

Abstract
Many pathogenic bacteria, including Streptococcus gordonii, possess a pathway for the cellular export of a single serine-rich-repeat protein that mediates the adhesion of bacteria to host cells and the extracellular matrix. This adhesin protein is O-glycosylated by several cytosolic glycosyltransferases and requires three accessory Sec proteins (Asp1–3) for export, but how the adhesin protein is processed for export is not well understood. Here, we report that the S. gordonii adhesin GspB is sequentially O-glycosylated by three enzymes (GtfA/B, Nss, and Gly) that attach N-acetylglucosamine and glucose to Ser/Thr residues. We also found that modified GspB is transferred from the last glycosyltransferase to the Asp1/2/3 complex. Crystal structures revealed that both Asp1 and Asp3 are related to carbohydrate-binding proteins, suggesting that they interact with carbohydrates and bind glycosylated adhesin, a notion that was supported by further analyses. We further observed that Asp1 also has an affinity for phospholipids, which is attenuated by Asp2. In summary, our findings support a model in which the GspB adhesin is sequentially glycosylated by GtfA/B, Nss, and Gly and then transferred to the Asp1/2/3 complex in which Asp1 mediates the interaction of the Asp1/2/3 complex with the lipid bilayer for targeting of matured GspB to the export machinery.
Keywords: bacterial pathogenesis, glycosylation, adhesin, crystallography, protein export, protein secretion, glycosyltransferase
Introduction
Adhesion proteins are instrumental for the pathogenicity of bacteria (1). Streptococci and staphylococci bacteria express serine-rich repeat (SRR)5 adhesins that are exported from the cell but remain associated with the cell wall and allow the bacteria to attach to the host cells and their extracellular matrix (2, 3). In addition, these adhesins may also mediate interactions between bacteria, facilitating biofilm formation and bacterial colonization (4). The biosynthesis of SRR adhesins is a promising target of novel antibiotics that could be used to treat diseases caused by streptococci and staphylococci, such as infective endocarditis, pneumococcal pneumonia, neonatal sepsis, and meningitis (3).
SRR adhesins use a dedicated pathway for their export from the cytosol, called the accessory Sec system (5, 6); most other proteins are exported from the bacterial cell by the canonical Sec pathway (7). In the canonical pathway, proteins are moved by the SecA ATPase through the protein-conducting SecY channel. In the accessory Sec pathway, export is mediated by distinct SecA and SecY proteins (SecA2 and SecY2). These components are encoded in an operon that also includes the adhesin substrate as well as several glycosyltransferases and accessory Sec system proteins (Asps) (5, 6). The glycosyltransferases attach sugar residues to adhesin before its export from the cytosol (2, 8), but the exact roles of the glycosyltransferases and Asps in the export pathway is not well defined.
The SRR adhesins are initially modified with N-acetylglucosamine (GlcNAc) at multiple Ser/Thr residues by the heterodimeric GtfA/B glycosyltransferase (9–14). The deletion of GtfA or GtfB results in non-glycosylated adhesins that are prone to degradation (11, 14, 15). Glycosylation is physiologically important as the deletion of GtfA also reduces the adhesion of bacteria to host cells (15, 16). Recent results show that GtfA is the catalytic subunit, whereas GtfB is involved in substrate binding (10). Most SRR adhesins are further modified by additional glycosyltransferases that are also encoded by the same operon (5, 6). In Streptococcus parasanguinis and Streptococcus pneumoniae, they modify adhesins in a sequential manner (17, 18). In Streptococcus gordonii, there are two such glycosyltransferases, Nss and Gly (5). Deletion of either enzyme results in compromised modification of the SRR adhesin GspB (9). Nss from related streptococcal species adds glucose to GlcNAc attached to Ser/Thr-containing peptides (19–21). It is unclear how Gly modifies the adhesin GspB and whether Nss and Gly act sequentially or have redundant functions.
S. gordonii encode three Asps (Asp1–3), which are conserved among different bacterial species that express SRR adhesins (5, 6). Deletion of any of the Asps blocks the export of the adhesin GspB and results in its intracellular accumulation (15, 22). An essential role for the Asps in the biogenesis of SRR adhesins has also been observed in other species (16, 23–25). Interactions among the Asps and of the Asps with substrate and SecA2 have been reported for both S. gordonii and S. parasanguinis (where the Asps are called Gaps) (22–24, 26–28), but it remains unclear how the Asps function in GspB export.
Here, we show that the modification of the adhesin GspB of S. gordonii by the glycosyltransferases occurs in a sequential manner. First, GlcNAc residues are attached to Ser/Thr residues in the SRR domains of GspB. Next, Nss adds glucose to GlcNAc, and finally, Gly adds glucose to previously attached glucose residues. Interestingly, Gly remains bound to the modified substrate. Release of modified GspB from Gly is caused by the complex of the three Asps (Asp complex). Crystal structures suggest that both Asp1 and Asp3 are indeed carbohydrate-binding proteins. Asp1 seems to be a catalytically inactive member of the GT-B family of glycosyltransferases, and Asp3 contains a carbohydrate-binding module also found in several glycosidases. Our results also show that Asp1 has an affinity for negatively charged phospholipids, which may facilitate substrate delivery to the membrane. Taken together, our results suggest a model for the pathway by which the adhesin is modified and targeted to the export machinery.
Results
Glycosyltransferases act in a sequential manner
To test the role of the glycosyltransferases in adhesin modification, we produced a fragment of the GspB substrate by in vitro translation in reticulocyte lysate in the presence of [35S]methionine. The GspB fragment (GspB-F; Fig. S1) contains residues 91–736, including the first Ser/Thr-rich domain (SRR1), an intervening sequence that normally binds to host cells (binding region), and the N-terminal part of the second Ser/Thr-rich domain (SRR2N). It lacks the N-terminal signal sequence. GspB-F with the signal sequence is glycosylated in S. gordonii cells and secreted with the same efficiency as full-length adhesin (29). In vitro translation of GspB-F generated non-glycosylated protein that could be visualized as a single band after SDS-PAGE and autoradiography (Fig. 1A, lane 1). As described previously, when a purified complex of GtfA and GftB and UDP-GlcNAc was added after translation, a size shift was observed, caused by modification of GspB-F with GlcNAc residues (G1 species; Fig. 1A, lane 2). Subsequent addition of purified Nss and UDP-Glc resulted in a further size shift (G2 species; Fig. 1A, lane 3). Nss did not function with UDP-GlcNAc (Fig. 1B, lane 3 versus lane 2). Finally, yet a larger species was generated when purified Gly was introduced (G3 species; Fig. 1A, lane 4). Nss did not attach Glc residues to unmodified GspB-F (Fig. 1C, lane 1 versus lane 3), indicating that it can only modify substrate after GtfA/B has added GlcNAc residues. The same is true for Gly (Fig. 1D, lane 1). Finally, modification by Gly was dependent on the prior action of Nss (Fig. 1E, lane 3 versus lane 4). Taken together, these results indicate that GtfA/B, Nss, and Gly function in a defined order; GtfA adds GlcNAc residues to Ser/Thr residues in the SRR domains, which are then further modified with Glc residues by the sequential action of Nss and Gly.
Figure 1.

Adhesin is sequentially O-glycosylated by three glycosyltransferases. A, fragment of the S. gordonii adhesin GspB (GspB-F) was synthesized in reticulocyte lysate in the presence of [35S]methionine. After translation, the glycosyltransferases GtfA/B, Nss, and Gly were added, as indicated, together with UDP-GlcNAc or UDP-Glc. The samples were analyzed by SDS-PAGE and autoradiography. G1, G2, and G3 indicate different glycosylated species. B, in vitro synthesized GspB-F was incubated with Nss and either UDP-Glc or UDP-GlcNAc. C, in vitro synthesized GspB-F was incubated with UDP-sugars and Nss in the absence or presence of GtfA/B. D, in vitro synthesized GspB-F was modified with GtfA/B or Gly in the presence of UDP-GlcNAc or UDP-Glc, respectively. E, in vitro synthesized GspB-F was modified with GtfA/B and further incubated in the presence of UDP-Glc with either Nss or Gly. F, left panel shows the domain organization of Gly. The right panel shows in vitro synthesized GspB-F that was incubated with GtfA/B, Nss, and either full-length Gly or its N- or C-terminal domains, together with UDP-Glc or UDP-GlcNAc.
To test whether the modification of GspB with multiple sugars occurs in vivo, we purified GspB-F secreted from S. gordonii and used mass spectrometry to analyze sugars released by β-elimination (Fig. S2, A and B). The major peaks were further analyzed by tandem mass spectrometry (Fig. S2C), and the monosaccharide composition of tryptic peptides was determined after hydrolysis by anion-exchange chromatography (Fig. S2D). These experiments indicated that the main glycan species were GlcNAc (mass 330.19), GlcGlcNAc (mass 534.29), and Glc2GlcNAc (mass 738.39). Similar results were obtained when GspB-F was expressed in Escherichia coli together with GtfA/B, Nss, and Gly, and the purified protein was subjected to glycan analysis (Fig. S3, A–D); again, GlcNAc, GlcGlcNAc, and Glc2GlcNAc were the major species. Although identification of modified GspB-F peptides by mass spectrometry was challenging, we identified with confidence an SRR1 peptide that contained a Ser modified by one N-acetylhexosamine (HexNAc) and two hexoses (Fig. S3E). Taken together, these results are consistent with the idea that Nss and Gly add glucose residues to GlcNAc attached by GtfA/B to Ser/Thr of GspB-F.
Nss consists of a single domain that has a typical GT-B glycosyltransferase fold (20, 21). Gly consists of three domains (Fig. 1F, left panel). The first two domains are predicted to have GT-A and GT-B glycosyltransferase folds, respectively, and the third domain has a GT-D fold (30). Of note, the isolated GT-D domain from a Gly homolog of S. parasanguinis has enzymatic activity for its substrate (30). We found that the isolated GT-D of S. gordonii Gly was capable of adding Glc residues to GspB-F pre-modified with GtfA/B and Nss (Fig. 1F, right panel, lane 6), whereas the isolated N-terminal fragment containing the GT-A and GT-B folds was inactive (lane 5). Thus, despite the fact that the N-terminal domains are sequence-related to glycosyltransferases, they seem to lack enzymatic activity.
Substrate binds to Gly and is released by the Asp complex
To our surprise, we noticed that the fully modified GspB-F remained associated with Gly, the last glycosyltransferase; essentially, all G3 species could be recovered with a fusion of Gly with glutathione S-transferase (Gly-GST), followed by binding to a glutathione resin (Fig. 2A, lane 2). In contrast, neither GtfA/B (10) nor Nss (Fig. 2A, lane 1) had appreciable affinity for the product of their modification reactions, as commonly seen for enzymes. The material bound to Gly-GST could be partially released from the beads with a 100-fold excess of full-length Gly (Fig. 2B, lane 4) but not with the isolated C-terminal GT-D domain (lane 2). These data suggest that the modified substrate is reversibly bound by the N-terminal GT-A/B domains. This conclusion is supported by the fact that the isolated GT-D domain can generate the G3 species but does not interact strongly with it, as demonstrated by the absence of competition with the full-length Gly protein (Fig. 2C, lane 1). This experiment also shows that the binding of Gly to its enzymatic product is separable from the modification reaction per se. In our system, product binding by the N-terminal GT-A/B domains does not interfere with the enzymatic activity of the C-terminal GT-D domain, because Gly is in large excess over substrate and the N-terminal domains binds reversibly to the product.
Figure 2.
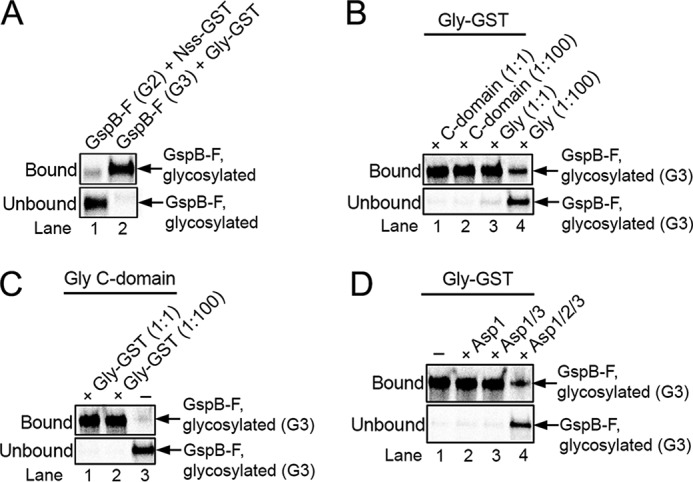
Glycosyltransferase Gly remains associated with its enzymatic product. A, Gsp-F was synthesized in vitro in the presence of [35S]methionine and then glycosylated with GtfA/B to generate the G1 species, followed by incubation with either a GST fusion to Nss (Nss-GST; lane 1) or wildtype Nss (lane 2) to generate the G2 species. The sample in lane 2 was further incubated with a GST fusion to Gly (Gly-GST) to generate the G3 species. The samples were then incubated with glutathione beads, and the bound and unbound fractions were analyzed by SDS-PAGE and autoradiography. The G2 and G3 species migrated relative to molecular weight markers as in Fig. 1A. B, G3 species was generated with GtfA/B, Nss, and GST-Gly and bound to glutathione beads. After washing, the beads were incubated with either the same amount (1:1) or a 100-fold excess (1:100) of His-tagged versions of either full-length Gly (Gly) or C-terminal Gly domain (C domain). The bound and unbound fractions were analyzed as in A. C, G3 species was generated with a His-tagged version of the C domain of Gly (Gly C domain). The sample was then mixed with either the same amount (1:1) or a 100-fold excess (1:100) of Gly-GST and incubated with glutathione beads. Bound and unbound fractions were analyzed as in A. D, G3 species was generated with Gly-GST and bound to glutathione beads. After washing, the beads were incubated with Asp1, Asp1/3 complex, or Asp1/2/3 complex, and the bound and unbound fractions were analyzed as in A.
Next, we tested the role of the Asps. Neither of the three Asps had an effect on the glycosylation reactions catalyzed by GtfA/B, Nss, or Gly (Fig. S4). However, the complex of the three Asps released the fully glycosylated G3 species from Gly-GST (Fig. 2D, lane 4). Asp1 alone or a complex of Asp1 and Asp3 (Asp1/3) was inactive in the release reaction (Fig. 2D, lanes 2 and 3). These results suggest that the complex of all three Asp proteins may be involved in the transfer of glycosylated substrate from the last glycosyltransferase to the next step in the export pathway.
Structures of the Asps
Because our data suggest that the Asp complex can accept fully glycosylated substrate from Gly, we suspected that it can interact with carbohydrates. To test this possibility, we determined the crystal structures of Asp1 alone (resolution of 2.77 Å) and of an Asp1/3 complex (resolution of 3.11 Å) (Table S1). The structure of Asp1 is similar to that of GtfA and GtfB (Fig. 3, A, B, and F)., Like GtfA or GtfB, Asp1 has two Rossmann-like folds (R-folds I and II), which are typical for the GT-B family of glycosyltransferases (Fig. 3A). In addition, it has the typical extended β-sheet domain (EBD). Together, these domains form a U-shaped structure. As in the enzymatically inactive GtfB protein, the cleft between R-folds I and II is negatively charged (Fig. 3B). In contrast, GtfA and other enzymatically active GT-B family members have a positively charged cleft that is required to bind UDP-sugars (Fig. 3B). Like GtfB, Asp1 lacks two positively charged residues in the active site and has a Gln residue at position 438 in place of an essential Glu residue (Fig. 3B, lower panels). The structure thus supports the idea that Asp1, like GtfB, is a carbohydrate-binding protein, rather than an active glycosyltransferase. Consistent with the postulated substrate-binding site, when two conserved Asp residues in the cleft between the R-folds were mutated to Arg, secretion of GspB-F from S. gordonii cells was abolished (Fig. 4, A and B; sequence alignment shown in Fig. S5).
Figure 3.

Crystal structures of Asp1 and Asp3. A, crystal structure of Asp1 alone. Shown is a ribbon diagram of the model embedded in a space-filling presentation. The helices and the β-sheets of Rossmann folds I and II (R-folds I and II) are shown in cyan and purple, respectively. The extended β-sheet domain (EBD) is shown in green. B, upper panels show space-filling models of Asp1, GtfB, and GtfA, with the electrostatic surface calculated with the Adaptive Poisson-Boltzmann Solver, as implemented in PyMOL, using a scale from −5.000 to 5.000 (bottom). The yellow circle indicates the cleft between the R-folds, which is negatively charged for Asp1 and GtfB and positively charged for GtfA. The lower panels show magnified views of the cleft in ribbon presentation. Residues in the active site of GtfA and the corresponding residues in the enzymatically inactive Asp1 and GtfB are shown in stick presentation. C, structure of the Asp1/3 complex. Shown is a ribbon diagram of the model, with Asp1 in green and Asp3 in blue. The right panel also shows a space-filling model of the interfaces between Asp1 and Asp3. D, comparison of Asp3 with two CBM families. The two CBMs shown bind their sugar substrates at different sites. E, comparison of the structures of Asp1 in isolation and when bound to Asp3 (brown and green, respectively). F, comparison of the open conformation of Asp1 in isolation with GtfA (left panel: brown and cyan, respectively), and of the closed conformation of Asp1 when bound to Asp3 with GtfB (right panel: green and yellow, respectively).
Figure 4.
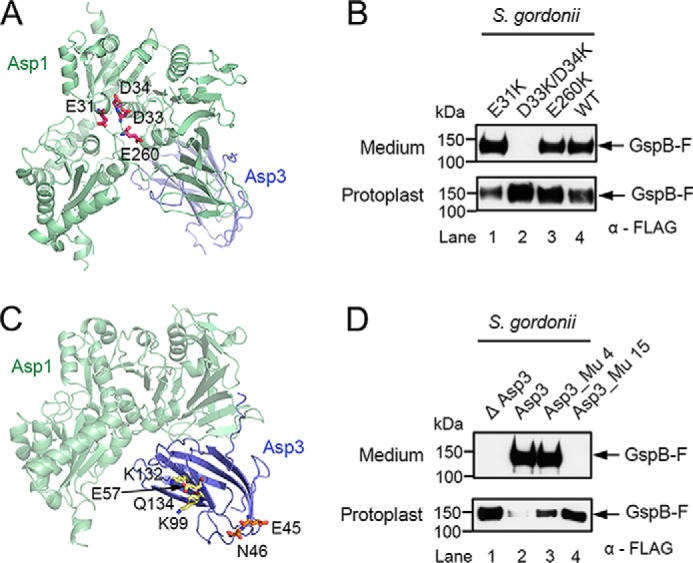
Mutations in Asp1 and Asp3 affect secretion of adhesin from S. gordonii cells. A, ribbon diagram of the Asp1/3 complex (Asp1 in green and Asp3 in blue), with mutated Asp1 residues in the cleft between R-folds I and II shown in stick presentation in magenta. B, endogenous Asp1 protein of S. gordonii was replaced with the indicated Asp1 mutants. Secretion of FLAG-tagged GspB-F from the cells was tested by subjecting the medium and protoplasts to SDS-PAGE, followed by immunoblotting with FLAG antibodies. C, as in A, but with mutated Asp3 residues. Mutations in the potential carbohydrate-binding regions, the concave surface of the β-sandwich, and at the tips of the β-strands are shown in stick presentation in yellow and orange, respectively. D, as in B, but with an S. gordonii strain lacking Asp3, expressing either wild type or mutant Asp3. Asp3_Mu4 containing mutations E57K, K132E, K99E, Q134R, and Asp3_Mu15 harbors the mutations E45K and N46K.
Asp1 forms a stable 1:1 complex with Asp3 (Fig. S6A). Asp3 could not be stably isolated on its own, suggesting that it has Asp1 as an obligatory partner. Consistent with this observation, deletion of the Asp1 homolog Gap1 in S. parasanguinis results in the degradation of the Asp3 homolog Gap3 (28). The Asp1/3 structure shows that Asp3 consists of two anti-parallel β-sheets (β-sandwich) (Fig. 3C). Asp3 uses two different regions to bind to Asp1 (interfaces I and II). Interface I binds to the EBD of Asp1, and interface II to both the EBD and the cleft between the R-folds (Fig. 3C). Asp3 is structurally related to carbohydrate-binding modules (CBM) in glycosidases (Fig. 3D) (31, 32). Interestingly, different CBMs bind their sugar ligands with different surfaces, some with the concave surface of the β-sandwich and others with the tips of the β-strands (Fig. 3D) (33, 34). In the case of Asp3, the latter binding site seems to be more important, as mutations of conserved residues in this area had a drastic effect on GspB-F secretion from S. gordonii, whereas mutations in the concave surface of the β-sandwich had only a small effect (Fig. 4, C and D). In the Asp1/3 complex, the Asp1 protein adopts a closed conformation, in which R-fold II moves toward the EBD (Fig. 3E). The two ends of the U-shaped structure of Asp1 are much closer in the closed conformation than in the open state (10 Å versus 20 Å; Fig. 3F). The open and closed conformations resemble those of GtfA and GtfB, respectively (Fig. 3F) (10). A conformational change from the open to the closed state has been observed for GtfA/B and is likely required for the binding of adhesin.
Asp1–3 co-migrated in gel filtration (Fig. S6B), but light-scattering experiments indicated that there was a mixture of monomeric and dimeric complexes that contain one copy each of Asp1, -2, and -3 (Fig. S6C). This heterogeneity is likely the explanation for why these complexes did not crystallize. We therefore attempted to obtain structural information by other means. Addition of trypsin to the Asp1/3 complex generated one Asp1 and one Asp3 peptide, which were not observed with the Asp1/2/3 complex (Fig. 5A, indicated by stars). The cleavage sites protected by Asp2 are Arg-430 of Asp1 and Arg-23 of Asp3 (Fig. 5B; Arg-23 is in an unstructured region, so the figure shows flanking residues). Thus, Asp2 seems to bind to both Asp1 and Asp3 at the open end of the U-shaped Asp1/3 complex. Next, we used negative-stain electron microscopy (EM) to analyze the Asp1/3 and Asp1/2/3 complexes (Fig. 5C and Fig. S7). To better locate the individual proteins in the images, we fused the maltose-binding protein (MBP) to Asp1, Asp2, or both. Complexes containing the MBP fusions were monomeric, indicating that the MBP domain interfered with dimerization (Fig. S6C). The results confirm that Asp2 sits at the open end of the Asp1/3 complex (Fig. 5C and Fig. S7). Negative-stain EM also confirmed that without MBP, the Asp1/2/3 complex consisted of a mixture of monomers and dimers. In the dimer, the Asp1/2/3 monomers associate in an anti-parallel fashion (Fig. 5C, 4th panel from left). It is unclear which form of the Asp complex is physiologically relevant.
Figure 5.

Domain organization of the Asp1/2/3 complex. A, purified Asp1/3 and Asp1/2/3 complexes were incubated with different concentrations of trypsin. The samples were analyzed by SDS-PAGE and staining with Coomassie Blue. Tryptic fragments seen only with Asp1/3 are indicated by stars. C, control without trypsin. B, approximate position of Asp2 in the Asp1/2/3 complex. The crystal structure of the Asp1/3 complex is shown as a ribbon diagram and Asp2 as a pink ellipse. Trypsin cleavage at residues Arg-430 and Arg-23 is prevented by Asp2. R430 is indicated as yellow balls. Arg-23 is located in a flexible region that is invisible in the crystal structure, so residues G12 and S32 in the flanking segments are shown (in orange). C, left three panels show the domain organization of Asp complexes that contain fusions of MBP to Asp1 and/or Asp2. Shown are 2D averages of particles analyzed by negative-stain electron microscopy. The MBP tag prevents dimerization of the Asp1/2/3 complex. The characteristic two-globule shape of MBP and the domain structure of Asp1/3, as deduced from a comparison with the crystal structure, were used to identify the proteins. The right two panels show the domain organization of dimeric and monomeric untagged Asp1/2/3 complex.
Substrate targeting to membranes by Asp1/3
After being released from Gly by the Asp complex, the glycosylated substrate needs to be targeted to the membrane, a process that might be mediated by the Asps. We therefore tested whether the Asps have an affinity for membranes. To this end, purified Asps were incubated with liposomes of different phospholipid composition. The samples were then subjected to flotation in a Nycodenz gradient, and fractions were analyzed by SDS-PAGE and Coomassie staining (Fig. 6A). With liposomes containing a high percentage of negatively charged lipids (dioleoylphosphatidylglycerol (DOPG)), Asp1 alone or Asp1/3 floated to the second fraction from the top (Fig. 6B), which is also the peak position of the lipids (Fig. S8). The Asp1/2/3 complex also bound to the liposomes, but it peaked at fraction 3, suggesting that Asp2 weakens the interaction with the liposomes. Indeed, when the percentage of negatively charged lipids was decreased, the binding of Asp1/2/3 was selectively reduced (Fig. 6, C and D). These results indicate that Asp1 and Asp1/3 have an affinity for negatively charged lipids. Given that Asp1 and Asp3 are always in a complex, Asp1 is likely responsible for membrane targeting of both proteins. Asp2 inhibits membrane interaction, suggesting that lipid headgroups and Asp2 may compete for interaction with the Asp1/3 complex. No interaction of Asp1 and Asp1/3 with polar lipids from E. coli was observed (Fig. 6E), consistent with the fact that E. coli contains a much lower percentage of negatively charged lipids than do streptococci (35–37).
Figure 6.

Interaction of the Asps with phospholipid bilayers. A, scheme of the binding assay. Liposomes containing different lipid compositions were mixed with Asps, and the samples were subjected to flotation in a Nycodenz gradient. Fractions were collected from the top and analyzed by Coomassie staining. Liposome-bound proteins (green) are expected to co-float with the lipids (white). Non-associated proteins (red) stay at the bottom. B–D, indicated combinations of Asps were tested for binding to liposomes containing a different ratio of the negatively charged lipid DOPG and the neutral lipid DOPE. Lipids peak in fraction 2. Bovine serum albumin (BSA) was used as a control. E, binding of the Asps to liposomes generated with E. coli polar lipids.
To test whether a glycosylated substrate can be targeted to the membrane by the Asps, we incubated glycosylated GspB-F with either Asp1, Asp1/3, or Asp1/2/3, followed by incubation with liposomes containing negatively charged lipids. The vesicles were then floated in a Nycodenz gradient and fractions analyzed by immunoblotting for a FLAG tag on GspB-F and Coomassie staining. With either Asp1 or Asp1/3, a small but reproducible fraction of the substrate floated with the liposomes (Fig. 7A). No co-flotation was observed with Asp1/2/3 or without the Asps. An Asp1/3 complex containing Asp3 mutations at the tip of the β-strands, which abolished GspB-F secretion in vivo (Fig. 4, C and D, mutant 15), was almost completely defective in substrate flotation (Fig. 7B). Asp3 mutations in the concave surface area had a more moderate effect, again consistent with in vivo results. These results show that a substrate can be recruited by the Asp1/3 complex to the membrane.
Figure 7.

Asps target adhesin to the membrane. A, glycosylated, FLAG-tagged GspB-F was purified from S. gordonii cells lacking Nss and Gly, but the glycans contain significant amounts of hexose attached to GlcNAc, probably because of other glycosyltransferases. The purified protein was incubated with different combinations of the Asps, as indicated, followed by incubation with liposomes containing 70% DOPG, 29.5% DOPE, and 0.5% Texas Red-DHPE. The liposomes were floated in a Nycodenz gradient, and fractions were analyzed by SDS-PAGE, followed by immunoblotting with FLAG antibodies and Coomassie Blue staining (upper and lower panels, respectively). The percentage of membrane-associated Gsp-B was quantified in three experiments and is given below the FLAG immunoblot as means and standard error of the means. B, as in A but with Asp1/3 complexes containing either wildtype or mutant Asp3. Asp3_Mu 1: E57K, K132E, Q134R; Asp3_Mu 4: E57K, K132E, K99E, Q134R; Asp3_Mu 12: K132E; Asp3_Mu 15: E45K and N46K. The quantification with the mutant complexes shows the means and standard error of two experiments.
Discussion
Our results suggest a model for the first steps in the export of an SRR adhesin from the pathogenic bacterium S. gordonii. The adhesin (GspB) is first made as an unmodified protein. It is then sequentially glycosylated by three glycosyltransferases (see model in Fig. 8, box 1). The first enzyme GftA/B adds GlcNAc residues to Ser/Thr residues in SRR domains (G1 species). Next, Nss adds Glc to the GlcNAc residues (G2 species), and finally, Gly adds further Glc residues to those attached by Nss (G3 species). The fully glycosylated substrate is then transferred to the Asp1/2/3 complex (Fig. 8, box 2). In the next step, the Asp1 protein would mediate the interaction of the Asp1/2/3 complex with the lipid bilayer (Fig. 8, box 2). The Asp1/2/3 complex has a relatively low affinity for the membrane, so we assume that it continuously cycles between the cytosol and membrane, with the majority staying in the cytosol. Membrane binding of the Asp complex probably requires a conformational change to expose a lipid interaction domain on Asp1, an interface that seems to be fully available for membrane interaction in Asp1 or the Asp1/3 complex. Once at the membrane, the substrate would be delivered to SecA2 and SecY2 for translocation across the membrane (Fig. 8, box 3). This is consistent with previous findings that the Asp1/2/3 localizes near SecA2 at the membrane (38).
Figure 8.

Model for the export of adhesin from S. gordonii. Box 1, adhesin GspB is synthesized and then sequentially O-glycosylated by three glycosyltransferases. The first enzyme is GtfA/B, a tetramer that adds GlcNAc residues to Ser/Thr residues (G1 species). Then, Nss adds Glc to GlcNAc (G2 species), and finally Gly adds Glc to Glc residues (G3 species). Gly remains bound to the G3 species until it is transferred to the Asp1/2/3 complex. All molecules, including GlcNAc and Glc, are shown in cartoon presentation. Box 2, although the Asp complex has a low affinity for the lipid bilayer, it probably continuously cycles between the cytosol and membrane. The Asp complex likely undergoes a conformational change, in which Asp2 moves away from a lipid-binding surface, allowing the Asp complex to deliver substrate GspB to the membrane. Box 3, GspB engages SecA2 and the SecY2 channel for its translocation across the membrane, and the Asp complex returns to the cytosol.
Our data show that the glycosyltransferases act in a strictly sequential manner. GtfA/B only modifies Ser/Thr residues and has specificity for GlcNAc, whereas Nss and Gly recognize GlcNAc and Glc residues, respectively. Gly is an unusual enzyme, as it has affinity for the product of the reaction it catalyzes. This is explained by the fact that the enzymatic reaction is catalyzed by the C-terminal GT-D domain, whereas the binding to the product is mediated by the N-terminal GT-A and -B domains. Despite their similarity with glycosyltransferases, the GT-A and -B domains seem to belong to the class of catalytically inactive carbohydrate-binding proteins, which also include GtfB and Asp1. Although the affinity of the GT-A and -B domains is high, glycosylated substrate can still dissociate from Gly, allowing its binding to the downstream Asp complex.
Surprisingly, both Asp1 and Asp3 are structurally related to carbohydrate-binding proteins. Asp1 appears to be a catalytically inactive member of the GT-B family of glycosyltransferases, and Asp3 is similar to the CBM domain of glycosidases. The presence of carbohydrate-binding motifs in Asp1 and Asp3 strongly supports the idea that they bind glycosylated adhesin. Indeed, our mutagenesis data provide evidence for substrate interaction with Asp3. Asp1 also seems to bind substrate, as it allows co-flotation with liposomes. The interactions of Asp1 and Asp3 with substrate are weak, as they do not survive in pulldown or gel filtration experiments (data not shown). Although we have not been able to show direct binding of the Asps to carbohydrates, substrate release from Gly (Fig. 2) and co-flotation experiments (Fig. 7) indicate that the Asps interact with substrate. The low-binding affinity is in fact typical for carbohydrate-binding proteins (39). Our results do not exclude the possibility that the Asps also interact with non-glycosylated adhesin. In fact, Asp1 has a similar structure as GtfB (Fig. 3B), which can bind non-glycosylated substrate (10), and Asp2 and Asp3 have been shown to bind non-glycosylated GspB (26). Such an interaction would explain why GspB is secreted in an Asp-dependent manner in glycosylation-defective S. gordonii strains, although in this situation, much of the substrate is degraded (22). It should also be noted that some bacterial species export SRR adhesins in an Asp-dependent manner, although they lack Gly and Nss (5). Asp1 also has an affinity for the lipid bilayer, which facilitates the recruitment of substrate to the membrane. Given that binding of Asp1 requires negatively charged phospholipids and is enhanced at higher salt concentrations (compare Fig. 6B (100 mm) with Fig. 7A (300 mm)), it seems that it is mediated by both hydrophobic and electrostatic interactions. Taken together, our data indicate that Asp1 and Asp3 are carbohydrate- and lipid-binding proteins. We favor a model in which the Asps facilitate the transfer of glycosylated substrate to the membrane (Fig. 8), rather than simply prevent their premature folding in the cytosol; the repetitive structure of the SRR domains and their extensive O-glycosylation would prevent the folding or aggregation of substrate even in the absence of the Asps. Indeed, under these conditions, glycosylated GspB accumulates in the cytosol as a soluble protein (15).
How the substrate is delivered to SecA2 and SecY2 remains to be clarified. The signal sequence of adhesin and the adjacent “accessory Sec transport” (AST) domain are required to target the precursor to the accessory Sec system and initiate translocation (41). Although not very hydrophobic (5), the signal sequence could still facilitate the interaction with the lipid bilayer. Once the substrate is bound to the membrane, it could associate with SecA2, a transfer that may be facilitated by an interaction between the Asp complex and SecA2 (22, 38). Based on a homology model, SecA2 has a pronounced positively charged surface patch (Fig. S9, A and B), which could mediate its interaction with negatively charged phospholipids in the membrane. No such basic surface patch is seen in a homology model for S. gordonii SecA1 (Fig. S9C). Because we were unsuccessful in purifying soluble SecA2, even in the presence of detergent, we speculate that SecA2 requires a lipid environment to maintain its native conformation and that it is permanently bound to the membrane, in contrast to SecA1, which cycles between the cytosol and membrane (7). According to the model, SecA2 would rely on the Asps to deliver substrate to the membrane where SecA2 could engage both the AST domain and the remainder of the mature domain, whereas in the canonical secretion system, SecA1 would do the job, with the chaperone SecB acting upstream for some substrates and bacteria (7). Once substrate has been recruited to the SecA2/SecY2 complex, it is likely translocated across the membrane by a mechanism similar to that of the canonical system.
Materials and methods
Purification of the glycosyltransferases GtfA/B, Nss, and Gly
All proteins were expressed in E. coli. The GtfA/B complex was prepared as described previously (10). Genes encoding S. gordonii Nss, Gly, Gly N domain (residue 1–411), and Gly C domain (residue 412–682) were amplified from S. gordonii genomic DNA by PCR and cloned into the pET21b vector. All constructs contain a His6 tag at the C terminus. E. coli BL21 (DE3) cells were transformed with each plasmid, and the expression of recombinant Nss and Gly was induced with 0.25 mm isopropyl β-d-1-thiogalactopyranoside (IPTG). Proteins were purified from cell lysates after sedimentation of membranes by Ni-NTA–affinity chromatography (Qiagen), followed by ion-exchange (HiTrap Q FF, GE Healthcare) chromatography and gel filtration (Superdex 200 10/300 GL, GE Healthcare). Nss and Gly were also overexpressed with a C-terminal GST tag and purified by glutathione-Sepharose 4B (GE Healthcare) chromatography, followed by gel filtration.
Purification of Asp1 and Asp1/3 for X-ray crystallography
S. gordonii Asp1 was cloned into the pET21b vector with a C-terminal GST tag and expressed in E. coli BL21 (DE3) cells. To co-express Asp1 and Asp3, DNA sequences encoding Asp1-GST and Asp3 were separately cloned downstream of the two promoters into the pCOLADUET-1 vector. The plasmid was transformed into E. coli BL21 (DE3) cells. Both Asp1-GST and Asp1-GST/Asp3 were isolated from cell lysates after sedimentation of membranes by glutathione-Sepharose 4B resin. The GST tag was subsequently removed by thrombin protease. Asp1 and Asp1/3 complex were further purified by ion-exchange and gel-filtration chromatography. Selenomethionine (Se-Met)-derivatized Asp1 and Asp1/3 complexes were obtained from cells grown in M9 minimal medium (Sigma) in the presence of Se-Met (Anatrace). 5 mm dithiothreitol (Sigma) was included during the purification.
Mutations were introduced into Asp1 or Asp3 by QuikChange mutagenesis, and Asp1/3 complexes carrying these mutations were purified in the same way as the wildtype complex.
Purification of Asp1/2/3 and Asp1/3 for electron microscopy
DNA sequences encoding Asp1 and Asp3 were cloned under the two T7 promoters separately into the pCOLADUET-1 vector. The Asp2 gene was cloned into the pMAL-c5x vector to generate Asp2 fused at its N terminus to the MBP, and into the pGEX-2T vector to generate Asp2 with a removable GST tag at the N terminus.
To co-express Asp1–3, either one of the two Asp2 plasmids was co-transformed with the pCOLADUET-1 vector, carrying the Asp1 and Asp3 genes, into E. coli BL21 (DE3) cells. Expression was induced by addition of 0.25 mm IPTG. Asp1/MBP–Asp2/Asp3 and Asp1/GST–Asp2/Asp3 complexes were isolated from cell lysates by amylose resin (New England Biolabs) and glutathione-Sepharose 4B resin chromatography, respectively, and further purified by ion-exchange chromatography and gel filtration.
To generate the Asp1/2/3 complex without a tag, the GST tag of Asp1/GST–Asp2/Asp3 complex was cleaved from Asp2 by thrombin protease, and it was subsequently removed by size-exclusion chromatography.
The pCOLADUET-1 vector carrying both Asp1 and Asp3 genes was also further engineered to include an MBP tag at the N terminus of Asp1. This plasmid was used for overexpression of MBP–Asp1/Asp3 and also for co-transformation with the pMAL-c5x vector containing the Asp2 gene to generate an MBP–Asp1/MBP–Asp2/Asp3 complex. MBP–Asp1/Asp3 and MBP–Asp1/MBP–Asp2/Asp3 were purified first with an amylose affinity resin, followed by ion-exchange chromatography and gel filtration.
In vitro glycosylation assays
GspB-F was generated by in vitro translation in reticulocyte lysate in the presence of [35S]methionine and analyzed as described previously (10). In vitro glycosylation was performed at 37 °C. To test glycosylation of GspB-F by GtfA/B, Nss, and Gly, 1.6 μm GtfA–GtfB complex and 10 mm UDP-GlcNAc were first mixed with 2 μl of GspB-F solution in a final volume of 5 μl. After incubation for 10 min, 6.4 μm Nss and 10 mm UDP-Glc were added to the reaction mixture. To start Gly modification, 6 μm Gly and 10 mm UDP-glucose were added to the reaction 20 min after initiation of glycosylation by Nss, and the reaction was continued at 37 °C for 1 h. In some experiments, Gly N domain or C domain proteins were added instead of Gly. 17 μm Asp1, Asp1/3, or Asp1/2/3 were also included in the reaction, where indicated.
Pulldown experiments
To test the binding of glycosyltransferases Nss and Gly to their modification products, GST-tagged enzymes were used in the glycosylation assay, and magnetic glutathione resins were added to the mixtures after glycosylation. The samples were incubated at 4 °C for 2 h. Bound and unbound fractions were separated by removing the beads with a magnet and analyzed by SDS-PAGE and autoradiography.
To test the dissociation of glycosylated GspB-F (G3) from Gly by Asp complexes, GspB-F (G2) modified by GtfA/B and Nss was incubated with 0.7 μm Gly-GST at 37 °C for 1 h. The reaction was then mixed with 50 μm Asp1, Asp1/3, or Asp123 before adding the glutathione magnetic beads. Bound and unbound fractions were analyzed by SDS-PAGE and autoradiography.
To test the dissociation of glycosylated GspB-F (G3) from Gly-GST, GtfA/B and Nss-modified GspB-F (G2) was incubated with 0.7 μm Gly-GST at 37 °C for 1 h. After glycosylation, Gly C domain or Gly without the GST tag were added to the mixtures at the indicated concentrations. After incubation with glutathione magnetic resin, the bound and unbound fractions were analyzed by SDS-PAGE and autoradiography.
To test the dissociation of glycosylated GspB-F (G3) from Gly C domain, GtfA/B and Nss-modified GspB-F (G2) was incubated with 0.7 μm Gly C domain at 37 °C for 1 h. The reaction mix was incubated with Gly-GST at the indicated concentrations before adding the glutathione magnetic beads. Bound and unbound fractions were analyzed by SDS-PAGE and autoradiography.
Crystallization and structure determination
Crystallization of native and Se-Met-substituted Asp1 and Asp1/3 complex was performed by the hanging-drop vapor-diffusion method at 22 °C. Asp1/3 was crystallized by mixing 12 mg/ml protein solution with an equal volume of well solution containing 220 mm Na2HPO4 and 20% (w/v) PEG3350. Tetragonal rods appeared after 2–3 days and grew to full size within a week. Optimal crystals of Asp1 were obtained by mixing the protein solution with an equal volume of 100 mm BisTris propane, pH 9.2, and 28% (w/v) PEG6000. Thin rod crystals grew to full size within 2 days. All crystals were equilibrated in well solution plus 10–15% glycerol (v/v) and flash-frozen in a liquid nitrogen stream. Both native and selenium SAD data sets were collected at beamline 24ID-C at the Argonne National Laboratory and processed with XDS (42) and autoPROC (43).
SAD data were obtained with Se-Met-containing Asp1/3 crystals at the peak wavelength for selenium. The positions of selenium atoms were determined, and phases were calculated using AutoSol Wizard in PHENIX (44). A partial initial model of Asp1 was built with AutoBuild in PHENIX. A complete model of Asp1/3 was built in Coot (45), facilitated by the position of Se-Met. The structure was refined with Phenix.refine (46). The final refined atomic model contains residues 1–186 and 189–526 of Asp1 followed by Val-Asp-Lys derived from the C-terminal tag, and residues 1–12, 32–46, and 48–154 of Asp3.
The structure of Asp1 was determined by molecular replacement using PHASER in PHENIX (44), with the Asp1 structure from Asp1/3 complex, lacking the R-fold II, as the initial search model. The model was modified in Coot (45) and refined with Phenix.refine (46). The final refined atomic model contains residues 1–115, 118–390, and 407–526, followed by a Val-Asp-Lys-Leu-Val-Pro-Arg peptide derived from the C-terminal tag.
Figures showing structures were prepared in PyMOL (Version 1.5.0.4 Schrödinger, LLC). All software packages were accessed through SBGrid (47).
Secretion of GspB-F from S. gordonii
Asp1 or Asp3 variants were cloned into plasmid pMSP3545 as described (15) and introduced into S. gordonii strain PS1242 (gspB::pB736flagC Δasp1::spec) or PS1244 (gspB::pB736flagC Δasp3::spec), respectively (22, 38), by natural transformation (48). Strains were grown at 37 °C for 18 h in Todd-Hewitt broth supplemented with 60 μg/ml erythromycin. The cultures were diluted 1:5 into fresh medium, and then incubated for additional 5 h at 37 °C. GspB-F secretion into the culture medium versus retention in the protoplast fraction was determined by immunoblotting with anti-FLAG monoclonal antibodies (Sigma) as described before (29).
Negative stain electron microscopy
Negatively stained specimens were prepared following an established protocol with minor modifications (49). Specifically, 2.5 μl of purified sample was applied to glow-discharged copper EM grids coated with a thin layer of continuous carbon film, and the grids were stained with 2% (w/v) uranyl formate. These grids were imaged on a Tecnai T12 electron microscope (FEI) operated at 120 kV at a nominal magnification of ×67,000 using a 4k x 4k CCD camera (UltraScan 4000, Gatan), corresponding to a calibrated pixel size of 1.68 Å on the specimen level. The images were processed as described previously (50).
Limited trypsin proteolysis
Asp1/3 or Asp1/2/3 (5.6 μg) was incubated with 6.6, 2.2, 0.73, 0.24, 0.08, or 0.03 μg of trypsin protease in 8 μl of buffer containing 20 mm Tris/HCl, pH 8.0, and 100 mm NaCl at 22 °C. After 20 min of incubation, 2× SDS-PAGE loading buffer (100 mm Tris/HCl, pH 6.8, 200 mm DTT, 4% SDS, 0.2% bromphenol blue, and 20% glycerol) was added to stop the reaction, and the samples were analyzed by SDS-PAGE and Coomassie Blue staining. To locate the tryptic sites that were protected by Asp2, tryptic fragments of Asp1 and Asp3 were subjected to analysis by MALDI-TOF mass spectrometry (Taplin Mass Spectrometry Core Facility, Harvard Medical School) and N-terminal sequencing (Tufts University Core Facility).
Liposome flotation assay
DOPG and DOPE lipids (Avanti) in chloroform were mixed at the ratios indicated. Texas Red-DHPE (Thermo Fisher Scientific) was also included in some experiments. E. coli polar lipids (Avanti) were also dissolved in chloroform. Lipid mixtures were dried in a SpeedVac and hydrated with buffer containing 20 mm HEPES, pH 7.5, 100 mm NaCl. Liposomes were prepared by extrusion of the mixtures through membranes with a 100-nm pore diameter.
Asp1, Asp1/3, Asp1/2/3, and BSA were mixed with liposomes at a 1:1300 molar ratio of protein to lipid. The samples were adjusted to a final concentration of 40% (w/v) Nycodenz in buffer containing 20 mm Tris/HCl, pH 7.5, 100 mm NaCl, and sequentially overlaid with 30% (w/v) Nycodenz, 15% (w/v) Nycodenz, and buffer lacking Nycodenz. Centrifugation was performed in a TLS-55 swinging bucket rotor (Beckman Coulter) at 48,000 rpm for 1 h. Fractions were collected from the top of the gradient and analyzed by SDS-PAGE followed by Coomassie Blue staining.
To test for co-flotation of the Asps with GspB-F, glycosylated GspB-F was isolated from S. gordonii cells as described before (10) and incubated with the indicated Asp complexes prior to the addition of liposomes. The Nycodenz gradient was prepared in buffer containing 20 mm Tris/HCl, pH 7.5, 300 mm NaCl. After centrifugation, fractions were subjected to both SDS-PAGE and immunoblot analysis using FLAG antibodies (Sigma).
Purification of secreted GspB-F from S. gordonii
S. gordonii strain carrying signal sequence-containing GspB-F in place of wildtype GspB was cultured in Todd Hewitt Broth (BD Biosciences) at 37 °C for 7 h. The medium was harvested, and glycosylated GspB-F was enriched by ammonium sulfate precipitation. The pellet was solubilized and dialyzed against buffer containing 20 mm Tris/HCl, pH 7.5, 150 mm NaCl. Glycosylated GspB-F was purified with an affinity resin containing succinylated wheat germ agglutinin, followed by gel filtration.
Purification of glycosylated GspB-F from E. coli
Genes encoding GspB-F, Nss, and Gly were cloned into the pCOLADUET-1 vector, and a DNA segment encoding GtfA/B was cloned into the pETDUET-1 vector. For co-expression of these proteins, the two plasmids were transformed into E. coli BL21 (DE3) cells. Glycosylated GspB-F was purified by Ni-NTA–affinity chromatography, followed by ion-exchange and size-exclusion chromatography.
Glycan and glycopeptide analysis by mass spectrometry
All reagents were purchased from Sigma unless otherwise mentioned. Mass spectrometric data acquisition was performed on a Thermo Fisher Scientific LTQ Orbitrap Fusion Tribrid mass spectrometer attached with a Dionex nano-LC system and on an AB SCIEX MALDI TOF/TOF 5800 (Applied Biosystem MDS Analytical Technologies) mass spectrometer. Data analysis was performed by using Data Explorer Version 4.5, Xcalibur 3.0, GlycoWorkbench 1.1, and Proteome DiscovererTM 1.4 software. Monosaccharides were analyzed by high performance anion-exchange chromatography using a Dionex ICS3000 system equipped with a gradient pump, an electrochemical detector, and an autosampler.
The overall workflow for glycan analysis included the following steps: β-elimination; permethylation; MALDI-MS; ESI-MS; and ESI-MS/MS. O-Glycans were β-eliminated by treatment of extracted tryptic peptides with NaOH/NaBH4 at 45 °C for 16 h. The samples were neutralized by 10% acetic acid, passed through a Dowex H+ ion-exchange column, and lyophilized. Borates were subsequently removed by the addition of 500 μl of methanol/acetic acid (9:1). The samples were dried under a nitrogen stream and stored at −30 °C.
The glycans were permethylated for analysis by MALDI-MS and ESI-MS. Briefly, the sample was dissolved in dimethyl sulfoxide (DMSO) and incubated with methyl iodide in a DMSO/NaOH mixture. The reaction was quenched with H2O, and permethylated O-glycans were extracted with methylene chloride. The sample was dissolved in methanol and crystallized with α-dihydroxybenzoic acid matrix for MALDI-MS analysis. ESI-MS and MS/MS analysis of permethylated glycans was performed on an Orbitrap Fusion mass spectrometer through an ESI probe. The MSn spectra (CID and HCD) of the glycans were acquired at high resolution (selected CID spectra are shown because HCD produced similar spectra). Assignment of glycan structures was done manually and by using GlycoWorkbench software.
For monosaccharide analysis, tryptic peptides were hydrolyzed with trifluoroacetic acid. A mixture of known amounts of neutral and amino sugar standards (Fuc, GalNAc, GlcNAc, Gal, Glc, and Man) was hydrolyzed and subjected to chromatography on a Dionex CarboPac analytical column with an amino trap, using nanopure H2O and NaOH as eluents. Four concentrations of the standard mixture were prepared and used for calibration. Identification and quantification of the monosaccharides are based on the sugar standards, using the retention time and peak area.
Glycopeptide analysis and site mapping were performed using LC-MS/MS and multiple fragmentation methods (CID, ETD, and HCD). Glycosylated GspB-F was subjected to SDS-PAGE, and the gel band was cut into small pieces of about 1 mm2. After destaining in 1:1 digestion buffer (50 mm NH4HCO3 plus acetonitrile) and 100% acetonitrile, 50 μl of digestion buffer was added to the gel pieces, and the protein was digested with 0.5 μg/μl sequencing-grade trypsin (Promega) at 37 °C for 12 h. The peptides were extracted with 5% formic acid in 1:2 water/acetonitrile, dried, and subsequently re-dissolved in 0.1% formic acid and stored at −30 °C.
Desalted glycopeptides were analyzed on an Orbitrap Fusion mass spectrometer equipped with a nanospray ion source and connected to a Dionex binary solvent system. Pre-packed nano-LC columns of 15-cm length with 75-μm internal diameter, filled with 2-μm C18 reverse-phase material were used for chromatographic separation of the samples. The precursor ion scan was acquired at 120,000 resolution in an Orbitrap analyzer- and precursors at a time frame of 3 s were selected for subsequent fragmentation using either a HCD product-triggered CID/ETD program or ETD with a charge-dependent varying reaction time program. The threshold for triggering an MS/MS event on the ion-trap was set to 500 counts. Charge-state screening was enabled, and precursors with unknown charge state or a charge state of +1 were excluded. Dynamic exclusion was enabled (exclusion duration of 30 s). The fragment ions were analyzed on an Orbitrap for HCD and CID at 30,000 resolution, and on an ion trap for ETD.
The LC-MS/MS spectra of tryptic peptides of glycosylated GspB-F were searched against the sequence of GspB-F using Proteome Discoverer 1.4 software. In the search, oxidation of methionine and O-glycan modification with HexNAc, HexHexNAc, and Hex2HexNAc were set as a variable modification. The LC-MS/MS spectra were also analyzed manually for glycopeptides using Xcalibur software. The HCD and CID MS2 spectra of glycopeptides were evaluated for glycan neutral loss patterns, oxonium ions, and glycopeptide fragmentation to assign the sequence and the presence of glycans. Peptide fragments from ETD spectra were analyzed for the localization of O-glycosylation sites.
SEC-MALS
SEC-MALS was performed as described before (10). 100 μl of 1 mg/ml protein solution were applied in each experiment. Light-scattering data were recorded and analyzed using Astra V software (Wyatt Technology).
Gel-filtration chromatography
Asp1/2/3 (0.3 mg) was applied to a Superdex 200 10/300 GL column (GE Healthcare) in buffer containing 10 mm Tris/HCl, pH 7.5, 150 mm NaCl, and 5 mm DTT. The peak fractions were analyzed by SDS-PAGE and Coomassie Blue staining.
Homology modeling
Models of S. gordonii SecA1 and SecA2 were generated by SWISS-MODEL (51–54), using Thermotoga maritima SecA (PDB code 3DIN) (55) and Bacillus subtilis SecA (PDB code 3JV2) (40) as templates, respectively. T. maritima SecA (PDB code 3DIN) shares 47.3% sequence identity with S. gordonii SecA1, and the coverage is 91%. B. subtilis SecA (PDB code 3JV2) covers 96% of the S. gordonii SecA2, and the sequence identity is 41.8%. The predicted models have Global Model Quality Estimation scores of 0.69 and 0.72, respectively.
Author contributions
Y. C. and T. A. R. conceptualization; Y. C., B. A. B., M. L., P. A., P. M. S., and T. A. R. data curation; Y. C., B. A. B., R. S., M. L., P. A., P. M. S., and T. A. R. formal analysis; Y. C. validation; Y. C., B. A. B., W. M., M. L., A. S., R. N. S., and P. A. investigation; Y. C., B. A. B., W. M., M. L., and A. S. visualization; Y. C., W. M., M. L., P. D. J., A. S., R. N. S., and P. A. methodology; Y. C. and T. A. R. writing-original draft; Y. C., B. A. B., P. M. S., and T. A. R. writing-review and editing; M. L., P. A., P. M. S., and T. A. R. supervision; T. A. R. funding acquisition.
Supplementary Material
Acknowledgments
We thank the staff at the Advanced Photon Source of the Northeastern Collaborative Access Team (NE-CAT) beamline for help with data collection. NE-CAT is supported by National Institutes of Health Grant P41 GM103403 from NIGMS. The Pilatus 6 M detector on 24-ID-C beam line is funded by National Institutes of Health ORIP HEI Grant S10 RR029205. This research used resources of the Advanced Photon Source, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
This work was supported in part by National Institutes of Health Grant GM052586 (to the T. A. R. laboratory). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains Figs. S1–S9 and Table S1.
The atomic coordinates and structure factors (codes 5VAE and 5VAF) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- SRR
- serine-rich repeat
- PDB
- Protein Data Bank
- SEC-MALS
- size-exclusion chromatography–multiangle laser light scattering
- MBP
- maltose-binding protein
- BisTris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- EBD
- extended β-sheet domain
- Se-Met
- selenomethionine
- DOPG
- dioleoylphosphatidylglycerol
- DOPE
- dioleoylphosphatidylethanolamine
- ESI-MS
- electrospray ionization-mass spectrometry
- GST
- glutathione S-transferase
- SAD
- single-wavelength anomalous diffraction
- Ni-NTA
- nickel-nitrilotriacetic acid
- AST
- accessory Sec transport
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- CBM
- carbohydrate-binding module
- CID
- collision-induced dissociation
- HCD
- high-energy collisional dissociation
- ETD
- electron-transfer dissociation.
References
- 1. Kline K. A., Fälker S., Dahlberg S., Normark S. and Henriques-Normark B. (2009) Bacterial adhesins in host-microbe interactions. Cell Host Microbe 5, 580–592 10.1016/j.chom.2009.05.011 [DOI] [PubMed] [Google Scholar]
- 2. Zhou M., and Wu H. (2009) Glycosylation and biogenesis of a family of serine-rich bacterial adhesins. Microbiology 155, 317–327 10.1099/mic.0.025221-0 [DOI] [PubMed] [Google Scholar]
- 3. Lizcano A., Sanchez C. J., and Orihuela C. J. (2012) A role for glycosylated serine-rich repeat proteins in Gram-positive bacterial pathogenesis. Mol. Oral Microbiol. 27, 257–269 10.1111/j.2041-1014.2012.00653.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sanchez C. J., Shivshankar P., Stol K., Trakhtenbroit S., Sullam P. M., Sauer K., Hermans P. W., and Orihuela C. J. (2010) The pneumococcal serine-rich repeat protein is an intra-species bacterial adhesin that promotes bacterial aggregation in vivo and in biofilms. PLoS Pathog. 6, e1001044 10.1371/journal.ppat.1001044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bensing B. A., Seepersaud R., Yen Y. T., and Sullam P. M. (2014) Selective transport by SecA2: an expanding family of customized motor proteins. Biochim. Biophys. Acta 1843, 1674–1686 10.1016/j.bbamcr.2013.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Feltcher M. E., and Braunstein M. (2012) Emerging themes in SecA2-mediated protein export. Nat. Rev. Microbiol. 10, 779–789 10.1038/nrmicro2874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Park E., and Rapoport T. A. (2012) Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Annu. Rev. Biophys. 41, 21–40 10.1146/annurev-biophys-050511-102312 [DOI] [PubMed] [Google Scholar]
- 8. Iwashkiw J. A., Vozza N. F., Kinsella R. L., and Feldman M. F. (2013) Pour some sugar on it: the expanding world of bacterial protein O-linked glycosylation. Mol. Microbiol. 89, 14–28 10.1111/mmi.12265 [DOI] [PubMed] [Google Scholar]
- 9. Takamatsu D., Bensing B. A., and Sullam P. M. (2004) Four proteins encoded in the gspB-secY2A2 operon of Streptococcus gordonii mediate the intracellular glycosylation of the platelet-binding protein GspB. J. Bacteriol. 186, 7100–7111 10.1128/JB.186.21.7100-7111.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen Y., Seepersaud R., Bensing B. A., Sullam P. M., and Rapoport T. A. (2016) Mechanism of a cytosolic O-glycosyltransferase essential for the synthesis of a bacterial adhesion protein. Proc. Natl. Acad. Sci. U.S.A. 113, E1190–E1199 10.1073/pnas.1600494113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bu S., Li Y., Zhou M., Azadin P., Zeng M., Fives-Taylor P., and Wu H. (2008) Interaction between two putative glycosyltransferases is required for glycosylation of a serine-rich streptococcal adhesin. J. Bacteriol. 190, 1256–1266 10.1128/JB.01078-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu R., Zhou M., and Wu H. (2010) Purification and characterization of an active N-acetylglucosaminyltransferase enzyme complex from streptococci. Appl. Environ. Microbiol. 76, 7966–7971 10.1128/AEM.01434-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee I.-C., van Swam I. I., Tomita S., Morsomme P., Rolain T., Hols P., Kleerebezem M., and Bron P. A. (2014) GtfA and GtfB are both required for protein O-glycosylation in Lactobacillus plantarum. J. Bacteriol. 196, 1671–1682 10.1128/JB.01401-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li Y., Huang X., Li J., Zeng J., Zhu F., Fan W., and Hu L. (2014) Both GtfA and GtfB are required for SraP glycosylation in Staphylococcus aureus. Curr. Microbiol. 69, 121–126 10.1007/s00284-014-0563-2 [DOI] [PubMed] [Google Scholar]
- 15. Takamatsu D., Bensing B. A., and Sullam P. M. (2004) Genes in the accessory sec locus of Streptococcus gordonii have three functionally distinct effects on the expression of the platelet-binding protein GspB. Mol. Microbiol. 52, 189–203 10.1111/j.1365-2958.2004.03978.x [DOI] [PubMed] [Google Scholar]
- 16. Zhou M., Peng Z., Fives-Taylor P., and Wu H. (2008) A conserved C-terminal 13-amino-acid motif of Gap1 is required for Gap1 function and necessary for the biogenesis of a serine-rich glycoprotein of Streptococcus parasanguinis. Infect. Immun. 76, 5624–5631 10.1128/IAI.00534-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhu F., Zhang H., Yang T., Haslam S. M., Dell A., and Wu H. (2016) Engineering and dissecting the glycosylation pathway of a streptococcal serine-rich repeat adhesin. J. Biol. Chem. 291, 27354–27363 10.1074/jbc.M116.752998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiang Y.-L., Jin H., Yang H.-B., Zhao R.-L., Wang S., Chen Y., and Zhou C.-Z. (2017) Defining the enzymatic pathway for polymorphic O-glycosylation of the pneumococcal serine-rich repeat protein PsrP. J. Biol. Chem. 292, 6213–6224 10.1074/jbc.M116.770446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou M., Zhu F., Dong S., Pritchard D. G., and Wu H. (2010) A novel glucosyltransferase is required for glycosylation of a serine-rich adhesin and biofilm formation by Streptococcus parasanguinis. J. Biol. Chem. 285, 12140–12148 10.1074/jbc.M109.066928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhu F., Erlandsen H., Ding L., Li J., Huang Y., Zhou M., Liang X., Ma J., and Wu H. (2011) Structural and functional analysis of a new subfamily of glycosyltransferases required for glycosylation of serine-rich streptococcal adhesins. J. Biol. Chem. 286, 27048–27057 10.1074/jbc.M110.208629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhu F., Zhang H., and Wu H. (2015) A conserved domain is crucial for acceptor substrate binding in a family of glucosyltransferases. J. Bacteriol. 197, 510–517 10.1128/JB.02267-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seepersaud R., Bensing B. A., Yen Y. T., and Sullam P. M. (2010) Asp3 mediates multiple protein-protein interactions within the accessory Sec system of Streptococcus gordonii. Mol. Microbiol. 78, 490–505 10.1111/j.1365-2958.2010.07346.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li Y., Chen Y., Huang X., Zhou M., Wu R., Dong S., Pritchard D. G., Fives-Taylor P., and Wu H. (2008) A conserved domain of previously unknown function in Gap1 mediates protein-protein interaction and is required for biogenesis of a serine-rich streptococcal adhesin. Mol. Microbiol. 70, 1094–1104 10.1111/j.1365-2958.2008.06456.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Echlin H., Zhu F., Li Y., Peng Z., Ruiz T., Bedwell G. J., Prevelige P. E. Jr., and Wu H. (2013) Gap2 promotes the formation of a stable protein complex required for mature Fap1 biogenesis. J. Bacteriol. 195, 2166–2176 10.1128/JB.02255-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Siboo I. R., Chaffin D. O., Rubens C. E., and Sullam P. M. (2008) Characterization of the accessory Sec system of Staphylococcus aureus. J. Bacteriol. 190, 6188–6196 10.1128/JB.00300-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yen Y. T., Seepersaud R., Bensing B. A., and Sullam P. M. (2011) Asp2 and Asp3 interact directly with GspB, the export substrate of the Streptococcus gordonii accessory sec system. J. Bacteriol. 193, 3165–3174 10.1128/JB.00057-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou M., Zhang H., Zhu F., and Wu H. (2011) Canonical SecA associates with an accessory secretory protein complex involved in biogenesis of a streptococcal serine-rich repeat glycoprotein. J. Bacteriol. 193, 6560–6566 10.1128/JB.05668-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhou M., Zhu F., Li Y., Zhang H., and Wu H. (2012) Gap1 functions as a molecular chaperone to stabilize its interactive partner Gap3 during biogenesis of serine-rich repeat bacterial adhesin. Mol. Microbiol. 83, 866–878 10.1111/j.1365-2958.2012.07970.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bensing B. A., Takamatsu D., and Sullam P. M. (2005) Determinants of the streptococcal surface glycoprotein GspB that facilitate export by the accessory Sec system. Mol. Microbiol. 58, 1468–1481 10.1111/j.1365-2958.2005.04919.x [DOI] [PubMed] [Google Scholar]
- 30. Zhang H., Zhu F., Yang T., Ding L., Zhou M., Li J., Haslam S. M., Dell A., Erlandsen H., and Wu H. (2014) The highly conserved domain of unknown function 1792 has a distinct glycosyltransferase fold. Nat. Commun. 5, 4339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boraston A. B., Bolam D. N., Gilbert H. J., and Davies G. J. (2004) Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem. J. 382, 769–781 10.1042/BJ20040892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shoseyov O., Shani Z., and Levy I. (2006) Carbohydrate binding modules: biochemical properties and novel applications. Microbiol. Mol. Biol. Rev. 70, 283–295 10.1128/MMBR.00028-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bae B., Ohene-Adjei S., Kocherginskaya S., Mackie R. I., Spies M. A., Cann I. K., and Nair S. K. (2008) Molecular basis for the selectivity and specificity of ligand recognition by the family 16 carbohydrate-binding modules from Thermoanaerobacterium polysaccharolyticum ManA. J. Biol. Chem. 283, 12415–12425 10.1074/jbc.M706513200 [DOI] [PubMed] [Google Scholar]
- 34. Montanier C., van Bueren A. L., Dumon C., Flint J. E., Correia M. A., Prates J. A., Firbank S. J., Lewis R. J., Grondin G. G., Ghinet M. G., Gloster T. M., Herve C., Knox J. P., Talbot B. G., Turkenburg J. P., et al. (2009) Evidence that family 35 carbohydrate binding modules display conserved specificity but divergent function. Proc. Natl. Acad. Sci. U.S.A. 106, 3065–3070 10.1073/pnas.0808972106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rosch J. W., Hsu F. F., and Caparon M. G. (2007) Anionic lipids enriched at the ExPortal of Streptococcus pyogenes. J. Bacteriol. 189, 801–806 10.1128/JB.01549-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Trombe M. C., Lanéelle M. A., and Lanéelle G. (1979) Lipid composition of aminopterin-resistant and sensitive strains of Streptococcus pneumoniae. Effect of aminopterin inhibition. Biochim. Biophys. Acta 574, 290–300 10.1016/0005-2760(79)90010-9 [DOI] [PubMed] [Google Scholar]
- 37. van der Does C., Swaving J., van Klompenburg W., and Driessen A. J. (2000) Non-bilayer lipids stimulate the activity of the reconstituted bacterial protein translocase. J. Biol. Chem. 275, 2472–2478 10.1074/jbc.275.4.2472 [DOI] [PubMed] [Google Scholar]
- 38. Yen Y. T., Cameron T. A., Bensing B. A., Seepersaud R., Zambryski P. C., and Sullam P. M. (2013) Differential localization of the streptococcal accessory sec components and implications for substrate export. J. Bacteriol. 195, 682–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nagae M., and Yamaguchi Y. (2014) Three-dimensional structural aspects of protein-polysaccharide interactions. Int. J. Mol. Sci. 15, 3768–3783 10.3390/ijms15033768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zimmer J., and Rapoport T. A. (2009) Conformational flexibility and peptide interaction of the translocation ATPase SecA. J. Mol. Biol. 394, 606–612 10.1016/j.jmb.2009.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bensing B. A., and Sullam P. M. (2010) Transport of preproteins by the accessory Sec system requires a specific domain adjacent to the signal peptide. J. Bacteriol. 192, 4223–4232 10.1128/JB.00373-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 12 10.1107/S09074449090473375–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vonrhein C., Flensburg C., Keller P., Sharff A., Smart O., Paciorek W., Womack T., and Bricogne G. (2011) Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 67, 293–302 10.1107/S0907444911007773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 10.1107/S0907444912001308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Morin A., Eisenbraun B., Key J., Sanschagrin P. C., Timony M. A., Ottaviano M., and Sliz P. (2013) Collaboration gets the most out of software. Elife 2, e01456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bensing B. A., and Sullam P. M. (2002) An accessory sec locus of Streptococcus gordonii is required for export of the surface protein GspB and for normal levels of binding to human platelets. Mol. Microbiol. 44, 1081–1094 10.1046/j.1365-2958.2002.02949.x [DOI] [PubMed] [Google Scholar]
- 49. Booth D. S., Avila-Sakar A., and Cheng Y. (2011) Visualizing proteins and macromolecular complexes by negative stain EM: from grid preparation to image acquisition. J. Vis. Exp. 2011, 3227 10.3791/3227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ru H., Chambers M. G., Fu T.-M., Tong A. B., Liao M., and Wu H. (2015) Molecular mechanism of V(D)J recombination from synaptic RAG1-RAG2 complex structures. Cell 163, 1138–1152 10.1016/j.cell.2015.10.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Gallo Cassarino T., Bertoni M., Bordoli L., and Schwede T. (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 10.1093/nar/gku340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Guex N., Peitsch M. C., and Schwede T. (2009) Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 30, S162–S173 10.1002/elps.200900140 [DOI] [PubMed] [Google Scholar]
- 53. Kiefer F., Arnold K., Künzli M., Bordoli L., and Schwede T. (2009) The SWISS-MODEL repository and associated resources. Nucleic Acids Res. 37, D387–D392 10.1093/nar/gkn750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Arnold K., Bordoli L., Kopp J., and Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 10.1093/bioinformatics/bti770 [DOI] [PubMed] [Google Scholar]
- 55. Zimmer J., Nam Y., and Rapoport T. A. (2008) Structure of a complex of the ATPase SecA and the protein-translocation channel. Nature 455, 936–943 10.1038/nature07335 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.