

Abstract
Diverse biochemical and biophysical experiments indicate that all proteins, regardless of size or origin, undergo a dynamic transition near 200 K. The cause of this shift in dynamic behavior, termed a “glass transition,” and its relation to protein function are important open questions. One explanation postulated for the transition is solidification of correlated motions in proteins below the transition. We verified this conjecture by showing that crambin's radius of gyration (Rg) remains constant below ≈180 K. We show that both atom position and dynamics of protein and solvent are physically coupled, leading to a novel cooperative state. This glassy state is identified by negative slopes of the Debye-Waller (B) factor vs. temperature. It is composed of multisubstate side chains and solvent. Based on generalization of Adam-Gibbs' notion of a cooperatively rearranging region and decrease of the total entropy with temperature, we calculate the slope of the Debye-Waller factor. The results are in accord with experiment.
Coupling of protein activity to solvent offers a unique opportunity to study basic physical phenomena. Solvent not only provides conditions for stabilizing native tertiary structure (structure-folding problem), but also, in its liquid state, provides enough flexibility for substrate diffusion, conformational changes, and enzymatic activity (function-protein dynamics).
The temperature dependence of the activity of most proteins is unusual. For these proteins, enzymatic function ceases below ≈220 K (1, 2), although the temperature at which function ceases varies with protein and solvent (3, 4). The transition in slope of root-mean-square fluctuations for proteins and glass-forming liquids was detected at comparable temperatures both experimentally by Mössbauer spectroscopy (5), neutron scattering (6), and x-ray scattering (7), and theoretically by computer simulations (8). It was described as a changeover from harmonic to anharmonic (4) protein dynamics (9, 10). These behaviors suggest that proteins form a glassy state (11–14). Indeed, their dynamics have been described as glass-like, arising from rearrangement of side chains to substates of nearly equal energy (15), as well as liquid-like, based on correlated motion of 5–8 Å (16, 17) for the protein side chains (18–20).
These results invoke a series of questions. What is the connection, at the atomic-molecular level, between protein glass-like properties and protein function? What is the physical origin of the transition and is this transition temperature a universal parameter for hydrated protein systems? Can solid state physics contribute to understanding the behavior of a system as complicated as a hydrated protein?
Water behavior may provide clues to answer these questions. Water by itself has a glass transition at ≈136 K (8). Recently, Angell (21) concluded that pure H2O is an unusual glass former and has strong or fragile glass character, respectively, below or above a 228 K transition temperature. The shift in glass transition temperature for pure water vs. hydrated protein suggests that new physical phenomena occur at the protein solvent interface.
In fact, protein solvent has been directly connected to the dynamic glass transition of proteins by solvent dependence of heat capacity increase at 170 K (22), a hydration-dependent protein glass transition at 180 K (6, 23), water coupling to low-frequency protein vibrational modes (24), and protein molecular dynamics simulations (25). Solid state NMR experiments show that water deuterium atoms in crystals of lysozyme (26), ribonuclease (26), and crambin (27) undergo a mobile to stationary transition at ≈180 K. Water dynamics in crambin crystals match these enzymes. Further, the hydrophobic/hydrophilic accessible surface area in crambin crystals is very close to that found in carboxypeptidase and myoglobin crystals (28). Thus, crambin and its water can elucidate function-related solvent dynamics in enzymes previously studied (1, 3).
To elucidate the solvent's role in protein dynamics and obtain an atomic-level picture of the glassy state, both of which can elucidate the role of water in protein function, we chose to study the water around the protein crambin. Crystals of crambin (MW 4720 D) diffract to at least 0.54 Å (29) and have remarkable water order (28). They provide a model for water structure and dynamics at all protein surfaces.
Materials and Methods
X-Ray Data Collection.
Five diffractometer data sets on a single crambin crystal (0.7 × 0.6 × 0.5 mm) were collected at 160 K, 180 K, 200 K, 220 K, and 240 K to 0.89 Å (Table 1). Change in cell dimensions with temperature was as expected 0.3–0.5% (10) without discontinuities.
Table 1.
Crambin crystal parameters and refinement results
| 100 K* | 130 K† | 160 K‡ | 180 K‡ | 200 K‡ | 220 K‡ | 240 K | 293 K§ | |
|---|---|---|---|---|---|---|---|---|
| a, Å | 40.69 | 40.76 | 40.84 | 40.87 | 40.86 | 40.88 | 40.90 | 40.96 |
| b, Å | 18.43 | 18.49 | 18.47 | 18.48 | 18.52 | 18.56 | 18.59 | 18.65 |
| c, Å | 22.27 | 22.33 | 22.34 | 22.37 | 22.39 | 22.41 | 22.4 | 22.52 |
| β, ° | 90.59 | 90.61 | 90.80 | 90.59 | 90.68 | 90.67 | 90.8 | 90.77 |
| V, Å3 | 16,698 | 16,828 | 16,850 | 16,896 | 16,941 | 17,005 | 17,030 | 17,201 |
| % ΔV from 293 K | 2.9 | 2.17 | 2.05 | 1.78 | 1.51 | 1.16 | 0.99 | 0.00 |
| Space group | P21 | |||||||
| Resolution, Å | 0.67 | 0.83 | 0.89 | 0.89 | 0.89 | 0.89 | 0.99 | 0.945 |
| Fo > 2σ(Fo) | 41,991¶ | 23,759 | 22,754 | 21,814 | 21,431 | 19,941 | 7,043 | 21,013 |
| R value, %‖ | (6.9/8.3)** | 10.5 | 14.5 | 13.7 | 13.6 | 15.2 | 9.7 | 11.0 |
| —‡‡ | (8.9)** | (7.5)** | (8.9)** | (8.3)** | (8.99)** | |||
| Rσ, %‖ | 4.0 | 7.0 | 5.9 | 6.5 | 7.2 | 7.9 | 13.9 | 4.1 |
Data were collected using λ = 0.54 Å synchrotron radiation and a 300-mm MarResearch imaging plate detector at the European Molecular Biology Laboratory BW7A beamline at the DORIS storage ring, Deutsches Elektronen-Synchrotron (DESY), Hamburg, Germany and are 97.6% complete.
From ref. 28.
Five data sets at 160 K, 180 K, 200 K, 220 K and 240 K were collected with one crystal (0.7 × 0.6 × 0.5 mm) using an AFC5 diffractometer, a Rigaku rotating anode x-ray generator (RU-200), and an MSC rigid tube low temperature system with ω continuous background-peak-background scans at 16°/min. The temperature was changed very slowly between data collections to minimize damage. Because absorption was less than 10% and check reflections invariant, no corrections were applied. The resulting 160 K structure was adopted as the initial model for 180 K structure refinement, and each subsequent one for the higher temperature. The 240 K data set showed evidence of deterioration due to water loss at the end of data collection (note larger Rsigma and fewer observed data). Refinement was with prolsq (30) using a 3-parameter anisotropic temperature factor.
Data were further refined to 0.945 Å (40).
Data greater than 4σ(Fo).
R value = 100⋅Σ(|Fo − Fc|)/Σ(Fo) and Rσ = 100⋅Σ(σ(Fo))/Σ(Fo).
Numbers in parenthesis are for shelxl-97 refinement on 4σ(Fo) data (32).
R values for shelxl refinement. The first is for 4σ(Fo) data and the second for all the data.
Refinement and Analysis.
All but the 100 K model were refined in prolsq with stereochemical restraints (30). The Pro-22/Leu-25 sequence structure at 150 K including solvent (31) was the initial model for the 160 K structure. Models at 160–220 K were also refined with shelxl-97 (32). The 100 K structure was only refined with shelxl-97.
Multiple substates were modeled by using 2Fo − Fc electron density maps at the 2σ level and 2(Fo − Fc) difference map at the 3σ level. Temperature factors were isotropic for substate modeling.
Debye-Waller factors were analyzed in excel. All waters with Debye-Waller factors above 30 Å2 and occupancy below 0.1 were considered unreliable and deleted. Radius of gyration (Rg), the root mean square distance to the center of mass, was calculated from the x-ray coordinates in x-plor (33). The Rg is also the fourth moment of the intramolecular correlation function. Water rings were analyzed with the program chain (34) on an SGI Indigo Elan and by local software.
Results and Discussion
Correlated Multisubstate Side Chain and Water Motion.
Crambin crystal models refined at eight temperatures (100 K, 130 K, 160 K, 180 K, 200 K, 220 K, 240 K, 293 K) to an average resolution of 0.89 Å produce parameters such as Debye-Waller factor and Rg with much lower errors than previously attainable (Table 1). Data from 160–240 K were collected on the same crystal to further minimize errors.
A significant portion (≈30%) of crambin residues have multiple conformational substates, many of which are correlated (31), and the substates account for all of the electron density (31, ¶). In the four water cavities in crambin crystals (28), 50–85% of the waters have multiple substates. Additionally, protein substates are structurally coupled to solvent substates (31) (Fig. 1). This coupling is at the core of the temperature behavior of crambin, and, we assert, of all proteins. We elucidate the connection between the glassy state of crambin and its water through analysis of the temperature dependence of water rings, of the Debye-Waller (B) factor, and of the Rg of the protein.
Figure 1.

A and B substates of the Asn-12 side chain in crambin coupled to the A and B water networks at 130 K. Note that the electron density for the water A and B substates are well separated for W105 and W119. Hydrogen bonds drawn as dotted lines are all between 2.75 and 3.11 Å. Electron density (2Fo − Fc) is contoured at the 2σ level.
Water Rings in Crambin Crystals Reflect Glassy State.
We find that water in crambin crystals at low temperatures adopts a ringed water structure, primarily composed of pentagons and hexagons, as predicted for glassy water (36). At 130 K, a cluster of 8 edge-sharing pentagon rings forms near a hydrophobic patch on the protein surface (28). Although anchored to the protein, the cluster makes more hydrogen bonds to itself than to protein (37). Six edge-sharing rings with water pentagons, hexagons, and heptagons are linked in crambin crystals (Fig. 2a). Fourteen more rings form with waters and protein atoms, and pentagons dominate these rings.
Figure 2.

Water rings in crambin at 160 K. (a) A stereodiagram of the extensive network of water rings from the largest crystal solvent region. Single letter codes (T = Thr, P = Pro, D = Asp) are used for the protein and symmetry-related molecules (residues with # after their number.) Electron density (2Fo − Fc) is contoured at the 1.5σ level. Water-water hydrogen bonds, as well as protein bonds, are shown in thick lines. One pentagon ring at the left (114BO-116BO-142O-165O-99O) is repeated at the right for 2-fold-screw-axis related waters (#). Water molecules with B in their label indicate that the ring arrays shown here are for the B substate and have partial occupancy. This cluster of rings contains two pentagon rings, three hexagon rings, and one heptagon ring. Hexagons can adopt a chair conformation when two waters in the ring hydrogen bond to the same carbonyl (97O# and 150#). Electron density (2Fo − Fc) is contoured at the 1.5σ level. (b) Number of rings vs. temperature for crambin water molecules in the largest water region. The number of rings is plotted for 130 K, 160 K, 200 K, 220 K, and 293 K. Pentagons are represented by filled triangles, hexagons by shaded squares, and heptagons by open circles. At 293 K, the remaining pentagons cluster near Leu-18 (37). Note that data for 160 K, 200 K, 220 K, and 240 K are on the same crystal.
The water-only rings become less numerous as the temperature is raised above 200 K (Fig. 2b). Decreasing the number of rings reflects a disruption of larger water aggregates at higher temperatures, as predicted by theoretical work (36). Edge sharing of rings enables formation of low energy clusters. Ring formation optimizes the hydrogen bond capacity of water with little distortion. Planar water pentagons are unstrained, with 108° angles close to ideal tetrahedral (109°). These observed rings below 200 K experimentally confirm Stillinger's hypothesis that supercooled water adopts relatively unstrained ring clusters linked by a random network, primarily pentagons and hexagons of water sharing faces or edges (36).
Experimentally Determined Debye-Waller Factor and Rg Change Temperature Behavior at 180 K.
For crambin crystals, analysis of average Debye-Waller factor (〈B〉) vs. temperature shows four characteristic features. The first is a change in slope ≈180 K‖ indicative of the glassy phase transition (Fig. 3a). Second are negative slopes for multisubstate (discretely disordered) side-chain atoms and water below ≈180 K (Fig. 3b). Third, there is an unexpected correlation in slopes between water and multisubstate atoms (Fig. 3 a and b). Finally, above the glass transition, the slope is the same for the ordered atoms and for oxygen atoms in hexagonal ice (Fig. 3b). The change in slope at around ≈180 K,‖ traditionally interpreted in other systems as a glassy phase transition (10), is present in crambin. The unexpected slope correlation between multisubstate atoms and water 〈B〉 (Fig. 3 a and b; refs. 32 and 39–41)) suggests that a new glassy state consists of these two components. The negative slope for both below ≈180 K indicates that water is dynamically coupled with multiconformer protein atoms, resulting in a new, highly cooperative physical state. In contrast, the slope for fully ordered atoms is positive. The fact that the slope for the ordered protein atoms above 180 K matches that for the oxygen atoms in hexagonal ice (Fig. 3b) suggests that, above ≈180 K, the ordered interior of the protein behaves dynamically like ice.
Figure 3.

Change in properties of crambin crystals as a function of temperature (K). In a and b, the equations for the lines above and below 180 K (y = mx + b) are indicated. Similar slopes (m) are found whether the break point is taken at 160 K. (a) Average B (Debye-Waller factor) in Å2 for all atoms, for protein and for water vs. temperature. Note the negative slope for water. B factors for water greater than 30 Å2 were judged unreliable and excluded. Refinement with prolsq (30) is designated by filled diamonds, by filled circles, or by filled triangles. The same data refined with shelxl (32) is shown with open diamonds, open circles, or with open triangles. (b) Average B factor in Å2 (Debye-Waller factor) for multisubstate (discretely disordered) side-chain atoms, for one-substate atoms in the protein, and for crystalline hexagonal ice water alone (41) vs. temperature. The equation of each line above and below 180 K is given, except for water alone, where the equation is only above 180 K. Note that, below 180 K, the negative slope for multisubstate atoms (m = −0.01) is close to that for the water in a (m = −0.03). Symbols are defined as in Fig. 3a above. Note that the slope for water above 180 K (m = 0.025) is the same as that for the one-substate protein atoms in b (m = 0.025). (c) Rg (Å) for the protein vs. temperature from 100 K to 293 K. The Rg is calculated with x-plor (33).
Finally, slope change in Rg plot vs. temperature (Fig. 3c; ref. 33) provides important experimental definition of the glassy “phase” transition temperature (more precisely than B-factor plots). It also provides physical evidence to support the proposed mechanism of enzyme function loss (42, 43). Namely, it suggests that solidification of the liquid glass at the transition temperature ≈180 K is responsible for cessation of enzymatic protein function below this temperature.
Cooperative Relaxation in Supercooled Liquids Elucidates the Debye-Waller Factor.
Although, from a theoretical analysis, the interior of a protein is consistent with a solid according to the Lindemann criterion (44), we have shown experimentally that the interfacial layers are liquid-like and correlated. Now we show that the variation in Debye-Waller factor with temperature relates to non-equilibrium characteristics of supercooled and glassy states and to coupling between the protein and the sequestered glassy water in cavities, including the interfacial layer.
We suggest that the entropy of supercooled water is the relevant thermodynamic variable. First, the rate of entropy decrease with temperature of hyperquenched glassy and vapor-deposited water gets smaller with decrease of temperature below 150 K (45). Second, the enthalpy of supercooled water is close to hexagonal ice above the apparent glass transition temperature Tg ≈ 136 K (46). Third, at the same temperature, the density of supercooled water is lower than hexagonal ice (47). Thus, the heat capacity difference Cp,vib,water − Cp,ice may become negative at temperatures where water density is lower than ice (47, 48).
To address cooperative phenomena that mitigate the Debye-Waller behavior, we invoke Adam-Gibbs' idea of a cooperative rearranging region (49). This area is in weak contact with the system and is capable of independent rearrangement because of enthalpy fluctuations (49, 50). Assuming the relaxation time for cooperative rearrangements in supercooled water is driven by decrease in entropy S(T) (51), we obtain τ(T) ≈ Aexp(Δμs*/kBTS(T). Here, s* is the “critical” entropy of the rearranging region, Δμ is the potential energy barrier per molecule, A has units of time and is weakly temperature dependent, and kB is the Boltzmann constant. This expression is a generalization of the Adam-Gibbs model that includes configurational and vibrational contributions to the entropy of the liquid (35).
From the temperature dependence of entropy and critical size of
cooperative rearranging regions in glass-forming liquids (see
Part 1, which is published as supporting information on the
PNAS web site, www.pnas.org), we calculate the critical number of
molecules participating in such a region. It is ≈8, 4.3, 2.8, and
1.9, at temperatures 136 K, 155 K, 185 K and 250 K, respectively. The
potential energy barrier height that hinders cooperative rearrangements
in supercooled water is Δμ ≈ C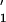 × 372.04
J/mol and corresponds to the hydrogen bond energy (5–10 kJ/mole;
refs. 47 and 48).
× 372.04
J/mol and corresponds to the hydrogen bond energy (5–10 kJ/mole;
refs. 47 and 48).
Estimating the Slope of the Debye-Waller Factor.
In Part 2 of the supporting information on the PNAS web site, we have derived an explicit expression for the slope of the mean-squared displacement with temperature. First, by putting reasonable limits on measurable experimental quantities, we estimate a bound for the slope of the Debye-Waller factor near 190 K to be .004 < d〈u2〉/dT < .07. Second, a similar, more tedious analysis at lower temperatures leads to the inequality −0.001 < d〈u2〉/dT < −0.025. Third, we find, in agreement with experiment, that the slope of the Debye-Waller factor is negative at low temperature but positive at higher temperature. Fourth, we show that the negative slope of the Debye-Waller factor may be attributed to the temperature dependence of water's excess vibrational entropy Sexc,vib for for T < 190 K (see Part 3 of the supporting information).
Next is the question of whether the protein-water system at low temperature behaves as a fragile or as a strong liquid. Considering the varied experimental evidence for both water (21) and the protein-water system (Part 4 of the supporting information), we conclude that the protein-water system exhibits fragile or strong characteristics depending on the physical property that is measured and the technique used to probe these degrees of freedom (21).
Conclusions
We asked three fundamental questions concerning (i) the connection between protein glass-like properties and protein function; (ii) the physical origin of the observed transition and its uniqueness; and (iii) whether solid state physics can clarify behavior of a hydrated protein. We used the structure of crambin at 0.89 Å resolution to elucidate these issues.
Crambin's structure provides an atomic description of structural protein to solvent correlations at atomic resolution (0.89Å) and supports the hypothesized glassy nature of proteins (52). Unstrained rings predicted by theory (36) characterize water's glassy “structure” at the protein surface. Dynamic coupling of protein and water is evident in the temperature behavior of water rings, the Debye-Waller factor (B), and the protein Rg. Water ring structure and dynamics provide an experimental measure of the protein-water glassy state that can stimulate developments in glassy state theory. From correlated slopes of B with temperature and temperature independence of Rg below ≈180 K, an interfacial glassy state of multiconformer side chains and solvent is identified. Because crambin's solvent dynamics and accessible surface area match enzymes, our conclusions should apply to enzymes where a glassy transition is linked to function cessation.
Physical Origin of Transition and Generality of Its Temperature.
The transition in dynamics for hydrated proteins has been attributed to clusters of rearranging atoms (53). We identify the clusters as multisubstate side chains linked to solvent. This coupling is the root cause of the change in slope of both Debye-Waller factor and Rg vs. temperature. The new interfacial glassy state links the thermodynamic behavior of this state to protein function. Interconversion of the interfacial liquid glassy state into a solid glassy state at the transition temperature (≈180 K) immobilizes the exterior of the protein and provides a direct molecular mechanism for loss of enzyme function. Protein surface may exert more influence below 180 K (25, 54). Our results also suggest that one glass transition temperature is not common to all proteins, but there is a proposed “slaved glass transition” (43). In fact, compositions both of the protein surface and of the solvent must influence the transition temperature (43, 55–57).
Statistical mechanics of supercooled fluids can address the unique nature of the interfacial layer. The Debye-Waller factor slope change with temperature can be predicted, based on a novel generalization of Adam-Gibbs' idea of cooperative rearranging regions in glass-forming liquids (49) and its connection to decrease of entropy with lowering of temperature. The negative slope of the Debye-Waller factor appears to be due to temperature dependence of water's excess vibrational entropy below 190 K. Further, the fragile vs. strong character of the protein-water system depends on the experimental probe.
Biological Implications.
Proteins use the solvent's role in protein dynamics to adapt to extreme environments, e.g., high temperature or salt. Sequence changes in exposed residues or solvent additives like sugars or salt modulate the glassy mobility of the interfacial layers and shift the glass transition temperature. This accommodation may explain the unique amino acid composition of thermophilic proteins and production of unusual small molecule osmolytes in extremophiles.
As previously noted (4, 43, 57), solute/solvent composition has a direct influence on protein function and the glass transition temperature. For instance, mixing protein with pure water raises water's glass transition temperature from 136 K to ≈180 K. A room-temperature glass raises the glassy kinetics of myoglobin to 300 K (58).
Future Directions.
Identification of a glassy interfacial state has consequences for quantitative models of hydrated biomolecules. They should describe (i) the bulk or interior of the biomolecule; (ii) the interfacial glassy state with transition ≈180 K, as modulated by solvent; and (iii) the bulk solvent glassy state usually with lower transition temperature (for water, ≈136 K).
Simulations suggest that protein docking is mediated by water substates (59). Further, protein-protein recognition seems to require protein substate sampling (60). Such substates were previously proposed as functionally important motions for proteins (61). The atomic dynamics of protein substates could be independently measured by structural/dynamic methods such as solid state NMR. Given assigned peaks (38), the correlated side chain and water dynamics with temperature can be determined. These experiments would provide a clearer picture of the rugged protein energy landscape and may identify additional functionally linked correlations.
Supplementary Material
Acknowledgments
We thank the National Science Foundation (Grant DMCB-9219857 to M.M.T.), the Keck Center for Computational Biology (National Library of Medicine Grant 2T15LM07093 to B.S.), and the Japan Society for Promotion of Science (U.M.) for partial support.
Abbreviations
- Rg
radius of gyration
- B
Debye-Waller factor
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes 1jxt, 1jxu, 1jxw, 1jxx, and 1jxy).
When the mixed sequence form of crambin, discussed in this paper, was purified, the electron densities of the pure sequence forms accounted for all the electron density of the mixed sequence form, showing discrete multi-substates.
The exact temperature of slope change (160 K vs. 180 K) is difficult to determine because the data from 160 K to 220 K were on a single crystal and different ones were used at 130 K and 100 K.
References
- 1.Rasmussen B F, Stock A M, Ringe D, Petsko G A. Nature (London) 1992;357:423–424. doi: 10.1038/357423a0. [DOI] [PubMed] [Google Scholar]
- 2.Ferrand M, Dianoux A J, Petry W, Zaccai G. Proc Natl Acad Sci USA. 1993;90:9668–9672. doi: 10.1073/pnas.90.20.9668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daniel R M, Smith J C, Ferrand M, Hery S, Dunn R, Finney J L. Biophys J. 1998;75:2504–2507. doi: 10.1016/S0006-3495(98)77694-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doster W. In: Hydration Processes in Biology. Bellisent-Funel M-C, Teixera J, editors. Amsterdam: IOS Press; 1998. [Google Scholar]
- 5.Hartmann H, Parak F, Steigemann W, Petsko G A, Ponzi D, Frauenfelder H. Proc Natl Acad Sci USA. 1982;79:4967–4971. doi: 10.1073/pnas.79.16.4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doster W, Cusack S, Petry W. Nature (London) 1989;338:754–756. doi: 10.1038/337754a0. [DOI] [PubMed] [Google Scholar]
- 7.Nienhaus G U, Heinzel J, Huenges E, Parak F. Nature (London) 1989;338:669–670. [Google Scholar]
- 8.Angell C A. Proc Natl Acad Sci USA. 1995;92:6675–6682. doi: 10.1073/pnas.92.15.6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parak F, Hartmann H, Aumann K D, Reuscher H, Rennekamp G, Bartunik H, Steigemann W. Eur Biophys J. 1987;15:237–249. doi: 10.1007/BF00577072. [DOI] [PubMed] [Google Scholar]
- 10.Tilton R F, Dewan J C, Petsko G A. Biochemistry. 1992;31:2468–2481. doi: 10.1021/bi00124a006. [DOI] [PubMed] [Google Scholar]
- 11.Goldanskii V I, Krupyanskii Y F. Q Rev Biophys. 1989;22:39–92. doi: 10.1017/s003358350000336x. [DOI] [PubMed] [Google Scholar]
- 12.Iben I E, Braunstein D, Doster W, Frauenfelder H, Hong M K, Johnson J B, Luck S, Ormos P, Schulte A, Steinbach P J, et al. Phys Rev Lett. 1989;62:1916–1919. doi: 10.1103/PhysRevLett.62.1916. [DOI] [PubMed] [Google Scholar]
- 13.Young R D. J Chem Phys. 1993;98:2488–2489. [Google Scholar]
- 14.Bryngleson J, Wolynes P. J Phys Chem. 1989;93:6902. [Google Scholar]
- 15.Frauenfelder H, Sligar S G, Wolynes P G. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 16.Caspar D L D, Clarage J B, Salunke D M, Clarage M S. Nature (London) 1988;332:659–662. doi: 10.1038/332659a0. [DOI] [PubMed] [Google Scholar]
- 17.Tengroth C, Borjesson L, Kagunya W W, Middendorf H D. Physica B. 1999;266:27–34. [Google Scholar]
- 18.Stec B, Zhou R, Teeter M M. Acta Cryst. 1995;D51:663–681. doi: 10.1107/S0907444994014484. [DOI] [PubMed] [Google Scholar]
- 19.Yamano A, Heo N H, Teeter M M. J Biol Chem. 1997;272:9597–9600. doi: 10.1074/jbc.272.15.9597. [DOI] [PubMed] [Google Scholar]
- 20.Andrews B K, Romo T, Clarage J B, Pettitt B M, Phillips G N. Structure Fold Des. 1998;6:587–594. doi: 10.1016/s0969-2126(98)00060-4. [DOI] [PubMed] [Google Scholar]
- 21.Angell C A. Nature (London) 1999;398:492–495. [Google Scholar]
- 22.Sartor G, Mayer E. Biophys J. 1994;67:1724–1732. doi: 10.1016/S0006-3495(94)80646-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doster W, Bachleitner A, Dunau R, Hiebl M, Luscher E. Biophys J. 1986;50:213–219. doi: 10.1016/S0006-3495(86)83455-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diehl M, Doster W, Petry W, Schober H. Biophys J. 1997;73:2726–2732. doi: 10.1016/S0006-3495(97)78301-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vitkup D, Ringe D, Petsko G A, Karplus M. Nat Struct Biol. 2000;7:34–38. doi: 10.1038/71231. [DOI] [PubMed] [Google Scholar]
- 26.Usha M G, Wittebort R J. Chem Phys. 1991;158:487–500. [Google Scholar]
- 27.Usha M G, Wittebort R J. J Mol Biol. 1989;208:669–678. doi: 10.1016/0022-2836(89)90157-5. [DOI] [PubMed] [Google Scholar]
- 28.Teeter M M, Roe S M, Heo N H. J Mol Biol. 1993;230:292–311. doi: 10.1006/jmbi.1993.1143. [DOI] [PubMed] [Google Scholar]
- 29.Jelsch C, Teeter M M, Lamzin V, Pichon-Pesme V, Blessing R, Lecomte C. Proc Natl Acad Sci USA. 2000;97:3171–3176. doi: 10.1073/pnas.97.7.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hendrickson W A, Konnert J H. Acta Crystallogr A. 1980;36:344–350. [Google Scholar]
- 31.Yamano A, Teeter M M. J Biol Chem. 1994;19:13956–13965. [PubMed] [Google Scholar]
- 32.Sheldrick G M, Schneider T R. In: Methods in Enzymology. Sweet R M, Carter C W, editors. Vol. 277. Orlando, FL: Academic; 1997. pp. 319–343. [PubMed] [Google Scholar]
- 33.Brünger A T. X-Plor Version 30: A System for Crystallography and NMR. New Haven, CT: Yale Univ. Press; 1992. [Google Scholar]
- 34.Sack J S. J Mol Graph. 1988;6:244–245. [Google Scholar]
- 35.Mohanty U, Craig N, Fourkas J T. J Chem Phys. 2001;114:10577–10578. [Google Scholar]
- 36.Stillinger F H. Science. 1980;209:451–457. doi: 10.1126/science.209.4455.451. [DOI] [PubMed] [Google Scholar]
- 37.Teeter M M. Proc Natl Acad Sci USA. 1984;81:6014–6018. doi: 10.1073/pnas.81.19.6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDermott A, Polenova T, Böckmann A, Zilm K W, Paulsen E K, Martin R W, Montelione G T. J Biomol NMR. 2000;16:209–219. doi: 10.1023/a:1008391625633. [DOI] [PubMed] [Google Scholar]
- 39.Hendrickson W A. In: Daresbury Study Weekend. Machin P A, Campbell J W, Elder M, editors. Daresbury, U.K.: Daresbury Lab.; 1980. [Google Scholar]
- 40.Hendrickson W A, Teeter M M. Nature (London) 1981;290:109–113. doi: 10.1038/290107a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuhs W F, Lehman M S. In: Water Science Review. Franks F, editor. Vol. 2. Cambridge, U.K.: Cambridge Univ. Press; 1986. pp. 1–65. [Google Scholar]
- 42.Doster W. Biophys Chem. 1983;17:97–103. doi: 10.1016/0301-4622(83)80002-7. [DOI] [PubMed] [Google Scholar]
- 43.Beece D, Eisenstein L, Frauenfelder H, Good M, Marden M C, Reinisch L, Reynolds A H, Sorensen L B. J Am Chem Soc. 1980;19:5147–5157. doi: 10.1021/bi00564a001. [DOI] [PubMed] [Google Scholar]
- 44.Zhou Y, Vitkup D, Karplus M. J Mol Biol. 1999;285:1371–1375. doi: 10.1006/jmbi.1998.2374. [DOI] [PubMed] [Google Scholar]
- 45.Mayer E. J. Mol. Struct. 2000. 520. [Google Scholar]
- 46.Sugisaki M, Suga H, Seiki H. Bull Chem Soc Jpn. 1968;41:259. [Google Scholar]
- 47.Eisenberg D, Kauzmann W. The Structure and Properties of Water. Oxford: Clarendon; 1969. [Google Scholar]
- 48.Johari G P. J Chem Phys. 2000;112:10957–10965. [Google Scholar]
- 49.Adam G, Gibbs J H. J Phys Chem. 1965;43:139–146. [Google Scholar]
- 50.Mohanty U. Adv Chem Phys. 1995;89:89–158. [Google Scholar]
- 51.Mohanty U. Physica A. 1991;177:345–355. [Google Scholar]
- 52.Frauenfelder H. In: Structure and Dynamics of Glasses and Glass Formers. Angel C A, Ngai K L, Kieffer J, Egami T, Nienhaus G U, editors. Vol. 456. Pittsburgh, PA: Mat. Res. Soc. Symp. Proc.; 1996. pp. 343–347. [Google Scholar]
- 53.Sokolov A P. Phil Mag B. 1999;77:349–355. [Google Scholar]
- 54.Lee A L, Wand A J. Nature (London) 2001;411:501–504. doi: 10.1038/35078119. [DOI] [PubMed] [Google Scholar]
- 55.Young R D, Frauenfelder H, Johnson J B, Lamb D C, Nienhaus G U, Philipp R, Scholl R. Chem Phys. 1991;158:315–327. [Google Scholar]
- 56.Doster W, Kleinert T, Post F, Settles M. In: Protein-Solvent Interactions. Gregory R B, editor. New York: Dekker; 1995. pp. 375–385. [Google Scholar]
- 57.Reat V, Dunn R, Ferrand M, Finney J L, Daniel P M, Smith J C. Proc Natl Acad Sci USA. 2000;97:9961–9966. doi: 10.1073/pnas.97.18.9961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Austin R H, Beeson K W, Eisenstein L, Frauenfelder H, Gunsalus I C. Biochemistry. 1975;14:5355–5373. doi: 10.1021/bi00695a021. [DOI] [PubMed] [Google Scholar]
- 59.Camacho C J, Kimura S R, DeLisi C, Vajda S. Biophys J. 2000;78:1094–1105. doi: 10.1016/S0006-3495(00)76668-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kimura S R, Brower R C, Vajda S, Camacho C J. Biophys J. 2001;80:635–642. doi: 10.1016/S0006-3495(01)76044-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ansari A, Berendzen J, Browne S F, Frauenfelder H, Eben I E T, Sauke T B, Shyamsunder E, Young R D. Proc Natl Acad Sci USA. 1985;82:5000–5004. doi: 10.1073/pnas.82.15.5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.