

Abstract
MicroRNAs (miRNAs) are post-transcriptional gene regulators that play important roles in the control of cell fitness, differentiation, and development. The CRISPR-Cas9 gene-editing system is composed of the Cas9 nuclease in complex with a single guide RNA (sgRNA) and directs DNA cleavage at a predetermined site. Several CRISPR-Cas9 libraries have been constructed for genome-scale knockout screens of protein function; however, few libraries have included miRNA genes. Here we constructed a miRNA-focused CRISPR-Cas9 library that targets 1594 (85%) annotated human miRNA stem–loops. The sgRNAs in our LX-miR library are designed to have high on-target and low off-target activity, and each miRNA is targeted by four to five sgRNAs. We used this sgRNA library to screen for miRNAs that affect cell fitness of HeLa or NCI-N87 cells by monitoring the change in frequency of each sgRNA over time. By considering the expression in the tested cells and the dysregulation of the miRNAs in cancer specimens, we identified five HeLa pro-fitness and cervical cancer up-regulated miRNAs (miR-31-5p, miR-92b-3p, miR-146b-5p, miR-151a-3p, and miR-194-5p). Similarly, we identified six NCI-N87 pro-fitness and gastric cancer up-regulated miRNAs (miR-95-3p, miR-181a-5p, miR-188-5p, miR-196b-5p, miR-584-5p, and miR-1304-3p), as well as three anti-fitness and down-regulated miRNAs (let-7a-3p, miR-100-5p, and miR-149-5p). Some of those miRNAs are known to be oncogenic or tumor-suppressive, but others are novel. Taken together, the LX-miR library is useful for genome-wide unbiased screening to identify miRNAs important for cellular fitness and likely to be useful for other functional screens.
Keywords: microRNA, CRISPR-Cas9, sgRNA library, functional screening, cancer
INTRODUCTION
MicroRNAs (miRNAs) are small (∼21 to 22 nucleotides [nt]) RNA molecules that post-transcriptionally regulate gene expression. Canonical miRNA biogenesis starts with transcription of the primary miRNA (pri-miRNA) by RNA polymerase II. The RNAse III–type enzyme Drosha (Lee et al. 2003) is then recruited to the pri-miRNA based partially on the stem–loop secondary structure of the RNA (Han et al. 2006) and cleaves the stem–loop out of the pri-miRNA to produce a precursor miRNA (pre-miRNA). After the excised pre-miRNA is exported out of the nucleus by Exportin 5 (Yi et al. 2003; Bohnsack et al. 2004), the top of the stem–loop is cleaved off by another RNAse III–type enzyme, Dicer (Bernstein et al. 2001; Hutvágner et al. 2001). The ∼22 base pair (bp) double-stranded RNA is then loaded into the RNA-induced silencing complex (RISC), and the passenger RNA strand is removed (Sontheimer 2005). The single-stranded mature miRNA in complex with RISC then binds the 3′-UTR of target mRNAs and degrades or represses translation of the mRNA. Several variations of this canonical biogenesis pathway have been identified in humans, including the Drosha-independent processing of miRNAs from short introns by splicing pathway components (Berezikov et al. 2007; Ruby et al. 2007) and the Dicer-independent processing of miR-451 by the RISC component AGO2 (Cifuentes et al. 2010; Yang et al. 2010).
MiRNAs may regulate up to 60% of all human protein-coding genes (Friedman et al. 2008) and play important roles in the regulation of many cellular processes including differentiation (Ivey and Srivastava 2010), tumorigenesis, metastasis, and proliferation (Nana-Sinkam and Croce 2014). In cancer, miRNAs can act as tumor suppressors or oncogenes (Ventura and Jacks 2009). Tumors often have a global down-regulation of miRNA expression (Lu et al. 2005; Thomson et al. 2006; Lee et al. 2008a) and an up-regulation of specific miRNAs (Volinia et al. 2006). The individual miRNAs involved in cancer vary based on the specific type of cancer, and some miRNAs may play different roles in different cancers (Sun et al. 2013). The availability of next-generation sequencing has allowed specific miRNA expression profiles to be associated with tumors of distinct origins and subtypes (Lu et al. 2005); however, the functional role of many miRNAs remains to be determined.
To determine the function of miRNAs in an unbiased and genome-wide manner, miRNA mimic and inhibitor arrays have previously been used. However, these arrays are expensive and miRNA mimics can have unintended effects on the miRNA processing machinery (Khan et al. 2009). With the recent development of the CRISPR (clustered regulatory interspaced short palindromic repeat)-Cas9 system for use in cells, precise genome editing can now be used to knock out miRNAs of interest in place of mimics and inhibitors. The CRISPR-Cas9 system, derived from the adaptive immune system of the bacterial species Streptococcus pyogenes, is composed of two components, a single guide RNA (sgRNA) and the Cas9 protein (Cong et al. 2013; Jinek et al. 2013; Mali et al. 2013). The sgRNA is an ∼100 nt single-stranded RNA molecule (Cho et al. 2013; Cong et al. 2013; Jinek et al. 2013), which includes a scaffold sequence necessary for Cas9 binding and a 20-nt guide sequence that is complementary to the desired target site. The CRISPR system can be used to target any 20-base sequence in the genome which is followed by a protospacer adjacent motif (PAM): NGG. The sgRNA forms a complex with the Cas9 protein, which binds to the PAM sequence and opens the DNA to allow for the guide sequence to form a duplex with the target site (Nishimasu et al. 2014; Jiang et al. 2015, 2016). Stable binding of the Cas9-sgRNA complex to a target sequence results in the creation of a DNA double-strand break (DSB) 3 bp upstream of the PAM sequence. This DSB can be repaired by the cell's normal DNA repair mechanisms, including homology-directed repair and nonhomologous end joining (NHEJ). NHEJ often results in insertions or deletions (indels) at the cleavage site, which can cause frame shift mutations in protein-coding genes. MicroRNA genes can also be inactivated by the formation of indels in the mature miRNA sequence or in regions necessary for the biogenesis of the miRNA (Jiang et al. 2014; Chen et al. 2015a; Chang et al. 2016).
The ease of targeting specific genomic sequences using the CRISPR-Cas9 system has allowed for genome-wide knockout screens to be performed. These screens are either pooled, where each cell in a pool of cells receives a different sgRNA, or arrayed (Schmidt et al. 2015), where cells are added to a multiwell plate containing a different sgRNA in each well. CRISPR-Cas9 pooled screens are conducted by transducing cells with a pool of sgRNAs and then subjecting the cells to either positive or negative selection. The selected cells are then sequenced to determine the change in frequency of each sgRNA relative to the nonselected pool of cells. Those sgRNAs with significant changes in frequency in the pool after selection presumably target genes involved in promoting or repressing the selected phenotype. Pooled screens have been used to identify essential genes (Shalem et al. 2014; Wang et al. 2014b, 2015; Hart et al. 2015) and genes that are involved in drug resistance (Koike-Yusa et al. 2014; Shalem et al. 2014; Wang et al. 2014b; Hart et al. 2015; Doench et al. 2016; Kurata et al. 2016), toxin susceptibility (Koike-Yusa et al. 2014; Zhou et al. 2014; Tao et al. 2016), and immune response (Parnas et al. 2015; Schmid-Burgk et al. 2016). More recent pooled screens have used single cell sequencing to determine the impact of each sgRNA on the transcriptome rather than examining a cellular phenotype (Adamson et al. 2016; Dixit et al. 2016; Jaitin et al. 2016; Datlinger et al. 2017).
The majority of pooled screens have solely investigated the role of protein-coding genes, and only a few have also investigated the role of miRNAs (Chen et al. 2015b; Wallace et al. 2016). Two studies used the GeCKOv2 library (Sanjana et al. 2014), which contains 123,411 sgRNAs, ∼6% of which targets miRNA genes. These screens identified a role for miR-152 and miR-345 in lung cancer metastasis (Chen et al. 2015b) and miR-155 in myeloid leukemia cell proliferation (Wallace et al. 2016). The large size of GeCKOv2 makes screening of microRNA with this library cumbersome and inefficient as tens of millions of cells are necessary to maintain representation of the library.
To facilitate the screening of miRNA function, we created a focused CRISPR-Cas9 library targeting human miRNA genes with sgRNAs optimized for on-target specificity and efficiency. We used this LX-miR library to screen for miRNAs involved in the fitness of HeLa cells and identified 44 pro-fitness miRNAs. The library was also used to identify 10 pro-fitness and 10 anti-fitness miRNAs in NCI-N87 gastric cancer cells. Many of the identified miRNAs have also been associated with cancer in patients.
RESULTS
miRNA-specific sgRNA library creation
There are 2588 mature miRNAs derived from 1881 primary miRNA that have been annotated in the human genome according to miRBase (Kozomara and Griffiths-Jones 2014). The goal was to design at least four unique sgRNAs for each primary miRNA to guard against false positives due to off-target events or false negatives due to inefficient cleavage. First, we identified all possible sgRNA target sites (X20NGG) in every miRNA stem–loop sequence in miRBase plus the 20 bp on either side of the stem–loop. The miRNA-targeting sgRNAs were then filtered to minimize possible off-target effects and maximize the probability of on-target cleavage (Supplemental Table S1). First, sgRNAs that also perfectly targeted one or more non-miRNA regions in the human genome were excluded. The sgRNAs targeting multiple primary miRNAs that produce the same mature miRNAs or miRNAs in the same family were retained (Fig. 1). Second, we used the CRISPR Design Tool (Hsu et al. 2013) to score each sgRNA based on the number of possible off-target sites in the human genome and removed those sgRNAs with scores below 0.2. Third, the sgRNAs with poly(T) sequences of four or longer, which is likely to result in premature RNA Pol III termination (Bogenhagen and Brown 1981; Matsuzaki et al. 1994), were excluded. Fourth, if the cleavage site was outside of the stem–loop region, the sgRNA was also removed, as indels outside of the stem–loop are less likely to affect the expression of the targeted miRNA. This filtering retained 1594 primary miRNAs (∼85%) that could be unambiguously and effectively targeted (Fig. 2A).
FIGURE 1.

Circos diagram of the human genome with miRNA and LX-miR library annotations. The outermost, first ring shows the genomic location of all annotated primary miRNA stem–loops, with primary miRNAs targeted by four to five sgRNAs marked in dark green, one to three sgRNAs in light green, and zero sgRNAs in black. The second and third rings show bar graphs of the log2 fold change of the sgRNA targeting its primary miRNA in the HeLa screen from replicate 1 (outer) or replicate 2 (inner). The internal links show the different genomic target locations for sgRNAs with multiple target sites. The blue links show sgRNAs that target two (dark blue) or more (light blue) primary miRNAs which produce the same mature miRNAs. The purple links show sgRNAs that target two (dark purple) or more (light purple) primary miRNAs which belong to the same miRNA family. The circos diagram shows the similarity of the two HeLa screen results even though the resolution of the diagram is limited by the pixel density.
FIGURE 2.

Creation of miRNA-targeting CRISPR library LX-miR. (A) Pie chart showing the reason primary miRNAs could not be targeted by at least four sgRNAs (see text for detail). The filtering results in 1594 primary miRNAs targeted by at least four sgRNAs. (B) Diagram showing the creation of the sgRNA library. Oligos were plate-synthesized, amplified and made double-stranded by PCR, and then Gibson-assembled into the pLX-sgRNA vector. The plasmid pool was then used to produce lentiviruses. (C–E) Comparisons between the LX-miR library and the miRNA-targeting portion of the GeCKOv2 library. (C) Box plot comparing the number of exact matches in the human genome of each sgRNA; dots represent outliers. (D) Box plot comparing off-target CRISPR Design Tool score of each sgRNA. (E) Box plot comparing on-target Azimuth score for each sgRNA.
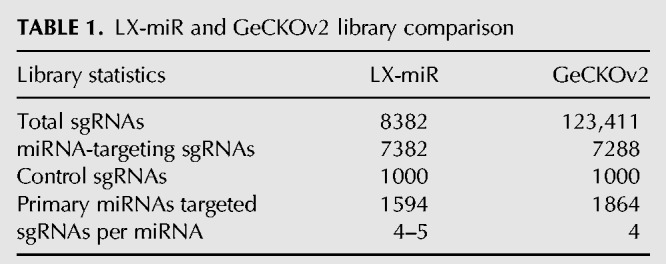
For those miRNAs with more than five possible sgRNAs after filtering, we selected the best four to five sgRNAs that have nonidentical seed sequences, as the seed region is critical for sgRNA targeting (Hsu et al. 2013; Jinek et al. 2013), as well as high predicted on-target activity using the Azimuth scoring method (Supplemental Table 1; Doench et al. 2016). The resulting library contains 7382 sgRNAs targeting 1594 annotated human primary miRNAs. Five additional miRNAs are targeted by the library with less than four sgRNAs due to the inclusion of sgRNAs which target multiple primary miRNAs. We also included 1000 nontargeting control sgRNAs from previously published libraries (Sanjana et al. 2014; Shalem et al. 2014; Wang et al. 2014b, 2015) which have no target sites in the human genome, for a final LX-miR library size of 8382 sgRNAs (Supplemental Table S2). Oligos for the final pool of 8382 sgRNAs were plate synthesized, PCR amplified, and cloned into the lentiviral pLX-sgRNA vector (Supplemental Fig. 1; Wang et al. 2014b). After cloning, the plasmid pool was deep sequenced and found to contain >99.9% of the library sgRNAs (Supplemental Fig. 2). The pLX-miR plasmid pool was then used to produce lentiviruses (Fig. 2B).
We compared our library with the GeCKOv2 sgRNA library, which includes miRNA-targeting sgRNAs in addition to sgRNAs targeting protein-coding genes (Table 1; Sanjana et al. 2014). Because the GeCKOv2 library was designed to target as many miRNAs as possible (1864/1881), some of the sgRNAs were not optimized. In fact, nearly 10% of the miRNA-targeting sgRNAs in GeCKOv2 have at least one other exact target sequence elsewhere in the human genome that is not relevant to miRNAs, and many of the sgRNAs target hundreds of sites (Fig. 2C). sgRNAs that cut at multiple sites may cause genomic instability and result in false positives during fitness screens (Aguirre et al. 2016; Munoz et al. 2016). Our design minimized multiple-targeting sgRNAs and focused on the 1594 “screenable” miRNAs. Furthermore, the sgRNAs in our library have higher predicted on-target activity and better off-target scores (Fig. 2D,E).
TABLE 1.
LX-miR and GeCKOv2 library comparison
HeLa LX-miR fitness screen
We used the LX-miR library to identify miRNAs that are important for HeLa cell fitness. To do this, we first created clonal HeLa-Cas9 cell lines which contain FLAG-tagged Cas9 under the control of a doxycycline inducible promoter (Wang et al. 2014b). A line was chosen that has no detectable Cas9 expression without doxycycline induction and produced increasing amounts of Cas9 with increasing concentrations of doxycycline (Fig. 3A).
FIGURE 3.

Creation of HeLa-Cas9 cell line and outline of LX-miR HeLa screen. (A) Western blot showing the expression of FLAG-tagged Cas9 without doxycycline and with increasing concentrations of doxycycline. (B) Outline of HeLa LX-miR screen. HeLa-Cas9 cells are transduced with lentivirus and then grown with and without doxycycline induction. Without induction, the proportion of cells with each sgRNA is expected to remain consistent. With induction, those cells in which pro-fitness miRNAs are knocked out (orange, purple, and green) are expected to decrease in the population. The number of cells with each sgRNA is determined by PCR amplifying the sgRNA-containing region of the genomic DNA and deep sequencing these amplicons. (C) Timeline of the HeLa LX-miR Screen.
These HeLa-Cas9 cells were then transduced with LX-miR lentivirus in two separate biological replicates. Both transductions were conducted at low multiplicity of infection (MOI < 0.2) to ensure each transduced cell would contain only one of the sgRNAs in the library. The LX-miR vector contains a blasticidin resistance gene, so successfully transduced cells were selected by treatment with blasticidin. More than 1.7 million blasticidin resistant cells were selected each time, which represents greater than 200-fold coverage of the library. The cells were split into two pools, one of which was treated with doxycycline to induce Cas9 expression and sgRNA-mediated mutations, while the other remained uninduced and unmutated (Fig. 3B). Both populations were grown in parallel for 14 d to allow for difference in the fitness after miRNA knockout to become apparent. Genomic DNA was collected from the initial population before dox induction (initial), the dox induced population, and the uninduced population (Fig. 3C). The relative frequency of each sgRNA in the population was determined by amplifying the integrated sgRNA region and deep sequencing these amplicons (Supplemental Table S3). We also examined the miRNA expression in the initial, induced, and uninduced pools by deep sequencing to determine if doxycycline treatment changed the miRNA expression profile of HeLa cells. Among the 1517 miRNAs with detectable levels of expression, only a single miRNA, miR-145-5p, had a significant change in expression upon doxycycline induction (Supplemental Fig. S3), indicating doxycycline treatment does not significantly change the miRNA expression profile of HeLa cells and is not likely to affect our fitness miRNA screens.
After the frequency of each sgRNA was determined, we examined the difference in sgRNA prevalence between the induced and the average of the initial and uninduced control pools. The change in prevalence for each sgRNA was similar between the biological replicates (Fig. 1; Supplemental Fig. S4). Many sgRNAs showed a significant decrease in representation in the induced pool (data from replicate 1 is shown in Fig. 4A, and a similar result was obtained from replicate 2, shown in Supplemental Fig. S5A). At the global level, the distribution of the log fold change of miRNA-targeting sgRNAs is shifted toward negative fold change compared to the distribution of nontargeting control sgRNAs (Fig. 4B; Supplemental Fig. S5B). The result suggested that many losses of a single miRNA in HeLa may decrease cell fitness, but very few losses of a single miRNA may increase cell fitness.
FIGURE 4.

Deep sequencing results from LX-miR HeLa screen. (A) Scatter plot of the induced versus uninduced/initial control read counts for each sgRNA in replicate 1. MiRNA-targeting sgRNAs are in blue, while nontargeting sgRNAs are in gray. The dotted line indicates no change between the populations. (B) The kernel density estimate of the log2 fold change between induced and uninduced/initial control reads per sgRNA in replicate 1. The blue line indicates the distribution of the nontargeting sgRNAs, and the green line indicates the distribution of the miRNA-targeting sgRNAs.
Identification of fitness-associated miRNAs
Since there are four to five sgRNAs for each primary miRNA, we can use the sgRNA changes to predict which miRNAs are likely true hits. Two algorithms were used to do this: Redundant siRNA Activity (RSA) (Konig et al. 2007) and Model-based Analysis of Genome-wide CRISPR/Cas9 Knockout (MAGeCK) (Li et al. 2014). Both rank the sgRNAs based on their change in frequency between the control and induced samples. Then the sgRNAs targeting a specific miRNA are examined to determine if they are significantly enriched among the high ranking sgRNAs or the low ranking sgRNAs, using robust rank aggregation (MAGeCK) or the hypergeometric distribution function (RSA). There was significant correlation between the P-values of both algorithms (Fig. 5A; Supplemental Fig. S6A). MAGeCK uses the control sgRNAs in the library to create the null distribution, while RSA uses the entire sgRNA population; as a result, MAGeCK produced a more asymmetric P-value distribution than RSA did (Fig. 5B). Nevertheless, miRNAs that showed significant changes were generally in agreement (Supplemental Fig. S6B–E), thus we collected those miRNAs that were identified using both algorithms. When both biological replicates were compared, approximately half of the pro-fitness primary miRNAs identified in each replicate were identified in both replicates, for 160 pro-fitness primary miRNAs (Fig. 5C). No anti-fitness miRNAs were found; this suggests that loss of a single miRNA may not be sufficient to enhance the fitness of the highly transformed and fast-growing HeLa cells.
FIGURE 5.

Fitness-associated miRNAs identified from HeLa LX-miR screen. (A) Scatter plot of MAGeCK and RSA −log10 P-values for each primary miRNA based on the decrease of sgRNAs targeting that primary miRNA. The P-values from both algorithms are highly correlated (Pearson correlation coefficient = 0.81–0.85; P-value = 0.0). Replicate 1 data shown in blue dots, replicate 2 in green Xs. (B) Median log2 fold change of all the sgRNAs targeting a primary miRNA plotted versus the −log10 P-value from RSA (green X) or MAGeCK (blue dot) for replicate 1. The dotted line indicates P-value = 0.05. (C) Venn diagram showing the overlap between significant pro-fitness miRNAs in replicate 1 and replicate 2. (D) Ranked bar plot of the mature miRNA read counts for significant pro-fitness miRNAs. Only miRNAs with at least 10 normalized reads (read per million) were plotted.
We then analyzed the miRNA-seq read counts of the 233 mature miRNAs produced from these 160 primary miRNAs, reasoning that a pro-fitness miRNA should be expressed at a detectable level in HeLa-Cas9 cells. There were 44 mature miRNAs with greater than 10 reads per million normalized reads (RPM) (Fig. 5D). The known functions of these 44 mature miRNAs in cellular proliferation and cancer as well as their targets are listed in the Supplemental Table S4. Among them, miR-31-5p (Zheng et al. 2015) and miR-3648 (Rashid et al. 2017) have previously been shown to promote the proliferation of HeLa cells, and miR-31-5p has also been shown to act as an oncogene in cervical cancer (Rao et al. 2012; Wang et al. 2014a; Zheng et al. 2015).
Overexpression of pro-fitness miRNAs in cervical cancer
We then wanted to determine if miRNAs identified as pro-fitness in cancer cell lines may also play pro-fitness roles in patient tumors. As HeLa cells are derived from cervical cancer, we looked for overlaps between our HeLa cell hits and those miRNAs found to be differentially expressed in cervical cancer. We examined published studies of differential miRNA expression in cervical cancer tumors or precancerous cervical intraepithelial neoplasia (CIN) versus normal cervical tissue (Supplemental Table S5; Martinez et al. 2007; Lee et al. 2008b; Wang et al. 2008; Pereira et al. 2010; Witten et al. 2010; Cheung et al. 2012; Lajer et al. 2012; Liu et al. 2012; Rao et al. 2012; Wilting et al. 2012). A total of 275 miRNAs were found up- or down-regulated in at least one of these studies (Supplemental Table S6). Among them, 19 miRNAs overlapped with the 44 putative pro-fitness miRNAs identified in our screen. Ten of the overlapping miRNAs were consistently up-regulated in cervical cancer or CIN samples, and five miRNAs were found up-regulated in multiple studies: miR-31-5p, miR-92b-3p, miR-146b-5p, miR151a-3p, and miR-194-5p (Table 2). The significant number of miRNAs identified in our screen that are also known to be overexpressed in cervical cancer tumors suggests many of the identified miRNAs may be important for cervical tumor cell growth in vivo, not just the proliferation of HeLa cells in vitro.
TABLE 2.
Pro-fitness miRNAs from this HeLa screen compared to dysregulated miRNAs in cervical cancer

Screen for fitness-associated miRNAs using LX-miR in NCI-N87 gastric cancer cells
To further test the utility of the LX-miR library, we also screened for miRNAs that may affect fitness in the NCI-N87 gastric cancer cell line. NCI-N87 cells are near diploid (Park et al. 1990), whereas HeLa cells are mostly polyploid (Macville et al. 1999). As with the HeLa screen, a clonal NCI-N87-Cas9 cell line with doxycycline-inducible Cas9 expression was created (Supplemental Fig. S7A) and transduced with the LX-miR library. The cells were grown for ∼14 cell duplication, with and without doxycycline (Supplemental Fig. S7B). Amplicon sequencing was performed to determine the frequency of each sgRNA in the control and doxycycline-induced samples (Fig. 6A). There was no significant shift between the overall fold-change distribution of the nontargeting control and the miRNA-targeting sgRNAs (Fig. 6B). This is different than the asymmetrical shift toward negative fold change seen in miRNA-targeting sgRNAs in HeLa cells (Fig. 4B) and suggests the loss of a single miRNA in NCI-N87 may affect cell fitness positively as well as negatively.
FIGURE 6.

LX-miR NCI-N87 Screen. (A) Scatter plot of the induced versus uninduced/initial control read counts for each sgRNA. The dotted line indicates no change between the populations. (B) The kernel density estimate of the log2 fold change between induced and uninduced/initial control reads per sgRNA. The blue line indicates the distribution of the nontargeting sgRNAs, and the green line indicates the distribution of the miRNA-targeting sgRNAs. (C) Median log2 fold change of all the sgRNAs targeting a primary miRNA plotted versus the −log10 P-value from RSA (green X) or MAGeCK (blue dot). The dotted line indicates P-value = 0.05. (D,E) Ranked bar plot of the mature miRNA read counts for significant (D) pro-fitness or (E) anti-fitness miRNAs. Only miRNAs with at least 10 normalized reads (read per million) were plotted.
We determined potential pro- and anti-fitness miRNAs by analyzing the frequency-change of sgRNAs using MAGeCK and RSA (Fig. 6C; Supplemental Table S7), and by considering mature miRNA expression (read counts >10 RPM). We identified 10 pro-fitness and 10 anti-fitness mature miRNAs in NCI-N87 cells (Fig. 6D,E). To determine if these miRNAs might be important for fitness of gastric cancer cells in vivo, we examined the TCGA data for 41 patients with paired tumor and normal gastric miRNA expression data. Six pro-fitness miRNAs identified in our screen (miR-95-3p, miR-181a-5p, miR-188-5p, miR-196b-5p, miR-584-5p, and miR-1304-3p) were significantly up-regulated in tumor samples, and three anti-fitness miRNAs (let-7a-3p, miR-100-5p, and miR-149-5p) were significantly down-regulated in tumor samples (Table 3).
TABLE 3.
NCI-N87 fitness-associated miRNAs gastric tumor versus normal tissue comparison in TGCA

We examined the literature for known roles of these fitness-associated miRNAs (Supplemental Table S8). Two pro-fitness miRNAs we identified, miR-181a-5p (Zhang et al. 2012) and miR-196b-5p (Tsai et al. 2010, 2014; Li et al. 2016b), have previously been shown to be gastric cancer oncogenes, and both are up-regulated in TCGA patient samples. Four of the anti-fitness miRNAs we identified have previously been shown as gastric cancer tumor suppressors: let-7a-5p (Tang et al. 2016), miR-100-5p (Shi et al. 2015; Yang et al. 2015), miR-149-5p (Wang et al. 2012), and miR-339-5p (Shen et al. 2015), and two of which are down-regulated in TCGA gastric tumor data. Thus, there is a strong correlation between miRNAs identified in our screen as fitness-associated and miRNAs dysregulated in patient data. Taken together, the LX-miR library we developed for genome-wide unbiased screening was able to identify miRNAs involved in cellular fitness of cancer cell lines, many of which have altered expression in tumors that is consistent with their potential fitness role.
DISCUSSION
To the best of our knowledge, this work represents the first CRISPR-Cas9 library created specifically for functional screening of human miRNAs. This LX-miR library is able to target 85% of the annotated human primary miRNAs with four to five sgRNAs per miRNA. We used the library to identify miRNAs involved in the fitness of two cancer cell lines. We found that miRNAs primarily play a pro-fitness role in HeLa cells and identified 44 putative pro-fitness mature miRNAs. Of these 44 miRNAs, 10 have previously been shown to be up-regulated in cervical tumors or CIN, emphasizing the clinical significance of the miRNAs identified. We also identified 10 pro-fitness and 10 anti-fitness miRNAs in NCI-N87 cells. Six of the pro-fitness and three of the anti-fitness miRNAs are also dysregulated in gastric cancer patient samples. Moreover, several of the pro-fitness miRNAs we identified have been reported as oncogenes, and likewise several of the anti-fitness miRNAs are known tumor suppressors. This indicates that the LX-miR screen is able to identify miRNAs with important roles in cancer.
Genome-wide screens of miRNA function have previously been conducted using miRNA mimic or inhibitor arrays. This involves multiwell plates with individual chemically modified mature miRNA mimics or inhibitors. After transfection with the mimic or inhibitor, the tested cells are then examined for changes in phenotypes, such as differentiation (Kamat et al. 2014), drug sensitivity (Lam et al. 2010), senescence (Kooistra et al. 2014), lipid metabolism (Goedeke et al. 2015), PI3K-Akt pathway changes (Bischoff et al. 2015), and viral infection (Yarbrough et al. 2014; Foo et al. 2016; Shim et al. 2016; Smith et al. 2017). There are several limitations to array approaches, however, as they are more expensive by fivefold or more, laborious to conduct, reliant on consistency between wells, can only be used in cell lines with high transfection efficiency, and may have cross-reactivity toward other miRNAs (Khan et al. 2009). A gene-knockout library circumvents many of these issues, as it can be conducted in a pooled manner, obtain complete inactivation of the miRNA, and be more target-specific; in addition, it would not suffer chemical toxicity associated with modified nucleotides. A miRNA-specific TALEN library has been elegantly designed and constructed (Kim et al. 2013); however, that library targets only 274 human miRNAs (∼15% of the annotated miRNAs) and is not intended for pool screens. The GeCKOv2 library (Sanjana et al. 2014) and our dedicated LX-miR library take advantage of the CRISPR-Cas9 technology and offer a sensitive and robust tool for identifying functional miRNAs. sgRNA screens could also be used to investigate the function of miRNAs in drug resistance, development, viral infection, and a variety of other cellular properties. The relatively small scale of the LX-miR library allows sgRNA representation to be maintained during the screen even in systems such as primary cells or in vivo.
A few considerations should be taken into account when using a gene editing knockout screen to identify functional miRNAs, as the majority of miRNAs (∼76%, Supplemental Fig. S8) overlap with other transcripts or regulatory elements. There could be some primary miRNAs for which sgRNA cleavage affects cellular fitness due to the impact of mutations on the overlapping transcripts or elements, either solely or in addition to the knockout of the miRNA. For example, miR-1226-3p and miR-320a were identified as putative pro-fitness miRNAs in our HeLa screen. The hsa-mir-1226 pre-miRNA is an intron in the putative RNA helicase DHX30 gene and is processed by the splicing machinery without the need for Drosha cleavage. Mutations that affect the processing of this miRNA could therefore also affect the splicing of its host gene, which has been shown to be a conditionally essential gene (Chen et al. 2017). hsa-mir-320a overlaps with the promoter of POLR3D, a subunit of RNA polymerase III and a conditionally essential gene (Chen et al. 2017). The fitness decrease seen after mutations to these miRNAs could be due to knocking out the miRNA or the effect of mutations on these essential genes or a combination of both. In addition, when performing a screen in cancer cell lines like HeLa, sgRNAs may cleave at multiple gene duplication sites resulting in DNA damage response and thus generate false readouts (Aguirre et al. 2016; Munoz et al. 2016). These considerations could also explain how some of the pro-fitness miRNAs identified in our screen have previously been shown to decrease HeLa cell proliferation: let-7a-5p (Wu et al. 2016), miR-139-3p (Huang et al. 2016), miR-143-3p (Liu et al. 2012), and miR-320a (Zhang et al. 2016). However, the discrepancy could also be due to differing effects of knocking out the miRNA completely versus the previous studies which knocked down or overexpressed the miRNA. It is also possible the 5p and the 3p miRNAs play opposite roles, and our screen directly mutated the DNA, which could affect the biogenesis of both 5p and 3p miRNAs. Despite these caveats, CRISPR-Cas9 screening is a useful tool for determining miRNA function and was able to identify several miRNAs that had additional evidence to support their role as fitness-relevant miRNAs.
Our screens identified 10 miRNAs that enhance fitness of HeLa cells and have been reported to be up-regulated in cervical cancer (Table 2). MiR-31 is reported to be pro-proliferative in HeLa cells and oncogenic in cervical cancer (Rao et al. 2012; Wang et al. 2014a; Zheng et al. 2015). We found knocking out miR-151a in HeLa cells caused up-regulation of cyclin-dependent kinase inhibitor p21 and growth disadvantage (JS Kurata and RJ Lin, in prep.). Our screens also identified six miRNAs that enhance fitness in NCI-N87 cells and are up-regulated in gastric cancer, as well as three miRNAs that impair fitness and are down-regulated (Table 3). In the two cell lines examined, no anti-fitness miRNAs were identified in HeLa cells whereas both pro- and anti-fitness miRNAs were identified in NCI-N87 cells. This could be due to ploidy of the cell lines, as HeLa cells are polyploidy (Macville et al. 1999) and NCI-N87 cells are almost diploid (Park et al. 1990). As previously discussed, multiple copies of a sgRNA-target site can result in a DNA damage response and decreased cellular proliferation (Aguirre et al. 2016; Munoz et al. 2016). Another possibility is that the fast growing HeLa cell line has already circumvented the mechanisms by which anti-fitness miRNAs control cell growth, while the slower growing NCI-N87 cell lines may still respond to these anti-fitness miRNAs.
By comparing the pro-fitness miRNAs identified in our HeLa and NCI-N87 screens and the previously reported MV4-11 myeloid leukemia cell pro-fitness miRNAs identified using the GeCKO v2 library (Wallace et al. 2016), a single miRNA, miR-1304-5p, was identified as pro-fitness in all three cell lines. The importance of this finding is not clear, since miR-1304-5p is shown to suppress the growth of non-small cell lung cancer cells (Li et al. 2016a); however, miR-1304 may function differently in other cancers. It is also possible that the targeting sgRNAs mutated an important regulatory region, as the active enhancer mark H3K4Me1 is high in this genomic region. The role of miR-1304 in cancer warrants further investigation.
The developed LX-miR library can also be improved using the latest research into CRISPR-Cas systems. For instance, several known HeLa fitness miRNAs were not identified in our screen, including miR-21 whose knockout in HeLa cells has been shown to decrease the rate of proliferation (Chen et al. 2015a). This could be due to low cleavage efficiency of the selected sgRNAs, which can be improved as more accurate computational models of sgRNA activity are introduced. The CRISPRpred tool for on-target activity prediction has recently been developed and shown to be an improvement over the Azimuth scoring method (Rahman and Rahman 2017). We are also unable to target 287 primary miRNAs partially because there are too few S. pyogenes Cas9-required NGG PAM sequences in these primary miRNAs, so the needed four to five sgRNAs could not be designed. The development of the CRISPR class 2 effector endonuclease Cpf1 recognizing a TTTN PAM sequence (Zetsche et al. 2015) and Staphylococcus aureus Cas9 recognizing a NNGRRT PAM sequence (Ran et al. 2015) could be helpful for targeting primary miRNAs with few GG sequences. The recent engineering of the xCas9 protein which has PAM sequences NG, GAA, and GAT (Hu et al. 2018) broadens the number of possible targets by a single Cas9 and should be able to expand the number of primary miRNAs targeted by the LX-miR library design.
In conclusion, our study indicates that a pooled fitness screen using a focused sgRNA library, combined with expression in the tested cell lines and dysregulation in clinical samples, is fruitful in identifying relevant, novel miRNAs. The developed LX-miR library could also be used to investigate the function of miRNAs in drug resistance, development, viral infection, and a variety of other cellular properties.
MATERIALS AND METHODS
miRNA-targeting sgRNA selection
The genomic location of each primary miRNA stem–loop was downloaded from miRBase v21 (Kozomara and Griffiths-Jones 2014). The stem–loop sequence plus 20 bp on either side was then fetched from the UCSC genome browser using the custom track tool. All possible sgRNAs (20 nt followed by NGG) within these sequences were identified using custom Python scripts. To allow for on-target activity scoring using the Azimuth (Doench et al. 2016) algorithm, the extended (NNNNsgRNANGGNNN) sequence for each sgRNA was fetched from the genome using the Ensembl REST API (Yates et al. 2015). The extended sgRNAs were then scored using the Azimuth API. The off-target activity of the sgRNAs was scored using the CRISPR design tool (mit.crispr.edu; [Hsu et al. 2013]). The number of exact matches in the genome for each sgRNA was determined by aligning the sgRNA sequence plus NGG to the human genome (hg19) using bowtie (Langmead et al. 2009) under settings which return all exact matches (-v 1 -a).
The sgRNAs were examined for identical seed (12 bp closest to the PAM) sequences. If multiple sgRNAs targeting the same miRNA had identical seed sequences, only the one with the highest Azimuth score was considered. The top five sgRNAs by Azimuth score per miRNA were then selected for inclusion in the pool. We then selected 1000 distinct control sgRNAs from previously published libraries (Sanjana et al. 2014; Shalem et al. 2014; Wang et al. 2014b, 2015). These sgRNAs were also aligned to the genome using bowtie to ensure no target sites were found (bowtie -v 0 -a). The 1000 control sgRNAs were then selected from among those which had no alignments. All of the code used to design the library can be found at https://github.com/jkurata/LX-miR_Design. Information about every miRNA-targeting sgRNA, including the scoring information and oligo designs, can be accessed at www.mirnalibrary.rjlinlab.com.
Vectors
pLX-sgRNA (Addgene plasmid # 50662) and pCW-Cas9 (Addgene plasmid # 50661) were a gift from Eric Lander and David Sabatini (Wang et al. 2014b). lentiCRISPRv2 was a gift from Feng Zhang (Addgene plasmid # 52961) (Sanjana et al. 2014).
We modified pLX-sgRNA to allow sgRNA insertion by Gibson assembly. Two BfuAI sites separated by 40 bp were inserted into pLX-sgRNA using overlap-extension PCR as described in the provided pLX-sgRNA protocol. In brief, the U6 promoter or the sgRNA scaffold containing regions of the vector were amplified using primers with BfuAI tails (pLX-F1 and pLX-R1-BfuAI or pLX-F2-BfuAI and pLX-R2; Supplemental Table S9). The two PCR products were then combined and a third round of PCR was performed using the pLX-F1 and pLX-R2 primers to amplify the entire region. The PCR product was then ligated between the XhoI and NheI sites of the pLX-sgRNA vector. To enable easy distinction between digested and undigested vector, the 2 kb stuffer from plentiCRISPR v2 was amplified by PCR using plenti-2k primers (Supplemental Table S9) and inserted between the BfuAI sites of the PCR linearized pLX-sgRNA-BfuAI vector (pLX-lin primers; Supplemental Table S9) using InFusion Cloning (Clontech) to create the pLX-sgRNA-BfuAI-2k vector.
LX-miR library cloning
For every sgRNA in the miRNA-targeting library, tails were added to allow for PCR amplification and Gibson assembly. The first nucleotide of the sgRNA was also replaced with G to allow for efficient RNA Pol III transcription. 85-mer ssDNA oligos of the form TCTTGTGGAAAGGACGAAACACCGX19GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCG were ordered from Custom Array Inc. The oligos (1 µL) were amplified using CloneAmp HiFi PCR Premix (TaKaRa) for 20 cycles with an annealing temperature of 56.5°C using pLX-gibson primers (Supplemental Table S9).
The PCR product was then gel purified using the NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel). Gibson assembly was performed using Gibson Assembly Master Mix (NEB) with gel purified BfuAI digested pLX-sgRNA-BfuAI-2k and the purified sgRNA PCR at a 1:10 vector to insert ratio. Fifteen transformations of 0.5 µL of the Gibson assembly product into 25 µL Endura ElectroCompetent cells (Lucigen) were performed. The transformation product was then plated on LB agar plus ampicillin (100 µg/mL) plates and incubated at 30°C for 16 h. Enough colonies for ∼50-fold representation of the library were collected by washing the plates with LB. Plasmid DNA was purified using the NucleoBond Xtra Maxi kit (Macherey-Nagel). To produce additional plasmid DNA, 2 µL pLX-miR plasmid (50 ng/µL) was transformed into Endura ElectroCompetent cells (Lucigen), diluted 1:100 and grown as above. After every transformation, the sgRNA frequency in the plasmid pool was determined by deep sequencing and the pool was verified to contain over 99.9% of the library sgRNAs.
Lentivirus production and transduction
To produce lentiviral particles, 10 cm plates of 293T cells were transfected with 15 µg lentiviral vector, 15 µg pC-GP-2, 5 µg pCMV-rev2, and 5 µg pCMV-G per plate using calcium phosphate transfection. After 6 h, 60 µL 0.6 M sodium butyrate was added to each plate. Medium was collected 72 h after transfection, centrifuged at 400g for 5 min and sterile filtered to remove any cells. The lentivirus was precipitated by incubating in 10% PEG at 4°C overnight. The precipitate was then collected by centrifugation at 2000g for 30 min and stored at −80°C.
For transduction, 50 µL lentivirus and 1.6 µL polybrene (10 mg/mL) were added to cells in 4 mL fresh medium. The virus was removed and medium changed after 24 h. Selection with the appropriate antibiotic was started 48 h after transduction.
Cell culture and HeLa-Cas9/NCI-N87-Cas9 creation
HeLa cells were grown in DMEM with 10% FBS at 37°C and 5% CO2. NCI-N87 cells were grown in RPMI with 10% FBS at 37°C and 5% CO2. To create the HeLa-Cas9 and NCI-N87-Cas9 cell lines, HeLa or NCI-N87 cells were transduced with pCW-Cas9 lentivirus. After a week (HeLa) or 2 wk (NCI-N87) of puromycin selection (1 µg/mL; InvivoGen), single colonies were selected and expanded. The HeLa-Cas9 or NCI-N87-Cas9 clone with no detectable background expression and the highest Cas9 expression level by western blot after induction was used for all experiments.
Western blot
Total cellular lysate was created by adding 100 µL Laemmli buffer per 106 cells. The lysate was then boiled for 5 min and either used directly or flash frozen and stored at −80°C. Ten or 20 µL of protein was separated by size on a SDS-PAGE gel and then transferred to a PVDF membrane. The membranes were blocked overnight at 4°C using 5% nonfat milk in 1x TBST and incubated with primary at room temperature for 1 h. The dilutions and primary antibodies used were 1:5000 mouse α-FLAG (GenScript A00187) and 1:5000 mouse α-GAPDH (Invitrogen 398600). The dilutions and secondary antibody used was 1:20,000 IRDye 800CW goat α-mouse (LiCor). All primary and secondary antibodies were diluted in 5% nonfat milk in 1× TBST. Membranes were imaged using an Odyssey imaging system (LiCor).
LX-miR screen
Ten plates of HeLa-Cas9 or NCI-N87-Cas9 cells were transduced with LX-miR lentivirus and selected using 5 µg/mL blasticidin (InvivoGen). The cells from all plates were combined and split every 2 to 3 d. After 7 d of selection, at least 1.6 million cells were collected to measure the frequency of each sgRNA in the initial pool. On day 8, 8 plates were treated with 2 µg/mL doxycycline. The cells were then grown for 14 cell duplication, 14 d for HeLa cells or 28 d for NCI-N87 cells, with all of the dox treated or untreated cells being collected and split every 2 to 3 d. Genomic DNA was extracted from 1–5 × 106 cells using DNeasy Blood and Tissue (Qiagen) and the provided protocol.
LX-miR deep sequencing
To determine the distribution of sgRNAs in the pool, the sgRNA region was amplified from the genomic DNA using Herculase II Fusion DNA Polymerase (Agilent). We used a nested PCR strategy adapted from Shalem et al. (2014) to increase the specificity of this PCR. To maintain 200× representation of our library, we used over 1.6 million genomic equivalents or 10.6 µg of genomic DNA in PCR1. Since too much DNA inhibits the PCR reaction, 24 50 µL reactions with 500 ng genomic DNA each were performed for PCR1 using the lentiC-dseq-2-F1 and pLX-dseq-R1 primers (Supplemental Table S9). After 25 cycles of PCR1 with an annealing temperature of 55°C, 2 µL of each PCR1 reaction were combined and 1 µL of the combined product was used per PCR2 reaction. Five PCR2 reactions were performed per sample for 10 cycles to add illumina adaptors using the lentiC-dseq-F1 and pLX-dseq-R2 primers (Supplemental Table S9). The PCR product was then purified using the illustra GFX PCR DNA and Gel Band Purification Kit (GE Healthcare Life Sciences). Deep sequencing was performed using HiSeq 2500 (Illumina) and produced 10 million to 20 million single end 50 bp reads per sample.
LX-miR data analysis
The raw fastq files were processed by removing the conserved vector sequences (5′: TGTGGAAAGGACGAAACACC; 3′: GTTTTAG) on either side of the sgRNA using Btrim (Kong 2011). Only those reads which were at least 16 nt long after trimming were kept (-l 16). The target sequences were then aligned to an index constructed from the sgRNAs sequences in the pLX-miR pool using Bowtie (Langmead et al. 2009) under settings which allow for a single mismatch between the expected sgRNA sequence and the read (-v 1). The number of aligned reads per sgRNA was then counted using a custom Python script.
sgRNA median ratio normalization
The number of reads per sgRNA were normalized using median ratio normalization (Li et al. 2014). This normalization takes the geometric mean of the read counts for each sgRNA across samples () and divides the read counts for that sgRNA in a specific sample to get a ratio.
The median of all of those ratios for all sgRNAs within a given sample is the size factor (sj) for that sample and all read counts within that sample are divided by the size factor to get the normalized read count ().
miRNA level analysis
Two different analysis methods Model-based Analysis of Genome-wide CRISPR/Cas9 Knockout (MAGeCK) (Li et al. 2014) and Redundant siRNA Activity (RSA) (Konig et al. 2007) were used to determine which miRNAs had significant impacts on fitness. MAGeCK was run using “mageck test -k <file name> -t <+dox sample name> -c <control sample names> -n <output prefix>”. RSA analysis requires fold change for each sgRNA to be calculated. Therefore, we median ratio normalized the data and added one to prevent errors when calculating fold change. The fold enrichment between the induced and the average of the uninduced and initial read counts was calculated for each sgRNA. RSA was run using “python RSA.py -o <output file> -s <column name> <enrichment file>” to determine which miRNAs had sgRNA decrease in the pool and “python RSA.py -r -l 1 -u 100 -o <output file> -s <column name> <enrichment file>” to determine which miRNAs had sgRNAs increase in the pool. Those miRNAs with P-value <0.05 were considered significant.
miRNA-sequencing
Total RNA, including miRNAs, was extracted from 106 HeLa-Cas9 LX-miR day 7 replicate 1, day 7 replicate 2, day 22 +dox replicate 1, and day 22 -dox replicate 1 cells using the miRNeasy Mini Kit (QIAGEN). For NCI-N87, RNA was extracted from NCI-N87 parental cells. 500 ng of RNA was prepared for sequencing using the Illumina protocol with minor modifications. In brief, small RNA libraries are prepared by 3′ adapter ligation, 5′ RT primer annealing, 5′ adapter ligation, reverse transcription, and PCR amplification. Libraries were then pooled in batches of 12 samples in equal amounts and clustered with a concentration of 10.5 pmol in one lane each of a single read flowcell using the cBot (Illumina). Sequencing of 50 cycles was performed on a HiSeq 2500 (Illumina). Demultiplexing of the raw sequencing data and generation of the fastq files were done using CASAVA v.1.8.2. The reads were then trimmed to remove the first 3 bases and the Illumina adaptor sequences using Btrim (Kong 2011) (-l 16 -3 -P -f 3). The trimmed reads were then aligned to hg38 using Bowtie (Langmead et al. 2009) under settings which allow for a single mismatch between the genome and the read (-v 1). The read counts per mature miRNA were determined using a custom Python script. The raw reads were read-per-million normalized using the formula: Raw Reads/Total Reads × 1,000,000 + 1 = Reads per million.
Differentially expressed tumor versus normal miRNAs
The read counts per miRNA of GSE20592 cervical cancer data set were generated by Witten et al. (2010) and downloaded from GEO (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE20592; 10/12/2016). The gastric cancer miRNA read counts (isoform expression quantification) were generated by the TCGA Research Network (http://cancergenome.nih.gov/) and processed using custom Python scripts. DESeq2 (Love et al. 2014) was used to determine which miRNAs are differentially expressed between tumor and normal samples.
DATA DEPOSITION
Raw and processed miRNA-sequencing data have been deposited in the NCBI Gene Expression Omnibus (GEO accession number GSE110784). Information about miRNA-targeting sgRNAs can be found at www.mirnalibrary.rjlinlab.com. The code used to construct the LX-miR library is available on GitHub (https://github.com/jkurata/LX-miR_Design).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jiing-Kuan Yee of City of Hope for plasmids pC-GP-2, pCMV-rev2, and pCMV-G; Dustin Schones and Xiwei Wu of City of Hope for help with computation and bioinformatics; and members of the Lin laboratory and J.S.K.’s dissertation committee for valuable suggestions and comments. J.S.K. was supported in part by the H.N. & Frances Berger Foundation Fellowship. This work was supported by Beckman Research Institute grants to R.J.L. and by National Institutes of Health grant P30-CA033572 for shared research core facilities at City of Hope.
Author contributions: J.S.K. and R.J.L. conceived the project and designed experiments; J.S.K. performed experiments; J.S.K. and R.J.L. analyzed the data and wrote the paper.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.066282.118.
REFERENCES
- Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, et al. 2016. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167: 1867–1882.e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang CZ, Ben-David U, Cook A, Ha G, Harrington WF, Doshi MB, et al. 2016. Genomic copy number dictates a gene-independent cell response to CRISPR/Cas9 targeting. Cancer Discov 6: 914–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezikov E, Chung WJ, Willis J, Cuppen E, Lai EC. 2007. Mammalian mirtron genes. Mol Cell 28: 328–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein E, Caudy AA, Hammond SM, Hannon GJ. 2001. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409: 363–366. [DOI] [PubMed] [Google Scholar]
- Bischoff A, Bayerlová M, Strotbek M, Schmid S, Beissbarth T, Olayioye MA. 2015. A global microRNA screen identifies regulators of the ErbB receptor signaling network. Cell Commun Signal 13: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogenhagen DF, Brown DD. 1981. Nucleotide sequences in Xenopus 5S DNA required for transcription termination. Cell 24: 261–270. [DOI] [PubMed] [Google Scholar]
- Bohnsack MT, Czaplinski K, Görlich D. 2004. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA 10: 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H, Yi B, Ma R, Zhang X, Zhao H, Xi Y. 2016. CRISPR/cas9, a novel genomic tool to knock down microRNA in vitro and in vivo. Sci Rep 6: 22312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Chen X, Wu X, Wang X, Wang Y, Lin TY, Kurata J, Wu J, Vonderfecht S, Sun G, et al. 2015a. Disruption of microRNA-21 by TALEN leads to diminished cell transformation and increased expression of cell–environment interaction genes. Cancer Lett 356: 506–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Sanjana NE, Zheng K, Shalem O, Lee K, Shi X, Scott DA, Song J, Pan JQ, Weissleder R, et al. 2015b. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 160: 1246–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WH, Lu G, Chen X, Zhao XM, Bork P. 2017. OGEE v2: an update of the online gene essentiality database with special focus on differentially essential genes in human cancer cell lines. Nucleic Acids Res 45: D940–D944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung TH, Man KNM, Yu MY, Yim SF, Siu NSS, Lo KWK, Doran G, Wong RRY, Wang VW, Smith DI, et al. 2012. Dysregulated microRNAs in the pathogenesis and progression of cervical neoplasm. Cell Cycle 11: 2876–2884. [DOI] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim JM, Kim JS. 2013. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31: 230–232. [DOI] [PubMed] [Google Scholar]
- Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, Ma E, Mane S, Hannon GJ, Lawson ND, et al. 2010. A novel miRNA processing pathway independent of dicer requires argonaute2 catalytic activity. Science 328: 1694–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datlinger P, Rendeiro AF, Schmidl C, Krausgruber T, Traxler P, Klughammer J, Schuster LC, Kuchler A, Alpar D, Bock C. 2017. Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods 14: 297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, et al. 2016. Perturb-Seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 167: 1853–1866.e1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. 2016. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34: 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo CH, Rootes CL, Cowley K, Marsh GA, Gould CM, Deffrasnes C, Cowled CJ, Klein R, Riddell SJ, Middleton D, et al. 2016. Dual microRNA screens reveal that the immune-responsive miR-181 promotes henipavirus entry and cell–cell fusion. PLoS Pathog 12: e1005974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KKH, Burge CB, Bartel DP. 2008. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedeke L, Rotllan N, Canfran-Duque A, Aranda JF, Ramirez CM, Araldi E, Lin C-S, Anderson NN, Wagschal A, De Cabo R, et al. 2015. MicroRNA-148a regulates LDL receptor and ABCA1 expression to control circulating lipoprotein levels. Nat Med 21: 1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, Sohn SY, Cho Y, Zhang BT, Kim VN. 2006. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell 125: 887–901. [DOI] [PubMed] [Google Scholar]
- Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown Kevin R, Macleod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, et al. 2015. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 163: 1515–1526. [DOI] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. 2013. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31: 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N, Zeina CM, Gao X, Rees HA, Lin Z, et al. 2018. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556: 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Xi J, Liu S. 2016. MiR-139–3p induces cell apoptosis and inhibits metastasis of cervical cancer by targeting NOB1. Biomed Pharmacother 83: 850–856. [DOI] [PubMed] [Google Scholar]
- Hutvágner G, Mclachlan J, Pasquinelli AE, Bálint É, Tuschl T, Zamore PD. 2001. A cellular function for the RNA-interference enzyme dicer in the maturation of the let-7 small temporal RNA. Science 293: 834–838. [DOI] [PubMed] [Google Scholar]
- Ivey KN, Srivastava D. 2010. MicroRNAs as regulators of differentiation and cell fate decisions. Cell Stem Cell 7: 36–41. [DOI] [PubMed] [Google Scholar]
- Jaitin DA, Weiner A, Yofe I, Lara-Astiaso D, Keren-Shaul H, David E, Salame TM, Tanay A, Van Oudenaarden A, Amit I. 2016. Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-seq. Cell 167: 1883–1896.e1815. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Meng X, Meng L, Chang N, Xiong J, Cao H, Liang Z. 2014. Small indels induced by CRISPR/Cas9 in the 5′ region of microRNA lead to its depletion and Drosha processing retardance. RNA Biol 11: 1243–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Zhou K, Ma L, Gressel S, Doudna JA. 2015. A Cas9–guide RNA complex preorganized for target DNA recognition. Science 348: 1477–1481. [DOI] [PubMed] [Google Scholar]
- Jiang F, Taylor DW, Chen JS, Kornfeld JE, Zhou K, Thompson AJ, Nogales E, Doudna JA. 2016. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science 351: 867–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. 2013. RNA-programmed genome editing in human cells. eLife 2: e00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat V, Paluru P, Myint M, French DL, Gadue P, Diamond SL. 2014. MicroRNA screen of human embryonic stem cell differentiation reveals miR-105 as an enhancer of megakaryopoiesis from adult CD34+ cells. Stem Cells 32: 1337–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AA, Betel D, Miller ML, Sander C, Leslie CS, Marks DS. 2009. Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs. Nat Biotechnol 27: 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Wee G, Park J, Kim J, Baek D, Kim JS, Kim VN. 2013. TALEN-based knockout library for human microRNAs. Nat Struct Mol Biol 20: 1458–1464. [DOI] [PubMed] [Google Scholar]
- Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera MDC, Yusa K. 2014. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol 32: 267–273. [DOI] [PubMed] [Google Scholar]
- Kong Y. 2011. Btrim: a fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics 98: 152–153. [DOI] [PubMed] [Google Scholar]
- Konig R, Chiang CY, Tu BP, Yan SF, Dejesus PD, Romero A, Bergauer T, Orth A, Krueger U, Zhou Y, et al. 2007. A probability-based approach for the analysis of large-scale RNAi screens. Nat Methods 4: 847–849. [DOI] [PubMed] [Google Scholar]
- Kooistra SM, Nørgaard LCR, Lees MJ, Steinhauer C, Johansen JV, Helin K. 2014. A screen identifies the oncogenic micro-RNA miR-378a-5p as a negative regulator of oncogene-induced senescence. PLoS One 9: e91034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. 2014. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42: D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurata M, Rathe SK, Bailey NJ, Aumann NK, Jones JM, Veldhuijzen GW, Moriarity BS, Largaespada DA. 2016. Using genome-wide CRISPR library screening with library resistant DCK to find new sources of Ara-C drug resistance in AML. Sci Rep 6: 36199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajer CB, Garnæs E, Friis-Hansen L, Norrild B, Therkildsen MH, Glud M, Rossing M, Lajer H, Svane D, Skotte L, et al. 2012. The role of miRNAs in human papilloma virus (HPV)-associated cancers: bridging between HPV-related head and neck cancer and cervical cancer. Br J Cancer 106: 1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam LT, Lu X, Zhang H, Lesniewski R, Rosenberg S, Semizarov D. 2010. A microRNA screen to identify modulators of sensitivity to BCL2 inhibitor ABT-263 (Navitoclax). Mol Cancer Ther 9: 2943–2950. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, et al. 2003. The nuclear RNase III Drosha initiates microRNA processing. Nature 425: 415–419. [DOI] [PubMed] [Google Scholar]
- Lee EJ, Baek M, Gusev Y, Brackett DJ, Nuovo GJ, Schmittgen TD. 2008a. Systematic evaluation of microRNA processing patterns in tissues, cell lines, and tumors. RNA 14: 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Choi CH, Choi JJ, Park YA, Kim SJ, Hwang SY, Kim WY, Kim TJ, Lee JH, Kim BG, et al. 2008b. Altered microRNA expression in cervical carcinomas. Clin Cancer Res 14: 2535–2542. [DOI] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love M, Zhang F, Irizarry R, Liu J, Brown M, Liu X. 2014. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15: 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CG, Pu MF, Li CZ, Gao M, Liu MX, Yu CZ, Yan H, Peng C, Zhao Y, Li Y, et al. 2016a. MicroRNA-1304 suppresses human non-small cell lung cancer cell growth in vitro by targeting heme oxygenase-1. Acta Pharmacol Sin 38: 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Wang W, Xu B, Gong H. 2016b. miR-196b regulates gastric cancer cell proliferation and invasion via PI3K/AKT/mTOR signaling pathway. Oncol Lett 11: 1745–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Yu X, Guo X, Tian Z, Su M, Long Y, Huang C, Zhou F, Liu M, Wu X, et al. 2012. miR-143 is downregulated in cervical cancer and promotes apoptosis and inhibits tumor formation by targeting Bcl-2. Mol Med Rep 5: 753–760. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, et al. 2005. MicroRNA expression profiles classify human cancers. Nature 435: 834–838. [DOI] [PubMed] [Google Scholar]
- Macville M, Schröck E, Padilla-Nash H, Keck C, Ghadimi BM, Zimonjic D, Popescu N, Ried T. 1999. Comprehensive and definitive molecular cytogenetic characterization of HeLa cells by spectral karyotyping. Cancer Res 59: 141–150. [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, Dicarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339: 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez I, Gardiner AS, Board KF, Monzon FA, Edwards RP, Khan SA. 2007. Human papillomavirus type 16 reduces the expression of microRNA-218 in cervical carcinoma cells. Oncogene 27: 2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki H, Kassavetis GA, Geiduschek EP. 1994. Analysis of RNA chain elongation and termination by Saccharomyces cerevisiae RNA polymerase III. J Mol Biol 235: 1173–1192. [DOI] [PubMed] [Google Scholar]
- Munoz DM, Cassiani PJ, Li L, Billy E, Korn JM, Jones MD, Golji J, Ruddy DA, Yu K, Mcallister G, et al. 2016. CRISPR screens provide a comprehensive assessment of cancer vulnerabilities but generate false-positive hits for highly amplified genomic regions. Cancer Discov 6: 900–913. [DOI] [PubMed] [Google Scholar]
- Nana-Sinkam SP, Croce CM. 2014. MicroRNA regulation of tumorigenesis, cancer progression and interpatient heterogeneity: towards clinical use. Genome Biol 15: 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu H, Ran FA, Hsu Patrick D, Konermann S, Shehata Soraya I, Dohmae N, Ishitani R, Zhang F, Nureki O. 2014. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156: 935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JG, Frucht H, Larocca RV, Bliss DP, Kurita Y, Chen TR, Henslee JG, Trepel JB, Jensen RT, Johnson BE, et al. 1990. Characteristics of cell lines established from human gastric carcinoma. Cancer Res 50: 2773–2780. [PubMed] [Google Scholar]
- Parnas O, Jovanovic M, Eisenhaure Thomas M, Herbst Rebecca H, Dixit A, Ye Chun J, Przybylski D, Platt Randall J, Tirosh I, Sanjana Neville E, et al. 2015. A genome-wide CRISPR screen in primary immune cells to dissect regulatory networks. Cell 162: 675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira PM, Marques JP, Soares AR, Carreto L, Santos MA. 2010. MicroRNA expression variability in human cervical tissues. PLoS One 5: e11780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman MK, Rahman MS. 2017. CRISPRpred: a flexible and efficient tool for sgRNAs on-target activity prediction in CRISPR/Cas9 systems. PLoS One 12: e0181943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, et al. 2015. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520: 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Q, Zhou H, Peng Y, Li J, Lin Z. 2012. Aberrant microRNA expression in human cervical carcinomas. Med Oncol 29: 1242–1248. [DOI] [PubMed] [Google Scholar]
- Rashid F, Awan H, Shah A, Chen L, Shan G. 2017. Induction of miR-3648 upon ER stress and its regulatory role in cell proliferation. Int J Mol Sci 18: 1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby JG, Jan CH, Bartel DP. 2007. Intronic microRNA precursors that bypass Drosha processing. Nature 448: 83–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F. 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11: 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, Hornung V. 2016. A Genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J Biol Chem 291: 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt T, Schmid-Burgk JL, Hornung V. 2015. Synthesis of an arrayed sgRNA library targeting the human genome. Sci Rep 5: 14987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343: 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Zhang Y, Yu S, Yuan Y, Zhong Y, Lu J, Feng J. 2015. MicroRNA-339, an epigenetic modulating target is involved in human gastric carcinogenesis through targeting NOVA1. FEBS Lett 589: 3205–3211. [DOI] [PubMed] [Google Scholar]
- Shi DB, Wang YW, Xing AY, Gao JW, Zhang H, Guo XY, Gao P. 2015. C/EBPα-induced miR-100 expression suppresses tumor metastasis and growth by targeting ZBTB7A in gastric cancer. Cancer Lett 369: 376–385. [DOI] [PubMed] [Google Scholar]
- Shim BS, Wu W, Kyriakis CS, Bakre A, Jorquera PA, Perwitasari O, Tripp RA. 2016. MicroRNA-555 has potent antiviral properties against poliovirus. J Gen Virol 97: 659–668. [DOI] [PubMed] [Google Scholar]
- Smith JL, Jeng S, Mcweeney SK, Hirsch AJ. 2017. A microRNA screen identifies the Wnt signaling pathway as a regulator of the interferon response during flavivirus infection. J Virol 91: e02388-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontheimer EJ. 2005. Assembly and function of RNA silencing complexes. Nat Rev Mol Cell Biol 6: 127. [DOI] [PubMed] [Google Scholar]
- Sun YM, Lin KY, Chen YQ. 2013. Diverse functions of miR-125 family in different cell contexts. J Hematol Oncol 6: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang R, Yang C, Ma X, Wang Y, Luo D, Huang C, Xu Z, Liu P, Yang L. 2016. MiR-let-7a inhibits cell proliferation, migration, and invasion by down-regulating PKM2 in gastric cancer. Oncotarget 7: 5972–5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao L, Zhang J, Meraner P, Tovaglieri A, Wu X, Gerhard R, Zhang X, Stallcup WB, Miao J, He X, et al. 2016. Frizzled proteins are colonic epithelial receptors for C. difficile toxin B. Nature 538: 350–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM. 2006. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev 20: 2202–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai KW, Hu LY, Wu CW, Li SC, Lai CH, Kao HW, Fang WL, Lin WC. 2010. Epigenetic regulation of miR-196b expression in gastric cancer. Genes Chromosomes Cancer 49: 969–980. [DOI] [PubMed] [Google Scholar]
- Tsai MM, Wang CS, Tsai CY, Chen CY, Chi HC, Tseng YH, Chung PJ, Lin YH, Chung IH, Chen CY, et al. 2014. MicroRNA-196a/-196b promote cell metastasis via negative regulation of radixin in human gastric cancer. Cancer Lett 351: 222–231. [DOI] [PubMed] [Google Scholar]
- Ventura A, Jacks T. 2009. MicroRNAs and cancer: short RNAs go a long way. Cell 136: 586–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et al. 2006. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci 103: 2257–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace J, Hu R, Mosbruger TL, Dahlem TJ, Stephens WZ, Rao DS, Round JL, O'Connell RM. 2016. Genome-wide CRISPR-Cas9 screen identifies microRNAs that regulate myeloid leukemia cell growth. PLoS One 11: e0153689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Tang S, Le SY, Lu R, Rader JS, Meyers C, Zheng ZM. 2008. Aberrant expression of oncogenic and tumor-suppressive microRNAs in cervical cancer is required for cancer cell growth. PLoS One 3: e2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zheng X, Zhang Z, Zhou J, Zhao G, Yang J, Xia L, Wang R, Cai X, Hu H, et al. 2012. MicroRNA-149 inhibits proliferation and cell cycle progression through the targeting of ZBTB2 in human gastric cancer. PLoS One 7: e41693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Zhou Y, Zheng L, Li H. 2014a. MiR-31 is an independent prognostic factor and functions as an oncomir in cervical cancer via targeting ARID1A. Gynecol Oncol 134: 129–137. [DOI] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, Lander ES. 2014b. Genetic screens in human cells using the CRISPR-Cas9 system. Science 343: 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, Sabatini DM. 2015. Identification and characterization of essential genes in the human genome. Science 350: 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilting SM, Snijders PJF, Verlaat W, Jaspers A, Van De Wiel MA, Van Wieringen WN, Meijer GA, Kenter GG, Yi Y, Le Sage C, et al. 2012. Altered microRNA expression associated with chromosomal changes contributes to cervical carcinogenesis. Oncogene 32: 106. [DOI] [PubMed] [Google Scholar]
- Witten D, Tibshirani R, Gu SG, Fire A, Lui W-O. 2010. Ultra-high throughput sequencing-based small RNA discovery and discrete statistical biomarker analysis in a collection of cervical tumours and matched controls. BMC Biol 8: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Chen X, Peng R, Liu H, Yin P, Peng H, Zhou Y, Sun Y, Wen L, Yi H, et al. 2016. Let-7a suppresses cell proliferation via the TGF-β/SMAD signaling pathway in cervical cancer. Oncol Rep 36: 3275–3282. [DOI] [PubMed] [Google Scholar]
- Yang J-S, Maurin T, Robine N, Rasmussen KD, Jeffrey KL, Chandwani R, Papapetrou EP, Sadelain M, O'Carroll D, Lai EC. 2010. Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proc Natl Acad Sci 107: 15163–15168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Gong Y, Wang Q, Wang Y, Zhang X. 2015. The role of miR-100-mediated Notch pathway in apoptosis of gastric tumor cells. Cell Signal 27: 1087–1101. [DOI] [PubMed] [Google Scholar]
- Yarbrough ML, Zhang K, Sakthivel R, Forst CV, Posner BA, Barber GN, White MA, Fontoura BMA. 2014. Primate-specific miR-576–3p sets host defense signalling threshold. Nat Commun 5: 4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates A, Beal K, Keenan S, Mclaren W, Pignatelli M, Ritchie GRS, Ruffier M, Taylor K, Vullo A, Flicek P. 2015. The ensembl REST API: ensembl data for any language. Bioinformatics 31: 143–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi R, Qin Y, Macara IG, Cullen BR. 2003. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev 17: 3011–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, Van der oost J, Regev A, et al. 2015. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163: 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Nie Y, Du Y, Cao J, Shen B, Li Y. 2012. MicroRNA-181a promotes gastric cancer by negatively regulating tumor suppressor KLF6. Tumor Biol 33: 1589–1597. [DOI] [PubMed] [Google Scholar]
- Zhang T, Zou P, Wang T, Xiang J, Cheng J, Chen D, Zhou J. 2016. Down-regulation of miR-320 associated with cancer progression and cell apoptosis via targeting Mcl-1 in cervical cancer. Tumor Biol 37: 8931–8940. [DOI] [PubMed] [Google Scholar]
- Zheng W, Liu Z, Zhang W, Hu X. 2015. miR-31 functions as an oncogene in cervical cancer. Arch Gynecol Obstet 292: 1083–1089. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhu S, Cai C, Yuan P, Li C, Huang Y, Wei W. 2014. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 509: 487–491. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.