 mM DTNB and 0.25 U GSH reductase. Just before reading, 0.6 mM NADPH was added to start the reaction. The formation of reduced DTNB was followed at 405 nm for 10 min using a VersaMax ELISA microplate reader (Molecular Devices). All standards and samples were measured in duplicate.
2.11. ROS production and cell death analysis
OSE‐C2 cells (empty, OX2‐SKD, OX5‐SKD, NRF2 cDNA) or A2780 cells (empty, OX2‐SKD) were seeded in 12‐wells plates. The next day, when cells were about 80% confluent, treatment was started. 24 h after starting treatment, floating cells were collected and remaining cells were incubated with 5 μM CellROX Deep Red Reagent (Life Technologies) for 30 min at 33 °C (OSE‐C2) or 37 °C (A2780) in complete medium. After 3 PBS washes, cells were harvested and combined with the floating cells. All together, the cells were stained with 5 μg/mL propidium iodide (PI) (Sigma–Aldrich) in PBS. After a 10 min incubation at 4 °C in the dark, PI fluorescence was measured using the FL‐3 channel and CellROX Deep Red fluorescence was measured in the FL‐4 channel of a FACScalibur flow cytometer (Beckton Dickenson Biosciences). MFI of CellROX Deep Red and percentage PI positive cells was determined with Kaluza 1.3 software (Beckman Coulter).
2.12. dsDNA breaks
The amount of dsDNA damage induced by PARP inhibitor (co‐)treatment was analyzed using intracellular γH2AX staining with FACS read‐out as described before (Li et al., 2010). In short, 12.5 × 104 A2780 cells (empty RLUC/BRCA1 siRNA, OX2‐SKD RLUC/BRCA1 siRNA) were seeded in 24‐wells plates. The next day, 1 μM olaparib or 0.1% DMSO treatment was started. 24 h later, cells were harvested, fixed with 4% formaldehyde for 10 min at 37 °C and permeabilized in 90% methanol for 30 min on ice. Cells were stained for 30 min at RT in the dark with 1:50 rabbit anti‐phospho‐histone H2A.X (ser139) conjugated Alexa Fluor 647 (20E3, Cell Signaling) and 100 μg/ml RNase A (Qiagen) in 0.5% BSA/PBS. After washing the cells with PBS, cells were stained with 5 μg/mL PI in PBS for 10 min at 4 °C in the dark. PI fluorescence was measured using the FL‐3 channel and γH2AX fluorescence was measured in the FL‐4 channel of a FACScalibur flow cytometer (Beckton Dickenson Biosciences). Cells in the subG1 population were excluded from analysis of dsDNA breaks in early/non‐apoptotic cells. Percentage γH2AX positive cells in the early/non‐apoptotic cell population was determined with Kaluza 1.3 software (Beckman Coulter). The cutoff for a γH2AX positive cell was set based on a background level of ∼3% positivity in the empty vector, RLUC siRNA, 0.1% DMSO treated control.
2.13. Colony forming assay
A2780 cells (empty RLUC/BRCA1 siRNA, OX2‐SKD RLUC/BRCA1 siRNA) were treated for 24 h with 1 μM olaparib or 0.1% DMSO. After treatment, for every condition 1000 cells were seeded in 6‐wells plates containing fresh media. After 6–7 days, media was replaced by Coomassie brilliant blue (Bio‐Rad). The colony forming potential was determined by counting the number of colonies (≥50 cells) using phase‐contrast microscopy. Data was generated in two independent experiments, performed in duplicate.
2.14. Statistical analysis
Statistical tests were performed with Graphpad Prism 5 software (GraphPad Software). All experiments were performed at least three times, unless stated otherwise. Data was evaluated by one‐way, two‐way analysis of variance and Student's t‐test. Multiple comparisons of ANOVA were followed with post‐hoc Bonferroni. A p‐value of <0.05 was considered statistically significant. All data are presented as the mean ± SEM.
3. Results
3.1. NRF2 expression can be modulated by NRF2‐targeting ATFs in both healthy and malignant ovarian epithelial cells
To modulate NRF2 expression, six different ZFP‐SKD constructs (OX1‐OX6), targeting the NRF2 promoter region (Figure 1A), were engineered and virally delivered in three ovarian cell lines: SKOV3, A2780 and OSE‐C2. Successful delivery was confirmed by GFP FACS (Suppl. Figure 1A). OX2‐SKD and OX5‐SKD modulated NRF2 expression about 1.5‐fold and up to twofold, respectively, in two out of three ovarian cell lines tested (A2780 and OSE‐C2) (Figure 1B). Unexpectedly, expression of OX5 fused to the gene repressor SKD, resulted in a twofold upregulation of NRF2 expression, in both A2780 and OSE‐C2 cells. To find a possible explanation for this unexpected finding that OX5 when fused to a transcriptional repressor (SKD) mediates activation, we performed a search for transcription factor binding sites in MatInspector (Genomatix). This search uncovered that binding of the transcription factor YY2 partly overlaps (33%) with the OX5 ZFP binding site. This opens up the possibility that YY2 binding or function can be affected by an OX5‐containing ATF.
Figure 1.
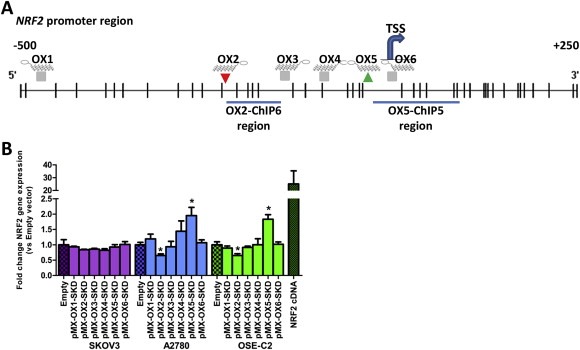
Modulation of NRF2 gene expression in normal and malignant ovarian epithelial cells by NRF2‐targeting ATFs. (A) Schematic overview of the NRF2 promoter region (transcript variant 1) containing the TSS (transcription start site, +1) and the target regions for the 6 engineered zinc finger proteins (ZFP OX1‐OX6). Histone modifications associated with ATF OX2 and OX5 were determined in ChIP region OX2‐ChIP6 and OX5‐ChIP5, respectively. CpGs are shown as vertical lines. (B) Relative NRF2 expression compared to empty vector control upon retroviral delivery of NRF2‐targeting ZFPs (OX1‐OX6) fused to the transcriptional repressor SKD in SKOV3, A2780 and OSE‐C2 cells. Relative NRF2 expression of the NRF2 cDNA control has only been determined in OSE‐C2 cells. Data is presented as mean ± SEM of at least three independent experiments. *p < 0.05.
To unravel the effects of OX2‐SKD and OX5‐SKD, validation experiments were performed in OSE‐C2 cells. ZFP binding to its predicted target region was first confirmed by HA‐tag ChIP (Figure 2A). Binding competition with other gene regulatory proteins was assessed by analyzing the effects of ZFPs only (NoED) and the ZFPs fused to the transcriptional activator VP64. Successful viral delivery of all these constructs was confirmed by GFP FACS (Suppl. Figure 1B). ZFPs without effector domain did not affect NRF2 expression (Figure 2B). OX2‐VP64 showed a twofold upregulation, whereas OX5‐VP64 showed a 1.5‐fold downregulation of NRF2 expression (Figure 2B). So, VP64 ATFs showed the same level of modulation as their SKD counterparts, but this time, in the opposite direction. The opposite effects of OX2 and OX5 were confirmed for the SKD fusions by NRF2 promoter luciferase reporter construct experiments (Figure 2C).
Figure 2.
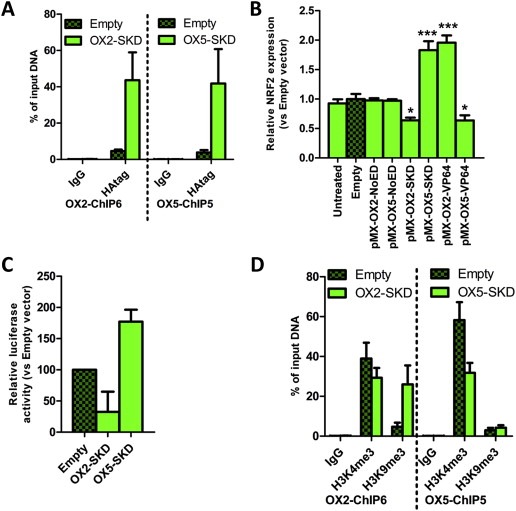
Validation of NRF2‐targeting ATFs in normal ovarian epithelial cells. OSE‐C2 cells were transduced to express OX2‐and OX5‐ZFP fusions and both ATFs were validated (A) for ZFP binding; (B) for effector domain‐specific effects; (C) for having a direct effect on the NRF2 promoter; (D) for epigenetic effects on their target region. (A) ZFP binding (HA‐tag) in the OX2‐ChIP6 region (left panel) and OX5‐ChIP5 region (right panel) was validated by quantitative ChIP, IgG was used as negative control. (B) Relative NRF2 expression compared to empty vector control upon retroviral delivery of NRF2‐targeting ZFPs (OX1‐OX6) fused to no effector domain (NoED), the transcriptional repressor SKD or the transcriptional activator VP64. (C) Relative NRF2 promoter activity as measured by the luciferase activity of pCpG‐NRF2 promoter‐luciferase of cells transduced to express OX2‐SKD and OX5‐SKD compared to cells expressing empty vector control. (D) Quantitative ChIP for an active histone modification (H3K4me3), a repressive histone modification (H3K9me3) and IgG control of the OX2‐ChIP6 region (left panel) and OX5‐ChIP5 region (right panel). Each bar represents the mean ± SEM of at least three independent experiments. *p < 0.05, ***p < 0.001.
To gain more insights into the native chromatin state as well as in the ATF‐induced changes of the OX2 and OX5 targeted region, a ChIP for the active histone mark H3K4me3 and repressive histone mark H3K9me3 was performed (Figure 2D). These experiments indicated that the NRF2 downregulating ATF OX2‐SKD induced the repressive histone mark H3K9me3, whereas the NRF2 upregulating ATF OX5‐SKD did not. Moreover, OX5‐SKD associated upregulation of NRF2 did not result in an increase of the active histone mark H3K4me3.
3.2. Gene‐targeted modulation of NRF2 by ATFs results in the modulation of downstream Nrf2 target genes
Before continuing to use the NRF2‐ATFs as tool to study the role of Nrf2 in ovarian carcinogenesis, they were further validated in healthy (OSE‐C2) and malignant (A2780) ovarian cell lines. Despite having about equal levels of GFP positive cells, the average infection efficiency per cell was at least 10 fold lower in A2780 compared to OSE‐C2 cells (data not shown). Therefore, in all subsequent experiments, average infection efficiency in A2780 cells was further improved by using lentivirally‐superinfected, GFP sorted cells. Modulation of NRF2 mRNA does not necessarily translate to functional changes, as many post‐translational factors regulate Nrf2 (van der Wijst et al., 2014), e.g. the cytoplasmic fraction of Nrf2 can be bound by Keap1, and subsequently degraded. Only when Nrf2 translocates to the nucleus, it can bind to downstream target genes, but also here, the outcome is determined by the balance between repressive (e.g. Bach1, Src kinases) and activating factors (Nrf2) (van der Wijst et al., 2014). Moreover, some Nrf2 target genes are known to be expressed only in certain tissues (Nakajima et al., 2011). A quick screen on the expression of known Nrf2 target genes in OSE‐C2 cells transduced to express NRF2 cDNA (Suppl. Figure 2A) or treated with the ARE‐inducer tBHQ (Suppl. Figure 2B), indicated that GCLC, an enzyme involved in glutathione synthesis, and the vitagene HMOX1, an enzyme involved in heme catabolism, but not the phase 2 detoxification enzyme NQO1, were potent downstream targets of Nrf2 in these cells. Subsequently, it was confirmed in OSE‐C2 cells, that the OX5‐SKD mediated upregulation of NRF2 mRNA (Figure 1B) resulted in activation of three well‐known Nrf2 target genes (GCLC, HMOX1, OGG1) (Malhotra et al., 2010; Singh et al., 2013). Indeed an increased expression of these downstream genes was observed in OX5‐SKD expressing cells compared to empty vector: GCLC was upregulated by 1.7‐fold, HMOX1 by 2.7‐fold and OGG1 by 1.6‐fold (Figure 3A). Vice‐versa, in A2780 cells the OX2‐SKD mediated downregulation of NRF2 mRNA (Figure 1B) resulted in decreased nuclear Nrf2 protein levels (detected at 110 kDa, see Suppl. Figure 3, as described before in (Lau et al., 2013; Malloy et al., 2013)) up to about 80% (Figure 3B). This translated to repression of Nrf2 target genes: GCLC was inhibited by 1.9 fold and HMOX1 by 1.7 fold (Figure 3C). Remarkably, despite the lack of robust NRF2 mRNA inhibition in A2780 cells by OX2‐SKD (Figure 1B), the mRNA inhibition was efficient enough to profoundly affect nuclear Nrf2 protein levels (Figure 3B) and downstream genes (Figure 3C). In addition to the NRF2 mRNA, many other factors can modulate the nuclear Nrf2 protein fraction (Chowdhry et al., 2013; Khalil et al., 2014; van der Wijst et al., 2014). These factors might explain the seemingly discrepancy between NRF2 mRNA and Nrf2 protein inhibition by OX2‐SKD.
Figure 3.
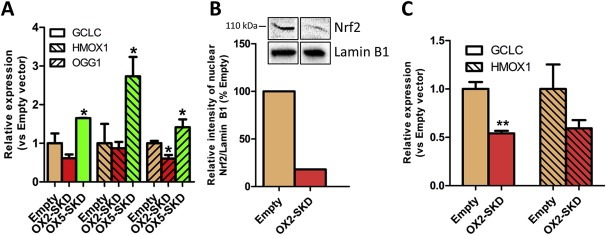
NRF2‐targeting ATFs modulate the expression of known downstream Nrf2 target genes. The effect of NRF2‐targeting ATFs on known downstream Nrf2 target genes was assessed in healthy (OSE‐C2) and malignant (A2780) ovarian cells. (A) Relative NRF2 expression of known Nrf2 target genes (GCLC, HMOX1, OGG1) compared to empty vector control upon expression of the NRF2‐targeting ATFs or NRF2 cDNA in OSE‐C2 cells. (B) Relative quantification of nuclear Nrf2 protein levels in NRF2‐inhibiting ATF OX2‐SKD expressing A2780 cells compared to empty vector control. Insert: western blot against nuclear Nrf2 and Lamin B1 (nuclear loading control) containing nuclear protein lysates of A2780 empty vector and OX2‐SKD expressing cells. (C) Relative expression compared to empty vector of known Nrf2 target genes (GCLC, HMOX1) upon expression of the NRF2‐inhibiting ATF OX2‐SKD in A2780 cells. Data values are mean ± SEM of at least three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
Although NRF2 mRNA was inhibited to the same extent by OX2‐SKD in A2780 and OSE‐C2 cells, this did not result in significant repression of Nrf2 downstream genes in OSE‐C2 cells (Figure 3A–B), with the exception of the 1.7‐fold reduction in OGG1 mRNA levels (Figure 3A). This gives us a first hint that healthy ovarian cells (OSE‐C2) might be less sensitive toward the effects of NRF2 inhibition compared to malignant ovarian cells (A2780), which would agree with current literature that cancer cells in general are often more dependent on a high (Nrf2‐induced) antioxidant capacity for their survival (Gorrini et al., 2013b).
3.3. Cytoprotective effect of NRF2 might be partly exerted by induction of ER UPR genes
Nrf2 is the master regulator of antioxidant and cytoprotective genes, and as such protects cells towards ROS. It is able to do so by activating downstream genes involved in the stimulation of antioxidant production (GCLC, HMOX1) and cytoprotection via the stimulation of the oxidative damage DNA repair enzyme OGG1 (Figure 3A). We wondered whether Nrf2 could also exert its cytoprotective function by inducing an ER UPR. To this end, four genes involved in this cytoprotective response were measured in OSE‐C2 cells: ERp72, HERP, Bip/GRP78, p58IPK. The expression of all these ER UPR genes was increased in OX5‐SKD expressing cells compared to empty vector: ERp72 was upregulated by 1.7‐fold, HERP by 1.9‐fold, Bip/GRP78 by 1.9‐fold and p58IPK by 1.8‐fold (Figure 4A–D). For all four genes a similar trend was visible for NRF2 cDNA. On the other hand, NRF2 inhibition by OX2‐SKD resulted in a trend towards reduction of gene expression. The increased expression of ER UPR genes in OX5‐SKD expressing cells did not affect cell growth (Suppl. Figure 4A).
Figure 4.
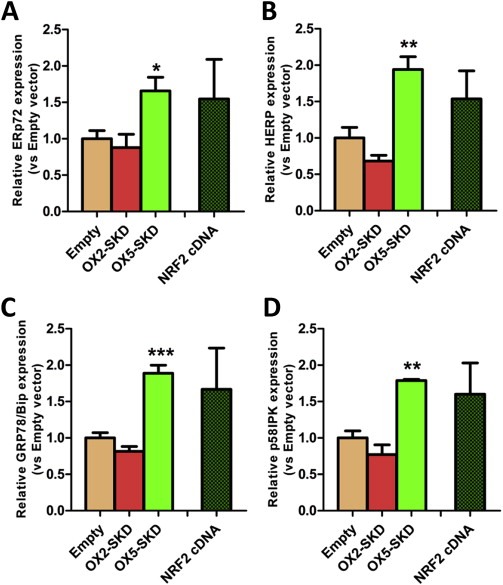
NRF2 ‐targeting ATFs modulate the expression of genes involved in the endoplasmatic reticulum unfolded protein response (ER UPR). Normal ovarian epithelial cells (OSE‐C2) were transduced to express NRF2‐targeting ATFs or NRF2 cDNA. (A–D) Relative expression of genes involved in the ER UPR (ERp72, HERP, Bip/GRP78, p58IPK) was determined. Data values are mean ± SEM of at least three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
3.4. Gene‐targeted modulation of NRF2 by ATFs affects the sensitivity toward ROS‐induced damage
As shown above, gene‐targeted modulation of NRF2 enabled us to modulate downstream target genes involved in the protection against ROS‐induced damage. Therefore, this is expected to influence endogenous ROS levels and as such protect (Nrf2 upregulation) or sensitize (Nrf2 downregulation) cells toward ROS‐induced cell death. To determine the influence of ATF‐mediated NRF2 modulation on ROS production in OSE‐C2 and A2780 cells, ROS levels were determined upon exposure to a lethal dose of ROS (30 μM N4Py). In OSE‐C2 cells harboring upregulated NRF2 expression either induced by OX5‐SKD or NRF2 cDNA, ROS levels were reduced by about 20% compared to empty vector (Figure 5A). On the other hand, OX2‐SKD mediated downregulation of NRF2 expression did not affect ROS production in these cells (Figure 5A). Similarly, in A2780 cells, no increase in ROS production was detected in NRF2 knockdown A2780 cells, as measured by the Cell Rox Deep Red ROS probe (Figure 5B). The absence of increased ROS production in NRF2 knockdown compared to empty A2780 cells, even in the presence of 30 μM N4Py, might be explained by the method of ROS induction. In order to test whether less acute ROS induction would make any difference, A2780 cells were depleted of glutathione by BSO. Glutathione depletion was confirmed in all tested BSO concentrations (0.5–10 mM BSO) (Suppl. Figure 5A). Two days after BSO‐induced glutathione depletion, cell death (Suppl. Figure 5B) and ROS production (Suppl. Figure 5C) were measured by FACS. Neither cell death, nor ROS production was affected by BSO treatment. Despite the absence of increased ROS production in both OSE‐C2 (Figure 5A) and A2780 cells (Figure 5B) upon ATF‐mediated NRF2 knockdown combined with exposure to lethal ROS levels, a 1.5‐fold higher level of ROS‐production in these A2780 compared to OSE‐C2 cells was observed (Figure 5C).
Figure 5.
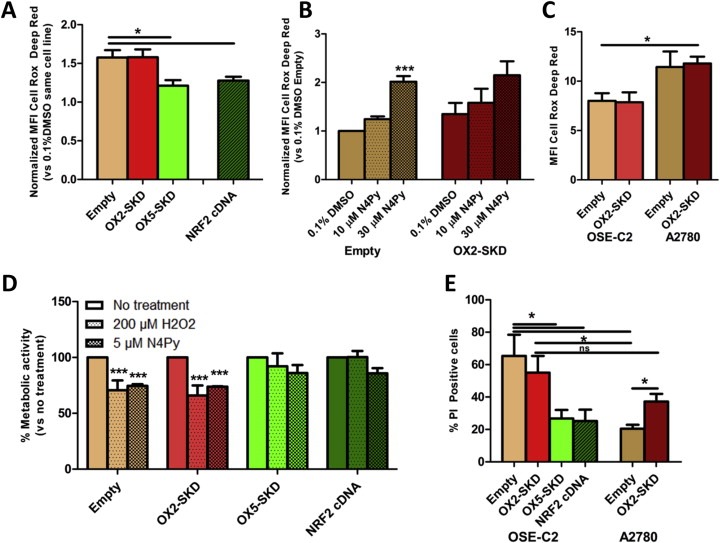
Modulation of NRF2 by ATFs affects the sensitivity of normal and malignant ovarian epithelial cells against non‐cytotoxic and cytotoxic levels of ROS. OSE‐C2 and A2780 cells were transduced to express NRF2‐ATFs and were exposed to either non‐cytotoxic (≤10 μM N4Py, 200 μM H2O2) or cytotoxic (30 μM N4Py) levels of ROS. The effect on ROS production compared to 0.1% DMSO control treatment was determined after a 24 h exposure to 30 μM N4Py in OSE‐C2 (A) and 10 μM or 30 μM N4Py in A2780 (B) cells. (C) For a direct comparison between both cell lines, the absolute ROS production in OSE‐C2 and A2780 cells was determined after a 24h treatment with 30 μM N4Py. (D) OSE‐C2 cells were exposed to either a bolus of 200 μM H2O2, 5 μM N4Py or no treatment. After 2 days, metabolic activity was measured by MTS and compared to no treatment of the same cell line. (E) OSE‐C2 and A2780 cells were treated for 24h with 30 μM N4Py and the percentage of late apoptotic/necrotic cells (PI positivity) was determined by FACS analysis. Each value shows the mean ± SEM of at least three independent experiments. *p < 0.05, ***p < 0.001.
Further experiments were conducted to reveal whether the observed effects on endogenous ROS levels also affect the sensitivity of OSE‐C2 and A2780 cells toward ROS‐induced damage. To start with, the effect of sublethal ROS levels, either induced by H2O2 or N4Py, was determined in OSE‐C2 cells that were transduced to express the NRF2‐ATFs. In cells expressing basal (empty vector) or lowered (OX2‐SKD) levels of NRF2, exposure to sublethal ROS levels resulted in lower metabolic activity: 35% decrease upon H2O2 and 26% decrease upon N4Py treatment compared to untreated cells (Figure 4D). Cells with increased expression of NRF2 were not significantly affected by the sublethal ROS levels; upon treatment with H2O2, metabolic activity was only slightly, but not significantly lowered in OX5‐SKD expressing cells (8%) compared to untreated cells, whereas metabolic activity was unchanged in cells expressing NRF2 cDNA (0%). Treatment with N4Py equally, but not significantly, affected OX5‐SKD and NRF2 cDNA expressing cells by reducing metabolic activity with 14% compared to untreated cells (Figure 5D). Similarly, when these cells were exposed to lethal levels of ROS (induced by 30 μM N4Py), cell death could be equally reduced by OX5‐SKD and NRF2 cDNA (Figure 5E). Compared to empty vector, cells harboring upregulated NRF2 expression showed a reduction of about 60% in cell death. So, the level of NRF2 upregulation (1.8‐fold for OX5‐SKD vs 25‐fold for NRF2 cDNA, Figure 1B) did not significantly affect the protective effect of high NRF2 expression against (sub)lethal ROS levels in OSE‐C2 cells (Figure 5D–E). In these cells, OX2‐SKD‐mediated downregulation of NRF2 expression did not result in increased sensitivity toward ROS‐induced cell death. On the contrary, when A2780 cells were exposed to lethal ROS levels (induced by 30 μM N4Py), OX2‐SKD‐mediated downregulation of NRF2 expression increased the sensitivity toward ROS‐induced cell death almost twice; the percentage PI positive cells was increased from 20% in empty vector control cells to 37% in ATF‐induced NRF2 knockdown cells (Figure 5E). These results indicate that upon ATF‐mediated NRF2 inhibition the sensitivity toward ROS‐induced damage can be further increased specifically in malignant ovarian cells (A2780), whereas the effects on healthy ovarian cells (OSE‐C2) seem to be unchanged. Despite this, baseline sensitivity toward ROS‐induced cell death was lower in empty vector control A2780 compared to OSE‐C2 cells: 20% versus 65% PI positive cells, respectively. Therefore, ATF‐mediated NRF2 knockdown could not significantly alter overall sensitivity toward ROS‐induced cell death between both cell lines: 37% PI positive cells in A2780 versus 55% in OSE‐C2 cells (Figure 5C). This finding highlights the importance of a combination therapy to further improve on the cancer‐selectivity of NRF2 inhibition.
3.5. Gene‐targeted downregulation of NRF2 by ATFs improves the cytotoxic effect of PARP inhibitors in BRCA1 knockdown A2780 ovarian cancer cells
Extensive validation of the NRF2‐targeting ATFs was performed by (1) confirming binding to the target region (Figure 2A); (2) confirming effective modulation at the mRNA level (Figure 1B) (3) and protein level (Figure 3B); (4) confirming functional downstream effects (3, 5A–E). This proved that NRF2‐ATFs can be potent tools to study the role of Nrf2 in ovarian carcinogenesis. Therefore, this tool was used to unravel the potency of Nrf2 inhibition as therapeutic target in ovarian cancer treatment. Moreover, it was determined whether combining knockdown of NRF2 with PARP inhibitors could further improve the effect of NRF2 inhibition on cancer cell growth of BRCA1 knockdown ovarian cancer cells. As shown in Suppl. Figure 6, BRCA1 protein was almost completely knocked down 3 and 4 days after transfection with BRCA1 siRNA compared to control RLUC siRNA. In this time frame, PARP inhibitors could only slightly affect metabolic activity (up to 17% reduction compared to untreated cells) when combined with NRF2 inhibition in BRCA1 knockdown A2780 cells (Figure 6A). To study the long term effects of PARP inhibitors in combination with NRF2 inhibition, first a dose–response curve was obtained to determine the lowest effective concentration of PARP inhibitor in A2780 cells with NRF2 and BRCA1 knockdown: 1 μM olaparib was chosen for subsequent colony forming assays (CFA) (Figure 6B). Next, it was confirmed that olaparib treatment, independent of Nrf2 expression, specifically affected the colony forming potential of BRCA1 knockdown and not of wild‐type cells (Figure 6B); PARP inhibitor treatment reduced the colony forming potential of BRCA1 knockdown A2780 cells by 62% compared to untreated, while no effect was observed for BRCA1 wildtype A2780 cells. BRCA1 can directly bind Nrf2, thereby interfere with Keap1 binding, and enhance the stability and activation of Nrf2 (Gorrini et al., 2013a). Therefore, BRCA1 inhibition might further inhibit the activity of Nrf2, and this could translate to a greater reduction in colony forming potential. Despite this, inhibition of NRF2 alone reduced the colony forming potential to the same extent in wild‐type (79%) and BRCA1 knockdown (76%) A2780 cells. In contrast, in healthy ovarian cells (OSE‐C2 cells) no effect on cell growth was observed upon inhibition of NRF2 (Suppl. Figure 4B), which reveals the potential therapeutic window for NRF2 inhibition treatment. Interestingly, in BRCA1 knockdown A2780 cells, the combination of 1 μM olaparib and ATF‐induced NRF2 inhibition almost completely diminished the colony forming potential (90% versus 62% without NRF2 inhibition). This data opens up the possibility to further increase the therapeutic window for NRF2 inhibition treatment by using a combination therapy with PARP inhibitors.
Figure 6.
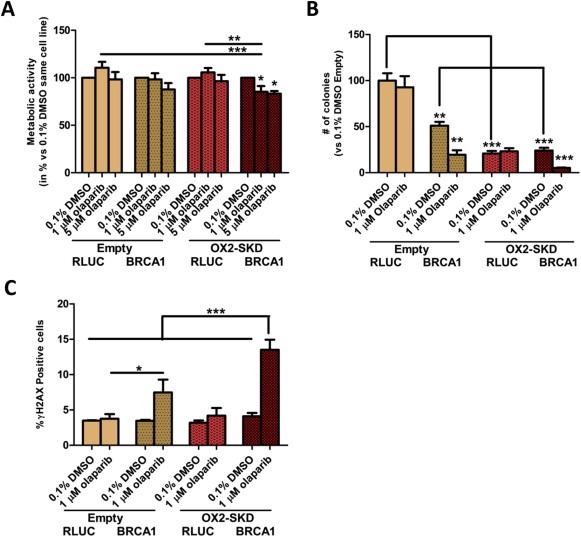
Downregulation of NRF2 improves the cytotoxic effect of PARP inhibitors in BRCA1 knockdown A2780 ovarian cancer cells. (A) A2780 cells with (OX2‐SKD) or without (empty) ATF‐mediated NRF2 downregulation, were transfected with BRCA1 siRNA or RLUC control siRNA. After 3 days, 1 μM or 5 μM olaparib (PARP inhibitor) or 0.1% DMSO treatment was started. 24 h later, metabolic activity was measured by the MTS assay. (B) After 24 treatment with either 1 μM olaparib or 0.1% DMSO, cells were seeded for the colony forming assay and colony formation was determined 6–7 days later. (C) After 24 h treatment, the percentage of cells with increased dsDNA breaks (γH2AX positive) was determined. *p < 0.05; **p < 0.01; ***p < 0.001.
In order to gain a better understanding of the mechanism by which the PARP‐1 inhibitor/NRF2 inhibition combination therapy in BRCA1 knockdown ovarian cancer cells acts, the effect of BRCA1 knockdown and 1 μM olaparib on ROS production was determined. Neither 1 μM olaparib, nor BRCA1 knockdown (or the combination of both) influenced ROS levels (Suppl. Figure 7). Inhibition of NRF2 is expected to sensitize cells toward ROS‐induced DNA damage, which is mainly repaired by BER (Mitra et al., 2001). As PARP inhibitors block BER, ROS‐induced DNA damage is expected to accumulate (Banerjee and Kaye, 2011; Fong et al., 2009). Upon cell division, these ssDNA breaks become dsDNA breaks. As BRCA1 impaired cells are unable to repair this damage, colony forming potential is expected to be reduced the most in these cells. Indeed, treatment of BRCA1 knockdown A2780 cells with 1 μM olaparib resulted in a 2.2‐fold increase in dsDNA breaks compared to wild‐type (Figure 6C). The amount of dsDNA breaks even further increased up to 3.3‐fold compared to wild‐type when NRF2 was inhibited in these cells. Without knockdown of BRCA1, inhibition of NRF2 did not increase the formation of dsDNA breaks, highlighting the selectivity of this combination treatment (6, 7).
Figure 7.
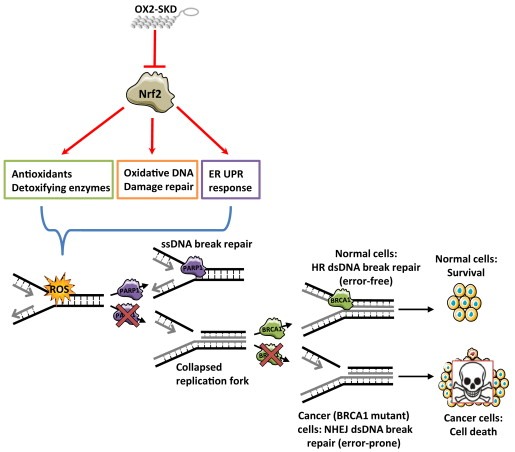
Hypothesis enhanced cytotoxic effect of combination therapy (PARP inhibitors and NRF2 inhibition) in BRCA1 mutant cancers. ATF‐mediated (OX2‐SKD) repression of NRF2 results in downregulation of downstream target genes involved in the protection against ROS. This results in enhanced sensitivity toward ROS‐induced DNA and protein damage. BER (base excision repair) is mainly involved in the repair of ROS‐induced DNA damage. When PARP inhibitors inhibit this DNA repair pathway, the ROS‐induced ssDNA breaks will turn into dsDNA breaks upon cell division. In normal, non‐BRCA1 defective cells, dsDNA breaks will be repaired by the error‐free HR (homologous recombination) DNA repair pathway. In contrast, in malignant, BRCA1 defective cells, dsDNA breaks cannot be repaired by HR, and therefore, the more error‐prone NHEJ (non‐homologous end joining) DNA repair pathway will take over. The end‐result is genomic instability and this will induce cell death.
4. Discussion
In this study, NRF2 targeting ZFP‐ATFs that are uniquely suited to study the dual role of Nrf2 in ovarian carcinogenesis, were constructed and validated. By the use of these NRF2 targeting ATFs, a chemopreventive role of Nrf2 in healthy cells was confirmed, whereas inhibition of Nrf2 had anti‐carcinogenic effects in malignant cells. With respect to the chemopreventive function of Nrf2, ATF‐induced upregulation of NRF2 was shown to increase the expression of genes involved in the protection against ROS‐induced damage, among which potentially new Nrf2 downstream genes involved in an adaptive ER UPR. This translated to increased protection against ROS‐induced cell death in healthy ovarian epithelial cells. With respect to the anti‐carcinogenic effects of Nrf2 inhibition, it was revealed that ATF‐induced downregulation of NRF2 resulted in an almost 80% reduction of colony forming potential in malignant ovarian epithelial cells. Interestingly, specifically in BRCA1 knockdown ovarian cancer cells, the effect of NRF2 inhibition on colony forming potential could be even further improved by co‐treating with PARP inhibitors: the colony forming potential was almost complete halted.
The rationale behind ROS‐targeted therapies, like inhibition of Nrf2, is based on the existence of a therapeutic window as healthy cells have a lower baseline level of ROS compared to cancer cells (Gorrini et al., 2013b). Here, it was confirmed that malignant (A2780) versus healthy (OSE‐C2) ovarian cells exhibit higher levels of ROS production. Therefore, to maintain redox balance, cancer cells are more dependent on ROS protective and neutralizing signaling, including Nrf2 signaling. Translated to our study, this would imply that exposure to ROS in combination with ATF‐mediated NRF2 inhibition, would result in a stronger increase in cell death in the malignant (A2780) compared to healthy (OSE‐C2) ovarian cells, which indeed was observed. However, despite specifically increasing sensitivity toward ROS‐induced cell death in malignant A2780 cells upon NRF2 downregulation, the reached level of cell death was comparable to that of healthy OSE‐C2 cells. This highlights the importance of a combination therapy that preferentially hits the tumor cells for further improvement in efficacy of Nrf2 inhibition in cancer.
We hypothesized that PARP inhibitor treatment could improve the therapeutic window of Nrf2 inhibition (Rouleau et al., 2010) specifically in BRCA1 mutant cancers, without acquiring the serious side‐effects of chemotherapy or radiation (Cao et al., 2012; Kim et al., 2014). In line with literature (Fong et al., 2009, 2010, 2010), the PARP inhibitor olaparib could only affect the colony forming potential of BRCA1 knockdown and not BRCA1 wt cells (independent of their Nrf2 status). Therefore, based on literature and our own data, healthy cells, which are BRCA1 wt, are not expected to be affected by PARP inhibition. BRCA1 siRNA enabled us to study the effect of BRCA1 expression in the same background. Here, it was shown that specifically in BRCA1 knockdown and not BRCA1 wt cells, the combination of PARP inhibitors and NRF2 inhibition resulted in an increase in dsDNA breaks, and this translated to an almost complete inability of the cells to form colonies. However, likely, also other unknown interactions have contributed to this effect, for example, via PARP‐1 its potential to affect transcription and chromatin modifications (Kim et al., 2005; Kraus and Lis, 2003). This successful combination of Nrf2 inhibition with PARP inhibitors opens up opportunities for a therapy in which the efficacy of Nrf2 inhibition can be increased without increasing the adverse effects of previous combinations with chemotherapy or radiation (Cao et al., 2012; Kim et al., 2014). Interestingly, recently, it was discovered that inhibition of the transcriptional, but not the enzymatic activity of PARP‐1, can inhibit the activity of PARP‐1 in serving as a transcriptional coactivator of Nrf2. As a transcriptional coactivator, PARP‐1 can stimulate the transcriptional activity of Nrf2 by enhancing the interaction among Nrf2, MafG, and the ARE (Wu et al., 2014). As such, it can be hypothesized that in addition to inhibiting the enzymatic activity of PARP‐1 by PARP inhibitors, inhibition of PARP‐1 expression would have beneficial effects when combined with Nrf2 inhibition. However, inhibiting all functions of PARP‐1 will probably also increase the adverse effects.
Exposure to oxidative stress can damage all kinds of biomolecules, such as proteins. Accumulation of damaged proteins can activate the ER UPR (Walter and Ron, 2011). One of the main activators of the ER UPR, PERK, has been shown to activate Nrf2 via site‐specific phosphorylation (Cullinan et al., 2003). However, the other way around, it is currently unknown whether Nrf2 can activate the ER UPR. Here, we gained the first insights into the capability of Nrf2 to (in)directly activate genes involved in the ER UPR; Upon ATF‐induced NRF2‐expression in OSE‐C2 cells, a similar expression pattern of ER UPR genes was observed as has been seen before in low ER stress adapted cells (Rutkowski et al., 2006). In these low ER stress adapted cells, an attenuation of the activation of the three main activators (IRE1, PERK and ATF6) was observed, whereas proteins such as Bip/GRP78 and p58IPK were kept stably activated at a low level. This resulted in an adaptive rather than a pro‐apoptotic ER stress response: cells did not undergo apoptosis, remained their proliferative capacity despite ER UPR activation and became desensitized by ROS‐induced stress. As our ATF‐induced NRF2‐expression OSE‐C2 cells showed a similar response and became desensitized to ROS‐induced stress, we assumed these cells had activated an adaptive rather than a pro‐apoptotic ER stress response. As protein homeostasis is strongly influenced by vitagenes, of which several are activated by Nrf2 (Calabrese et al., 2010), and is closely linked with health and life span of the organism (Calabrese et al., 2010), activation of the adaptive ER UPR response either directly by Nrf2 or indirectly via induction of vitagenes would be a new mechanism in which Nrf2 could stimulate longevity and cancer protection. Despite the inability of OX2‐SKD to downregulate (known) Nrf2 downstream genes, a trend towards inhibition of these downstream genes was seen. Therefore, it was unlikely that these findings could be contributed to the viral transduction, the expression of a zinc finger protein or off‐target effects of OX5‐SKD.
Remarkably, NRF2 cDNA did not activate ER UPR to a higher extent than the ATF OX5‐SKD, despite being able to upregulate NRF2 mRNA more than 10 fold higher compared to OX5‐SKD. Although unexpected, Nrf2 might be involved in an adaptive rather than a pro‐apoptotic ER stress response: To prevent overactivation of the ER stress response, and as such prevent a pro‐apototic ER stress response, the activation of ER UPR genes by Nrf2 should only occur to a certain extent. It can be envisioned that simultaneously with Nrf2‐induced ER UPR activation other proteins are counteracting this process, and as such prevent overactivation of the ER UPR genes. This would explain that independent of the level of Nrf2 activation, ER UPR genes are activated to about the same extent.
The KRAB domain in SKD is a transcriptional repressor which has been frequently fused to ZFPs to downregulate endogenous gene expression (Huisman et al., 2013; Sera, 2009; Stolzenburg et al., 2012). Surprisingly, in our study, ATF OX5‐SKD resulted in increased expression of NRF2. KRAB (co‐factor)‐mediated transcriptional activation has been seen before in specific cases for naturally occuring KRAB‐containing ZFPs (Chang et al., 1998; Rambaud et al., 2009; Rooney and Calame, 2001). Genome‐wide ChIP‐seq analysis combined with RNA‐seq expression analysis revealed relations between binding events and expressional changes for an engineered ZF‐SKD fusion protein (Grimmer et al., 2014). The ATF binding translated to expression changes in only a minority (∼3%) of the bound regions, of which about one‐third was upregulated. Previously, another engineered ZFP fused to SKD had been described to induce upregulation of its target gene OCT4 (Juarez‐Moreno et al., 2013). Interestingly, in this study no effect on OCT4 expression was observed with the ZFP fused to the transcriptional activator VP64, which is in contrast to our data obtained with the OX5 ZFP. The opposite effects of OX2 and OX5 ZFP when fused to VP64 compared to SKD were confirmed by a NRF2‐promoter luciferase reporter, suggesting a direct effect on the NRF2 promoter. Interestingly, the binding of transcription factor YY2, a close‐relative of YY1 with both activation and repression domains (Nguyen et al., 2004), partly overlaps with the OX5 ZFP binding region in the NRF2 promoter. As the function of YY2 might be (in)directly affected by OX5‐SKD, this could provide an explanation for our findings. Direct binding competition of the ZFP OX5 with YY2 could be excluded, as OX5‐NoED did not have any effect on NRF2 expression. The effector domain, however, could attract other proteins that affect the function of YY2 as shown before for its close‐relative YY1 (Yao et al., 2001).
In light of current literature, our results underline Nrf2 as a promising chemopreventive or therapeutic target in (ovarian) carcinogenesis. However, caution should be taken as the function of Nrf2 is context dependent (Sporn and Liby, 2012). To exploit Nrf2 inhibition as anti‐cancer treatment, it is essential to prevent a pro‐carcinogenic outcome in normal cells. Therefore, the therapeutic window of Nrf2 inhibition should be further broadened by combining Nrf2 inhibition with other treatments, such as was studied here with PARP inhibitor treatment. The main advantage of a combination with PARP inhibitor treatment over the combination with radiation or chemotherapy, is the fact that severe side‐effects are not expected (Liu et al., 2014). We speculate that this combination therapy of PARP inhibitors with Nrf2 inhibition is not only effective in BRCA1 defective cells (as shown in this study), but can also be translated to all “BRCAness” tumors. In conclusion, as hyperactivation of Nrf2 is a common phenomenon in cancer (Bauer et al., 2013; Jiang et al., 2010; Stacy et al., 2006; Wang et al., 2010), Nrf2 can be a potent target to combat not only ovarian, but also other cancer types.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Acknowledgments
We gratefully acknowledge Roelof Jan van der Lei for his help with cell sorting, Jelleke Dokter for assistance with cell culturing, Arjan Geersing for preparing and providing us with N4Py and Dr. Richard Edmondson for the conditionally immortalized ovarian epithelial cells (OSE‐C2). This work was supported by the Netherlands Organization for Scientific Research (NWO) through a CHEMTHEM grant, Grant no. 728.011.101 and NWO/VIDI grant, Grant no. 91786373.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2015.03.003.
van der Wijst Monique G.P., Huisman Christian, Mposhi Archibold, Roelfes Gerard, Rots Marianne G., (2015), Targeting Nrf2 in healthy and malignant ovarian epithelial cells: Protection versus promotion, Molecular Oncology, 9, doi: 10.1016/j.molonc.2015.03.003.
Contributor Information
Monique G.P. van der Wijst, Email: m.g.p.van.der.wijst@umcg.nl
Christian Huisman, Email: kriztian.huisman@gmail.com.
Archibold Mposhi, Email: a.mposhi@umcg.nl.
Gerard Roelfes, Email: j.g.roelfes@rug.nl.
Marianne G. Rots, Email: m.g.rots@umcg.nl
References
- Audeh, M.W. , Carmichael, J. , Penson, R.T. , 2010. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet 376, 245–251. [DOI] [PubMed] [Google Scholar]
- Banerjee, S. , Kaye, S. , 2011. PARP inhibitors in BRCA gene-mutated ovarian cancer and beyond. Curr. Oncol. Rep. 13, 442–449. [DOI] [PubMed] [Google Scholar]
- Bauer, A.K. , Hill, T. , Alexander, C.M. , 2013. The involvement of NRF2 in lung cancer. Oxid Med. Cell Longev. 2013, 746432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkman, U. , Ekholm, R. , 1995. Hydrogen peroxide degradation and glutathione peroxidase activity in cultures of thyroid cells. Mol. Cell Endocrinol. 111, 99–107. [DOI] [PubMed] [Google Scholar]
- Calabrese, V. , Cornelius, C. , Dinkova-Kostova, A.T. , Calabrese, E.J. , Mattson, M.P. , 2010. Cellular stress responses, the hormesis paradigm, and vitagenes: novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid. Redox Signal 13, 1763–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, B. , Que, L. , Zhang, Y. , 2012. Association analysis of the expression level of Nrf2 mRNA in peripheral blood nucleated cells and the severity of chemotherapy-induced myelosuppression. Tumor 32, 124–129. [Google Scholar]
- Chang, C.J. , Chen, Y.L. , Lee, S.C. , 1998. Coactivator TIF1beta interacts with transcription factor C/EBPbeta and glucocorticoid receptor to induce alpha1-acid glycoprotein gene expression. Mol. Cell Biol. 18, 5880–5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Zhang, L. , Hao, Q. , 2013. Olaparib: a promising PARP inhibitor in ovarian cancer therapy. Arch. Gynecol. Obstet. 288, 367–374. [DOI] [PubMed] [Google Scholar]
- Cho, J.M. , Manandhar, S. , Lee, H.R. , Park, H.M. , Kwak, M.K. , 2008. Role of the Nrf2-antioxidant system in cytotoxicity mediated by anticancer cisplatin: implication to cancer cell resistance. Cancer Lett. 260, 96–108. [DOI] [PubMed] [Google Scholar]
- Chowdhry, S. , Zhang, Y. , McMahon, M. , Sutherland, C. , Cuadrado, A. , Hayes, J.D. , 2013. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 32, 3765–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornblatt, B.S. , Ye, L. , Dinkova-Kostova, A.T. , 2007. Preclinical and clinical evaluation of sulforaphane for chemoprevention in the breast. Carcinogenesis 28, 1485–1490. [DOI] [PubMed] [Google Scholar]
- Cullinan, S.B. , Zhang, D. , Hannink, M. , Arvisais, E. , Kaufman, R.J. , Diehl, J.A. , 2003. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell Biol. 23, 7198–7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, B.R. , Steele, I.A. , Edmondson, R.J. , 2003. Immortalisation of human ovarian surface epithelium with telomerase and temperature-sensitive SV40 large T antigen. Exp. Cell Res. 288, 390–402. [DOI] [PubMed] [Google Scholar]
- Fong, P.C. , Boss, D.S. , Yap, T.A. , 2009. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 361, 123–134. [DOI] [PubMed] [Google Scholar]
- Fong, P.C. , Yap, T.A. , Boss, D.S. , 2010. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 28, 2512–2519. [DOI] [PubMed] [Google Scholar]
- Gaj, T. , Gersbach, C.A. , Barbas, C.F. , 2013. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31, 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gersbach, C.A. , Perez-Pinera, P. , 2014. Activating human genes with zinc finger proteins, transcription activator-like effectors and CRISPR/Cas9 for gene therapy and regenerative medicine. Expert Opin. Ther. Targets 18, 835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrini, C. , Baniasadi, P.S. , Harris, I.S. , 2013. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J. Exp. Med. 210, 1529–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrini, C. , Harris, I.S. , Mak, T.W. , 2013. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 12, 931–947. [DOI] [PubMed] [Google Scholar]
- Grimmer, M.R. , Stolzenburg, S. , Ford, E. , Lister, R. , Blancafort, P. , Farnham, P.J. , 2014. Analysis of an artificial zinc finger epigenetic modulator: widespread binding but limited regulation. Nucleic Acids Res. 42, (16) 10856–10868. 10.1093/nar/gku708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma, S. , Ishii, Y. , Morishima, Y. , 2009. Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clin. Cancer Res. 15, 3423–3432. [DOI] [PubMed] [Google Scholar]
- Huisman, C. , Wisman, G.B. , Kazemier, H.G. , 2013. Functional validation of putative tumor suppressor gene C13ORF18 in cervical cancer by Artificial Transcription Factors. Mol. Oncol. 7, 669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, T. , Chen, N. , Zhao, F. , 2010. High levels of Nrf2 determine chemoresistance in type II endometrial cancer. Cancer Res. 70, 5486–5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juarez-Moreno, K. , Erices, R. , Beltran, A.S. , 2013. Breaking through an epigenetic wall: re-activation of Oct4 by KRAB-containing designer zinc finger transcription factors. Epigenetics 8, 164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kansanen, E. , Jyrkkanen, H.K. , Levonen, A.L. , 2012. Activation of stress signaling pathways by electrophilic oxidized and nitrated lipids. Free Radic. Biol. Med. 52, 973–982. [DOI] [PubMed] [Google Scholar]
- Khalil, H.S. , Goltsov, A. , Langdon, S.P. , Harrison, D.J. , Bown, J. , Deeni, Y. , 2014. Quantitative analysis of NRF2 pathway reveals key elements of the regulatory circuits underlying antioxidant response and proliferation of ovarian cancer cells. J. Biotechnol 10.1016/j.jbiotec.2014.09.027 pii: S0168-1656(14)00941-9 [DOI] [PubMed] [Google Scholar]
- Kim, J.H. , Thimmulappa, R.K. , Kumar, V. , 2014. NRF2-mediated Notch pathway activation enhances hematopoietic reconstitution following myelosuppressive radiation. J. Clin. Invest. 124, 730–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, M.Y. , Zhang, T. , Kraus, W.L. , 2005. Poly(ADP-ribosyl)ation by PARP-1: 'PAR-laying' NAD+ into a nuclear signal. Genes Dev. 19, 1951–1967. [DOI] [PubMed] [Google Scholar]
- Konstantinopoulos, P.A. , Spentzos, D. , Fountzilas, E. , 2011. Keap1 mutations and Nrf2 pathway activation in epithelial ovarian cancer. Cancer Res. 71, 5081–5089. [DOI] [PubMed] [Google Scholar]
- Kou, X. , Kirberger, M. , Yang, Y. , Chen, N. , 2013. Natural products for cancer prevention associated with Nrf2–ARE pathway. Food Sci. Hum. Wellness 2, 22–28. [Google Scholar]
- Kraus, W.L. , Lis, J.T. , 2003. PARP goes transcription. Cell. 113, 677–683. [DOI] [PubMed] [Google Scholar]
- Lau, A. , Tian, W. , Whitman, S.A. , Zhang, D.D. , 2013. The predicted molecular weight of Nrf2: it is what it is not. Antioxid. Redox Signal 18, 91–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, A. , Villeneuve, N.F. , Sun, Z. , Wong, P.K. , Zhang, D.D. , 2008. Dual roles of Nrf2 in cancer. Pharmacol. Res. 58, 262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q. , van den Berg, T.A. , Feringa, B.L. , Roelfes, G. , 2010. Mononuclear Fe(II)-N4Py complexes in oxidative DNA cleavage: structure, activity and mechanism. Dalton Trans. 39, 8012–8021. [DOI] [PubMed] [Google Scholar]
- Liao, H. , Zhou, Q. , Zhang, Z. , 2012. NRF2 is overexpressed in ovarian epithelial carcinoma and is regulated by gonadotrophin and sex-steroid hormones. Oncol. Rep. 27, 1918–1924. [DOI] [PubMed] [Google Scholar]
- Lister, A. , Nedjadi, T. , Kitteringham, N.R. , 2011. Nrf2 is overexpressed in pancreatic cancer: implications for cell proliferation and therapy. Mol. Cancer 10, 37–45. 98-10-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J.F. , Barry, W.T. , Birrer, M. , 2014. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised phase 2 study. Lancet Oncol. 15, 1207–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak, K.J. , Schmittgen, T.D. , 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Ma, X. , Zhang, J. , Liu, S. , Huang, Y. , Chen, B. , Wang, D. , 2012. Nrf2 knockdown by shRNA inhibits tumor growth and increases efficacy of chemotherapy in cervical cancer. Cancer Chemother. Pharmacol. 69, 485–494. [DOI] [PubMed] [Google Scholar]
- Malhotra, D. , Portales-Casamar, E. , Singh, A. , 2010. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 38, 5718–5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malloy, M.T. , McIntosh, D.J. , Walters, T.S. , Flores, A. , Goodwin, J.S. , Arinze, I.J. , 2013. Trafficking of the transcription factor Nrf2 to promyelocytic leukemia-nuclear bodies: implications for degradation of NRF2 in the nucleus. J. Biol. Chem. 288, 14569–14583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell, J.G. , Barbas, C.F. , 2006. Zinc Finger Tools: custom DNA-binding domains for transcription factors and nucleases. Nucleic Acids Res. 34, W516–W523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez, V.D. , Vucic, E.A. , Thu, K.L. , Pikor, L.A. , Hubaux, R. , Lam, W.L. , 2014. Unique pattern of component gene disruption in the NRF2 inhibitor KEAP1/CUL3/RBX1 E3-ubiquitin ligase complex in serous ovarian cancer. Biomed. Res. Int. 2014, 10 10.1155/2014/159459 Article ID 159459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra, S. , Boldogh, I. , Izumi, T. , Hazra, T.K. , 2001. Complexities of the DNA base excision repair pathway for repair of oxidative DNA damage. Environ. Mol. Mutagen 38, 180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima, H. , Nakajima-Takagi, Y. , Tsujita, T. , 2011. Tissue-restricted expression of Nrf2 and its target genes in zebrafish with gene-specific variations in the induction profiles. PLoS One 6, e26884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, N. , Zhang, X. , Olashaw, N. , Seto, E. , 2004. Molecular cloning and functional characterization of the transcription factor YY2. J. Biol. Chem. 279, 25927–25934. [DOI] [PubMed] [Google Scholar]
- Rambaud, J. , Desroches, J. , Balsalobre, A. , Drouin, J. , 2009. TIF1beta/KAP-1 is a coactivator of the orphan nuclear receptor NGFI-B/Nur77. J. Biol. Chem. 284, 14147–14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney, J.W. , Calame, K.L. , 2001. TIF1beta functions as a coactivator for C/EBPbeta and is required for induced differentiation in the myelomonocytic cell line U937. Genes Dev. 15, 3023–3038. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Rouleau, M. , Patel, A. , Hendzel, M.J. , Kaufmann, S.H. , Poirier, G.G. , 2010. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 10, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer, Y. , Menu, C. , Liu, X. , Constantinescu, S.N. , 2004. High-throughput gateway bicistronic retroviral vectors for stable expression in mammalian cells: exploring the biologic effects of STAT5 overexpression. DNA Cell Biol. 23, 355–365. [DOI] [PubMed] [Google Scholar]
- Rutkowski, D.T. , Arnold, S.M. , Miller, C.N. , 2006. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. Plos Biol. 4, e374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sera, T. , 2009. Zinc-finger-based artificial transcription factors and their applications. Adv. Drug Deliv. Rev. 61, 513–526. [DOI] [PubMed] [Google Scholar]
- Sessa, C. , 2011. Update on PARP1 inhibitors in ovarian cancer. Ann. Oncol. 22, (Suppl 8) viii72–viii76. [DOI] [PubMed] [Google Scholar]
- Singh, A. , Boldin-Adamsky, S. , Thimmulappa, R.K. , 2008. RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Res. 68, 7975–7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, B. , Chatterjee, A. , Ronghe, A.M. , Bhat, N.K. , Bhat, H.K. , 2013. Antioxidant-mediated up-regulation of OGG1 via NRF2 induction is associated with inhibition of oxidative DNA damage in estrogen-induced breast cancer. BMC Cancer 13, 253-2407-13-253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa, V. , Moline, T. , Somoza, R. , Paciucci, R. , Kondoh, H. , LLeonart, M.E. , 2013. Oxidative stress and cancer: an overview. Ageing Res. Rev. 12, 376–390. [DOI] [PubMed] [Google Scholar]
- Sporn, M.B. , Liby, K.T. , 2012. NRF2 and cancer: the good, the bad and the importance of context. Nat. Rev. Cancer 12, 564–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacy, D.R. , Ely, K. , Massion, P.P. , 2006. Increased expression of nuclear factor E2 p45-related factor 2 (NRF2) in head and neck squamous cell carcinomas. Head Neck 28, 813–818. [DOI] [PubMed] [Google Scholar]
- Stolzenburg, S. , Rots, M.G. , Beltran, A.S. , 2012. Targeted silencing of the oncogenic transcription factor SOX2 in breast cancer. Nucleic Acids Res. 40, 6725–6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tietze, F. , 1969. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 27, 502–522. [DOI] [PubMed] [Google Scholar]
- Tutt, A. , Robson, M. , Garber, J.E. , 2010. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 376, 235–244. [DOI] [PubMed] [Google Scholar]
- van der Wijst, M.G. , Brown, R. , Rots, M.G. , 2014. Nrf2, the master redox switch: the Achilles' heel of ovarian cancer?. Biochim. Biophys. Acta 1846, 494–509. [DOI] [PubMed] [Google Scholar]
- Walter, P. , Ron, D. , 2011. The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. [DOI] [PubMed] [Google Scholar]
- Wang, J. , Zhang, M. , Zhang, L. , 2010. Correlation of Nrf2, HO-1, and MRP3 in gallbladder cancer and their relationships to clinicopathologic features and survival. J. Surg. Res. 164, e99–105. [DOI] [PubMed] [Google Scholar]
- Wang, X.J. , Sun, Z. , Villeneuve, N.F. , 2008. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 29, 1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, T. , Wang, X.J. , Tian, W. , Jaramillo, M.C. , Lau, A. , Zhang, D.D. , 2014. Poly(ADP-ribose) polymerase-1 modulates Nrf2-dependent transcription. Free Radic. Biol. Med. 67, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, Y.L. , Yang, W.M. , Seto, E. , 2001. Regulation of transcription factor YY1 by acetylation and deacetylation. Mol. Cell Biol. 21, 5979–5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data