

Abstract
Aims: Vascular stiffness, structural elastin abnormalities, and increased oxidative stress are hallmarks of hypertension. Lysyl oxidase (LOX) is an elastin crosslinking enzyme that produces H2O2 as a by-product. We addressed the interplay between LOX, oxidative stress, vessel stiffness, and elastin.
Results: Angiotensin II (Ang II)-infused hypertensive mice and spontaneously hypertensive rats (SHR) showed increased vascular LOX expression and stiffness and an abnormal elastin structure. Mice over-expressing LOX in vascular smooth muscle cells (TgLOX) exhibited similar mechanical and elastin alterations to those of hypertensive models. LOX inhibition with β-aminopropionitrile (BAPN) attenuated mechanical and elastin alterations in TgLOX mice, Ang II-infused mice, and SHR. Arteries from TgLOX mice, Ang II-infused mice, and/or SHR exhibited increased vascular H2O2 and O2.− levels, NADPH oxidase activity, and/or mitochondrial dysfunction. BAPN prevented the higher oxidative stress in hypertensive models. Treatment of TgLOX and Ang II-infused mice and SHR with the mitochondrial-targeted superoxide dismutase mimetic mito-TEMPO, the antioxidant apocynin, or the H2O2 scavenger polyethylene glycol-conjugated catalase (PEG-catalase) reduced oxidative stress, vascular stiffness, and elastin alterations. Vascular p38 mitogen-activated protein kinase (p38MAPK) activation was increased in Ang II-infused and TgLOX mice and this effect was prevented by BAPN, mito-TEMPO, or PEG-catalase. SB203580, the p38MAPK inhibitor, normalized vessel stiffness and elastin structure in TgLOX mice.
Innovation: We identify LOX as a novel source of vascular reactive oxygen species and a new pathway involved in vascular stiffness and elastin remodeling in hypertension.
Conclusion: LOX up-regulation is associated with enhanced oxidative stress that promotes p38MAPK activation, elastin structural alterations, and vascular stiffness. This pathway contributes to vascular abnormalities in hypertension. Antioxid. Redox Signal. 27, 379–397.
Keywords: : extracellular matrix, free radicals, microvascular, mitochondria, NADPH oxidases, tissue repair and remodeling
Introduction
Hypertension is associated with vascular remodeling and increased vascular stiffness, processes that are associated with cardiovascular events and target-organ damage (9, 34, 47, 52). Importantly, it has also been suggested that arterial stiffening itself may be a causal factor in essential hypertension (42, 58). Different factors and mechanisms have been implicated in increased vascular stiffness in hypertension (6, 34, 42). Of these, abnormalities in elastin, a key determinant of vascular mechanics in large and small arteries (7, 13, 58), may be particularly important.
Innovation.
Vascular remodeling, stiffness, and oxidative stress are hallmarks of vascular damage in hypertension. We provide new insights into mechanisms underlying these processes by focusing on the extracellular matrix (ECM) crosslinking enzyme lysyl oxidase (LOX). We demonstrate that LOX overexpression increases vascular stiffness, elastin alterations, and oxidative stress, and that LOX inhibition decreases hypertension-associated vascular remodeling, stiffness, elastin alterations, and oxidative stress. Moreover, we observed that p38 mitogen-activated protein kinase (p38MAPK), a redox-sensitive kinase, is a downstream LOX target that is involved in vascular stiffness and elastin structure. In summary, we identify vascular LOX as a novel regulator of redox-sensitive processes underlying vascular structural and mechanical alterations and remodeling in hypertension.
Previous studies from our group (7, 8) and others (3, 23) demonstrated that alterations in 3D elastin structure are responsible, at least in part, for the increased vascular stiffness in hypertension, and they precede the development of hypertension in spontaneously hypertensive rats (SHR) (22). Moreover, elastin alterations might participate in vascular remodeling and hypertension development, since adult elastin knockout mice (Eln+/−) have smaller and thinner arteries, increased vessel stiffness, and hypertension (20). Importantly, re-expression of elastin reverses the hypertension and cardiovascular changes associated with the vascular phenotype in Eln+/− mice (28).
ECM crosslinking determines the mechanical properties of connective tissues and is critically regulated by LOX. This enzyme promotes the formation of intra- and intermolecular covalent crosslinkages in collagen and elastin fibers by an oxidative deamination of lysine and hydroxilysine residues, while generating H2O2 as a by-product. A disturbance in LOX expression has been reported in cardiovascular diseases (49, 50), and an increase in vascular LOX activity has been described in experimental models of hypertension (43, 55). Interestingly, the enhanced production of H2O2 has been involved in some of the biological activities carried out by LOX, such as the control of cell migration or vascular smooth muscle cell (VSMC) chemotaxis (38, 45). However, the contribution of LOX to vascular oxidative stress and the abnormal vascular structure and mechanics in the setting of hypertension are not well established.
NADPH oxidase and mitochondria are key sources of vascular oxidative stress in hypertension (5, 15, 33). Redox-dependent crosstalk between mitochondria and NADPH oxidases has been recently described in the context of hypertension, with H2O2 having a central role (16, 17). Reactive oxygen species (ROS) are modulators of vascular function and structure in hypertension (5, 15, 33). In addition, positive correlations between plasma oxidative stress markers and vascular stiffness (14, 41, 46) have been reported. However, to our knowledge, no studies have directly evaluated the contribution of ROS to elastin alterations in resistance arteries in hypertension and its impact on vascular stiffness.
In this context, we hypothesized that LOX-derived ROS might be an important regulator of vascular oxidative stress that mediates vascular remodeling, stiffness, and structural elastin abnormalities, contributing to the vascular damage associated with hypertension. Because of the involvement of p38MAPK in ROS signaling, we also evaluated the role of this kinase in LOX-associated vascular alterations.
Results
Role of LOX in vascular remodeling, stiffness, and elastin structure
Because LOX plays a critical role in ECM maturation, we assessed whether this enzyme could impact vascular remodeling and stiffness in an animal model that over-expresses LOX in VSMC (44). VSMC from TgLOX mice display higher levels of LOX activity (44) and accordingly, these cells deposit more insoluble elastin, which corresponds with higher elastin assembly, than those isolated from wild-type (WT) mice (Fig. 1A, B). Transgenic mice showed similar blood pressure (WT: 101 ± 3, n = 9; TgLOX: 102 ± 2 mmHg, n = 10) and wall/lumen (Fig. 1C) values than their non-transgenic littermates. However, mesenteric resistance arteries (MRA) and aortas from TgLOX mice exhibited an enhanced vascular stiffness (demonstrated by the leftward shift of the stress-strain relationship and the increased β value) compared with WT mice (Fig. 1D and Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/ars). The increased vascular stiffness in TgLOX mice was associated with a decrease in fenestrae size and number in the internal elastic lamina (IEL) that was determined in mesenteric arteries (Fig. 1E).
FIG. 1.

TgLOX mice exhibit higher vascular stiffness and structural elastin abnormalities. (A) Elastin staining (green) in non-permeabilized vascular smooth muscle cells (VSMC) isolated from wild-type (WT) and TgLOX mice. Nuclei were stained with Hoechst 33342 (blue). Bar size: 20 μm. (B) Levels of insoluble elastin in VSMC from WT and TgLOX mice after [3H]-valine supplementation. Data were normalized per DNA content in each individual well. (C) Wall/lumen-pressure curves and (D) stress-strain curves in mesenteric resistance arteries (MRAs) from WT, TgLOX, or TgLOX mice receiving BAPN. β-values (slopes of the stress-strain relationships) are also shown. (E) Maximal projections of the internal elastic lamina (IEL) and fenestrae area and number of MRAs from WT, TgLOX, or TgLOX mice receiving BAPN. Projections were obtained from serial optical sections that were captured with a fluorescence confocal microscope ( × 63 oil immersion objective). Image size: 59.5 × 59.5 μm. Results are represented as mean ± SEM (n = 4–10; *p < 0.05, ***p < 0.001 vs. WT; +p < 0.05, ++p < 0.01 vs. untreated TgLOX mice). BAPN, β-aminopropionitrile; SEM, standard error of the mean. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Inhibition of LOX activity with β-aminopropionitrile (BAPN) did not modify vessel structure but normalized the increased vessel stiffness and the smaller fenestrae size in TgLOX mice, without affecting fenestrae number (Fig. 1C–E). Together, these findings suggest that the increase in VSMC LOX activity is a key determinant of the higher vascular stiffness and the disturbance in elastin structure exhibited by TgLOX mice.
We next aimed at determining the role of LOX in vascular remodeling and stiffness in pathological situations potentially associated with an up-regulation of LOX such as hypertension. LOX expression was higher in arteries from both SHR (Fig. 2A and Supplementary Fig. S2A) and Angiotensin II (Ang II)-infused mice (Fig. 2B and Supplementary Fig. S2B). Treatment of SHR with BAPN did not modify blood pressure levels (Table 1). However, BAPN treatment partially reduced the increase in wall/lumen (Fig. 2C) and in vascular stiffness (Fig. 2D) and ameliorated the changes in elastin structure (Fig. 2E) observed in the SHR group. Interestingly, BAPN partially limited the increase in blood pressure (Table 1) and prevented the enhanced wall/lumen, vascular stiffness, and the structural elastin abnormalities induced by Ang II in mice (Fig. 2F–H).
FIG. 2.

LOX inhibition prevents vascular stiffness and structural elastin abnormalities in hypertension models. LOX protein levels in vascular lysates from WKY and SHR (A) or control and Ang II-infused mice (B). Results were normalized by GADPH expression. Wall/lumen-pressure curves (C, F) and stress-strain curves (D, G) in MRAs from WKY, SHR, or SHR treated with BAPN (C, D) and in control or Ang II-infused mice pretreated or not with BAPN (F, G). Maximal projections of the IEL and fenestrae area and number of MRAs from WKY, SHR, or SHR plus BAPN (image size: 119 × 119 μm) (E) and control, Ang II, and Ang II plus BAPN mice (image size: 59.5 × 59.5 μm) (H). Data are represented as mean ± SEM (n = 4–12; *p < 0.05, **p < 0.01, ***p < 0.001 vs. WKY or control animals; +p < 0.05, ++p < 0.01, +++p < 0.001 vs. untreated SHR or Ang II-infused animals). Ang II, angiotensin II; LOX, lysyl oxidase; SHR, spontaneously hypertensive rats; WKY, Wistar Kyoto. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Table 1.
Effect of BAPN on Systolic Blood Pressure (mmHg) in SHR- and Ang II-Infused Mice
| WKY | SHR | Untreated C57BL/6J | Ang II C57BL/6J | |
|---|---|---|---|---|
| Untreated | 152 ± 3 | 220 ± 8a | 96 ± 3 | 157 ± 7a |
| BAPN | — | 212 ± 3a | — | 130 ± 4a,b |
Data are expressed as mean ± SEM. n = 6–8.
p < 0.05 versus WKY or untreated C57BL/6J mice.
p < 0.05 versus Ang II.
Ang II, angiotensin II; BAPN, β-aminopropionitrile; SEM, standard error of the mean; SHR, spontaneously hypertensive rats; WKY, Wistar Kyoto.
We next questioned whether Ang II might affect blood pressure and vascular stiffness in TgLOX mice. As shown in Supplementary Figure S3, blood pressure was similarly enhanced by Ang II in both transgenic and WT mice. Moreover, WT mice showed the expected Ang II-dependent increase in vascular stiffness. However, in the already stiff TgLox arteries, Ang II-infusion was unable to further build up this parameter, likely suggesting that LOX transgenesis triggers such a degree of long-lasting vascular rigidity that Ang II cannot increase it further.
Next, we aimed at characterizing the mechanisms that are responsible for the increased LOX expression in hypertension. Arteries from prehypertensive SHR exhibited similar LOX expression levels to age-matched Wistar Kyoto (WKY) (Supplementary Fig. S4A, D). Further, treatment of Ang II-infused mice with a combination of hydralazine and hydrochlorothiazide, which successfully decreased blood pressure, reduced the increased LOX expression observed in this hypertension model (Supplementary Fig. S4B, C and E). Together, these results suggest that LOX expression is modulated, at least in part, by the hemodynamic effect induced by high blood pressure.
Role of LOX in vascular oxidative stress and its effect on vascular stiffness and elastin alterations
Recent evidence suggests that H2O2 is a key driver of the crosstalk between mitochondrial ROS and NADPH oxidases in the vascular wall in the setting of hypertension (16, 17, 21). Because H2O2 is generated as a by-product of LOX activity, we questioned whether LOX might be a potential source of vascular ROS. As shown in Figure 3, arteries from TgLOX mice showed increased levels of H2O2 and O2.−, enhanced NADPH oxidase activity, and diminished mitochondrial membrane potential (a marker of mitochondrial dysfunction). Accordingly, NADPH oxidase activity was also enhanced in VSMC isolated from TgLOX mice compared with those from WT animals (Supplementary Fig. S5).
FIG. 3.

LOX is a source of vascular oxidative stress. Aortic H2O2 production (A, E), vascular NADPH oxidase activity (B, F), vascular superoxide anion production (C, G), and mitochondrial membrane potential (D) evaluated in wild-type (WT) and TgLOX mice treated or not with mito-TEMPO (mito-TP) or PEG-catalase (PEG-cat). Representative images of dihydroethidium staining are shown. Image size 238 × 238 μm. Data are represented as mean ± SEM (n = 4–10; *p < 0.05, **p < 0.01, vs. WT; +p < 0.05, ++p < 0.01 vs. untreated TgLOX mice). polyethylene glycol-conjugated catalase (PEG-catalase). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
We next questioned whether LOX transgenesis could also evoke changes in the expression of superoxide dismutases (SOD) and NADPH oxidase isoforms (NOX) that might account for the increased vascular H2O2 production in TgLOX mice. Vascular mRNA levels of the three SOD isoforms were similar in both WT and transgenic animals (Supplementary Fig. S6A). The expression of H2O2-producing NOX-4 and that of NOX-2 was not altered in arteries from TgLOX mice (Supplementary Fig. S6B). However, higher vascular NOX-1 expression was detected in TgLOX compared with WT mice (Supplementary Fig. 6B).
Interestingly, treatment with the mitochondria-targeted antioxidant mito-TEMPO, which did not modify blood pressure (data not shown), limited the rise in vascular oxidative stress detected in TgLOX mice (Fig. 3A–D). Similarly, the H2O2 scavenger polyethylene glycol-conjugated catalase (PEG-catalase) attenuated the higher vascular production of H2O2 and O2.− exhibited by transgenic mice, although this treatment did not modify the increase in NADPH oxidase activity detected in these animals (Fig. 3E–F). Together, these findings suggest that both H2O2-dependent and -independent mechanisms would be responsible for the increased oxidative stress status in the vascular wall from TgLOX mice.
We then evaluated whether the higher expression of LOX in hypertension could also be a potential source of oxidative stress. As shown in Figure 4A and B, we detected an increase in the levels of H2O2 and in NADPH oxidase activity in arteries from SHR, effects that were decreased by BAPN. Similarly, vessels from Ang II-infused mice exhibited an enhanced production of both H2O2 and O2.−, an increased NADPH oxidase activity, and a reduction in mitochondrial membrane potential effects that were attenuated by LOX inhibition with BAPN (Fig. 4C–G). In agreement, in Ang II-stimulated VSMCs, increased mitochondrial ROS production and NADPH oxidase activity were abolished by BAPN, as well as by the H2O2 scavenger catalase and/or mito-TEMPO (Supplementary Fig. S7A, B). Furthermore, Ang II failed to significantly increase NADPH oxidase activity in LOX-silenced cells (Supplementary Fig. S7C). Similarly, mesenteric arteries stimulated with Ang II showed increased NADPH oxidase activity and decreased mitochondrial membrane potential, all of which were abolished by BAPN, catalase, mito-TEMPO, and/or ML171 (NOX-1/2 inhibitor) (Supplementary Fig. S7D, E). Together, these findings suggest that LOX up-regulation in hypertension is responsible, at least in part, for the enhanced production of ROS from different sources, including mitochondria and NADPH oxidase.
FIG. 4.

LOX inhibition reduces vascular oxidative stress in hypertension animal models. Aortic H2O2 production (A) and vascular NADPH oxidase activity (B) in WKY, SHR, and SHR treated with BAPN. Aortic H2O2 production (C), vascular superoxide anion production evaluated by dihydroetidium fluorescence (D) and by 2-OH-E+ fluorescence HPLC (E), vascular NADPH oxidase activity (F), and mitochondrial membrane potential (G) were analyzed in control or Ang II-infused mice pretreated or not with BAPN. Representative images of dihydroethidium staining (image size 238 × 238 μm) and HPLC representative traces are shown. Data are represented as mean ± SEM (n = 4–10; *p < 0.05, **p < 0.01, ***p < 0.001 vs. WKY or control animals; +p < 0.05, ++p < 0.01 vs. untreated SHR or Ang II-infused animals). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
We then evaluated the role of LOX-derived oxidative stress in vascular stiffness in TgLOX mice. As shown in Figure 5, mito-TEMPO normalized the increased vascular stiffness and the altered elastin structure observed in mesenteric arteries from TgLOX mice. Likewise, PEG-catalase normalized the increased vascular stiffness in mesenteric arteries (Fig. 6A) and aorta (Supplementary Fig. S1) and the diminished fenestrae number (Fig. 6B). In addition, catalase ameliorated the enhanced elastin maturation exhibited by VSMC from TgLOX mice (Fig. 6C).
FIG. 5.

Oxidative stress underlies the higher vascular stiffness and structural elastin abnormalities detected in TgLOX mice. Stress-strain curves (A) and fenestrae area and number (B) in MRAs from wild-type (WT) and TgLOX mice treated or not with mito-TEMPO (mito-TP). Maximal projections of the IEL of MRAs are shown. Projections were obtained from serial optical sections that were captured with a fluorescence confocal microscope ( × 63 oil immersion objective). Image size: 59.5 × 59.5 μm. Data are represented as mean ± SEM (n = 6–10; **p < 0.01, ***p < 0.001 vs. WT; ++p < 0.01 vs. untreated TgLOX mice). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 6.

H2O2 production underlies the higher vascular stiffness and structural elastin abnormalities detected in TgLOX mice. Stress-strain curves (A) and fenestrae area and number (B) in MRAs from wild-type (WT) and TgLOX mice treated or not with PEG-catalase (PEG-cat). Maximal projections of the IEL of MRAs are shown. Projections were obtained from serial optical sections that were captured with a fluorescence confocal microscope ( × 63 oil immersion objective). Image size: 59.5 × 59.5 μm. (C) Elastin staining (green) in non-permeabilized VSMC isolated from TgLOX mice that were incubated or not with catalase (TgLOX/Cat). Nuclei were stained with Hoechst 33342 (blue). Bar size: 20 μm. Data are represented as mean ± SEM (n = 6–10; *p < 0.05 vs. WT; +p < 0.05 ++p < 0.01 vs. untreated TgLOX mice or cells). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Role of p38MAPK in vascular stiffness and elastin alterations
p38MAPK is a well-known redox-sensitive kinase that is involved in the enhanced vascular stiffness induced by Ang II (59). Therefore, we examined whether this kinase plays a role in LOX-dependent regulation of vascular stiffness. Ang II increased the activation of vascular p38MAPK, an effect that was prevented by BAPN (Fig. 7A and Supplementary Fig. S8A). TgLOX mice showed increased vascular p38MAPK activation compared with WT mice, which was normalized by mito-TEMPO (Fig. 7B, and Supplementary Fig. S8B) and PEG-catalase (Fig. 7C, and Supplementary Fig. S8C). Treatment of TgLOX mice with the p38MAPK inhibitor SB203580 did not affect vascular structure but limited the increased vascular stiffness and the altered elastin structure observed in the mesenteric arteries of these mice (Fig. 7D–F).
FIG. 7.

p38MAPK contributes to the LOX-dependent disturbances in vascular stiffness and elastin alterations. Activation of p38MAPK (p-p38) in vessels from control or Ang II-infused mice treated or not with BAPN (A) and in wild-type (WT) and TgLOX mice treated or not with mito-TEMPO (mito-TP) (B) or PEG-catalase (PEG-cat) (C). Results were normalized by total p38 levels. (D) Wall/lumen-pressure curves, (E) stress-strain curves, and (F) fenestrae area and number in MRAs from WT, TgLOX, or TgLOX mice receiving SB 203580 (SB). Maximal projections of the IEL of MRAs are shown. Projections were obtained from serial optical sections that were captured with a fluorescence confocal microscope ( × 63 oil immersion objective). Image size: 59.5 × 59.5 μm. Data are represented as mean ± SEM (n = 3–8; *p < 0.05, **p < 0.01, ***p < 0.001 vs. WT; +p < 0.05, ++p < 0.01 vs. untreated TgLOX mice). p38MAPK, p38 mitogen-activated protein kinase. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Role of oxidative stress in vascular remodeling, stiffness, and elastin alterations in hypertension
To unambiguously demonstrate the role of oxidative stress in vascular remodeling, stiffness, and altered 3D elastin structure in hypertension, we treated SHR and Ang II-infused mice with apocynin or mito-TEMPO. Both antioxidants decreased the augmented oxidative stress observed in arteries from both hypertensive models (Supplementary Fig. S9). However, neither the antioxidant apocynin nor mito-TEMPO affected the high blood pressure or the augmented wall/lumen in SHR (Fig. 8A, and Table 2). Both treatments partially prevented the high blood pressure induced by Ang II (40), but only apocynin prevented the increase in mesenteric wall/lumen induced by Ang II (Fig. 8B). Interestingly, apocynin and mito-TEMPO ameliorated the higher stiffness of mesenteric arteries and partially restored the abnormalities in elastin structure observed in arteries from both hypertensive models (Fig. 8C–F). Together, these findings demonstrate that oxidative stress, presumably from NADPH oxidase and mitochondria, underlies the increase in vascular stiffness and the anomalous elastin structure characteristic of Ang II-dependent models of hypertension.
FIG. 8.

Oxidative stress underlies vascular remodeling, stiffness, and elastin alterations in hypertensive animal models. Wall/lumen (A,B) stress-strain curves (C, D) and fenestrae area and number (E, F) in MRAs from WKY, SHR, and SHR treated with apocynin (APO) or mito-TEMPO (mito-TP) (A, C) and (E) or from control, Ang II-infused, and Ang II-infused mice treated with apocynin or mitoTEMPO (B, D) and (F). Maximal projections of the IEL of MRAs from WKY, SHR, or SHR plus antioxidants (image size: 119 × 119 μm) (E) and control, Ang II, and Ang II plus antioxidant-treated mice (image size: 59.5 × 59.5 μm) (F). Data are represented as mean ± SEM (n = 6–15; *p < 0.05, **p < 0.01, ***p < 0.001 vs. WKY or control animals; +p < 0.05, ++p < 0.01, +++p < 0.001 vs. untreated WKY or Ang II-infused animals). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Table 2.
Effect of Antioxidants on Systolic Blood Pressure (mmHg) in SHR
Data are expressed as mean ± SEM. n = 7–8.
p < 0.05 versus WKY.
Mito-TP, Mito-TEMPO.
Discussion
Four major novel findings emerge from this study: (1) LOX is a novel source of vascular ROS and contributes to vascular oxidative stress in hypertension; (2) LOX-derived ROS activate p38MAPK that critically influences elastin structure and vessel stiffness; (3) LOX is a key determinant of elastin structure and vascular stiffness and remodeling in hypertension; and (4) ROS are important contributors to the structural elastin abnormalities and the increased vessel stiffness in hypertension.
Previous studies reported increased LOX mRNA or activity levels in some models of hypertension (19, 43, 48). We confirm these observations and demonstrate that the up-regulation of vascular LOX protein levels observed in SHR and Ang II-infused mice relies, at least in part, on the hemodynamic effects induced by high blood pressure. Due to the lack of viable LOX knockout mice (39), BAPN, a well-known specific and irreversible inhibitor of LOX activity (51, 56), emerges as a useful pharmacological tool to characterize LOX-dependent vascular responses. In addition, our study using a transgenic mouse model that over-expresses LOX specifically in VSMC (44) provides unique information about the critical role of LOX in vascular stiffness and elastin remodeling and provides mechanistic insights regarding its pathophysiological impact.
Independent studies have suggested that an increased crosslinking is responsible for a higher stiffness of elastin (58) that could promote vessel stiffness (3, 7, 22). The increased insoluble elastin deposition in VSMC from TgLOX mice and the fact that BAPN normalized not only 3D elastin structural alterations but also vascular stiffness in both TgLOX mice and hypertensive animals strongly support that the enhanced elastin crosslinking is a novel mechanism that is responsible for vascular stiffness evoked by LOX up-regulation. However, we cannot exclude that the higher LOX-dependent collagen crosslink recently reported in TgLOX (44) and Ang II infused mice (19) could also contribute to vascular stiffness. Importantly, LOX inhibition also limited the enhanced wall/lumen in SHR and Ang II infused mice, suggesting that under pathological conditions, LOX has a role in vascular remodeling by mechanisms that might include altered mechanics and ECM deposition.
LOX catalyzes the exchange of an amine to an aldehyde group on peptidyl lysines of collagen or elastin fibers, producing H2O2 as a by-product (50). TgLOX mice exhibited elevated H2O2 vascular levels, in agreement with the increased LOX catalytic activity previously reported in this model (44) and without an apparent contribution of SODs or NOX-4 (the H2O2-producing NOX). More importantly, the increased vascular H2O2 levels detected in two hypertensive models were decreased by BAPN, suggesting, for the first time, the active participation of LOX in the enhanced H2O2 production in hypertension.
H2O2 is a stable and membrane-permeable ROS that has multiple effects at vascular level, including a role in the crosstalk between NADPH oxidases and mitochondria in hypertension (16, 17, 21). Moreover, H2O2 not only can influence other ROS sources but also a mitochondrial amplification of H2O2 microdomain signaling has been recently described in VSMC (11). In this context, it should be emphasized that LOX inhibition (either with BAPN or a specific siRNA) reduced NADPH oxidase activation, mitochondrial dysfunction, and/or O2.− production in the models analyzed (SHR, Ang II-infused mice, Ang II-stimulated VSMC, and/or Ang II-stimulated mesenteric arteries) and accordingly, LOX transgenesis enhances NADPH oxidase activation, O2.− production and causes mitochondrial dysfunction.
The relative contribution of each specific ROS source to the LOX-dependent oxidative stress is difficult to ascertain, particularly in the context of hypertension. However, treatment of TgLOX mice with mito-TEMPO or PEG-catalase reduced different parameters of vascular oxidative stress, suggesting the existence of a complex crosstalk between different ROS sources that might act in a reciprocal relationship, thereby mutually perpetuating (Fig. 9). Of note, the increased NADPH activity exhibited by TgLOX mice seem to be independent of LOX-derived H2O2 and might rely, at least in a part, on NOX-1 up-regulation. The molecular mechanisms involved in this relationship in vivo have not been characterized, and this is a limitation of our study.
FIG. 9.

LOX induces oxidative stress that promotes p38MAPK activation, elastin remodeling, and vascular stiffness. (A) LOX transgenesis enhances vascular H2O2 levels, NADPH oxidase activity, and NOX1 expression; causes mitochondrial dysfunction; induces O2.− production; activates p38MAPK signaling; disturbs elastin structure; and increases vascular stiffness. The H2O2 scavenger PEG-catalase (in pink) and the superoxide dismutase targeted antioxidant mitoTEMPO (in blue) decrease the enhanced O2.− production and p38MAPK activation and improve elastin structure and vascular stiffness (symbolized by respective colored arrows). Accordingly, p38 MAPK inhibition by SB203580 (in grey) reduces elastin alterations and vascular stiffness (gray arrows). Further, LOX blockade with BAPN (in red) limited elastin abnormalities and vascular stiffness. (B) Vascular LOX expression is induced in animal models of hypertension (Ang II-infused mice and SHR). In both hypertensive models, LOX up-regulation partially relies on high blood pressure, although additional mechanisms could be involved (indicated with a dashed arrow). In hypertension, BAPN (in red) reduces the enhanced vascular H2O2 levels, NADPH oxidase activation, mitochondrial dysfunction, O2.− production, and/or p38MAPK activation and further ameliorates elastin abnormalities and vascular stiffness (symbolized by red arrows). Mito-TEMPO (in blue) and the antioxidant apocynin (in green) diminish oxidative stress and normalize the altered elastin structure and the increased vascular stiffness observed in hypertensive models (indicated with respective colored arrows). Note that in the vascular wall, the LOX-dependent H2O2 production would contribute to an ROS amplification mechanism involving NADPH oxidase and mitochondria. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Previous studies suggest that oxidative stress might be a key determinant of vascular stiffness in aging (60) or DOCA-salt hypertension (12). Here, we provide new evidence demonstrating that excessive LOX-derived ROS have functional consequences on vessel stiffness and elastin structural abnormalities, since in LOX transgenic animals both parameters were normalized by mito-TEMPO and PEG-catalase. Importantly, BAPN and the antioxidants apocynin and mito-TEMPO also decreased vessel stiffness and improved elastin structure in two hypertensive models, supporting the relevance of this pathway in hypertension. Of note, treatment of SHR with the non-targeted SOD mimetic TEMPOL yielded similar results (unpublished observations), suggesting the key role of ROS, irrespective of the underlying source.
Our data on the relationship between oxidative stress, vascular stiffness, and elastin structure might have clinical implications, as suggested by a recent report in patients with Williams Syndrome (32), where increased vascular stiffness is directly affected by the number of copies of the NCF1 gene that encodes for the p47phox subunit of the NADPH oxidase. Together, these findings point to the possible beneficial effects of antioxidants in conditions associated with increased LOX expression/activity, vascular stiffness, and elastin alterations.
The decreased fenestrae size and number observed in arteries from hypertensive animals previously reported (7, 8, 26), and confirmed here, could simply be an adaptative process to maintain an adequate level of vascular stress in response to high blood pressure (3) and therefore, lowering blood pressure should be paralleled by a restoration of elastin structure. In line with this hypothesis, effective anti-hypertensive treatments in Ang II-infused mice (BAPN, apocynin, or mito-TEMPO) (present study and 57) normalized elastin structure and improved vessel stiffness. However, normotensive TgLOX mice show typical fenestrae alterations and increased stiffness and BAPN-, apocynin-, or mito-TEMPO-treated SHR evidenced improved elastin alterations and decreased stiffness in the absence of significant changes in blood pressure. Therefore, in addition to hemodynamic forces, other mechanisms such as oxidative stress could be involved in the altered elastin structure and vascular stiffness associated with increased LOX activity.
The relationship between vascular stiffness and hypertension is an area of active research and today, no clear answer on whether stiffness is a cause or a consequence of high blood pressure has been provided (2, 34). We cannot establish exactly the contribution of vascular stiffness to high blood pressure in our experimental models. Indeed, in Ang II-infused WT animals, vascular stiffness increases together with blood pressure and different treatments that partially prevent Ang II-induced vascular stiffness (BAPN, mito-TEMPO, and apocynin) also attenuated hypertension. However, in TgLOX mice, animals with stiff vessels, Ang II did not further enhance vascular stiffness, whereas blood pressure was increased similarly to WT mice, suggesting that in this model vascular stiffness would not be the only determinant of high blood pressure.
Similarly, despite the beneficial effects of LOX and ROS blockade on vascular stiffness in SHR, additional mechanisms are likely involved in hypertension development. Interestingly, antioxidants did not affect wall/lumen, another parameter that might influence blood pressure, of resistance arteries in SHR. The short duration of treatments or the long exposure to high blood pressure in SHR might also explain why antioxidants failed to restore wall/lumen and blood pressure, despite an improvement in vessel stiffness. Our data support the concept that LOX inhibition and antioxidant therapy decrease vessel stiffness, and this might have beneficial effects to protect from cardiovascular events in the long-term in hypertension, even without affecting blood pressure levels. In agreement, recent experimental and clinical evidence demonstrates that some pharmacological tools that improve vessel stiffness, such as statins, do not necessarily decrease blood pressure, while reducing cardiovascular risk (8, 29).
p38MAPK is a key downstream pathway of ROS, including H2O2 (10, 24). Because inhibition of p38MAPK with SB2035801 prevents the increased vascular stiffness and fibrosis induced by Ang II in aorta (59), attenuation of Ang II-induced p38MAPK phosphorylation by BAPN might be one of the downstream mechanisms underlying the protective effect of this drug, not only on vessel stiffness but also in elastin remodeling. This is reinforced by the higher vascular p38MAPK activation observed in TgLOX mice and by the restoration of vascular stiffness and elastin structure in transgenic mice treated with SB203580. Although p38MAPK can be activated by mechanical stretch induced by hypertension (59), TgLOX mice are normotensive, further supporting the involvement of additional blood pressure-independent mechanisms that are likely driven by ROS. In agreement, both mito-TEMPO and PEG-catalase decreased the augmented p38MAPK activity observed in TgLOX mice.
In conclusion, we have identified novel mechanisms that are responsible for vascular structural and mechanical alterations in hypertension. We demonstrated by using complementary gain of function and LOX inhibition approaches that in the vascular wall LOX up-regulation is associated with enhanced oxidative stress from different sources, which promotes p38MAPK activation, elastin structural alterations, and vascular stiffness. This pathway participates in vascular remodeling and stiffness in hypertension and might contribute to high blood pressure and hypertension-related end-organ damage.
Future perspectives
Our data further build on the concept of the critical role of elastin in vascular stiffness and remodeling in hypertension suggested by Faury et al. (20) and strengthened by studies in human patients with elastin deficiencies, showing that increased vascular stiffness precedes systolic blood pressure elevation (37) and predisposes to hypertension (54). In animal models, several pharmacological strategies that improve vessel stiffness have been described (6, 58). However, at the clinical level, there is less clear evidence of appropriate pharmacological tools to improve vessel stiffness, except for inhibitors of the renin-angiotensin-aldosterone system and calcium channel blockers (4, 18, 35, 53). Therefore, identification of novel pharmacological targets to prevent and/or ameliorate vessel stiffness in different cardiovascular diseases is awaited. Our study suggests that targeting LOX may be an interesting strategy. However, because long-term inhibition of LOX might interfere with normal crosslinking of collagen fibers and, therefore, compromise arterial strength (58), chronic studies to assess long-term effects of LOX inhibitors are warranted. Alternatively, attenuation of oxidative stress might represent an attractive perspective in pathological situations that are associated with increased LOX activity or altered elastin organization and increased vascular stiffness.
Materials and Methods
Animal models
Six-month- and 4-week-old male WKY and SHR rats were used. Adult SHR rats were divided into untreated groups and groups treated with BAPN (100 mg/kg/day; Sigma-Aldrich Co., St Louis, MO; 5 weeks, v.o), apocynin (1.5 mM −17 days, v.o; Sigma-Aldrich Co.), or mito-TEMPO (0.7 mg/Kg/day −17 days, i.p; Santa Cruz Biotechnology, Inc., Dallas, TX).
Three-month-old male C57BL/6J mice were infused with Ang II (1.44 mg/Kg/day, 2 weeks; Sigma-Aldrich Co.) with subcutaneously implanted Alzet osmotic minipumps (Durect Corp., Cupertino, CA) and pretreated or not with BAPN (100 mg/kg/day, i.p), apocynin (1.5 mM, v.o), mito-TEMPO (0.7 mg/Kg/day, i.p), or the combination of hydralazine plus hydrochlorothiazide (Hyd: 20 mg/Kg/day; HCTZ: 6 mg/Kg/day i.p; Sigma-Aldrich Co. both compounds). Untreated mice were used as controls. All treatments started 24 h before Ang II infusion.
Studies were also performed in a transgenic mouse model that over-expresses LOX in VSMC (TgLOX) in a C57BL/6J genetic background (44). TgLOX male mice (10 weeks) were divided into untreated groups and groups treated with BAPN (100 mg/kg/day, i.p, 15 days), mito-TEMPO (0.7 mg/Kg/day - i.p, 15 days), the H2O2 detoxifying enzyme PEG-catalase (40,000 U/Kg every 4 days, iv, 15 days; Sigma-Aldrich Co.), or the p38MAPK inhibitor SB203580 (10 mg/Kg/day, i.p, 15 days; SelleckChemicals, Houston, TX). A group of wild-type and Ang II-infused mice was also included as described earlier.
Blood pressure was measured by tail-cuff pletysmography. Animals were first habituated to the measurement device for 1 week before starting treatments.
All animal handling procedures were performed in compliance with the principles and guidelines established by the American Physiological Society for animal research. All procedures were reviewed and approved by the Ethical Committees at the Universidad Autónoma de Madrid and the Centro de Investigación Cardiovascular. Animals were taken care of and used according to the Spanish Policy for Animal Protection RD53/2013, which meets the European Union Directive 2010/63/UE on the protection of animals used for experimental and other scientific purposes.
Tissue preparation
Animals were euthanized by CO2 inhalation and aorta, and first- and third-order branches of the mesenteric artery were dissected free of fat and connective tissue and maintained in cold (4°C) Krebs-Henseleit solution (KHS) (115 mM NaCl, 25 mM NaHCO3, 4.7 mM KCl, 1.2 mM MgSO4.7H2O, 2.5 mM CaCl2, 1.2 mM KH2PO4, 11.1 mM glucose, and 0.01 mM Na2EDTA) that was bubbled with a 95% O2–5% CO2 mixture. Analysis of vascular structure and mechanics was done on the same day. For fluorescence studies, arterial segments were placed in phosphate-buffered saline (PBS) containing 30% sucrose for 20–50 min, transferred to a cryomold-containing Tissue Tek OCT embedding medium (Sakura Finetek Europe B.V., Alphen aan Den Rijn, The Netherlands), and frozen in liquid nitrogen. Tissues were kept at −70°C until O2.− determination studies. Other vascular segments were immediately frozen in liquid nitrogen and kept at −70°C until further processing for gene expression, NADPH oxidase activity, H2O2 production, mitochondrial membrane potential measurements or HPLC, and Western blot studies.
In some experiments, mesenteric arteries were stabilized in Krebs-Ringer HEPES solution (130 mM NaCl, 5.6 mM KCl, 2 mM CaCl2, 0.24 mM MgCl2, 8.3 mM HEPES, 11 mM glucose, pH 7.4) at 37°C for 30 min. Thereafter, segments were treated (30 min, 37°C) with mito-TEMPO (0.5 μM), BAPN (0.5 mM), the NOX-1/2 inhibitor, ML171 (0.5 μM; Sigma-Aldrich Co), and the H2O2-detoxifying enzyme, catalase (1000 U/ml; Sigma-Aldrich Co.). Then, Ang II (0.1 μM) was added to the arteries for 2 h at 37°C. Afterward, arteries were frozen (−80°C) for further determination of NADPH oxidase activity and mitochondrial membrane potential.
Pressure myography
The structural and mechanical properties of third-order (for rats) and first-order (for mice) MRA were studied with a pressure myograph (Danish Myo Tech., Model P100, J.P. Trading I/S, Aarhus, Denmark) as previously described (7). Briefly, vessels were placed on two glass microcannulae. The segment was set to a pressure of 45 mmHg in mice and 70 mmHg in rats, and allowed to equilibrate for 30 min at 37°C in calcium-free KHS (0Ca2+; omitting calcium and adding 1 mM EGTA) gassed with a mixture of 95% O2 and 5% CO2. A pressure-diameter curve was then obtained by increasing intraluminal pressure in 20 mmHg steps from 3 to 120 mmHg in mice and from 3 to 140 mmHg in rats. Internal and external diameters were continuously measured under passive conditions (Di0Ca, De0Ca). Finally, the artery was set to 45 mmHg in mice and to 70 mmHg in rats in 0Ca2+-KHS, then pressure-fixed with 4% paraformaldehyde in 0.2 M phosphate buffer, pH 7.2–7.4 at 37°C for 60 min, and kept in 4% paraformaldehyde at 4°C for confocal microscopy studies.
Calculation of passive structural and mechanical parameters
From internal and external diameter measurements, the following structural and mechanical parameters were calculated:
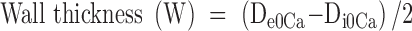 |
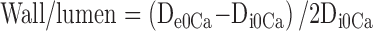 |
Circumferential wall strain (ɛ) = (Di0Ca−D00Ca)/D00Ca, where D00Ca is the internal diameter at 3 mmHg and Di0Ca is the observed internal diameter for a given intravascular pressure, both of which are measured in 0Ca2+ medium.
Circumferential wall stress (σ) = (P × Di0Ca)/(2W), where P is the intraluminal pressure (1 mmHg = 1.334 × 103 dynes·cm−2) and W is the wall thickness at each intraluminal pressure in 0Ca2+-KHS.
Arterial stiffness independent of geometry is determined by the Young's elastic modulus (E = stress/strain). The stress-strain relationship is non-linear; therefore, it is more appropriate to obtain a tangential or incremental elastic modulus (Einc) by determining the slope of the stress-strain curve (Einc = δσ/δɛ). Einc was obtained by fitting the stress-strain data from each animal to an exponential curve by using the equation: σ = σorigeβɛ, where σorig is the stress at the original diameter (diameter at 3 mmHg). Taking derivatives in the equation, we determine that Einc = βσ. For a given σ-value, Einc is directly proportional to β. An increase in β implies an increase in Einc, which means an increase in stiffness.
Aortic stiffness determination
Mice aortic stiffness was determined by using a wire myograph (Danish Myo Tech) as previously described (31) with some modifications. After a 30-min equilibration period in oxygenated 0Ca2+-KHS, passive vascular properties were assessed by exposure to stepwise increases in stretching while recording the developed force.
The vessels were assumed to be cylinders, the length of which did not vary with changes in radius. Since the vessels were found to be flat between the wires, the internal circumference (L) and hence the diameter of each vessel segment at each corresponding pressure were calculated by using the following equation, where d is the diameter of the mounting wires and s is the separation of the wires from each other's inner surface.
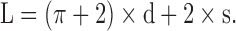 |
The circumferential wall force per unit length is the passive wall tension and was calculated from the following equation, which took into account the fact that the forces produced by a ring are equal to twice the force that would have been produced by an equivalent aortic strip, where F is the force exerted by the vessel on the tension transducer and g is the vessel length:
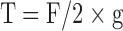 |
Effective transmural pressure P was calculated from Laplace's equation, where L is the internal circumference corresponding to the wall tension:
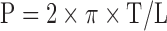 |
Assuming that the wall cross-sectional area remains practically constant through a range of intraluminal pressures, this parameter was calculated from individual histological transverse sections once the stretching curve was performed. External diameter was then extrapolated from the formula:
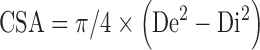 |
where Di is the internal diameter calculated from the internal circumference at each pressure:
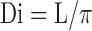 |
Wall thickness, circumferential wall stress and strain, and individual Young's elastic modulus were then calculated as stated earlier.
Organization of IEL
The elastin organization within the IEL was studied in segments of MRA, using fluorescence confocal microscopy based on the autofluorescent properties of elastin (excitation wavelength at 488 nm and emission wavelength at 500–560 nm), as previously described (7). Briefly, the experiments were performed in intact pressure-fixed segments with a Leica TCS SP2 confocal system (Leica Microsistemas S.L.U., Barcelona, Spain). Serial optical sections from the adventitia to the lumen (z step = 0.5 μm) were captured with a X63 oil objective, by using the 488 nm line of the confocal microscope. A minimum of two stacks of images of different regions were captured in each arterial segment. Quantitative analysis of the IEL was performed with Metamorph Image Analysis Software (Molecular Devices, Sunnyvale, CA), as previously described (6). From each stack of serial images, individual projections of the IEL were reconstructed, and total fenestrae number and mean fenestrae area were measured.
Western blot analysis
To obtain protein lysates, arteries were harvested and mixed gently in RIPA buffer containing: 50 mM Tris pH 7.5, 150 mM NaCl, 1 mM MgCl2, 1 mM EDTA, 1% Nonidet-P40 (NP-40), 0.5% deoxicolate Na, 1% sodium dodecyl sulfate (SDS), a protease inhibitor cocktail (Roche Applied Science, Barcelona, Spain), and a mix of phosphatase inhibitors (1 mM orthovanadate, 20 mM β-glycerophosphate, 10 mM NaF from Sigma-Aldrich Co.). Samples were centrifuged for 10 min at 13,000 rpm, and protein content in the supernatants was determined with Lowry (Bio-Rad Laboratories, Hercules, CA).
Total protein equivalents of each sample (30 μg) were separated on a 7.5% SDS-polyacrylamide gel and electrophoretically transferred to polyvinylidene difluoride membranes (Amersham, GE Healthcare, Buckinghamshire, United Kingdom) After blocking, membranes were incubated with antibodies for LOX (1:1000; Novus Biologicals LLC, Littleton, CO), p-p38 (1:1000) and p38 (1:1000) MAPK (Cell Signalling Technology, Inc., Danvers, MA), or glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 1:50,000; EMD Millipore, Billerica, MA) overnight at 4°C. Membranes were thoroughly washed and incubated with horseradish peroxidase-coupled secondary antibodies (1:2000; Bio-Rad) for 1 h at room temperature. Bands were detected by using the Luminata Forte (Millipore) detection system. GAPDH expression was used as a loading control.
Real-time polymerase chain reaction
Total RNA was isolated by using the Trisure Isolation Reagent (Roche Diagnostics, Indianapolis, IN) following the manufacturer's instructions. Total RNA (500 ng) was reverse transcribed into cDNA by using the High-Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA) with random hexamers. Quantification of mRNA levels was performed by real-time polymerase chain reaction (PCR) using an ABI PRISM 7900HT sequence detection system (Applied Biosystems, Foster City, CA), and specific primers and probes provided by Applied Biosystems (Assay-on-Demand system) or Integrated DNA technologies (Coralville, IA) as follows: NADPH oxidase 1 (NOX-1; Mm00549170_m1), NOX-2-(Mm01287743_m1), NOX-4 (Mm00479246_m1), SOD1 (Mm.PT.58.12368303), SOD2 (Mm.PT.58.14276358), and SOD3 (Mm.PT.58.12048855). GAPDH (Mm99999915_g1) was used as an endogenous control. Relative mRNA levels were determined by using the 2−ΔΔCt method.
Cell culture
Primary cultures of aortic VSMC were obtained from cleaned rat aortas and grown in DMEM-F12 medium that was supplemented with 10% fetal bovine serum containing 100 U/ml of penicillin, 100 μg/ml of streptomycin (all from Sigma-Aldrich Co.), as previously reported (1). Cell cultures were used between passages 2 and 4. VSMC were grown on glass coverslips and starved in serum-free media for 24 h. Then, VSMCs were stimulated or not with Ang II (0.1 μM, 4 h, or 24 h) in the absence and in the presence of BAPN (0.5 mM), catalase (1000 U/ml), or mito-TEMPO (0.5 μM).
Aortic VSMC from wild-type and TgLOX mice were obtained by the explant technique as previously described (44). Mouse VSMC were routinely subcultured in DMEM (ThermoFisher Scientific, Grand Island, NY) that was supplemented with 10% FCS, antibiotics, and 2 mM L-glutamine (ThermoFisher Scientific).
Quantitative assessment of insoluble elastin
VSMC from wild-type and TgLOX mice were seeded on 35 mm dishes in quadruplicate and grown to confluence for 3 days. Then, fresh media supplemented with [3H]-valine (10 μCi) were added to each well. VSMC were incubated for 7 days, and the cell layer was extensively washed with PBS. Cell remnants and deposited insoluble ECM were scraped and boiled in 500 μl of 0.1 N NaOH for 30 min to solubilize all matrix components except elastin. Pellets containing insoluble elastin were collected, solubilized by boiling in 200 μl of 5.7 N HCl for 1 h, and subjected to scintillation counting as previously described (27). Data were normalized per DNA content in each individual culture.
Immunocytochemistry
VSMC from wild-type and TgLOX mice were plated onto 35 mm μ-Dishes (Ibidi, Planegg/Martinsried, Germany) and allowed to grow for 10 days in the presence or in the absence of catalase (500 U/ml). Media were changed every 2 days. Afterward, cells were fixed with 4% paraformaldehyde and non-permeabilized VSMC were incubated with a rabbit polyclonal antibody against Elastin (ab21607; Abcam, Cambridge, United Kingdom) overnight. Then, dishes were washed and incubated for 1 h with a fluorescence-conjugated secondary antibody (goat anti-rabbit Alexa Fluor 488; ThermoFisher Scientific). Nuclei were stained with Hoechst 33342 (ThermoFisher Scientific) (25). Fluorescence images were acquired with a Leica DMIRE2 confocal microscope by using Leica Confocal Software (Leica Microsistemas S.L.U.). Quantitative analysis was performed by using Image J software.
siRNA transfection
The ON-TARGET plus SMART pool siRNA system (Dharmacon; GE Healthcare United Kingdom Buckinghamshire, United Kingdom) was used in knockdown experiments. Cells were transfected with 25 nM of an siRNA against LOX (E-091115–01) or with the ON TARGET plus non-targeting Control pool (siRandom; D-001810-10-05), using 7.5 μl of Lipofectamine RNAiMax Reagent (Thermo Fisher Scientific, Inc., Waltham, MA). After transfection (8 h), cells were stimulated with Ang II and finally processed for NADPH oxidase activity. Gene knockdown was verified by real-time PCR (not shown).
NADPH oxidase activity
The lucigenin-enhanced chemiluminescence assay was used to determine the NAD(P)H oxidase activity in total protein vascular homogenates. Tissues were homogenized in a lysis buffer (in mM: KH2PO4 50, ethylene glycol tetraacetic acid 1, sucrose 150, pH 7.4). The reaction was started by the addition of NAD(P)H (0.1 mM) to the suspension containing sample, lucigenin (5 μM), and assay phosphate buffer. The luminescence was measured in a plate luminometer (Auto-Lumat LB 953, Berthold Technologies GmbH, Bad Wildbad, Germany). Buffer blank was subtracted from each reading. Activity was expressed as relative light units/mg protein. Samples from nontreated animals were used as controls, and variations of NAD(P)H oxidase activity were calculated as the amount relative to controls.
In situ detection of vascular O2.− production
The oxidative fluorescent dye dihydroethidium (DHE, Sigma-Aldrich Co.) was used to evaluate O2.− production in situ. Arterial sections were equilibrated for 30 min at 37°C in Krebs-HEPES buffer (in mM: 130 NaCl, 5.6 KCl, 2 CaCl2, 0.24 MgCl2, 8.3 HEPES, 11 glucose, pH = 7.4). Fresh buffer containing DHE (2 μM) was topically applied onto each tissue section, cover-slipped, incubated for 30 min in a light-protected humidified chamber at 37°C, and viewed with a fluorescent laser scanning confocal microscope (Leica TCS SP2 equipped with a krypton/argon laser, × 40 objective; Leica Microsistemas S.L.U.). Fluorescence was detected with a 568 nm long-pass filter by using the same imaging settings for all experimental conditions. For quantification, two-three rings per animal were sampled for each experimental condition and averaged. The mean fluorescence densities in the target region were calculated. To minimize laser fluctuations from one day to another, data were expressed as% of signal in control arteries.
Measurement of O2.− production by high-performance liquid chromatography
O2.− production was determined by HPLC as previously described (36). Aortic samples were homogenized in acetonitrile (500 μl), sonicated, and centrifuged and the supernatant was collected and dried. Pellets were resuspended in Krebs-HEPES-DPTA 25 μM. For HPLC analysis, equal amounts of protein were resuspended in 70 μl Krebs-HEPES-DPTA, and a volume of 50 μl was injected. Samples were analyzed by HPLC (Varian 920 LC series, Agilent Technologies, Santa Clara, CA), by using a 4 μm C-18 reverse-phase column (Synergi 150 × 4.6 mm, Phenomenex, Torrance, CA) and a gradient of solutions A (pure acetonitrile) and B (water/10% acetonitrile/0.1% trifluoroacetic acid, v/v/v) at a flow rate of 1 ml/min, and run as described (34). Ethidium and 2-hydroxyethidium (2-OH-E+) were monitored by fluorescence detection with excitation at 510 nm and emission at 595 nm. The 2-OH-E+ peak reflects the amount of O2.− formed in the tissues. A positive control of 2-OH-Etidium+ was also used (Noxygen Science Transfer and Diagnostics GmbH, Elzach, Germany).
Measurement of H2O2
H2O2 measurements in aortic segments were made by using the horseradish peroxidase-linked Amplex Red fluorescence assay. Briefly, Amplex Red (ThermoFisher Scientific; 100 μM) and horseradish peroxidase type II (0.2 U/ml) were added to the aortic homogenates. Fluorescence readings were made in triplicate in a 96-well plate at Ex/Em = 530/580 nm by using 50-μl samples of media. H2O2 concentration was calculated by using a standard curve and normalized to cellular protein.
Mitochondrial ROS determination
Intact cells cultured on coverslips were incubated with fluorescent probes in Krebs-HEPES buffer for 20 min at 37°C in a CO2 incubator. Production of mitochondrial O2·− was visualized by using the fluorescent probe MitoSOX (2 μM; ThermoFisher Scientific; excitation (Ex)/emission (Em), 510/480 nm). Mitochondrial localization was ensured by mitoTracker Green (0.1 μM; ThermoFisher Scientific; Ex/Em 488/520 nm). Nuclei were stained with Hoescht 33342 (0.01 mg/ml; Sigma-Aldrich Co; Ex/Em, 364/461 nm).
Mitochondrial membrane potential (ΔΨ)
Mitochondrial membrane potential was monitored with the fluorescent probe tetramethylrhodamine methyl ester (TMRE, Ex/Em, 543/573 nm; ThermoFisher Scientific) by using a modification of the method previously described (30) and as previously performed (21). Vessels were incubated in oxygenated Krebs-HEPES buffer (37°C). After 30 min of stabilization, arteries were incubated with Krebs-HEPES buffer containing TMRE (0.1 μM) for 20 min at 37°C. After washing, fluorescence intensity was measured with a spectrofluorimeter (FLUOstar OPTIMA BMG Labtech, Cary, NC). Fluorescence intensity values were normalized by the amount of proteins, and values were expressed relative to controls.
Drugs and solutions
Unless specifically stated, reagent-grade general chemicals were obtained from different suppliers. All drugs were dissolved in distilled water, except ML171, which was dissolved in 75% ethanol, and SB203580, which was dissolved in 50% DMSO. Further dilutions were in distilled water.
Data analysis and statistics
All data are expressed as mean ± standard error of the mean of the number of animals used in each experiment or independent cell culture-based experiments. Results were analyzed by using paired or unpaired Student's t-test or one-way or two-way ANOVA followed by Bonferroni's post hoc test using the GraphPad Prism 5 software (San Diego, CA). A p < 0.05 was considered significant.
Supplementary Material
Abbreviations Used
- Ang II
angiotensin II
- BAPN
β-aminopropionitrile
- ECM
extracellular matrix
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HCTZ
hydrochlorothiazide
- IEL
internal elastic lamina
- KHS
Krebs-Henseleit solution
- LOX
lysyl oxidase
- MRA
mesenteric resistance arteries
- NOX
NADPH oxidase isoforms
- p38MAPK
p38 mitogen-activated protein kinase
- PBS
phosphate-buffered saline
- PEG-catalase
polyethylene glycol-conjugated catalase
- ROS
reactive oxygen species
- SB
SB203580
- SDS
sodium dodecyl sulfate
- SHR
spontaneously hypertensive rats
- SOD
superoxide dismutase
- TMRE
tetramethylrhodamine methyl ester
- VSMC
vascular smooth muscle cells
- WKY
Wistar Kyoto rats
- W
wall thickness
- WT
wild-type
Acknowledgments
The authors thank Laura García Redondo and Silvia Aguiló for their excellent technical assistance. They also thank Drs. Carmen Martínez and Manuel Gómez-Guzman for their help with some experiments and Dr. Jo de Mey for his suggestions on aortic stiffness measurements.
This project was supported by the Spanish Ministerio de Economía y Competitividad (MINECO) (SAF2012-36400; SAF2015-64767-R and SAF2016-80305-P), Instituto de Salud Carlos III (ISCIII)-Fondo Europeo de Desarrollo Regional (FEDER) a way to build Europe (PI13/01488; PI15/01016, RD12/0042/0024, RD12/0042/0053, CIBER de Enfermedades Cardiovasculares), COST BM1301, ĹOréal-FWIS-Spain and Sociedad Espa ola de Arterioesclerosis (Beca SEA 2016 para investigación básica). AMB was supported by the Ramón y Cajal Program (RYC-2010–06473). RMT was supported through grants from the Canadian Institutes of Health Research and the British Heart Foundation. A part of the study was presented at the 20th Annual Meeting of the European Council for Cardiovascular Research (ECCR), Poiano, Lake Garda, Italy (abstract was published in High Blood Pressure & Cardiovascular Prevention, Volume 23, Issue 4, pp 405–421).
ola de Arterioesclerosis (Beca SEA 2016 para investigación básica). AMB was supported by the Ramón y Cajal Program (RYC-2010–06473). RMT was supported through grants from the Canadian Institutes of Health Research and the British Heart Foundation. A part of the study was presented at the 20th Annual Meeting of the European Council for Cardiovascular Research (ECCR), Poiano, Lake Garda, Italy (abstract was published in High Blood Pressure & Cardiovascular Prevention, Volume 23, Issue 4, pp 405–421).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Aguado A, Galán M, Zhenyukh O, Wiggers GA, Roque FR, Redondo S, Peçanha F, Martín A, Fortuño A, Cachofeiro V, Tejerina T, Salaices M, and Briones AM. Mercury induces proliferation and reduces cell size in vascular smooth muscle cells through MAPK, oxidative stress and cyclooxygenase-2 pathways. Toxicol Appl Pharmacol 268: 188–200, 2013 [DOI] [PubMed] [Google Scholar]
- 2.AlGhatrif M. and Lakatta EG. The conundrum of arterial stiffness, elevated blood pressure, and aging. Curr Hypertens Rep 17: 12, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arribas SM, Hinek A, and González MC. Elastic fibres and vascular structure in hypertension. Pharmacol Ther 111: 771–791, 2006 [DOI] [PubMed] [Google Scholar]
- 4.Bonadei I, Vizzardi E, D'Aloia A, Sciatti E, Raddino R, and Metra M. Role of aliskiren on arterial stiffness and endothelial function in patients with primary hypertension. J Clin Hypertens (Greenwich) 16: 202–206, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Briones AM. and Touyz RM. Oxidative stress and hypertension: current concepts. Curr Hypertens Rep 12: 135–142, 2010 [DOI] [PubMed] [Google Scholar]
- 6.Briones AM, Arribas SM, and Salaices M. Role of extracellular matrix in vascular remodeling of hypertension. Curr Opin Nephrol Hypertens 19: 187–194, 2010 [DOI] [PubMed] [Google Scholar]
- 7.Briones AM, González JM, Somoza B, Giraldo J, Daly CJ, Vila E, González MC, McGrath JC, and Arribas SM. Role of elastin in spontaneously hypertensive rat small mesenteric artery remodelling. J Physiol 552: 185–195, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Briones AM, Rodríguez-Criado N, Hernanz R, García-Redondo AB, Rodrigues-Díez RR, Alonso MJ, Egido J, Ruiz-Ortega M, and Salaices M. Atorvastatin prevents angiotensin II-induced vascular remodeling and oxidative stress. Hypertension 54: 142–149, 2009 [DOI] [PubMed] [Google Scholar]
- 9.Buus NH, Mathiassen ON, Fenger-Grøn M, Præstholm MN, Sihm I, Thybo NK, Schroeder AP, Thygesen K, Aalkjær C, Pedersen OL, Mulvany MJ, and Christensen KL. Small artery structure during antihypertensive therapy is an independent predictor of cardiovascular events in essential hypertension. J Hypertens 31: 791–797, 2013 [DOI] [PubMed] [Google Scholar]
- 10.Cai H. NAD(P)H oxidase-dependent self-propagation of hydrogen peroxide and vascular disease. Circ Res 96: 818–822, 2005 [DOI] [PubMed] [Google Scholar]
- 11.Chaplin NL, Nieves-Cintrón M, Fresquez AM, Navedo MF, and Amberg GC. Arterial smooth muscle mitochondria amplify hydrogen peroxide microdomains functionally coupled to L-Type calcium channels. Circ Res 117: 1013–1023, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen QZ, Han WQ, Chen J, Zhu DL, Chen-Yan , and Gao PJ. Anti-stiffness effect of apocynin in deoxycorticosterone acetate-salt hypertensive rats via inhibition of oxidative stress. Hypertens Res 36: 306–312, 2013 [DOI] [PubMed] [Google Scholar]
- 13.Clifford PS, Ella SR, Stupica AJ, Nourian Z, Li M, Martinez-Lemus LA, Dora KA, Yang Y, Davis MJ, Pohl U, Meininger GA, and Hill MA. Spatial distribution and mechanical function of elastin in resistance arteries: a role in bearing longitudinal stress. Arterioscler Thromb Vasc Biol 31: 2889–2896, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delles C, Zimmerli LU, McGrane DJ, Koh-Tan CH, Pathi VL, McKay AJ, Steedman T, Dargie HJ, Hamilton CA, and Dominiczak AF. Vascular stiffness is related to superoxide generation in the vessel wall. J Hypertens 26: 946–955, 2008 [DOI] [PubMed] [Google Scholar]
- 15.Dikalov SI. and Nazarewicz RR. Angiotensin II-induced production of mitochondrial reactive oxygen species: potential mechanisms and relevance for cardiovascular disease. Antioxid Redox Signal 19: 1085–1094, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dikalov SI. and Ungvari Z. Role of mitochondrial oxidative stress in hypertension. Am J Physiol Heart Circ Physiol 305: H1417–H1427, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, and Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res 107: 106–116, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duprez DA. Is vascular stiffness a target for therapy? Cardiovasc Drugs Ther 24: 305–310, 2010 [DOI] [PubMed] [Google Scholar]
- 19.Eberson LS, Sanchez PA, Majeed BA, Tawinwung S, Secomb TW, and Larson DF. Effect of lysyl oxidase inhibition on angiotensin II-induced arterial hypertension, remodeling, and stiffness. PLoS One 10: e0124013, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faury G, Pezet M, Knutsen RH, Boyle WA, Heximer SP, McLean SE, Minkes RK, Blumer KJ, Kovacs A, Kelly DP, Li DY, Starcher B, and Mecham RP. Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J Clin Invest 112: 1419–1428, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.García-Redondo AB, Briones AM, Martínez-Revelles S, Palao T, Vila L, Alonso MJ, and Salaices M. c-Src, ERK1/2 and Rho kinase mediate hydrogen peroxide-induced vascular contraction in hypertension: role of TXA2, NAD(P)H oxidase and mitochondria. J Hypertens 33: 77–87, 2015 [DOI] [PubMed] [Google Scholar]
- 22.González JM, Briones AM, Somoza B, Daly CJ, Vila E, Starcher B, McGrath JC, González MC, and Arribas SM. Postnatal alterations in elastic fiber organization precede resistance artery narrowing in SHR. Am J Physiol Heart Circ Physiol 291: H804–H812, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Greenwald SE. Ageing of the conduit arteries. J Pathol 211:157–172, 2007. 17200940 [Google Scholar]
- 24.Griendling KK, Sorescu D, Lassègue B, and Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol 20: 2175–2183, 2000 [DOI] [PubMed] [Google Scholar]
- 25.Guadall A, Orriols M, Rodríguez-Calvo R, Calvayrac O, Crespo J, Aledo R, Martínez-González J, and Rodríguez C. Fibulin-5 is up-regulated by hypoxia in endothelial cells through a hypoxia-inducible factor-1 (HIF-1α)-dependent mechanism. J Biol Chem 286: 7093–7103, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hernanz R, Martínez-Revelles S, Palacios R, Martín A, Cachofeiro V, Aguado A, García-Redondo L, Barrús MT, de Batista PR, Briones AM, Salaices M, and Alonso MJ. Toll-like receptor 4 contributes to vascular remodelling and endothelial dysfunction in angiotensin II-induced hypertension. Br J Pharmacol 172: 3159–3176, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hinek A, Smith AC, Cutiongco EM, Callahan JW, Gripp KW, and Weksberg R. Decreased elastin deposition and high proliferation of fibroblasts from Costello syndrome are related to functional deficiency in the 67-kD elastin-binding protein. Am J Hum Genet 66: 859–872, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirano E, Knutsen RH, Sugitani H, Ciliberto CH, and Mecham RP. Functional rescue of elastin insufficiency in mice by the human elastin gene: implications for mouse models of human disease. Circ Res 101: 523–531, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Kanaki AI, Sarafidis PA, Georgianos PI, Kanavos K, Tziolas IM, Zebekakis PE, and Lasaridis AN. Effects of low-dose atorvastatin on arterial stiffness and central aortic pressure augmentation in patients with hypertension and hypercholesterolemia. Am J Hypertens 26: 608–616, 2013 [DOI] [PubMed] [Google Scholar]
- 30.Katakam PV, Gordon AO, Sure VN, Rutkai I, and Busija DW. Diversity of mitochondria-dependent dilator mechanisms in vascular smooth muscle of cerebral arteries from normal and insulin-resistant rats. Am J Physiol Heart Circ Physiol 307: H493–503, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kingwell BA, Arnold PJ, Jennings GL, and Dart AM. Spontaneous running increases aortic compliance in Wistar-Kyoto rats. Cardiovasc Res 35: 132–137, 1997 [DOI] [PubMed] [Google Scholar]
- 32.Kozel BA, Danback JR, Waxler JL, Knutsen RH, de Las Fuentes L, Reusz GS, Kis E, Bhatt AB, and Pober BR. Williams syndrome predisposes to vascular stiffness modified by antihypertensive use and copy number changes in NCF1. Hypertension 63: 74–79, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lassègue B, San Martín A, and Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res 110: 1364–1390, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laurent S. and Boutouyrie P. The structural factor of hypertension: large and small artery alterations. Circ Res 116: 1007–1021, 2015 [DOI] [PubMed] [Google Scholar]
- 35.Laurent S. and Boutouyrie P. Vascular Mechanism Collaboration. Dose-dependent arterial destiffening and inward remodeling after olmesartan in hypertensives with metabolic syndrome. Hypertension 64: 709–716, 2014 [DOI] [PubMed] [Google Scholar]
- 36.Laurindo FR, Fernandes DC, and Santos CX. Assessment of superoxide production and NADPH oxidase activity by HPLC analysis of dihydroethidium oxidation products. Methods Enzymol 441: 237–260, 2008 [DOI] [PubMed] [Google Scholar]
- 37.Le VP, Knutsen RH, Mecham RP, and Wagenseil JE. Decreased aortic diameter and compliance precedes blood pressure increases in postnatal development of elastin-insufficient mice. Am J Physiol Heart Circ Physiol 301: H221–H229, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li W, Liu G, Chou IN, and Kagan HM. Hydrogen peroxide-mediated, lysyl oxidase-dependent chemotaxis of vascular smooth muscle cells. J Cell Biochem 78: 550–557, 2000 [PubMed] [Google Scholar]
- 39.Mäki JM, Räsänen J, Tikkanen H, Sormunen R, Mäkikallio K, Kivirikko KI, and Soininen R. Inactivation of the lysyl oxidase gene Lox leads to aortic aneurysms, cardiovascular dysfunction, and perinatal death in mice. Circulation 106: 2503–2509, 2002 [DOI] [PubMed] [Google Scholar]
- 40.Martínez-Revelles S, Avendaño MS, García-Redondo AB, Alvarez Y, Aguado A, Pérez-Girón JV, García-Redondo L, Esteban V, Redondo JM, Alonso MJ, Briones AM, and Salaices M. Reciprocal relationship between reactive oxygen species and cyclooxygenase-2 and vascular dysfunction in hypertension. Antioxid Redox Signal 18: 51–65, 2013 [DOI] [PubMed] [Google Scholar]
- 41.Masugata H, Senda S, Murao K, Inukai M, Himoto T, Hosomi N, Okada H, and Goda F. Association between urinary 8-hydroxydeoxyguanosine, an indicator of oxidative stress, and the cardio-ankle vascular index in hypertensive patients. J Atheroscler Thromb 19: 747–755, 2012 [PubMed] [Google Scholar]
- 42.Mitchell GF. Arterial stiffness and hypertension: chicken or egg? Hypertension 64: 210–214, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ooshima A. and Midorikawa O. Increased lysyl oxidase activity in blood vessels of hypertensive rats and effect of beta-aminopropionitrile on arteriosclerosis. Jpn Circ J 41: 1337–1340, 1977 [DOI] [PubMed] [Google Scholar]
- 44.Orriols M, Guadall A, Galán M, Martí-Pàmies I, Varona S, Rodríguez-Calvo R, Briones AM, Navarro MA, de Diego A, Osada J, Martínez-González J, and Rodríguez C. Lysyl oxidase (LOX) in vascular remodelling. Insight from a new animal model. Thromb Haemost 112: 812–824, 2014 [DOI] [PubMed] [Google Scholar]
- 45.Payne SL, Fogelgren B, Hess AR, Seftor EA, Wiley EL, Fong SF, Csiszar K, Hendrix MJ, and Kirschmann DA. Lysyl oxidase regulates breast cancer cell migration and adhesion through a hydrogen peroxide-mediated mechanism. Cancer Res 65: 11429–11436, 2005 [DOI] [PubMed] [Google Scholar]
- 46.Plantinga Y, Ghiadoni L, Magagna A, Giannarelli C, Franzoni F, Taddei S, and Salvetti A. Supplementation with vitamins C and E improves arterial stiffness and endothelial function in essential hypertensive patients. Am J Hypertens 20: 392–397, 2007 [DOI] [PubMed] [Google Scholar]
- 47.Rizzoni D, Porteri E, Boari GE, De Ciuceis C, Sleiman I, Muiesan ML, Castellano M, Miclini M, and Agabiti-Rosei E. Prognostic significance of small-artery structure in hypertension. Circulation 108: 2230–2235, 2003 [DOI] [PubMed] [Google Scholar]
- 48.Rodgers RL, Breault PH, Sessa WC, Jr., and Chichester CO. Depressor effect of diabetes in spontaneously hypertensive rat: role of vascular reactivity and prolyl hydroxylase and lysyl oxidase activities. Life Sci 37: 2237–2247, 1985 [DOI] [PubMed] [Google Scholar]
- 49.Rodríguez C, Alcudia JF, Martínez-González J, Guadall A, Raposo B, Sánchez-Gómez S, and Badimon L. Statins normalize vascular lysyl oxidase down-regulation induced by proatherogenic risk factors. Cardiovasc Res 83: 595–603, 2009 [DOI] [PubMed] [Google Scholar]
- 50.Rodríguez C, Rodríguez-Sinovas A, and Martínez-González J. Lysyl oxidase as a potential therapeutic target. Drug News Perspect 21: 218–224, 2008 [DOI] [PubMed] [Google Scholar]
- 51.Rodríguez C, Martínez-González J, Raposo B, Alcudia JF, Guadall A, and Badimon L. Regulation of lysyl oxidase in vascular cells: lysyl oxidase as a new player in cardiovascular diseases. Cardiovasc Res 79: 7–13, 2008 [DOI] [PubMed] [Google Scholar]
- 52.Safar ME, Blacher J, and Jankowski P. Arterial stiffness, pulse pressure, and cardiovascular disease-is it possible to break the vicious circle? Atherosclerosis 218: 263–271, 2011 [DOI] [PubMed] [Google Scholar]
- 53.Safar ME, Nilsson PM, Blacher J, and Mimran A. Pulse pressure, arterial stiffness, and end-organ damage. Curr Hypertens Rep 14: 339–344, 2012 [DOI] [PubMed] [Google Scholar]
- 54.Salaymeh KJ. and Banerjee A. Evaluation of arterial stiffness in children with Williams syndrome: does it play a role in evolving hypertension? Am Heart J 142: 549–555, 2001 [DOI] [PubMed] [Google Scholar]
- 55.Sheridan PJ, Kozar LG, and Benson SC. Increased lysyl oxidase activity in aortas of hypertensive rats and effect of beta-aminopropionitrile. Exp Mol Pathol 30: 315–324, 1979 [DOI] [PubMed] [Google Scholar]
- 56.Tang SS, Trackman PC, and Kagan HM. Reaction of aortic lysyl oxidase with beta-aminopropionitrile. J Biol Chem 258: 4331–4338, 1983 [PubMed] [Google Scholar]
- 57.Virdis A, Neves MF, Amiri F, Touyz RM, and Schiffrin EL. Role of NAD(P)H oxidase on vascular alterations in angiotensin II-infused mice. J Hypertens 22: 535–542, 2004 [DOI] [PubMed] [Google Scholar]
- 58.Wagenseil JE. and Mecham RP. Elastin in large artery stiffness and hypertension. J Cardiovasc Transl Res 5: 264–273, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L, Madhur MS, Chen W, and Harrison DG. Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circ Res 114: 616–625, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou RH, Vendrov AE, Tchivilev I, Niu XL, Molnar KC, Rojas M, Carter JD, Tong H, Stouffer GA, Madamanchi NR, and Runge MS. Mitochondrial oxidative stress in aortic stiffening with age: the role of smooth muscle cell function. Arterioscler Thromb Vasc Biol 32: 745–755, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.