

Abstract
Notch signaling is aberrantly activated in approximately one third of non-small cell lung cancers (NSCLC). We characterized the interaction between BMS-906024, a clinically relevant Notch gamma secretase inhibitor (GSI), and front-line chemotherapy in preclinical models of NSCLC. Chemosensitivity assays were performed on 14 human NSCLC cell lines. There was significantly greater synergy between BMS-906024 and paclitaxel than BMS-906024 and cisplatin (mean CI value = 0.54 and 0.85, respectively, P = 0.01). On an extended panel of 31 NSCLC cell lines, 25 of which were adenocarcinoma, the synergy between BMS-906024 and paclitaxel was significantly greater in KRAS- and BRAF-wildtype than KRAS- or BRAF-mutant cells (mean CI = 0.43 vs. 0.90, respectively; P = 0.003). Paclitaxel-induced Notch1 activation was associated with synergy between BMS-906024 and paclitaxel in the KRAS- or BRAF-mutant group. Knockdown of mutant KRAS increased the synergy between BMS-906024 and paclitaxel in heterozygous KRAS-mutant cell lines. Among KRAS- or BRAF-mutant NSCLC, there was a significant correlation between synergy and mutant or null TP53 status, as well as between synergy and a low H2O2 pathway signature. Exogenous overexpression of activated Notch1 or Notch3 had no effect on the enhanced sensitivity of NSCLC to paclitaxel by BMS-906024. In vivo studies with cell line- and patient-derived lung adenocarcinoma xenografts confirmed enhanced antitumor activity for BMS-906024 plus paclitaxel versus either drug alone via decreased cell proliferation and increased apoptosis. These results show that BMS-906024 sensitizes NSCLC to paclitaxel, and that wildtype KRAS and BRAF status may predict better patient response to the combination therapy.
Keywords: gamma secretase inhibitor, Notch, KRAS, patient-derived xenograft, non-small cell lung cancer
Introduction
Lung cancer is responsible for nearly one-fourth of all cancer deaths in the United States (1). Non-small cell lung cancer (NSCLC) is the most common subtype, accounting for 85% of lung cancer cases. Although there have been substantial improvements in early detection screening programs and lung cancer prevention (2–4), the majority of NSCLC patients are diagnosed at an advanced stage when potentially curative surgery is no longer an option (5). Fortunately, several breakthroughs have changed how NSCLC is treated. Immune checkpoint inhibitors targeting PD-1 have recently been approved for treating NSCLC patients with high PD-L1 expression following demonstrated success in the clinic (6). Moreover, up to 14% of lung adenocarcinoma (LUAD) patients with tumors harboring EGFR mutations or ALK or ROS1 fusions are treated with targeted therapy (7). As the majority of patients with Stage IV NSCLC are not candidates for these treatments, they receive standard therapy consisting of systemic platinum-based doublet chemotherapy with or without subsequent radiation therapy. Most patients either do not respond or relapse with chemoresistant tumors (8). This underscores the need to identify novel compounds that may improve the antitumor response of NSCLC to conventional cytotoxic agents.
Gamma secretase inhibitors (GSIs) are a class of small molecule Notch inhibitors that have been used recently in cancer clinical trials as single agents or in combination with chemotherapy. GSIs were originally developed with the intent to treat Alzheimer’s disease by decreasing the formation of beta-amyloid in the brain, though clinical trials failed due to high toxicity in this population and/or poor response (9, 10). However, GSIs can be administered safely with tolerable toxicities in cancer patients as shown by phase I clinical trials in solid tumors (11, 12).
Notch signaling is highly deregulated in T-cell acute lymphoblastic leukemia (T-ALL), as well as in solid tumors (13–17). Gain-of-function NOTCH1 mutations have been described in NSCLC (14), which enable ligand-independent release of the active Notch1 intracellular domain (ICD) or disrupt the C-terminal PEST domain, resulting in Notch1 ICD stabilization and sustained Notch signaling. Additionally, 30% of NSCLC cases downregulate Numb expression, a negative regulator of Notch, resulting in extended Notch activation (14). Notch is considered a NSCLC driver because it can contribute directly to lung carcinogenesis. For example, when activated Notch1 is conditionally expressed in the murine alveolar epithelium, it causes pulmonary adenoma formation and significantly hastens Myc-induced adenocarcinoma development (18). Moreover, Notch1 is required for tumor initiation via suppression of p53-mediated apoptosis in KRAS-induced lung adenocarcinoma (19).
BMS-906024 is a potent and selective gamma secretase inhibitor, and prevents the activation of all four Notch receptors with a high oral bioavailability. BMS-906024 demonstrated a low nanomolar half maximal concentration (IC50) in in vitro enzyme assays and cellular Notch reporter assays (20, 21). Oral administration of BMS-906024 in mice bearing T-ALL or breast cancer xenografts resulted in dose-dependent inhibition of tumor growth, with no overt toxicity observed in dosages up to 6 mg/kg (20). Three Phase 1 clinical trials utilizing BMS-906024 for leukemia and solid tumors have completed the enrollment of patients (clinicaltrials.gov: NCT01292655, NCT01363817, and NCT01653470) and the safety, tolerability and efficacy are being assessed.
Cisplatin and paclitaxel are widely used cytotoxic chemotherapies for the treatment of advanced-stage NSCLC. Here we report that combination therapy with paclitaxel and the GSI BMS-906024 showed a high degree of synergy in in vitro assays, and significantly limited the tumor growth rate of NSCLC cell line- and patient-derived xenografts in vivo. Moreover, tumors that were wildtype (WT) for KRAS and BRAF were particularly sensitive to the treatment regimen. The reduction in tumor growth was accompanied by a decrease in expression of the cell proliferation marker Ki67 and an increase in apoptosis. This preclinical work demonstrates that BMS-906024 can elicit a potent enhancement of tumor sensitivity to paclitaxel, and suggests that lung adenocarcinoma patients whose tumors are KRAS- and BRAF-WT are more likely to benefit from this combination therapy.
Materials and Methods
Drugs
BMS-906024 was provided by Bristol-Myers Squibb, RO4929097 was purchased from Selleck Chemicals, and cisplatin and paclitaxel were obtained from the Rutgers Cancer Institute of New Jersey Pharmacy. GSIs were dissolved in DMSO for in vitro studies. BMS-906024 was formulated in 10% vitamin E TPGS, 10% ethanol, and 80% PEG300 for in vivo studies. Cisplatin and paclitaxel were diluted in 0.9% sodium chloride for in vivo use.
Cell culture and reagents
Cell lines used in this study, their source, year obtained, and KRAS, BRAF, EGFR and TP53 mutational statuses are listed in Supplementary Table S1. Cells were cultured as described (22), used in experiments within 2 months after thawing, and were authenticated in January 2017 by short tandem repeat (STR) profiling at the Rutgers New Jersey Medical School Molecular Resource Facility (Newark, NJ), using the GenePrint 24 System (Promega). Data were interpreted as described (23). Cell lines were routinely confirmed to be free of Mycoplasma using the MycoAlert Mycoplasma Detection Kit (Lonza).
Patient-derived xenografts
We generated a patient-derived xenograft (PDX) tumor bank from de-identified NSCLC patient samples collected by the Rutgers Cancer Institute of New Jersey Biospecimen Repository Service through a non-human subjects protocol approved by the Institutional Review Board of Rutgers University (24). PDX T-042, tested at passages 3 and 4, was derived from a human lung adenocarcinoma and confirmed to be WT for KRAS and BRAF. T-042 retained a TP53R273L mutation present in the original patient tumor (24).
Plasmids and transfections
The pcDNA3-Notch1 ICD and pcDNA3.1 control plasmids were kind gifts from Lucio Miele (25). The pCLE-Notch3 ICD and empty vector plasmids were a gift from Nicholas Gaiano (26) (Addgene plasmid #26894, #17703). Cells were transfected using FuGENE HD (Promega) according to the manufacturer’s instructions. Twenty-four hours post-transfection, cells were plated for chemosensitivity MTS assays, while an aliquot was plated for immunoblotting to verify gene overexpression.
siRNA targeting the KRAS-G12C mutation, previously validated (27), and siGENOME Non-Targeting siRNA #2 were purchased from Dharmacon. Cells were transfected with 60 nM siRNA using Lipofectamine RNAiMAX (ThermoFisher) according to the manufacturer’s protocol. Twenty-four hours post-transfection, cells were plated for MTS assays.
Immunoblotting
Western blotting was performed as previously described (28). Xenograft tumor samples were snap-frozen in liquid nitrogen and stored at −80°C. Frozen samples were grinded in liquid N2, lysed and sonicated. Antibody information is provided in Supplementary Table S2. Protein band quantification was done using ImageJ software (NIH).
RNA extraction and real-time reverse transcription PCR (qRT-PCR)
Cells were treated with 100 nM BMS-906024 for 24 hours. One hour prior to RNA extraction, the GSI was replenished after the cells were washed once with calcium-free phosphate-buffered saline. Total RNA was isolated with TRIzol (Invitrogen) according to the manufacturer’s instructions. cDNA was synthesized using the QuantiTect Reverse Transcription Kit (Qiagen). qRT-PCR was conducted using Power SYBR Green PCR Master Mix (Applied Biosystems) on a Stratagene Mx3005p qPCR system (Agilent Technologies) (22). Reactions were done in triplicate.
RT-PCR and restriction fragment length polymorphism (RFLP) analysis
To detect WT or mutant KRAS transcripts, RT-PCR-RFLP was performed as described (27). The sense primer was designed to introduce a base substitution that created a BstNI recognition site in the WT codon 12 (GGT), but not in mutant codon 12 (AGT). PCR products were digested with BstNI (New England Biolabs). GAPDH PCR products and KRAS digestion products were visualized on 3% agarose gels stained with SYBR Safe (ThermoFisher). For the WT KRAS transcript, digestion of the 186-bp PCR product produced 156- and 30-bp fragments, while the mutant KRAS PCR product remained uncleaved.
In vitro chemosensitivity MTS assays
Sensitization to chemotherapy by BMS-906024 was determined by the CellTiter 96 Aqueous Non-Radioactive Cell Proliferation MTS Assay (Promega). Cells were plated in 96-well flat bottom plates and grown for 24 hours, then treated with 0.25x, 0.5x, 1x, 2x, 4x, and 8x IC50 concentrations of cisplatin or paclitaxel or with 0.25x, 0.5x, 1x, 2x, 4x, and 8x 100 nM of BMS-906024 or the combination of cisplatin or paclitaxel with 100 nM BMS-906024 for 72 hours. Cells were incubated in the MTS reagent until wells containing untreated cells had an absorbance at 490 nm of approximately 1.0 after background subtraction (Perkin Elmer 1420 multilabel counter VICTOR3 plate reader). Chemosensitivity was measured by mean values of cytotoxicity, calculated as a percent of cell viability relative to untreated cells. IC50 values were calculated using Microsoft Excel (Microsoft Corporation) and dose-response curves were generated using GraphPad Prism version 4 (GraphPad Software Inc). Synergy between BMS-906024 and cisplatin or paclitaxel was assessed via the Chou-Talalay method to estimate the combination index (CI) using CalcuSyn version 2 software (Biosoft) (29). Because BMS-906024 was not cytotoxic as a single agent, enhanced sensitivity rather than conventional synergy was calculated (30). To estimate the CI, we set the fraction of cells affected to 0.002 at 50 – 800 nM BMS-906024, and to 0.001 at 25 nM BMS-906024. CI values < 0.7 were considered synergistic, with decreasing CI values indicating greater synergy.
Xenograft establishment and in vivo studies
Animal studies were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) guidelines of Rutgers University. Six to 12-week-old female NOD scid gamma (NSG) mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ; The Jackson Laboratory) were utilized. We conducted the experiments with female mice to ensure the conditions were consistent. HCC44 (2 × 106 cells), SKLU1 (2 × 106 cells), H838 (4 × 106 cells), or A549 (0.5 × 106 cells) were injected subcutaneously into both dorsal flanks of NSG mice with 50% Matrigel Basement Membrane Matrix (Corning). PDX T-042 (~2 mm3 fragment) was surgically implanted into both dorsal flanks with 50 μL of Matrigel. Tumors were measured twice weekly using calipers and tumor volume was calculated using the formula V = (width2 × length)/2. When the mean tumor volume was approximately 100 mm3, mice were randomized (6–8 mice per group) and treatment was initiated: (i) vehicles control, (ii) BMS-906024 at 8.5 mg/kg by oral gavage (p.o.), days 1 through 4 of each week for 3 weeks, (iii) cisplatin by intraperitoneal injection (i.p.), day 1 of each week for 3 weeks, (iv) paclitaxel i.p., day 1 of each week for 3 weeks, (v) combination of BMS-906024 and cisplatin or (vi) BMS-906024 and paclitaxel at the aforementioned doses and schedules. Body weight was measured on each treatment day. To avoid drug-drug interactions, BMS-906024 was administered at least one hour after the chemotherapy on the days that required administration of both compounds. Mice were euthanized once tumor size reached IACUC standards (> 1.5 cm), unless otherwise specified.
The BMS-906024 tolerability study consisted of non-tumor bearing NSG mice (4–5 per group) treated with (i) vehicle or BMS-906024 at (ii) 3.5 mg/kg, (iii) 4.5 mg/kg, (iv) 5.5 mg/kg, (v) 6.5 mg/kg, or (vi) 7.5 mg/kg p.o. on days 1 through 4 of each week for 3 weeks. Given that no dose-limiting toxicity was seen in any of the treatment groups, along with the lack of toxicity observed in the combination studies that included BMS-906024 at 8.5 mg/kg, we adopted the 8.5 mg/kg dosage for experiments.
To determine in vivo chemotherapy dosages, xenograft-bearing mice were randomized (3–5 mice per group) and treated with vehicle or varying doses of cisplatin or paclitaxel i.p. on day 1 of each week for 3 weeks. The dosage that decreased tumor volume compared to vehicle control by approximately 50% after 3 weeks of treatment was utilized in future experiments.
Pharmacodynamic studies, histology and immunohistochemistry (IHC)
Harvested xenografts were snap-frozen or formalin fixed and paraffin embedded. Hematoxylin and eosin (H&E) staining and IHC were performed by the Rutgers Cancer Institute of New Jersey Histopathology Shared Resource using an anti-Ki67 antibody (Abcam ab15580, dilution 1:1000). Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was done with a commercial apoptosis detection kit according to manufacturer’s instructions (R&D Systems). The number of cells stained positive for Ki67 or TUNEL were counted from 5 random fields and averaged from the tumors of 3 separate mice for each treatment group under 200X magnification.
Statistical analysis
Data were expressed as the mean ± standard error of the mean (SEM) unless otherwise indicated. Statistical analysis was performed using STATA version 12.0 software (Stata Corporation), GraphPad Prism 4.0, or Microsoft Excel. A Fisher’s exact test was used to compare synergy between groups when the number of cell lines being compared in any quadrant group dropped below five. Otherwise, comparisons between mean synergy levels were done using a Wilcoxon Mann-Whitney test. Comparisons between pathway activity scores were also performed using a Wilcoxon Mann-Whitney test. Correlations between CI values and Notch1 ICD protein levels were assessed by the Pearson correlation coefficient. An unpaired, two-tailed Student’s t-test was also used for comparisons of tumor growth inhibition in the in vivo studies. A paired, two-tailed Student’s t-test was used to compare the IC50 of chemotherapy in cell lines with versus without the GSI or to compare changes in CI values in overexpression experiments. Kaplan-Meier survival analysis and log-rank tests were used to determine and compare the progression-free survival (defined as tumor size < 500 mm3) between groups treated with paclitaxel alone versus paclitaxel plus BMS-906024. A P value less than 0.05 was considered statistically significant.
Results
BMS-906024 potently impairs activation of Notch1 in NSCLC
It was previously reported that BMS-906024 potently inhibits the activation of all four Notch receptors (20). To determine the optimal concentration of BMS-906024 that inhibits the presenilin gamma secretase complex in NSCLC, we measured activated Notch1 ICD protein levels. BMS-906024 reduced Notch1 ICD levels in all six lung cancer cell lines tested at concentrations as low as 5 nM, with maximal depletion at 50 – 100 nM (Fig. 1A, Supplementary Fig. S1A). BMS-906024 at 100 nM, had no effect on total Notch1 (Fig. 1B), and down-regulated Hes1 transcript (Fig. 1C), confirming that Notch signaling was functionally inhibited. We compared the potency of BMS-906024 to RO4929097, another gamma secretase inhibitor used in clinical trials (31–33). BMS-906024 treatment at 50 – 100 nM was functionally similar to RO4929097 treatment at 0.5 – 1 μM (Supplementary Fig. S1B), the concentration of RO4929097 previously reported to maximally inhibit Notch1 activation (34). Therefore, we chose 100 nM of BMS-906024 for subsequent studies.
Figure 1.
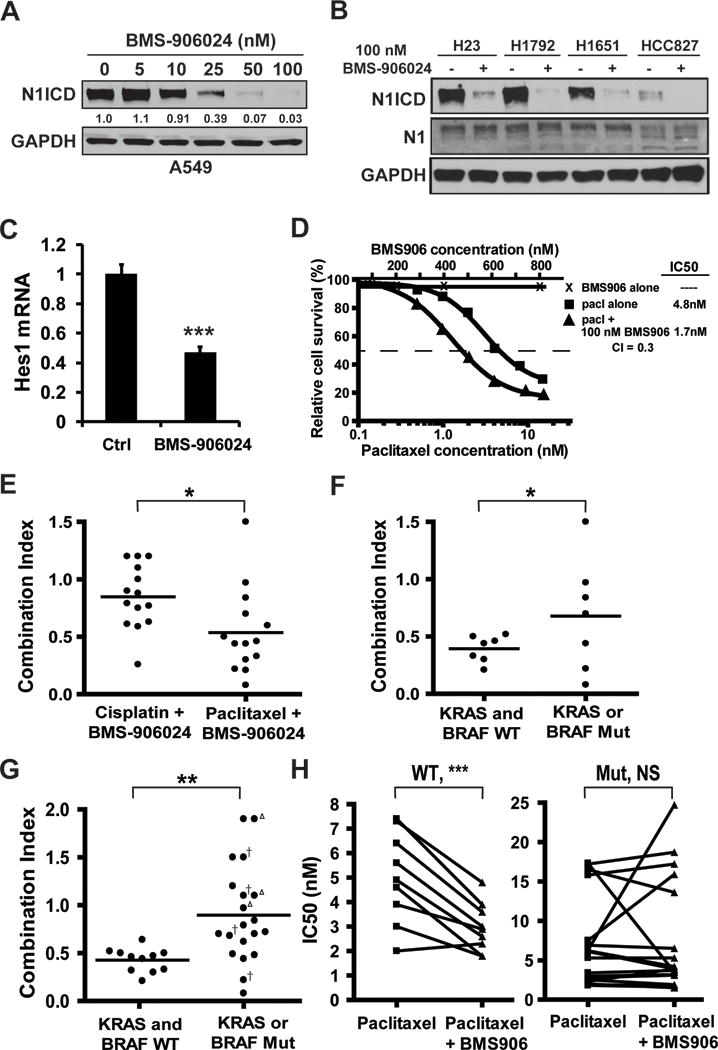
BMS-906024 is a potent inhibitor of Notch activation and enhances the anti-tumor activity of paclitaxel in vitro. A, BMS-906024 treatment of NSCLC cell lines for 72 hrs decreases levels of Notch1 intracellular domain (N1ICD). The change in N1ICD protein levels relative to untreated cells (0 nM) and normalized to GAPDH is indicated below the immunoblot. B, treatment with 100 nM of BMS-906024 for 72 hrs maximally decreases N1ICD without affecting total uncleaved Notch1 (N1). C, downregulation of Hes1 mRNA in A549 NSCLC cells treated with BMS-906024 for 24 hrs. Error bars represent standard deviation. D, chemosensitivity MTS assay performed on H2228 NSCLC cells treated for 72 hrs with BMS-906024 (BMS906), paclitaxel (pacl), or 100 nM BMS-906024 and paclitaxel. E, Combination Index (CI) values from 14 NSCLC cell lines treated with 100 nM BMS-906024 and either cisplatin or paclitaxel for 72 hrs. F, CI values at the IC50 for BMS-906024 and paclitaxel in 14 NSCLC cell lines grouped by KRAS/BRAF status. G, CI values at the IC50 for BMS-906024 and paclitaxel in 31 NSCLC cell lines (including the 14 shown in E) grouped by KRAS/BRAF status. Open triangle, KRAS-dependent cell line; †, KRAS-independent cell line. H, shift in IC50 values for the KRAS- and BRAF-WT (left) or the KRAS- or BRAF-mutant (right) cell lines when treated for 72 hrs with paclitaxel alone or paclitaxel and 100 nM BMS-906024. Outlier cell lines that were paclitaxel-resistant were removed from these graphs (WT: H1838, H1693; mutant: H1395, H2405). *, P < 0.05; **, P < 0.01; ***, P < 0.001. NS, not significant.
BMS-906024 enhances the anti-tumor activity of paclitaxel in vitro
To measure the extent by which BMS-906024 sensitizes lung cancer to paclitaxel or cisplatin, we performed MTS assays on an initial set of 14 NSCLC cell lines, 11 of which were lung adenocarcinoma or bronchioloalveolar carcinoma. BMS-906024 was not cytotoxic in vitro in these cell lines at concentrations as high as 800 nM (Fig. 1D). The IC50 of cisplatin ranged from 0.87 to 15.1 μM (median, 4.2 μM) and the IC50 of paclitaxel ranged from 2.0 to 17.3 nM (median, 5.6 nM), with the exception of one outlier that was 23.1 μM (Supplementary Table S1). We next determined the CI for combined drug treatments based on the method of Chou-Talalay (29), except that synergy was conservatively defined as a CI value below 0.7 rather than 1.0. A representative example of the cytotoxicity curves for the chemosensitivity assays is shown in Fig. 1D. Synergy between cisplatin and BMS-906024 was present in four cell lines and synergy between paclitaxel and BMS-906024 was present in ten cell lines. Significantly more robust synergy was observed between BMS-906024 and paclitaxel than with cisplatin (mean CI value = 0.54 and 0.85, respectively, P = 0.01) (Fig. 1E). Thus, subsequent studies focused on the ability of BMS-906024 to sensitize cells to paclitaxel.
Synergy between BMS-906024 and paclitaxel correlates with KRAS and BRAF status
We explored if the synergy observed between paclitaxel and BMS-906024 is restricted to a specific genetic subtype. We grouped the cell lines by EGFR, TP53 or KRAS/BRAF status. Out of the 14 cell lines, two were EGFR-mutant (CI values were 0.33 and 0.44), and two were TP53-WT (CI values were 0.46 and 1.5). Thus, correlations between synergy and EGFR or TP53 status were not calculated. Synergy (CI < 0.7) was observed in all seven KRAS- and BRAF-WT cell lines, whereas there was synergy in only three of the seven cell lines with KRAS or BRAF gain-of-function mutations (P = 0.03) (Fig. 1F).
To further examine the association between KRAS/BRAF status and synergy between paclitaxel and BMS-906024, we tested 17 additional NSCLC cell lines in which the experimenter was blinded to the genetic background of the samples. As with the first set, BMS-906024 was ineffective as a single agent in every cell line, even at concentrations as high as 800 nM. Out of the 31 samples, there was significantly stronger synergy in KRAS- and BRAF-WT cells (mean CI = 0.43) as compared to KRAS- or BRAF-mutant cells (mean CI = 0.90, P = 0.003) (Fig. 1G). BMS-906024 was synergistic with paclitaxel in all 11 KRAS- and BRAF-WT cell lines, but in only seven of the 20 KRAS- or BRAF-mutant cell lines. Moreover, synergy among cell lines that had a KRAS or BRAF gain-of-function mutation did not depend on “KRAS addiction” status as previously defined (35) (Fig. 1G), nor was there a pattern regarding the specific mutation and drug combination CI value (Supplementary Table S1). The IC50 of paclitaxel generally decreased when combined with 100 nM BMS-906024 in the KRAS- and BRAF-WT subgroup (P = 0.0008), but not in the KRAS or BRAF-mutant cell lines (Fig. 1H). Thus BMS-906024 sensitizes lung adenocarcinoma cells to paclitaxel in vitro, particularly in KRAS- and BRAF-WT samples, suggesting that KRAS/BRAF status may serve as a biomarker for tumor response.
Mechanisms of synergy between BMS-906024 and paclitaxel in NSCLC
Activated Notch1 plays a role in chemoresistance and inhibition of Notch3 cleavage by GSIs has anti-tumor activity in NSCLC (36). Thus, we hypothesized that there would be an association between basal levels of Notch1 ICD or cleaved Notch3 levels and synergy between paclitaxel and BMS-906024. Notch1 ICD and cleaved Notch3 were detectable in 24 (77%) and 17 (55%) of the cell lines, respectively (Fig. 2A). There were no apparent patterns between levels of cleaved Notch3 and synergy. However, in KRAS- and BRAF-WT cell lines, samples with the highest Notch1 ICD levels tended to have more robust synergy (Fig. 2B). In contrast, in the KRAS- or BRAF-mutant cell lines, samples with undetectable or low Notch1 ICD expression levels trended toward having better synergy (Fig. 2C), though overall, the Pearson correlation coefficients were low.
Figure 2.
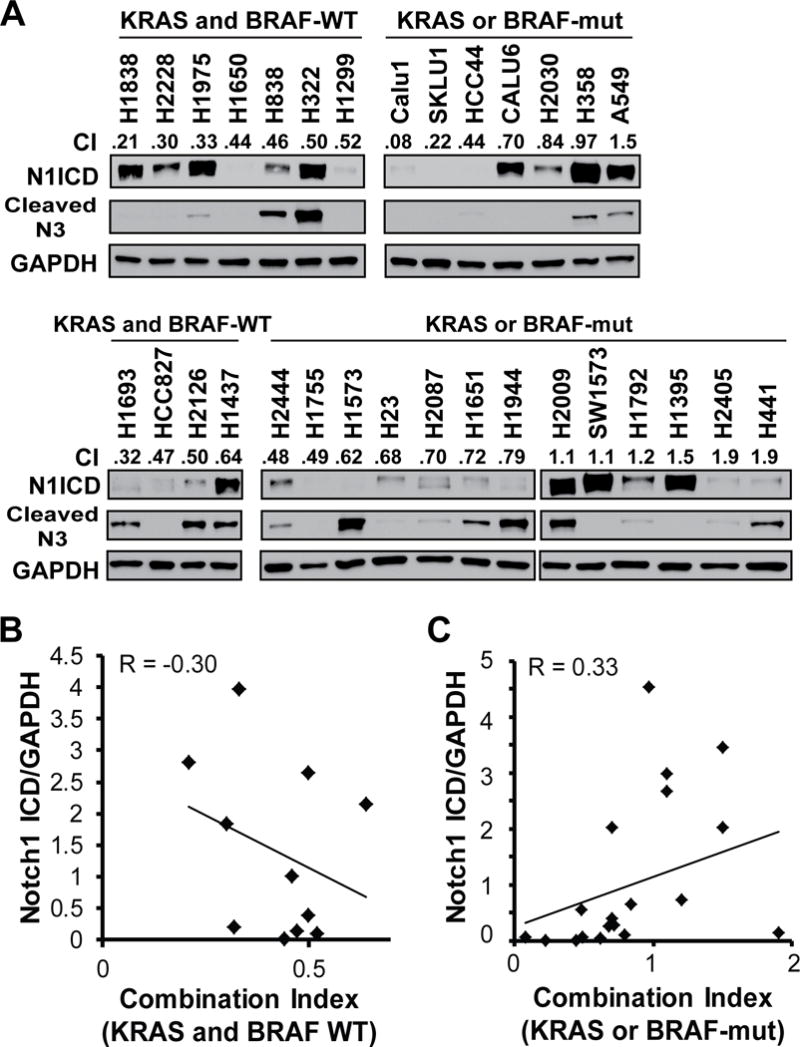
Correlation between activated Notch1 intracellular domain (N1ICD) or cleaved Notch3 and combination index (CI) values for treatment with BMS-906024 and paclitaxel. A, western blot of basal levels of N1ICD and cleaved N3 for all 31 untreated NSCLC cell lines used in the study grouped by KRAS/BRAF status (top: initial set of 14 lines; bottom: additional 17 lines). CI value at the IC50 for paclitaxel and 100 nM BMS-906024 are shown. B, Linear regression of CI values for paclitaxel plus BMS-906024 versus levels of Notch1 ICD normalized to GAPDH in the KRAS and BRAF-WT cell lines. R, Pearson correlation coefficient C, Linear regression of CI values for paclitaxel plus BMS-906024 versus levels of Notch1 ICD normalized to GAPDH in the KRAS or BRAF-mut cell lines. R, Pearson correlation coefficient.
It was previously reported that treatment of colon cancer with the platinum-based drug, oxaliplatin, induced the expression of Notch1 ICD (37). Thus, we surmised that exposure to paclitaxel might induce the activation of Notch1. There was no pattern regarding the effect of paclitaxel on Notch1 ICD in the KRAS- and BRAF-WT cell lines (Supplementary Fig. S2A). However, in the KRAS- or BRAF-mutant background, paclitaxel maintained or induced Notch1 ICD expression in cell lines in which BMS-906024 and paclitaxel combination treatment were synergistic (Supplementary Fig. S2B). In contrast, paclitaxel treatment resulted in Notch1 ICD depletion in cell lines in which the drugs were not synergistic, with the exception of H1792 cells (Supplementary Fig. S2C). These data suggest that the induction of Notch1 ICD by paclitaxel could potentially be a biomarker for enhanced antitumor response for the combination therapy in the setting of KRAS- or BRAF-mutant NSCLC.
We further examined if Notch1 modulates the sensitization of NSCLC to paclitaxel by BMS-906024. If Notch1 is the GSI target responsible for the synergy between the drugs, then exogenous introduction of active Notch1 ICD should abrogate the sensitization of NSCLC to paclitaxel by BMS-906024. To test this, we transiently transfected activated Notch1 ICD then repeated the chemosensitivity MTS assays in cell lines in which the drugs were synergistic, two of which were KRAS- and BRAF-WT and two of which were KRAS- or BRAF-mutant. Surprisingly, exogenous Notch1 ICD expression did not decrease the synergy between BMS-906024 (Supplementary Fig. S3A, B). We also introduced exogenous Notch3 ICD in two cell lines, but Notch3 ICD did not alter the observed synergy (Supplementary Fig. S3C, D). In total, these data suggest that Notch1 and Notch3 are likely not individually responsible for sensitization of a subset of NSCLC cell lines to paclitaxel, though this does not rule out the possibility that a combination of Notch receptors might be responsible for enhanced sensitivity to paclitaxel by BMS-906024.
Notch signaling increases EGFR expression in a p53-dependent manner, leading to increased activation of the KRAS/MAPK pathway (15, 38–40). If paclitaxel decreases KRAS levels, then downregulation of EGFR expression by gamma secretase inhibition could further limit KRAS signaling. Paclitaxel treatment decreased KRAS protein levels in both KRAS-mutant and KRAS-WT cell lines (Fig. 3A). We postulated that silencing of mutant KRAS would enhance the synergy between BMS-906024 and paclitaxel, particularly in KRAS-mutant cell lines in which synergy was not originally observed. When mutant KRAS was knocked down with a G12C-specific siRNA (Fig. 3B), there was increased synergy between paclitaxel and BMS-906024 in heterozygous KRASG12C-mutant cell lines (Fig. 3C), suggesting that, in certain tumors, mutant KRAS might interfere with the ability of BMS-906024 to enhance the antitumor effects of paclitaxel.
Figure 3.

Effect of modulating mutant KRAS expression on synergy between paclitaxel and BMS-906024. A, western blot of Notch1 intracellular domain (N1ICD) and KRAS following 72 hrs of treatment with control (ctrl), 100 nM BMS-906024 (BMS906), cell line-specific IC50 for cisplatin (cispl) or paclitaxel (pacl), or the combination of 100 nM BMS-906024 (BMS) with IC50 for cisplatin or paclitaxel. The change in KRAS protein levels relative to the control and normalized to GAPDH is indicated below the immunoblot. B, Agarose gel verification showing RT-PCR and RFLP analysis of knockdown of mutant KRAS expression in H2030 cells 48 and 72 hrs post-transfection with 60 nM siRNA against mutant KRAS-G12C (siKRAS-G12C). The change in mutant KRAS transcript levels relative to the non-targeting siRNA control and normalized to GAPDH is indicated below the gel. Parental H2030 is shown as an untransfected control. H1299 is shown as a KRAS-WT control. C, CI values at the IC50 for paclitaxel and 100 nM BMS-906024 in two KRAS-G12C mutant NSCLC cell lines in which mutant KRAS was knocked down by siRNA (siKRAS) as compared to a non-targeting siRNA control (Ctrl).
Biomarkers of synergy between BMS-906024 and paclitaxel in NSCLC with gain-of-function KRAS or BRAF mutations
Because there was a wide range of CI values among cell lines with KRAS or BRAF gain-of-function mutations, we set out to identify biomarkers that are associated with synergy within this genetic subgroup. Of note, all 7 KRAS- or BRAF-mutant cell lines in which paclitaxel and BMS-906024 were synergistic were also TP53-mut or -null, whereas all 4 KRAS- or BRAF-mutant cell lines that were TP53-WT showed no synergy between paclitaxel and BMS-906024 (P = 0.04) (Fig. 4A).
Figure 4.
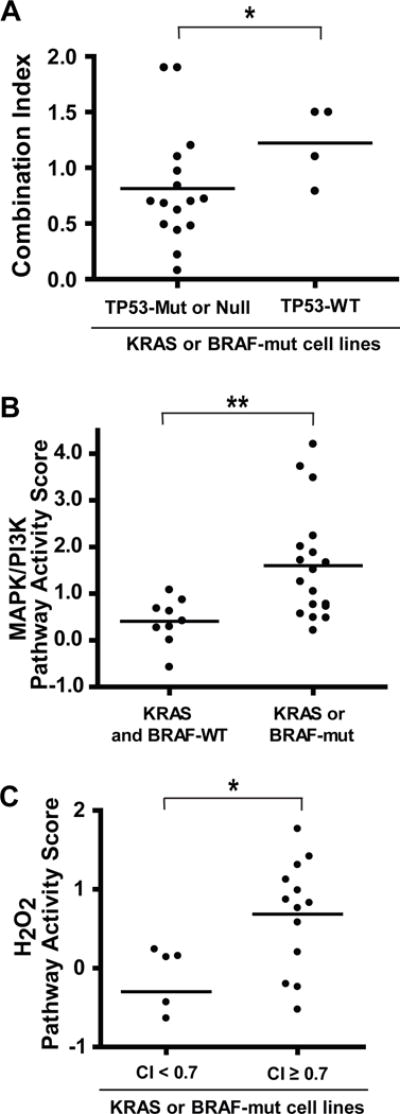
Potential biomarkers for synergy between BMS-906024 and paclitaxel in KRAS- or BRAF-mutant NSCLC. A, combination index (CI) values for BMS-906024 and paclitaxel in the 20 NSCLC cell lines that had KRAS or BRAF gain-of-function mutations, grouped into those that were mutant (Mut) or null versus wildtype (WT) for TP53. B, MAPK/PI3K pathway activity scores for 27 NSCLC cell lines grouped by KRAS/BRAF mutational status. C, H2O2 pathway activity scores among 18 KRAS- or BRAF-mutant cell lines grouped by synergy level. *, P < 0.05, **, P < 0.01.
We also investigated the genomic and basal gene expression data for 27 cell lines in our study. These cell lines had been previously profiled (41), including SPEED pathway activity scores for 11 signaling pathways (42). Each score represents a set of genes that are differentially expressed when a pathway is perturbed and are calculated using independent activation-response experiments. As expected, we observed significantly higher MAPK-PI3K activity in KRAS or BRAF-mutant versus wile-type cell lines (P = 0.005) (Fig. 4B). Amongst the 18 mutant cell lines, we noted lower H2O2 signaling pathway activity in five synergistic cell lines compared to the others (P = 0.035) (Fig. 4C). However, we did not find other genetic markers of response when we examined point mutations, small indels, and copy number variations in genes involved in the Notch pathway or other frequently mutated genes in NSCLC cell lines.
BMS-906024 enhances the antitumor activity of paclitaxel in NSCLC in vivo
We next tested if BMS-906024 sensitizes NSCLC to chemotherapy in vivo. We determined the dose of cisplatin and paclitaxel that reduced tumor growth by 50% (Supplementary Fig. S4). We reasoned that if the chemotherapy was either ineffective or too effective, sensitization of the tumors to the chemotherapy by the GSI could be over- or underestimated, respectively.
In a KRAS- and BRAF-WT PDX-T42, BMS-906024 significantly enhanced the tumor growth inhibition of paclitaxel, but had no significant effect on cisplatin treatment (Fig. 5A). Similarly, in the KRAS- and BRAF-WT H838 xenograft, BMS-906024 significantly enhanced the tumor growth inhibition of paclitaxel, but did not significantly affect the antitumor activity of cisplatin (Fig. 5), consistent with in vitro results. A Kaplan–Meier analysis of tumor progression demonstrated that the paclitaxel group had a significantly shorter event-free survival than the paclitaxel plus BMS-906024 groups (log-rank test, P < 0.0001for T42 and H838; Fig. 5). Tumor growth resumed after treatment stopped (Supplementary Fig. S5A), which was expected since the chemotherapy doses were chosen to examine enhanced tumor growth inhibition rather than overall cure rate. We also performed in vivo tumor growth inhibition assays on three cell lines with gain-of-function KRAS mutations in which the in vitro MTS assays revealed synergy between BMS-906024 and a) both cisplatin and paclitaxel (SKLU1), b) paclitaxel but not cisplatin (HCC44), or c) neither paclitaxel nor cisplatin (A549). In each case, BMS-906024 significantly enhanced the antitumor activity of the chemotherapy in a manner consistent with what was observed in vitro (Supplementary Fig. S5B). The drug combinations were tolerable and did not cause significant weight loss (Supplementary Fig. S5C) or other signs of toxicity, such as diarrhea or distress. Given the high concordance between the in vitro and in vivo data, these results support the conclusion that BMS-906024 enhances the antitumor activity of standard chemotherapy, particularly that of paclitaxel in KRAS- and BRAF-WT NSCLC.
Figure 5.
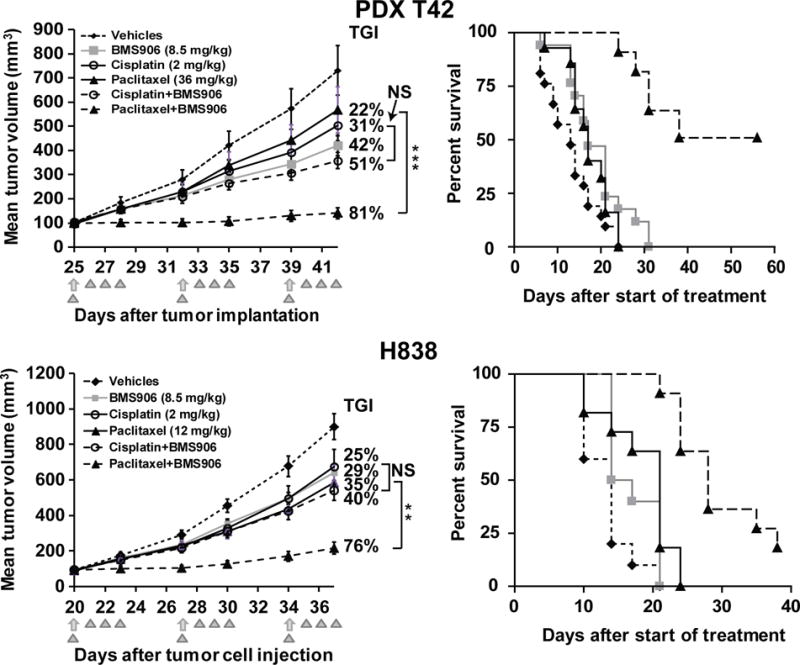
BMS-906024 enhances the anti-tumor activity of chemotherapy in vivo. NSCLC cell line- or patient-derived xenografts (PDX) were treated with vehicles, BMS-906024 (BMS906; triangles), paclitaxel or cisplatin (arrows), or the noted drug combinations as indicated in the insets. Left panels show tumor growth inhibition (TGI). Error bars represent the data range. *, P < 0.05; **, P < 0.01; ***, P < 0.001. NS, not significant. Right panels show Kaplan–Meier survival analysis of vehicles, BMS-906024, paclitaxel and BMS-906024 plus paclitaxel-treated groups in PDX-T42 and H838 xenografts (log rank test for paclitaxel vs. paclitaxel plus BMS-906024-treated groups, P < 0.0001 for both). N = 6–8 mice per group for each experiment.
BMS-906024 enhances the antitumor activity of paclitaxel through increased apoptosis and decreased proliferation
To gain an understanding of the mechanisms by which BMS-906024 sensitizes NSCLC cells to paclitaxel in vivo, we harvested tumors from HCC44 xenograft-bearing mice. As expected, BMS-906024 generally decreased levels of activated Notch1 ICD, as well as the Notch1 target gene SOX9 (22), in the tumors (Fig. 6A). Moreover, BMS-906024 plus paclitaxel induced a 54% reduction in Ki67 positivity and a more than ten-fold increase in TUNEL positivity compared to xenografts that were treated with paclitaxel only (Fig. 6B, C). To further analyze the underlying mechanism mediating reduced tumor growth upon treatment with BMS-906024 and paclitaxel, we examined protein levels of p21 and p57, previously shown to be positively or negatively regulated by Notch signaling, respectively (43, 44). P21 levels were largely unchanged by the treatments; p57 levels decreased in tumors treated with paclitaxel or cisplatin, but p57 expression was not further modified by BMS-906024 (Supplementary Figure S6). Overall, these results indicate that BMS-906024 enhances paclitaxel-mediated cytotoxicity in vivo in NSCLC through a combination of inhibiting proliferation and promoting apoptosis, in a p21 and p57-independent manner.
Figure 6.
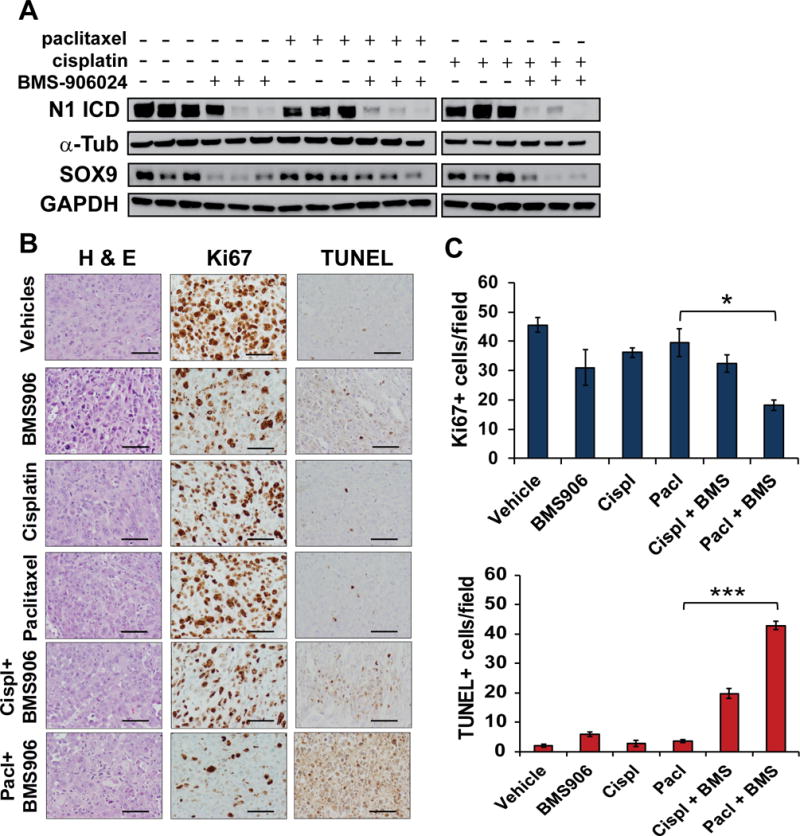
BMS-906024 enhances paclitaxel-induced apoptosis and proliferation inhibition. A, western blot of Notch1 intracellular domain (N1 ICD) and SOX9 protein levels from HCC44 xenografts harvested after four days of treatment. α-Tub and GAPDH were the loading controls for N1 ICD and SOX9, respectively (α-Tub = α-tubulin). B, representative images from hematoxylin and eosin (H & E), Ki67 and TUNEL staining of harvested HCC44 xenografts following one round of treatment with vehicles, BMS-906024 (BMS906), cisplatin (cispl), paclitaxel (pacl), or the combination of BMS-906024 (BMS) and cisplatin or paclitaxel. 200X magnification. Scale bars, 200 μm. C, quantification of Ki67 (top) or TUNEL (bottom) staining. The average number of positive cells per 200X field from five random areas of three tumors each is shown. Error bars represent standard deviation. *, P < 0.05; ***, P < 0.001.
Discussion
Our study evaluated GSI BMS-906024 alone and in combination with cisplatin or paclitaxel, two cytotoxic chemotherapeutic agents widely used in the treatment of NSCLC. Our comprehensive screen of 31 NSCLC cell lines, 25 of which were adenocarcinomas, demonstrated that BMS-906024 sensitizes lung adenocarcinoma cells to paclitaxel in vitro, particularly in KRAS- and BRAF-WT tumors, suggesting that KRAS/BRAF mutation status may serve as a biomarker for tumor response. The wide range of CI values among the KRAS- or BRAF-mutant cell lines suggests that there may be additional biomarkers that could be associated with synergy for the combination of paclitaxel and BMS-906024 in this genetic subgroup. We noted that TP53 status and an H2O2 pathway signature correlated with synergy in the KRAS- or BRAF-mutant group.
Growth factor stimulation of EGFR activates the KRAS/MAPK pathway, which may play a role in chemoresistance (45) and Notch1 signaling regulates EGFR expression as well as EGFR-dependent ERK activation (15, 39, 40). Consequently, inhibition of Notch activation by BMS-906024 may result in the downregulation of EGFR, which can cause decreased MAPK signaling in KRAS- and BRAF-WT samples and possibly contribute to sensitization to paclitaxel. However, KRAS- or BRAF-mutant cells might be less affected by GSI-mediated Notch inhibition and its influence on ERK activation, which may result in a lack of synergy for the BMS-906024 and paclitaxel combination in certain cases.
If Notch signaling were the main driver of paclitaxel chemoresistance in NSCLC, then inhibition of Notch activation would be responsible for the BMS-906024-induced sensitization of cells to paclitaxel. We expected endogenous activated Notch levels to correlate with GSI sensitivity such that cell lines with higher endogenous activated Notch expression would have greater synergy for the combination of BMS-906024 and paclitaxel compared to samples with undetectable or low Notch ICD. While there was a positive trend in KRAS- and BRAF-WT cell lines, there was an inverse correlation in the KRAS- or BRAF-mutant samples. We ruled out the possibility that ineffectiveness of BMS-906024 to inhibit Notch activation in KRAS- or BRAF-mutant cell lines caused the lack of synergy with paclitaxel by demonstrating maximal inhibition of Notch1 ICD formation in multiple mutant non-synergistic cell lines. Thus, there is no conclusive evidence that basal levels of activated Notch1 or Notch3 predict the degree of synergy between BMS-906024 and paclitaxel.
Chemotherapy treatment can induce Notch activation, which contributes to chemoresistance (37, 46). Consequently, we would expect paclitaxel-induced upregulation of activated Notch expression to correlate with GSI sensitivity. However, paclitaxel treatment maintained or decreased Notch1 ICD levels in the KRAS- and BRAF-WT samples displaying synergy for the combination therapy. In contrast, in the KRAS- or BRAF-mutant background, paclitaxel treatment caused a reduction of Notch1 ICD in some cell lines in which BMS-906024 and paclitaxel combination treatment were not synergistic, while paclitaxel maintained or induced Notch1 ICD expression in cell lines in which the drugs were synergistic. These data suggest that the modulation of Notch1 ICD by paclitaxel could be a biomarker for antitumor response in the setting of KRAS- or BRAF-mutant NSCLC. Given that paclitaxel-induced upregulation of activated Notch expression occurred in mutant cell lines that lacked synergy, additional samples should be tested as well as evaluated for the other Notch receptors in order to investigate this matter further. Aside from the four Notch receptors, there are 87 other known gamma secretase substrates (47) whose GSI-mediated cleavage inhibition may also contribute to enhanced chemosensitivity to paclitaxel. Assessment of the expression levels of each of these gamma secretase targets before and after BMS-906024 plus paclitaxel combination treatment would need to be performed in order to evaluate their role in enhanced chemosensitization and synergy prediction.
BMS-906024 is currently in phase 1 clinical trials to evaluate safety and tolerability as a monotherapy as well as in combination with front-line chemotherapy in both hematological malignancies and solid tumors. Nonetheless, in order to be successful in phase 2 trials, both an understanding of the mechanism behind GSI-induced sensitization to chemotherapy as well as identification of biomarkers that predict a high likelihood of success for this combination treatment is needed. Such knowledge will aid in selection of the patient population, critical for the success of targeted therapy clinical trials. Specifically, our preclinical studies found that BMS-906024 enhanced the antitumor activity of paclitaxel in lung adenocarcinoma models, particularly in KRAS- and BRAF-WT samples, suggesting that KRAS/BRAF status may serve as a biomarker for tumor response. Our data supports the continued development of BMS-906024 combined with cytotoxic chemotherapy for lung adenocarcinoma patients.
Supplementary Material
Acknowledgments
We thank Chrise’s Dare to Dream for their contributions for our research. We thank Lucio Miele and Nicholas Gaiano for their kind gifts of plasmids. We thank Erin Michaud from Bristol-Myers Squibb for her advice regarding in vitro chemosensitivity MTS assays and in vivo drug combination studies. We thank Irina Teplova for breeding the mice. We are appreciative of the Rutgers Cancer Institute of New Jersey Shared Resources, including the Biospecimen Repository Service for providing de-identified patient tumor samples, Functional Genomics for running the ThunderBolts Cancer Panel, and Histopathology for H&E and IHC staining. We are appreciative to Philip Tedeschi, Sonia Dolfi, Aparna Kareddula, Monica Bartucci, and Whitney Petrosky, for their advice throughout this study.
Support: This research was supported by the National Cancer Institute (NCI), National Institutes of Health (NIH) (K22CA140719 and R01CA190578 to SRP, F30CA196103 to KMM), by Bristol-Myers Squibb (to SRP), and Chrise’s Dare to Dream Research Fund (SRP). The Shared Resources of Rutgers Cancer Institute of New Jersey are supported by a Cancer Center Support Grant from the NCI (P30CA072720).
Reference List
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Aberle DR, DeMello S, Berg CD, Black WC, Brewer B, Church TR, et al. Results of the two incidence screenings in the National Lung Screening Trial. N Engl J Med. 2013;369:920–31. doi: 10.1056/NEJMoa1208962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kensler TW, Spira A, Garber JE, Szabo E, Lee JJ, Dong Z, et al. Transforming Cancer Prevention through Precision Medicine and Immune-oncology. Cancer Prev Res (Phila) 2016;9:2–10. doi: 10.1158/1940-6207.CAPR-15-0406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rzyman W, Mulshine JL. Lung cancer screening moving forward. Ann Transl Med. 2016;4:149. doi: 10.21037/atm.2016.04.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas A, Liu SV, Subramaniam DS, Giaccone G. Refining the treatment of NSCLC according to histological and molecular subtypes. Nat Rev Clin Oncol. 2015;12:511–26. doi: 10.1038/nrclinonc.2015.90. [DOI] [PubMed] [Google Scholar]
- 6.Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med. 2016 doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 7.Lindeman NI, Cagle PT, Beasley MB, Chitale DA, Dacic S, Giaccone G, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Mol Diagn. 2013;15:415–53. doi: 10.1016/j.jmoldx.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 8.Tsao AS, Scagliotti GV, Bunn PA, Jr, Carbone DP, Warren GW, Bai C, et al. Scientific Advances in Lung Cancer 2015. J Thorac Oncol. 2016;11:613–38. doi: 10.1016/j.jtho.2016.03.012. [DOI] [PubMed] [Google Scholar]
- 9.Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med. 2013;369:341–50. doi: 10.1056/NEJMoa1210951. [DOI] [PubMed] [Google Scholar]
- 10.Coric V, Salloway S, van Dyck CH, Dubois B, Andreasen N, Brody M, et al. Targeting Prodromal Alzheimer Disease With Avagacestat: A Randomized Clinical Trial. JAMA Neurol. 2015;72:1324–33. doi: 10.1001/jamaneurol.2015.0607. [DOI] [PubMed] [Google Scholar]
- 11.Aster JC, Blacklow SC. Targeting the Notch pathway: twists and turns on the road to rational therapeutics. J Clin Oncol. 2012;30:2418–20. doi: 10.1200/JCO.2012.42.0992. [DOI] [PubMed] [Google Scholar]
- 12.Yuan X, Wu H, Xu H, Xiong H, Chu Q, Yu S, et al. Notch signaling: an emerging therapeutic target for cancer treatment. Cancer Lett. 2015;369:20–7. doi: 10.1016/j.canlet.2015.07.048. [DOI] [PubMed] [Google Scholar]
- 13.Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–71. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 14.Westhoff B, Colaluca IN, D’Ario G, Donzelli M, Tosoni D, Volorio S, et al. Alterations of the Notch pathway in lung cancer. Proc Natl Acad Sci U S A. 2009;106:22293–8. doi: 10.1073/pnas.0907781106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haruki N, Kawaguchi KS, Eichenberger S, Massion PP, Olson S, Gonzalez A, et al. Dominant-negative Notch3 receptor inhibits mitogen-activated protein kinase pathway and the growth of human lung cancers. Cancer Res. 2005;65:3555–61. doi: 10.1158/0008-5472.CAN-04-3132. [DOI] [PubMed] [Google Scholar]
- 16.Pece S, Serresi M, Santolini E, Capra M, Hulleman E, Galimberti V, et al. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol. 2004;167:215–21. doi: 10.1083/jcb.200406140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Purow BW, Haque RM, Noel MW, Su Q, Burdick MJ, Lee J, et al. Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res. 2005;65:2353–63. doi: 10.1158/0008-5472.CAN-04-1890. [DOI] [PubMed] [Google Scholar]
- 18.Allen TD, Rodriguez EM, Jones KD, Bishop JM. Activated Notch1 induces lung adenomas in mice and cooperates with Myc in the generation of lung adenocarcinoma. Cancer Res. 2011;71:6010–8. doi: 10.1158/0008-5472.CAN-11-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Licciulli S, Avila JL, Hanlon L, Troutman S, Cesaroni M, Kota S, et al. Notch1 Is Required for Kras-Induced Lung Adenocarcinoma and Controls Tumor Cell Survival via p53. Cancer Res. 2013 doi: 10.1158/0008-5472.CAN-13-1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gavai AV, Quesnelle C, Norris D, Han WC, Gill P, Shan W, et al. Discovery of Clinical Candidate BMS-906024: A Potent Pan-Notch Inhibitor for the Treatment of Leukemia and Solid Tumors. ACS Med Chem Lett. 2015;6:523–7. doi: 10.1021/acsmedchemlett.5b00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ran Y, Hossain F, Pannuti A, Lessard CB, Ladd GZ, Jung JI, et al. gamma-Secretase inhibitors in cancer clinical trials are pharmacologically and functionally distinct. EMBO Mol Med. 2017;9:950–66. doi: 10.15252/emmm.201607265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Capaccione KM, Hong X, Morgan KM, Liu W, Bishop JM, Liu L, et al. Sox9 mediates Notch1-induced mesenchymal features in lung adenocarcinoma. Oncotarget. 2014;5:3636–50. doi: 10.18632/oncotarget.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Capes-Davis A, Reid YA, Kline MC, Storts DR, Strauss E, Dirks WG, et al. Match criteria for human cell line authentication: where do we draw the line? Int J Cancer. 2013;132:2510–9. doi: 10.1002/ijc.27931. [DOI] [PubMed] [Google Scholar]
- 24.Morgan KM, Riedlinger GM, Rosenfeld J, Ganesan S, Pine SR. Patient-Derived Xenograft Models of Non-Small Cell Lung Cancer and Their Potential Utility in Personalized Medicine. Front Oncol. 2017;7:2. doi: 10.3389/fonc.2017.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, De Marco MA, Graziani I, Gazdar AF, Strack PR, Miele L, et al. Oxygen concentration determines the biological effects of NOTCH-1 signaling in adenocarcinoma of the lung. Cancer Res. 2007;67:7954–9. doi: 10.1158/0008-5472.CAN-07-1229. [DOI] [PubMed] [Google Scholar]
- 26.Dang L, Yoon K, Wang M, Gaiano N. Notch3 signaling promotes radial glial/progenitor character in the mammalian telencephalon. Dev Neurosci. 2006;28:58–69. doi: 10.1159/000090753. [DOI] [PubMed] [Google Scholar]
- 27.Sunaga N, Shames DS, Girard L, Peyton M, Larsen JE, Imai H, et al. Knockdown of oncogenic KRAS in non-small cell lung cancers suppresses tumor growth and sensitizes tumor cells to targeted therapy. Mol Cancer Ther. 2011;10:336–46. doi: 10.1158/1535-7163.MCT-10-0750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hong X, Liu W, Song R, Shah JJ, Feng X, Tsang CK, et al. SOX9 is targeted for proteasomal degradation by the E3 ligase FBW7 in response to DNA damage. Nucleic Acids Res. 2016;44:8855–69. doi: 10.1093/nar/gkw748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–6. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 30.Chou TC. Preclinical versus clinical drug combination studies. Leuk Lymphoma. 2008;49:2059–80. doi: 10.1080/10428190802353591. [DOI] [PubMed] [Google Scholar]
- 31.Strosberg JR, Yeatman T, Weber J, Coppola D, Schell MJ, Han G, et al. A phase II study of RO4929097 in metastatic colorectal cancer. Eur J Cancer. 2012;48:997–1003. doi: 10.1016/j.ejca.2012.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee SM, Moon J, Redman BG, Chidiac T, Flaherty LE, Zha Y, et al. Phase 2 study of RO4929097, a gamma-secretase inhibitor, in metastatic melanoma: SWOG 0933. Cancer. 2015;121:432–40. doi: 10.1002/cncr.29055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pan E, Supko JG, Kaley TJ, Butowski NA, Cloughesy T, Jung J, et al. Phase I study of RO4929097 with bevacizumab in patients with recurrent malignant glioma. J Neurooncol. 2016;130:571–9. doi: 10.1007/s11060-016-2263-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luistro L, He W, Smith M, Packman K, Vilenchik M, Carvajal D, et al. Preclinical profile of a potent gamma-secretase inhibitor targeting notch signaling with in vivo efficacy and pharmacodynamic properties. Cancer Res. 2009;69:7672–80. doi: 10.1158/0008-5472.CAN-09-1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15:489–500. doi: 10.1016/j.ccr.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Konishi J, Kawaguchi KS, Vo H, Haruki N, Gonzalez A, Carbone DP, et al. Gamma-secretase inhibitor prevents Notch3 activation and reduces proliferation in human lung cancers. Cancer Res. 2007;67:8051–7. doi: 10.1158/0008-5472.CAN-07-1022. [DOI] [PubMed] [Google Scholar]
- 37.Meng RD, Shelton CC, Li YM, Qin LX, Notterman D, Paty PB, et al. gamma-Secretase inhibitors abrogate oxaliplatin-induced activation of the Notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer Res. 2009;69:573–82. doi: 10.1158/0008-5472.CAN-08-2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Purow BW, Sundaresan TK, Burdick MJ, Kefas BA, Comeau LD, Hawkinson MP, et al. Notch-1 regulates transcription of the epidermal growth factor receptor through p53. Carcinogenesis. 2008;29:918–25. doi: 10.1093/carcin/bgn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dai J, Ma D, Zang S, Guo D, Qu X, Ye J, et al. Cross-talk between Notch and EGFR signaling in human breast cancer cells. Cancer Invest. 2009;27:533–40. doi: 10.1080/07357900802563036. [DOI] [PubMed] [Google Scholar]
- 40.Baumgart A, Seidl S, Vlachou P, Michel L, Mitova N, Schatz N, et al. ADAM17 regulates epidermal growth factor receptor expression through the activation of Notch1 in non-small cell lung cancer. Cancer Res. 2010;70:5368–78. doi: 10.1158/0008-5472.CAN-09-3763. [DOI] [PubMed] [Google Scholar]
- 41.Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell. 2016;166:740–54. doi: 10.1016/j.cell.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parikh JR, Klinger B, Xia Y, Marto JA, Bluthgen N. Discovering causal signaling pathways through gene-expression patterns. Nucleic Acids Res. 2010;38:W109–W117. doi: 10.1093/nar/gkq424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo D, Ye J, Dai J, Li L, Chen F, Ma D, et al. Notch-1 regulates Akt signaling pathway and the expression of cell cycle regulatory proteins cyclin D1, CDK2 and p21 in T-ALL cell lines. Leuk Res. 2009;33:678–85. doi: 10.1016/j.leukres.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 44.Giovannini C, Gramantieri L, Minguzzi M, Fornari F, Chieco P, Grazi GL, et al. CDKN1C/P57 is regulated by the Notch target gene Hes1 and induces senescence in human hepatocellular carcinoma. Am J Pathol. 2012;181:413–22. doi: 10.1016/j.ajpath.2012.04.019. [DOI] [PubMed] [Google Scholar]
- 45.Zhao Y, Shen S, Guo J, Chen H, Greenblatt DY, Kleeff J, et al. Mitogen-activated protein kinases and chemoresistance in pancreatic cancer cells. J Surg Res. 2006;136:325–35. doi: 10.1016/j.jss.2006.06.031. [DOI] [PubMed] [Google Scholar]
- 46.Liu YP, Yang CJ, Huang MS, Yeh CT, Wu AT, Lee YC, et al. Cisplatin selects for multidrug-resistant CD133+ cells in lung adenocarcinoma by activating Notch signaling. Cancer Res. 2013;73:406–16. doi: 10.1158/0008-5472.CAN-12-1733. [DOI] [PubMed] [Google Scholar]
- 47.Haapasalo A, Kovacs DM. The many substrates of presenilin/gamma-secretase. J Alzheimers Dis. 2011;25:3–28. doi: 10.3233/JAD-2011-101065. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.