

Summary
Calcineurin inhibitors (CNIs) have been used off‐label for the treatment of refractory Kawasaki disease (KD). However, it remains unknown whether CNIs show protective effects against the development of coronary artery lesions in KD patients. To investigate the effects of CNIs on coronary arteries and the mechanisms of their actions on coronary arteritis in a mouse model of KD, we performed experiments with FK565, a ligand of nucleotide‐binding oligomerization domain‐containing protein 1 (NOD1) in wild‐type, severe combined immunodeficiency (SCID), caspase‐associated recruitment domain 9 (CARD9)–/– and myeloid differentiation primary response gene 88 (MyD88)–/– mice. We also performed in‐vitro studies with vascular and monocytic cells and vascular tissues. A histopathological analysis showed that both cyclosporin A and tacrolimus exacerbated the NOD1‐mediated coronary arteritis in a dose‐dependent manner. Cyclosporin A induced the exacerbation of coronary arteritis in mice only in high doses, while tacrolimus exacerbated it within the therapeutic range in humans. Similar effects were obtained in SCID and CARD9–/– mice but not in MyD88–/– mice. CNIs enhanced the expression of adhesion molecules by endothelial cells and the cytokine secretion by monocytic cells in our KD model. These data indicated that both vascular and monocytic cells were involved in the exacerbation of coronary arteritis. Activation of MyD88‐dependent inflammatory signals in both vascular cells and macrophages appears to contribute to their adverse effects. Particular attention should be paid to the development of coronary artery lesions when using CNIs to treat refractory KD.
Keywords: calcineurin inhibitors, coronary arteritis, Kawasaki disease, mouse model, MyD88
Introduction
Kawasaki disease (KD) is an acute febrile illness of unknown aetiology characterized by a systemic vasculitis of small‐ and medium‐sized arteries, particularly coronary arteries 1. The standard therapy for KD consists of aspirin and high‐dose intravenous immunoglobulin (IVIG). Approximately 10–30% of KD patients are resistant to IVIG 2, 3, 4, 5, and some develop coronary artery lesions (CALs) such as coronary aneurysms, a clinically important problem. Although therapeutic options for KD patients refractory to high‐dose IVIG include high‐dose corticosteroid, infliximab and plasma exchange, no definite treatments for such patients have been established.
Calcineurin inhibitors (CNIs), such as cyclosporin A (CsA) and tacrolimus (Tac), have been used as off‐label drugs to treat KD 6, 7, 8, 9, 10. Inositol 1,4,5‐trisphosphate 3 kinase C (ITPKC), a susceptibility gene of KD, encodes a kinase that regulates the intracellular Ca2+ level negatively and inhibits calcineurin (CN)‐dependent activation of nuclear factor of activated T cells (NFAT) by phosphorylating inositol trisphosphate. Given the inhibitory effects on the CN/NFAT pathway, CNIs are expected to be useful as an alternative treatment for KD patients. In several observational studies 7, 8, the anti‐pyretic and anti‐inflammatory effects of CsA have been described in refractory KD patients; however, it remains to be determined whether or not CNIs exert any effect on coronary arteries.
We recently established a mouse model of KD by the administration of FK565, a synthetic ligand of nucleotide‐binding oligomerization domain‐containing protein 1 (NOD1) 11. The histopathological features of FK565‐induced coronary arteritis in mice are similar to those of CALs in acute‐phase KD 11, 12. We also reported that vascular cells and cardiac CD11c+ macrophages play a pivotal role in the pathogenesis of acute coronary arteritis 13.
Using a NOD1‐mediated KD animal model, we investigated the effects of CNIs on coronary arteries and examined the mechanism of CNI‐induced exacerbation of coronary arteritis.
Materials and methods
Animals
C57BL6/N [wild‐type (WT)] and CB‐17 severe combined immunodeficiency (SCID) mice were purchased from KBT Oriental (Charles River Grade, Saga, Japan). Myeloid differentiation primary response gene 88 (MyD88) knockout (MyD88–/–) mice in a C57BL/6 background were purchased from Oriental Yeast (Tokyo, Japan). Caspase‐associated recruitment domain 9 (CARD9) knock‐out (CARD9–/–) mice were generated as described 14. Calcineurin subunit B1 (Cnb1)flox/flox and Cd11c‐Cre mice in a C57BL/6 background were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). All mice were 5–7 weeks old, and were kept under specific pathogen‐free conditions. All animal care and handling procedures were approved by the Institutional Animal Care and Use Committee of Kyushu University (protocol number: A‐27‐154), and followed the Guideline for Proper Conduct of Animal Experiments, Science Council of Japan.
Administration of innate immune ligands and CNIs
FK565, a synthetic NOD1 ligand, was supplied initially by Astellas Pharma (Tokyo, Japan) and prepared later by Shimoyama and Fukase (see the Supporting information). CsA and Tac were purchased from Wako Pure Chemical Industries (Osaka, Japan) and Enzo Life Sciences (Lausen, Switzerland), respectively. Muramyl dipeptide (MDP; NOD2 ligand) and lipopolysaccharide [LPS; Toll‐like receptor (TLR)‐4 ligand] were purchased from Lonza (Basel, Switzerland) and InvivoGen (Toulouse, France), respectively.
CsA and Tac were dissolved in endotoxin‐free dimethylsulphoxide (DMSO) (Hybri‐MaxTM; Sigma‐Aldrich, St Louis, MO, USA), and we confirmed that these solutions were endotoxin‐free with a ToxinSensorTM Chromogenic LAL endotoxin assay kit (GenScript, Piscataway, NJ, USA).
The protocol to induce arteritis in mice was as follows: mice were administered 100 μg of FK565 orally, which was dissolved in sterile distilled H2O once daily for 5 consecutive days, or administered 10, 100 or 500 μg of FK565 subcutaneously on days 0 and 3. Simultaneously, CsA (4, 12, 40 or 120 mg/kg body weight per dose) or Tac (0·2, 0·6, 2 or 6 mg/kg body weight per dose), dissolved in 10% DMSO, was administered intraperitoneally for 5 consecutive days. MDP (500 μg), dissolved in sterile distilled H2O, was administered intraperitoneally on days 0 and 3. LPS (10 μg), dissolved in sterile distilled H2O, was administered intraperitoneally twice (days 0 and 3) or once (day −1 or 0).
Histological analyses
After mice were euthanized, the hearts were dissected on day 5. They were 4% paraformaldehyde‐fixed and paraffin‐embedded for histological analyses. Cross‐sections of the aortic roots were prepared, with the three aortic valve cusps and coronary arteritis assessed by haematoxylin and eosin (HE) staining, as described previously 11. For scoring inflammatory cell infiltration, the lesion area and cell infiltration around the coronary arteries were measured in one section per heart using the Image J software program (National Institutes of Health, Bethesda, MD, USA) 13.
Blood concentrations of CsA and Tac
CsA (4, 12, 40 or 120 mg/kg body weight per dose) or Tac (0·2, 0·6, 2 or 6 mg/kg body weight per dose) was administered intraperitoneally to 5‐week‐old mice once daily for 5 consecutive days. Blood samples were collected from mice 0 (trough), 2 and 4 h after drug administration on day 5. Blood concentrations of CsA and Tac were measured by liquid chromatography (LC)–tandem mass spectrometry (MS)/MS, as described previously 15, with minor modifications. Briefly, all whole blood samples (150 μl) were transferred to glass tubes and spiked with 25 μl of ascomycin (20 μg/ml; Sigma‐Aldrich), which served as the internal standard. Then, 600 μl of water and 2 ml of extraction solution (methyl‐t‐butyl ether/cyclohexane, 1 : 3 v/v) were added to the glass tubes. Each tube was capped securely, mixed on a horizontal shaker for 15 min and centrifuged at 1610 ×g for 15 min. The organic layer was transferred to a new tube and evaporated using an Automatic Environmental Speed Vac® System (Thermo Fisher Scientific Inc., Waltham, MA, USA). Each sample was reconstituted with 200 μl of 50% methanol solution and then mixed with a vortex mixer for 1 min. A 20‐μl aliquot of each sample was injected into the LC‐MS/MS system. Each sample was analysed by high‐performance liquid chromatography (HPLC, Eksigent® ekspertTM ultra LC SYSTEM100XL; AB Sciex, Framingham, MA, USA) on an analytical column (Inertsil‐ODS3, 150 × 2·1 mm i.d.; GL Sciences, Inc., Tokyo, Japan) and an MS/MS detector (QTRAP® 4500; AB Sciex). The mobile phase consisted of a multiple gradient of solvent A (1 mM ammonium acetate) and solvent B (methanol/1 mM ammonium acetate). The flow rate was set at 250 μl/min, the column was operated at 60°C and the eluent was introduced directly into the electrospray ion source of the mass spectrometer. Selected reaction monitoring transitions in the positive ion mode were m/z 1220 → m/z 1203 for CsA, m/z 821 → m/z 768 for Tac and m/z 809 → m/z 756 for ascomycin. CsA, Tac and their metabolites were detected as ammonium adducts (m+ NH4). Peak areas were linear from 0·5 to 30 ng/ml for CsA, Tac and their metabolites.
Organ culture and protein determination
The aortic roots isolated sterilely from C57CL/6 mice were cultured for 24 h in a 96‐well plate with endothelial basal medium (EBM)‐2 with EGM‐2MV (Lonza) in a CO2 (5%) incubator at 37°C, as described previously 11. The protein concentrations of aortic root tissues were measured by a Bio‐Rad protein assay (BioRad, Hercules, CA, USA) after homogenization with phosphate‐buffered saline containing Cell Culture Lysis Reagent (Promega, Madison, WI, USA) and Protease Inhibitor Cocktail (Nacalai Tesque, Kyoto, Japan). Whole protein contents of aortic root tissue were measured to calculate the chemokine (C‐C motif) ligand 2 (CCL2)/interleukin (IL)‐6 levels per tissue protein content.
Cells
Murine bone marrow cells were harvested from the femur and tibia. Bone marrow‐derived macrophages (BMDMs) were prepared from bone marrow cells cultured in RPMI‐1640 (Gibco Laboratories, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; MP Biomedicals, Santa Ana, CA, USA) and 10% L929 culture supernatant (source of macrophage colony‐stimulating factor) for 7–10 days 13. Bone marrow‐derived dendritic cells (BMDCs) were prepared from bone marrow cells cultured in RPMI‐1640 supplemented with 10% FBS and 10% MGM‐5 culture supernatant (source of granulocyte–macrophage colony‐stimulating factor) for 7–10 days 16. BMDCs were stained with phycoerythrin‐conjugated anti‐CD11c antibody (BD Biosciences, San Jose, CA, USA) and analysed using an EC800 Analyzer (Sony Biotechnology, Tokyo, Japan).
Human coronary artery endothelial cells (HCAECs) and human coronary artery smooth muscle cells (HCASMCs) derived from healthy donors were purchased from Lonza. Murine monocyte/macrophage cell line RAW264.7 (RCB0535) was obtained from Riken Cell Bank (Tsukuba, Japan). HCAECs and HCASMCs were cultured in EBM‐2 medium with EGM‐2MV and smooth muscle basal medium (SmBM) with SmGM‐2 (Lonza), respectively. RAW264.7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Wako) with 10% FBS (MP Biomedicals). These cells were incubated in a 5% CO2 incubator at 37°C.
Mouse heart endothelial cells (MHECs) were isolated from murine heart using CD31 microbeads (Miltenyi Biotec, Auburn, CA, USA) and cultured as described previously 13. Briefly, diced heart tissues were treated with collagenase II (Worthington Biochemical, Freehold, NJ, USA) for 1 h at 37°C. Cell suspensions were incubated with rat anti‐mouse CD31, and CD31‐positive cells were purified using MACS (Miltenyi). CD31‐positive cells were cultured in DMEM containing endothelial cell growth supplement (Sigma‐Aldrich) for 7 days and used for subsequent analyses.
Flow cytometric analyses
HCAECs stimulated with FK565 (10 μg/ml) in the presence or absence of CsA (10 μM = 12.03 μg/ml) or Tac (10 μM = 8.22 μg/ml) for 24 h were collected and stained with fluorescein isothiocyanate (FITC)‐conjugated anti‐CD54 [intercellular adhesion molecule‐1 (ICAM‐1)] monoclonal antibody or IgG1 isotype control (Beckman Coulter, Miami, FL, USA), and phycoerythrin (PE)‐conjugated anti‐CD106 [vascular cell adhesion molecule‐1 (VCAM‐1)] monoclonal antibody or IgG1 isotype control (BD). The expression of ICAM‐1 and VCAM‐1 was analysed using an EPICS XL flow cytometer (Beckman Coulter). The data were analysed with the Kaluza software program, version 1.5 (Beckman Coulter).
Quantitative real‐time–polymerase chain reaction
HCAECs stimulated with FK565 (10 μg/ml) in the presence or absence of CsA (10 μM) or Tac (10 μM) for 6 h were collected, and the total RNA was extracted using RNeasy Micro Kit (Qiagen, Hilden, Germany). Complementary DNA was synthesized using a high‐capacity RNA to cDNA Kit (Life Technologies, Gaithersburg, MD, USA) in accordance with the manufacturer's protocol. A quantitative real‐time–polymerase chain reaction (qRT–PCR) was performed using Fast SYBR Green Master Mix and StepOnePlus (Life Technologies). Human β‐actin (ACTB) was used as an internal control gene. The sequences of the gene‐specific primers were as follows: (F: forward, R: reverse primers): ICAM‐1: 5′‐CGGCCAGCTTATACACAAGAAC‐3′ (F) and 5′‐AATTTTCTGGCCACGTCCAG‐3′ (R), VCAM‐1: 5′‐AAGGCAGAGTACGCAAACAC‐3′ (F) and 5′‐ATTTTCGGAGCAGGAAAGCC‐3′ (R), E‐selectin: 5′‐TGAGGAAGGCTTCATGTTGC‐3′ (F) and 5′‐TGTGCACTGGAAAGCTTCAC‐ 3′ (R), and ACTB: 5′‐CACCCTGAAGTACCCCATCG‐3′ (F) and 5′‐TGCCAGATTTTCTCCATGTCG‐3′ (R).
To evaluate the expression of NOD1 in the murine vascular tissue, aorta tissues (from arch to abdominal aorta) isolated from wild‐type C57BL/6 mice were stimulated with DMSO (control), CsA (0·1, 1 or 10 µM) or Tac (0·1, 1 or 10 µM) for 48 h and collected. Then, total RNA was extracted using the RNeasy Mini Kit (Qiagen), and complementary DNA was synthesized. A qRT–PCR was performed using predesigned PrimeTime® qPCR assay (Integrated DNA Technologies, Coralville, IA, USA) for mouse NOD1, TLR‐4 and ACTB.
The PCR conditions were 95°C (20 s), 40 cycles of 95°C (3 s) and 60°C (30 s). The relative gene expression was calculated by the ddCt method and presented as 2^[Ct(ACTB) – CT(gene)] 17.
Cell stimulation and cytokine assay
BMDMs/BMDCs/RAW264.7 (1·0 × 105 cells/well) or HCAEC/HCASMC (3200 cells/well) were seeded into 96‐well plates. The next day, the medium was changed and the cells were stimulated with each of the following reagents: PolyI:C (TLR‐3 ligand; Invivogen), LPS or plate‐coated trehalose‐6,6'‐dimycolate (TDM; C‐type lectin Mincle ligand; Sigma‐Aldrich) as positive controls. The supernatants were collected for assay 24 h after stimulation.
The concentrations of tumour necrosis factor (TNF), IL‐6, IL‐8 and CCL2 were measured using a BD™ Cytometric Bead Array Flex Set System, Mouse Inflammation Kit and Human Inflammatory Cytokine kit (BD Biosciences). The cytokines were analysed using an EC800 Analyzer (Sony Biotechnology).
Statistical analysis
The data were analysed using Student's t‐test, Dunnett's test or the Tukey–Kramer test using the statistical software program JMP® version 9.0 (SAS Institute, Cary, NC, USA). Values of P < 0·05 were considered statistically significant.
Results
CNIs exacerbate NOD1‐mediated coronary arteritis
To investigate whether or not CNIs exert anti‐inflammatory effects on coronary arteritis in a NOD1‐mediated KD animal model 11, we administered CsA or Tac to mice in combination with FK565 (Fig. 1a). In the preliminary experiments, we determined the dose and duration of FK565 administration to induce coronary arteritis in mice (Supporting information, Fig. S1A). Unexpectedly, CNIs exacerbated NOD1‐mediated coronary arteritis in a dose‐dependent manner (Fig. 1b). In the quantitative evaluation of inflammatory lesions by number and infiltration area of inflammatory cells, coronary arteritis was exacerbated significantly by the administration of CsA 120 mg/kg or Tac 2 and 6 mg/kg (Fig. 1c,d). No coronary arteritis was induced by CNIs alone (Supporting information, Fig. S2).
Figure 1.
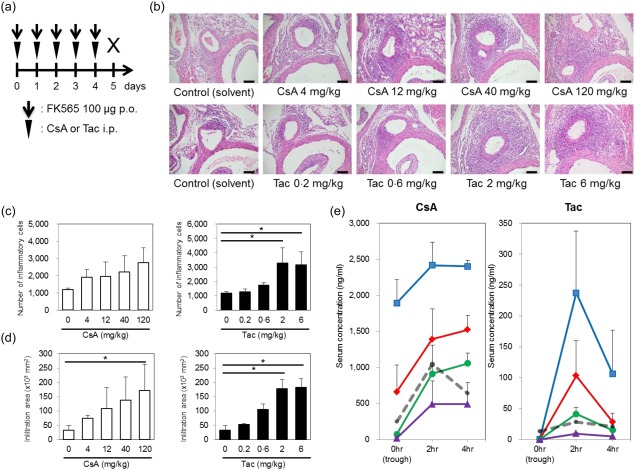
Exacerbation of coronary arteritis by calcineurin inhibitors (CNIs). (a) The experimental protocol; p.o.: per os, i.p.: intraperitoneal injection. The histological evaluation of coronary arteritis (b) induced by cyclosporin A (CsA) or tacrolimus (Tac) and 100 µg of FK565 in wild‐type mice (scale bar, 100 μm). The numbers (c) and infiltration areas (d) of inflammatory cells are shown. The data are presented as the mean ± standard deviation (s.d.) (n = 3). *P < 0·05 (Dunnett's test). (e) After CsA (4, 12, 40 or 120 mg/kg body weight per dose) or Tac (0·2, 0·6, 2 or 6 mg/kg body weight per dose) was administered i.p. once daily for 5 consecutive days, blood concentrations of CsA and Tac at 0 (trough), 2 and 4 h after drug administration on day 5. The data are presented as the mean ± s.d. (n = 3). Cyclosporin A (CsA) (120 mg/kg) and 6 mg/kg of Tac (blue squares), 40 mg/kg of CsA and 2 mg/kg of Tac (red diamonds), 12 mg/kg of CsA and 0·6 mg/kg of Tac (green circles), and 4 mg/kg of CsA and 0·2 mg/kg of Tac (purple triangles). Grey dashed lines represent the blood concentrations of human therapeutic doses (5·26 mg/kg of CsA and 0·16 mg/kg of Tac from Refs [ 18, 20]). [Colour figure can be viewed at wileyonlinelibrary.com]
To determine the doses at which CNIs exacerbated coronary arteritis, we measured the blood concentrations of CNIs before and after 5‐day administration. When mice were administered 120 or 40 mg/kg of CsA, the trough levels were above the toxic level of CsA in humans (300 ng/ml) 18. The trough levels after the administration of 6 mg/kg of Tac did not reach the toxic level in humans (20 ng/ml) 19 (Fig. 1e). The blood levels after administration of 12 mg/kg of CsA and 0·6 mg/kg of Tac in mice were close to those after administration of standard therapeutic doses of 5·26 mg/kg of CsA 18 and 0·16 mg/kg of Tac 20 in humans, respectively, as shown in Fig. 1e. These results indicated that coronary arteritis might be exacerbated by high concentrations of CsA and at therapeutic concentrations of Tac in humans.
Non‐T, non‐B cells are associated with exacerbation of coronary arteritis by CNIs
To examine which cells were associated with the exacerbation of coronary arteritis by CNIs, we repeated the same experiments as above using SCID mice. Exacerbation of coronary arteritis by CNIs was also found in the SCID mice, suggesting that T and B cells were not essential for the exacerbation of arteritis (Fig. 2a–c).
Figure 2.
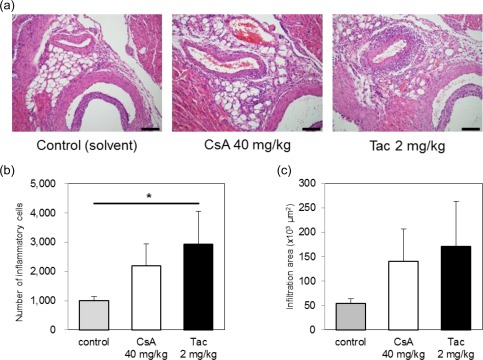
Exacerbation of coronary arteritis by calcineurin inhibitors (CNIs) in CB‐17 severe combined immunodeficiency (SCID) mice. CB‐17 mice were administered 40 mg/kg of cyclosporin A (CsA), 2 mg/kg of tacrolimus (Tac) or a solvent control [10% dimethylsulphoxide (DMSO)] intraperitoneally with 100 μg of FK565 [per os (p.o.)] once daily for 5 consecutive days. A histological evaluation of coronary arteritis (a) induced by CsA or Tac and 100 μg of FK565 in CB‐17 SCID mice (scale bar, 100 μm). The numbers (b) and infiltration areas (c) of inflammatory cells are shown. The data are presented as the mean ± standard deviation (s.d.) (n = 3). *P < 0·05 (Dunnett's test). [Colour figure can be viewed at wileyonlinelibrary.com]
Effects of CNIs on vascular and monocytic cells
We reported that monocytic and vascular cells play an essential role in the pathogenesis of NOD1‐mediated coronary arteritis 13. First, to examine the effects of CNIs on murine vascular cells in vitro, we stimulated MHECs with CNIs and FK565 and assayed the production of cytokines. When a high concentration of Tac (10 μM) was added, the production of IL‐6 and CCL2 was enhanced in the absence of FK565 stimulation but not in the presence of FK565 (Fig. 3a). In contrast, the production of these cytokines was suppressed with high concentrations (1 and 10 μM) of CsA in the presence or absence of FK565 (Fig. 3a).
Figure 3.
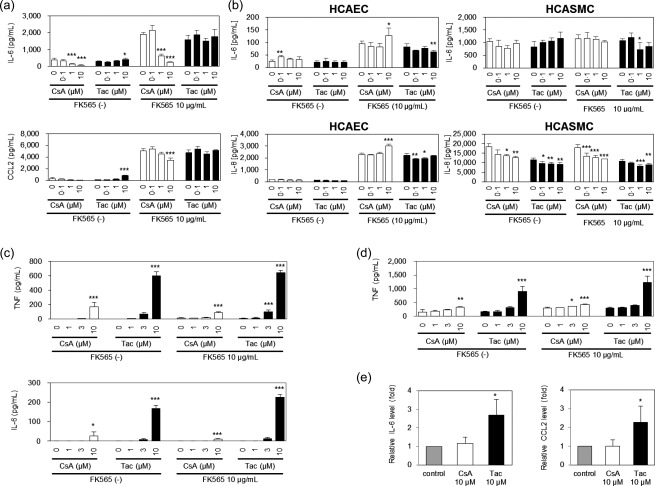
The effects of calcineurin inhibitors (CNIs) on vascular and monocytic cells. The culture supernatants of mouse heart endothelial cells (MHECs) (a), human coronary artery endothelial cells (HCAECs) and human coronary artery smooth muscle cells (HCASMCs) (b) stimulated with cyclosporin A (CsA) or tacrolimus (Tac) in the presence of FK565 were assayed for interleukin (IL)‐6, chemokine (C‐C motif) ligand 2 (CCL2) and IL‐8. (c) The culture supernatants of bone marrow‐derived macrophages (BMDMs) from wild‐type mice stimulated with CsA or Tac in the presence or absence of FK565 were assayed for tumour necrosis factor (TNF) and IL‐6. (d) The culture supernatants of RAW264.7 monocytic cells stimulated with CsA or Tac in the presence or absence of FK565 were assayed for TNF. (e) The culture supernatants of aortic root tissues stimulated with CsA or Tac and 10 μg/ml FK565 were assayed for IL‐6 and CCL2. The data are presented as the mean ± standard deviation (n = 4). *P < 0·05; **P < 0·01, ***P < 0·001 compared to controls (0 μM CsA or Tac) of each group (Dunnett's test).
To investigate the effects of CNIs on human vascular cells, we stimulated HCAECs and HCASMCs with CNIs in the presence or absence of FK565 and examined the cytokine production. The stimulation of HCAECs and HCASMCs with Tac did not increase cytokine production in the presence or absence of FK565 (Fig. 3b). The production of IL‐6 and IL‐8 was increased in the presence of 10 μM of CsA with FK565 in HCAECs, but was somewhat suppressed in HCASMCs under these conditions. Taken together, these data showed that the co‐administration of CNIs with FK565 resulted in variable responses of endothelial cells and smooth muscle cells in the production of proinflammatory cytokines.
We next performed experiments focusing on monocytes/macrophages. To investigate the effect of CNIs on monocytes/macrophages in vitro, we stimulated BMDMs and RAW264.7 cells with CNIs in the presence or absence of FK565 and examined the production of cytokines. We found that CNIs enhanced the spontaneous and FK565‐induced production of TNF and IL‐6 in a concentration‐dependent manner, and the effect of Tac was stronger than that of CsA in BMDMs and RAW264.7 cells (Fig. 3c,d).
Effects of CNIs on the cytokine production in vascular tissues
We have demonstrated previously that NOD1 ligands enhanced the production of inflammatory cytokines from aortic roots ex vivo 11. To examine the direct effect of CNIs on the aortic roots, we cultured the aortic root tissues isolated from C57BL/6 mice with FK565 and CNIs and measured the IL‐6 and CCL2 levels in the culture supernatant. In the presence of FK565, the production of IL‐6 and CCL2 was increased in the presence of high concentrations of Tac (Fig. 3e). In contrast, 10 μM CsA did not enhance the IL‐6 and CCL2 production from aortic tissues. These data recapitulated the differential effects of CsA and Tac on FK565‐associated coronary arteritis (Fig. 1) and confirmed that Tac exaggerated the cytokine releases from aortic roots.
CNIs increase the expression of adhesion molecules on endothelial cells
As CNIs did not necessarily enhance the production of cytokines in endothelial cells, we examined the effects of CNIs on the adhesion of leucocytes to endothelial cells in the presence of FK565. First, we performed an endothelial cell–leucocyte adhesion assay using HCAECs and U937 cells (human monocyte‐like cell line) 21. We found that CNIs did not enhance the adhesion between endothelial cells and leucocytes significantly under stimulation with FK565 (data not shown).
Next, to examine whether or not CNIs increase the expression of adhesion molecules on endothelial cells, we stimulated HCAECs with CNIs in the presence or absence of FK565 and analysed the expression of ICAM‐1, VCAM‐1 and E‐selectin by flow cytometry and qRT–PCR. We found that CsA and Tac enhanced FK565‐induced ICAM‐1 expression on HCAECs (Fig. 4a and Supporting information, Fig. S3). In the quantitative real‐time PCR, CsA increased ICAM‐1 and E‐selectin expression in the absence of FK565 (Fig. 4b). Although there were some discrepancies between the expression of ICAM1 mRNA and the cell‐surface ICAM‐1 protein, we interpreted the dissociated results as a sequential time–course of the whole‐cell expression of ICAM1 mRNA in HCAECs, followed by the post‐transcriptional regulation and post‐translational modification of the ICAM1 protein towards its cell‐surface expression. Taken together, these results suggested that CNIs increased the expression of adhesion molecules on vascular endothelial cells, which may lead to the migration of inflammatory cells, such as macrophages, and induce the progression of coronary arteritis.
Figure 4.
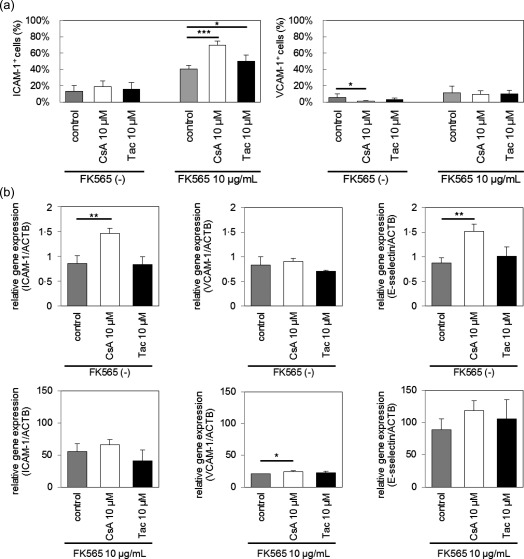
Effect of calcineurin inhibitors (CNIs) and FK565 on adhesion molecule expression on human coronary artery endothelial cells (HCAECs). (a) HCAECs were stimulated with cyclosporin A (CsA) or tacrolimus (Tac) and FK565. The frequencies of intercellular adhesion molecule‐1 (ICAM‐1)‐ and vascular cell adhesion molecule‐1 (VCAM‐1)‐positive cells on HCAECs were measured by fluorescence activated cell sorter (FACS). The data are presented as the mean ± standard deviation (s.d.) (n = 6). (b) HCAECs were stimulated with CsA or Tac and FK565. The mRNA expression of ICAM‐1, VCAM‐1 and E‐selectin in HCAECs was measured by a quantitative real‐time–polymerase chain reaction (PCR) analysis, and were normalized to those of β‐actin. The data are presented as the mean ± s.d. (n = 3). *P < 0·05, **P < 0·01, ***P < 0·001 (Dunnett's test).
The role of CN in the CNI‐induced cytokine production of monocytic cells
To examine the functional role of CN in the CNI‐induced cytokine production of monocytic cells, we employed the Cre‐loxP system to obtain CN‐deficient cells, as CN‐knock‐out was lethal to mouse embryos 22, 23. CD11c‐positive macrophages play a critical role in NOD1‐mediated coronary arteritis 13; therefore, we used CD11c‐positive cell‐specific Cnb1 subunit conditional knock‐out mice (Cd11c‐Cre+Cnb1flox/flox; Cnb1 CKO) 24. First, we ensured that Tac increased TNF production, regardless of the absence or presence of FK565 in WT BMDCs, the majority of which expressed CD11c (Fig. 5a).
Figure 5.
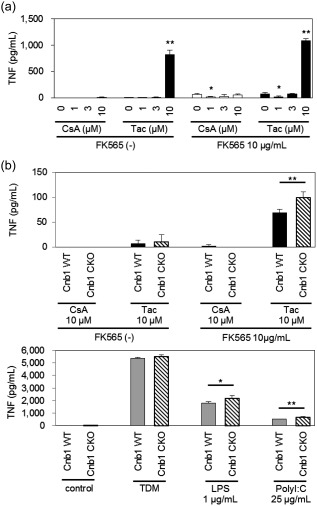
Tumour necrosis factor (TNF) production enhanced by calcineurin inhibitors (CNIs) was calcineurin‐independent. (a) The culture supernatants of bone marrow‐derived dendritic cells (BMDCs) from wild‐type (WT) mice stimulated with cyclosporin A (CsA) or tacrolimus (Tac) in the presence or absence of FK565 were assayed for TNF production. The data are presented as the mean ± standard deviation (s.d.) (n = 4). *P < 0·05; **P < 0·001 compared to the controls (0 μM CsA or Tac) of each group (Dunnett's test). (b) The culture supernatants of BMDCs from Cnb1flox/flox (Cnb1 WT) and Cd11c‐Cre+Cnb1flox/flox (Cnb1 CKO) mice stimulated with CsA or Tac and FK565, and with plate‐coated trehalose‐6,6'‐dimycolate (TDM), lipopolysaccharide (LPS) or PolyI:C as positive controls were assayed for TNF. The data are presented as the mean ± s.d. (n = 4). *P < 0·05 **P < 0·001 (Student's t‐test).
To investigate a possible role of CN in the effects of CNIs, we performed stimulation experiments using BMDCs from Cnb1 CKO and Cnb1flox/flox mice (Cnb1 WT). We found that the production of TNF in Cnb1 CKO mice induced by Tac and FK565 was significantly higher than that in Cnb1 WT mice (Fig. 5b, upper). In addition, the TNF production of CD11c‐positive macrophages from Cnb1 CKO mice after stimulation with LPS (TLR‐4 ligand) and PolyI:C (TLR‐3 ligand) was slightly higher than that in control mice. In contrast, no increase in cytokine production was observed after stimulation with TDM (ligand of Mincle). These results were consistent with the previous finding that CN regulates TLR‐mediated activation pathways negatively 25.
The exacerbation of coronary arteritis by CNIs depends on MyD88
KD is characterized by the activation of the innate immune system 26, 27, 28, 29. A previous report showed that CNIs activated the TLR‐4–MyD88 signal pathway, an innate immune pathway, and increased the production of proinflammatory cytokines, such as TNF, from macrophages 25. MyD88 is the adapter molecule downstream of TLRs. In contrast, CARD9 is a signal transducer of the non‐TLR innate immune pathway. We therefore investigated whether or not a deficiency of MyD88 or CARD9 affected the exacerbation of NOD1‐mediated coronary arteritis by CNIs.
To create the experimental conditions to induce mild coronary arteritis, we optimized the treatment doses of FK565 at 100 μg for WT and CARD9–/– mice and 500 μg for MyD88–/– mice (Fig. 6a and Supporting information, Fig. S1B). In WT and CARD9–/– mice, 0·6 mg/kg of Tac exacerbated NOD1‐mediated coronary arteritis similarly (Fig. 6b–d). In contrast, Tac was unable to exacerbate coronary arteritis in MyD88–/– mice (Fig. 6e–g). As the Nod2 ligand MDP, exacerbated NOD1‐mediated coronary arteritis in WT mice 11, we administered MDP to MyD88–/– mice as a positive control, and the coronary arteritis was exacerbated by MDP as expected (Fig. 6e–g). Therefore, we concluded that CNIs exacerbated NOD1‐mediated coronary arteritis by activating the MyD88‐dependent inflammatory signals, whereas CARD9 was not essential in the exacerbation process.
Figure 6.
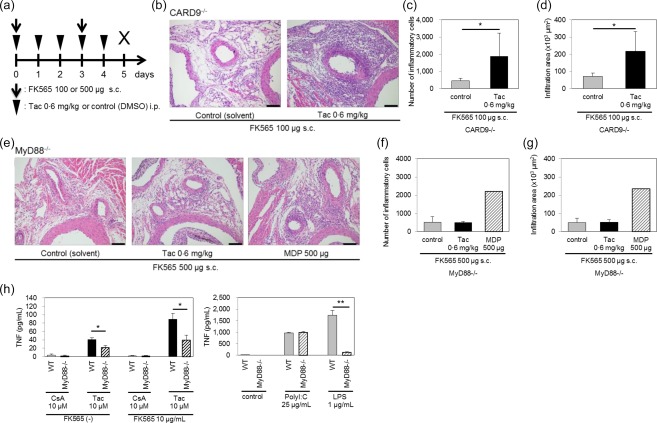
The coronary arteritis exacerbation and macrophage activation by calcineurin inhibitors (CNIs) were myeloid differentiation primary response gene 88 (MyD88)‐dependent. (a) The experimental protocol; subcutaneous injection (s.c.); intraperitoneal injection (i.p.). A histological evaluation of coronary arteritis (b) induced by tacrolimus (Tac) and 100 μg of FK565 in caspase‐associated recruitment domain 9 (CARD9)–/– mice (scale bar, 100 μm). The numbers (c) and infiltration areas (d) of inflammatory cells are shown. The data are presented as the mean ± standard deviation (s.d.) (n = 5). *P < 0·05 (Dunnett's test). A histological evaluation of coronary arteritis (e) induced by Tac or muramyl dipeptide (MDP) and 500 μg of FK565 in MyD88–/– mice (scale bar, 100 μm). The numbers (f) and infiltration areas (g) of inflammatory cells are shown. The data are presented as the mean ± s.d. (control or Tac: n = 3, MDP; n = 1). (h) The culture supernatants of bone marrow‐derived macrophages (BMDMs) from wild‐type (WT) and MyD88–/– mice stimulated with cyclosporin A (CsA) or Tac in the presence or absence of FK565, and with PolyI:C and lipopolysaccharide (LPS) as positive controls were assayed for tumour necrosis factor (TNF). The data are presented as the mean ± s.d. (n = 4). *P < 0·01; **P < 0·001 (Student's t‐test). [Colour figure can be viewed at wileyonlinelibrary.com]
We examined further whether or not a MyD88 deficiency might reduce the macrophage activation by CNIs in vitro. We stimulated BMDMs from MyD88–/– and WT mice with CNIs in the absence or presence of FK565 (Fig. 6h). We confirmed that the TNF production in BMDMs from MyD88–/– mice was not enhanced by LPS but was enhanced by polyI:C (Fig. 6h, right). These data were consistent with the fact that LPS is a stimulator of the TLR‐4–MyD88 signalling pathway, whereas polyI:C is a stimulator of the MyD88‐independent TLR‐3 signalling pathway 30. TNF production by Tac and FK565 was suppressed by 56% in MyD88–/– BMDMs compared with WT BMDMs (P < 0·01), suggesting that the activation of macrophages by CNIs was partially MyD88‐dependent (Fig. 6h, left).
CsA exacerbates coronary arteritis in the presence of LPS
When BMDMs from WT mice were stimulated with various doses of CsA in the absence or presence of LPS in vitro, the TNF production by BMDMs was increased in a dose‐dependent manner (Fig. 7a). These data prompted us to explore in‐vivo evidence for the additive actions of LPS and CsA on coronary arteritis, as LPS was shown previously to enhance NOD1‐mediated coronary arteritis 11. When we administered 10 μg of FK565 and 10 μg of LPS to induce mild coronary arteritis, 4 mg/kg of CsA, which is equivalent to the therapeutic dose in humans (Fig. 1e), enhanced NOD1‐mediated coronary arteritis (Fig. 7b–d). These results therefore suggested that doses of CsA similar to those usually used in humans might enhance coronary arteritis in the presence of innate immune pathogen‐associated molecular patterns such as LPS.
Figure 7.
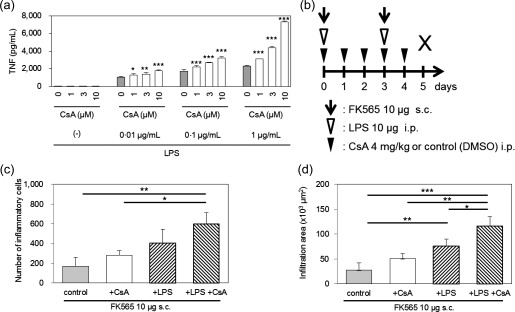
The combination of cyclosporin A (CsA) and lipopolysaccharide (LPS) further activated macrophages and exacerbated coronary arteritis. (a) The culture supernatants of bone marrow‐derived macrophages (BMDMs) from wild‐type (WT) mice stimulated with CsA in the presence or absence of LPS were assayed for tumour necrosis factor (TNF). The data are presented as the mean ± standard deviation (s.d.) (n = 4). *P < 0·05; **P < 0·01; **P < 0·001 compared to controls [0 μM CsA] of each group (Dunnett's test). (b) The experimental protocol. We set the treatment protocol as 10 μg of FK565 for 2 days with or without 4 mg/kg of CsA for 5 consecutive days. This protocol induced only mild coronary arteritis in the WT mice. Injections of LPS (10 μg) for 2 days were added to this mild coronary arteritis‐inducing protocol. The numbers (c) and infiltration areas (d) of inflammatory cells are shown. The data are presented as the mean ± s.d. (n = 3–4). *P < 0·05; **P < 0·01; ***P < 0·001 (Tukey–Kramer test).
Discussion
In this report, we first demonstrated that both CsA and Tac exacerbated NOD1‐mediated coronary arteritis in a mouse model of KD 11. CsA induced the exacerbation of coronary arteritis in mice only in high doses, the trough levels of which exceeded the toxic level in humans. In contrast, Tac exacerbated it even when administered at a dose within the therapeutic range in humans.
CsA and Tac bind to CN and inhibit its kinase activity, and thus regulate the activation of the downstream transcription factor, NFAT 31, 32, 33, 34. Although CNIs are known as potent immunosuppressants, they affect not only T cells but also many other cells, including B cells, neutrophils, macrophages, vascular endothelial cells and smooth muscle cells 35, 36, 37, 38, 39, 40, 41.
In our KD model, vascular cells and cardiac CD11c+ macrophages play a critical role in the pathogenesis of acute coronary arteritis 13. In this study, we also found that the exacerbation of coronary arteritis by CsA and Tac involved both vascular and monocytic cells, but not T cells/B cells, by the experiments using SCID mice. Tac was sufficient to increase the cytokine secretion by vascular tissues ex vivo. Furthermore, both CsA and Tac enhanced the surface expression of adhesion molecules on endothelial cells but not the cytokine secretion from endothelial cells or smooth muscle cells. As NOD1 expression of vascular tissue was not elevated by CNIs (Supporting information, Fig. S4), we speculate that the exacerbation of coronary arteritis due to CNI was not related directly to the change in the expression levels of NOD1.
Further experiments revealed that the cytokine secretion by monocytic cells (macrophages, RAW264.7 cells and dendritic cells) was enhanced by CsA and Tac in a dose‐dependent manner. Controversies exist concerning the effects of CsA and Tac, especially on monocytic cells. Some reports have argued that CsA and Tac inhibit TNF production from monocytic cells 35, 40, 41 and others insist that CsA and Tac enhance cytokine production, such as TNF from macrophages 35, 40, 41. Kang et al. 25 reported that CN regulated the TLR‐mediated activation pathways of macrophages negatively. Their data appear to support ours, as CNIs (CsA and Tac) activated the TLR–MyD88 and nuclear factor (NF)‐κB‐associated pathways, thereby enhancing the TNF production by macrophages. With regard to other cell types, several studies have demonstrated experimental evidence for the enhanced production of inflammatory cytokines by CNIs using fibroblasts 36, aortic endothelial cells 42 and smooth muscle cells 39. Thus, CsA and Tac show either immunosuppressive or immunostimulatory effects, depending on the dose, cell type and interaction with endothelial cells 21, 43, 44.
A recent report showed that CNIs (CsA and Tac) induced endothelial adhesion molecule synthesis and ex‐vivo vascular inflammation by activation of NF‐kΒ/p65, oxidative stress and reactive oxygen species (ROS) in vascular tissue through the TLR‐4–MyD88 signalling pathway 42. We also demonstrated that the exacerbation of coronary arteritis by CNI (Tac) in mice was MyD88‐dependent and CARD9‐independent, using MyD88–/– and Card9–/– mice. Although our findings have overlapped partly with those in the previous report by Rodrigues‐Diez et al. 42, the fundamental distinction between our and their reports is that our conclusion was based on not only ex‐vivo and in‐vitro data, but also in‐vivo experiments. It was useful because we could evaluate the effect of CNIs directly by the analysis of severity of vasculitis, which is accompanied with marked cellular infiltration around the coronary artery. Thus, to our knowledge, our report has first shown the histological exacerbation of coronary arteritis caused by CNIs.
Furthermore, one of the important additional findings is that the combination of CsA and LPS exacerbated coronary arteritis further (Fig. 7). As LPS priming was as effective as LPS‐CsA simultaneous administration in exacerbation of the coronary arteritis (Supporting information, Fig. S5), it was speculated that there might be a higher inflammatory alert status of the cells in terms of the additive effect of LPS on CNI. However, it remains to be determined whether the priming effect was due to the utilization of the same receptor or a general activation of the cells.
The effect of CNI (Tac) on TNF production by dendritic cells was enhanced in the absence of CN, consistent with the negative regulatory effect of CN on the TLR‐mediated activation pathways 25. Another report showed that the proinflammatory activity of CNIs in endothelial cells was partially dependent upon CN 42, whereas other reports showed that CNIs (CsA and Tac) also have CN‐independent effects and do not reproduce the phenotype obtained upon CN gene deletion 45, 46, 47. Thus, it is likely that the CN‐independence of the proinflammatory activity of CNIs varies, depending on the cell type.
CsA exerts anti‐pyretic and anti‐inflammatory effects in most refractory KD patients. However, clinical studies have suggested that CsA at the therapeutic concentration has no strong protective or promotive effect on the development of coronary aneurysms. Suzuki et al. reported that CsA was not able to suppress the progression of CAL in two cases with CAL among 28 IVIG‐refractory cases, and two of 26 cases without CAL developed CAL during CsA treatment 7. Hamada et al. reported that, among 19 IVIG refractory cases who were treated by CsA, one of 14 CsA responders (afebrile within 5 days) and two of five CsA non‐responders developed CAL 9. Tremoulet et al. reported that among 10 IVIG‐resistant KD cases, three of nine CsA‐treated cases and the one Tac‐treated case developed coronary aneurysms 8. Currently, a Phase III multi‐centre, randomized, open‐label, blinded end‐point trial is in progress to evaluate the efficacy and safety of immunoglobulin plus CsA in patients with severe KD (KAICA trial) 48. With the primary end‐point set as the occurrence of CAL, this prospective study will clarify the favourable or unfavourable effects of the CsA on the CAL development in KD patients.
The present study was based mainly on animal and cell experiments, whereas these data might not necessarily reflect the exacerbating effect of CNIs on coronary artery lesions in humans. Taking the above reports 7, 8, 9 into account, we speculate that the results of this study may be related partly to the mechanism of exacerbation of human coronary artery lesions. As even a small amount of CsA can exacerbate coronary arteritis in the presence of other innate immune stimulants such as LPS in mice, it would be safer that the use of CNIs is monitored carefully, especially for severe forms (KD shock syndrome, etc.) of KD patients, who might also be receiving certain innate immune stimulants.
In summary, the present study revealed the exacerbation of coronary arteritis by CNIs in a NOD1‐mediated KD murine model. CNIs appear to exert their effects through the activation of vascular cells and macrophages via the MyD88 pathway (Fig. 8). CsA may still be used in the treatment of refractory KD provided that particular care is taken with respect to the coronary arteries.
Figure 8.
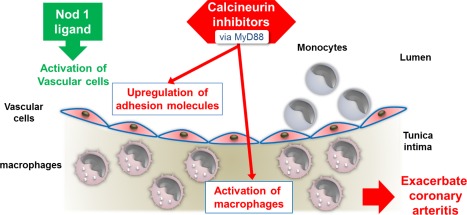
Graphical abstract. A nucleotide‐binding oligomerization domain‐containing protein 1 (NOD1) ligand, FK565, activates vascular cells, which produce large amounts of chemokines. Chemokine receptor‐expressing monocytes in the peripheral blood are recruited to FK565‐activated vascular cells. This process subsequently induces the differentiation of the monocytes into cardiac macrophages, which play a pivotal role in the pathogenesis of acute coronary arteritis. Calcineurin inhibitors exacerbate NOD1‐mediated coronary arteritis in a dose‐dependent manner in a murine model of Kawasaki disease through the activation of vascular cells and macrophages via the myeloid differentiation primary response gene 88 (MyD88) pathway. [Colour figure can be viewed at wileyonlinelibrary.com]
Disclosure
None.
Author contributions
S. Y. and T. H. designed the study; K. M., Y. M., T. T., S. K., T. Y., M. O. A. S. and H. N. performed animal and laboratory experiments; H. T., S. M., K. F., M. O. and H. H. analysed the results and interpreted the data; K. M., H. N. Y. S. and T. H. wrote the paper; S. O. and T. H. supervised the study and scrutinized the paper.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. FK565‐induced coronary arteritis after oral or subcutaneous administration. (a) Histological evaluation of coronary artery on the following day after oral administration of 100 μg of FK565 or distilled H2O (solvent of FK565) for 5 or 7 days (scale bar, 100 μm). (b) Histological evaluation of coronary artery on the following day after subcutaneous administration of 100 or 500 μg of FK565 twice (days 0 and 3) (scale bar, 100 μm).
Fig. S2. No coronary arteritis was induced only by calcineurin inhibitors (CNIs). A histological evaluation of the coronary artery after 5 days' administration of cyclosporin A (CsA), tacrolimus (Tac) and distilled H2O (solvent of FK565) (scale bar, 100 µm).
Fig. S3. Histogram of intercellular adhesion molecule‐1 (ICAM)‐1 and vascular adhesion molecule‐1 (VCAM‐1) expression in human coronary artery endothelial cells (HCAECs). Histograms show flow cytometric analysis of ICAM‐1 and VCAM‐1 expression in HCAECs after stimulation of FK565 with cyclosporin A (CsA) or tacrolimus (Tac). Respective isotype controls are shown as light grey area.
Fig. S4. Effect of calcineurin inhibitors (CNIs) on nucleotide‐binding oligomerization domain‐containing protein 1 (NOD1) expression in human coronary artery endothelial cells (HCAECs). HCAECs were stimulated with cyclosporin A (CsA) or tacrolimus (Tac). NOD1 and TLR4 mRNA expression levels in HCAECs were measured by a quantitative real‐time–PCR analysis, normalized to those of β‐actin, and compared to controls (relative levels in untreated HCAECs). The data are presented as the mean ± standard deviation (s.d.) (n = 3). *P < 0·01 (Dunnett's test).
Fig. S5. Comparison of coronary arteritis exacerbation between simultaneous administration and priming of lipopolysaccharide (LPS). (a) The experimental protocol. We set the treatment protocol as 10 μg of FK565 for 2 days and 4 mg/kg of cyclosporin A (CsA) for 5 consecutive days with or without single dose of LPS (10 μg). LPS was administrated simultaneously with calcineurin inhibitor (CNI)/FK565 or primed 1 day prior to CNI/FK565. (b–d) The histological evaluation of coronary arteritis (b) induced by CsA and FK565 with LPS (simultaneous or priming treatment) in wild‐type (WT) mice (scale bar, 100 μm). The numbers (c) and infiltration areas (d) of inflammatory cells are shown. The data are presented as the mean ± standard deviation (s.d.) (n = 3). *P < 0·05; n.s.: not significant (Tukey–Kramer test).
Preparation of FK565
Acknowledgements
We thank Hiroshi Fujii, Department of Pathology, Kyushu University for their technical assistance, Junji Kishimoto, Department of Research and Development of Next Generation Medicine, Faculty of Medical Sciences, Kyushu University for help with the statistical analyses, Brian Quinn, Editor‐in‐Chief of Japan Medical Communication, for English editing, and Takashi Imai and Kenji Toyonaga, Division of Molecular Immunology, Research Center for Infectious Diseases, Medical Institute of Bioregulation, Kyushu University for engaging in discussion and providing mice. This work was supported by the elucidation of the pathogenesis and pathophysiology of KD and the development of its novel therapy from Japan Agency for Medical Research and development (AMED), grants from the Japan Society for Promotion of Science, the Health and Labour Sciences Research Grants from the Japanese Ministry of Health, Labour and Welfare, a grant for Kawasaki Disease Research from Japan Blood Products Organization, and MEXT KAKENHI, Grant number 16K19652.
References
- 1. Kawasaki T, Kosaki F, Okawa S, Shigematsu I, Yanagawa H. A new infantile acute febrile mucocutaneous lymph node syndrome (MLNS) prevailing in Japan. Pediatrics 1974; 54:271–6. [PubMed] [Google Scholar]
- 2. Burns JC, Capparelli EV, Brown JA, Newburger JW, Glode MP. Intravenous gamma‐globulin treatment and retreatment in Kawasaki disease. Pediatr Infect Dis J 1998; 17:1144–8. [DOI] [PubMed] [Google Scholar]
- 3. Durongpisitkul K, Soongswang J, Laohaprasitiporn D, Nana A, Prachuabmoh C, Kangkagate C. Immunoglobulin failure and retreatment in Kawasaki disease. Pediatr Cardiol 2003; 24:145–8. [DOI] [PubMed] [Google Scholar]
- 4. Uehara R, Belay ED, Maddox RA et al Analysis of potential risk factors associated with nonresponse to initial intravenous immunoglobulin treatment among Kawasaki disease patients in Japan. Pediatr Infect Dis J 2008; 27:155–60. [DOI] [PubMed] [Google Scholar]
- 5. Makino N, Nakamura Y, Yashiro M et al Descriptive epidemiology of Kawasaki disease in Japan, 2011–2012: from the results of the 22nd nationwide survey. J Epidemiol 2015; 25:239–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuijpers TW, Biezeveld M, Achterhuis A et al Longstanding obliterative panarteritis in Kawasaki disease: lack of cyclosporin A effect. Pediatrics 2003; 112:986–92. [DOI] [PubMed] [Google Scholar]
- 7. Suzuki H, Terai M, Hamada H et al Cyclosporin A treatment for Kawasaki disease refractory to initial and additional intravenous immunoglobulin. Pediatr Infect Dis J 2011; 30:871–6. [DOI] [PubMed] [Google Scholar]
- 8. Tremoulet AH, Pancoast P, Franco A et al Calcineurin inhibitor treatment of intravenous immunoglobulin‐resistant Kawasaki disease. J Pediatr 2012; 161:506–12 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hamada H, Suzuki H, Abe J et al Inflammatory cytokine profiles during cyclosporin treatment for immunoglobulin‐resistant Kawasaki disease. Cytokine 2012; 60:681–5. [DOI] [PubMed] [Google Scholar]
- 10. Okada S, Azuma Y, Suzuki Y et al Adjunct cyclosporine therapy for refractory Kawasaki disease in a very young infant. Pediatr Int 2016; 58:295–8. [DOI] [PubMed] [Google Scholar]
- 11. Nishio H, Kanno S, Onoyama S et al NOD1 ligands induce site‐specific vascular inflammation. Arterioscler Thromb Vasc Biol 2011; 31:1093–9. [DOI] [PubMed] [Google Scholar]
- 12. Takahashi K, Oharaseki T, Naoe S, Wakayama M, Yokouchi Y. Neutrophilic involvement in the damage to coronary arteries in acute stage of Kawasaki disease. Pediatr Int 2005; 47:305–10. [DOI] [PubMed] [Google Scholar]
- 13. Motomura Y, Kanno S, Asano K et al Identification of pathogenic cardiac CD11c+ macrophages in NOD1‐mediated acute coronary arteritis. Arterioscler Thromb Vasc Biol 2015; 35:1423–33. [DOI] [PubMed] [Google Scholar]
- 14. Hara H, Ishihara C, Takeuchi A et al The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM‐associated and Toll‐like receptors. Nat Immunol 2007; 8:619–29. [DOI] [PubMed] [Google Scholar]
- 15. Hosohata K, Uesugi M, Hashi S et al Association between CYP3A5 genotypes in graft liver and increase in tacrolimus biotransformation from steroid treatment in living‐donor liver transplant patients. Drug Metab Pharmacokinet 2014; 29:83–9. [DOI] [PubMed] [Google Scholar]
- 16. Ishikawa T, Itoh F, Yoshida S et al Identification of distinct ligands for the C‐type lectin receptors Mincle and Dectin‐2 in the pathogenic fungus Malassezia. Cell Host Microbe 2013; 13:477–88. [DOI] [PubMed] [Google Scholar]
- 17. Ohkubo K, Sakai Y, Inoue H et al Moyamoya disease susceptibility gene RNF213 links inflammatory and angiogenic signals in endothelial cells. Sci Rep 2015; 5:13191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. David‐Neto E, Araujo LM, Brito ZM et al Sampling strategy to calculate the cyclosporin‐A area under the time‐concentration curve. Am J Transplant 2002; 2:546–50. [DOI] [PubMed] [Google Scholar]
- 19. Hooper DK, Fukuda T, Gardiner R et al Risk of tacrolimus toxicity in CYP3A5 nonexpressors treated with intravenous nicardipine after kidney transplantation. Transplantation 2012; 93:806–12. [DOI] [PubMed] [Google Scholar]
- 20. Ragette R, Kamler M, Weinreich G, Teschler H, Jakob H. Tacrolimus pharmacokinetics in lung transplantation: new strategies for monitoring. J Heart Lung Transplant 2005; 24:1315–9. [DOI] [PubMed] [Google Scholar]
- 21. Rafiee P, Johnson CP, Li MS et al Cyclosporine A enhances leukocyte binding by human intestinal microvascular endothelial cells through inhibition of p38 MAPK and iNOS. Paradoxical proinflammatory effect on the microvascular endothelium. J Biol Chem 2002; 277:35605–15. [DOI] [PubMed] [Google Scholar]
- 22. Mesaeli N, Nakamura K, Zvaritch E et al Calreticulin is essential for cardiac development. J Cell Biol 1999; 144:857–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Papp S, Dziak E, Opas M. Embryonic stem cell‐derived cardiomyogenesis: a novel role for calreticulin as a regulator. Stem Cells 2009; 27:1507–15. [DOI] [PubMed] [Google Scholar]
- 24. Neilson JR, Winslow MM, Hur EM, Crabtree GR. Calcineurin B1 is essential for positive but not negative selection during thymocyte development. Immunity 2004; 20:255–66. [DOI] [PubMed] [Google Scholar]
- 25. Kang YJ, Kusler B, Otsuka M et al Calcineurin negatively regulates TLR‐mediated activation pathways. J Immunol 2007; 179:4598–607. [DOI] [PubMed] [Google Scholar]
- 26. Foell D, Ichida F, Vogl T et al S100A12 (EN‐RAGE) in monitoring Kawasaki disease. Lancet 2003; 361:1270–2. [DOI] [PubMed] [Google Scholar]
- 27. Popper SJ, Shimizu C, Shike H et al Gene‐expression patterns reveal underlying biological processes in Kawasaki disease. Genome Biol 2007; 8:R261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ikeda K, Yamaguchi K, Tanaka T et al Unique activation status of peripheral blood mononuclear cells at acute phase of Kawasaki disease. Clin Exp Immunol 2010; 160:246–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hara T, Nakashima Y, Sakai Y, Nishio H, Motomura Y, Yamasaki S. Kawasaki disease: a matter of innate immunity. Clin Exp Immunol 2016; 186:134–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Akira S, Takeda K. Toll‐like receptor signalling. Nat Rev Immunol 2004; 4:499–511. [DOI] [PubMed] [Google Scholar]
- 31. Clipstone NA, Crabtree GR. Identification of calcineurin as a key signalling enzyme in T‐lymphocyte activation. Nature 1992; 357:695–7. [DOI] [PubMed] [Google Scholar]
- 32. Wallemacq PE, Reding R. FK506 (tacrolimus), a novel immunosuppressant in organ transplantation: clinical, biomedical, and analytical aspects. Clin Chem 1993; 39:2219–28. [PMC free article] [PubMed] [Google Scholar]
- 33. Ruhlmann A, Nordheim A. Effects of the immunosuppressive drugs CsA and FK506 on intracellular signalling and gene regulation. Immunobiology 1997; 198:192–206. [DOI] [PubMed] [Google Scholar]
- 34. Steinbach WJ, Reedy JL, Cramer RA Jr, Perfect JR, Heitman J. Harnessing calcineurin as a novel anti‐infective agent against invasive fungal infections. Nat Rev Microbiol 2007; 5:418–30. [DOI] [PubMed] [Google Scholar]
- 35. Svensson U, Holst E, Sundler R. Cyclosporin‐sensitive expression of cytokine mRNA in mouse macrophages responding to bacteria. Mol Immunol 1995; 32:157–65. [DOI] [PubMed] [Google Scholar]
- 36. Conboy IM, Manoli D, Mhaiskar V, Jones PP. Calcineurin and vacuolar‐type H+‐ATPase modulate macrophage effector functions. Proc Natl Acad Sci USA 1999; 96:6324–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev 2000; 80:1483–521. [DOI] [PubMed] [Google Scholar]
- 38. Jeanmart H, Malo O, Carrier M, Nickner C, Desjardins N, Perrault LP. Comparative study of cyclosporine and tacrolimus vs newer immunosuppressants mycophenolate mofetil and rapamycin on coronary endothelial function. J Heart Lung Transplant 2002; 21:990–8. [DOI] [PubMed] [Google Scholar]
- 39. Murakami R, Kambe F, Mitsuyama H et al Cyclosporin A enhances interleukin‐8 expression by inducing activator protein‐1 in human aortic smooth muscle cells. Arterioscler Thromb Vasc Biol 2003; 23:2034–40. [DOI] [PubMed] [Google Scholar]
- 40. Yoshino T, Nakase H, Honzawa Y et al Immunosuppressive effects of tacrolimus on macrophages ameliorate experimental colitis. Inflamm Bowel Dis 2010; 16:2022–33. [DOI] [PubMed] [Google Scholar]
- 41. Lopez‐Flores R, Bojalil R, Benitez JC et al Consecutive low doses of cyclosporine A induce pro‐inflammatory cytokines and accelerate allograft skin rejection. Molecules 2011; 16:3969–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rodrigues‐Diez R, Gonzalez‐Guerrero C, Ocana‐Salceda C et al Calcineurin inhibitors cyclosporine A and tacrolimus induce vascular inflammation and endothelial activation through TLR4 signaling. Sci Rep 2016; 6:27915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Escolano A, Martinez‐Martinez S, Alfranca A et al Specific calcineurin targeting in macrophages confers resistance to inflammation via MKP‐1 and p38. EMBO J 2014; 33:1117–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Badiwala MV, Guha D, Tumiati L et al Epidermal growth factor‐like domain 7 is a novel inhibitor of neutrophil adhesion to coronary artery endothelial cells injured by calcineurin inhibition. Circulation 2011; 124:S197–203. [DOI] [PubMed] [Google Scholar]
- 45. Kanoh S, Kondo M, Tamaoki J, Kobayashi H, Motoyoshi K, Nagai A. FK506 inhibits Cl‐ secretion in airway epithelium via calcineurin‐independent mechanism. Eur J Pharmacol 2001; 419:121–6. [DOI] [PubMed] [Google Scholar]
- 46. Childs EW, Tharakan B, Nurudeen S et al Cyclosporine A–protection against microvascular hyperpermeability is calcineurin independent. Am J Surg 2010; 199:542–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sachewsky N, Hunt J, Cooke MJ et al Cyclosporin A enhances neural precursor cell survival in mice through a calcineurin‐independent pathway. Dis Model Mech 2014; 7:953–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Aoyagi R, Hamada H, Sato Y et al Study protocol for a phase III multicentre, randomised, open‐label, blinded‐end point trial to evaluate the efficacy and safety of immunoglobulin plus cyclosporin A in patients with severe Kawasaki disease (KAICA Trial). BMJ Open 2015; 5:e009562. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. FK565‐induced coronary arteritis after oral or subcutaneous administration. (a) Histological evaluation of coronary artery on the following day after oral administration of 100 μg of FK565 or distilled H2O (solvent of FK565) for 5 or 7 days (scale bar, 100 μm). (b) Histological evaluation of coronary artery on the following day after subcutaneous administration of 100 or 500 μg of FK565 twice (days 0 and 3) (scale bar, 100 μm).
Fig. S2. No coronary arteritis was induced only by calcineurin inhibitors (CNIs). A histological evaluation of the coronary artery after 5 days' administration of cyclosporin A (CsA), tacrolimus (Tac) and distilled H2O (solvent of FK565) (scale bar, 100 µm).
Fig. S3. Histogram of intercellular adhesion molecule‐1 (ICAM)‐1 and vascular adhesion molecule‐1 (VCAM‐1) expression in human coronary artery endothelial cells (HCAECs). Histograms show flow cytometric analysis of ICAM‐1 and VCAM‐1 expression in HCAECs after stimulation of FK565 with cyclosporin A (CsA) or tacrolimus (Tac). Respective isotype controls are shown as light grey area.
Fig. S4. Effect of calcineurin inhibitors (CNIs) on nucleotide‐binding oligomerization domain‐containing protein 1 (NOD1) expression in human coronary artery endothelial cells (HCAECs). HCAECs were stimulated with cyclosporin A (CsA) or tacrolimus (Tac). NOD1 and TLR4 mRNA expression levels in HCAECs were measured by a quantitative real‐time–PCR analysis, normalized to those of β‐actin, and compared to controls (relative levels in untreated HCAECs). The data are presented as the mean ± standard deviation (s.d.) (n = 3). *P < 0·01 (Dunnett's test).
Fig. S5. Comparison of coronary arteritis exacerbation between simultaneous administration and priming of lipopolysaccharide (LPS). (a) The experimental protocol. We set the treatment protocol as 10 μg of FK565 for 2 days and 4 mg/kg of cyclosporin A (CsA) for 5 consecutive days with or without single dose of LPS (10 μg). LPS was administrated simultaneously with calcineurin inhibitor (CNI)/FK565 or primed 1 day prior to CNI/FK565. (b–d) The histological evaluation of coronary arteritis (b) induced by CsA and FK565 with LPS (simultaneous or priming treatment) in wild‐type (WT) mice (scale bar, 100 μm). The numbers (c) and infiltration areas (d) of inflammatory cells are shown. The data are presented as the mean ± standard deviation (s.d.) (n = 3). *P < 0·05; n.s.: not significant (Tukey–Kramer test).
Preparation of FK565