

Summary
Dopamine is a neurotransmitter that has been implicated in processes as diverse as reward, addiction, control of coordinated movement, metabolism and hormonal secretion. Correspondingly, dysregulation of the dopaminergic system has been implicated in diseases ranging from schizophrenia, Parkinson’s disease, depression, attention deficit hyperactivity disorder, nausea and vomiting, among others. Dopamine’s actions are mediated by a family of five G-protein coupled receptors (GPCRs) (viz. D1, D2, D3, D4 and D5)1. The D2 dopamine receptor (DRD2) is the primary target for both typical2 and atypical3,4 antipsychotic drugs, and for Parkinson’s disease drugs. Unfortunately, many drugs targeting DRD2 frequently suffer from serious and potentially life-threatening side effects due to promiscuous activities against related receptors4,5. Accordingly, a molecular understanding of DRD2 structure and function could provide a template for the design of safer and more effective medications. Here we provide the crystal structure of DRD2 in complex with the widely prescribed atypical antipsychotic drug risperidone. The DRD2-risperidone structure reveals an unexpected mode of antipsychotic drug binding to dopamine receptors, and illuminates structural determinants essential for the actions of risperidone and related drugs at DRD2.
The D2 dopamine receptor (DRD2) is essential for mediating the actions of antipsychotic drugs2,3,4,6, and also for medications used to treat Parkinson’s disease, hyperprolactinemia, nausea and vomiting, among many other disorders1,7,8. DRD2 has also been implicated in the actions of several drugs of abuse including amphetamines, cocaine and opioids9. Although DRD2 was cloned nearly 30 years ago10,11,12 and has been subject to extensive pharmacological13, mutagenesis14 and molecular modeling studies15, we lack high resolution structures of DRD2 in complex with ligands, impeding a molecular understanding of the receptor’s function. Meanwhile, a 3.2 Å crystal structure of the related D3 dopamine (DRD3) was reported 7 years ago16 while 1.95 and 2.2 Å structures of the D4 dopamine (DRD4) receptors have been more recently reported17. The DRD3 and DRD4 ligand complexes–obtained with the substituted benzamides eticlopride and nemonapride, respectively–revealed distinctive extended binding sites16,17. Given the importance of DRD2-targeted drugs, and recent successes in leveraging GPCR structures for the structure-guided discovery of new probes and drug-leads18,19, the structure of a DRD2 complexed with non-benzamides ligands will not only clarify the specificity determinants of the family, but will also expand our understanding of how different scaffolds interact with dopamine receptors. We anticipate that the ligand discovery enabled by DRD2 structures would thus inform both basic and translational neuroscience20.
Structural studies were carried out using an engineered human DRD2 construct, which included three thermostabilizing mutations (I1223.40A, L3756.37A and L3796.41A), and T4 lysozyme (T4L) fused into intracellular loop 3 (Extended Data Figs.1a-b and METHODS). This construct was purified and crystallized in complex with the atypical antipsychotic risperidone. The binding affinities of multiple antipsychotics were similar with this DRD2 construct versus the wild-type receptor (Extended Data Table 1), suggesting that the engineered alterations which facilitate crystallization do not perturb ligand binding. The crystal structure of the DRD2/risperidone complex was determined at 2.9 Å resolution (Extended Data Table 2 and Extended Data Figs. 1c-h).
Compared with DRD4 (PDB codes: 5WIU and 5WIV) and DRD3 (PDB code: 3PBL), DRD2 displays substantial structural differences in extracellular loops 1 and 2 (EL1 and EL2) and the extracellular ends of Transmembrane helices (TM) V, VI and VII (Figs. 1a-c). Unexpectedly, the largest extracellular loop of DRD2–EL2–flips away from the top of the receptor core (Fig. 1c) when compared with DRD3 and DRD4. Importantly, the highly conserved hydrophobic residue of EL2, which is two residues away from the conserved cysteine of EL2 in all extant aminergic GPCR structures and is represented by Ile184 in DRD2, points towards the receptor core (Extended Data Fig. 2). This residue has been implicated in the on- and off-rate kinetics and in β-arrestin biased signaling for some ligands at DRD2 and other receptors19,20,21. However, because of the rearrangement of EL2 and its formation of a small helical turn (residues 182–185) in the DRD2/risperidone structure (Fig. 1c and Extended Data Fig. 2c), Ile184 does not directly interact with this ligand, as does the analogous residue in some aminergic structures (Extended Data Figs. 2a-l). Instead, Ile184 points across the binding pocket to interact with Trp100 in EL1, forming a network of hydrophobic residues near the binding pocket’s orifice (Extended Data Fig. 2m). We note that interactions between T4L with part of EL1 and EL2 in the crystal lattice may further stabilize this conformation (Extended Data Figs. 1c-e). While crystal contacts may influence sidechain rotamers on protein surfaces, their low binding energies are unlikely to induce the observed conformation.
Figure 1. Structural details of DRD2 and comparison with DRD3 and DRD4.
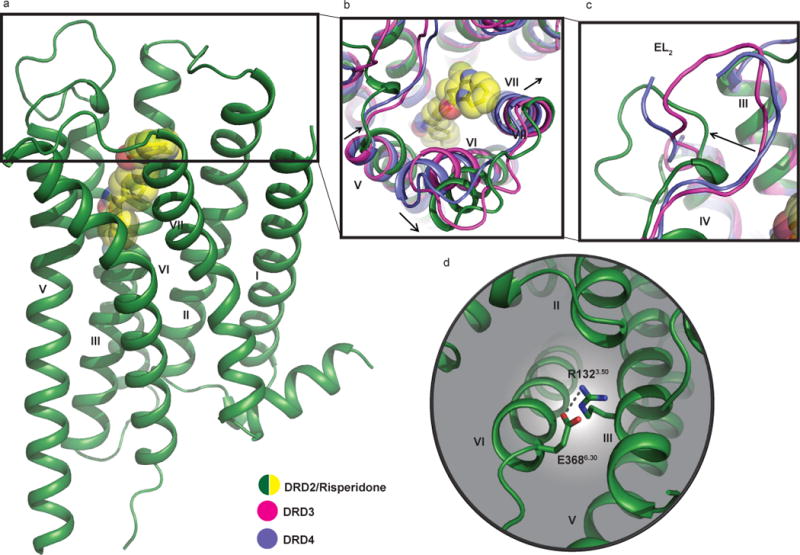
In all panels, dopamine receptor structures are shown aligned to the DRD2, with the DRD2 colored in green; DRD3 in magenta (PDB code 3PBL), and DRD4 in blue (PDB code 5WIU). Risperidone (yellow) is shown in sphere representation. a, Overall structure of the DRD2/Risperidone complex. b, c, Comparison of the view from the extracellular side. d, Cytoplasmic surface showing salt-bridge interaction (grey dotted line) between R1323.50 and E3686.30. In all panels, the Ballesteros-Weinstein numbering is shown as superscript.
Another difference observed between DRD2 and the other two D2-like dopamine receptors is that the extracellular tip of TM V is shifted towards the transmembrane bundle, while the extracellular tips of TM VI and TM VII move away from the receptor core by approximately 5.8/7.3 Å and 1.4/2.1 Å (Fig. 1b), respectively, versus DRD3 and DRD4. As in DRD3, an inter-helical hydrogen bond forms between Tyr7.35 and His6.55 (Extended Data Fig. 3a-d), which in DRD3 is important for regulating constitutive activity17. The side-chain conformations for DRD2, DRD316 and DRD417 at residues Tyr/Val7.35 and His6.55 (Extended Data Figs. 3a-c) are also distinct17. Specifically, the side chain of Tyr7.35 in DRD2 is rotated 52° compared to DRD3 to accommodate risperidone (Extended Data Fig. 3d). Together, these differences may further stabilize the outward movement of TM VI.
Like most antipsychotics, risperidone is a DRD2 inverse agonist22, and the DRD2/risperidone complex appropriately reflects an inactive state conformation. The most notable difference between active and inactive state GPCR structures is the extent to which the cytoplasmic tip of TM VI moves away from the transmembrane helical bundle to accommodate transducer binding23. A comparison of DRD2/risperidone with the active and inactive β2 adrenergic receptor (β2AR) or adenosine A2A receptor (A2AR) structures reveals no substantial outward movement of the intracellular end of TM VI (Extended Data Figs. 3e-f)–a finding consistent with an inactive-state structure. Another important structural feature of GPCR activation is the rearrangement of side chains in the highly conserved microswitches D(E)/RY (TM III) and NPxxY (TM VII)23. Here, Tyr7.53 from the NPxxY motif and Arg3.50 from the DRY motif, adopt almost identical positions with homologous residues in the β2AR and A2AR inactive structures (Extended Data Figs. 3g-j). Moreover, a key inactive-state salt-bridge interaction, the ‘ionic lock’ between the conserved Arg3.50 and Glu6.30 24,25,26 is maintained in the DRD2/risperidone structure (Fig. 1d).
Risperidone–a benzisoxazole27–displays a unique mode of dopamine receptor binding versus those of the substituted benzamides eticlopride at DRD3 and nemonapride at DRD4 (Fig. 2). The benzisoxazole moiety of risperidone extends into a deep binding pocket defined by the side chains of helices III, V and VI (Figs. 2a and 2d), interacting with Cys1183.36, Thr1193.37, Ser1975.46, Phe1985.47, Phe3826.44, Phe3906.52 and Trp3866.48 which form a subpocket below the orthosteric site (Fig. 2d). Additionally, another hydrophobic pocket above the orthosteric site encloses risperidone’s tetrahydropyridopyrimidinone moiety while Asp1143.32 forms a salt-bridge with risperidone’s tertiary amine (Fig. 2d). Alanine mutagenesis of many of these contact residues reduce risperidone’s affinity at DRD2 (Fig. 2d and Extended Data Table 3). In the DRD3 and DRD4 structures, neither eticlopride nor nemonapride engage this deeper hydrophobic pocket (Figs. 2b and 2c). Importantly, alanine substitutions of the equivalent residues in this deeper hydrophobic pocket do not substantially alter [3H]-nemonapride binding affinity, except for Trp3866.48 and Phe3906.52, which are large enough that mutagenesis-induced alterations in helical packing alone might explain the observed effects (Extended Data Table 3).
Figure 2. Comparison of the ligand binding pocket across the D2-like family receptors.
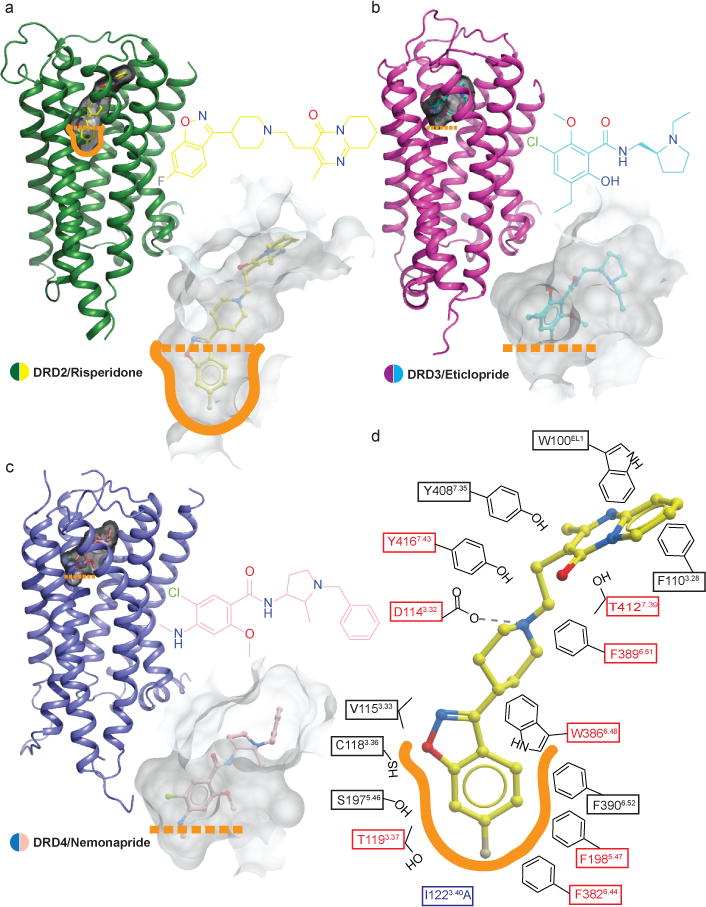
a, b, c, Surface representations of the ligand binding pockets of DRD2 (a), DRD3 (b) (PDB code 3PBL) and DRD4 (c) (PDB code 5WIU) are shown in transparent gray. d, A schematic representation of risperidone binding interactions at a 4.0 Å cut-off is shown. Hydrogen bonds are shown in grey dashed lines. Mutations of the amino acid in the red boxes reduces risperidone binding affinity by more than tenfold. The thermo-stabilizing mutation (I1223.40A) colored in blue. The outline of deeper hydrophobic pocket is colored as orange.
Comparison of the overall ligand binding pocket of DRD2 with DRD3 and DRD4 structures revealed striking differences around residues Val/Phe2.61, TrpEL1, Phe/Leu3.28 and Tyr/Val7.35, which help to define a DRD2 extended binding pocket (DRD2-EBP) (Figs. 3a and b). Indeed, previous studies16,17 on DRD3 and DRD4 revealed a selective EBP for each receptor. The DRD3-EBP is formed by the junction of EL1 and EL2 and the interface of helices II, III and VII and towards the EL1 (Fig. 3c). While the DRD4-EBP is elsewhere, reaching deep into a cleft between TMs II and III in this receptor, defined by Phe912.61/Leu1113.28 (Fig. 3d); the structural definition of this DRD4-EBP enabled the structure-based discover of agonists highly specific for this receptor17. Unique to the DRD2, the DRD2-EBP extends toward the extracellular part of TM VII consisting of EL1 and the junction of helices I, II and VII (Fig. 3b).
Figure 3. Different extended binding pockets revealed across D2-like family receptors.
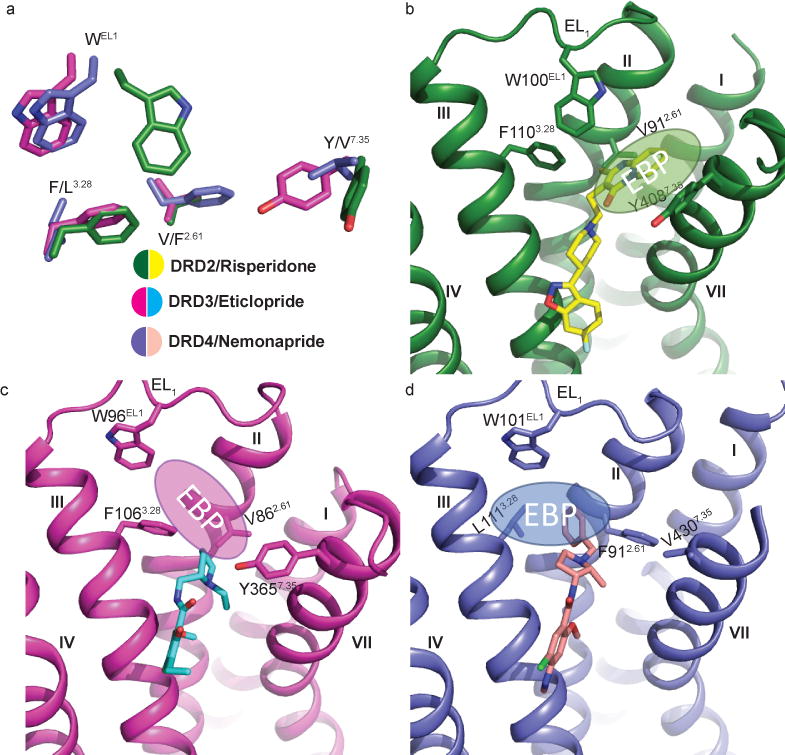
a, The distinctive selective extended binding pocket (EBP) defined by four key residues in the D2-like family receptors are delineated. The residues of DRD2 (green), DRD3 (pink) (PDB code 3PBL) and DRD4 (blue) (PDB code 5WIU) are shown as sticks. b, c, d, Structural differences in the EBPs of DRD2 (b), DRD3 (c) and DRD4 (d) are evident. Residues and ligands are colored as in (a). The position of each EBP is shown as an ellipse with the Ballesteros-Weinstein numbering as superscript.
There are four distinctive features of the DRD2-EBP: (1) Compared with the DRD3 structure, part of the EL1 loop rotates to move the conserved residue TrpEL1 to the top of the binding pocket at DRD2 (Figs. 3a-c and Extended Data Fig. 4), thereby disrupting what would be the DRD3-EBP (Figs. 3a-c). In all published aminergic receptor structures, only DRD2’s TrpEL1 adopts this unique conformation (Extended Data Fig. 4). (2) Compared with the DRD4 structure, the residue phenylalanine is located at 3.28 of DRD2, not 2.61, thereby eliminating the extended pocket as it exists in DRD4 (Figs. 3a, b and d). (3) Meanwhile, the side chain of Tyr4087.35 rotates towards the side chain of His3936.55 thereby avoiding clashes with risperidone (Extended Data Figs. 3a and 3d). (4) Finally, an outward movement of the extracellular tip of TM VII (Fig. 1b) makes additional space for the DRD2-EBP.
Compared to the conformation risperidone adopts when crystalized by itself4, in complex with DRD2, risperidone’s tetrahydropyridopyrimidinone ring rotates by ~90° (Extended Data Fig. 5a). This ring interacts with a hydrophobic patch formed by the side-chains of Trp100EL1, Ile184EL2, and Leu942.64. Although the electron density for Leu942.64 is weaker than for the other residues, it appears that the observed conformation of Trp100EL1 is stabilized by any rotamer of Leu942.64 that would fit the density.
In the DRD2/risperidone structure, the side chain of Trp100EL1 forms extensive contacts with the tetrahydropyridopyrimidinone ring, wedging it into the DRD2-EBP (Figs. 3b, 4a and Extended Data Fig. 5b). Besides these hydrophobic contacts between Trp100EL1 and risperidone, Trp100EL1 is also stabilized by contacts with Ile184EL2 and, perhaps Leu942.64, though the side-chain of Leu942.64 lacks electron density (Fig. 4b and Extended Data Fig. 5c). The observed configuration of risperidone is likely driven by the DRD2 binding pocket, and the conservation of key pocket residues such as Trp100EL1 implies that risperidone could bind other aminergic receptors (e.g. 5-HT2A or the α1A adrenergic receptor) in a similar binding mode, though further structures will be needed to test this notion.
Figure 4. The hydrophobic “patch” of the DRD2 binding pocket.
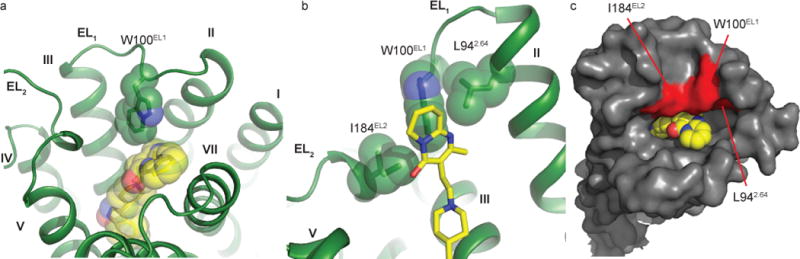
a, Risperidone (yellow) bound to DRD2 (green) orthosteric pocket viewed from extracellular space. b, The W100EL1 side chain forms extensive hydrophobic contacts with residues L942.64 and I184EL2. c, The residues L942.64, W 100EL1 and I184EL2 form a patch (red, with other residues in grey) that narrows DRD2’s binding pocket.
Intriguingly, molecular docking of risperidone to homology models of the DRD2, templated on either the DRD3 or DRD4 structures, failed to reproduce the unique pose of risperidone (Extended Data Figs. 5d-h). Rather, docking places the ligand higher in the binding site, in a space analogous to where both eticlopride and nemonapride are observed in the DRD3 and DRD4 structures (Figs. 2b-c), respectively. This is a direct consequence of the conformational rearrangements in DRD2 concomitant with accommodating risperidone–mainly movement of TMs V, VI, VII, and the relocation of Trp100EL1, which consequently affects the size and shape of the ligand binding pocket, allowing risperidone to engage a deep binding pose and DRD2-EBP. Moreover, the docked conformation of risperidone resembles that of the receptor-free risperidone crystal structure4, not that adopted in the receptor-bound complex (Extended Data Figs. 5d-h). This is not a problem of conformational sampling on the part of docking–the receptor-free structure is, after all, a low energy structure, and docking does capture it, but rather it reflects the incorrect modeling of Trp100EL1, owing to the lack of an analogous configuration in templates used in the modeling. Accordingly, docking did not predict the ~90° rotation of the tetrahydropyridopyrimidinone ring of risperidone in the DRD2-complex. This unique DRD2 binding pocket, and the unusual risperidone conformation that is accommodated by it, are unexpected features of this structure, with implications for our understanding of ligand recognition by this receptor, and the future design of new ligands to modulate it.
Compared with the DRD3 and DRD4 structures, the rearrangement of the extracellular surface and movement of Trp100EL1 not only allows it to interact with risperidone, but also forms, together with Ile184EL2 and Leu942.64, a hydrophobic patch that potentially narrows DRD2’s binding pocket (Figs. 4b-c). We hypothesized that these residues prevent risperidone’s egress from the binding pocket and found that Trp100EL1Phe, Trp100EL1Leu and Trp100EL1Ala mutants decreased risperidone residence time from 233 min (WT) to 59, 23, 28 min (Table 1 and Extended Data Figs. 6a-d), respectively. Interestingly, these kinetic effects of the Trp100EL1Phe, Trp100EL1Leu and Trp100EL1Ala mutants on residence time were shared with other tested antipsychotics, including N-methylspiperone, nemonapride and aripiprazole (Extended Data Figs. 6h-k and 6o-p and Extended Data Table 4). Similar to the findings obtained for the I184ECL2A/L942.64A double mutation (Table 1 and Extended Data Fig. 6g), this double mutation also reduces risperidone residence time to 6 min, as it does for other antipsychotics (Table 1, Extended Data Figs.6n, 6q and 6r and Extended Data Table 4). In summary, L942.64, Trp100EL1 and I184ECL2 form hydrophobic contacts which contribute to risperidone’s slow DRD2 dissociation.
Table 1.
Risperidone dissociation and association rates at wild-type and mutant DRD2.
| Receptor | Residence Time, min (koff ± SEM) min−1 |
kon ± SEM, M−1 min−1 |
Kd, nM (pKd ± SEM) |
|---|---|---|---|
| DRD2 wild-type | 233 (0.0043 ± 0.0003) | 1.65 × 106 ± 1.7 × 105 | 2.51 (8.65 ± 0.21) |
|
|
|||
| DRD2 W100 EL1A | 28 (0.036 ± 0.0022)*p=0.007 | 5.63 × 106 ± 3.2 × 105 | 6.74 (8.17 ± 0.04) |
|
|
|||
| DRD2 W100EL1L | 23 (0.043 ± 0.004) *p=0.06 | 6.32 × 106 ± 5.5× 105 | 6.77 (8.17 ± 0.002) |
|
|
|||
| DRD2 W100EL1F | 59 (0.017 ± 0.002) *p=0.01 | 3.12 × 106 ± 1.8 × 105 | 5.30 (8.28 ± 0.02) |
|
|
|||
| DRD2 L942.64A | 139 (0.0072 ± 0.0029)n.s. | 1.43 × 107 ± 2.3 × 106 | 0.48 (9.33 ± 0.12) |
|
|
|||
| DRD2 I184EL2A | 185 (0.0054 ± 0.002) n.s. | 9.84 × 106 ± 1.4 × 106 | 0.54 (9.28 ± 0.10) |
|
|
|||
| DRD2 L942.64A/I184EL2A | 6 (0.16 ± 0.05) *p=0.005 | 2.36 × 107 ± 7.8 × 106 | 7.01 (8.15 ± 0.02) |
|
|
|||
Data were acquired by association and dissociation kinetic experiments conducted in parallel at room temperature using [3H]-N-methylspiperone (0.8–1.0 nM). Estimates of koff, kon, and Kd were obtained from four independent experiments. Residence time was calculated as 1/koff. All data are the mean
SEM of four independent assays (n = 4 independent experiments). Asterisks indicate statistically significant differences between WT and mutant receptors (n.s.=Not significant, p values are indicated, unpaired two-tailed Student’s t-test).
Among the most dramatic and serious side-effects of antipsychotics are extra pyramidal symptoms (EPS). In patients with EPS, a consistent finding is DRD2 occupancy > 80% in the central nervous system, as demonstrated by positron emission tomography (PET)28. It has been suggested that differential binding kinetics29,30 as well as the relatively higher affinity of atypical antipsychotic drugs for 5-HT2A serotonin receptors3,4, contribute to the lower incidence of EPS with atypical antipsychotic drugs like risperidone versus typical antipsychotics. Relevant to these hypotheses we note that Trp100EL1 regulates both the association and dissociation kinetics for risperidone, and that many of the residues essential for risperidone binding to DRD2 are shared with 5-HT2A serotonin and other biogenic amine receptors. Thus, although our findings do not definitively resolve these hypotheses, they do provide the initial underpinnings for molecularly-derived models of antipsychotic drug actions at dopamine and other receptors. Finally, given recent successes in leveraging crystal structures of GPCRs for ligand discovery17,18,19, we anticipate that the DRD2/risperidone complex structure will accelerate the search for novel antipsychotic drugs targeting DRD2.
Methods
Protein engineering for structural studies
To facilitate expression, purification and crystallography, a human DRD2 (D2 long receptor variant31) construct was generated with several modifications. T4L residues 2–16132 were fused into third intracellular loop of DRD2 (V223–R361) with truncations of the N termini residues1–34. The DRD2-T4L gene was further modified by introducing three mutations I1223.40A, L3756.37A and L3796.41A -identified by alanine scanning - to improve protein thermo-stability. In brief, alanine scanning was used to identify thermo-stabilization mutations (see Radioligand Binding Assay for details; Extended Data Fig. 1a). The receptor chimeric sequences were then subcloned into a modified pFastBac1 vector (Invitrogen), designated as pFastBac1-833100, which contained an expression cassette with a haemagglutinin (HA) signal sequence followed by a Flag tag, a 10×His tag, and a TEV protease recognition site at the N terminus before the receptor sequence.
Protein expression and purification
The modified DRD2-T4L protein was expressed in Spodoptera frugiperda (Sf9) cells (Expression Systems) using Bac-to-Bac Baculovirus Expression System (Invitrogen) for 48 h. The insect cells were lysed by repeated washing and centrifugation, with hypotonic buffer with low (10 mM HEPES, pH 7.5, 10 mM MgCl2, 20 mM KCl and EDTA-free complete protease inhibitor cocktail tablets (Roche)) (one time) and high salt (1.0 M NaCl, 10 mM HEPES, pH 7.5, 10 mM MgCl2, 20 mM KCl) (three times). The washed membranes were suspended in buffer containing 10 mM HEPES, pH 7.5, 10 mM MgCl2, 20 mM KCl, 150 mM NaCl, 20 μM risperidone, and EDTA-free complete protease inhibitor cocktail tablets, and incubated at room temperature for 1 h. And, incubated at 4 °C for 30 min before solubilization. The membranes were then solubilized in 10 mM HEPES, pH 7.5, 150 mM NaCl, 1% (w/v) n-dodecyl-β-D-maltopyranoside (DDM, Anatrace), 0.2% (w/v) cholesteryl hemisuccinate (CHS, Sigma) for 2 hr at 4 °C.
The supernatant was isolated by centrifugation at 150,000 × g for 30 min, followed by incubation in 20 mM buffered imidazole (pH 7.5), 800m M NaCl with TALON IMAC resin (Clontech) at 4 °C, overnight. The resin was then washed with 10 column volumes (cv) of Wash Buffer I (50 mM HEPES, pH 7.5, 800 mM NaCl, 0.1% (w/v) DDM, 0.02% (w/v) CHS, 20 mM imidazole, 10% (v/v) glycerol, and 10 μM risperdone, followed by 10 cv of Wash Buffer II (25 mM HEPES, pH 7.5, 150 mM NaCl, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 10% (v/v) glycerol, and 10 μM risperidone). The protein was then eluted in 3–4 column volumes of Elution Buffer (50 mM HEPES (pH 7.5), 50 μM risperidone, 500 mM NaCl, 10% (v/v) glycerol, 0.05% (w/v) DDM, 0.01% (w/v) CHS, and 250 mM imidazole). A PD MiniTrap G-25 column (GE Healthcare) was used to remove imidazole. The protein was then treated overnight with His-tagged TEV protease and His-tagged PNGase F (NEB) to remove the N-terminal His-tag, Flag-tag and deglycosylate the receptor. His-tagged TEV protease, His-tagged PNGase F, cleaved His-tag and uncleaved protein were removed from the sample by passing the sample over equilibrated TALON IMAC resin (Clontech). The receptor was then concentrated to 40–50 mg ml−1 with a 100 kDa molecular mass cut-off Vivaspin 500 centrifuge concentrator (Sartorius Stedim).
Lipidic cubic phase crystallization
Protein samples of DRD2 in complex with risperidone were reconstituted into the lipidic cubic phase (LCP) by mixing 40% of ~60 mg/ml with 60% lipid (10%(w/w)cholesterol, 90%(w/w)monoolein) using the twin-syringe method33. Crystallization trials were performed in glass sandwich plates (Marienfeld GmbH) using a handheld dispenser (Art Robbins Instruments), dispensing 45 nL of protein-laden LCP and 1 μl precipitant solution per well. Plates were then incubated at 20 °C. Crystals were obtained from precipitant conditions containing 100 mM Tris/HCl pH 7.8, 230 mM Lithium nitrate, 25% PEG400, 4% (±)-1,3-Butanediol. Crystals grew to maximum size of 40 μm ×40 μm ×10 μm within two weeks and were harvested directly from the LCP matrix using MiTeGen micromount and flash frozen in liquid nitrogen.
Data collection, structure solution and refinement
Crystallographic diffraction data collection were performed at the 23ID-B and 23ID-D beamlines (GM/CA CAT) at the Advanced Photon Source, Argonne, IL using a 10 μm minibeam at a wavelength of 1.0330 Å and a Dectris Eiger-16m or MarMosaic 300 CCD or Pilatus3 6M detector, respectively. The crystals were exposed to 0.5 s of unattenuated beam using 0.5° oscillation per frame. A 97.3 % complete data set at 2.90 Å resolution of DRD2/risperidone from 20 crystals were integrated, scaled and merged using HKL300034. Initial phase information was obtained by molecular replacement (MR) with the program PHASER35 using two independent search models - a receptor portion of the DRD4/nemonapride complex (PDB code: 5WIU), and the T4L portions of β2AR-T4L (PDB code: 2RH1) as initial models. Refinement was performed with PHENIX36 and REFMAC followed by manual examination and rebuilding of the refined coordinates in the program COOT37 using |2Fo| - |Fc|, |Fo| - |Fc|, and omit maps.
Radioligand Binding Assay
Binding assays were performed using Sf9 membrane fractions expressing the crystallization construct DRD2-T4L (I1223.40A, L3756.37A and L3796.41A) or HEK293 T membrane preparations transiently expressing DRD2 (D2 long receptor) and different mutants. HEK293 T cells (ATCC CRL-11268; 59587035; mycoplasma free) were transfected and membrane preparation and radioligand binding assays were set up in 96-well plates as described previously38. All binding assays were conducted in standard binding buffer (50 mM Tris, 10 mM MgCl2, 0.1 mM EDTA, 0.1% BSA, pH 7.4). For displacement experiments, increasing concentrations of compounds were incubated with membrane and radioligands (0.8–1.0 nM [3H]-N-methylspiperone or 0.1–0.5 nM [3H]-nemonapride) (PerkinElmer) for 2 hr at room temperature in the dark. To determine the affinity of nemonapride in DRD2 and different mutants, all assays utilized at least 2 concentrations of [3H]-nemonapride. The reaction was terminated by rapid vacuum filtration onto chilled 0.3% PEI-soaked GF/A filters followed by three quick washes with cold washing buffer (50 mM Tris HCl, pH 7.4) and quantified as described previously8. Results (with or without normalization) were analyzed using GraphPad Prism using one-site shift models where indicated.
Radioligand Based Thermostability Assay
Membranes from HEK293 T cells expressing wild-type or mutant human DRD2 were resuspended in binding buffer (50 mM Tris, 10 mM MgCl2, 0.1 mM EDTA, 0.1% BSA, pH 7.4). [3H]-N-methylspiperone added to the membranes to give a final concentration of 1 nM. The samples were incubated at room temperature for 1 hr, then were aliquoted into PCR strips. Samples were heated to the desired temperature for exactly 30 min, then cooled down to 25 °C for 30 min. The samples were terminated by rapid vacuum filtration onto chilled 0.3% PEI-soaked GF/A filters followed by three quick washes with cold washing buffer (50 mM Tris HCl, pH 7.4) and quantified as described previously8. Results were analyzed using GraphPad Prism. Apparent Tm values were derived from sigmoidal dose–response analysis. Results represent the mean ± SEM of three independent experiments.
Differential Scanning Fluorimetry Based Thermostability Assay
The thermal stability of purified protein was determined by measuring fluorescence of thiol-reactive dye BODIPY FL-L-cystine (Invitrogen). The standard assay conditions were 20 mM HEPES (pH 7.5), 200 mM NaCl, 0.025% DDM and 10 mM risperidone with protein concentrations 1 mg/mL and BODIPY FL-L-cystine concentrations 1 μM. The melting experiments were performed on a StepOnePlus real-time PCR System from Applied Biosystems. The melting curve experiments were conducted (1 °C/min) and recorded using StepOne software from Applied Biosystems. Results were analyzed using GraphPad Prism. Apparent Tm values were derived from sigmoidal dose–response analysis. Results represent the mean ± SEM of three independent experiments.
Ligand Association and Dissociation Radioligand Binding Assays
Binding assays were performed using HEK293 T membrane preparations transiently expressing DRD2 (D2 long receptor) and different mutants at room temperature. Radioligand dissociation and association assays were performed in parallel utilizing the same concentrations of radioligand, membrane preparations, and binding buffer (50 mM Tris, 10 mM MgCl2, 0.1 mM EDTA, 0.1% BSA, pH 7.4). All assays utilized at least 2 concentrations of radioligand ([0.5–1.0 nM [3H]-N-methylspiperone; 0.5–2.0 nM [3H]-nemonapride). For dissociation assays, membranes were incubated with radioligand for at least 2 hr at room temperature before the addition of 10 μL of 10 μM excess cold ligand to the 200 μL membrane suspension at designated time points. For association experiments, 100 μL of radioligand was added to 100 μL membrane suspensions at designated time points. Time points spanned 1 minutes to 7 hr, depending on experimental conditions and radioligand. For the determination of kon and koff for unlabeled risperidone or aripiprazole, membranes of either wild type or mutants were incubated with [3H]-methylspiperone and several concentrations of risperidone or aripiprazole. Non-specific binding was determined by addition of 10 μM nemonapride. Immediately at time = 0 min, plates were harvested by vacuum filtration onto 0.3% polyethyleneimine pre-soaked 96-well filter mats (Perkin Elmer) using a 96-well Filtermate harvester, followed by three washes of cold wash buffer (50 mM Tris pH 7.4). Scintillation cocktail (Meltilex, Perkin Elmer) was melted onto dried filters and radioactivity was counted using a Wallac Trilux MicroBeta counter (PerkinElmer). Data were analyzed using “Dissociation-One phase exponential decay” or “Association kinetics-Two or more concentrations of hot radioligand” in Graphpad Prism 5.0. The previously determined [3H]-N-methylspiperone kon and koff rates of DRD2 or mutants was used to estimate the kon and koff rates of risperidone and aripiprazole using the “Kinetics of competitive binding” equation in Graphpad Prism 5.0 as put forth by Motulsky and Mahan39.
Homology modeling of DRD2
Sequence alignment for construction of the DRD2 homology models was generated with PROMALS3D40, using sequences of human DRD2 (Uniprot accession number: P14416), DRD3 (P35462) and DRD4 (P21917), as well as sequences of available DRD2-family X-ray structures [DRD3 - PDB: 3PBL (chain A)41 and DRD4 - 5WIU (chain A)42]. The alignment was manually edited to remove the amino and carboxy termini which extended past the template structures, and to remove the engineered T4 lysozyme (3PBL) or apocytochrome b562 RIL (BRIL; 5WIU) from the template sequences. MODELLER-9v1543 was then used to generate (1) a total of 1000 homology models of DRD2, based on the crystal structure of DRD4 in complex with Nemonapride as the template, and (2) a set of 500 models based on the crystal structure of DRD3 in complex with Eticlopride. We then evaluated the models for their ability to enrich known DRD2 ligands over property-matched decoys through docking to the orthosteric binding site, using DOCK 3.744 (as detailed below). While sharing physical properties of known ligands, decoy molecules are topologically distinct and so unlikely to bind the receptor, thus controlling for the enrichment of molecules by physical properties alone. Thirty-two known DRD2 antagonists with MW < 420 were extracted from the IUPHAR database45, and 1836 property-matched decoys were generated using the DUD-E server46. The models were then ranked on the basis of their adjusted logAUC. The selected best scoring model in terms of ligand enrichment was further optimized through minimization with the AMBER protein force field and the GAFF ligand force field supplemented with AM1BCC charges47.
Molecular docking of risperidone
Risperidone was docked to the orthosteric binding site of the DRD2 homology models based on the DRD3 or DRD4 crystal structures, using DOCK3.744. DOCK3.7 places pre-generated flexible ligands into the binding site by superimposing atoms of each molecule on matching spheres, representing favorable positions for individual ligand atoms. Forty-five matching spheres were used here, based on the pose of the corresponding x-ray ligand (eticlopride/nemonapride) in the template structure. The resulting docked ligand poses were scored by summing the receptor-ligand electrostatics and van der Waals interaction energies, and corrected for context-dependent ligand desolvation. Receptor structures were protonated using Reduce48. Partial charges from the united-atom AMBER47 force field were used for all receptor atoms. Grids which evaluate the different energy terms of the DOCK scoring function were precalculated using AMBER47 for the van der Waals term, QNIFFT49,50 (an adaptation of DELPHI) for electrostatics, and ligand desolvation51. Ligands were protonated with Marvin (version 15.11.23.0, ChemAxon, 2015; http://www.chemaxon.com), at pH 7.4. Each protomer was rendered into 3D using Corina52 (Molecular Networks GmbH) and conformationally sampled using Omega53 (OpenEye Scientific Software). Ligand charges and initial solvation energies were calculated using AMSOL54,55.
Data availability
Atomic coordinates and structure factor files for the DRD2/Risperidone structure have been deposited in the RCSB Protein Data Bank with identification code 6C38. All other data are available from the corresponding authors upon reasonable request.
Extended Data
Extended Data Figure 1. Thermo-stability of DRD2 constructs, crystal packing of the DRD2/Risperidone complex and representative electron density of the DRD2 structure.
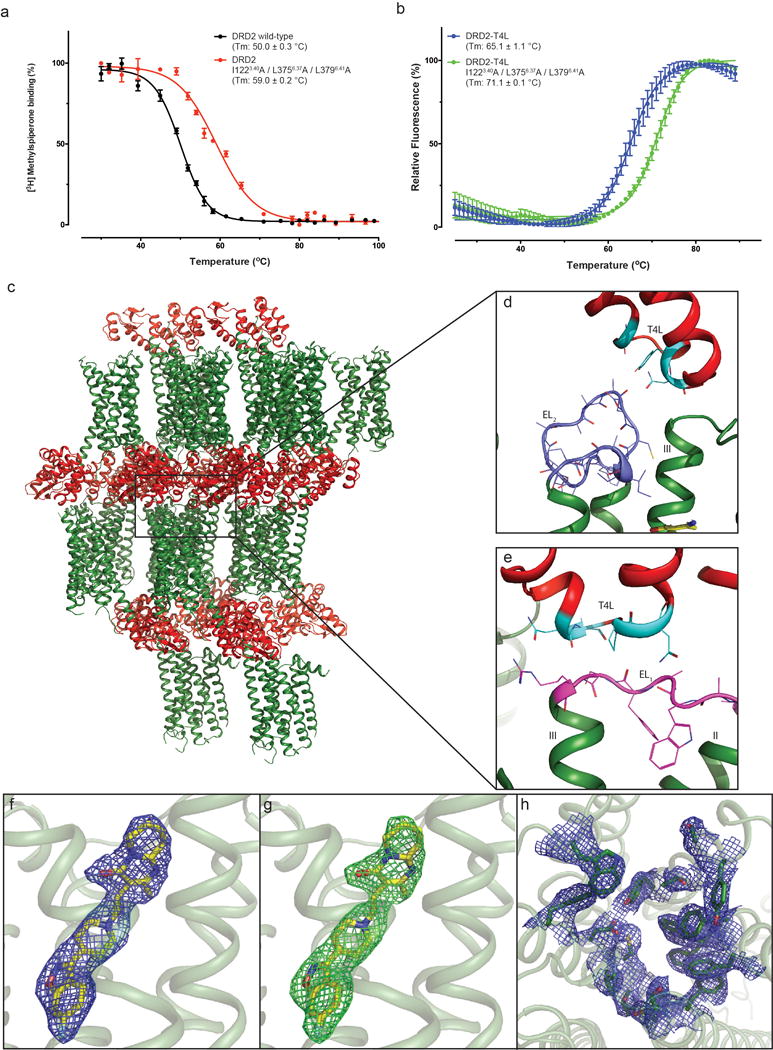
a, DRD2 or thermo-stability mutation membrane with 1nM [3H]-N-methylspiperone were heated for 30 min, the amount of [3H]-ligand bound determined. b, Purified DRD2-T4L (with or without thermo-stability mutation) protein with 10 μM risperidone and 1 μM BODIPY FL L-cystine dye were heated by a temperature gradient and the amount of dye bound to unfolding protein determined. Data were analyzed by nonlinear regression and apparent Tm values (transition temperature where 50% of the receptor is inactive) were determined from analysis of the sigmoidal dose-response curves. All data in a-b are the mean SEM of three independent assays. Error bars in a-b denote SEM from three independent assays. c, d, e, Packing of the DRD2/Risperidone complex crystallized in the P212121 spacegroup. The DRD2 is shown in green and T4L fusion protein is shown in red or cyan (interact with DRD2). EL1 and EL2 of DRD2 were shown in magenta and blue, respectively. f, 2Fo-Fc electron density map (blue mesh) of risperidone (yellow) contoured at 1σ. g, Fo-Fc omit map (green mesh) contoured at 3.0σ of risperidone (yellow). h, 2Fo-Fc electron density map of DRD2 binding pocket residues (blue mesh) contoured at 1σ.
Extended Data Figure 2. Conserved hydrophobic residue of EL2 in all available aminergic receptor structures.
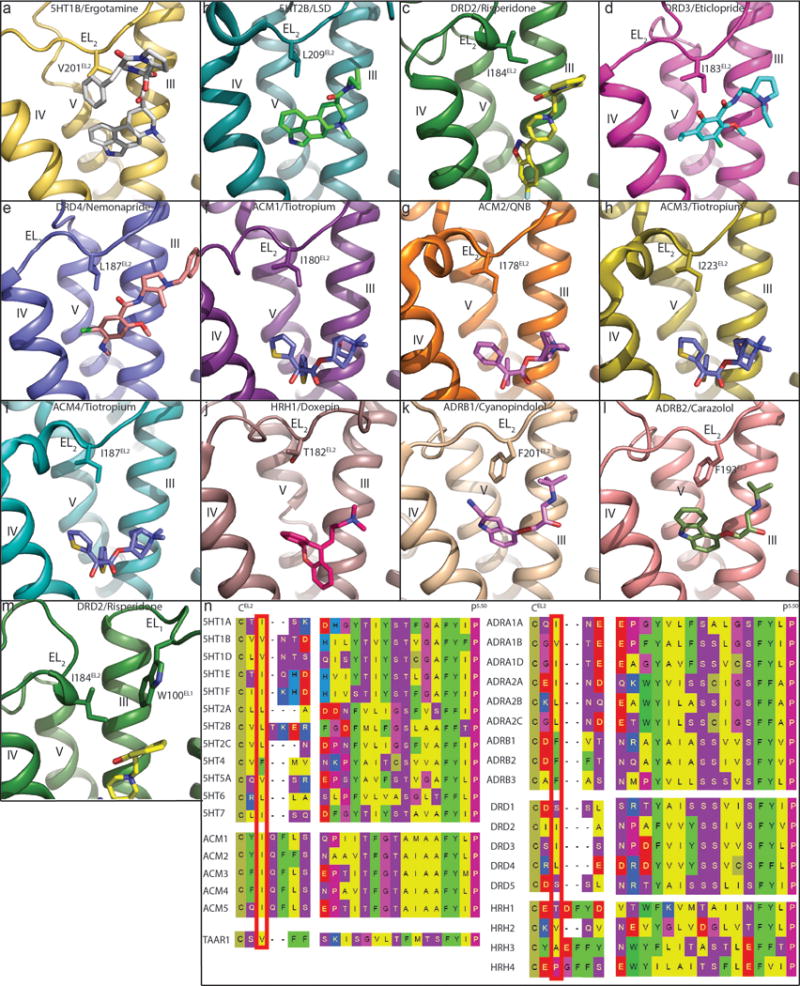
In all panels, receptors are shown as cartoon. Ligands and residues are shown as sticks. a, 5HT1B (PDB code 4IAR). b, 5HT2B (PDB code 5TVN). c, DRD2. d, DRD3 (PDB code 3PBL). e, DRD4 (PDB code 5WIU). f, ACM1 (PDB code 5CXV). g, ACM2 (PDB code 3UON). h, ACM3 (PDB code 4ADJ). i, ACM4 (PDB code 4DSG). j, HRH1 (PDB code 3RZE). k, ADRB1 (PDB code 2VT4). l, ADRB2 (PDB code 2RH1). m, DRD2. n, Conserved EL2 hydrophobic residues (red box) are located two residues away from conserved cysteine that forms a disulfide bridge between EL2 and helix III. Notable exceptions to the presence of a hydrophobic residue are DRD1 and DRD5, which contain a serine, and HRH1 and HRH4, which contain a threonine and proline, respectively.
Extended Data Figure 3. Comparison of D2 receptors view from the extracellular side and structural alignment with the β2 adrenergic receptor (β2AR) and A2A adenosine (A2AAR) reveals inactive-state of the DRD2.
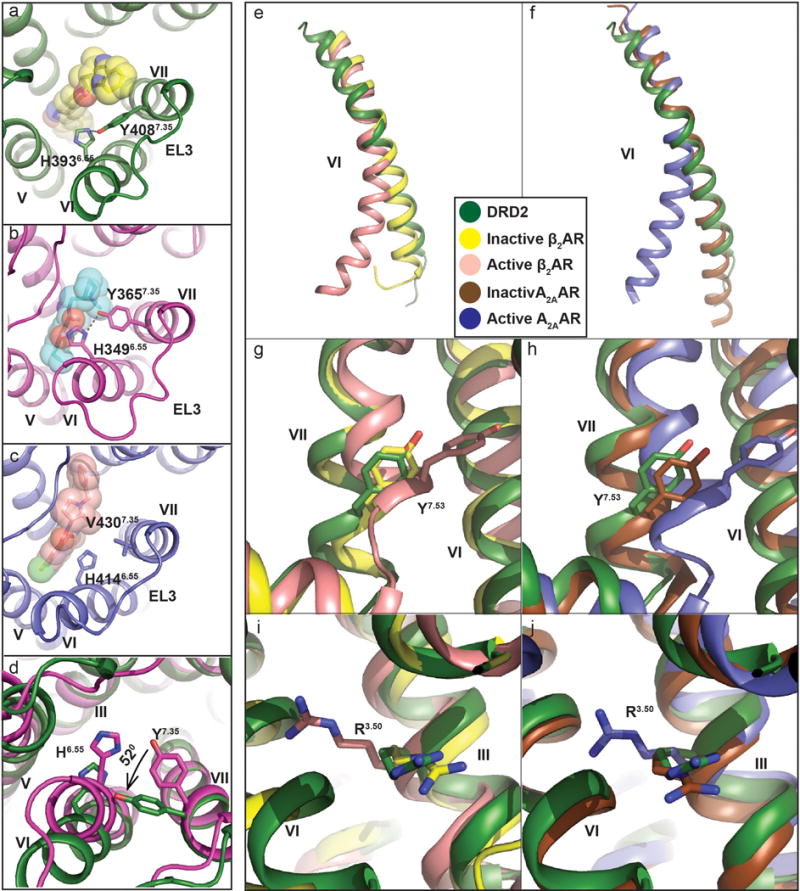
In a-d panels, the DRD2 colored in green; DRD3 in magenta (PDB code 3PBL), and DRD4 in blue (PDB code 5WIU). Risperidone (yellow), eticlopride (cyan) and nemonapride (lightpink) are shown sticks and spheres. a, b, c, Displacements of H6.55 and Y/V7.35 are shown at DRD2 (a), DRD3 (b) and DRD4 (c). d, The views from extracellular side of DRD2 and DRD3. e, f, Superposition of TM VI at DRD2 (green), inactive β2AR (yellow) (PDB code: 2RH1), active β2AR (lightpink) (PDB code: 3SN6), inactive A2AAR (brown) (PDB code: 3REY) and active A2AAR (blue) (PDB code: 5G53) aligned through helices I-IV. g, h, i, j, Cytoplasmic view of an alignment between DRD2 and active/inactive β2AR (g, h) or A2AAR (i, j). Rearrangements of two highly conserved residues (Y7.53 and R3.50) within the core of the receptor are shown as sticks. Ligands are omitted for clarity, hydrogen bonds are shown as grey dotted line and the Ballesteros-Weinstein numbering is shown as superscript.
Extended Data Figure 4. Conserved Trp of EL1 in all available aminergic receptor structures illustrates unique position in DRD2/risperidone.
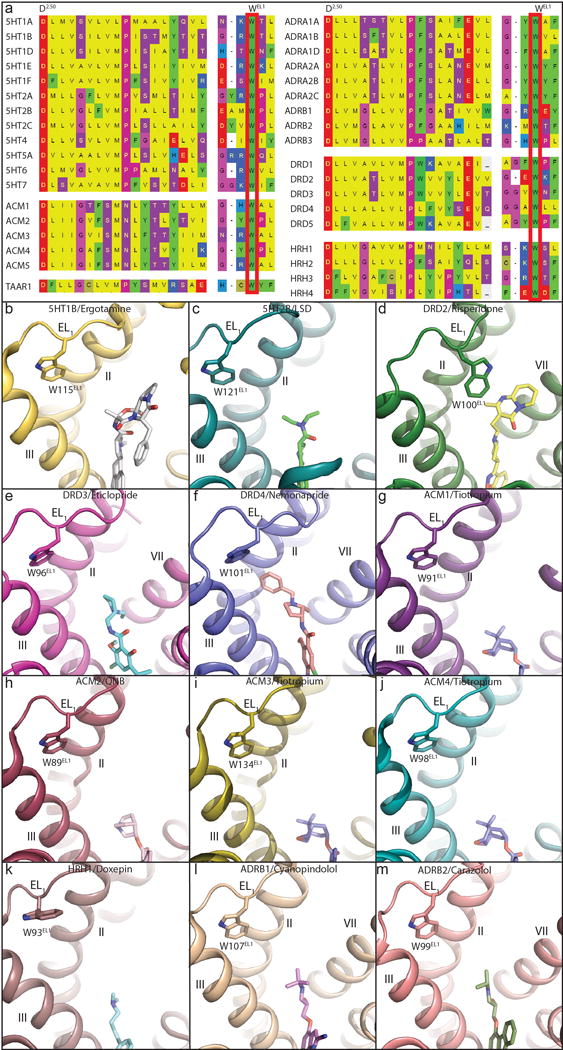
In all panels, receptors are shown as cartoon. Ligands and residues are shown as sticks. a, Conserved Trp of EL1 was show in red box. b, 5HT1B (PDB code 4IAR). c, 5HT2B (PDB code 5TVN). d, DRD2. e, DRD3 (PDB code 3PBL). f, DRD4 (PDB code 5WIU). g, ACM1 (PDB code 5CXV). h, ACM2 (PDB code 3UON). i, ACM3 (PDB code 4ADJ). j, ACM4 (PDB code 4DSG). k, HRH1 (PDB code 3RZE). l, ADRB1 (PDB code 2VT4). m, ADRB2 (PDB code 2RH1).
Extended Data Figure 5. Risperidone has distinct poses in solution and in complex with DRD2 and comparison of x-ray structure and model of DRD2.
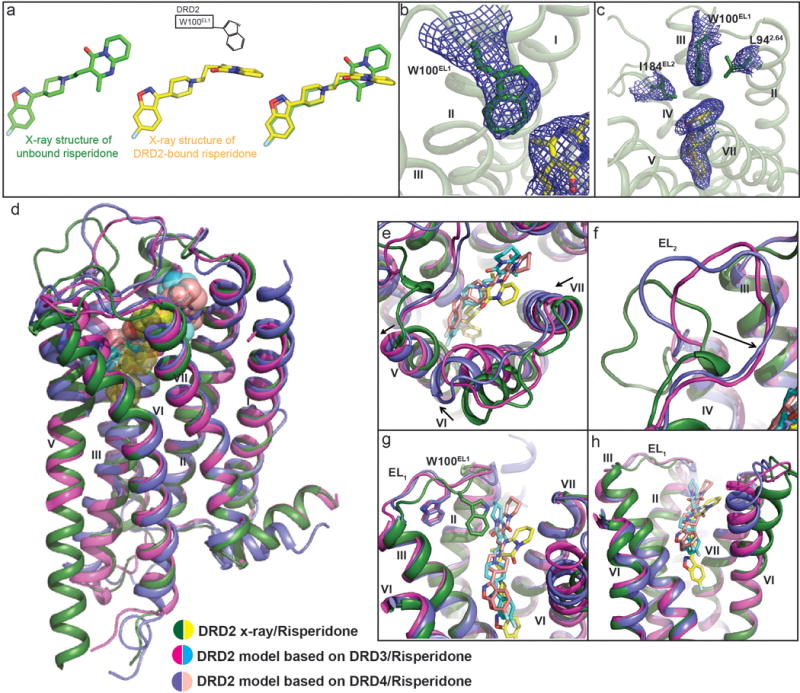
a, Trp100EL1 determines the configuration of the tetrahydropyridopyrimidinone moiety of risperidone. Structure of unbound risperidone colored in green and DRD2-bound risperidone in yellow. b, Electron density (2Fo-Fc maps, blue mesh) for W100EL1 in the DRD2/Risperidone complex (contoured at 1.0σ). c, 2Fo-Fc electron density map (blue mesh) of Leu942.64, Trp100EL1, Ile184EL2 and risperidone (yellow) contoured at 0.8σ. Ballesteros-Weinstein numbering is shown as superscript. d, Overall view of DRD2/Risperidone x-ray structure and model. e, f, g, h, Comparison of x-ray structure and model of DRD2. In d-h panels, DRD2 x-ray structure and model is shown as cartoons, with the x-ray structure colored in green and model in magenta or blue. Risperidone in x-ray structure is shown as yellow spheres or sticks and model as cyan or lightpink.
Extended Data Figure 6. Patch residues of the DRD2 orthosteric pocket impair the dissociation rates of risperidone, aripiprazole, N-methylspiperone and nemonapride.
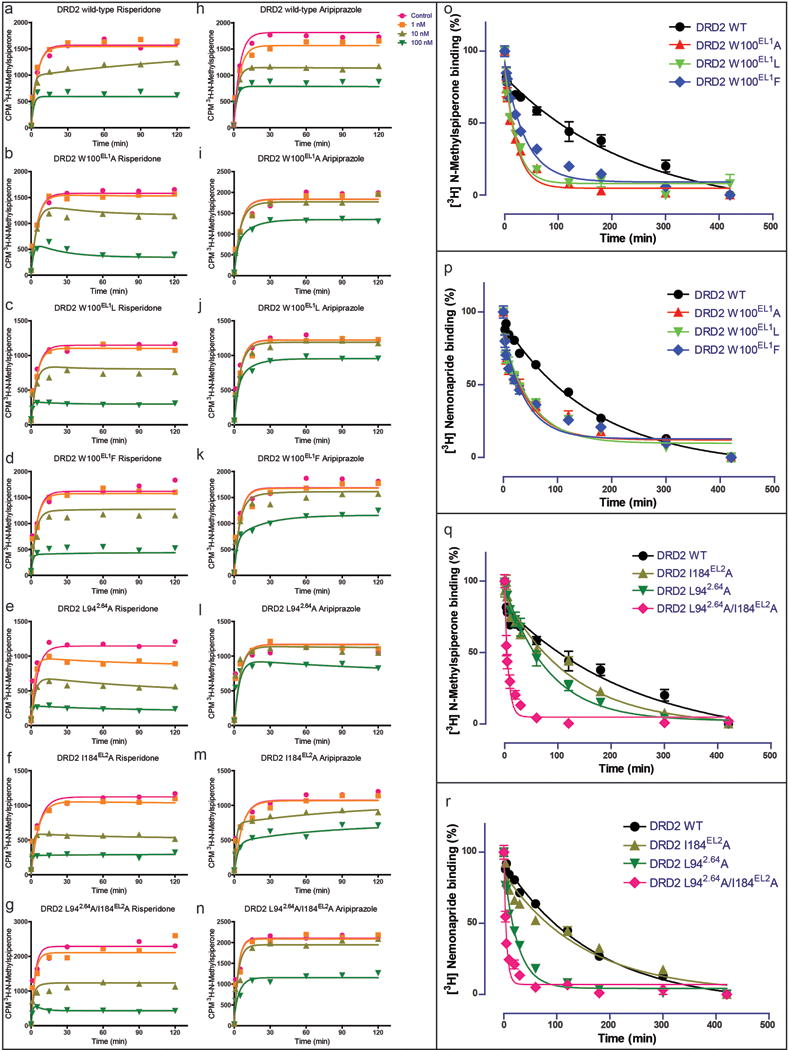
a, b, c, e, f, g, Comparison of risperidone dissociation from wild-type DRD2 (a) and W100EL1A (b), W100EL1L (c), W100EL1F (d), L942.64 A (e), I184EL2A (f) or L942.64A /I184EL2A (g) mutants. h, i, j, k, l, m, n, Comparison of aripiprazole dissociation from wild-type DRD2 (h) and W100EL1A (i), W100EL1L (j), W100EL1F (k), L942.64 A (l), I184EL2A (m) or L942.64A /I184EL2A (n) mutants. o, p, Comparison of N-methylspiperone (o) or nemonapride (p) dissociation from wild-type DRD2 and W100EL1A, W100EL1L or W100EL1F mutants(n=3). q, r, Comparison of N-methylspiperone (q) or nemonapride (r) dissociation from wild-type DRD2 and L942.64 A, I184EL2A or L942.64A /I184EL2A mutants. All data are the mean SEM of four independent assays (n = 4 independent experiments). Error bars in o-r denote SEM from four independent assays. Ballesteros-Weinstein numbering is shown as superscript.
Extended Data Table 1. Affinities of antipsychotic drugs for thermostabilized mutant and wild-type RDR2.
Data represent mean Ki (pKi ± SEM) for competition binding experiments using [3H]-N-methylspiperone (0.8–1.0 nM) as radioligand. All data are the mean SEM of three independent assays (n = 3 independent experiments).
| Receptor Kj, nM (pKi±SEM) | Risperidone | Aripiprazole | N-Methylspiperone | Nemonapride | Bifeprunox |
|---|---|---|---|---|---|
| DRD2 wild-type |
1.91 (8.84±0.19) |
6.28 (8.21±0.05) |
0.04 (11.06±0.18) |
0.03 (11.06±0.10) |
1.04 (9.52±0.38) |
| DRD2 I1223.40A/L3756.37A/L3796.41A |
1.86 (9.10±0.38) |
1.25 (8.91±0.04) |
0.09 (11.01±0.11) |
0.05 (11.03±0.06) |
0.24 (9.62±0.02) |
| DRD2-T4L(Sf9) I1223.40A/L3756.37A/L3796.41A |
3.13 (8.57±0.18) |
1.88 (8.73±0.02) |
0.06 (11.02±0.04) |
0.09 (11.03±0.33) |
0.57 (9.25±0.03) |
Extended Data Table 2. Data collection and refinement statistics.
Highest resolution shell is shown in parentheses.
| Structure | Human DRD2 (ΔN/ΔICL3T4L/ΔC)-Risperidone complex |
|---|---|
|
| |
| Data Collection | APS, GMCA/CAT 23ID-B/D, 10 μm microfocus beam |
|
| |
| Crystals | 20 |
| Resolution range | 30.00 - 2.90 (2.99 - 2.90) |
| Space group | P212121 |
| Unit cell Dimensions a, b, c (Å) | 50.98 72.52 151.31 |
| Unique reflections | 12826 (889) |
| Multiplicity | 5.5 (2.5) |
| Completeness (%) | 97.3 (86.9) |
| Mean 1/σ(l) | 15.2 (1.0) |
| Rmerge (%) | 13.4 (73.8) |
| CC1/2(%) | 99.4 (53.5) |
|
Refinement Statistics | |
| Reflections used in refinement | 12826 (889) |
| Reflections used for R-free | 622 (40) |
| R-work (%) | 22.6 (37.4) |
| R-free (%) | 24.9 (34.1) |
|
Number of Atoms | |
| DRD2 | 1948 |
| T4L | 1176 |
| Risperidone | 30 |
| Lipid and other | 82 |
|
Overall B-factors (Å2) | |
| Receptor | 84.1 |
| T4L | 97 |
| Risperidone | 75.8 |
| Lipids, water, other | 86.8 |
|
Model Statistics | |
| RMSD-bonds (Å) | 0.004 |
| RMSD-angles (°) | 0.56 |
| Ramachandran favored (%)# | 97.36 |
| Ramachandran allowed (%)# | 2.64 |
| Ramachandran outliers (%)# | 000 |
| Rotamer outliers (%)# | 0.67 |
| Clashscore* | 399 |
Rmerge = Σhkl |I(hkl) − <I(hkl)>|/Σhkl(hkl), where <I(hkl)> is the mean of the symmetry equivalent reflections of I(hkl).
As defined in MolProbity.
Extended Data Table 3. Affinity of risperidone and nemonapride at ligand binding pocket mutants of the D2 dopamine receptor.
Data represent mean Ki (pKi ± SEM) for competition binding experiments and Kd (pKd ± SEM) for homologous competition binding experiments using [3H]-nemonapride (0.1–0.5 nM) as radioligand. All data are the mean SEM of three independent assays (n = 3 independent experiments).
| Receptor | Risperidone | Nemonapride | ||
|---|---|---|---|---|
| Ki, nM (pKi ± SEM) |
ΔpKi (mutant-WT) |
Kd, nM (pKd ± SEM) |
ΔpKd (mutant-WT) |
|
| DRD2 wild-type |
4.50 (8.41 ±0.07) |
– | 0.21 (9.69 ±0.06) |
|
| DRD2 W100EL1A |
8.14 (8.19 ±0.13) |
−0.21 | 1.97 (8.71 ±0.02) |
−0.98 |
| DRD2 F1103.28A |
36.89 (7.48±0.09) |
−0.93 | 0.17 (9.77 ±0.03) |
0.08 |
| DRD2 D1143.32A |
>10000 | – | 8.10 (8.09 ±0.04) |
−1.60 |
| DRD2 V1153.33A |
3.07 (8.52 ±0.04) |
0.11 | 0.84 (9.08 ±0.02) |
−0.61 |
| DRD2 C1183.36A |
4.84 (8.32 ±0.01) |
−0.09 | 0.40 (9.40 ±0.02) |
−0.29 |
| DRD2 T1193.37A |
177.19 (6.83 ±0.12) |
−1.58 | 0.43 (9.38 ±0.06) |
−0.31 |
| DRD2 I1223.40A |
13.87 (7.97 ±0.13) |
−0.44 | 0.30 (9.52 ±0.01) |
−0.17 |
| DRD2 S1975.46A |
1.22 (8.92 ±0.03) |
0.51 | 043 (9.37 ±0.01) |
−0.32 |
| DRD2 F1985.47A |
41.95 (7.38 ±0.03) |
−1.02 | 0.76 (9.12 ±0.02) |
−0.57 |
| DRD2 F3826.44A |
57.70 (7.25 ±0.05) |
−1.16 | 030 (9.53 ±0.05) |
−0.16 |
| DRD2 W3866.48A |
>10000 | – | 402 (8.40 ±0.04) |
−1.29 |
| DRD2 F3896.51A |
2992 (5.65 ±0.17) |
−2.76 | 4.70 (8.35 ±0.08) |
−1.34 |
| DRD2 F3906.52A |
31.20 (7.61 ±0.15) |
−0.80 | 1.30 (8.89 ±0.03) |
−0.80 |
| DRD2 Y4087.35A |
13.63 (7.95±0.13) |
−0.46 | 0.18 (9.76 ±0.02) |
0.07 |
| DRD2 T4127.39A |
102.68 (7.02 ± 0.08) |
−1.77 | 4.92 (8.33 ±0.10) |
−1.36 |
| DRD2 Y4167.43A |
2772 (5.61 ±0.15) |
−2.80 | 088 (9.06 ±0.01) |
−0.63 |
Extended Data Table 4. Compound dissociation and association rates at wild-type and mutant DRD2.
Data were acquired by association and dissociation kinetic experiments conducted in parallel at room temperature using [3H]-N-methylspiperone (0.8–1.0 nM) for Aripiprazole and N-methylspiperone or [3H]-nemonapride (0.8–1.0 nM) for nemonapride. Estimates of koff, kon, and Kd were obtained from four independent experiments. Residence time was calculated as 1/koff. All data are the mean SEM of four independent assays (n = 4 independent experiments). Asterisks indicate statistically significant differences between WT and mutant receptors (n.s.=Not significant. p values are indicated, unpaired two-tailed Student’s t-test).
| Compound | Receptor | Residence Time, min (koff ± SEM) min−1 | kon ± SEM, M−1, min−1 | kdr nM (pKd ± SEM) |
|---|---|---|---|---|
| Aripiprazole | DRD2 wild-type | 154 (0.0065 ± 0.0004) |
7.68 × 105 ± 4.94 × 105 | 9.43(8.03 ± 0.07) |
| DRD2 W100EL1A | 15 (0.067 ± 0.015)⊥p=0.006 |
2.48 × 105 ± 6.5 × 104 | 273(6.56 ± 0.02) | |
| DRD2 W100EL1L | 14 (0.071 ± 0.0007)⊥p=0.006 |
1.89 × 105 ± 3.7 × 104 | 387(6.42 ± 0.08) | |
| DRD2 W100EL1F | 26 (0.038 ± 0.007)⊥p=0.008 |
6.32 × 105 ± 8.5 × 104 | 62.8(7.22 ± 0.14) | |
| DRD2 L942.64A | 59 (0.017 ± 0.002)n.s. |
1.23 × 106 ± 1.06 × 106 | 49.8(7.56 ± 0.52) | |
| DRD2 I184EL2A | 100 (0010 ± 0.001)n.s. |
6.65 × 105 ± 5.1 × 104 | 15.5(7.81 ± 0.03) | |
| DRD2 L942.64A/I184EL2A | 3 (0.32 ± 0.06)⊥p=0.005 |
2.93 × 106 ± 2.58 × 106 | 413(6.64 ± 0.52) | |
| N-Methyl spiperone | DRD2 wild-type | 250 (0.004 ± 0.0003) |
2.34 × 108 ± 6 × 107 | 0.018(10.75 ± 0.08) |
| DRD2 W100 EL1A | 21 (0.048 ± 0.0079) ⊥p=0.0073 |
1.65 × 108 ± 6 × 107 | 0.31(9.51 ± 0.08) | |
| DRD2 W100EL1L | 20 (0.050 ± 0.0064)⊥p=0.0072 |
1.72 × 108 ± 4 × 107 | 0.29(9.53 ± 0.03) | |
| DRD2 W100EL1F | 38 (0.026 ± 0.00003)⊥p=0.0083 |
2.08 × 108 ± 5 × 107 | 0.13(9.89 ± 0.10) | |
| DRD2 L942.64A | 77 (0.013 ± 0.0047)n.s. |
2.08 × 108 ± 4 × 107 | 0.062(10.21 ± 0.08) | |
| DRD2 I184EL2A | 128 (0.0078 ± 0.00004)n.s. |
1.70 × 108 ± 3 × 107 | 0.048(10.33 ± 0.08) | |
| DRD2 L942.64A/I184EL2A | 6 (0.170 ± 0.063)⊥p=0.064 |
1.62 × 108 ± 1 × 107 | 1.02(9.01 ± 0.14) | |
| Nemonaptide | DRD2 wild-type | 167 (0.006 ± 0.0002) |
2.0 × 108 ± 5 × 107 | 0.031(10.52 ± 0.09) |
| DRD2 W100EL1A | 43 (0.023 ± 0.001)⊥p =0.002 |
1.17 × 108 ± 2 × 107 | 0.19(9.75 ± 0.14) | |
| DRD2 W100EL1L | 45 (0.022 ± 0.0018)⊥p =0.003 |
1.07 × 108 ± 3 × 107 | 0.20(9.70 ± 0.02) | |
| DRD2 W100EL1F | 40 (0.025 ± 0.0019)⊥p =0.002 |
2.03 × 108 ± 6 × 107 | 0.13(9.90 ± 0.10) | |
| DRD2 L942.64A | 26 (0.039 ± 0.0033)⊥p =0.0015 |
2.97 × 108 ± 5 × 107 | 0.13(9.88 ± 0.03) | |
| DRD2 I184EL2A | 149 (0.0067 ± 0.0004)n.s. |
9.60 × 107 ± 7 × 106 | 0.07(10.16 ± 0.06) | |
| DRD2 L942.64A/I184EL2A |
5 (0.20 ± 0.0048)⊥p=0.0009 |
2.89 × 108 ± 1 × 108 | 0.82(9.12 ± 0.19) |
Supplementary Material
Acknowledgments
This work was supported by NIH Grants RO1MH61887, U19MH82441, the NIMH Psychoactive Drug Screening Program Contract and the Michael Hooker Chair for Protein Therapeutics and Translational Proteomics (to B.L.R.) and by GM59957 (to B.K.S.). We gratefully acknowledge J. Sondek and S. Endo-Streeter for providing independent structure quality control analysis; M. J. Miley and the UNC macromolecular crystallization core for advice and use of their equipment for crystal harvesting and transport, which is supported by the National Cancer Institute under award number P30CA016086; B.E. Krumm for advice on data processing and help with thermostabilization assays, and the staff of GM/CA@APS, which has been funded with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Footnotes
Author Contributions
S.W. designed experiments, developed the DRD2 construct and purification; expressed, purified and crystallized the receptor; collected diffraction data; solved and refined the structure; analyzed the structure, performed radioligand binding, and prepared the manuscript. T.C. performed radioligand binding, analyzed the data, and assisted with preparing the manuscript. A.L. conducted the homology modeling and docking and helped edit the manuscript. B.K.S. supervised the modeling and docking and helped prepare the manuscript. D.W. refined and analyzed the structure, supervised the structure determination and assisted with preparing the manuscript. B.L.R. supervised the overall project and management and prepared the manuscript.
Author Information
Atomic coordinates and structure factors for the reported crystal structure have been deposited in the PDB under accession 6C38.
Readers are welcome to comment on the online version of the paper. Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The authors declare no competing financial interests.
References
- 1.Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- 2.Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192:481–483. doi: 10.1126/science.3854. [DOI] [PubMed] [Google Scholar]
- 3.Meltzer HY, Matsubara S, Lee JC. Classification of typical and atypical antipsychotic drugs on the basis of dopamine D-1, D-2 and serotonin2 pKi values. J Pharmacol Exp Ther. 1989;251:238–246. [PubMed] [Google Scholar]
- 4.Roth BL, Sheffler DJ, Kroeze WK. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov. 2004;3:353–359. doi: 10.1038/nrd1346. [DOI] [PubMed] [Google Scholar]
- 5.Roth BL. Drugs and valvular heart disease. N Engl J Med. 2007;356:6–9. doi: 10.1056/NEJMp068265. [DOI] [PubMed] [Google Scholar]
- 6.Seeman P, Lee T. Antipsychotic drugs: direct correlation between clinical potency and presynaptic action on dopamine neurons. Science. 1975;188:1217–1219. doi: 10.1126/science.1145194. [DOI] [PubMed] [Google Scholar]
- 7.Sibley DR, Monsma FJ., Jr Molecular biology of dopamine receptors. Trends Pharmacol Sci. 1992;13:61–69. doi: 10.1016/0165-6147(92)90025-2. [DOI] [PubMed] [Google Scholar]
- 8.Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- 9.Volkow ND, Fowler JS, Wang GJ, Swanson JM, Telang F. Dopamine in drug abuse and addiction: results of imaging studies and treatment implications. Arch Neurol. 2007;64:1575–1579. doi: 10.1001/archneur.64.11.1575. [DOI] [PubMed] [Google Scholar]
- 10.Bunzow JR, et al. Cloning and expression of a rat D2 dopamine receptor cDNA. Nature. 1988;336:783–787. doi: 10.1038/336783a0. [DOI] [PubMed] [Google Scholar]
- 11.Grandy DK, et al. Cloning of the cDNA and gene for a human D2 dopamine receptor. Proc Natl Acad Sci U S A. 1989;86:9762–9766. doi: 10.1073/pnas.86.24.9762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monsma FJ, Jr, McVittie LD, Gerfen CR, Mahan LC, Sibley DR. Multiple D2 dopamine receptors produced by alternative RNA splicing. Nature. 1989;342:926–929. doi: 10.1038/342926a0. [DOI] [PubMed] [Google Scholar]
- 13.Allen JA, et al. Discovery of beta-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc Natl Acad Sci U S A. 2011;108:18488–18493. doi: 10.1073/pnas.1104807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Javitch JFD, Chen J, Karlin A. Mapping the binding-site crevice of the dopamine D2 receptor by the substituted-cysteine accessibility method. Neuron. 1995;14:825–831. doi: 10.1016/0896-6273(95)90226-0. [DOI] [PubMed] [Google Scholar]
- 15.Ballesteros JA, Shi L, Javitch JA. Structural mimicry in G protein-coupled receptors: implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Mol Pharmacol. 2001;60:1–19. [PubMed] [Google Scholar]
- 16.Chien EY, et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang S, et al. D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science. 2017;358:381–386. doi: 10.1126/science.aan5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manglik A, et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature. 2016;537:185–190. doi: 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wacker D, Stevens RC, Roth BL. How Ligands Illuminate GPCR Molecular Pharmacology. Cell. 2017;170:414–427. doi: 10.1016/j.cell.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCorvy JD, et al. Structure-inspired design of beta-arrestin-biased ligands for aminergic GPCRs. Nat Chem Biol. doi: 10.1038/nchembio.2527. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Free RB, et al. Discovery and characterization of a G protein-biased agonist that inhibits beta-arrestin recruitment to the D2 dopamine receptor. Mol Pharmacol. 2014;86:96–105. doi: 10.1124/mol.113.090563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts DJ, Strange PG. Mechanisms of inverse agonist action at D2 dopamine receptors. Br J Pharmacol. 2005;145:34–42. doi: 10.1038/sj.bjp.0706073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rasmussen SG, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shapiro DA, Kristiansen K, Weiner DM, Kroeze WK, Roth BL. Evidence for a model of agonist-induced activation of 5-HT2A serotonin receptors which involves the disruption of a strong ionic interaction between helices 3 and 6. J Biol Chem. 2002;18:18. doi: 10.1074/jbc.M111675200. [DOI] [PubMed] [Google Scholar]
- 25.Ballesteros JA, et al. Activation of the beta 2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J Biol Chem. 2001;276:29171–29177. doi: 10.1074/jbc.M103747200. [DOI] [PubMed] [Google Scholar]
- 26.Palczewski K, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 27.Janssen PA, et al. Pharmacology of risperidone (R 64,766) a new antipsychotic with serotonin-S2 and dopamine-D2 antagonistic properties. Journal of Pharmacology and Experimental Therapeutics. 1988;244:685–693. [PubMed] [Google Scholar]
- 28.Kapur S, Zipursky R, Jones C, Remington G, Houle S. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry. 2000;157:514–520. doi: 10.1176/appi.ajp.157.4.514. [DOI] [PubMed] [Google Scholar]
- 29.Kapur S, Seeman P. Does fast dissociation from the dopamine d(2) receptor explain the action of atypical antipsychotics?: A new hypothesis. Am J Psychiatry. 2001;158:360–369. doi: 10.1176/appi.ajp.158.3.360. [DOI] [PubMed] [Google Scholar]
- 30.Sykes DA, et al. Extrapyramidal side effects of antipsychotics are linked to their association kinetics at dopamine D2 receptors. Nat Commun. 2017;8:763. doi: 10.1038/s41467-017-00716-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monsma FJ, Jr, McVittie LD, Gerfen CR, Mahan LC, Sibley DR. Multiple D2 dopamine receptors produced by alternative RNA splicing. Nature. 1989;342:926–929. doi: 10.1038/342926a0. [DOI] [PubMed] [Google Scholar]
- 32.Rosenbaum DM, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 33.Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat Protoc. 2009;4:706–731. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 35.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica Section D, Biological crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allen JA, et al. Discovery of beta-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc Natl Acad Sci U S A. 2011;108:18488–18493. doi: 10.1073/pnas.1104807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Motulsky HJ, Mahan LC. The kinetics of competitive radioligand binding predicted by the law of mass action. Mol Pharmacol. 1984;25:1–9. [PubMed] [Google Scholar]
- 40.Pei J, Grishin NV. PROMALS3D: multiple protein sequence alignment enhanced with evolutionary and three-dimensional structural information. Methods Mol Biol. 2014;1079:263–271. doi: 10.1007/978-1-62703-646-7_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chien EY, et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang S, et al. D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science. 2017;358:381–386. doi: 10.1126/science.aan5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Webb B, Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Bioinformatics. 2014;47:561–5632. doi: 10.1002/0471250953.bi0506s47. [DOI] [PubMed] [Google Scholar]
- 44.Coleman RG, Carchia M, Sterling T, Irwin JJ, Shoichet BK. Ligand pose and orientational sampling in molecular docking. PLoS One. 2013;8:e75992. doi: 10.1371/journal.pone.0075992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Southan C, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res. 2016;44:D1054–1068. doi: 10.1093/nar/gkv1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mysinger MM, Carchia M, Irwin JJ, Shoichet BK. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem. 2012;55:6582–6594. doi: 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Case DA, et al. AMBER. 2015;2015 [Google Scholar]
- 48.Word JM, Lovell SC, Richardson JS, Richardson DC. Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation1. Journal of Molecular Biology. 1999;285:1735–1747. doi: 10.1006/jmbi.1998.2401. [DOI] [PubMed] [Google Scholar]
- 49.Gallagher K, Sharp K. Electrostatic contributions to heat capacity changes of DNA-ligand binding. Biophys J. 1998;75:769–776. doi: 10.1016/S0006-3495(98)77566-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharp KA. Polyelectrolyte electrostatics: Salt dependence, entropic, and enthalpic contributions to free energy in the nonlinear Poisson–Boltzmann model. Biopolymers. 1995;36:227–243. [Google Scholar]
- 51.Mysinger MM, Shoichet BK. Rapid context-dependent ligand desolvation in molecular docking. J Chem Inf Model. 2010;50:1561–1573. doi: 10.1021/ci100214a. [DOI] [PubMed] [Google Scholar]
- 52.Sadowski J, Gasteiger J, Klebe G. Comparison of Automatic Three-Dimensional Model Builders Using 639 X-ray Structures. Journal of Chemical Information and Computer Sciences. 1994;34:1000–1008. [Google Scholar]
- 53.Hawkins PC, Skillman AG, Warren GL, Ellingson BA, Stahl MT. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model. 2010;50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chambers CC, Hawkins GD, Cramer CJ, Truhlar DG. Model for Aqueous Solvation Based on Class IV Atomic Charges and First Solvation Shell Effects. The Journal of Physical Chemistry. 1996;100:16385–16398. [Google Scholar]
- 55.Li J, Zhu T, Cramer CJ, Truhlar DG. New Class IV Charge Model for Extracting Accurate Partial Charges from Wave Functions. The Journal of Physical Chemistry A. 1998;102:1820–1831. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factor files for the DRD2/Risperidone structure have been deposited in the RCSB Protein Data Bank with identification code 6C38. All other data are available from the corresponding authors upon reasonable request.