

Abstract
Fungal glucosylceramide (GlcCer) is a plasma membrane sphingolipid in which the sphingosine backbone is unsaturated in carbon position 8 (C8) and methylated in carbon position 9 (C9). Studies in the fungal pathogen, Cryptococcus neoformans, have shown that loss of GlcCer synthase activity results in complete loss of virulence in the mouse model. However, whether the loss of virulence is due to the lack of the enzyme or to the loss of the sphingolipid is not known. In this study, we used genetic engineering to alter the chemical structure of fungal GlcCer and studied its effect on fungal growth and pathogenicity. Here we show that unsaturation in C8 and methylation in C9 is required for virulence in the mouse model without affecting fungal growth in vitro or common virulence factors. However, changes in GlcCer structure led to a dramatic susceptibility to membrane stressors resulting in increased cell membrane permeability and rendering the fungal mutant unable to grow within host macrophages. Biophysical studies using synthetic vesicles containing GlcCer revealed that the saturated and unmethylated sphingolipid formed vesicles with higher lipid order that were more likely to phase separate into ordered domains. Taken together, these studies show for the first time that a specific structure of GlcCer is a major regulator of membrane permeability required for fungal pathogenicity.
Keywords: Sphingolipid, fungi, infectious disease, plasma membrane, Cryptococcus, glucosylceramide
Graphical Abstract
1. INTRODUCTION
Cryptococcus species are environmental fungi that are capable of causing life-threatening neurological infections. Cryptococcal cells are able to survive and replicate at both neutral and acidic pH environments, making them capable of living both intracellularly, inside the phagolysosome of infected macrophages, and extracellularly, in the bloodstream (1). Invasive cryptococcal infections, primarily caused by Cryptococcus neoformans and Cryptococcus gattii, can lead to fungal meningitis, which particularly threatens HIV/AIDS patients. Recent studies have shown that sphingolipid pathways play a prominent role in regulating the virulence of cryptococcal species. Specific sphingolipid metabolizing enzymes regulate cryptococcal growth at acidic and neutral pH, thereby affecting cryptococcal virulence (2–4). In addition to C. neoformans, sphingolipids play a role in the pathogenicity of other fungi. Sphingolipids regulate hyphal growth and elongation in Candida albicans (5,6) and play an important role in germination and hyphal growth of Aspergillus fumigatus (7).
GlcCer is a sphingolipid primarily localized in the cell membrane (8). Studies using an engineered C. neoformans strain lacking GlcCer synthase 1 (Δgcs1), and thus GlcCer, have shown that this mutant is not capable of causing disease because it cannot grow at neutral/alkaline pH at physiologically relevant CO2 concentrations (2). The native GlcCer structure in fungi is a 9-methyl-4,8-sphingadienine in an amide linkage to 2-hydroxyoctadecanoic acid (9,10). Compared to mammalian GlcCer, fungal GlcCer is unsaturated in position 8 and methylated in position 9 of the sphingosine backbone. In previous studies, a mutant C. neoformans lacking the enzyme that methylates ceramide (Δsmt1), and thus accumulating unmethylated GlcCer, showed a partial growth defect at neutral/alkaline pH at physiologically relevant CO2 concentrations and a significant attenuation in its ability to establish virulence in the mouse model (11). These studies suggest a connection between GlcCer structure and cryptococcal virulence, the exact mechanisms of which are yet unknown.
In this study, we deleted the enzyme that desaturates ceramide in position 9 of the sphingosine backbone and studied the effect of this mutation on cryptococcal virulence. The biophysical properties of synthetic membranes containing purified GlcCer, or total lipid extracts from the wild-type, Δsmt1, and Δsld8 mutants were also studied. It was observed that a cryptococcal strain that accumulates GlcCer with a saturated sphingosine backbone at position 8 (Δsld8) completely loses its ability to establish virulence in the mouse model without showing a defect in its growth profile or common virulence factors. An increased sensitivity to membrane stressors and increased permeability of the plasma membrane was also observed resulting in a significant defect in intracellular growth and fungal pathogenicity. Biophysical characterization of vesicles containing purified wild-type or mutated forms of GlcCer revealed changes in membrane biophysical properties of synthetic vesicles. These observations demonstrate that the chemical structure of GlcCer is an important regulator of fungal pathogenicity and changes in GlcCer chemical structure alter membrane physical properties.
2. EXPERIMENTAL PROCEDURES
2.1. Strains, plasmids and growth conditions
C. neoformans var. grubii H99 (serotype A), also referred to as wild-type (WT) (kind gift from Dr. John Perfect, Duke University), was used in this study to generate the mutant strains. The Δsmt1 strain and its reconstruct were engineered from this strain as previously described (11). The Δsld8 strain was generated using the same wild-type strain, and SLD8 was reconstituted back into the Δsld8 background (sld8+SLD8). Escherichia coli strain DH5α (Invitrogen, Carlsbad, CA) was used for all plasmid propagations. Plasmids pCR 2.1- TOPO (Invitrogen) and pBluescript SKII (Stratagene, San Diego, CA) were used as recipient vectors for the sld8 deletion and reconstitution cassettes, respectively. Yeast Peptone Dextrose (YPD) (BD Biosciences, San Jose, CA) was used for routine propagation of strains at 30 °C. YPD supplemented with nourseothricin or hygromycin was used to select for transformants (see sections 2 and 3). Yeast Nitrogen Base (YNB) containing all amino acids and 2% dextrose (BD), buffered with HEPES or succinic acid (Sigma-Aldrich, St. Louis, MO) was used for spot assays. Dulbecco’s Modified Eagle Media (DMEM) buffered with HEPES at pH 4.0 and 7.4 was used for monitoring growth curve. Laura-Bertani (LB) medium supplemented with 50 μg/mL ampicillin (Sigma) was used for plasmid propagation.
2.2. Deletion of SLD8
The SLD8 gene locus (CNAG_03870.2) is located on chromosome 2. The deletion cassette was generated as described in Supplementary Figure 2A, containing 922 bp of the 5′ untranslated and 889 bp of the 3′ untranslated regions. H99 genomic DNA was used as the template for PCR amplification of the 5′UTR and 3′UTR. Amplified products were individually cloned into the pCR 2.1 vector (Invitrogen) following the manufacturer’s instructions and confirmed by restriction enzyme and sequence analyses. The 5′ UTR was then released by BamHI and SpeI digestion and cloned upstream of the nourseothricin acetyl transferase 1 gene conferring resistance to the antibiotic nourseothricin (Werner BioAgents, Germany), in pTOPO (pTOPO-NAT1).(12,13) Similarly, the 3′UTR was released by EcoRV and XhoI digestion and introduced downstream of the NAT1 gene in the pTOPO-5′UTR-NAT1 vector to generate the final vector pΔsld8. The wild-type strain was used as a recipient for biolistic delivery of pΔsld8(14) and transformants were selected on YPD plates containing 100 μg/mL nourseothricin. Resulting transformants were picked and purified by serial passage over YPD nourseothricin plates. Genomic DNA was isolated from the transformants and southern blot analysis was carried out using probes specific for either the 5′UTR, NAT1, or SLD8 ORF.(15) Transformant 61, showing the deletion of the SLD8 ORF and the presence of the NAT1, was named the Δsld8 strain and selected for future analyses.
2.3. Reconstitution of SLD8
SLD8 was reconstituted back into its locus in the Δsld8 strain. A 3909 bp fragment of SLD8 containing the 2128 bp of the ORF and 1383 bp of the 5′ and 398 bp of the 3′ UTR was amplified from genomic DNA and introduced into pSK vector carrying the hygromycin resistance gene under the cryptococcal actin promoter to generate pSK-SLD8-HYG. This was biolistically delivered into the Δsld8 strain and transformants were selected on YPD + 200 μg/mL hygromycin plates. Genomic DNA was extracted and southern hybridization was carried out with SLD8 and HYG1 probes. Reconstituted mutants 2, 5, and 51 were analyzed by mass spectrometry and mutant 2 was designated as the Δsld8+SLD8 strain.
2.4. Lipid extraction and mass spectrometry analysis
Sphingolipids were extracted as described earlier (15). YPD cultures were set up from single colonies of WT, Δsld8 and Δsld8+SLD8 strains and grown at 30 °C for 22 hours. Cells were washed with sterile water, counted and 5×108 cells were taken per tube. Lipids were extracted following the methods of Mandala (16) and Bligh and Dyer (17). Aliquots were taken out for inorganic phosphate analysis. Mass Spectroscopy analyses were carried out on the Thermo Finnigan TSQ7000 triple quadrupole mass spectrometer with electrospray ionization as described (18).
2.5. Metabolic labeling of lipids
YPD cultures of WT, Δsld8, Δsld8+SLD8 were counted and resuspended to 5×108 cells per tube in the overnight conditioned media. Thirty μCi of 3H-palmitate was added to each tube and tubes were incubated for 2 hours at 30 °C. Lipid extractions were carried out as described in the previous section and the dried lipids were subjected to alkaline hydrolysis. Vacuum dried extracts were resuspended in 2:1 (v:v) chloroform/methanol mixture and resolved by thin layer chromatography against a soy glucosylceramide standard (Avanti Polar Lipids, Alabaster, AL) in a 173:75:13 (v:v) mixture of chloroform/methanol/water solvent. The plate was dried to evaporate the solvent, placed in a cassette and exposed to film.
2.6. In Vitro growth assay
Overnight cultures of WT, Δsld8, Δsld8+SLD8 were grown in YNB at 30 °C. Cultures were washed twice with sterile water, counted and diluted to104 cells/mL in DMEM pH 4.0 and pH 7.4 buffered with 50 mM HEPES. Aliquots of 200 μL were placed in a 96-well plate in quadruplicates and the plate was incubated at 37 °C in the presence of 5% CO2. At designated time points, 100 μL aliquots were withdrawn, serially diluted and plated onto YPD agar plates. Colony forming units (CFU) were enumerated after 48 hours of incubation and used to plot the growth curve.
2.7. Sytox green uptake assay
The Sytox green uptake assay was performed as previously described (11). Briefly, cells were grown in YNB at pH 4.0 or 7.4 in a shaker incubator at 37 °C in the presence of 5% CO2 for 20 hours. Cells were washed and resuspended in the same media at a concentration of 108 cells/mL. Hundred microliters of this suspension was added to a 96 well plate containing equal amount of YNB in the presence or absence of NO and H2O2. After 2 hours cells were pelleted and medium was decanted. Sytox green (2 mM) in PBS was added to each cell and incubated in the dark for 10 minutes. Fluorescence was measured using a Spectrofluorometer (Perkin Elmer, Waltham, MA) at an excitation wavelength 488 nm and emission wavelength of 535 nm. Fluorescence of the medium was taken as background and subtracted from the measured values.
2.8. Spot assay
Overnight cultures of WT, Δsld8, Δsld8+SLD8 and Δgcs1 were set up in either YPD or YNB, washed and counted. Five microliters containing 10-fold dilutions of cells starting from 105 cells were spotted onto YPD plates containing 0.05% SDS or 0.025% Triton X-100. Plates were incubated at 37 °C in the presence of 5% CO2 and growth was monitored for 48 h.
2.9. Virulence studies in a mouse model of cryptococcosis
Four-week old female CBA/J mice (Jackson Laboratories, Bar Harbor, Maine) were used for virulence studies. Ten mice were used per strain. Mice were anaesthetized with intraperitoneal injections of 60 μL ketamine-xylazine mixture containing 95 mg ketamine and 5 mg xylazine per kg of body weight. Strains WT, Δsld8 and Δsld8+SLD8 were grown for 20 hours in YPD at 30 °C under shaking conditions. Cultures were washed twice with PBS, counted and diluted to 2.5×107 cells/mL in PBS. Anaesthetized mice were intranasally inoculated with 20 μL (5×105 cells) of each strain. Mice were fed ad libitum and their survival was monitored on a daily basis. On signs of sickness, mice were sacrificed by CO2 inhalation followed by cervical dislocation. All procedures were approved by Stony Brook University Institutional Animal Care and Use Committee and followed the guidelines of the American Veterinary Medical Association.
2.10. Tissue burden and histological analysis
The 10 mice that survived infection with the Δsld8 strain were used for tissue burden and histological analyses 90 days post-infection. Major organs (lungs, brains, livers, kidneys, and spleens) were extracted from five mice and homogenized in 10 mL PBS using a Stomacher 80 (Seward, United Kingdom) for two minutes at high setting. The homogenates were serially diluted and dilutions were plated on YPD. Plates were incubated at 30 °C for 48 hours and colonies were enumerated. For histological analyses, lungs and brains were also extracted from the other five mice, placed in cassettes and fixed for 24 h in 10 % formalin. Cassettes were then transferred into PBS and stained with haematoxylin and eosin to visualize the host inflammatory response (19), and mucicarmine to stain the C. neoformans capsule (20).
2.11. Quantification of secreted GXM
For analysis of GXM secretion, one single colony of each strain was grown in a defined minimal media (29.4 mM KH2PO4, 10 mM MgSO4, 13mM glycine, 15 mM dextrose, and 3 μM thiamine-HCl) for 24 h or 48 h in each temperature: 30 °C, 37 °C, 37 °C with 5% of CO2 atmosphere. At each time point, an aliquot was taken and GXM quantification was done by ELISA as described by Casadevall and colleagues (21). Briefly, the supernatants were diluted 1:50 and 1:100 in a 96-well plate and incubated for 1h at 37 °C. The wells were blocked with PBS-BSA 1% for 1 h at 37 °C and then coated with 1 μg/mL of monoclonal antibody 18B7 (in PBS-BSA 1%) for 1h at 37 °C. The plate was then washed with PBS-Tween three times and coated with 50 μL of anti-mouse-horseradish peroxidase (HRP) (1:5,000 in PBS-BSA 1%). After incubation at 37 °C for 1h, liquids were discarded and the wells were washed with PBS-Tween three times. Fifty microliters of 3,3′,5,5′-Tetramethylbenzidine (TMB) was then added. Reaction was stopped by adding 50 μL sulfuric acid (0.16 M) to each well and readings were performed at 450 nm.
2.12. Urease activity
The rapid urease broth was used as described by Roberts and colleagues(22) with modifications. The media was prepared 2× concentrated with 4 g urea, 0.02 g of yeast extract, 2 mg phenol red, 0.273 g KH2PO4, 0.285 g Na2HPO4. The media was dissolved in 100 mL of distilled water, pH adjusted to 6.8, and filtered. To check the urease activity, a full loop of yeast cells grown for 48 h at 30 °C on YPD was suspended in 2 mL of distilled water. One mL of cells (concentration of 108 cell/mL) in water was added to 1 mL of 2× urea broth and incubated at 37 °C with shaking for 1, 3, 4, and 6 hours. The magenta red color was considered a positive reaction and orange-yellow color was considered a negative reaction. The absorbance was measured at 570 nm every hour and readings over 0.3 were considered positive (23).
2.13. C. neoformans pigmentation assay
The melanization assay was performed in minimal medium agar (15 mM glucose, 10 mM MgSO4, 29.4 mM KH2PO4, 13 mM glycine, and 3.0 μM thiamine-HCl plus 2% agar) with addition of 1 mM of L-3,4-dihydroxyphenylalanine (L-DOPA). Fungal cells were grown in YPD broth for 24 h, adjusted to different cell concentrations (107, 106, 105, and 104 cell/mL) and then plated on minimal media agar plates at 30 °C, 37 °C, 37 °C with 5% CO2 for 72 h. After this time, the plates with melanized cells were photographed. Laccase activity was measured by monitoring the oxidation of 2,2′-azinodi-3-ethyl-benzothiazoline-6-sulfuric acid (ABTS) at 450 nm and quantified using Enzymatic Units (EU) (24). The reaction mixture contained 1 mM ABTS, 0.5 mM sodium acetate (pH 5.0), and 107 cell/mL of each strain in water bath at 37 °C. At each time point, absorbance was measured at 4503nm in a spectrophotometer against a suitable blank.
2.14. Calcofluor White staining
The Calcofluor White stain containing 1 g/L Calcofluor White and 0.5 g/L Evans blue was purchased from Sigma-Aldrich (St. Louis, MO) and staining was performed according to the manufacturer’s instructions. Briefly, cryptococcal cells were grown overnight, washed in sterile PBS, and then placed on a clear glass slide. One drop of Calcofluor White Stain and one drop of 10% Potassium Hydroxide was added to the cell suspension and allowed to incubate for approximately one minute. Imaging was performed using an upright confocal fluorescence microscope (Zeiss LSM 510, Zeiss, Thornwood, NY).
2.15. Measurement of phospholipase activity
Lysophospholipase (LPL) activity assay was performed by monitoring the rate of loss of radiolabelled 1,2-di[1-14C]palmitoyl-phosphatidylcholine (PC) (30,000 dpm) or dipalmitoyl-[2-palmitoyl-9,10-3H(N)] PC (30,000 dpm) after the addition of culture supernatant. Lysophospholipase transacylase (LPTA) activity assay was performed by measuring the rate of radioblabelled PC production from 1-[14C]palmitoyl-lyso-PC. Both assays were performed according to the methods of Chen and colleagues (25) with no modifications.
2.16. GlcCer purification
GlcCer purification was performed according to the methods of Rodrigues and colleagues with minor modifications (9). Briefly, the wild-type, Δsmt1, and Δsld8 strains were grown in 1.5 liters YPD (5 flasks of 300 mL each). The cells were washed and counted and their lipids were extracted following the methods of Mandala and Bligh and Dyer. Extracted lipids were then dissolved in 4 mL of chloroform:acetic acid (99:1 (v:v)) and ran through a 12 cc, silica, Sep-Pak column (Waters, Milford, MA), which was prewashed with 90 mL of chloroform. After running the sample, the column was eluted with 60 mL of acetone. The acetone containing lipids of interest was dried in a SpeedVac (Savant™ SPD 2010 Speed Vac, ThermoScientific, Pittsburgh, PA) and was subject to base hydrolysis and then redissolved in 4 mL of chloroform:acetic acid (99:1) and ran through another pre-washed, 12 cc, silica, Sep-Pak column. The column was eluted sequentially with the following mixtures: chloroform-methanol (99:1, 95:5, 90:10 (v:v)), and methanol alone. Purified GlcCer fraction was obtained in the chloroform-methanol 9:1 (v:v) fraction and its purity and structure was confirmed with TLC and mass spectrometry.
2.17. Vesicle preparation
Multilamellar vesicles (MLVs) or large unilamellar vesicles were prepared according to the methods of Pathak and London (26). Briefly, lipids from each solution were pipetted into glass tubes and dried under nitrogen. To ensure complete mixing, the dried lipids were redissolved in 20 μL chloroform and redried under nitrogen. Lipids were then further dried under high vacuum for 2 hours, and dispersed in 70 °C phosphate-buffered saline (PBS, pH 7.4). Final samples contained 100 μM lipids. Large unilamellar vesicles (LUVs) were prepared by subjecting MLVs to freeze-thawing and passing through a mini-extruder (Avanti Polar Lipids).
2.18. Fluorescence anisotropy
DPH fluorescence anisotropy measurements were made using a SPEX automated Glan-Thompson polarizer accessory (Horiba Scientific, Edison, NJ). Anisotropy values were calculated according to the methods of Bakht and colleagues.(27) Anisotropy as a function of temperature was measured for MLV samples containing 0.1 mol % DPH and 100 μM lipid. The samples were incubated at room temperature for 1 hour and then cooled to 16 °C. Samples were then heated in steps of approximately 4 °C and anisotropy was measured at each step once the temperature stabilized.
2.19. Forster resonance energy transfer (FRET)
FRET experiments were performed according to the methods of Pathak and London (26). DPH 0.1 mol% was used as donor while rhodamine-DOPE 2 mol% was used as acceptor in F samples. The F0 samples contained only unlabeled lipids and FRET donor. Background samples for F0 (containing only unlabeled lipid) and for F (containing unlabeled lipid plus FRET acceptor) samples were also prepared. Samples were prepared at 70 °C, incubated at room temperature for 1 hour, and then cooled to 16 °C. Samples were then heated in steps of approximately 4 °C and fluorescence intensity was measured at each step once the temperature stabilized. Background fluorescence was also measured and subtracted from the FRET sample values. The ratio of fluorescence intensity in the presence of acceptor to its absence (F/F0) was calculated. The domain detection midpoint temperature (Tm) was calculated for each curve. Tm was defined as a point of maximum slope of a sigmoidal fit of F/F0 data.
2.20. Preparation and imaging of giant unilamellar vesicles (GUV)
GUVs were prepared according to the methods of Lin and London (28). Briefly, total lipid extracts from cryptococcal cells or a mixture of GlcCer, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, and ergosterol were dried under N2, and then dissolved in chloroform at a concentration of 10 mg/mL. A small volume (1–2 μL) was then spread on ITO-coated coverslips. The solvent was then completely evaporated by drying under vacuum for 2 hours. The lipid-containing coverslip and a blank coverslip were then positioned in a home-built flow chamber at a distance of ~2 mm from each other. Afterwards, 200–300 μL trehalose solution was added to the chamber. Next, a voltage of 1.2 V at 10 Hz was applied for at least 2 hours at room temperature. GUVs were imaged using Zeiss LSM 510 META NLO Two-Photon Laser Scanning Confocal Microscope System (Zeiss, Thornwood, NY).
2.21. Vesicle Permeability Assays
LUVs containing 5(6)-carboxyfluorescein (Sigma-Aldrich) were prepared according to the method of Zhang and colleagues (29) and were used in vesicle permeability studies. Briefly, MLVs were prepared using the methods described above, except that 5(6)-carboxyfluorescein was dissolved in phosphate-buffered saline (PBS, pH 7.4) at a concentration of 80 mM before hydration of the lipids. LUVs were then prepared by subjecting the MLVs to 7 freeze-thaw cycles followed by passage through a 100 nm polycarbonate filter (Avanti Polar Lipids, Alabaster, AL) 11 times in order to obtain uniform vesicle size. Non-encapsulated 5(6)-carboxyfluorescein was removed using a PD-10 desalting column (GE Healthcare Life Sciences, Pittsburgh, PA). A fresh vesicle solution was used for each experiment.
Leakage experiments were performed on a Beckman Coulter DTX880 plate reader with excitation and emission wavelength filters of 485 nm and 535 nm, respectively. LUVs were incubated in a 96-well quartz microplate at 25 °C. The fluorescence signal of 5(6)-carboxyfluorescein was continuously measured during the course of each experiment. The maximum leakage for totally disrupted vesicles (Fmax) was measured by adding the detergent Triton X-100 to a final concentration of 0.2% (vol:vol). The percent leakage is calculated from: Percent Leakage = 100 × [F(T) − Fbaseline]/(Fmax − Fbaseline), where F(T) is the fluorescence intensity at time T, Fmax is the fluorescence intensity when all of the vesicles have been disrupted and Fbaseline is the base line fluorescence observed at time zero.
2.22. Statistical Analysis
Animal studies were performed by assigning mice randomly to treatment groups. Group sizes were chosen such that a statistical power of at least 80% was reached. No samples or animals were excluded from the analysis. The results of mouse survival studies were compared for statistical significance using the Student-Newman-Keuls t-test or the Kruskal-Wallis test. Statistical analysis for tissue burden was performed using the analysis of variance (ANOVA). Further statistical analysis were performed using unpaired t tests using the GraphPad Prism (La Jolla, CA, USA) software package. All data were reported as mean ± standard deviation and results were considered significant at P ≤ 0.05.
3. RESULTS
3.1. Growth and virulence profile of the Δsld8 strain
To create a C. neoformans strain with a saturated GlcCer structure the SLD8 gene was deleted from the wild-type, C. neoformans H99. Southern hybridization analysis confirmed the generation of the Δsld8 mutant strain, which was then used to reintroduce the SLD8 gene and create the reconstituted Δsld8+SLD8 strain (Supplementary Figures 1 and 2). Our previous studies in engineered C. neoformans strains that did not produce GlcCer (Δgcs1) (2), or produced GlcCer without a methyl group on carbon position 9 (Δsmt1) (11), had revealed defects in C. neoformans growth in DMEM at neutral pH, 37 °C, and 5% CO2. However, the Δsld8 strain did not show any growth defect in acidic (pH=4) or neutral/alkaline (pH=7.4) in DMEM at 37 °C, and 5% CO2 (Figure 1A and B), and its growth profile was similar to the wild-type and the reconstituted Δsld8+SLD8 strains. The role of GlcCer chemical structure in cryptococcal virulence was investigated using in vivo infection studies in a mouse model of cryptococcosis. CBA/J mice were infected with 5×105 cryptococcal cells and their survival was monitored over 90 days. It was observed that while the mice infected with the wild-type and the reconstituted Δsld8+SLD8 strains succumbed to infection in less than 40 days, all the mice infected with the Δsld8 mutant strain survived the infection (Figure 1C). Quantitation of the fungal burden in the lungs and the brain of strains infected with the Δsld8, the wild-type, and the Δsld8+SLD8 strains revealed that while all strains led to a significant number of colony forming units (CFU) in the lungs, the number of CFU of the Δsld8 strain in the lungs were reduced over time (Figure 1D). In addition, the Δsld8 strain was unable to establish meningitis as evidenced by a lack of CFUs in the brain (Figure 1E). The loss of virulence of the Δsld8 strain in mice was a surprising phenotype, given that this strain did not show any growth defects. Therefore, common virulence factors of this strain were characterized to elucidate the underlying mechanisms for loss of virulence. Analysis of capsule size, cell wall, glucuronoxylomannan (GXM) secretion, melanization at different temperatures, urease and laccase activity, and the release and activity of phospholipase B1 (Plb1) revealed no significant differences between the Δsld8 strain and the wild-type (Supplementary Figure 3). Taken together, these analyses suggest that the Δsld8 strain is not significantly different from the wild- type in its virulence traits, despite being avirulent in the mouse model.
Figure 1. The Δsld8 strain does not show a growth defect at acidic or neutral pH, but is completely avirulent.

The mutant and wildtype strains were examined for their growth profile in DMEM at A) pH=4.0 and B) pH=7.4 at 37 °C in the presence of 5% CO2. Growth was assayed by drawing 100 μL aliquots, serial dilution, and plating on YPD agar to enumerate CFU (three independent experiments were performed for each growth condition). C) Four weeks old immunocompetent CBA/J mice were infected with wild-type, Δsld8, and Δsld8+SLD8 mutant strains and monitored for survival (10 were used per strain). Mice infected with the wild-type and reconstituted strain succumbed to the infection in an average of 25.5 and 31.5 days, respectively. Mice infected with the Δsld8 strain remained alive for the duration of the experiment (90 days post infection, p value=0.0001). Fungal burden was quantified in D) the lungs and E) the brain of five mice per condition. The fungal burden values were statistically significantly different compared to the wild-type (shown by *) or the reconstituted strain (shown by $). A p-value of 0.05 was used for all analysis.
3.2. GlcCer structure regulates plasma membrane permeability and intracellular survival
Since GlcCer is primarily localized at the cell membrane (8), the effects of changes in GlcCer chemical structure on plasma membrane permeability and resistance to environmental stress were investigated. Plasma membrane permeability was examined using a Sytox green uptake assay. Sytox green shows significant increase in fluorescence upon binding to nucleic acids and can be used as a measure of membrane permeability (30). Sytox green uptake assay at acidic and neutral/alkaline pH revealed a significant increase in the uptake of the dye in the Δsld8 strain compared to the wild-type and the reconstituted strains (Figure 2A). Interestingly, this increased uptake was observed even in the absence of oxidative and nitrosative stressors and was significantly higher in acidic compared to neutral/alkaline pH. While oxidative stress by H2O2 did not increase membrane sensitivity, nitrosative stress by 1 mM NO resulted in a dramatic increase in Sytox green uptake (Figure 2A). These results demonstrate that the Δsld8 strain has a more permeable membrane compared to the wild-type similar to the increased permeability previously reported for the Δsmt1 strain (11). The Δsld8 strain also showed increased sensitivity to ionic (0.05% SDS) and non-ionic (0.025% Triton X-100) detergents (Figure 2B), suggesting that changes in GlcCer structure lead to defects in the fungal cell membrane.
Figure 2. The Δsld8 mutant strain exhibits increased membrane sensitivity and reduced ability to survive inside alveolar macrophages.

A) Fluorescence intensity of Sytox Green after exposing Cryptococcus cells to the dye in acidic (left) or neutral/alkaline pH conditions (right) under oxidative or nitrosative stress (average of three experiments). B) Five microliter spots of serial dilutions starting from 105 cells were placed onto YPD agar plates with or without 0.05% SDS and 0.025% Triton X-100. Plates were incubated at 37 °C in the presence of 5% CO2 and growth monitored (representative images from three experiments). C) Intracellular growth of the wild-type (WT), Δsld8, and Δsld8+SLD8 strains inside primary alveolar macrophages from CBA/J. Two hours post-infection, the cells were washed, fixed with ice-cold methanol, stained with the Giemsa stain for microscopy analysis. A minimum of 100 internalized cryptococcal cells was analyzed for budding to calculate intracellular growth (%). Experiments were performed a minimum of three times and statistical analysis were performed to determine whether changes observed in experiments with the mutant strain were statistically significant compared to the wild-type (shown by *) or the reconstituted strain (shown by $). A p-value of 0.05 was used for all analysis.
To further investigate the mechanisms of loss of virulence in the Δsld8 strain, the ability of this strain to grow inside the acidic environment of activated macrophages was investigated. To this aim, primary alveolar macrophages were isolated from CBA/J mice and infected with opsonized wild-type, Δsld8, or Δsld8+SLD8 strains with a multiplicity of infection (MOI) of 1 and the ability of intracellular fungal cells to form buds was investigated by counting the buds in at least 100 intracellular Cryptococcus cells. While the Δsld8 strain was able to form buds intracellularly, its intracellular growth was significantly slower compared to the wild-type (p=0.0036) and the Δsld8+SLD8 (p=0.0057) reconstituted strains (Figure 2C).
3.3. GlcCer purification and biophysical characterization
Given the changes observed in the cell membrane, biophysical methods were employed to further investigate the effects of GlcCer on cell membrane properties. It was hypothesized that changes in GlcCer structure affect membrane stability and organization and contribute to the attenuated virulence in Δsmt1 and Δsld8 mutant strains. To investigate this hypothesis, GlcCer was purified from the wild-type, Δsmt1, and Δsld8 strains using column chromatography. The purified lipids were loaded on thin layer chromatography (TLC) plates and migrated the same distance as a GlcCer standard purified from soy lipids (Supplementary Figure 4). Liquid Chromatography-Mass Spectrometry (LC-MS) analysis confirmed the lack of methyl group on carbon position 9 in GlcCer purified from the Δsmt1 strain, and the lack of methyl group on carbon position 9 and a double-bond in carbon position 8, in the Δsld8 strain (Supplementary Figure 4). Purified lipids were used for biophysical characterization studies. Vesicles were made out of each purified GlcCer species and fluorescence anisotropy was used to investigate lipid order as a function of temperature using the lipophilic fluorophore, diphenyl hexatriene (DPH). At low temperature, all GlcCer structures showed high anisotropy as is common for lipids that tend to form ordered domains (31,32). Reduced anisotropy was observed with increasing temperature; however, different GlcCer structures showed differences in temperature dependence. The more saturated structures showed higher anisotropy at temperatures close to, or slightly higher than, physiologically relevant values (Figure 3A). The temperature at which the lipids transitioned from ordered to disordered state (transition temperature or Tm) was found by fitting a sigmoidal curve to the anisotropy data and showed a similar dependence on lipid chemical structure (Tm=34.7 ± 0.1 °C for GlcCer from the wild-type, Tm=40.7 ± 1.1 °C for GlcCer from the Δsmt1 strain, and 43.2 ± 1.1 °C for GlcCer from the Δsld8 strain), suggesting that GlcCer from the mutant strains are likely to increase the lipid order in the cell membrane at physiologically relevant temperatures.
Figure 3. Biophysical characterization of vesicles containing fungal GlcCer.

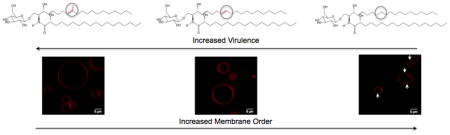
A) Fluorescence anisotropy vs. temperature of synthetic vesicles composed of GlcCer from the wild-type (left), Δsmt1 (middle), and Δsld8 strains (right). A sigmoid function was fit to the anisotropy profiles and the chain melting temperature (Tm) was defined as the inflection point of that sigmoid. Error bars represent the standard deviation from three experiments for the wildtype and two experiments for the mutant strains. B) Images of giant unilamellar vesicles (GUVs), synthesized from GlcCer, POPC, and ergosterol (Erg), in a 1:1:1 molar ratio. GlcCer purified from the wild-type strain (left), Δsmt1 strain (middle), and Δsld8 strain (right) was mixed with equimolar concentrations of POPC and Erg. Rhodamine-DOPE (0.01 mol%), localizing in disordered lipid phases, was used as the fluorescence probe.
The tendency of GlcCer structures to promote lipid phase segregation and form ordered domains in simple plasma membrane models was investigated by imaging giant unilamellar vesicles (GUVs). Mixtures of a high Tm lipid, a low Tm lipid, and a sterol have been suggested as a simple model of the plasma membrane (33–38). To mimic the fungal cell membrane, a ternary mixture of equimolar concentrations of GlcCer (as the high Tm lipid), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC, as the low Tm lipid), and ergosterol (henceforth referred to as G:P:E) was used as a simple model of the plasma membrane. Although this system does not mimic the physiological concentration of GlcCer, it is close to the level of glycosphingolipids in the lipid rafts of pathogenic fungi (39). GUVs were made from the G:P:E mixtures and were doped with 0.01 mol% rhodamine-DOPE, which localizes to the disordered lipid phase. Microscopy studies revealed that only the GlcCer purified from the Δsld8 strain formed large ordered domains that could be visualized with confocal microscopy (Figure 3B). It should be noted that GlcCer was able to form ordered domains in synthetic membrane mixtures with POPC and ergosterol even in its wild-type, most unsaturated, structure as demonstrated by Förster Resonance Electron Transfer (FRET) assays, which can detect submicroscopic domains (Supplementary Figure 5). Thus, we conclude the size of the domains in GlcCer purified from the wild-type (and likely the Δsmt1 strain) were too small to be observed using microscopy.
3.4. Whole cell lipid biophysical characterization and leakage assay
The increased lipid order and domain formation in vesicles containing pure GlcCer, motivated further studies to examine whether changes in lipid order will also be observed in whole cell lipid extracts. To this aim, DPH anisotropy was used to examine lipid order in vesicles synthesized from whole cell lipid extracts. As expected, all strains showed lower anisotropy values at 37 °C compared to 23 °C, as temperature increase is expected to increase molecular motion and reduce membrane order (Figure 4A). However, no significant differences were observed, at either temperature, in the anisotropy of vesicles synthesized from the lipids of different strains, revealing no significant changes in lipid order.
Figure 4. Biophysical characterization of vesicles synthesized from total lipid extracts.

A) Fluorescence anisotropy of synthetic vesicles composed of total lipid extracts from the wild-type, Δsmt1, Δsld8 strains, and their respective reconstructs at 23 °C and 37 °C. B) Time-course of carboxyfluorescein leakage from synthetic vesicles composed of total lipid extracts from the wild-type, Δsmt1, Δsld8 strains, and their respective reconstructs in PBS buffer, pH 7.4 at room temperature. C) Images of GUVs, synthesized from total lipid extracts from the wild-type, Δsmt1, Δsld8 strains. Rhodamine-DOPE (0.01 mol%), localizing in disordered lipid phases, was used as the fluorescence probe.
Since an increased uptake of Sytox green was observed in both Δsmt1 (11) and Δsld8 (Fig 2A) strains, the leakage of the fluorescent dye 5(6)-carboxyfluorescein from inside the vesicles was also investigated. The fluorescence of 5(6)-carboxyfluorescein is self-quenched at high concentrations. Thus, the fluorescence intensity is reduced when high concentrations of this dye are encapsulated inside the vesicles. However, upon release from the vesicles, the dilution of the dye results in increased fluorescence, which can be used as a measure of leakage from the vesicles (29). To investigate leakage from the vesicles synthesized from whole cell lipid extracts, 5(6)-carboxyfluorescein was encapsulated in vesicles made from the lipids of the wild-type strain as well as the Δsmt1 and Δsld8 mutant strains. All vesicles were allowed to incubate in buffer at pH=7.4 at room temperature and the fluorescence of 5(6)-carboxyfluorescein was recorded. Experiments up to 24 hours, revealed no differences in leakage from the vesicles (Figure 4B), suggesting no changes in membrane permeability of the vesicles synthesized from the whole cell extract lipids of different strains. Similarly, GUVs synthesized from the whole cell extract lipids did not show large-scale phase segregation in any of the strains (Figure 4C).
4. DISCUSSION
The results of the current study suggest that there is a direct connection between the unsaturation of the sphingosine backbone in GlcCer and the ability of C. neoformans to establish virulence. The connection between GlcCer, saturated at carbon 8 and unmethylated at carbon 9, and loss of virulence is not due to changes in common virulence factors, but is likely due to changes in membrane properties. While our previous studies demonstrate a 70% loss of virulence in the Δsmt1 strain that produces GlcCer without a methyl group, the current study reveals that the Δsld8, with more saturated GlcCer completely loses its virulence despite being able to grow normally at both acidic and neutral/alkaline pH environments. This strain also did not exhibit significant changes in common cryptococcal virulence factors such as capsule formation, secretion of GXM, melanization, laccase, Plb1, and urease activity.
Several lines of evidence suggest changes in membrane structure in the Δsld8 strain: this strain showed increased membrane permeability and increased sensitivity to detergents. Interestingly, the membrane permeability of the Δsld8 strain is increased in acidic pH. This explains the reduced intracellular growth, as the fungus is subject to acidic environment as well as oxidative and nitrosative stressors inside macrophages. The changes in cryptococcal cell membrane structure appear to be linked, directly or indirectly, to GlcCer chemical structure as both the Δsmt1 and the Δsld8 strains showed increased uptake of Sytox green. This is further supported by the observed changes in the Tm of the GlcCer structures purified from mutants and the presence of ordered domains in GUVs containing GlcCer from the Δsld8 strain (Figure 3). The increased sensitivity to stressors in acidic conditions is also important in the context of the extracellular growth of the Δsld8 strain. The pH of infected tissue is acidic; thus, the fungus is more likely to be vulnerable to immune response extracellularly, in such acidic environment. Taken together, these observations suggest that changes in membrane structure, and not necessarily changes in virulence factors, might have been the cause for loss of virulence the Δsld8 strain.
It should be noted that the physical behavior of membranes made out of pure cerebrosides (either GlcCer or galactosylceramide) as a function of temperature is unusual and complex. Prior studies have reported, in addition to solid to liquid melting at very high temperature, they from two crystal-like phases, and there is a thermal transition between them that depends on the thermal history of the samples (40,41). The melting temperature detected by differential scanning calorimetry in these studies was much higher than we have detected for GlcCer by DPH anisotropy. This may be because previous studies were carried out in “fully hydrated” samples that are in fact mostly lipid by weight, while those in this report were carried out in very dilute vesicles, at a lipid concentration over 1000-fold more dilute. Interbilayer interactions that may influence lipid behavior are unlikely to be present under our conditions. It is unlikely the difference is due to structural differences between Δsld8 mutant GlcCer and mammalian GlcCer, as, we see similar DPH anisotropy vs. temperature for these species (data not shown). In any case, it is the difference between thermal transition temperatures of different GlcCer species in the wild type and mutants, and not absolute transitions temperatures, that is of most interest in this study.
Changes observed in the biophysical properties of GlcCer-containing vesicles, depending on GlcCer structure, were not reflected in vesicles synthesized from whole cell lipids of each strain (Figure 4A and 4C). The permeability of these strains also remained unchanged (Figure 4B), which was not in accordance with the observations using live cells (Figure 2A), clearly showing increased membrane permeability. The discrepancy between the in vitro and in vivo permeability may be due to the inability of the liposome preparation to reproduce the lipid composition of the plasma membrane. In fact, while using whole cell lipids reflects changes in the lipid composition of the cell that might have occurred as a result of changes in GlcCer pathway, one inherent flaw in biophysical characterization of vesicles synthesized from whole cell lipids is that these lipids are “contaminated” by other, intracellular membrane lipids and thus the final composition is different from the plasma membrane lipids. It is also possible that changes in GlcCer structure might affect membrane physical properties through changes in membrane protein expression and localization We have reported previously that changes in the chemical structure of cryptococcal GlcCer leads to significant changes in the expression of six membrane-localized proteins, especially iron and sugar transporters (42). It is possible that another mechanism exists: GlcCer structure leads to minor changes in membrane biophysical properties, detected in synthetic but not in whole cell extract vesicles, and these minor changes affect the expression and/or localization of these or other transporter proteins leading to the disruption of transmembrane signaling complex.
While avirulent mutants of C. neoformans have been constructed before, the loss of virulence has been associated with an inability to grow at certain conditions (2,3), changes in virulence factors (43), or accumulation of immunomodulatory compounds (44). The Δsld8 strain generated in this study does not show any evidence of the previously found mechanisms for loss of virulence, but is more sensitive to oxidative and nitrosative stress and is likely to have altered plasma membrane biophysical properties. Increased sensitivity to stressors and changes to membrane properties can both contribute to changes in virulence. Increased membrane sensitivity, particularly in acidic conditions, can lead to defective growth in the intracellular environment, such as that seen in Figure 2C, allowing the immune system to contain the infection in the lungs. In addition, increased sensitivity to nitric oxide will reduce the ability of the fungus to survive inside the macrophages. This will strip the fungus of the possibility to utilize the macrophages as a “Trojan Horse” for extrapulmonary dissemination. The Trojan Horse mechanism is known to contribute to cryptococcal infection of the brain (45). We have previously shown that depletion of alveolar macrophages reduces the dissemination of the fungal cells to the central nervous system following intranasal infection, further confirming this mechanism (46). Alternatively, it is also possible that increased order and thus rigidity of the plasma membrane, as suggested by anisotropy and GUV imaging experiments, can result in loss of virulence by affecting the ability of fungal cells to travel through the capillaries in the body. This hypothesis is supported by parallels observed in Gaucher disease, caused by accumulation of mammalian GlcCer in red blood cells (47). In Gaucher disease, abnormal cell shapes are observed in red blood cells (48). These abnormal shapes are a result of GlcCer accumulation and cells regain their normal shape after treatment with glucocerebrosidase, which restores normal GlcCer levels. GlcCer accumulation and morphological changes in red blood cells have been suggested to hamper cell deformability, which prevents the circulation of red blood cells in capillaries (48). The structure of the GlcCer accumulated in the Δsld8 strain is identical to the saturated, mammalian GlcCer, which accumulates in red blood cells in Gaucher disease (49). It is likely that the accumulation of a “mammalian” GlcCer on fungal membrane leads to a rigid membrane, which cannot sufficiently deform to travel through the capillaries and enter brain microvasculature, thereby preventing the establishment of a virulent and invasive cryptococcosis. Fungal burden studies revealed the presence of fungal cells in the lungs, but not in the brain, suggesting that the pathogen is unable to infiltrate the brain. Being unable to travel within the microvasculature, the Δsld8 is captured by macrophages where it cannot survive due to the leakiness of its membrane, leading to the loss of pathogenicity.
Supplementary Material
Highlights.
C. neoformans strain (Δsld8) producing saturated glucosylceramide was generated.
The Δsld8 strain loses its ability to establish virulence in the mouse model.
Glucosylceramide from Δsld8 forms more ordered vesicles compared to the wild-type.
The plasma membrane of the Δsld8 strain is hyper-sensitive to stressors.
Changes in glucosylceramide affect virulence by affecting membrane properties.
Acknowledgments
The authors would like to thank Johnna St. Clair for her help with GUV imaging studies. This work was supported by NIH grants AI116420, AI125770, and by a Merit Review grant I01BX002624 from the Veterans Affairs Program in Biomedical Laboratory Research and Development to MDP, NIH grants GM099892 and GM122493 to EL, NIH grant GM078114 to DPR. AF acknowledges financial support from the Russ College of Engineering and Technology and the Department of Chemical and Biomolecular Engineering at Ohio University as well as funding from the Ohio University Foundation Board of Trustees (1804 Fund). Maurizio Del Poeta is Burroughs Wellcome Investigator in Infectious Diseases.
Footnotes
CONFLICT OF INTEREST
Dr. Maurizio Del Poeta is a Co-Founder and Chief Scientific Officer (CSO) of MicroRid Technologies Inc. All other authors have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.García-Rodas R, Zaragoza O. Catch me if you can: phagocytosis and killing avoidance by Cryptococcus neoformans. FEMS Immunology & Medical Microbiology. 2012;64:147–161. doi: 10.1111/j.1574-695X.2011.00871.x. [DOI] [PubMed] [Google Scholar]
- 2.Rittershaus PC, Kechichian TB, Allegood JC, Merrill AH, Hennig M, Luberto C, Del Poeta M. Glucosylceramide synthase is an essential regulator of pathogenicity of Cryptococcus neoformans. The Journal of Clinical Investigation. 2006;116:1651–1659. doi: 10.1172/JCI27890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shea JM, Kechichian TB, Luberto C, Del Poeta M. The cryptococcal enzyme inositol phosphosphingolipid-phospholipase C confers resistance to the antifungal effects of macrophages and promotes fungal dissemination to the central nervous system. Infection and Immunity. 2006;74:5977–5988. doi: 10.1128/IAI.00768-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mor V, Rella A, Farnoud AM, Singh A, Munshi M, Bryan A, Naseem S, Konopka JB, Ojima I, Bullesbach E. Identification of a new class of antifungals targeting the synthesis of fungal sphingolipids. MBio. 2015;6:e00647–00615. doi: 10.1128/mBio.00647-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oura T, Kajiwara S. Candida albicans sphingolipid C9-methyltransferase is involved in hyphal elongation. Microbiology. 2010;156:1234–1243. doi: 10.1099/mic.0.033985-0. [DOI] [PubMed] [Google Scholar]
- 6.Martin SW, Konopka JB. Lipid raft polarization contributes to hyphal growth in Candida albicans. Eukaryotic Cell. 2004;3:675–684. doi: 10.1128/EC.3.3.675-684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levery SB, Momany M, Lindsey R, Toledo MS, Shayman JA, Fuller M, Brooks K, Doong RL, Straus AH, Takahashi HK. Disruption of the glucosylceramide biosynthetic pathway in Aspergillus nidulans and Aspergillus fumigatus by inhibitors of UDP-Glc: ceramide glucosyltransferase strongly affects spore germination, cell cycle, and hyphal growth. FEBS letters. 2002;525:59–64. doi: 10.1016/s0014-5793(02)03067-3. [DOI] [PubMed] [Google Scholar]
- 8.Rhome RM, Singh A, Kechichian T, Drago M, Morace G, Luberto C, Del Poeta M. Surface Localization of Glucosylceramide During Cryptococcus Neoformans Infection Allows Targeting as a Potential Anti-fungal. 2011 doi: 10.1371/journal.pone.0015572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodrigues ML, Travassos LR, Miranda KR, Franzen AJ, Rozental S, de Souza W, Alviano CS, Barreto-Bergter E. Human antibodies against a purified glucosylceramide from Cryptococcus neoformans inhibit cell budding and fungal growth. Infection and immunity. 2000;68:7049–7060. doi: 10.1128/iai.68.12.7049-7060.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Del Poeta M, Nimrichter L, Rodrigues ML, Luberto C. Synthesis and biological properties of fungal glucosylceramide. PLoS Pathogens. 2014;10:e1003832. doi: 10.1371/journal.ppat.1003832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh A, Wang H, Silva LC, Na C, Prieto M, Futerman AH, Luberto C, Del Poeta M. Methylation of glycosylated sphingolipid modulates membrane lipid topography and pathogenicity of Cryptococcus neoformans. Cellular Microbiology. 2011;14:500–516. doi: 10.1111/j.1462-5822.2011.01735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heung LJ, Luberto C, Plowden A, Hannun YA, Del Poeta M. The sphingolipid pathway regulates Pkc1 through the formation of diacylglycerol in Cryptococcus neoformans. J Biol Chem. 2004;279:21144–21153. doi: 10.1074/jbc.M312995200. [DOI] [PubMed] [Google Scholar]
- 13.Heung LJ, Kaiser AE, Luberto C, Del Poeta M. The role and mechanism of diacylglycerol-protein kinase C1 signaling in melanogenesis by Cryptococcus neoformans. J Biol Chem. 2005;280:28547–28555. doi: 10.1074/jbc.M503404200. [DOI] [PubMed] [Google Scholar]
- 14.Toffaletti DL, Rude TH, Johnston SA, Durack DT, Perfect JR. Gene transfer in Cryptococcus neoformans by use of biolistic delivery of DNA. J Bacteriol. 1993;175:1405–1411. doi: 10.1128/jb.175.5.1405-1411.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh A, Qureshi A, Del Poeta M. Quantitation of cellular components in Cryptococcus neoformans for system biology analysis. Methods Mol Biol. 2011;734:317–333. doi: 10.1007/978-1-61779-086-7_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mandala SM, Thornton RA, Frommer BR, Curotto JE, Rozdilsky W, Kurtz MB, Giacobbe RA, Bills GF, Cabello MA, Martin I, et al. The discovery of australifungin, a novel inhibitor of sphinganine N-acyltransferase from Sporormiella australis. Producing organism, fermentation, isolation, and biological activity. J Antibiot (Tokyo) 1995;48:349–356. doi: 10.7164/antibiotics.48.349. [DOI] [PubMed] [Google Scholar]
- 17.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 18.Bielawski J, Pierce JS, Snider J, Rembiesa B, Szulc ZM, Bielawska A. Comprehensive quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods Mol Biol. 2009;579:443–467. doi: 10.1007/978-1-60761-322-0_22. [DOI] [PubMed] [Google Scholar]
- 19.Lazcano O, Speights V, Jr, Strickler J, Bilbao J, Becker J, Diaz J. Combined histochemical stains in the differential diagnosis of Cryptococcus neoformans. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 1993;6:80–84. [PubMed] [Google Scholar]
- 20.Lazcano O, Speights VO, Jr, Bilbao J, Becker J, Diaz J. Combined Fontana-Masson-mucin staining of Cryptococcus neoformans. Arch Pathol Lab Med. 1991;115:1145–1149. [PubMed] [Google Scholar]
- 21.Casadevall A, Mukherjee J, Scharff MD. Monoclonal antibody based ELISAs for cryptococcal polysaccharide. Journal of immunological methods. 1992;154:27–35. doi: 10.1016/0022-1759(92)90209-c. [DOI] [PubMed] [Google Scholar]
- 22.Roberts G, Horstmeier C, Land G, Foxworth J. Rapid urea broth test for yeasts. Journal of clinical microbiology. 1978;7:584–588. doi: 10.1128/jcm.7.6.584-588.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwon-Chung K, Wickes BL, Booth J, Vishniac HS, Bennett JE. Urease inhibition by EDTA in the two varieties of Cryptococcus neoformans. Infection and immunity. 1987;55:1751–1754. doi: 10.1128/iai.55.8.1751-1754.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alvarado-Ramírez E, Torres-Rodríguez JM, Sellart M, Vidotto V. Laccase activity in Cryptococcus gattii strains isolated from goats. Revista iberoamericana de micologia. 2008;25:150. doi: 10.1016/s1130-1406(08)70035-4. [DOI] [PubMed] [Google Scholar]
- 25.Chen S, Wright LC, Santangelo RT, Muller M, Moran VR, Kuchel PW, Sorrell TC. Identification of extracellular phospholipase B, lysophospholipase, and acyltransferase produced by Cryptococcus neoformans. Infection and immunity. 1997;65:405–411. doi: 10.1128/iai.65.2.405-411.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pathak P, London E. Measurement of lipid nanodomain (raft) formation and size in sphingomyelin/POPC/cholesterol vesicles shows TX-100 and transmembrane helices increase domain size by coalescing preexisting nanodomains but do not induce domain formation. Biophysical journal. 2011;101:2417–2425. doi: 10.1016/j.bpj.2011.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bakht O, London E. Cholesterol precursors stabilize ordinary and ceramide-rich ordered lipid domains (lipid rafts) to different degrees Implications for the Bloch hypothesis and sterol biosynthesis disorders. Journal of Biological Chemistry. 2006;281:21903–21913. doi: 10.1074/jbc.M600395200. [DOI] [PubMed] [Google Scholar]
- 28.Lin Q, London E. Ordered raft domains induced by outer leaflet sphingomyelin in cholesterol-rich asymmetric vesicles. Biophysical journal. 2015;108:2212–2222. doi: 10.1016/j.bpj.2015.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang X, St Clair JR, London E, Raleigh DP. Islet Amyloid Polypeptide Membrane Interactions: Effects of Membrane Composition. Biochemistry. 2017 doi: 10.1021/acs.biochem.6b01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roth BL, Poot M, Yue ST, Millard PJ. Bacterial viability and antibiotic susceptibility testing with SYTOX green nucleic acid stain. Applied and environmental microbiology. 1997;63:2421–2431. doi: 10.1128/aem.63.6.2421-2431.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Megha L, Erwin Ceramide Selectively Displaces Cholesterol from Ordered Lipid Domains (Rafts) Implications for lipid raft structure and function. Journal of Biological Chemistry. 2004;279:9997–10004. doi: 10.1074/jbc.M309992200. [DOI] [PubMed] [Google Scholar]
- 32.Gidwani A, Holowka D, Baird B. Fluorescence anisotropy measurements of lipid order in plasma membranes and lipid rafts from RBL-2H3 mast cells. Biochemistry. 2001;40:12422–12429. doi: 10.1021/bi010496c. [DOI] [PubMed] [Google Scholar]
- 33.de Almeida RF, Fedorov A, Prieto M. Sphingomyelin/phosphatidylcholine/cholesterol phase diagram: boundaries and composition of lipid rafts. Biophysical Journal. 2003;85:2406–2416. doi: 10.1016/s0006-3495(03)74664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Veatch SL, Keller SL. Separation of liquid phases in giant vesicles of ternary mixtures of phospholipids and cholesterol. Biophysical Journal. 2003;85:3074–3083. doi: 10.1016/S0006-3495(03)74726-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan C, Furlong J, Burgos P, Johnston LJ. The size of lipid rafts: an atomic force microscopy study of ganglioside GM1 domains in sphingomyelin/DOPC/cholesterol membranes. Biophysical journal. 2002;82:2526–2535. doi: 10.1016/S0006-3495(02)75596-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahmed SN, Brown DA, London E. On the origin of sphingolipid/cholesterol-rich detergent-insoluble cell membranes: physiological concentrations of cholesterol and sphingolipid induce formation of a detergent-insoluble, liquid-ordered lipid phase in model membranes. Biochemistry. 1997;36:10944–10953. doi: 10.1021/bi971167g. [DOI] [PubMed] [Google Scholar]
- 37.Schroeder R, London E, Brown D. Interactions between saturated acyl chains confer detergent resistance on lipids and glycosylphosphatidylinositol (GPI)-anchored proteins: GPI-anchored proteins in liposomes and cells show similar behavior. Proceedings of the National Academy of Sciences. 1994;91:12130–12134. doi: 10.1073/pnas.91.25.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silvius JR, del Giudice D, Lafleur M. Cholesterol at different bilayer concentrations can promote or antagonize lateral segregation of phospholipids of differing acyl chain length. Biochemistry. 1996;35:15198–15208. doi: 10.1021/bi9615506. [DOI] [PubMed] [Google Scholar]
- 39.Tagliari L, Toledo MS, Lacerda TG, Suzuki E, Straus AH, Takahashi HK. Membrane microdomain components of Histoplasma capsulatum yeast forms, and their role in alveolar macrophage infectivity. Biochimica et Biophysica Acta (BBA)-Biomembranes. 2012;1818:458–466. doi: 10.1016/j.bbamem.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 40.Ruocco M, Atkinson D, Small D, Skarjune R, Oldfield E, Shipley G. X-ray diffraction and calorimetric study of anhydrous and hydrated N-(palmitoylgalactosylsphingosine cerebroside) Biochemistry. 1981;20:5957–5966. doi: 10.1021/bi00524a006. [DOI] [PubMed] [Google Scholar]
- 41.Freire E, Bach D, Correa-Freire M, Miller I, Barenholz Y. Calorimetric investigation of the complex phase behavior of glucocerebroside dispersions. Biochemistry. 1980;19:3662–3665. doi: 10.1021/bi00557a004. [DOI] [PubMed] [Google Scholar]
- 42.Singh A, Rella A, Schwacke J, Vacchi-Suzzi C, Luberto C, Poeta M. Transmembrane transporter expression regulated by the glucosylceramide pathway in Cryptococcus neoformans. BMC research notes. 2015;8:681. doi: 10.1186/s13104-015-1613-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cox GM, McDade HC, Chen SC, Tucker SC, Gottfredsson M, Wright LC, Sorrell TC, Leidich SD, Casadevall A, Ghannoum MA. Extracellular phospholipase activity is a virulence factor for Cryptococcus neoformans. Molecular microbiology. 2001;39:166–175. doi: 10.1046/j.1365-2958.2001.02236.x. [DOI] [PubMed] [Google Scholar]
- 44.Rella A, Mor V, Farnoud AM, Singh A, Shamseddine AA, Ivanova E, Carpino N, Montagna MT, Luberto C, Del Poeta M. Role of Sterylglucosidase 1 (Sgl1) on the pathogenicity of Cryptococcus neoformans: potential applications for vaccine development. Frontiers in microbiology. 2015:6. doi: 10.3389/fmicb.2015.00836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Charlier C, Nielsen K, Daou S, Brigitte M, Chretien F, Dromer F. Evidence of a role for monocytes in dissemination and brain invasion by Cryptococcus neoformans. Infection and immunity. 2009;77:120–127. doi: 10.1128/IAI.01065-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kechichian TB, Shea J, Del Poeta M. Depletion of alveolar macrophages decreases the dissemination of a glucosylceramide-deficient mutant of Cryptococcus neoformans in immunodeficient mice. Infection and immunity. 2007;75:4792–4798. doi: 10.1128/IAI.00587-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nilsson O, Håkansson G, Dreborg S, Groth CG, Svennerhoim L. Increased cerebroside concentration in plasma and erythrocytes in Gaucher disease: significant differences between type I and type III. Clinical genetics. 1982;22:274–279. doi: 10.1111/j.1399-0004.1982.tb01445.x. [DOI] [PubMed] [Google Scholar]
- 48.Bratosin D, Tissier JP, Lapillonne H, Hermine O, de Villemeur TB, Cotoraci C, Montreuil J, Mignot C. A cytometric study of the red blood cells in Gaucher disease reveals their abnormal shape that may be involved in increased erythrophagocytosis. Cytometry Part B: Clinical Cytometry. 2011;80:28–37. doi: 10.1002/cyto.b.20539. [DOI] [PubMed] [Google Scholar]
- 49.Rella A, Farnoud AM, Del Poeta M. Plasma membrane lipids and their role in fungal virulence. Progress in lipid research. 2016;61:63–72. doi: 10.1016/j.plipres.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.