

Summary
Aging is commonly defined as the loss of global homeostasis, which results from progressive alteration of all organs function. This model is currently challenged by recent data showing that interventions that extend lifespan do not always increase the overall fitness of the organism. These data suggest the existence of tissue‐specific factors that regulate the pace of aging in a cell‐autonomous manner. Here, we investigated aging of Caenorhabditis elegans striated muscles at the subcellular and the physiological level. Our data show that muscle aging is characterized by a dramatic decrease in the expression of genes encoding proteins required for muscle contraction, followed by a change in mitochondria morphology, and an increase in autophagosome number. Myofilaments, however, remain unaffected during aging. We demonstrated that the conserved transcription factor UNC‐120/SRF regulates muscle aging biomarkers. Interestingly, the role of UNC‐120/SRF in the control of muscle aging can be dissociated from its broader effect on lifespan. In daf‐2/insulin/IGF1 receptor mutants, which exhibit a delayed appearance of muscle aging biomarkers and are long‐lived, disruption of unc‐120 accelerates muscle aging but does not suppress the lifespan phenotype of daf‐2 mutant. Conversely, unc‐120 overexpression delays muscle aging but does not increase lifespan. Overall, we demonstrate that UNC‐120/SRF controls the pace of muscle aging in a cell‐autonomous manner downstream of the insulin/IGF1 receptor.
Keywords: biomarkers, Caenorhabditis elegans, DAF‐2/insulin‐IGF‐1 receptor, muscle, transcription factor, UNC‐120/SRF
1. INTRODUCTION
The survival of multicellular organisms relies on their adaptation to environmental and nutritional variations through complex and permanent interactions between tissues. Thus, increasing the lifespan of an organism is thought to reflect the maintenance of overall tissues integrity and function at advanced age (Hansen & Kennedy, 2016; Laplante & Sabatini, 2012; Rando & Chang, 2012; Russell & Kahn, 2007). Based on this concept, assessing aging in model organisms relies mostly on lifespan measurement. However, recent studies demonstrated that the span of good health is not necessarily proportional to the lifespan among strains with distinct genotypes (Podshivalova, Kerr & Kenyon, 2017) and even among isogenic individuals within a population (Zhang et al., 2016).
These data raise the question of a tissue‐specific impact for the numerous genes identified as modifiers of Caenorhabditis elegans lifespan. Indeed, while C. elegans is commonly used as an aging model, as illustrated by hundreds of genes identified that influence its lifespan, aging at the tissue scale has been largely overlooked (Herndon et al., 2002; Liu et al., 2013; McGee et al., 2011). To further delineate factors that modulate the pace of physiological tissue aging, we aimed at defining the subcellular changes that take place during aging of muscle tissue. We chose the body‐wall muscle cells (BWM) of C. elegans that are functionally equivalent to vertebrate skeletal muscles and are required for locomotion (Gieseler, Qadota & Benian, 2016). Similar to mammals, C. elegans display progressive loss of mobility that involves muscle (Herndon et al., 2002) and neurotransmission defects (Liu et al., 2013).
Our data show that the loss of physical performance with age is associated with gradual subcellular changes taking place at different time points in muscle and include a drastic transcriptional downregulation of specific muscle genes, progressive mitochondria fragmentation, and autophagic vesicle accumulation while actin–myosin network remains intact. This sequence of events can be accelerated or delayed upon inhibition or upregulation of the conserved transcription factor UNC‐120/SRF, respectively, and this without necessarily affecting lifespan. Finally, we showed that loss of daf‐2 delays the pace of muscle aging, at least in part, through the upregulation of unc‐120 transcripts levels.
2. RESULTS
2.1. Time course of muscle changes associated with loss of mobility during aging
To identify potential cellular hallmarks of muscle aging, we analyzed worms at different time points during aging, starting from young adults until day 12, when around 50% of worms showed loss of locomotion (Herndon et al., 2002 and Figure S1).
First, we analyzed mitochondria morphology using transgenic worms that express a mitochondria‐targeted GFP specifically in BWM (Benedetti, Haynes, Yang, Harding & Ron, 2006). As previously published (Regmi, Rolland & Conradt, 2014), we observed the progressive fragmentation of muscle mitochondria with age with a significant decrease in the proportion of animals showing tubular mitochondria as soon as day 2 (Figures 1a and S2a). To analyze whether mitochondria network morphology mirrors the functional status of muscle cells, we separated 9‐day‐old worms according to their locomotion phenotype and scored the mitochondria phenotype in mobile vs. immobile worms. At this age, loss of locomotion was observed in about 20% of worms (Figure S1). Interestingly, the fragmented pattern was clearly restricted to worms showing a loss of locomotion, and tubular mitochondria could only occasionally be observed. By contrast, mobile worms retained either a “young” tubular interconnected or an intermediate pattern (about 40% and 60%, respectively, Figure 1b). These data strongly suggest that mitochondria fragmentation is reflecting loss of muscle function rather than just chronological age.
Figure 1.
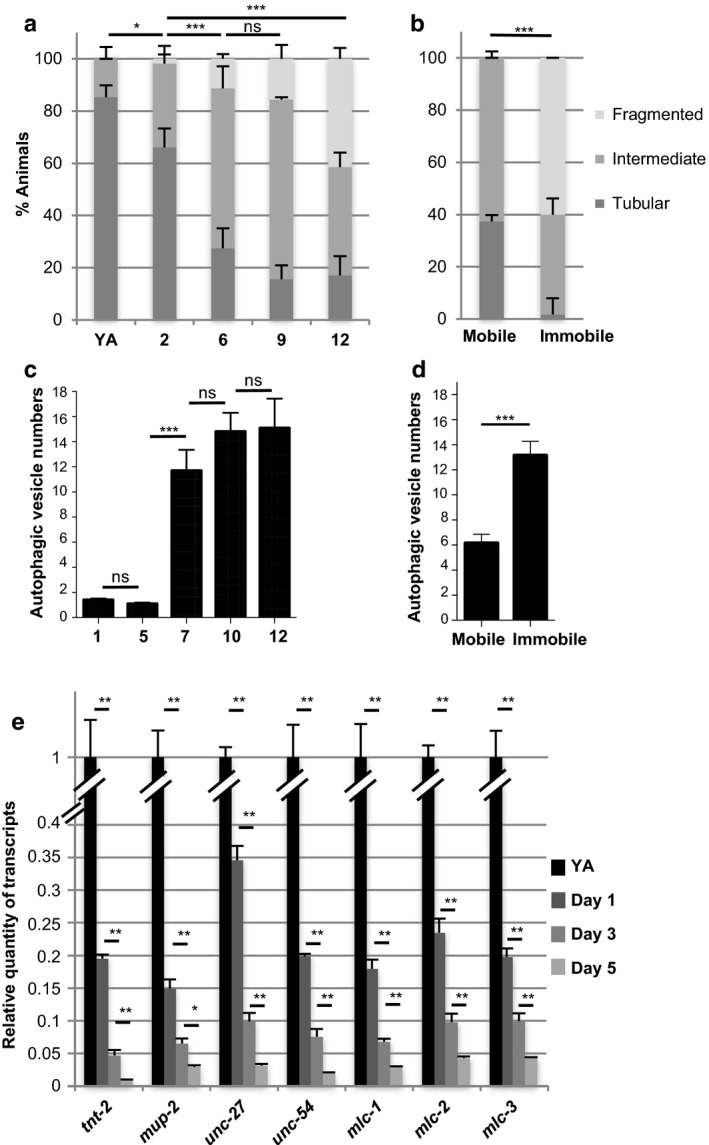
Time course quantification of muscle changes associated with loss of mobility with aging. (a–b) Quantification of animals with the indicated muscle mitochondria phenotypes at different ages from the young adult (YA) stage (n = 41, 56, 62, 45, and 94 for YA, days 2, 6, 9, and 12, respectively) (a) or at day 9, in worms which have been sorted according to their mobility phenotype (n = 59 and 60 for mobile and immobile worms, respectively) (b). Bars are mean ± SEM, chi‐square test. (c–d) Quantification of autophagic vesicles number per muscle cell at the indicated ages (n = 35, 40, 48, 63, and 41 for days 1, 5, 7, 10, and 12, respectively) (c) or at day 9 of adulthood, in worms which have been sorted according to their mobility phenotype (n = 38 and 27 for mobile and immobile worms, respectively) (d). Bars are mean ± SEM, (c) Kruskal–Wallis and Dunn's post hoc test or (d) Mann–Whitney test. (e) Relative quantity of muscle‐specific gene transcripts at different ages during adulthood. For each gene, levels were normalized to the YA value. Bars are mean ± SEM (n = 6), Mann–Whitney test
Second, we analyzed autophagy in aging muscle cells. Previous studies reported the beneficial impact of autophagy on lifespan in C. elegans and other species (Madeo, Zimmermann, Maiuri & Kroemer, 2015), and inhibition of autophagy was recently shown to enhance mitochondria dysfunction associated with muscle aging in mice (Carnio et al., 2014). To determine whether the autophagic process was altered during muscle aging, we generated transgenic worms expressing GFP::LGG‐1 under the control of the muscle‐specific dyc‐1S promoter (Lecroisey et al., 2008) (Figure S2b). LGG‐1, the C. elegans orthologue of LC3, changes from diffuse to punctate pattern when autophagy is induced (Melendez et al., 2003). To validate this marker, we used starvation to induce autophagy (Melendez et al., 2003). The mean number of autophagosomes per muscle cell increased by 56% in starved worms compared to well‐fed control animals (Figure S3a), thus validating this transgene as a tool to monitor autophagy in muscle cells. Furthermore, we checked that the lifespan of transgenic worms was not different as compared to wild type (Figure S3b). Under standard conditions, the average number of autophagic vesicles per muscle cells was stable during the first 5 days of adulthood and subsequently increased by more than sixfold between day 5 and day 7 of adulthood (Figure 1c). When 9‐day‐old worms were separated according to their locomotion phenotype, we observed twice as much vesicles in immobile worms compared to worms that were still mobile (Figure 1d). To ensure that the GFP puncta correspond to autophagic vesicles rather than GFP::LGG‐1 aggregates, we constructed a transgenic line expressing a mutant form of GFP::LGG‐1(G116A) which is no longer targeted to autophagosomes (Manil‐Segalen et al., 2014), specifically in muscle.
Worms carrying this transgene showed a diffuse cytoplasmic GFP staining of muscle cells at all ages, thus excluding the possibility that the GFP::LGG‐1 puncta observed in aged worms would correspond to aggregates triggered by general proteostasis imbalance (Figure S3c). Altogether these data show that the increase in autophagosome number correlates with the loss of mobility but appears at later ages as compared to mitochondria network fragmentation.
We next investigated structural features of muscle cells with age by observing actin and myosin myofilaments, which are essential for muscle contraction. Disorganization of actin thin filaments is a hallmark of muscle degeneration occurring under pathological conditions in C. elegans (Gieseler, Grisoni & Segalat, 2000). Interestingly, actin filaments did not show any obvious defects in wild‐type animals up to day 13 (n = 82, Figure S2), while at this age, 50% of the animals are immobile (Figure S1).
We also examined the pattern of the myosin heavy‐chain protein MYO‐3, one of the main components of BWM thick filaments. Several previous studies using the RW1596 strain expressing a Pmyo‐3:: GFP::myo‐3 reporter transgene (Campagnola et al., 2002) described alteration in thick filaments integrity within the first week of adulthood. However, careful analysis of these transgenic animals by the Moerman laboratory revealed myofilaments instability as early as L4 stage, probably as a result of GFP‐tagged myosin overexpression (Meissner et al., 2009). To monitor thick filament integrity in a more physiological context, we generated a GFP::MYO‐3 knock‐in strain (KAG420). Using this line, we did not observe any changes in myosin filament organization as far as day 13 (n = 88, Figure S2). This suggests that the loss of mobility is not the consequence of myofilament disruption.
In order to extend our analysis to transcriptional changes, we used the results of a microarray study performed in our laboratory to identify genes whose expression level varies with life expectancy (see Supporting information). Focusing on genes predominantly expressed in BWMs according to Wormbase, we identified 28 evolutionarily conserved genes with expression levels varying from twofold to 20‐fold between the YA stage (before egg production started) and day 7 of adulthood (when worms just stopped to lay eggs). The majority of these genes was downregulated (27 out of 28) and encode proteins required for muscle contraction, including core components of the sarcomere such as the subunits of the troponin complex (six of seven encoded by the C. elegans genome: tnt‐2; mup‐2; unc‐27; tni‐1; tni‐3, and pat‐10), the tropomyosin (lev‐11), muscle myosin regulatory light chains (three of five predicted: mlc‐1; mlc‐2; mlc‐3), the two myosin heavy chains (myo‐3 and unc‐54), the paramyosin unc‐15, but also nonsarcomeric proteins such as the potassium channel twk‐18, the calsequestrin csq‐1 (Figure S4a). Microarray results were validated for 13 genes randomly picked among those showing between fivefold and 20‐fold variation in transcript levels (Figure S4b). Notably, variations did not seem to result from a general sickness of muscle cells as other muscle‐expressed genes did not show significant variations of their transcript levels (Figure S4b). We then asked whether expression levels started to decrease before day 7 of adulthood. To this aim, we used rrf‐3(pk1426) mutants which are sterile at 25°C (thus precluding contamination from the embryonic transcriptome) while harboring wild‐type lifespan (Masse et al., 2008) and analyzed transcript levels for seven genes (among those showing the strongest variations). We observed a significant downregulation as early as day 1 of adulthood for all tested genes (Figure 1e). Thus, the downregulation of sarcomeric gene expression could be an early hallmark of muscle aging and may play a causal role in muscle aging.
2.2. Role of myogenic transcription factors in the regulation of muscle gene expression during aging
We hypothesized that myogenic transcription factors may be responsible for the transcript variation observed during early adulthood. The three transcription factors UNC‐120/SRF, HLH‐1/MYOD, and HND‐1/HAND1 are required for BWM differentiation during embryogenesis (Fukushige, Brodigan, Schriefer, Waterston & Krause, 2006; Kuntz, Williams, Sternberg & Wold, 2012; Lei et al., 2010). UNC‐120/SRF and HLH‐1/MYOD remain expressed at adult stage, but their role at this age is unknown (McKay et al., 2003). We thus asked whether their inactivation during adulthood might also affect muscle gene expression with age. To this end, we treated worms with RNAi targeting either hlh‐1 or unc‐120 genes starting at pre‐adult/L4 stage. After 24 hr, both hlh‐1 and unc‐120 transcripts levels were reduced by 40%, and at 7 days of adulthood, they were reduced by 90% and 50%, respectively (Figure S5a,b). In unc‐120 RNAi‐treated worms, we consistently observed a twofold to fivefold downregulation of muscle transcript levels at day 7 of adulthood, when compared to age‐matched control (Figure 2a) for which we checked that the downregulation in transcript levels with age was still observed (Figure S6). Conversely, hlh‐1 RNAi conditions did not cause significant differences with controls except for four genes that showed a weak trend (0.6 fold) toward increased expression (Figure 2b).
Figure 2.

Downregulation of unc‐120 expression induces precocious appearance of muscle aging markers. (a–b) Relative quantity of transcripts measured in 7‐day‐old animals fed either control or (a) unc‐120 or (b) hlh‐1 RNAi bacteria. For each gene, results were normalized to their respective control. Bars are mean ± SEM (n = 6), Mann–Whitney test. (c–d) Relative quantity of (c) unc‐120 or (d) hlh‐1 transcripts in wild‐type young adults (YA) and 7‐day‐old animals. For each gene, results were normalized to the respective YA stage values. Bars are mean ± SEM (n = 6), Mann–Whitney test. (e) Quantification of animals with the indicated muscle mitochondria phenotypes, at days 2, 6, and 9 of adulthood animals expressing a mitochondria‐targeted GFP in muscle cells (see Experimental procedures section), fed control or unc‐120 RNAi bacteria. n = 52, 57, and 54 for control worms and n = 58, 63, and 70 for unc‐120 RNAi‐treated worms at days 2, 6, and 9, respectively. Data are presented as mean ± SEM, chi‐square test. (f) Number of autophagic vesicles per muscle cell at different days of adulthood in animals expressing a GFP marker of autophagic vesicles in body‐wall muscles (see Experimental procedures section), fed control or unc‐120 RNAi bacteria. Number of animals scored: n = 40, 48, 63, and 41 for control worms; n = 44, 50, 50, and 10 for unc‐120 RNAi‐treated worms at days 2, 7, 10, and 12, respectively. Bars are mean ± SEM, Kruskal–Wallis and Dunn's post hoc test. (g) Body bend frequency at different adult ages of animals fed control or unc‐120 RNAi bacteria. n = 37, 131, 92, and 66 for control worms and n = 34, 126, 94, and 70 for unc‐120 RNAi‐treated animals at YA, days 3, 7, and 10, respectively. Bars are mean ± SEM, Kruskal–Wallis and Dunn's post hoc test. (h) Survival curves of worms fed control or unc‐120 RNAi bacteria. The corresponding average lifespans were 16.9 ± 0.4 days (n = 137) and 14.1 ± 0.3 days (n = 121) for control and unc‐120 RNAi conditions, respectively
We then asked whether unc‐120 and hlh‐1 expression varies with age and we indeed observed a twofold decrease in unc‐120 transcripts during the first week of adulthood while hlh‐1 expression levels showed a weak but significant increase (Figure 2c,d). Overall these data suggested that UNC‐120 might play a physiological role in the maintenance of muscle gene expression during adulthood, while HLH‐1 does not seem to have much impact.
2.3. Downregulation of unc‐120 expression induced earlier appearance of muscle aging markers
We next asked whether later markers of muscle aging such as mitochondrial network morphology and autophagosome increase were also affected by unc‐120 RNAi. At day 6 of adulthood, unc‐120 RNAi animals already exhibited altered mitochondria morphology with a decreased fraction of worms with interconnected mitochondria and the appearance of animals with fragmented mitochondria (Figure 2e). Thus, the mitochondrial pattern of 6‐day‐old worms treated with unc‐120 RNAi was more similar to 9‐day‐old than to age‐matched control worms. At day 9 of adulthood, the proportion of worms with fragmented mitochondria was increased by threefold in unc‐120 RNAi‐treated worms as compared to age‐matched controls. Quantification of autophagic vesicles in muscle also revealed a stronger increase in vesicles number in unc‐120 RNAi‐treated worms compared to age‐matched control worms (Figure 2f), while worms expressing the GFP::LGG‐1(G116A) mutant did not show aggregate formation under the same experimental condition (n = 19, 37, and 51 at days 3, 7, and 10, respectively, data not shown).
These data strongly suggest that unc‐120 RNAi causes precocious muscle aging, which might cause the decline of muscle function at younger age during adulthood. To address this hypothesis, we evaluated the capacity of worms to perform body bends in liquid medium. As previously reported (Bansal, Zhu, Yen & Tissenbaum, 2015), control worms showed a progressive age‐related decline in body bend frequency. This decline was enhanced in unc‐120 RNAi worms, starting from day 7 of adulthood (Figure 2g). We finally asked whether unc‐120 downregulation affects longevity and we indeed observed a reduction in average lifespan by 16% (Figure 2h).
Overall our results showed that inhibition of unc‐120 expression accelerates the appearance of several markers of muscle aging and strongly suggested that unc‐120 is required for the maintenance of muscle integrity and function during aging.
2.4. Muscle‐specific unc‐120 overexpression is sufficient to delay markers of muscle aging without extending lifespan
Because unc‐120 downregulation reduces lifespan unc‐120 might regulate the pace of muscle aging through systemic mechanisms and/or in a tissue‐specific manner. To further explore UNC‐120 function in muscle aging, we characterized its tissue expression during adulthood using a GFP::unc‐120 knock‐in line (Supporting information). GFP::UNC‐120 expression was observed in the 95 BWM that run along the anterior–posterior axis, but also in the 20 gonadal sheath cells and in 55 cells in the head (37 pharyngeal muscle cells, nine marginal cells, and nine pharyngeal epithelial cells, Figure 3a,b). In agreement with unc‐120 transcript variations with age (Figure 2c), we observed a decrease in UNC‐120 protein levels during aging (Figure S7).
Figure 3.
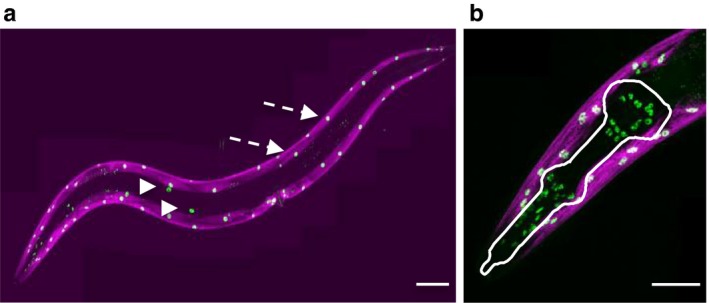
GFP::UNC‐120 expression pattern. (a) Confocal images of a transgenic gfp::unc‐120 expressing animal. The GFP::UNC‐120 protein is detected in the nuclei of all striated body‐wall muscles cells (two are indicated by dashed arrows) and of the gonadal sheath cells (arrowheads). Body‐wall muscles also express a troponin::SL2::RFP knock‐in reporter transgene (red) (see Experimental Procedure). Scale bar: 100 μm. (b) Enlargement of the head region showing the body‐wall muscle nuclei at the periphery and cells nuclei in the pharyngeal region (delineated in white, see text for detailed description). Scale bar: 50 μm
We then investigated whether unc‐120 over‐expression in BWM only was sufficient to modulate muscle aging. unc‐120 over‐expression (OE) in BWM (see Supporting information, Figures 4a and S8a) induced an increase in the expression of muscle genes in 7‐day‐old adults by at least twofold for most of the genes tested (Figures 4b and S8).
Figure 4.
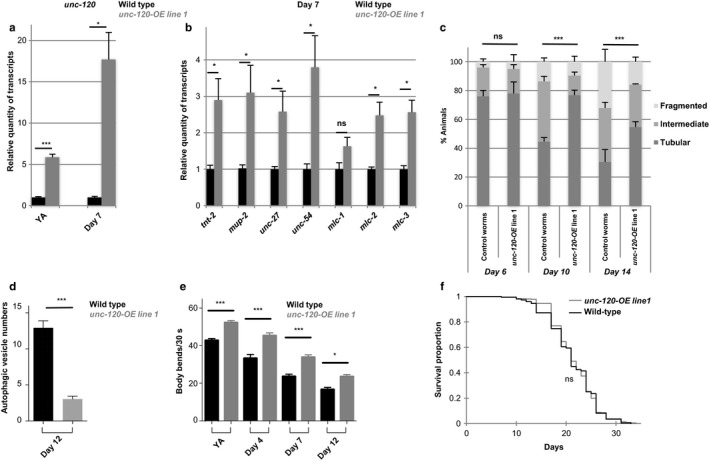
Muscle‐specific unc‐120 over‐expression is sufficient to delay muscle aging without extending lifespan. (a–b) Relative quantity of (a) unc‐120 or (b) muscle genes transcripts measured in wild‐type (WT) or unc‐120 overexpressing worms (unc‐120‐OE) at young adult (YA) stage and at day 7 of adulthood. For each gene, results were normalized to the WT level and expressed in relative units. Bars are mean ± SEM (n = 8 and 4, respectively, for YA and day 7 of adulthood), Mann–Whitney test. (c) Quantification of animals with the indicated muscle mitochondria phenotypes among WT or unc‐120 overexpressing worms (unc‐120‐OE) at different ages during adulthood. n = 100, 123, and 93 for control worms and n = 100, 117, and 121 for unc‐120‐OE worms, at day 6, 10, and 14, respectively. Data are presented as mean ± SEM, chi‐square test. (d) Number of autophagic vesicles per muscle cell at day 12 of adulthood in WT or unc‐120 overexpressing worms (unc‐120‐OE). Number of animals scored: n = 77 and 79 for WT and unc‐120‐OE worms, respectively. Data are mean ± SEM, Mann–Whitney test. (e) Body bend frequency of WT or unc‐120 overexpressing worms (unc‐120‐OE) at different ages during adulthood. n = 140, 58, 108, and 112 for control worms and n = 123, 86, 152, and 130 for unc‐120‐OE worms, at YA stage and days 4, 7, and 12, respectively. Bars are mean ± SEM, Kruskal–Wallis and Dunn's post hoc test. (f) Survival curves of WT or unc‐120 overexpressing worms (unc‐120‐OE). The corresponding average lifespans were 21.2 ± 0.3 days (n = 245) and 21.6 ± 0.2 days (n = 372) for WT and unc‐120‐OE worms, respectively. Five independent experiments have been pooled
Furthermore, mitochondria morphology remained tubular for longer time and accumulation of autophagic vesicles was prevented in those animals (Figure 4c,d). unc‐120 OE in muscle was also sufficient for improving worms physical performance until late age as revealed by a 40% increase in the frequency of body bends in 12‐day‐old worms as compared to control (Figures 4e and S8c).
We then asked whether the beneficial effect of unc‐120 OE on muscle aging markers could result from a broader impact on lifespan (Demontis, Piccirillo, Goldberg & Perrimon, 2013). Remarkably, independent transgenic strains showed either no impact (Figure 4f) or even a 20% reduction in average lifespan (Figure S8d) while they all demonstrated a beneficial impact of unc‐120 overexpression on muscle aging markers. Overall these results showed that unc‐120 functions in a cell‐autonomous manner to control muscle aging independently of lifespan regulation.
2.5. UNC‐120 is a novel effector of the DAF‐2 insulin/IGF‐1 receptor pathway
The DAF‐2 insulin/IGF‐1 receptor pathway is the most universal regulator of lifespan across species. We thus asked whether muscle aging biomarkers were delayed in long‐lived daf‐2(e1370) mutants. We observed a delay in the loss of physical performance after the first week of adulthood in those mutants compared to wild‐type (Figure S9a) in agreement with previously published data (Hahm et al., 2015; Mulcahy, Holden‐Dye & O'Connor, 2013). We next assessed muscle‐specific changes. The expression of sarcomeric genes was increased by two to eightfold at day 5 of adulthood as compared to wild‐type. This increase was observed as early as day 1 of adulthood, while at young adult stage, the mRNA levels of all but one (tnt‐2) assessed muscle‐specific genes were not affected in daf‐2 mutants (Figure S9b–d). Analysis of muscle mitochondria pattern demonstrated a delayed mitochondria fragmentation and only a modest increase in autophagic vesicle number with age in daf‐2 mutants (Figure S8e–f). Overall, these observations support the hypothesis that daf‐2 mutations delay the pace of muscle aging.
We then asked whether the beneficial effect of daf‐2 mutation may be dependent on unc‐120. Inhibition of unc‐120 expression in daf‐2 mutants caused a dramatic decreased of physical performance at day 18 (Figure 5a). This reduction can unlikely be explained by a systemic effect of unc‐120 inactivation because at that stage, 100% of the daf‐2(e1370); unc‐120 RNAi worms are alive (Figure 5b). We thus addressed whether unc‐120 downregulation also impacts the mitochondria and autophagy phenotypes with age. We indeed observed a significant decrease in the percentage of worms with interconnected mitochondria (Figure 5c) and a significant increase in autophagic vesicles number (Figure 5d). These data thus strongly suggest that unc‐120 is required, at least in part, in daf‐2 mutants to delay the loss of mobility while daf‐2 and unc‐120 may act independently to regulate lifespan. As unc‐120 expression is downregulated with physiological aging, we asked whether daf‐2 mutation may prevent its downregulation. We indeed observed a twofold increase in unc‐120 transcripts levels in daf‐2 mutants as early as day 1 of adulthood (Figure 5e) suggesting that the genetic interaction between daf‐2 and unc‐120 in the control of muscle aging relies at least in part on unc‐120 transcriptional regulation.
Figure 5.
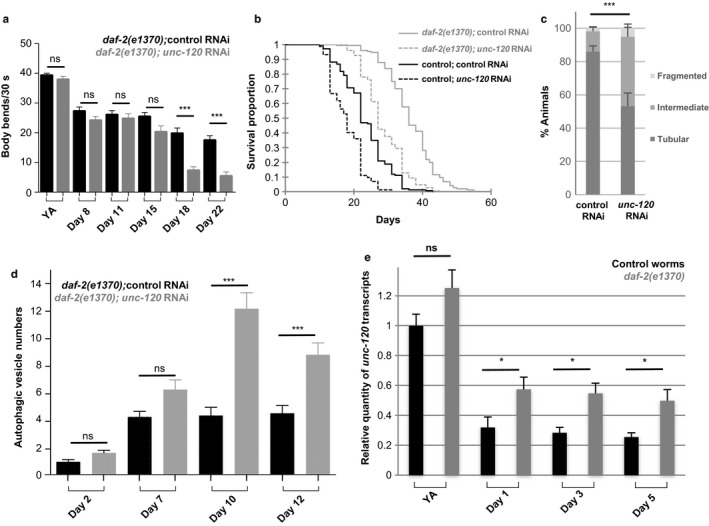
Inhibition of unc‐120 expression counteracts daf‐2 beneficial impact on muscle aging. (a) Body bend frequency of rrf‐3(pk1426); daf‐2(e1370) mutant worms fed either control or unc‐120 RNAi bacteria. Data from two independent experiments have been pooled (number of animals scored at young adult [YA] stage and at days 8, 11, 15, 18, and 22 of adulthood: 66, 65, 77, 70, 63, and 72 for control RNAi; 56, 53, 60, 50, 68, and 45 for unc‐120 RNAi, respectively) and presented as mean ± SEM, Kruskal–Wallis and Dunn's post hoc test. (b) Survival curves of control (black) and daf‐2(e1370) mutant (grey) worms fed control or unc‐120 RNAi bacteria. Two independent experiments have been pooled. The corresponding average lifespans were 23.5 ± 0.6 days (n = 143) and 17.9 ± 0.4 days (n = 145) for control worms on control or unc‐120 RNAi bacteria, respectively, and 36.5 ± 0.6 days (n = 146) and 28.5 ± 0.5 days (n = 149) for daf‐2(e1370) worms on control or unc‐120 RNAi bacteria, respectively. (c) Quantification of animals with the indicated muscle mitochondria phenotypes at day 9 of adulthood among daf‐2(e1370) worms fed control or unc‐120 RNAi bacteria. Number of animals scored: n = 74 and 91 for worms on control and unc‐120 RNAi, respectively. Data are presented as mean ± SEM, chi‐square test. (d) Number of autophagic vesicles per muscle cell at different days of adulthood in daf‐2(e1370) worms expressing a GFP marker of autophagic vesicles in muscle, fed control, or unc‐120 RNAi bacteria. Number of animals scored: n = 42, 48, 63, and 41 for control worms; n = 48, 54, 60, and 50 for unc‐120 RNAi‐treated worms at days 2, 7, 10, and 12, respectively. Data are presented as mean ± SEM, Kruskal–Wallis and Dunn's post hoc test. (e) Relative quantity of unc‐120 transcripts measured in rrf‐3(pk1426) control or rrf‐3(pk1426); daf‐2(e1370) worms at YA stage and at different days of adulthood. For each gene, results were normalized to the respective control. Bars are mean ± SEM (n = 6), Mann–Whitney test
3. DISCUSSION
A number of factors have been proposed to be involved in muscle aging in mammals (including mitochondria defects and imbalanced proteostasis (Marzetti et al., 2013; Carnio et al., 2014)). However, studies analyzed experimentally induced muscle atrophy models, which may imply different mechanisms responsible for muscle dysfunction as compared to physiological aging. Our study establishes a sequence of subcellular and molecular events that occur during muscle aging and that are associated with the loss of mobility with age.
3.1. Mitochondria alteration precedes autophagy deregulation in aging muscle
Age‐dependent muscle degeneration involves a stereotyped sequence of events in which mitochondria modification constitutes the first morphological modification observed. Mitochondria are dynamic organelles whose morphology depends on different factors including their movement along cytoskeletal tracks, their fusion and fission (Otera & Mihara, 2011). Increased fragmentation of mitochondrial network is usually associated with impaired mitochondria that will be targeted for degradation by autophagy (Twig & Shirihai, 2011). Our data showed that mitochondria fragmentation appeared between day 2 and day 6 of adulthood when the decrease in physical performance was first observed (Figure 2g). Similar changes in mitochondria morphology have been reported by Regmi et al. (2014) using a different reporter strain, while they also showed that the mitochondria pattern at day 5 or day 7 of adulthood was not predictive of animal lifespan. Interestingly, our data strongly suggest that it is rather predictive of muscle health span, as mitochondria fragmentation correlated with loss of mobility (Figure 1b), and that it can be considered as an early marker of muscle fitness.
Our data show that autophagy is deregulated in muscle with age and that the increase in autophagosomes is more important in immobile vs. mobile worms of the same age. This increase could be due to an enhancement or a blockade of autophagy. Recent results (Chang, Kumsta, Hellman, Adams & Hansen, 2017) described a similar kinetics in the increase in autophagosomes number with age, using a line in which mCherry::GFP::LGG‐1 was expressed under the control of its own promoter. Interestingly, in the presence of bafilomycin A (an inhibitor of lysosomal acidification which blocks autophagy flux), the pool size of autophagic vesicles did not increase much in day 7 and older animals as compared to younger adults, thus strongly suggesting that a blockade of the autophagy flux takes place with aging (Chang et al., 2017).
Yet the increase in autophagosomes number is taking place several days after the onset of physical performance decline. In agreement with those data, a previous study reported that in muscle tissue, the rupture of protein homeostasis was not detectable before day 12 of adulthood, as revealed by aggregates formation of a conformation unstable firefly luciferase mutant (Gupta et al., 2011). Overall those observations suggest that alteration of autophagy does not appear as a primary cause but rather as a consequence of muscle aging.
3.2. Early downregulation of muscle gene expression
The earliest muscle‐specific change that we observed is the dramatic downregulation in the expression of genes encoding proteins required for muscle contraction, as soon as day 1 of adulthood. These results provide mechanistic insights that substantiate the proteomic data previously published by Liang et al. (2014) who used an unbiased global proteomics approach to investigate proteomic variations in worms with age. They reported that between day 1 and day 5 of adulthood the amount of numerous sarcomeric proteins decreases, including myosin heavy‐chain isoforms (MYO‐1, MYO‐2, MYO‐3, UNC‐54), myosin light‐chain isoforms (MLC‐1, MLC‐3), troponin T (MUP‐2), tropomyosin (LEV‐11), and paramyosin (UNC‐15), mitochondrial creatine kinase (W10C8.5) and calsequestrin (CSQ‐1), which we all found to be transcriptionally downregulated. Interestingly, a second proteomic study reported an increased quantity of several of those proteins in daf‐2 mutants (Depuydt et al., 2013). In light of our results, those changes in protein levels may result at least in part from the impact of daf‐2 on transcripts levels of the corresponding genes (Figure S8b–d). Furthermore, proteins turnover with age was also recently investigated by Dhondt et al. (2016) and revealed that, while some proteins like the myosin heavy‐chain UNC‐54 exhibited an extremely long half‐live (298 hr), others, such as the paramyosin UNC‐15, showed much higher turnover (66 hr). Therefore, the substantial transcriptional changes that we reported during early adulthood will most probably have different consequence on muscle integrity depending on the stability and function of the corresponding protein.
How this sequence of events is related to pathologic muscle degeneration? Progressive muscle degeneration is also observed in the context of genetic disorders such as muscular dystrophy. Interestingly, daf‐2 mutation was shown to counteract muscle defects associated with dystrophin mutation (Oh & Kim, 2013). Furthermore, mitochondria fragmentation has also been involved in muscle degeneration in a model of Duchenne muscular dystrophy in C. elegans, which combines mutations of both hlh‐1/myoD and dys‐1/dystrophin (Giacomotto et al., 2013; Gieseler et al., 2000). However, in this genetic context, mitochondria fragmentation occurs well before any obvious sign of degeneration can be detected (Giacomotto et al., 2013). Additionally, dystrophic mutants are also characterized by a strong disorganization of actin cytoskeleton, early accumulation of autophagosomes, and the loss of muscle cells (Brouilly et al., 2015). In contrast during physiological aging of wild‐type worms, actin and myosin filaments remain essentially intact in worms with complete loss of mobility and accumulation of autophagosomes is a late event. Thus, although similar subcellular features are observed in pathologic and age‐dependent muscle degeneration, the sequence of events that take place during muscle aging is different. Those observations strongly suggest that dystrophic models and muscle aging involve distinct mechanisms.
3.3. UNC‐120/SRF is required for muscle maintenance during adulthood
Our data uncover a novel role for unc‐120 in muscle maintenance during adulthood. Interestingly, while myogenesis requires hlh‐1 and involves the direct positive transcriptional regulation of unc‐120 by HLH‐1, HLH‐1 does not seem to play an essential role in the regulation of the different muscle aging biomarkers (Figure 2b and data not shown). Conversely, unc‐120 transcript levels decrease with age and precocious downregulation of unc‐120 expression further decreases sarcomeric genes expression, accelerates mitochondria fragmentation, autophagic vesicle accumulation, and is associated with premature loss of mobility. Furthermore, unc‐120 overexpression is sufficient to delay muscle aging.
The function of UNC‐120 in muscle aging may involve the regulation of the expression of genes required for mitochondria dynamics and autophagy activity. Likewise in Drosophila, FOXO muscle‐specific overexpression is sufficient to delay muscle functional decay, and this effect relies at least in part on the increased expression of several core components of autophagosomes which appears to be sufficient for maintaining basal autophagy with age (Demontis & Perrimon, 2010). Further investigation of UNC‐120 transcriptional targets should help to decipher its mechanism of action.
UNC‐120 is homologous to the mammalian MADS box family transcription factor SRF (for serum response factor). Similar to UNC‐120, SRF is essential for muscle development and muscle‐specific gene expression in mice (Fukushige et al., 2006; Li et al., 2005). Selective SRF knockout in adult postmitotic myofibers is responsible for premature muscle atrophy associated with different features that are reminiscent of defects observed in aging skeletal muscle. Age‐associated decrease in SRF expression was also observed in both mice and human muscles (Lahoute et al., 2008). Our data suggest that the age‐associated physiological decrease in UNC‐120/SRF expression contributes to different muscle changes associated with muscle aging and that this mechanism may be conserved through species.
3.4. Role of the DAF‐2/insulin‐IGF‐1 pathway in muscle aging
Our data show that daf‐2 mutants exhibit delayed appearance of different biomarkers of muscle aging and support the hypothesis that the health span of this tissue is extended in these mutants. The dramatic increase in autophagy vesicles observed during aging is strongly suppressed in daf‐2 mutants. Chang et al. (2017) also reported an absence of increase in autophagy vesicles in old daf‐2 mutants, although the number of vesicles was high during early adulthood. This apparent discrepancy might be related to the different promoters used to express LGG‐1 reporters in the two studies. Our results also strongly suggest that daf‐2 impacts muscle aging at least in part through the control of unc‐120 transcript levels. Indeed, daf‐2 mutants show higher levels of unc‐120 transcripts, and inhibiting unc‐120 expression by RNAi in daf‐2 mutants restores wild‐type levels of autophagic vesicles numbers, mitochondria fragmentation, and mobility.
However, under the same experimental conditions, downregulating unc‐120 expression in daf‐2 mutants is not sufficient to recapitulate wild‐type lifespan. Thus, our data strongly suggest that the control of muscle fitness by DAF‐2 through UNC‐120 can be dissociated from its function in overall lifespan. In agreement with this hypothesis, unc‐120 overexpression increases mobility at late age without extending lifespan.
Several reports described the cell nonautonomous impact of different genes on lifespan and support a model where aging rates between tissues are interdependent (Chang et al., 2017; Durieux, Wolff & Dillin, 2011; Laplante & Sabatini, 2012; Rando & Chang, 2012; Russell & Kahn, 2007).
Furthermore, genes that delay motility decline also affect lifespan in both C. elegans, such as hpa‐1 and hpa‐2 (Iwasa, Yu, Xue & Driscoll, 2010), and in Drosophila as exemplified by FOXO muscle‐specific overexpression (Demontis & Perrimon, 2010).
Our results, however, strongly suggest that the pace of aging of a specific organ does not necessarily impact overall lifespan. Large‐scale screening for genetic modifiers of aging was essentially based on the C. elegans lifespan phenotype. Our results, which dissociate the regulation of muscle aging from lifespan, show that future screen focused on tissue‐specific features should uncover novel aging regulators.
4. EXPERIMENTAL PROCEDURES
4.1. C. elegans strains and media
All strains were maintained at 20°C (unless otherwise indicated) on nematode growth medium agar plates freshly poured and seeded with Escherichia coli strain OP50 culture. The wild‐type reference strain was C. elegans N2 Bristol. Previously generated lines used in this study were as follows: NL2099 rrf‐3(pk1426) (Simmer et al., 2002), CB1370 daf‐2(e1370) (Kenyon, Chang, Gensch, Rudner & Tabtiang, 1993), and SJ4103 zcIs14 [Pmyo‐3::GFP(mitochondria)] (Benedetti et al., 2006). The transgenic lines created for this work are described in supporting information.
Processing of transgenic strains for mitochondria, autophagic vesicles, and myofilament analysis was performed as described in supporting information.
4.2. RNAi feeding and lifespan assays
Bacterial feeding RNAi experiments and lifespan assay were carried out as described previously (Masse et al., 2008). Further details are in supporting information.
4.3. Body bend frequency assays
Body bend frequency assay was performed as described in supporting information.
4.4. Reverse transcription–qPCR
Transcript‐level analysis was performed as described in supporting information.
4.5. Identification of muscle‐specific genes whose expression varies with lifespan expectancy
Muscle genes whose expression varies with lifespan expectancy were identified from a microarray approach described in supporting information.
4.6. Statistical analysis
Statistical analyses were performed as described in supporting information.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
A.MdL., L.M., and F.S. conceived, designed the study, performed the experimental works, and wrote the manuscript. A.MdL., L.M., F.S., and J.L.B analyzed the data. L. P. and M.C.M. generated and validated the muscle‐specific autophagy reporter strains. A.MdL., L.M., K.G., J.L.B., and F.S. edited the manuscript.
Supporting information
ACKNOWLEDGMENTS
We thank the Caenorhabditis Genetic Center (which is funded by NIH Office of Research Infrastructure Programs, P40 OD010440) for strains. A.MdL was funded by the Region Rhône‐Alpes and L.P. by the Ministère de l′Education Nationale, de l'Enseignement Supérieur et de la Recherche (MENESR). This work was supported by the Région Rhône‐Alpes (Projet cible, FS, No 3319), Association pour la Recherche sur le Cancer (ARC, FS, No. 076751), AFM (AFM, KG N°16109), the Fédération pour la Recherche sur le Cerveau (AO “Espoir en tête” 2013), the Programme and ERC (JLB, N°69595). Avenir Lyon Saint‐Etienne. We are grateful to S.Croze, N. Nazaret, and C. Rey from ProfileXpert, Plateforme de Génomique et Microgénomique, Université Lyon1, SFR santé LYON‐EST, UCBL‐INSERM US 7‐CNRS UMS 3453 for gene expression analysis.
Mergoud dit Lamarche A, Molin L, Pierson L, et al. UNC‐120/SRF independently controls muscle aging and lifespan in Caenorhabditis elegans . Aging Cell. 2018;17:e12713 https://doi.org/10.1111/acel.12713
REFERENCES
- Bansal, A. , Zhu, L. J. , Yen, K. , & Tissenbaum, H. A. (2015). Uncoupling lifespan and healthspan in Caenorhabditis elegans longevity mutants. Proceedings of the National Academy of Sciences of the United States of America, 112, E277–E286. https://doi.org/10.1073/pnas.1412192112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti, C. , Haynes, C. M. , Yang, Y. , Harding, H. P. , & Ron, D. (2006). Ubiquitin‐like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics, 174, 229–239. https://doi.org/10.1534/genetics.106.061580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouilly, N. , Lecroisey, C. , Martin, E. , Pierson, L. , Mariol, M. C. , Qadota, H. , … Gieseler, K. (2015). Ultra‐structural time‐course study in the C. elegans model for Duchenne muscular dystrophy highlights a crucial role for sarcomere‐anchoring structures and sarcolemma integrity in the earliest steps of the muscle degeneration process. Human Molecular Genetics, 24, 6428–6445. https://doi.org/10.1093/hmg/ddv353 [DOI] [PubMed] [Google Scholar]
- Campagnola, P. J. , Millard, A. C. , Terasaki, M. , Hoppe, P. E. , Malone, C. J. , & Mohler, W. A. (2002). Three‐dimensional high‐resolution second‐harmonic generation imaging of endogenous structural proteins in biological tissues. Biophysical Journal, 82, 493–508. https://doi.org/10.1016/S0006-3495(02)75414-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnio, S. , LoVerso, F. , Baraibar, M. A. , Longa, E. , Khan, M. M. , Maffei, M. , … Sandri, M. (2014). Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Reports, 8, 1509–1521. https://doi.org/10.1016/j.celrep.2014.07.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, J. T. , Kumsta, C. , Hellman, A. B. , Adams, L. M. , Hansen, M. (2017). Spatiotemporal regulation of autophagy during Caenorhabditis elegans aging. Elife 6, pii: e18459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demontis, F. , & Perrimon, N. (2010). FOXO/4E‐BP signaling in Drosophila muscles regulates organism‐wide proteostasis during aging. Cell, 143, 813–825. https://doi.org/10.1016/j.cell.2010.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demontis, F. , Piccirillo, R. , Goldberg, A. L. , & Perrimon, N. (2013). Mechanisms of skeletal muscle aging: Insights from Drosophila and mammalian models. Disease Models & Mechanisms, 6, 1339–1352. https://doi.org/10.1242/dmm.012559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depuydt, G. , Xie, F. , Petyuk, V. A. , Shanmugam, N. , Smolders, A. , Dhondt, I. , … Braeckman, B. P. (2013). Reduced insulin/insulin‐like growth factor‐1 signaling and dietary restriction inhibit translation but preserve muscle mass in Caenorhabditis elegans . Molecular & Cellular Proteomics: MCP, 12, 3624–3639. https://doi.org/10.1074/mcp.M113.027383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhondt, I. , Petyuk, V. A. , Cai, H. , Vandemeulebroucke, L. , Vierstraete, A. , Smith, R. D. , … Braeckman, B. P. (2016). FOXO/DAF‐16 activation slows down turnover of the majority of proteins in C. elegans . Cell Reports, 16, 3028–3040. https://doi.org/10.1016/j.celrep.2016.07.088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieux, J. , Wolff, S. , & Dillin, A. (2011). The cell‐non‐autonomous nature of electron transport chain‐mediated longevity. Cell, 144, 79–91. https://doi.org/10.1016/j.cell.2010.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushige, T. , Brodigan, T. M. , Schriefer, L. A. , Waterston, R. H. , & Krause, M. (2006). Defining the transcriptional redundancy of early bodywall muscle development in C. elegans: Evidence for a unified theory of animal muscle development. Genes & Development, 20, 3395–3406. https://doi.org/10.1101/gad.1481706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomotto, J. , Brouilly, N. , Walter, L. , Mariol, M. C. , Berger, J. , Segalat, L. , … Gieseler, K. (2013). Chemical genetics unveils a key role of mitochondrial dynamics, cytochrome c release and IP3R activity in muscular dystrophy. Human Molecular Genetics, 22, 4562–4578. https://doi.org/10.1093/hmg/ddt302 [DOI] [PubMed] [Google Scholar]
- Gieseler, K. , Grisoni, K. , & Segalat, L. (2000). Genetic suppression of phenotypes arising from mutations in dystrophin‐related genes in Caenorhabditis elegans . Current Biology, 10, 1092–1097. https://doi.org/10.1016/S0960-9822(00)00691-6 [DOI] [PubMed] [Google Scholar]
- Gieseler, K. , Qadota, H. , Benian, G. M. (2016). Development, structure, and maintenance of C. elegans body wall muscle. WormBook, 2016, 1–59. https://doi.org/10.1895/wormbook.1.81.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, R. , Kasturi, P. , Bracher, A. , Loew, C. , Zheng, M. , Villella, A. , … Raychaudhuri, S. (2011). Firefly luciferase mutants as sensors of proteome stress. Nature Methods, 8, 879–884. https://doi.org/10.1038/nmeth.1697 [DOI] [PubMed] [Google Scholar]
- Hahm, J. H. , Kim, S. , DiLoreto, R. , Shi, C. , Lee, S. J. , Murphy, C. T. , & Nam, H. G. (2015). C. elegans maximum velocity correlates with healthspan and is maintained in worms with an insulin receptor mutation. Nature Communications, 6, 8919 https://doi.org/10.1038/ncomms9919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, M. , & Kennedy, B. K. (2016). Does longer lifespan mean longer healthspan? Trends in Cell Biology, 26, 565–568. https://doi.org/10.1016/j.tcb.2016.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herndon, L. A. , Schmeissner, P. J. , Dudaronek, J. M. , Brown, P. A. , Listner, K. M. , Sakano, Y. , … Driscoll, M. (2002). Stochastic and genetic factors influence tissue‐specific decline in ageing C. elegans . Nature, 419, 808–814. https://doi.org/10.1038/nature01135 [DOI] [PubMed] [Google Scholar]
- Iwasa, H. , Yu, S. , Xue, J. , & Driscoll, M. (2010). Novel EGF pathway regulators modulate C. elegans healthspan and lifespan via EGF receptor, PLC‐gamma, and IP3R activation. Aging Cell, 9, 490–505. https://doi.org/10.1111/j.1474-9726.2010.00575.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon, C. , Chang, J. , Gensch, E. , Rudner, A. , & Tabtiang, R. (1993). A C. elegans mutant that lives twice as long as wild type. Nature, 366, 461–464. https://doi.org/10.1038/366461a0 [DOI] [PubMed] [Google Scholar]
- Kuntz, S. G. , Williams, B. A. , Sternberg, P. W. , & Wold, B. J. (2012). Transcription factor redundancy and tissue‐specific regulation: Evidence from functional and physical network connectivity. Genome Research, 22, 1907–1919. https://doi.org/10.1101/gr.133306.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahoute, C. , Sotiropoulos, A. , Favier, M. , Guillet‐Deniau, I. , Charvet, C. , Ferry, A. , … Daegelen, D. (2008). Premature aging in skeletal muscle lacking serum response factor. PLoS ONE, 3, e3910 https://doi.org/10.1371/journal.pone.0003910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante, M. , & Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell, 149, 274–293. https://doi.org/10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecroisey, C. , Martin, E. , Mariol, M. C. , Granger, L. , Schwab, Y. , Labouesse, M. , … Gieseler, K. (2008). DYC‐1, a protein functionally linked to dystrophin in Caenorhabditis elegans is associated with the dense body, where it interacts with the muscle LIM domain protein ZYX‐1. Molecular Biology of the Cell, 19, 785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei, H. , Fukushige, T. , Niu, W. , Sarov, M. , Reinke, V. , & Krause, M. (2010). A widespread distribution of genomic CeMyoD binding sites revealed and cross validated by ChIP‐Chip and ChIP‐Seq techniques. PLoS ONE, 5, e15898 https://doi.org/10.1371/journal.pone.0015898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S. , Czubryt, M. P. , McAnally, J. , Bassel‐Duby, R. , Richardson, J. A. , Wiebel, F. F. , … Olson, E. N. (2005). Requirement for serum response factor for skeletal muscle growth and maturation revealed by tissue‐specific gene deletion in mice. Proceedings of the National Academy of Sciences of the United States of America, 102, 1082–1087. https://doi.org/10.1073/pnas.0409103102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, V. , Ullrich, M. , Lam, H. , Chew, Y. L. , Banister, S. , Song, X. , … Nicholas, H. R. (2014). Altered proteostasis in aging and heat shock response in C. elegans revealed by analysis of the global and de novo synthesized proteome. Cellular and Molecular Life Sciences, 71, 3339–3361. https://doi.org/10.1007/s00018-014-1558-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. , Zhang, B. , Lei, H. , Feng, Z. , Liu, J. , Hsu, A. L. , & Xu, X. Z. (2013). Functional aging in the nervous system contributes to age‐dependent motor activity decline in C. elegans . Cell Metabolism, 18, 392–402. https://doi.org/10.1016/j.cmet.2013.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeo, F. , Zimmermann, A. , Maiuri, M. C. , & Kroemer, G. (2015). Essential role for autophagy in life span extension. The Journal of Clinical Investigation, 125, 85–93. https://doi.org/10.1172/JCI73946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manil‐Segalen, M. , Lefebvre, C. , Jenzer, C. , Trichet, M. , Boulogne, C. , Satiat‐Jeunemaitre, B. , & Legouis, R. (2014). The C. elegans LC3 acts downstream of GABARAP to degrade autophagosomes by interacting with the HOPS subunit VPS39. Developmental Cell, 28, 43–55. https://doi.org/10.1016/j.devcel.2013.11.022 [DOI] [PubMed] [Google Scholar]
- Marzetti, E. , Csiszar, A. , Dutta, D. , Balagopal, G. , Calvani, R. , & Leeuwenburgh, C. (2013). Role of mitochondrial dysfunction and altered autophagy in cardiovascular aging and disease: From mechanisms to therapeutics. American Journal of Physiology. Heart and Circulatory Physiology, 305, H459–H476. https://doi.org/10.1152/ajpheart.00936.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masse, I. , Molin, L. , Mouchiroud, L. , Vanhems, P. , Palladino, F. , Billaud, M. , & Solari, F. (2008). A novel role for the SMG‐1 kinase in lifespan and oxidative stress resistance in Caenorhabditis elegans . PLoS ONE, 3, e3354 https://doi.org/10.1371/journal.pone.0003354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee, M. D. , Weber, D. , Day, N. , Vitelli, C. , Crippen, D. , Herndon, L. A. , … Melov, S. (2011). Loss of intestinal nuclei and intestinal integrity in aging C. elegans . Aging Cell, 10, 699–710. https://doi.org/10.1111/j.1474-9726.2011.00713.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay, S. J. , Johnsen, R. , Khattra, J. , Asano, J. , Baillie, D. L. , Chan, S. , … Moerman, D. G. (2003). Gene expression profiling of cells, tissues, and developmental stages of the nematode C. elegans . Cold Spring Harbor Symposia on Quantitative Biology, 68, 159–169. https://doi.org/10.1101/sqb.2003.68.159 [DOI] [PubMed] [Google Scholar]
- Meissner, B. , Warner, A. , Wong, K. , Dube, N. , Lorch, A. , McKay, S. J. , … Moerman, D. G. (2009). An integrated strategy to study muscle development and myofilament structure in Caenorhabditis elegans . PLoS Genetics, 5, e1000537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez, A. , Talloczy, Z. , Seaman, M. , Eskelinen, E. L. , Hall, D. H. , & Levine, B. (2003). Autophagy genes are essential for dauer development and life‐span extension in C. elegans . Science, 301, 1387–1391. https://doi.org/10.1126/science.1087782 [DOI] [PubMed] [Google Scholar]
- Mulcahy, B. , Holden‐Dye, L. , & O'Connor, V. (2013). Pharmacological assays reveal age‐related changes in synaptic transmission at the Caenorhabditis elegans neuromuscular junction that are modified by reduced insulin signalling. Journal of Experimental Biology, 216, 492–501. https://doi.org/10.1242/jeb.068734 [DOI] [PubMed] [Google Scholar]
- Oh, K. H. , & Kim, H. (2013). Reduced IGF signaling prevents muscle cell death in a Caenorhabditis elegans model of muscular dystrophy. Proceedings of the National Academy of Sciences of the United States of America, 110, 19024–19029. https://doi.org/10.1073/pnas.1308866110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otera, H. , & Mihara, K. (2011). Molecular mechanisms and physiologic functions of mitochondrial dynamics. Journal of Biochemistry, 149, 241–251. https://doi.org/10.1093/jb/mvr002 [DOI] [PubMed] [Google Scholar]
- Podshivalova, K. , Kerr, R. A. , & Kenyon, C. (2017). How a mutation that slows aging can also disproportionately extend end‐of‐life decrepitude. Cell Reports, 19, 441–450. https://doi.org/10.1016/j.celrep.2017.03.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando, T. A. , & Chang, H. Y. (2012). Aging, rejuvenation, and epigenetic reprogramming: Resetting the aging clock. Cell, 148, 46–57. https://doi.org/10.1016/j.cell.2012.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regmi, S. G. , Rolland, S. G. , & Conradt, B. (2014). Age‐dependent changes in mitochondrial morphology and volume are not predictors of lifespan. Aging, 6, 118–130. https://doi.org/10.18632/aging.100639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, S. J. , & Kahn, C. R. (2007). Endocrine regulation of ageing. Nature Reviews Molecular Cell Biology, 8, 681–691. https://doi.org/10.1038/nrm2234 [DOI] [PubMed] [Google Scholar]
- Simmer, F. , Tijsterman, M. , Parrish, S. , Koushika, S. P. , Nonet, M. L. , Fire, A. , … Plasterk, R. H. (2002). Loss of the putative RNA‐directed RNA polymerase RRF‐3 makes C. elegans hypersensitive to RNAi. Current Biology, 12, 1317–1319. https://doi.org/10.1016/S0960-9822(02)01041-2 [DOI] [PubMed] [Google Scholar]
- Twig, G. , & Shirihai, O. S. (2011). The interplay between mitochondrial dynamics and mitophagy. Antioxidants & Redox Signaling, 14, 1939–1951. https://doi.org/10.1089/ars.2010.3779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W. B. , Sinha, D. B. , Pittman, W. E. , Hvatum, E. , Stroustrup, N. , & Pincus, Z. (2016). Extended Twilight among isogenic C. elegans causes a disproportionate scaling between lifespan and health. Cell Systems, 3, 333–345.e334. https://doi.org/10.1016/j.cels.2016.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials