

Abstract
Pioneering research studies, beginning with those using Drosophila, have identified several molecular and cellular mechanisms for active forgetting. The currently known mechanisms for active forgetting include neurogenesis-based forgetting, interference-based forgetting, and intrinsic forgetting, the latter term describing the brain’s chronic signaling systems that function to slowly degrade molecular and cellular memory traces. The best-characterized pathway for intrinsic forgetting includes “forgetting cells” that release dopamine onto engram cells, mobilizing a signaling pathway that terminates in the activation of Rac1/cofilin to effect changes in the actin cytoskeleton and neuron/synapse structure. Intrinsic forgetting may be the default state of the brain, constantly promoting memory erasure and competing with processes that promote memory stability like consolidation. A better understanding of active forgetting will provide insights into the brain’s memory management system and human brain disorders that alter active forgetting mechanisms.
Keywords: active forgetting, memory, Rac1, hippocampal neurogenesis, consolidation, dopamine, intrinsic forgetting
Introduction
The human brain has the remarkable capacity to acquire, store, and recall information across decades of time. The acquisition of information, or learning, alters the physiological state of certain neurons in ways that encode memory (Figure 1). These state changes, or molecular and cellular memory traces, can be, in principle, any change in the activity of the cell that is induced by learning that becomes part of the neural code for that memory. For instance, changes can occur in the expression or function of ion channels that cause neurons to be more or less excitable and therefore more or less capable of conducting action potentials or other electrical signals. Learning may mobilize neuronal growth processes that establish new connections, or neurite retraction to remove existing connections. The changes may include adaptations in cell signaling that alter the neuron’s overall ability to integrate inputs from different types of cues, and morphological or functional changes in synapses that increase or decrease the neuron’s ability to stimulate its synaptic partners. The collection of all molecular and cellular memory traces that are induced by learning across all neurons engaged by the learning event together comprise the overall memory engram (Squire, 1987; Dudai, 2002; Davis, 2011) that can guide behavior upon subsequent retrieval.
Figure 1. The Engram, Engram Cells, Molecular and Cellular Memory Traces, and Constructs of Forgetting from Experimental Psychology.
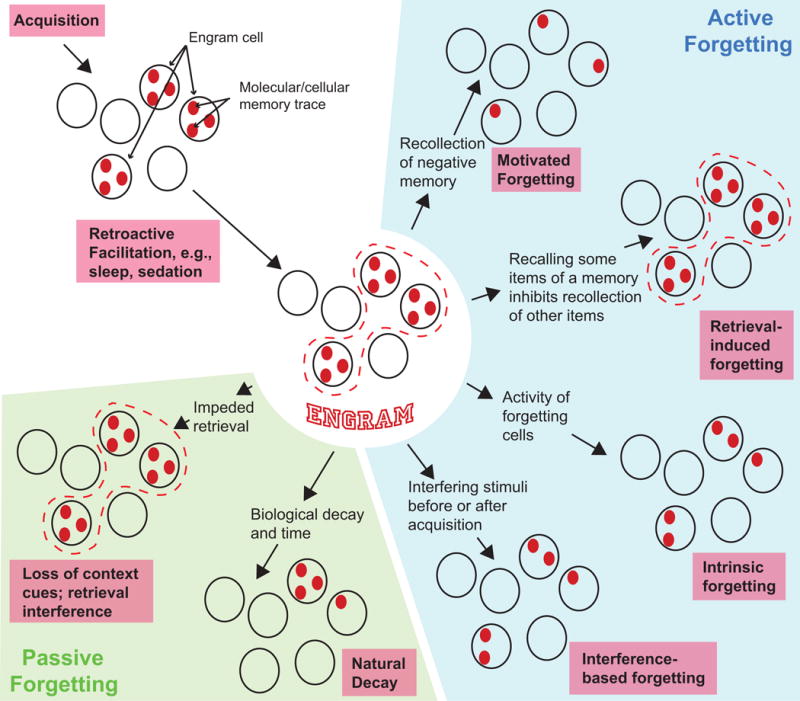
Acquisition leads to the formation of molecular and cellular memory traces (red ovals) within a set of neurons – the engram cells – that store learned information. Sleep or sedation, after acquisition, can protect from interfering stimuli and thus strengthen memory traces (center of figure). The memory engram is envisioned as the composite of all molecular and cellular memory traces that form during acquisition within the population of engram cells (dashed red line) that encode a memory. Memory encoding can lead to weak or strong memory engrams depending on the strength and nature of the learned information. Passive forgetting (lower left of figure) is postulated to occur through at least three separate mechanisms: (1) loss of context cues across time that make retrieval difficult, (2) interference during retrieval from other similar memories accumulated across time, and (3) the “natural” decay of memory traces from the general instability of biological materials and the passage of time. Loss of context cues and retrieval interference may leave the engram intact, whereas natural decay would lead to a loss of its integrity. At least three major forms of active forgetting have been postulated from research in experimental psychology (right side of figure). Interference-based forgetting posits that other competing information or activities before (proactive) or after (retroactive) the learning event accelerate the decay of memory traces. Motivated forgetting occurs when cognitive mechanisms are voluntarily engaged to weaken memory traces, often because the memory has some unpleasant quality. Retrieval-induced forgetting, offered here as an example for contrast, occurs when some aspects of a memory are recalled that suppress the recall of other aspects related to the recalled memory. This type of forgetting may disrupt the retrieval of a relatively intact memory engram. We introduce in this Perspective the concept of intrinsic forgetting – one type of active forgetting – that involves the activity of forgetting cells and the brain’s intrinsic biochemical and molecular pathways to degrade molecular and cellular memory traces.
The number of “engram neurons” (Tonegawa et al., 2015) that form a specific memory and the number of molecular and cellular memory traces that form in these neurons due to acquisition is unknown but must depend on the nature of the memory formed and its strength. But both numbers are likely to be vast. It is also unknown how many memories are formed and stored by the human brain on an average day and how many memory engrams accumulate across an 80–90 year lifetime. But again, the number must be extraordinarily large. It seems possible that the brain might simplify each memory engram after initial encoding into “index” engram cells, like the perceptual concept of grandmother cells (Gross, 2002) to streamline the logistics of memory management. Alternatively, the cells comprising the engram might become interconnected at acquisition, losing their independence, so that a retrieval stimulus reactivates the circuit of interconnected cells (Tonegawa et al., 2015). But irrespective of whether the brain simplifies memory engrams and their traces, it must have an extremely efficient memory management system to catalog each engram so that retrieval is seamless and efficient. In addition, because of the extraordinary large number of memory engrams that can accumulate in the brain across time, it seems logical that the brain must have, as one part of its memory management system, a mechanism or series of mechanisms to remove memories that become unused. These mechanisms make up what we term as “active forgetting.”
Traditional studies in the neuroscience of learning and memory have emphasized other aspects of the brain’s memory management system, such as the major processes of acquisition and consolidation (Figure 2; Squire, 1987; Menzel and Muller, 1996; McGaugh, 2000; Dudai, 2004; Davis, 2005; Shema et al., 2007; Jonides et al., 2008; Wang and Morris, 2010; Johansen et al., 2011; Kandel et al., 2014; Dudai et al., 2015). Extinction, reconsolidation, and generalization can also be considered processes that comprise the brain’s memory management system (Seger and Miller, 2010; Censor, 2013; Giustino and Maren, 2015; Dunsmoor et al., 2015; Helmstedt et al., 2017). Much has been learned about several of these processes. For instance, seminal studies from multiple researchers revealed that consolidation into protein synthesis-dependent memory involves a molecular cascade initiated by changes in gene expression at acquisition within engram cells through cAMP-dependent mechanisms that include Creb, C/EBP, and other transcription factor families (Yin et al., 1994; Bailey et al., 1996; Dudai, 2004; Kandel et al., 2015; Alberini and Kandel, 2015). The net effect of this molecular cascade is to produce long lasting changes in engram cell physiology, including synaptic plasticity, that underlie protein synthesis-dependent long-term memory. The progress in understanding the molecular pathways for acquisition and consolidation has led to significant efforts to enhance memory using drugs acting on molecules involved in these processes (Scott et al., 2002; Barco et al., 2003; Lynch et al., 2011). However, we argue that forgetting, the flip side of acquisition and consolidation is equally important and that the traditional emphasis on mechanisms of acquisition and consolidation needs to be rebalanced with more effort to understand the biology of active forgetting (Figure 2). This is the focus of this Perspective. Although forgetting can occur due to failed retrieval of an intact engram, we emphasize active forgetting through the likely biological degradation of molecular and cellular memory traces or the engram cell circuit. Active forgetting may eliminate all traces and engram cells for a given memory, but it is more likely that forgetting occurs initially from erosion of only some of the molecular and cellular memory traces, or when a fraction of the engram cells become disconnected from the engram circuit. This would make the memory engram incomplete and unresponsive to recall mechanisms.
Figure 2. Balance between Acquisition/Consolidation Forces and Forgetting.
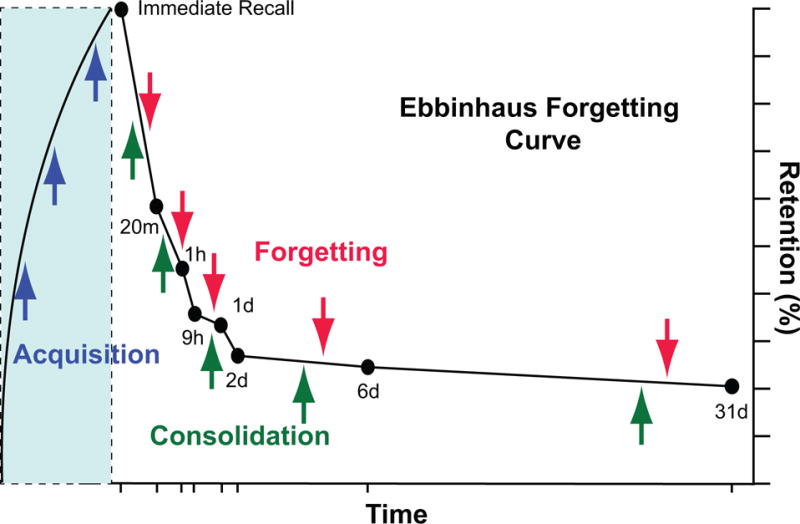
Graph using Ebbinhaus’s famous forgetting curve to display the balance between the strengthening forces of acquisition and consolidation and the weakening forces of forgetting in forming a memory engram. Acquisition leads to molecular and cellular memory traces in engram cells that encode memory (Figure 1). These are stabilized, and evolve, by the process of consolidation, leading to a rather stable memory shown in this case to occur at 2 days after acquisition. Active forgetting provides a balance to these biological processes by eroding memory traces. Traditional studies in the neuroscience of learning and memory have emphasized processes underlying acquisition and consolidation. More recently, biological insights into the mechanisms for active forgetting have been made.
There are at least two other factors that weigh into any consideration of active forgetting. First, memory comes in different temporally and mechanistically distinct forms that are represented by distinct engrams. Acquisition leads to short-term memories (STM) that persist for seconds or minutes, intermediate-term memories that persist for a few hours, and long-term memory (LTM) that can persist for years or decades. An early declarative memory might be encoded by a group of hippocampal neurons but the engram becomes distributed to engram cells in other brain regions through the process of systems consolidation (Kandel et al., 2014; Dudai et al., 2015; Rubin et al., 2015; Attardo et al., 2015). Although rudimentary, our current knowledge of active forgetting suggests that mechanisms for active forgetting for different temporal forms of memory may be distinct. Second, there are multiple types of memory that can overlap temporally but are mechanistically different. These include working memory and short-term episodic and classical conditioning memory to name a few. It also seems likely based on current evidence discussed below that different mechanisms will be employed to erode these distinct types of memory.
Constructs of forgetting from experimental psychology
Experimental psychologists have debated for more than 50 years whether forgetting occurs through an active involvement of external or internal factors or whether it occurs through passive mechanisms. Although the constructs for forgetting from psychological studies proposed by different researchers have fuzzy edges, the term passive forgetting has often been used to describe the biological decay of memory traces due to constitutive molecular turnover (natural decay); the incidental accumulation across time of similar memory traces that impede proper retrieval of the wanted one; and changes in the memory’s context between acquisition and retrieval that impairs efficient recall (Figure 1; Wixted, 2004; Anderson and Hanslmayr, 2014; Baddley et al., 2015; Ricker et al., 2015). The psychological viewpoint of passive forgetting does not consider the brain as having the capacity to actively degrade the substrates of memory, even though it is widely accepted as the biological machine that forms and stores memory.
An alternative point of view is that forgetting is active and triggered by defined external or internal factors (Figure 1; Wixted, 2004; Ricker et al., 2015). Interference-based forgetting has been widely studied in experimental contexts and posits that brain activity due to new information presented prior to the learning event (proactive interference) or after the learning event (retroactive interference) attenuates memory expression. Wixted (2004) generalized this idea with the notion that normal mental activity and memory formation utilize the resources of the hippocampus and interfere with the process of consolidation, providing an explanation for retroactive interference based on the failure to develop, maintain or transfer the appropriate memory traces. This forgetting mechanism is active in the sense that it requires a trigger, the retroactive interfering stimulus, but does not embrace the idea that the brain is built with biological mechanisms to erode memory traces. Rather, it emphasizes a possible process that interferes with the forces of consolidation to stabilize memories.
Nevertheless, the majority of the psychological literature has focused on the model whereby interfering stimuli create a problem with retrieval due to accumulated and competing memory traces (Figure 1; Bahrick et al., 1991; Anderson and Neely, 1996; Anderson, 2003), placing it in the passive, “retrieval interference” category as noted above. For instance, one might fail to retrieve on a particular day the location of one’s car in the parking lot used daily due to competition from similar memory traces accumulated across the prior month. There is no clear active process underlying this failure of memory retrieval and so it can be considered passive in nature. However, competing memories formed shortly before or after acquisition of another may also interfere actively with memory storage as discussed above. Thus, forgetting through interference mechanisms can be viewed as both passive and active depending on when the interfering material was learned and whether it maintains or erodes memory traces (Figure 1).
Motivated forgetting refers to forgetting processes under our own cognitive control and ones that generally contain an emotional motive, such as processes imposed on memories that threaten a positive self-image, run counter to strongly-held beliefs or attitudes, or evoke sadness, guilt, or embarrassment (Anderson and Green, 2001; Anderson and Hanslmayr, 2014; Baddeley et al., 2015). If such memories are recalled during encoding or consolidation, suppressing the memories voluntarily may disrupt the process of forming a stable engram, leading to forgetting (Fawcett et al., 2016). Top-down suppression can also occur during the retrieval of stable memories. Retrieval can be suppressed but the effect is time-limited, suggesting that in this case retrieval processes of an intact memory engram are just temporarily disrupted.
Retrieval-induced forgetting is produced by interference and can be illustrated after learning a series of items in different categories such as orange and lemon within fruits, and vodka and rum within drinks (Anderson and Bjork, 1994; Murayama et al., 2014). The experimental observation made is that practiced recall of selected items from a category weakens the recall the non-practiced items from the same category, and to a greater degree than items from non-practiced categories. This type of forgetting has important implications when extended from the experimental world to everyday life, since it suggests that the mere act of retrieving some memories can be deleterious to others. There is debate over whether retrieval-induced forgetting occurs from inhibition overlaid on the non-practiced items by the recall of target items from the same category (Murayama et al., 2014; Anderson and Hanslmayr, 2014; Wimber et al., 2015), competition from memory traces associated with the same cue during attempted recall (Verde, 2013), strengthening of the memory traces associated with target items during practice recall (Raaijmakers et al., 2012), or changes in the context during the procedure that disables the recall of the non-target items (Jonker et al., 2013). However, a recent and impressive fMRI study of word:picture associations showed a weakening of the blood-flow based memory traces for the non-practiced items, providing support for the hypothesis of forgetting by overlaid inhibition (Wimber et al., 2015). Irrespective of the mechanism for retrieval-induced forgetting, spontaneous recovery can occur with time (Miguez et al., 2014), indicating that a retrieval failure of perhaps an intact engram may often underlie this phenomenon. This does not preclude the possibility that overlaid inhibition may produce a retrieval failure and this failure might subsequently erode the suppressed engram.
These observations and those discussed below lead us to speculate that there exist many different mechanisms for forgetting, some of which affect the integrity of the memory engram and others that disrupt retrieval of relatively intact memory engrams. Although forgetting due to retrieval errors is of high interest, we focus our discussion on forgetting mechanisms that likely disrupt the integrity of the memory engram. We posit that unlike the active forgetting due to retroactive interference that potentially disrupts consolidation described above, that the brain also has the inherent biological capacity to erode memory traces using signaling systems like those used for acquisition and consolidation (Kasai et al., 2010). We term this type of active forgetting, which is unrecognized in the experimental psychology literature, as “intrinsic forgetting” because it functions through molecular signaling systems and neuronal circuits that are intrinsic to the brain (Figure 1, and below). We note, however, that forgetting currently is generally monitored through behavior, and neuroscience-based studies are just nearing the point of being able to fully address the issue of whether memory traces are actually eroded for some types of forgetting. Nevertheless, in some of the discussed cases the accumulated evidence is supportive of the idea that memory traces may be eroded and cause behavioral forgetting. Furthermore, although studies from experimental psychology have provided some concepts about the types of forgetting (Figure 1; Wixted, 2004; Anderson and Hansylmayr, 2014; Ricker et al., 2015; Baddeley et al., 2015), resolving the biological basis for forgetting and providing a robust delineation of each type will only emerge after elucidating the underlying circuit and cellular mechanisms.
The phenomenon of retroactive facilitation (Figure 1) plays an important role in our discussion and account for active forgetting. Several different experiences, including an episode of sleep or rest, or sedation produced by alcohol consumption or benzodiazepine administration, can facilitate the memory of events learned just prior to the sleep or drug experience (Wixted, 2004). Although the consensus of thought has accepted the idea that sleep enhances the process of consolidation (Cipolli et al., 2013), and perhaps plays an active role in selecting the memories to be consolidated (Stickgold and Walker, 2013), an alternative and recent suggestion is that sleep suppresses intrinsic forgetting, leading to enhanced memory expression after an episode of sleep (Berry et al., 2015). Thus, it is possible that facilitating effects of sleep on memory may arise from combined effects of reduced forgetting due to blocking retroactive interference, and enhanced stabilization and/or consolidation.
The viewpoint of non-forgetting of memory traces
Data from several types of experiments are consistent with the concept of non-forgetting of memory traces, with forgetting being due only to retrieval failures. First, many experiments have been reported showing that memory as assayed behaviorally, forgotten due to the passage of time or by treatment with amnestic agents to disrupt consolidation, can be reinstated by “reminders” of weak training trials, exposure to context, or administration of stimulants (Riccio and Richardson, 1984; Deweer and Sara, 1984; Dekeyne et al., 1987; Sara, 2000; de Hoz et al., 2004; Clem and Schiller, 2016). For instance, rats forget the location of food rewards located in a six-place maze three weeks after training. However, this behavioral forgetting is alleviated by a brief exposure to the context in which training took place just before the final test (Deweer and Sara, 1984). Administration of the stimulant amphetamine or cocaine just before testing also facilitates retrieval (Sara, 2000). Observations of this type have led to the conclusion that the engram or some of its components remain sufficiently intact across time to be reconstituted by a brief reminder, despite the complete loss of behavioral performance without a reminder. A counterargument to this conclusion is that a residual engram may exist at the time of testing, weakened by active forgetting such that it is too weak to drive behavioral recall by itself, but strong enough to be strengthened by a reminder. Waiting for a longer period before testing may deteriorate the engram further so that a reminder no longer strengthens it.
Similar observations have been made using extinction training. Extinction after classical conditioning can be induced by multiple presentations of the conditioned stimulus without the unconditioned stimulus. Such treatment extinguishes the conditioned response, but this response often spontaneously resurfaces later, albeit in a weakened form, or it can be renewed by changing the context or reinstated by presenting the unconditioned stimulus (Dunsmoor et al., 2015; Maren, 2015; Izquierdo et al., 2016). These observations suggest that the original engram, or most of it, remains generally intact but is suppressed by extinction training, or practiced retrieval of selected items, and is consistent with the idea that engrams can remain intact despite the lack of behavioral performance. The extreme extension of these observations is that all memory engrams remain intact and forgetting occurs only from the inability to retrieve them properly.
Second, some evidence indicates that biochemical memory traces persist beyond the time at which memory can be successfully retrieved. Li and Richardson (2013) trained 17 day-old rat pups in a tone:electric shock conditioning paradigm. Injection of antagonists to the NMDA receptor prior to such conditioning blocks the acquisition of tone:shock memory. After two weeks, the pups had forgotten the cues as assayed by behavior, but memory could be reacquired in a second round of conditioning. Interestingly, the reacquisition became independent of NMDA receptor activity, consistent with the possibility that some aspect of the NMDA receptor biochemical memory trace instilled during initial conditioning persisted across the forgetting time window. Kim et al., (2012) measured phosphorylated mitogen-activated protein kinase levels in the amygdala in postnatal day 16 and 23 rats after tone:electric shock conditioning. Although the behavioral memory of this conditioning decayed in the P16 rats two days later relative to the P23-trained rats, the elevation in pMAPK levels did not fall. If this biochemical memory trace is a true component of the engram, this would indicate that at least this trace persists in the absence of behavioral expression. Although these results, which are relatively weak relative to the issue at hand, might be interpreted as supporting the idea that memory engrams are not forgotten, they do not distinguish between the possibilities that the engrams are fully intact and unretrievable or only partially intact due to active forgetting.
Third, there are reports of unconscious behaviors that might reflect an intact engram despite the absence of memory revealed by behavioral tests. As one example, Càmara and Fuentemilla (2014) designed a spatial location task in which a series of pictures appeared in one of four locations on a computer screen concomitant with a unique auditory cue. At the time of testing, the human participants when prompted with the auditory cue indicated they had no recollection of the correct location for each picture, yet their eye movements tended to dwell over the correct location. These results can be interpreted as suggesting that a memory engram remained intact, providing guidance over eye movements to the correct location, despite no self-expressed knowledge of the correct location.
The extreme viewpoint of non-forgetting envisions that all memory engrams once represented in the brain continue to be represented, with forgetting being due only to unsuccessful recall. Successful recall probably depends on context, mood state, energy state, the strength of competing mental activities, and other unknown factors. Nevertheless, forgetting will not be understood until we have unambiguous assays (imaging or otherwise) for multiple engrams that can be used to query their integrity across time in parallel with behavioral memory.
The viewpoint of active biological forgetting of memory traces
Although the studies discussed above make clear that interference and other mechanisms cause some memory failure, it is doubtful that they account for all. Indeed, in addition to the discoveries of biological mechanisms for active forgetting discussed in the next section, logic dictates that the brain must be endowed with inherent mechanisms to erase engrams from existence.
First, biological processes, in general, operate with separate and dedicated pathways for synthesis and degradation. Using this logic and the acceptance of biological pathways for the acquisition of memory (Figure 2), it follows that there should be biological pathways for the erasure of memory. Consider the opposing processes of mitosis for cell birth and apoptosis for programmed cell death. The complexity and coordination required for proper cell birth is breathtaking and yet, when too many cells are born, they are not removed by reversing the process of mitosis nor do they deteriorate from the simple passage of time. The complex process of apoptosis, which occurs via several different biochemical pathways, each involving numerous molecular steps, actively removes the extra cells (Green and Reed, 1998). Consider also the cellular process of protein biosynthesis (Zaher and Green, 2009). This cytoplasmic process is extraordinarily complex, consisting of initiation, elongation, and termination, and requires the coordinated interactions of ribosomes, mRNA, aminoacyl tRNAs, and numerous other regulatory components. Protein biosynthesis is balanced by protein degradation, also extraordinarily complex, and consists of targeting unwanted proteins to the lysosome or proteasome for generally non-selective or selective proteolysis, respectively (Hoff et al., 2004; Luzio et al., 2014). Finally, consider the process of protein phosphorylation. This very common post-translational modification represents an important mode of regulating active vs inactive states of proteins and occurs via the action of hundreds of protein kinases. But again, a separate pathway of dephosphorylation via protein phosphatases opposes the forward pathway of phosphorylation (Woolfrey and Dell’Acqua, 2015). Thus, the alignment of biological pathways into separate synthetic and degradative arms offers sound logic to the premise that forgetting is active and occurs through biological pathways that are distinct from those involved in acquisition and consolidation.
Second, the concept of homeostasis of biological systems provides additional logic for the notion of intrinsic forgetting processes. Biological systems, in general, are tuned to function within a specified range and when events bump them away from the normal range, homeostatic mechanisms are engaged to return the system to the proper state. For instance, mammals actively regulate their core body temperature (Morrison, 2016). When the core body temperature falls below a certain level, blood flow to the skin is reduced as one mechanism to conserve heat. When elevated, sweat glands are activated to cool the skin along with other homeostatic mechanisms. Other homeostatic signaling mechanisms are known to counterbalance neural plasticity to stabilize neuronal function (Turrigiano, 2011; Davis, 2013). A conserved and bidirectional form of homeostatic signaling has been documented at the neuromuscular junction in both vertebrates and invertebrates (Davis, 2013). Inhibiting postsynaptic receptor function leads to a compensatory increase in presynaptic release; whereas, manipulations that increase the amount of neurotransmitter packaged into vesicles is counterbalanced by a decreased probability of presynaptic release.
We view intrinsic forgetting in this same biological perspective. Even across a single day, the brain is bombarded with new information that increases excitatory processes for learning. Intrinsic forgetting can be viewed as a homeostatic mechanism to bring the brain back to its basal state. From this perspective, forgetting as mediated by intrinsic forgetting mechanisms may be the default state of the brain; intrinsic forgetting may operate chronically at a low level to slowly remove each newly acquired memory, although its strength may be regulated by internal or external factors. Consolidation would play the role of the judge, allowing memories that are deemed important and worthwhile to remain while allowing the irrelevant ones to be removed by intrinsic forgetting. We can therefore view consolidation as being in constant competition with a “homeostatic” signal from intrinsic forgetting, with the fate of newly acquired memories being determined by the winner. This does not necessarily mean that consolidated memories are immune from intrinsic forgetting. Rather, we imagine that these stabilized memories would be just much more resistant to degradation.
Finally, there are several broad and non-biological based arguments for active forgetting. We do not have a clear understanding of the human brain’s capacity for memory and if limiting, it would make sense that mechanisms would exist to remove irrelevant memories. The clearance of irrelevant memories would open new storage space and facilitate retrieval, making the brain more efficient for error-free retrieval (Rosenzweig et al., 2002). Moreover, it seems intuitive that some level of active forgetting would offer increased fitness by reducing recurring and unwanted situations or unrewarded perseveration (Kraemer and Golding, 1997; Storm, 2011; Brea et al., 2014). One strain of Drosophila named rovers explore their environment actively whereas an allelic variant, named sitters, are much more stationary. Interestingly, rover flies exhibit robust reversal learning-induced forgetting, whereas sitter flies show reduced levels of this type of forgetting (Reaume et al., 2011). The increased forgetting in rover flies may reflect a greater ability to update memory for more flexible food-searching strategies. Finally, it has been speculated that adaptive forgetting might be a trait of creative individuals (Storm and Patel, 2014), allowing them to move quickly from unsuccessful ideas and pursuits to those that are more successful.
The neuroscience of active forgetting
Recent pioneering studies using Drosophila have begun to reverse the focus on acquisition and consolidation with directed dissection of the molecular and cellular biology of active forgetting (Shuai et al., 2010; Shuai et al., 2011a; Berry et al., 2012; Berry et al., 2015; Cervantes-Sandoval et al., 2016). Although this research area is still in its infancy, a few principles have begun to emerge across organisms and a range of memory tasks.
Rac1-dependent forgetting
Rac is a member of the Rho GTPase family of small G proteins (Etienne-Manneville and Hall, 2002; Luo, 2000). In developing neurons, the activation state of Rac influences lamellipodial extensions of growth cones while in adult neurons, it influences the size and shape of synaptic spines by regulating actin polymerization (Heasman and Ridley, 2008). Through such influences, Rac has been reported to alter synaptic plasticity and behavioral memory in several preparations (Haditsch et al., 2009; Oh et al., 2010; Haditsch et al., 2013; Gao et al., 2015; Tejada-Simon, 2015).
In Drosophila, evidence suggests that Rac1 mediates at least four types of active forgetting of olfactory memories, including intrinsic forgetting, interference-induced forgetting, trace memory forgetting, and reversal learning-activated forgetting (Figure 1; Shuai et al., 2010; Shuai et al., 2011a; Dong et al., 2016). The fundamental observations are that expressing a transgene carrying a dominant negative form of Rac1 in the mushroom body neurons (MBn) inhibits intrinsic forgetting of odor:shock pairing, whereas expressing a constitutively active form in the same neurons accelerates forgetting (Shuai et al., 2010). The term “intrinsic forgetting” is used to refer to an active and inherent biological signaling system that likely erodes memory traces in engram cells. In addition, the same authors demonstrated that inhibiting Rac1 activity impairs forgetting caused by retroactive interference from learning an additional odor paired with the electric shock before testing for the original contingency; and reversal learning, which is reversing the contingency between shock and CS+/CS− odor pairs. Since the CS+/CS− odors are reversed from the first to an immediate and subsequent second training session, reversal learning offers a valuable assay for testing the ability of the organism to update memory. Trace conditioning, in which the US is presented at delayed intervals after the CS, is also enhanced in flies expressing a Rac1 dominant negative in the MBn (Shuai et al., 2011a), consistent with a role for this molecule to normally erode the sensory memory for the odor CS.
The interpretation of the experiments described above that employ in vivo expression of transgenes carrying variants of Rac1 is that this molecule itself is a participant in intrinsic forgetting. However, overexpression of transgenes of this family can influence the signaling patterns of other Rho GTPases (Boulter et al., 2010). Although this issue leaves room for some debate about the identity of the Rho family GTPase involved, Rac1 is currently thought to be the major player.
The seminal discovery of Rac1-dependent active forgetting was unexpected in light of conventional thought at the time and offers several important lessons. First, conditioning activates signaling pathways for both the acquisition of memory and for active forgetting. Biochemical experiments demonstrate that training protocols used to instill memory also activate Rac1, with a maximum activation level detected 1 hour after training (Shuai et al., 2010). Conditioning thus establishes a race between the mechanisms for the formation of stable memory and those for active forgetting (Figure 2), with the winner determined by the magnitude and time courses of the competing forces. Rac1-susceptible memories last for several hours and are intrinsically unstable, destined to decay away because learning trials activate Rac1 to erase them. Second, memory formation and forgetting are independent. Forgetting does not occur by reversing the acquisition pathway; acute manipulation of Rac1 has no effect on memory formation but functions specifically on forgetting. Third, more robust STM is not automatically consolidated into LTM. Inhibition of Rac1 activity extends the time course for STM by several-fold. Surprisingly, this extended STM remains sensitive to anesthesia, a signature of STM. Thus, factors other than the STM magnitude and duration dictate the switch to a consolidated form. Finally, this initial discovery put a molecular handle (Rac1 activity) on multiple types of active forgetting previously measured only behaviorally.
Rac1 is known to regulate actin-filament turnover and actin polymerization through multiple pathways (Heasman and Ridley, 2008). Shuai et al., (2010) extended the Rac1-based forgetting pathway by showing that the Rac1-regulated molecules, PAK/LIMK/cofilin, also participate in active forgetting. Constitutively active cofilin slows memory decay and overexpression of constitutively active Rac1 carrying a mutation that prevents its binding to PAK fails to accelerate memory decay. More recently, the Scribble protein was identified through biochemical approaches as a scaffolding protein that assembles a signaling complex in the MBn that includes Rac1, PAK3, and cofilin for mediating active forgetting (Figure 3; Cervantes-Sandoval et al., 2016). When Scribble expression is genetically reduced, memory is enhanced, consistent with its role in active forgetting. In addition, the Scribble signaling complex is genetically downstream of dopamine (DA) inputs into the MBn mediated by the DA receptor, DAMB, and upstream of Rac1 (Cervantes-Sandoval et al., 2016). Importantly, mutations in the DAMB receptor have little effect on memory immediately after conditioning, but dramatically enhance memory a few hours afterwards (Berry et al., 2012). Furthermore, Berry et al., (2012) identified the DAn inputs to the MB that are responsible for active forgetting and showed that these neurons are chronically active, but modulated in intensity (see below) providing an “ongoing” signal for forgetting (Figure 3). Stimulating this chronically active but weak signal after acquisition but before testing causes forgetting; blocking it inhibits forgetting (Berry et al., 2012). This prompts speculation that forgetting is the default pathway, with the brain designed to slowly forget memories that are encoded unless consolidation intervenes and solidifies the memories deemed valuable. In addition, the discovery of DAn that initiate the forgetting signal indicates that “forgetting cells” are part of normal memory circuits. Their function is to stimulate molecular forgetting pathways in engram cells (MBn) (Figure 3). However, the designation of at least a subset of DAn as forgetting cells should not be construed to mean that spurring the process of forgetting is their sole function, just as engram cells and other neurons in the central nervous system have multiple functions (Muller et al., 1987; Krashes et al., 2009; Li et al., 2014). Together, these studies identify a signaling pathway from the DA forgetting cells, to the DAMB receptor expressed on MBn, and to the Scribble scaffolding complex of Rac1/PAK/cofilin to mediate active forgetting.
Figure 3. The Dopamine Neuron/Rac1/Cofilin Signaling Pathway for Active Forgetting.
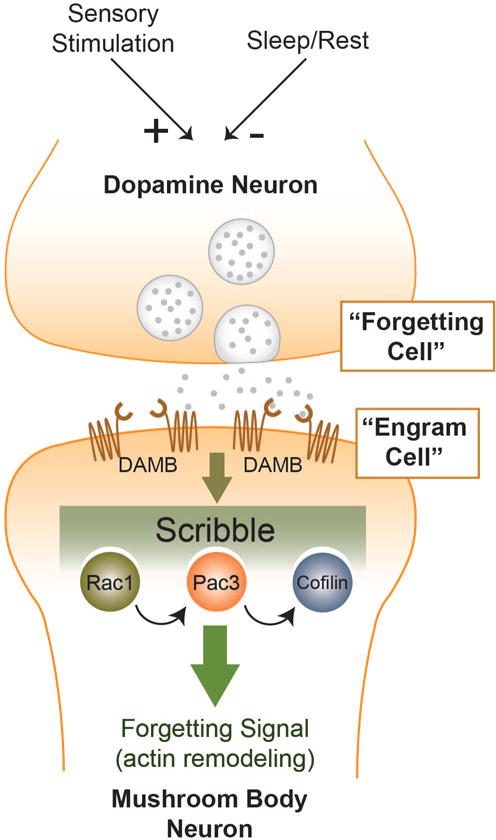
A subset of dopamine neurons that innervate the mushroom body neurons promote forgetting. Although these neurons exhibit chronic activity, they are modulated by external and internal factors with increased sensory stimulation increasing their activity and sleep/rest decreasing their activity. The signal from the dopamine neurons is received by the DAMB receptor expressed on mushroom body neurons. This signal is communicated through a signaling complex scaffolded by the protein Scribble and containing Rac1, Pac3, and Cofilin. Cofilin regulates the remodeling of the actin cytoskeleton to mediate active forgetting. As conceptualized in Figure 1, the mushroom body neurons in this case are “engram cells.” Neurons that promote forgetting (dopamine neurons) in the engram cells are thus termed “forgetting cells.”
Neuronal circuits that activate Rac1-dependent forgetting
So far, studies of Rac1-dependent forgetting in Drosophila have been confined to olfactory memories. Prior studies identified the MBn as critical for the formation and storage of olfactory memories (Heisenberg, 2003; Davis, 2005; Margulies et al., 2005). However, the intrinsic Kenyon cells of the MB are divided into three major types, including the α/β, α′/β′, and γ MBn, and their axonal projections occupy three distinct neuropil regions named lobes. The current evidence suggests that STM relies primarily on the γ MBn while LTM relies on the α/β MBn (Zars et al., 2000; Isabel et al., 2004; Akalal et al., 2006; Blum et al., 2009; Huang et al., 2012; Huang et al., 2013; Xie et al., 2013). Rac1-dependent forgetting of anesthesia-sensitive memory has also been mapped to the α/β and γ MB neurons (Shuai et al., 2010; Berry et al., 2012; Cervantes-Sandoval et al., 2016).
Multiple circuits are capable of activating Rac1-dependent forgetting in the MBn. Among 15 clusters of DAn that exist in the adult fly brain, the PPL1 cluster of 12 neurons contains five distinct types with stereotyped innervation of zones of the MB lobes (Mao and Davis, 2009). This cluster is necessary for acquisition of aversive olfactory memories (Schwaerzel et al., 2003) and artificial stimulation of these neurons in the presence of an odor is sufficient to form aversive olfactory memory (Schroll et al., 2006; Claridge-Chang et al., 2009; Aso et al., 2012). A subset of the same cluster of PPL1 neurons, after learning, regulates the rate of forgetting of both aversive and appetitive olfactory memories (Berry et al., 2012). At present, it is unknown how MBn distinguish a DA-based “acquisition” signal from a “forgetting” signal. Two distinct DA receptors, dDA1 and DAMB, are highly expressed in the MB lobes. The dDA1 receptor is required for both aversive and appetitive olfactory memory formation in adult flies (Kim et al., 2007), whereas DAMB is required nearly exclusively for forgetting (Berry et al., 2012). It may be that different signaling pathways are activated by the two different receptors to mediate acquisition or forgetting. Alternatively, the PPL1 cluster of DAn may include some neurons that promote acquisition and others that promote forgetting, as suggested by the observation that only 3 of the 12 in the cluster are required for active forgetting (Berry et al., 2012). A third possibility is that the signals for acquisition vs forgetting are quantitatively or qualitatively different from one another.
Two additional small clusters of neurons that promote forgetting were identified by screening for enhanced memory retention after inhibiting neural activity among a diverse set of MB extrinsic neurons that connect the MB lobes with other brain regions (Shuai et al., 2015). These include a small cluster of DAn (PAM-β′1) and a pair of glutamatergic neurons that terminate in distinct subdomains of the MB lobes. These neurons are not required for memory formation or retrieval, but appear to be devoted to the regulation of forgetting. Increasing their activity accelerates forgetting and silencing them retards forgetting. Most interestingly, these two clusters of forgetting neurons have more restricted forgetting functions, regulating only intrinsic forgetting without influencing interference-induced forgetting, trace memory forgetting, and forgetting due to reversal learning. This observation emphasizes the conclusion that many forgetting cells must exist dedicated to regulating the active forgetting of different types of memories. The TIR-1/JNK-1-pathway mutants of C. elegans prolong memory, proposed as being due to a blockade in the secretion of forgetting signals from upstream neurons that activate forgetting in engram neurons (Inoue et al., 2013). Thus, forgetting neurons are not unique to Drosophila.
Internal states and external factors that stimulate or inhibit forgetting cells
Some and perhaps all of the neural circuits that promote forgetting are regulated by external factors and internal states, including activity, stress, and sleep (Shuai et al., 2011b; Berry et al., 2015). It is well established that memory is enhanced by a sleep or rest episode after learning in humans and in a wide range of other organisms (Diekelmann and Born, 2010), including in fruit flies (Donlea et al., 2011). However, the underlying mechanisms of sleep-dependent memory enhancement are still being defined. One accepted mechanism is that sleep enhances memory consolidation (Diekelmann and Born, 2010; Stickgold and Walker, 2013). Another possibility, not mutually exclusive with the first, is that sleep and rest inhibit active forgetting (Berry et al., 2015). Indeed, Berry et al., (2015), demonstrated that sleep after learning enhances olfactory memory in Drosophila while suppressing the strength of the forgetting signal imposed on MBn. In addition, they demonstrated that the forgetting-related DA signal is increased robustly with locomotor activity. In the context of the psychological constructs for forgetting (Figure 1), the results from this study indicate that activity and other sensory stimuli increase the activity of DA forgetting cells and presumably Rac1 activity in the downstream MBn, providing a biological explanation for interference-based forgetting. Sleep and rest inhibit the DA forgetting cells, providing a biological explanation for how sleep retroactively facilitates memory by protecting from interference (Figure 1).
Cdc42-dependent forgetting
In Drosophila, a single trial of aversive olfactory conditioning yields at least two operationally distinguishable memory components, anesthesia-sensitive memory (ASM) that persists for a few hours and anesthesia-resistant memory (ARM) that persists for more than 24 hours (Figure 4; Heisenberg, 2003; Davis, 2005; Margulies et al., 2005). Rac1- dependent forgetting regulates the decay of ASM without affecting ARM. Active forgetting of ARM is regulated by another small G protein, Cdc42 (Zhang et al., 2016), a member in the same Rho GTPase family along with Rac. As with Rac1-dependent forgetting, the activity of Cdc42 is increased by conditioning, such that conditioning promotes both acquisition and forgetting. Manipulating Cdc42 activity has no effect on the formation of ARM, but decay of ARM is accelerated with expression of a constitutively active mutant of Cdc42. Conversely, ARM becomes prolonged with expression of a dominant negative mutant Cdc42 in the MBn. Manipulating Cdc42 activity has no effect on the formation or decay of ASM. These observations offer yet another example of memory-type specific forgetting mechanisms.
Figure 4. Small G-protein Involvement in the Active Forgetting of Memory Types in Drosophila.

Memory retention after aversive olfactory classical conditioning is depicted as the solid line and is composed of at least two types of memory, anesthesia-sensitive memory (ASM) and anesthesia-resistant (ARM) memory. Experimental studies have revealed that Rac1 is involved in the biochemistry of forgetting ASM whereas cdc42 is involved in the biochemistry of forgetting ASM.
Cdc42 is essential for filopodia extensions in developing neurons (Heasman and Ridley, 2008) and may regulate the number of dendritic spines in adult animals (Kim et al., 2013; Moutin et al., 2016). Postnatal deletion of Cdc42 leads to deficits in synaptic plasticity and in remote memory recall (Kim et al., 2014). In Aplysia, serotonin stimulates Cdc42 activity to initiate the reorganization of the presynaptic actin network, leading to outgrowth of filopodia for the learning-related formation of new varicosities (Udo et al., 2005).
Rac1-dependent forgetting in vertebrates
Although Rac1 has been reported to regulate synaptic plasticity and memory in vertebrates, it is difficult to align these studies among themselves and with the Drosophila studies because vertebrate memory studies have often failed to measure acquisition, focused on different forms of memory that engage different brain regions, and have employed training protocols extending over several days. For instance, Haditsh et al., (2009) report that removing Rac1 in adult pyramidal neurons using a CaMKIIα>Cre recombinase transgene impairs hippocampal LTP and reduces the rate of acquiring spatial memory across a week of training in the water maze, but this occurred without impairing memory tested subsequently in probe trials. In contrast, Oh et al., (2010) report that genetically enhancing basal Rac1 activity impairs the acquisition of spatial memory and subsequent performance in probe trials. Gao et al., (2015) tested auditory fear conditioning in mice deficient in Rac1 activity in the basolateral amygdala and discovered impaired STM and LTM without altering acquisition. Jiang et al., (2016) have reported that inhibiting Rac in the hippocampus increases memory after massed, contextual fear conditioning, while activating Rac weakens memory produced by spaced, contextual fear conditioning.
A more recent study provided data that are consistent with a conserved role for Rac1 in the active forgetting. Increased activation of Rac1 in the mouse hippocampus through targeted expression of constitutively active mutant Rac1 accelerates decay of object-recognition memory by about 3-fold (Liu et al., 2016). Conversely, inhibition of Rac1 activity by expressing a dominant-negative mutant Rac1 significantly prolongs memory. Inhibiting Rac1 activity also completely blocks interference-induced forgetting. Surprisingly, these perturbations of hippocampal Rac1 activity were without effect on contextual fear conditioned memory or cued (tone:shock) trace memory. This study is consistent with the conclusion that Rac1-dependent forgetting functions in vertebrate organisms. However, the data do hint at an undiscovered complexity of active forgetting. Active forgetting will likely prove to employ multiple signaling systems and cellular processes in cell- and memory-type specific ways.
Neurogenesis-based forgetting
Throughout life, neurons are continuously added to the dentate gyrus with the rate of neurogenesis being modulated with stimulation from enriched environments, physical activity, and other factors. Neurogenesis remodels hippocampal circuits and has been suggested as a mechanism for clearing memory traces (Feng et al., 2001; Deisseroth et al., 2004). Indeed, increasing neurogenesis after memory formation promotes forgetting in adult mice. Infant mice have high levels of hippocampal neurogenesis and exhibit rapid forgetting; reducing the rate of neurogenesis attenuates forgetting (Akers et al., 2014). These studies establish hippocampal neurogenesis as one mechanism for forgetting, seemingly distinct from Rac1- and actin-dependent changes in synaptic structure. Nevertheless, Rac1 may also be involved in neurogenesis-based forgetting. Rac1 is required for a learning-evoked increase in the rate of neurogenesis (Haditsch et al., 2013). Loss of Rac1 exerts no effect on the basal level of neurogenesis, nor does it impact the experience-evoked increase in survival of young neurons, but the number of new neurons formed due to learning is reduced. This learning-evoked, and Rac1-dependent mechanism for neurogenesis may serve as a mechanism for clearing hippocampal memory engrams.
The actin cytoskeleton as the target for Rac1-dependent forgetting
The DAn➔Rac1➔cofilin signaling pathway for active forgetting in MBn may work by modifying the actin cytoskeleton in synaptic structures. One study supporting this idea came from analyzing forgetting of chemotaxis-based associative learning in C. elegans (Hadziselimovic et al., 2014). This associative memory persists for tens of hours, with forgetting depending on the expression of members of Arp2/3 complex, which promotes branching of the actin cytoskeleton. Increasing in the expression of the Arp2/3 complex stimulates actin polymerization leading to the enlargement of synapses and prolonged memory retention. Conversely, reducing expression leads to reduced branching, actin filament depolymerization, synapse shrinkage, and memory loss.
Conditioned place preference memory associated with the addictive stimulant methamphetamine is also lost by disrupting actin depolymerization with the toxin, Latrunculin A, injected into the basolateral amygdala complex after conditioning (Young et al., 2014). This result suggests that methamphetamine-associated memory is maintained by actin-based structural changes of the synapse. A model that the actin cytoskeleton mediates synapse size and Rac1- dependent behavioral forgetting is further supported by a spectacular study of optical erasure of synaptic memory traces in the mouse motor cortex (Hayashi-Takagi et al., 2015). A novel synaptic optoprobe, AS-PaRac1 (activated synapse targeting photoactivatable Rac1) was developed to label and tag learning-potentiated dendritic spines with photoactivatable Rac1 so that they can be shrunk by illumination with light. In vivo imaging of the AS-PaRac1 probe revealed that a motor learning task induces substantial synaptic remodeling in a small subset of neurons. Remarkably, optically activating Rac1 and shrinking the potentiated spines disrupted the acquired motor learning.
AMPA receptor endocytosis-based forgetting
GluA2-dependent AMPA receptor endocytosis has been proposed to be an active process underlying the intrinsic forgetting of long-term potentiation (LTP) and the forgetting of relevant memories (Hardt et al., 2013; Dong et al., 2015). Inhibiting PKMζ in the dorsal hippocampus with the inhibitory peptide, ZIP, abolishes long-term recognition memory for where objects are located without altering recognition memory for object identity (Hardt et al., 2010). This effect seems to be due to the loss of GluA2 receptors by endocytosis, since PKMζ activity maintains synaptic localization of these receptors, hippocampal-based memory is correlated with the abundance of post-synaptic GluA2 receptors, and blocking the internalization of GluA2 after conditioning inhibits normal forgetting (Migues et al., 2010; Migues et al., 2016). Thus, the endocytosis of GluA2 receptors offers an attractive mechanism for active forgetting, despite the controversy around the activity of the ZIP peptide and PKMζ (Sacktor, 2011; Lee et al., 2013; Volk et al., 2013; Tsokas et al., 2016). However, the signaling systems mobilized to remove surface GluA2 receptors remain to be identified. Participants in this may include the activation calcium-dependent protein phosphatases or kinases and insulin stimulation (Beattie et al., 2000; Widago et al., 2015).
Active forgetting, autism spectrum disorders and other brain disorders
Many brain disorders impair cognitive function, including intellectual disability, autism spectrum disorders (ASD), and Alzheimer’s disease (AD) to name but a few. Although each disorder has some unique pathophysiology, alterations in synaptic structure and function seem to be a common thread connecting the disorders. We speculate that altered active forgetting will prove to be fundamental to several of these disorders, but limit our discussion in this perspective to ASD.
Many ASD-risk loci have been identified through population case:control, GWAS, or family based studies (Neale et al., 2012). The genes tentatively associated with these loci function in many different cellular processes, including synaptic organization, synaptic transmission, intracellular signaling, and the regulation of protein synthesis/degradation (Brandler and Sebat, 2015). One systems biology study based on gene-expression analysis of cerebellar samples concluded that Rac1 plays a critical role in the neuropathological events associated with ASD (Zeidán-Chuliá F, et al., 2013). Another recent study found that autism is associated with genetic variation and copy number deletion of P-Rex1, which encodes phosphatidylinositol-3,4,5-trisphosphate-dependent Rac exchange factor 1 (Li et al., 2015). Genetic deletion or knockdown of P-Rex1 in the CA1 region of the mouse hippocampus produces ASD-like behaviors, including impaired social interactions, reversal learning deficits in the water maze, and extinction-resistant memory of fear conditioning. These behavioral changes could be due to impaired active forgetting or in a broader sense, as a failure to update memories.
Dong et al., (2016) probed the question of whether ASD risk genes confer impairments in Rac1-dependent active forgetting. Drosophila mutants in five ASD-associated genes, including Fmr1 (Fragile X mental retardation 1), Ube3a (Ubiquitin-protein ligase E3A), Nrx-1 (Neurexin-1), Nlg4 (Neuroligin 4), and Tsc1 (Tuberous sclerosis complex 1), were chosen to measure behavioral flexibility using reversal learning tests. Although these mutants impair diverse biochemical and cellular functions, they exhibited the common phenotype of behavioral inflexibility in reversal learning tests, attributed by the authors to impairment in Rac1-dependent forgetting. This phenotype may be associated with a subset of ASD risk genes within the spectrum of the disorder, providing a way to distinguish and delineate the spectrum.
Summary and perspectives
One major perspective offered here is that the brain has the inherent capacity to erase memories through the actions of forgetting cells that stimulate molecular forgetting cascades in post-synaptic engram cells. Active forgetting, along with attention, acquisition, and consolidation, is thus part of the brain’s biological system for managing memories. We offer a second perspective that forgetting cells are likely to be part of the circuitry used for the formation of many different types of memory. We accept the existence of cells that form memory traces to store the engram, along with neurons in circuits that convey the essential information for memory formation to these engram cells. We speculate the most of these “learning circuits” will have associated forgetting cells whose function is to erode the memory traces formed in engram cells. A third major perspective offered is that dozens of molecular and cellular pathways likely exist to erode memories. The forgetting pathway(s) operant on a memory of interest may depend on its phase (STM, LTM, etc.) and nature (aversive, appetitive, episodic, ARM, ASM, etc.). The best characterized forgetting pathway, the DAn➔Rac1➔Cofilin pathway functions on early memories formed after olfactory classical conditioning that are sensitive to anesthesia (Figure 5). This pathway along with the cdc42 pathway that functions on early memories that are anesthesia resistant, make up the process we term intrinsic forgetting. These two pathways also promote forgetting due to interfering stimuli presented after conditioning. Neurogenesis, which may also engage Rac activity, promotes forgetting by removing hippocampal neurons that comprise part of a memory engram (Figure 5). And AMPA receptor endocytosis offers yet another mechanism for forgetting although the signaling system for this physiological process has not yet been fully defined. The nature and potency of each forgetting system will influence how fast a memory is forgotten as it competes with stabilizing forces such as consolidation. A fourth perspective offered is that deficits in active forgetting are part of the pathophysiology of human brain disorders. Evidence has emerged suggesting that some types of ASD in both human subjects and mouse genetic models are associated with reversal learning or active forgetting deficits (Micheau et al., 2014; D’Cruz et al., 2016). Such deficits may extend to the strong and disabling memories associated with post-traumatic stress disorder, and the perseveration associated with schizophrenia and other psychiatric disorders. In addition, dissecting the biology of active forgetting promises a wealth of information and a renewed understanding on how the brain manages the incomprehensible amount of information that it processes daily. Finally, it offers the promise of identifying new molecular targets for nootropics, or cognitive enhancers. Antagonists to molecular targets within the DAn➔Rac1➔cofilin or cdc42 pathways, or other more promising pathways yet to be discovered may attenuate forgetting due to intrinsic forgetting or interference.
Figure 5. Biological Mechanisms for Active Forgetting.
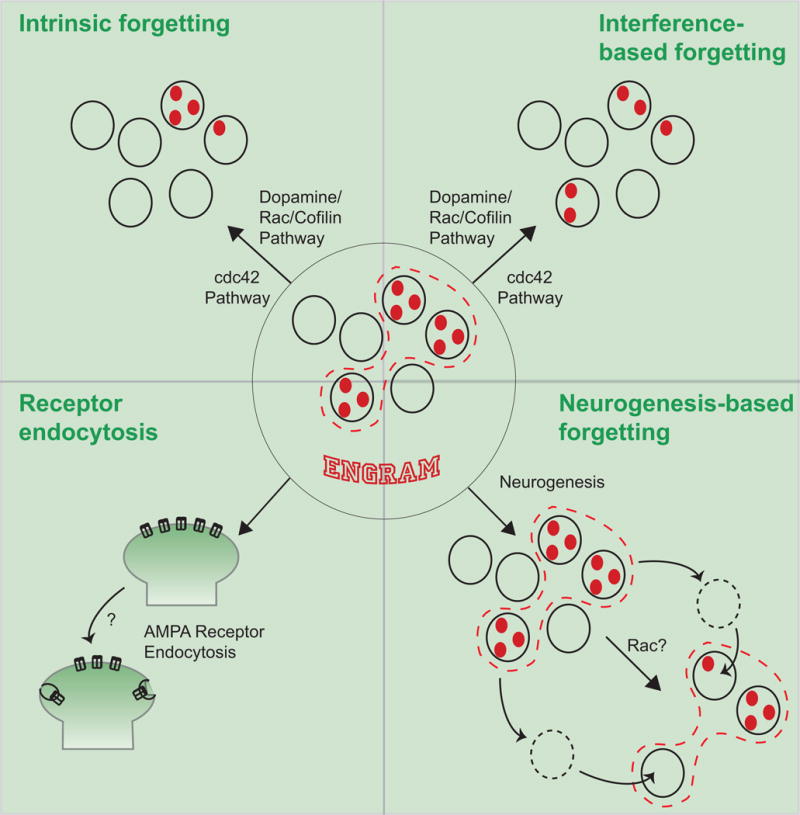
Recent neuroscience studies have identified several biological mechanisms for active forgetting. Two different biochemical pathways, one including Dopamine➔Rac1➔Cofilin and the other involving cdc42 are involved in both intrinsic forgetting and interference-based active forgetting. Although it is likely that both pathways lead to the erosion of the memory engram, this has not yet been shown experimentally. Hippocampal-based memories are subject to active forgetting through neurogenesis degrading existing memory engrams, which may also involve the activity of Rac1. AMPA receptor endocytosis has been hypothesized to offer a fourth active forgetting mechanism operating on some types of memory.
Acknowledgments
We thank Michael Anderson (University of Cambridge) for discussions regarding experimental psychology studies on forgetting. Research in the Zhong laboratory is supported by grants 2013cb835100 from the 973 program of the Ministry of Science and Technology of China, 91332207 from the National Science Foundation of China, and No. Z161100002616010 from Beijing Municipal Science & Technology Commission. Research on forgetting in the Davis laboratory is supported by grants 4R37NS019904, 5R01NS052351 and 1R35NS097224 from the NINDS to R.L.D.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Davis and Zhong review the neuroscience mechanisms for active forgetting, embedding these within types of forgetting established from experimental psychology. Intrinsic forgetting, one type of active forgetting that chronically erodes memory traces may be the default state of the brain.
References
- Akalal DBG, Wilson CF, Zong L, Tanaka NK, Ito K, Davis RL. Roles for Drosophila mushroom body neurons in olfactory learning and memory. Learn Mem. 2006;13:659–668. doi: 10.1101/lm.221206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akers KG, Martinez-Canabal A, Restivo L, Yiu AP, De Cristofaro A, Hsiang HL, Wheeler AL, Guskjolen A, Niibori Y, Shoji H, et al. Hippocampal neurogenesis regulates forgetting during adulthood and infancy. Science. 2014;344:598–602. doi: 10.1126/science.1248903. [DOI] [PubMed] [Google Scholar]
- Alberini CM, Kandel ER. The regulation of transcription in memory consolidation. Cold Spring Harb Perspect Biol. 2015;7:a021741. doi: 10.1101/cshperspect.a021741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MC. Rethinking interference theory: Executive control and the mechanisms of forgetting. J Mem Lang. 2003;49:415–445. [Google Scholar]
- Anderson MC, Bjork RA. In: Mechanisms of Inhibition in Long-Term Memory In Inhibitory Processes in Attention, Memory, and Language. Dagenbach D, Carr T, editors. Academic Press; 1994. pp. 265–325. [Google Scholar]
- Anderson MC, Green C. Suppressing unwanted memories by executive control. Nature. 2001;410:366–369. doi: 10.1038/35066572. [DOI] [PubMed] [Google Scholar]
- Anderson MC, Hanslmayr S. Neural mechanisms of motivated forgetting. Trends Cogn Sci. 2014;6:279–292. doi: 10.1016/j.tics.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MC, Neely JH. Interference and inhibition in memory retrieval. In: Bjork EL, Bjork RA, editors. Memory. Academic Press; 1996. pp. 237–311. [Google Scholar]
- Aso Y, Herb A, Ogueta M, Siwanowicz I, Templier T, Friedrich AB, Ito K, Scholz H, Tanimoto H. Three dopamine pathways induce aversive odor memories with different stability. PLoS Genet. 2012;8:e1002768. doi: 10.1371/journal.pgen.1002768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attardo A, Fitzgerald JE, Schnitzer MJ. Impermanence of dendritic spines in live adult CA1 hippocampus. Nature. 2015;523:592–596. doi: 10.1038/nature14467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baddeley A, Eysenck MW, Anderson MC. Memory. Second. London and New York: Psychology Press; 2015. [Google Scholar]
- Bahrick HP, Hall LK. Preventative and corrective maintenance of access to knowledge. Applied Cognitive Psychology. 1991;5:1–18. [Google Scholar]
- Bailey CH, Bartsch D, Kandel ER. Toward a molecular definition of long-term memory storage. Proc Natl Acad Sci USA. 1996;93:13445–13452. doi: 10.1073/pnas.93.24.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco A, Pittenger C, Kandel ER. CREB, memory enhancement and the treatment of memory disorders: promises, pitfalls and prospects. Expert Opin Ther Targets. 2003;7:101–114. doi: 10.1517/14728222.7.1.101. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka RC. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci. 2000;3:1291–1300. doi: 10.1038/81823. [DOI] [PubMed] [Google Scholar]
- Berry JA, Cervantes-Sandoval I, Chakraborty M, Davis RL. Sleep facilitates memory by blocking dopamine neuron-mediated forgetting. Cell. 2015;161:1656–1667. doi: 10.1016/j.cell.2015.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry JA, Cervantes-Sandoval I, Nicholas EP, Davis RL. Dopamine is required for learning and forgetting in Drosophila. Neuron. 2012;74:530–542. doi: 10.1016/j.neuron.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum AL, Li W, Cressy M, Dubnau J. Short- and long-term memory in Drosophila require cAMP signaling in distinct neuron types. Curr Biol. 2009;19:1341–1350. doi: 10.1016/j.cub.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulter E, Garcia-Mata R, Guilluy C, Dubash A, Rossi G, Brennwald PJ, Burridge K. Regulation of Rho GTPase crosstalk, degradation and activity by RhoGDI1. Nat Cell Biol. 2010;12:477–83. doi: 10.1038/ncb2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandler WM, Sebat J. From de novo mutations to personalized therapeutic interventions in autism. Annu Rev Med. 2015;66:487–507. doi: 10.1146/annurev-med-091113-024550. [DOI] [PubMed] [Google Scholar]
- Brea J, Urbanczik R, Senn W. A normative theory of forgetting: lessons from the fruit fly. PLoS Comput Biol. 2014;10(6):e1003640. doi: 10.1371/journal.pcbi.1003640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camara E, Fuentemilla L. Accessing forgotten memory traces from long-term memory via visual movements. Front Hum Neurosci. 2014;8:930. doi: 10.3389/fnhum.2014.00930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Censor N. Generalization of perceptual and motor learning: a causal link with memory encoding and consolidation? Neuroscience. 2013;250:201–207. doi: 10.1016/j.neuroscience.2013.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes-Sandoval I, Chakraborty M, MacMullen C, Davis RL. Scribble scaffolds a signalosome for active forgetting. Neuron. 2016;90:1230–1242. doi: 10.1016/j.neuron.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolli C, Mazzetti M, Plazzi G. Sleep-dependent memory consolidation in patients with sleep disorders. Sleep Med Rev. 2013;17:91–103. doi: 10.1016/j.smrv.2012.01.004. [DOI] [PubMed] [Google Scholar]
- Claridge-Chang A, Roorda RD, Vrontou E, Sjulson L, Li H, Hirsh J, Miesenbock G. Writing memories with light-addressable reinforcement circuitry. Cell. 2009;139:405–415. doi: 10.1016/j.cell.2009.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem RL, Schiller D. New learning and unlearning: strangers or accomplices in threat memory attenuation? Trends Neurosci. 2016;39:340–351. doi: 10.1016/j.tins.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW. Homeostatic signaling and the stabilization of neural function. Neuron. 2013;80:718–728. doi: 10.1016/j.neuron.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL. Olfactory memory formation in Drosophila: From molecular to systems neuroscience. Annu Rev Neurosci. 2005;28:275–302. doi: 10.1146/annurev.neuro.28.061604.135651. [DOI] [PubMed] [Google Scholar]
- Davis RL. Traces of Drosophila memory. Neuron. 2011;70:8–19. doi: 10.1016/j.neuron.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Cruz AM, Mosconi MW, Ragozzino ME, Cook EH, Sweeney JA. Alterations in the functional neural circuitry supporting flexible choice behavior in autism spectrum disorders. Transl Psychiatry. 2016;6:e916. doi: 10.1038/tp.2016.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoz L, Martin SJ, Morris RG. Forgetting, reminding, and remembering: The retrieval of lost spatial memory. PLoS Biol. 2004;2:1233–1242. doi: 10.1371/journal.pbio.0020225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisseroth K, Singla S, Toda H, Monje M, Palmer TD, Malenka RC. Excitation-neurogenesis coupling in adult neural stem/progenitor cells. Neuron. 2004;42:535–552. doi: 10.1016/s0896-6273(04)00266-1. [DOI] [PubMed] [Google Scholar]
- Dekeyne A, Deweer B, Sara SJ. Background stimuli as a reminder after spontaneous forgetting: Potentiation by stimulation of the mesencephalic reticular formation. Psychobiology. 1987;2:161–166. [Google Scholar]
- Deweer B, Sara SJ. Background stimuli as a reminder after spontaneous forgetting: Role of duration of cuing and cuing-test interval in Animal Learning and Behavior. Psychonomic Society, Inc.; 1984. pp. 238–247. [Google Scholar]
- Diekelmann S, Born J. The memory function of sleep. Nat Rev Neurosci. 2010;11:114–126. doi: 10.1038/nrn2762. [DOI] [PubMed] [Google Scholar]
- Dong T, He J, Wang S, Wang L, Cheng Y, Zhong Y. Inability to activate Rac1-dependent forgetting contributes to behavioral inflexibility in mutants of multiple autism-risk genes. Proc Natl Acad Sci U S A. 2016;113:7644–7649. doi: 10.1073/pnas.1602152113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Han H, Li H, Bai Y, Wang W, Tu M, Peng Y, Zhou L, He W, Wu X, Tan T, Liu M, Wu X, Zhou W, Jin W, Zhang S, Sacktor TC, Li T, Song W, Wang YT. Long-term potentiation decay and memory loss are mediated by AMPAR endocytosis. J Clin Invest. 2015;125:234–247. doi: 10.1172/JCI77888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlea JM, Thimgan MS, Suzuki Y, Gottschalk L, Shaw PJ. Inducing sleep by remote control facilitates memory consolidation in Drosophila. Science. 2011;332:1571–1576. doi: 10.1126/science.1202249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudai Y. Memory, from A to Z. Oxford University Press; New York: 2002. [Google Scholar]
- Dudai Y. The neurobiology of consolidations, or, how stable is the engram? Annu Rev Psychol. 2004;55:51–86. doi: 10.1146/annurev.psych.55.090902.142050. [DOI] [PubMed] [Google Scholar]
- Dudai Y, Karni A, Born J. The consolidation and transformation of memory. Neuron. 2015;88:20–32. doi: 10.1016/j.neuron.2015.09.004. [DOI] [PubMed] [Google Scholar]
- Dunsmoor JE, Niv Y, Daw N, Phelps EA. Rethinking extinction. Neuron. 2015;88:47–63. doi: 10.1016/j.neuron.2015.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- Fawcett JM, Lawrence MA, Taylor TL. The representational consequences of intentional forgetting: Impairments to both the probability and fidelity of long-term memory. J Exp Psychol Gen. 2016;145:56–81. doi: 10.1037/xge0000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng R, Rampon C, Tang YP, Shrom D, Jin J, Kyin M, Sopher B, Martin GM, Kim SH, Langdon RB, Sisodia SS, Tsien JZ. Deficient neurogenesis in forebrain-specific Presenilin-1 knockout mice is associated with reduced clearance of hippocampal memory traces. Neuron. 2001;32:911–926. doi: 10.1016/s0896-6273(01)00523-2. [DOI] [PubMed] [Google Scholar]
- Gao Q, Yao W, Wang J, Yang T, Liu C, Tao Y, Chen Y, Liu X, Ma L. Post-training activation of Rac1 in the basolateral amygdala is required for the formation of both short-term and long-term auditory fear memory. Front Mol Neurosci. 2015;8:65. doi: 10.3389/fnmol.2015.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Gross CG. Geneaology of the “grandmother cell”. Neuroscientist. 2002;8:512–518. doi: 10.1177/107385802237175. [DOI] [PubMed] [Google Scholar]
- Giustino TF, Maren S. The role of the medial prefrontal cortex in conditioning and extinction of fear. Front Behav Neurosci. 2015;9:298. doi: 10.3389/fnbeh.2015.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haditsch U, Anderson MP, Freewoman J, Cord B, Babu H, Brakebusch C, Palmer TD. Neuronal Rac1 is required for learning-evoked neurogenesis. J Neurosci. 2013;33:12229–12241. doi: 10.1523/JNEUROSCI.2939-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haditsch U, Leone DP, Farinelli M, Chrostek-Grashoff A, Brakebusch C, Mansuy IM, McConnell SK, Palmer TD. A central role for the small GTPase Rac1 in hippocampal plasticity and spatial learning and memory. Mol Cell Neurosci. 2009;41:409–419. doi: 10.1016/j.mcn.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadziselimovic N, Vukojevic V, Peter F, Milnik A, Fastenrath M, Fenyves BG, Hieber P, Demougin P, Vogler C, de Quervain DJ, et al. Forgetting is regulated via Musashi-mediated translational control of the Arp2/3 complex. Cell. 2014;156:1153–1166. doi: 10.1016/j.cell.2014.01.054. [DOI] [PubMed] [Google Scholar]
- Hardt O, Migues PV, Hastings M, Wong J, Nader K. PKMζ maintains 1-day- and 6-day-old long-term object location but not object identity memory in dorsal hippocampus. Hippocampus. 2010;20:691–695. doi: 10.1002/hipo.20708. [DOI] [PubMed] [Google Scholar]
- Hardt O, Nader K, Nadel L. Decay happens: the role of active forgetting in memory. Trends Cogn Sci. 2013;17:111–120. doi: 10.1016/j.tics.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Hayashi-Takagi A, Yagishita S, Nakamura M, Shirai F, Wu YI, Loshbaugh AL, Kuhlman B, Hahn KM, Kasai H. Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature. 2015;525:333–338. doi: 10.1038/nature15257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- Heisenberg M. Mushroom body memoir: From maps to models. Nat Rev Neurosci. 2003;4:266–275. doi: 10.1038/nrn1074. [DOI] [PubMed] [Google Scholar]
- Helmstedt TJ, Lattal KM, Wood MA. Reconsolidation and extinction: Using epigenetic signatures to challenge conventional wisdom. Neurobiol Learn Mem. 2017:S1071–7427. doi: 10.1016/j.nlm.2017.01.007. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff H, Zhang H, Sell C. Protein degradation by the proteasome. Methods Mol Biol. 2004;285:79–92. doi: 10.1385/1-59259-822-6:079. [DOI] [PubMed] [Google Scholar]
- Huang C, Wang P, Xie Z, Wang L, Zhong Y. The differential requirement of mushroom body alpha/beta subdivisions in long-term memory retrieval in Drosophila. Protein Cell. 2013;4:512–519. doi: 10.1007/s13238-013-3035-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Zheng X, Zhao H, Li M, Wang P, Xie Z, Wang L, Zhong Y. A permissive role of mushroom body alpha/beta core neurons in long-term memory consolidation in Drosophila. Curr Biol. 2012;22:1981–1989. doi: 10.1016/j.cub.2012.08.048. [DOI] [PubMed] [Google Scholar]
- Inoue A, Sawatari E, Hisamoto N, Kitazono T, Teramoto T, Fujiwara M, Matsumoto K, Ishihara T. Forgetting in C. elegans is accelerated by neuronal communication via the TIR-1/JNK-1 pathway. Cell Rep. 2013;3:808–819. doi: 10.1016/j.celrep.2013.02.019. [DOI] [PubMed] [Google Scholar]
- Isabel G, Pascual A, Preat T. Exclusive consolidated memory phases in Drosophila. Science. 2004;304:1024–1027. doi: 10.1126/science.1094932. [DOI] [PubMed] [Google Scholar]
- Izquierdo I, Furini CR, Myskiw JC. Fear memory. Physiol Rev. 2016;96:695–750. doi: 10.1152/physrev.00018.2015. [DOI] [PubMed] [Google Scholar]
- Jiang L, Mao R, Zhou Q, Yang Y, Cao J, Ding Y, Yang Y, Zhang X, Li L, Xu L. Inhibition of Rac1 activity in the hippocampus impairs the forgetting of contextual fear memory. Mol Neurobiol. 2016;53:1247–1253. doi: 10.1007/s12035-015-9093-6. [DOI] [PubMed] [Google Scholar]
- Johansen JP, Cain CK, Ostroff LE, LeDoux JE. Molecular mechanisms of fear learning and memory. Cell. 2011;147:509–524. doi: 10.1016/j.cell.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonides J, Lewis RL, Nee DE, Lustig CA, Berman MG, Moore KS. The mind and brain of short-term memory. Annu Rev Psychol. 2008;59:193–224. doi: 10.1146/annurev.psych.59.103006.093615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonker TR, Seli P, MacLeod CM. Putting retrieval-induced forgetting in context: an inhibition-free, context-based account. Psychol Rev. 2013;120:852–872. doi: 10.1037/a0034246. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Dudai Y, Mayford MR. The molecular and systems biology of memory. Cell. 2014;157:163–186. doi: 10.1016/j.cell.2014.03.001. [DOI] [PubMed] [Google Scholar]
- Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, Noguchi J. Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 2010;33:121–129. doi: 10.1016/j.tins.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Kim IH, Wang H, Soderling SH, Yasuda R. Loss of Cdc42 leads to defects in synaptic plasticity and remote memory recall. Elife. 2014;3 doi: 10.7554/eLife.02839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Li S, Hamlin AS, McNally GP, Richardson R. Phosphorylation of mitogen-activated protein kinase in the medial prefrontal cortex and the amygdala following memory retrieval or forgetting in developing rats. Neurobiol Learn Mem. 2012;97:59–68. doi: 10.1016/j.nlm.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Kim Y, Ha CM, Chang S. SNX26, a GTPase-activating protein for Cdc42, interacts with PSD-95 protein and is involved in activity-dependent dendritic spine formation in mature neurons. J Biol Chem. 2013;288:29453–29466. doi: 10.1074/jbc.M113.468801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YC, Lee HG, Han KA. D1 dopamine receptor dDA1 is required in the mushroom body neurons for aversive and appetitive learning in Drosophila. J Neurosci. 2007;27:7640–7647. doi: 10.1523/JNEUROSCI.1167-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer PJ, Golding JM. Adaptive forgetting in animals. Psychon Bull Rev. 1997;4:480–491. [Google Scholar]
- Krashes MJ, DasGupta S, Vreede A, White B, Armstrong JD, Waddell S. A neural circuit mechanism integrating motivational state with memory expression in Drosophila. Cell. 2009;139:416–427. doi: 10.1016/j.cell.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AM, Kanter BR, Wang D, Lim JP, Zou ME, Qiu C, McMahon T, Dadgar J, Fischbach-Weiss SC, Messing RO. Prkcz null mice show normal learning and memory. Nature. 2013;493:416–419. doi: 10.1038/nature11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Chai A, Wang L, Ma Y, Wu Z, Yu H, Mei L, Lu L, Zhang C, Yue W, Xu L, Rao Y, Zhang D. Synaptic P-Rex1 signaling regulates hippocampal long- term depression and autism-like social behavior. Proc Natl Acad Sci U S A. 2015;112:E6964–6972. doi: 10.1073/pnas.1512913112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Richardson R. Traces of memory: reacquisition of fear following forgetting is NMDAr-independent. Learn Mem. 2013;20:174–182. doi: 10.1101/lm.029504.112. [DOI] [PubMed] [Google Scholar]
- Li Z, Liu J, Zheng M, Xu XZS. Encoding of both analog- and digital-like behavioral outputs by one C. elegans interneuron. Cell. 2014;159:751–765. doi: 10.1016/j.cell.2014.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Du S, Lv L, Lei B, Shi W, Tang Y, Wang L, Zhong Y. Hippocampal activation of Rac1 regulates the forgetting of object-recognition memory. Curr Biol. 2016;26:2351–2357. doi: 10.1016/j.cub.2016.06.056. [DOI] [PubMed] [Google Scholar]
- Luo L. Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci. 2000;1:173–180. doi: 10.1038/35044547. [DOI] [PubMed] [Google Scholar]
- Luzio JP, Hackmann Y, Dieckmann NM, Griffiths GM. The biogenesis of lysomsomes and lysosome-related organelles. Cold Spring Harb Perspect Biol. 2014;6(9):a016840. doi: 10.1101/cshperspect.a016840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G, Palmer LC, Gall CM. The likelihood of cognitive enhancement. Pharmacol Biochem Behav. 2011;99:116–129. doi: 10.1016/j.pbb.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Davis RL. Eight different types of dopaminergic neurons innervate the Drosophila mushroom body neuropil: anatomical and physiological heterogeneity. Front Neural Circuits. 2009;3:5. doi: 10.3389/neuro.04.005.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S. Out with the old and in with the new: Synaptic mechanisms of extinction in the amygdala. Brain Res. 2015;1621:231–238. doi: 10.1016/j.brainres.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies C, Tully T, Dubnau J. Deconstructing memory in Drosophila. Curr Biol. 2005;15:R700–R713. doi: 10.1016/j.cub.2005.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaugh JL. Memory–a century of consolidation. Science. 2000;287:248–251. doi: 10.1126/science.287.5451.248. [DOI] [PubMed] [Google Scholar]
- Menzel R, Muller U. Learning and memory in honeybees: from behavior to neural substrates. Annu Rev Neurosci. 1996;19:379–404. doi: 10.1146/annurev.ne.19.030196.002115. [DOI] [PubMed] [Google Scholar]
- Micheau J, Vimeney A, Normand E, Mulle C, Riedel G. Impaired hippocampus-dependent spatial flexibility and sociability represent autism-like phenotypes in GluK2 mice. Hippocampus. 2014;24:1059–69. doi: 10.1002/hipo.22290. [DOI] [PubMed] [Google Scholar]
- Migues PV, Hardt O, Wu DC, Gamache K, Sacktor TC, Wang YT, Nader K. PKMζ maintains memories by regulating GluR2-dependent AMPA receptor trafficking. Nat Neurosci. 2010;13:630–634. doi: 10.1038/nn.2531. [DOI] [PubMed] [Google Scholar]
- Migues PV, Liu L, Archbold GE, Einarsson EO, Wong J, Bonasia K, Ko SH, Wang YT, Hardt O. Blocking synaptic removal of GluA2-containing AMPA receptors prevents the natural forgetting of long-term memories. J Neuro. 2016;36:3481–3494. doi: 10.1523/JNEUROSCI.3333-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miguez G, Mash LE, Polack CW, Miller RR. Failure to observe renewal following retrieval-induced forgetting. Behav Processes. 2014;103:43–51. doi: 10.1016/j.beproc.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SF. Central control of body temperature [version 1; referees: 3 approved] F1000Research 1–10. 2016 doi: 10.12688/f1000research.7958.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutin E, Nikonenko I, Stefanelli T, Wirth A, Ponimaskin E, De Roo M, Muller D. Palmitoylation of cdc42 promotes spine stabilization and rescues spine density deficit in a mouse model of 22q11.2 Deletion Syndrome. Cereb Cortex. 2016 doi: 10.1093/cercor/bhw183. [DOI] [PubMed] [Google Scholar]
- Muller RJ, Kubie JL, Ranck JB. Spatial firing patterns of hippocampal complex-spike cells in a fixed environment. J Neurosci. 1987;7:1935–1950. doi: 10.1523/JNEUROSCI.07-07-01935.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama K, Miyatsu T, Buchli D, Storm BC. Forgetting as a consequence of retrieval: a meta-analytic review of retrieval-induced forgetting. Psychol Bull. 2014;140:1383–1409. doi: 10.1037/a0037505. [DOI] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, Polak P, Yoon S, Maguire J, Crawford EL, Campbell NG, Geller ET, Valladares O, Schafer C, Liu H, Zhao T, Cai G, Lihm J, Dannenfelser R, Jabado O, Peralta Z, Nagaswamy U, Muzny D, Reid JG, Newsham I, Wu Y, Lewis L, Han Y, Voight BF, Lim E, Rossin E, Kirby A, Flannick J, Fromer M, Shakir K, Fennell T, Garimella K, Banks E, Poplin R, Gabriel S, DePristo M, Wimbish JR, Boone BE, Levy SE, Betancur C, Sunyaev S, Boerwinkle E, Buxbaum JD, Cook EH, Jr, Devlin B, Gibbs RA, Roeder K, Schellenberg GD, Sutcliffe JS, Daly MJ. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newey SE, Velamoor V, Govek EE, Van Aelst L. Rho GTPases, dendritic structure, and mental retardation. J Neurobiol. 2005;64:58–74. doi: 10.1002/neu.20153. [DOI] [PubMed] [Google Scholar]
- Oh D, Han S, Seo J, Lee JR, Choi J, Groffen J, Kim K, Cho YS, Choi HS, Shin H, et al. Regulation of synaptic Rac1 activity, long-term potentiation maintenance, and learning and memory by BCR and ABR Rac GTPase-activating proteins. J Neurosci. 2010;30:14134–14144. doi: 10.1523/JNEUROSCI.1711-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaijmakers JG, Jakab E. Retrieval-induced forgetting without competition: testing the retrieval specificity assumption of the inhibition theory. Mem Cognit. 2012;40:19–27. doi: 10.3758/s13421-011-0131-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaume CJ, Sokolowski MB, Mery F. A natural genetic polymorphism affects retroactive interference in Drosophila melanogaster. Proc Biol Sci. 2011;278:91–98. doi: 10.1098/rspb.2010.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio DC, Richardson R. The status of memory following experimentally induced amnesia: Gone, but not forgotten. Physiol Psychology. 1984;12:59–72. [Google Scholar]
- Ricker TJ, Vergauwe E, Cowan N. Decay theory of immediate memory: from Brown (1958) to today (2014) Q J Exp Psychol (Hove) 2015:1–27. doi: 10.1080/17470218.2014.914546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig ES, Barnes CA, McNaughton BL. Nat Neurosci. 2002;5:6–8. doi: 10.1038/nn0102-6. [DOI] [PubMed] [Google Scholar]
- Rubin A, Geva N, Sheintuch L, Ziv Y. Hippocampal ensemble dynamics timestamp events in long-term memory. Elife. 2015 doi: 10.7554/eLife.12247. pii: e12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacktor TC. How does PKMzeta maintain long-term memory? Nat Rev Neurosci. 2011;12:9–15. doi: 10.1038/nrn2949. [DOI] [PubMed] [Google Scholar]
- Sara SJ. Retrieval and reconsolidation: toward a neurobiology of remembering. Learn Mem. 2000;7:73–84. doi: 10.1101/lm.7.2.73. [DOI] [PubMed] [Google Scholar]
- Schwaerzel M, Monastirioti M, Scholz H, Friggi-Grelin F, Birman S, Heisenberg M. Dopamine and octopamine differentiate between aversive and appetitive olfactory memories in Drosophila. J Neurosci. 2003;23:10495–10502. doi: 10.1523/JNEUROSCI.23-33-10495.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott R, Bourtchuladze R, Gossweiler S, Dubnau J, Tully T. CREB and the discovery of cognitive enhancers. J Mole Neurosci. 2002;19:171–177. doi: 10.1007/s12031-002-0029-z. [DOI] [PubMed] [Google Scholar]
- Schroll C, Riemensperger T, Bucher D, Ehmer J, Völler T, Erbguth K, Gerber B, Hendel T, Nagel G, Buchner E, Fiala A. Light-induced activation of distinct modulatory neurons triggers appetitive or aversive learning in Drosophila larvae. Curr Biol. 2006;16:1741–1747. doi: 10.1016/j.cub.2006.07.023. [DOI] [PubMed] [Google Scholar]
- Seger CA, Miller EK. Category learning in the brain. Ann Rev Neurosci. 2010;33:203–219. doi: 10.1146/annurev.neuro.051508.135546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shema R, Sacktor TC, Dudai Y. Rapid erasure of long-term memory associates in the cortex by an inhibitor of PKM zeta. Science. 2007;317:951–953. doi: 10.1126/science.1144334. [DOI] [PubMed] [Google Scholar]
- Shuai Y, Hirokawa A, Ai Y, Zhang M, Li W, Zhong Y. Dissecting neural pathways for forgetting in Drosophila olfactory aversive memory. Proc Natl Acad Sci U S A. 2015;112:6663–6672. doi: 10.1073/pnas.1512792112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuai Y, Hu Y, Qin H, Campbell RA, Zhong Y. Distinct molecular underpinnings of Drosophila olfactory trace conditioning. Proc Natl Acad Sci U S A. 2011a;108:20201–20206. doi: 10.1073/pnas.1107489109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuai Y, Lu B, Hu Y, Wang L, Sun K, Zhong Y. Forgetting is regulated through Rac activity in Drosophila. Cell. 2010;140:579–589. doi: 10.1016/j.cell.2009.12.044. [DOI] [PubMed] [Google Scholar]
- Shuai Y, Zhang Y, Gao L, Zhong Y. Stress resistance conferred by neuronal expression of dominant-negative Rac in adult Drosophila melanogaster. J Neurogenet. 2011b;25:35–39. doi: 10.3109/01677063.2011.573891. [DOI] [PubMed] [Google Scholar]
- Shuai YC, Lu BY, Hu Y, Wang LZ, Sun K, Zhong Y. Forgetting Is Regulated through Rac Activity in Drosophila. Cell. 2010;140:579–589. doi: 10.1016/j.cell.2009.12.044. [DOI] [PubMed] [Google Scholar]
- Squire LR. Memory and Brain. Oxford University Press; New York: 1987. [Google Scholar]
- Stickgold R, Walker MP. Sleep-dependent memory triage: evolving generalization through selective processing. Nat Neurosci. 2013;16:139–145. doi: 10.1038/nn.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm BC. The Benefit of Forgetting in Thinking and Remembering. Curr Dir Psych Sci. 2011;20:219–295. [Google Scholar]
- Storm BC, Patel TN. Forgetting as a consequence and enabler of creative thinking. J Exp Psychol Learn Mem Cogn. 2014;40:1594–1609. doi: 10.1037/xlm0000006. [DOI] [PubMed] [Google Scholar]
- Tejada-Simon MV. Modulation of actin dynamics by Rac1 to target cognitive function. J Neurochem. 2015;133:767–779. doi: 10.1111/jnc.13100. [DOI] [PubMed] [Google Scholar]
- Tonegawa S, Liu X, Ramirez S, Redondo R. Memory engram cells have come of age. Neuron. 2015;87:918–931. doi: 10.1016/j.neuron.2015.08.002. [DOI] [PubMed] [Google Scholar]
- Tsokas P, Hsieh C, Yao Y, Lesburgueres E, Wallace EJ, Tcherepanov A, Jothianandan D, Hartley BR, Pan L, Rivard B, et al. Compensation for PKMzeta in long-term potentiation and spatial long-term memory in mutant mice. Elife. 2016;5 doi: 10.7554/eLife.14846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu Rev Neurosci. 2011;34:89–103. doi: 10.1146/annurev-neuro-060909-153238. [DOI] [PubMed] [Google Scholar]
- Udo H, Jin I, Kim JH, Li HL, Youn T, Hawkins RD, Kandel ER, Bailey CH. Serotonin-induced regulation of the actin network for learning-related synaptic growth requires Cdc42, N-WASP, and PAK in Aplysia sensory neurons. Neuron. 2005;45:887–901. doi: 10.1016/j.neuron.2005.01.044. [DOI] [PubMed] [Google Scholar]
- Verde MF. Retrieval induced forgetting in recall: Competitor interference revisited. J Exp Psychol Learn Mem Cogn. 2013;39:1433–1448. doi: 10.1037/a0032975. [DOI] [PubMed] [Google Scholar]
- Volk LJ, Bachman JL, Johnson R, Yu Y, Huganir RL. PKM-zeta is not required for hippocampal synaptic plasticity, learning and memory. Nature. 2013;493:420–423. doi: 10.1038/nature11802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SH, Morris RG. Hippocampai-neocortical interactions in memory formation, consolidation, and reconsolidation. Annu Rev Psychol. 2010;61:49–79. doi: 10.1146/annurev.psych.093008.100523. [DOI] [PubMed] [Google Scholar]
- Widagdo J, Chai YJ, Ridder MC, Chau YQ, Johnson RC, Sah P, Huganir RL, Anggono V. Activity-dependent ubiquitination of GluA1 and GluA2 regulates AMPA receptor intracellular sorting and degradation. Cell Rep. 2015;10:783–795. doi: 10.1016/j.celrep.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimber M, Alink A, Charest I, Kriegeskorte N, Anderson MC. Retrieval induces adaptive forgetting of competing memories via cortical pattern suppression. Nat Neurosci. 2015;18:582–589. doi: 10.1038/nn.3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wixted JT. The psychology and neuroscience of forgetting. Annu Rev Psychol. 2004;55:235–269. doi: 10.1146/annurev.psych.55.090902.141555. [DOI] [PubMed] [Google Scholar]
- Woolfrey KM, Dell’Acqua ML. Coordination of protein phosphorylation and dephosophorylation in synaptic plasticity. J Biol Chem. 2015;290:28604–28612. doi: 10.1074/jbc.R115.657262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Huang C, Ci B, Wang L, Zhong Y. Requirement of the combination of mushroom body gamma lobe and alpha/beta lobes for the retrieval of both aversive and appetitive early memories in Drosophila. Learn Mem. 2013;20:474–481. doi: 10.1101/lm.031823.113. [DOI] [PubMed] [Google Scholar]
- Yin JCP, Wallach JS, Del Vecchio M, Wilder EL, Zhou H, Quinn WG, Tully T. Induction of a dominant negative CREB transgene specifically blocks long-term memory in Drosophila. Cell. 1994;79:49–58. doi: 10.1016/0092-8674(94)90399-9. [DOI] [PubMed] [Google Scholar]
- Young EJ, Aceti M, Griggs EM, Fuchs RA, Zigmond Z, Rumbaugh G, Miller CA. Selective, retrieval-independent disruption of methamphetamine-associated memory by actin depolymerization. Biol Psychiatry. 2014;75:96–104. doi: 10.1016/j.biopsych.2013.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaher HS, Green R. Fidelity at the molecular level: lessons from protein synthesis. Cell. 2009;136:746–762. doi: 10.1016/j.cell.2009.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zars T, Fischer M, Schulz R, Heisenberg M. Localization of a short-term memory in Drosophila. Science. 2000;288:672–675. doi: 10.1126/science.288.5466.672. [DOI] [PubMed] [Google Scholar]
- Zeidán-Chuliá F, Rybarczyk-Filho JL, Salmina AB, de Oliveora BH, Noda M, Moreira JC. Exploring the multifactorial nature of autism through computational systems biology: Calcium and the Rho GTPase RAC1 under the spotlight. Neuromolecular Med. 2013;15:364–383. doi: 10.1007/s12017-013-8224-3. [DOI] [PubMed] [Google Scholar]
- Zhang X, Li Q, Wang L, Liu Z, Zhong Y. Cdc42-dependent forgetting regulates repetition effect in prolonging memory retention. Cell Rep. 2016;16:1–9. doi: 10.1016/j.celrep.2016.06.041. [DOI] [PubMed] [Google Scholar]