

Abstract
Signaling by H2S is proposed to occur via persulfidation, a posttranslational modification of cysteine residues (RSH) to persulfides (RSSH). Persulfidation provides a framework for understanding the physiological and pharmacological effects of H2S. Due to the inherent instability of persulfides, their chemistry is understudied. In this review, we discuss the biologically relevant chemistry of H2S and the enzymatic routes for its production and oxidation. We cover the chemical biology of persulfides and the chemical probes for detecting them. We conclude by discussing the roles ascribed to protein persulfidation in cell signaling pathways.
Graphical abstract

1. INTRODUCTION
Hydrogen sulfide (H2S) is inextricably tied to the emergence of life on Earth. The recent discovery of unanalyzed samples from Miller’s 1958 experiment confirmed that sulfur-containing molecules (including the amino acids cysteine and methionine) could have been formed under the atmospheric conditions of early Earth from H2S, which is released in volcanic emissions and from other geothermal activity.1 It is postulated that RNA, protein, and lipid precursors have common origins in a cyanosulfidic protometabolism.2 It is possible to create nucleic acid precursors on metal centers starting with hydrogen cyanide, H2S, and ultraviolet light. Furthermore, the conditions that produced nucleic acid precursors likely also created the starting materials for natural amino acids and lipids suggesting that a simple set of reactions could have given rise to most of life’s building blocks.2
Early life forms likely thrived in an H2S-rich environment. H2S would have been useful for synthetic purposes but also as a source of metabolic energy. One of the first reported examples of lithothrophy, i.e., the ability to utilize inorganic substrates for energy generation, is of the H2S-oxidizing bacterium Beggiatoa, discovered by Winogradsky.3 In this organism, H2S provides the reducing power for CO2 fixation via the Calvin cycle. Furthermore, green and purple sulfur bacteria use H2S as an electron donor for photosynthetic CO2 reduction. High levels of H2S are lethal to most animals, but a few like pupfish, poeciliids, molluscs, and giant tubeworms are specialized to flourish in H2S-rich habitats like marshes and deep-sea hydrothermal vents.
In medicine, perhaps the earliest reference to H2S, even before its identity was established, was in a 1713 publication titled De Morbis Artificum Diatribes (Disease of Workers) by the Italian physician, Bernardino Ramazzini.4 He described an occupation hazard that manifested as a painful inflammation in the eye in workers who were chronically exposed to an unknown “acidic vapor” while cleaning privies and cesspits. Ramazzini also noted that this acidic vapor was responsible for coating silver and copper coins in the pockets of ill workers with a black substance (presumably silver sulfide and copper sulfide).4 While experimenting with pyrite ore (FeS2) and mineral acid, the Swedish pharmacist Carl Wilhelm Scheele generated H2S, which he described as “sulfur air” (Schwefelluft) in 1777.5
The historical reputation of H2S as a poisonous gas endured until 1996, when Abe and Kimura first demonstrated that H2S plays a role as an endogenous neuromodulator.6 Kimura’s group was also the first to report that H2S acts as a smooth muscle relaxant,7 although the beneficial effects of H2S on blood vessels had been known for a while. Thus, several Russian publications in the 1960s reported the beneficial effects of H2S baths on coronary vasodilation and peripheral blood circulation after reconstructive operations on major arteries,8–10 and the effect of H2S on isolated rabbit aorta was reported by Kruszina and colleagues in 1985.11 The vasodilatory property of endogenously generated H2S was demonstrated in mice lacking γ-cystathionine (CSE),12 as they developed profound age-related hypertension, with some parallels to another gas signaling molecule, nitric oxide (NO•). These early observations propelled the current explosion of research on H2S biology and signaling.
Another serendipitous discovery that put H2S in the spotlight was the report that exposure of mice to subtoxic H2S levels (20–80 ppm) decreased energy expenditure within a few minutes and induced a suspended animation-like state.13 The body temperature dropped by almost 20 °C, and the respiration rate decreased to 10% of normal. Remarkably, these effects were completely reversible, and the animals showed no apparent deficits upon recovery.13 This observation has spurred interest in the potential therapeutic development of H2S to “buy time” for treating trauma patients.14
The past decade has witnessed a burgeoning literature on the physiological effects of H2S and its role in many disease states, which are covered in several excellent reviews.15–17 The proposal that signaling by H2S involves postranslational modification of cysteine residues (i.e., Cys-SSH) provided a framework for understanding its physiological and pharmacological effects.18,19 Protein persulfidation (erroneously described as sulfhydration) is also involved in biosynthetic pathways that require sulfur transfer, e.g., iron–sulfur clusters, biotin, thiamine, lipoic acid, molybdopterin, and sulfur-containing bases in RNA. The presence of the persulfide modification at a proteomic level was first examined only recently.18–20
Due to its inherent instability, persulfide chemistry remains understudied. In this review, we introduce the biologically relevant chemistry of H2S, cover the enzymatic routes for its production and oxidation, discuss the chemical biology of persulfides and review progress on the development of chemical probes for persulfide labeling and visualization. We conclude by discussing how persulfidation can control protein function and cell signaling pathways.
2. CHEMICAL PROPERTIES OF H2S
H2S is a flammable gas with the smell of rotten eggs. The water–H2S system strictly obeys Henry’s law.21–23 Some basic physicochemical properties of H2S are given in Table 1. H2S is a highly toxic gas. The human nose is considered to be one of the most sensitive H2S sensors with a detection threshold of 0.02–0.03 ppm.24,25 At 10 ppm, H2S leads to eye soreness;26 20 ppm is the maximal allowable concentration for a daily 8 h exposure,27 while exposure to 50 ppm of H2S lead to conjunctival and mild respiratory irritation.24,27,28 At 100 ppm, H2S leads to olfactory loss within 3–15 min,27 150 ppm to olfactory nerve paralysis,24,27,28 and exposure to 300–500 ppm represents an imminent threat to life leading to pulmonary edema.29 Exposure to ≥500 ppm of H2S leads to rapid loss of consciousness, cessation of respiration, and death.30
Table 1.
Basic Physicochemical and Thermodynamic Properties of H2S
| dipole moment | 0.97 D |
|---|---|
| boling temperature | −60 °C |
| solubility (in H2O) | 110 mM/atm, 25 °C |
| 210 mM/atm, 0 °C | |
| boiling temperature | −60.2 °C |
| density (25 °C, 1 atm) | 1.36 kg/m3 |
| IRa | ν1 2525, 2536 cm−1 |
| ν2 1169, 1184, 1189 cm−1 | |
| ν3 2548 cm−1 | |
| 1H NMRb | 0.52 ppm |
| pK1 | 6.98 |
| pK2 | >17 at 25 °C |
| λmax (HS−) | 230 nm |
| ε | 8 × 103 M−1 cm−1 |
| Henry’s law coefficient (298 K) | 0.087135 mol solute/mol water atom |
| detection threshold by human nose | 0.02–0.03 ppm |
| lethal dose | >500 ppm |
| ΔfG°(H2S) | −28 kJ/mol |
| ΔfG°(HS−) | +12 kJ/mol |
| ΔfG°(S2−) | +86 kJ/mol |
| E°′(S•−, H+/HS−) | +0.91 Vc |
| E°′(HS2−, H+/2HS−) | −0.23 Vc |
Values are for the crystalline phase III.
Value obtained from crude sulfane oil.
Versus SHE.
2.1. Nomenclature
Formerly called hydrosulfuric acid or sulfhydric acid due to the acidic nature of its aqueous solutions, dihydrogen sulfide and sulfane are now the names recommended for H2S by IUPAC. For HS−, the IUPAC recommended names are sulfanide or hydrogen(sulfide)(1−); for S2−, sulfide(2−), or sulfanediide. The term “H2S” is used in this review for the gas and for the mixture of H2S and HS− in aqueous solution at a certain pH, unless otherwise specified.
The term “sulfanes”, according to the IUPAC Gold Book, includes polysulfanes, hydropolysulfides, and polysulfides, but its use is discouraged to avoid confusion with the newer systematic name sulfane for H2S and the names derived therefrom. In the literature on the biological effects of H2S, the term sulfane sulfur, sometimes abbreviated S0, is used to refer to a sulfur atom that is covalently bonded to two or more sulfur atoms (e.g., RS(S)nSR, where (S)n represents sulfane sulfurs) or to a sulfur atom and an ionizable hydrogen (e.g., Cys-SSH).31,32 Some compounds containing sulfane sulfur are thiosulfate (S2O32− or −S-SO3−), persulfides (RSSH), inorganic and organic polysulfanes (HSSnSH, RSSnSR, and RSSnH), polythionates (−SO3–Sn–SO3−), and cyclooctasulfur (S8). Sulfane sulfur has six valence electrons in contrast to sulfide sulfur, which has eight, and is incorrectly referred to as “zero valence” sulfur, although it is always attached to other sulfur atoms or to an ionizable hydrogen. Sulfane sulfur can also be defined as sulfur that can tautomerize to the thiosulfoxide form (i.e., RSSH to RS(S)H). Sulfane sulfur usually has an oxidation state of zero.33,34 It can be transferred to cyanide (CN−) to form thiocyanate (SCN−) and it can be reduced to H2S by thiols (RSH). In this review, the term sulfane sulfur will be used to refer to a sulfur covalently bonded to two or more sulfur atoms or to a sulfur atom and an ionizable hydrogen. Elemental sulfur, that can be present in many different allotropic states of which the most abundant is S8, will be abbreviated Sn.
2.2. Physicochemical Properties of H2S
H2S is a covalent hydride. Its structure is analogous to that of water, the hydride that is formed with oxygen, the companion to sulfur in the chalcogen group together with selenium and tellurium. However, the bond angles in H2S are smaller than in water (93 versus 104°).35 The frontier orbitals for the bent H2S molecule are well described.35,36 The molecular orbitals for H2S result from the linear combination of the 1s orbital of the hydrogen atom and the 3s and 3p orbitals of the sulfur atom.35,37 The energies of orbitals for H2S versus HS− are an important feature that defines the differences in their reactivity. For example the HOMO orbital of HS− is less stable (−2.37 eV calculated and −2.31 eV measured)38,39 than of H2S (−10.47 eV) indicating that HS− is more nucleophilic and basic than H2S, which is consistent with their known reactivities. Because the HOMO is so stable, H2S is not an excellent one-electron donor. The LUMO orbital for H2S (+0.509 eV calculated and −1.1 eV based on electron affinity data) suggests that H2S can be an excellent electron acceptor.35,37 However, because LUMO is an antibonding orbital in the bent H2S, electrons added to this orbital cause a weakening of both S–H bonds.35
Interestingly, H2S forms relatively strong hydrogen bonds with HS− in aprotic solvents. At low temperatures H2S reacts with triethylammonium hydrosulfide and with tetramethylammonium hydrosulfide to form complexes containing, respectively, 2 and 3 mol of H2S per mole of salt. The energy of the hydrogen bond in HSH⋯SH− is greater than 29 kJ/mol and possibly as large as 58 kJ/mol.40 At pressures above 90 GPa (Gigapascal), H2S becomes a metallic conductor of electricity. When subjected to extremely high pressures (∼1.5 million atmospheres (150 GPa)) and cooled below 203 K, H2S displays the classic hallmarks of superconductivity: zero electrical resistance and a phenomenon known as the Meissner effect. The Meissner effect occurs when a superconducting material is placed in an external magnetic field and there is no field inside the sample, unlike in normal materials.41
H2S has three low-temperature (ambient pressure) thermodynamic crystalline phases. IR spectra of crystalline phase III shows low-frequency bending vibration ν2 at 1169, 1184, and 1189 cm−1, higher frequency stretching vibrations, a symmetric stretch, ν1 at 2525 and 2536 cm−1, and an asymmetric stretch ν3 at 2548 cm−1.42
Sulfur is larger than oxygen (covalent radius of 105 against 66), has a lower electronegativity (2.58 against 3.44 in the Pauling scale), and is more polarizable. As a consequence, the dipolar moment is lower for H2S than for water (0.97 versus 1.85 D) and the intermolecular interactions are weaker. Thus, H2S is a gas at room temperature and normal pressure, while water is a liquid (boiling points of −60 °C versus 100 °C). Nevertheless, H2S has relatively high solubility in water (110 mM atm−1 at room temperature and 210 mM atm−1 at 0 °C).43,44
H2S is a weak diprotic acid (eqs 1 and 2).
| (1) |
| (2) |
In aqueous solution, the pKa valuesof the first dissociation (eq 1) are 6.98 and 6.76 at 25 and 37 °C, respectively. Different values for the second dissociation constant for HS− have been reported. The original data indicated a pK2 value at 25 °C ranging from 12.5 to 15.43,45–49 However, Giggenbach pointed out that polysulfides formed at higher pH due to the oxidation of HS− interfere with the determination of pK2.50,51 Based on optical spectra of highly alkaline, oxygen free, HS− solution, Giggenbach estimated pK2 to be 17 ± 0.1 at 24 °C.51 Meyer et al., confirmed this based on Raman spectroscopic monitoring of the H−S stretch in an oxygen-free HS− solution with sodium hydroxide concentrations ranging from 5 to 22 M.52 Licht and Mansen proposed 17.3 for pK2 of H2S, based on pH measurements of highly alkaline K2S solutions.53 Using weak acid theory, which predicts a difference of 12.3 between pK1 and pK2 for an acid in which the negative charge resulting from the first dissociation step is localized on the same atom to which the second proton is bonded, Myers calculated a pK2 value of 19 ± 2.54 Extrapolating from the thermodynamic data for the dissociation of polysulfides and avoiding the experimental and theoretical difficulties associated with measurements in highly alkaline HS− solutions, Schoonen and Barnes calculated pK2 to be 18.51 ± 0.56.55 Thus, although reference to the original values for pK2 (12–15) persists in the biochemical literature, it is in fact higher (17–19).
At the physiological pH of 7.4 and at 37 °C, H2S is in fast equilibrium with HS−, and the proportions of HS− and H2S are 81 and 19%, respectively. The concentration of S2− is negligible (1.7 × 10−12 M) but still sufficient to cause precipitation of metal sulfides, due to very low product solubility constants. Solutions of H2S in water are mildly acidic with a pH of ∼4, solutions of NaHS are alkaline, and solutions of Na2S are strongly alkaline. This information needs to be taken into consideration when adding H2S or its salts to chemical or biological assays. The concentration of the HS− anion can be determined from its absorption at 230 nm,56 using a molar absorptivity of 8000 M−1 cm−1. However, air oxidation and polysulfide formation can complicate accurate determination.
2.3. Concentration in Membranes and Permeation of H2S
The signaling actions of H2S in compartments where it is not generated will be greatly influenced by its ability to concentrate in and diffuse across membranes. The partition coefficients (i.e., the ratio of its concentration in organic solvent/buffer) of H2S between the organic solvents octanol or hexane and water are 2.1 ± 0.2 and 1.9 ± 0.5, respectively, at 25 °C and pH 3.8, a pH where the diprotonated H2S form predominates.57 These values indicate that H2S is slightly hydrophobic since it is twice as soluble in organic solvents as in water. When the pH is increased to the more physiological value of 7.4, the partition coefficient decreases to 0.64 ± 0.05 (for octanol), due to the ionization of H2S to HS− in the aqueous phase.57 Consistent with the values in organic solvents, the partition coefficient between dilauroylphosphatidylcholine liposomes and water is 2.0 ± 0.6 (pH 3.8, 25 °C).57 This relatively high solubility in a membrane model is consistent with the high permeability of H2S across biological membranes. Experimental estimates, comparison with other molecules, and molecular dynamics studies suggest that membrane permeability is as high as 11.9 cm s−1 and that aquaporins or other protein facilitators are not needed for H2S to cross membranes.57–59 Nevertheless, according to mathematical models, biological membranes are expected to slow down H2S transport resulting in local increases at sites of formation.57
2.4. Reactivity of H2S
The ability of HS− to donate a pair of electrons and form a covalent bond, i.e., its nucleophilicity, is very good. This can be explained by its negative charge, by its high polarizability, and by the relatively low electronegativity of sulfur. Furthermore, HS− is highly available at neutral pH due to the pK1 value of H2S being ∼7.
A generic reaction of HS− with an electrophile (E1+) is represented in eq 3. In contrast to the analogous reactions of thiolates (RS−, where the sulfur is bound to a carbon), the product formed from the reaction of HS− with an electrophile can ionize and react with a second electrophile (E2+) leading to a distinct product (eq 4). This differential reactivity between HS− and thiolates is the basis of several methods for H2S detection (see section 3).
| (3) |
| (4) |
The measure of nucleophilicity is a kinetic one and is estimated by comparing reaction rates, i.e. the faster the reaction, the greater the nucleophilicity. In this regard, it is interesting to compare the nucleophilicity of HS− with that of alkyl thiolates (RS−). This comparison is biochemically relevant, since thiolates are abundant in biological systems. The rate constants of the reactions of HS− with different disulfides (eq 5) are about 1 order of magnitude smaller than the corresponding reactions of thiolates (eq 6).60
| (5) |
| (6) |
Equation 6 represents a thiol disulfide exchange reaction. These reactions occur through a concerted mechanism in which the attack of HS− or RS− on one of the sulfurs in the disulfide is accompanied by the release of the other as a thiolate. The lower rate constants in the case of HS− versus RS− can be attributed to the lack of an inductive effect by the adjacent methylene, to differences in polarizability, or to solvation effects.60 Accordingly, computational calculations show that the energy of the highest occupied Kohn–Shan orbital, an indicator of nucleophilicity, is lower for HS− than for thiolates, while the chemical hardness is higher.60 The reactivity of HS− toward hydrogen peroxide and peroxynitrite is also lower than that of thiolates.61,62
The two-electron reduction potential E°′(HS2−, H+/2HS−) is −0.23 V (versus SHE), which means that H2S is a strong reductant.63,64 The value is similar to the potentials for the cysteine and glutathione redox couples.63,64 Importantly, the reaction of H2S with two-electron oxidants such as hydroperoxides does not yield a disulfide (HSSH/HSS−) directly. Instead, sulfenic acid (HSOH) is formed as an intermediate (see section 5).
The one-electron reduction potential E°′(S•−, H+/HS−) is estimated to be +0.91 V based on the thermodynamic parameters for these two species: ΔfG°(S•−) = +140 kJ/mol, ΔfG°(HS−) = +12 kJ/mol, pKa (HS•) = 3.464 and is identical to the experimentally determined value of 0.92 V.65 The value compares well with the values for thiols (E°′(RS•, H+/RSH) = +0.96 V).63,64 Given that pK1 of H2S is ∼7, the Gibbs energies of formation of H2S and HS− are identical at pH 7. The bond dissociation energy of H2S is 90 kcal mol−1 or 377 kJ mol−1.66 For comparison, the bond dissociation energy of H2O is 118 kcal mol−1 or 494 kJ mol−1 and the one-electron reduction potential is E°′(HO•, H+/H2O) = +2.32 V.67
The one-electron oxidation of H2S to the sulfyil radical (HS•) by biological oxidants is expected to be difficult given the high reduction potential of the S•−/HS− couple. Yet, H2S is known to decay in air and is also oxidized by metals. The discrepancy is explained by the high reactivity of the resulting HS•/S•− radicals. HS• (λmax = 240 nm) dimerizes to give H2S2,65,68 which in turn, readily decomposes to give Sn and H2S (eq 7, 8),69,70 both of which are removed from the system pulling the redox equilibrium in the direction of H2S oxidation.
| (7) |
| (8) |
At pH > 5, •SH/S•− reacts with HS− (kf = 4 × 109 M−1 s−1, kr = 5 × 105 s−1) to form disulfanuidyl (or dihydrogen disulfide radical anion), HSSH•−/HSS•2− (λmax = 380 nm) (eq 9).65,68
| (9) |
Both HS•/S•− and HSSH•−/HSS•2− react with oxygen (eq 10–12). HSS•2− is a weaker oxidant than S•− (E°′(HSS•2−, H+/HS−) = +0.67 V). EPR studies have identified the •SH radical in irradiated glassy solutions of sulfides and determined that its reaction with O2 leads to formation of OSO•− (λmax = 255 nm) and not −SOO•.71
| (10) |
| (11) |
| (12) |
A major technical problem while working with H2S solutions is the propensity for autoxidation. When H2S or Na2S are added to oxygen-free water a clear solution is formed. If the solution contains oxygen and trace metals in the pH range 6–9, a yellow-green color develops. The intensity of the color depends upon the concentration of elemental sulfur, Sn. Upon acidification, a whitish colloidal sulfur suspension forms.
Although the one-electron reduction of oxygen by HS− (eq 13) is not thermodynamically favored, as reflected by the reduction potential E°′(O2/O2•−) = −0.35 V (−0.18 V for a 1 M O2 standard state), H2S oxidation nonetheless occurs, albeit slowly.72–76
| (13) |
The reaction between H2S and O2 is expected to be very slow from a kinetic point of view due to a spin barrier. O2 has a triplet electronic ground state and a diradical character, which promotes its reactions with species with unpaired electrons but slows its reactions with species with paired electrons as in H2S. The oxidation of H2S (HS− and S2−) by O2 has a complicated stoichiometry with an array of products and metastable intermediates being produced. Products of all sulfur oxidation states have been reported: polysulfide ions (S42− and S52−), sulfur (colloidal or orthotrombic), S4O62−), S2O32−, SO32−, S2O62−and SO42−. Elemental sulfur, sulfite, sulfate, and thiosulfate are the major product observed in many studies and are usually formed in the stoichiometries shown in eqs 14–17:72–76
| (14) |
| (15) |
| (16) |
| (17) |
Transition metal ions and complexes are effective catalysts as they are able to lower the activation energy for redox reactions.77 This chemistry is exploited during industrial removal of H2S, which is a corrosive gas, from sour waters and wastewaters, from gaseous streams, and from raw oil. For large industrial scale cleaning applications (especially for sour gases), the Claus process is employed, converting H2S to Sn in two steps. In the first step, H2S gets oxidized to SO2, which symproportionates in a second reaction with another mole of H2S to elemental sulfur of high purity (>99.5%).78,79
For smaller applications and quantities, diverse setups and methods have been employed and patented. Some well-described applications include H2S removal by bacteria, ultrasonic irradiation80 bare iron or iron oxide surfaces,81,82 iron solutions (Fe2(SO4)3), or iron chelated agents (e.g., EDTA and CDTA).83–85 Interestingly, addition of the heavy metal ion chelator DTPA (diethylenetriaminepentaacetic acid), which unlike EDTA completely chelates iron, stabilizes H2S in solution and prevents its oxidation. The use of DTPA is highly recommended when working with H2S (Na2S or NaHS) solutions.86
Inorganic polysulfides and sulfur formed during H2S oxidation could have biological effects of their own. Inorganic polysulfides and their biological effects are covered in detail in section 8.4.
The chemistry of sulfur has been reviewed extensively.87–89 Sulfur exists in many allotropic forms of which cyclic S8 is the most stable.87–89 In polar solvents such as methanol or acetonitrile, S8 is partially transformed to S7 and S6 at ambient temperatures.90 In aprotic solvents, hydroxide ion reacts with elemental sulfur S8 to give the trisulfur anion radical S3•− as the major product.91 S3•− is highly reactive (see section 8.5). The allotropic composition of elemental sulfur is further perturbed by light.92 Sulfur can also exist as polymeric sulfur (S∞). During oxidation of H2S solutions, sulfur sol can arise, which consists of a sulfur core with hydrophilic polythionate (Sx(SO3−3)2) tails that enhance solubility and give rise to the characteristic yellow color.87–89 Sulfur also has biological effects akin to H2S. Intravenous injection of sulfur in rabbits led to the immediate detection of H2S in breath.93 Addition of colloidal sulfur to liver extracts led to its reduction to H2S and to increased oxygen uptake.94 Red blood cells can reduce sulfur in an NADPH- and glutathione-dependent manner, leading to H2S release.95 Some elemental sulfur preparations have entered preclinical trials recently, as H2S donors.96
3. WORKING WITH H2S
Several methods are available for the qualitative and/or quantitative detection of H2S. Before describing the different analytical methods, important considerations such as H2S source, handling and safety precautions, and possible interferences in the measurements are discussed.
3.1. Handling Precautions
Although H2S is relatively soluble in water and the pH dependent ionization to HS− and S2− increases the concentration of total H2S species in the aqueous phase, solutions of H2S or its salts, NaHS and Na2S, lose H2S to the gas phase. This loss is more significant when solutions are acidic rather than alkaline (pKa of H2S = 6.98, 25 °C) and when containers have large headspaces. Therefore, it is necessary to use sealed vials and to transfer H2S-containing solutions using gastight syringes.97 In addition, since H2S tends to oxidize, particularly in the presence of metal ion contaminants, it is necessary to prepare solutions in anaerobic water or buffers, free of trace metals.97 Storage of H2S solutions is not recommended and solutions should be prepared immediately before use.
H2S is highly toxic and should be handled in fume hoods. Investigators should not rely on their sense of smell for monitoring H2S, because, although it can be detected at concentrations as low as 0.02 ppm, the inability to smell H2S is one of the first signs of H2S toxicity. Before discarding H2S-containing solutions, a quenching solution containing zinc acetate (30 g/L), sodium citrate (9 g/L), and NaOH (12 g/L) can be used that results in insoluble ZnS formation. The safety aspects of working with H2S have been reviewed recently.86,98
3.2. Inorganic Sources of H2S for Reference and Experiments
H2S from commercially available gas cylinders can be a very pure source of this gas. Solutions are usually prepared by dissolving the gas in a deoxygenated solvent. A saturated solution containing ∼0.1 M H2S and with a pH of ∼4 can be prepared in water. HS− solutions can be prepared in buffer solutions with pH ∼9. The gas flow-through should be trapped as ZnS to avoid H2S release into the atmosphere.86 Alternatively, H2S solutions can be prepared by mixing sulfide salts with acid in variations of Kipp’s apparatus for the preparation of gases.99 In addition, concentrated solutions of H2S (0.8 M) in tetrahydrofuran are commercially available.
Sulfide salts rather than H2S gas are in fact often used for practical reasons. The salts that are usually used are sodium hydrogen sulfide (NaSH·xH2O) and disodium sulfide, either anhydrous (Na2S) or nonahydrate (Na2S·9H2O). The purity of the salts is an important consideration, particularly in the case of the NaSH salts.86,97,98,100,101 Being highly hygroscopic, these salts bind water from air and become liquid with time if kept outside of a glovebox. Of particular concern is the use of salts with unidentified numbers of water molecules (commercially available as NaSH·xH2O, where x = 1–10), as it is unclear how researchers can calculate H2S concentrations without a defined molecular weight. Recently the preparation of tetrabutylammonium hydrosulfide (NBu4SH) was reported, which is a potentially useful source of HS− in experiments performed in organic solvents.102 However, NBu4SH is very hygroscopic and should also be handled with care in a glovebox. Frequent impurities found in all these sources are water, elemental sulfur, polysulfides, and other oxidation products such as sulfite and thiosulfate. Another concern is the alkaline pH of the solutions of the salts, particularly Na2S, in water. The salts should be white, anhydrous powders and should be stored in desiccators under vacuum or in an argon box. It is convenient to wash the crystals with distilled water to remove oxidation products from the surface.86 To eliminate sulfane sulfur contaminants, stock solutions can be treated with immobilized phosphines103 or with cyanide.100,101
Several natural and synthetic compounds can slowly liberate H2S to potentially simulate its formation in biological systems. Various chemical groups and release mechanisms are involved, and this subject has been extensively reviewed.104–110 NaSH and Na2S salts are sometimes incorrectly referred to as H2S donors; slow release of H2S from them cannot be invoked. The acid base equilibration of these salts is extremely fast and consequently, the corresponding concentrations of HS− and H2S exist in solution with their ratio depending on the pH.
3.3. H2S Donors
Recent advances in H2S donor design have led to the development of several classes of donors that are showing very promising pharmacological effects. One main difference between these donors and sulfide salts is the slow release of H2S, potentially mimicking physiological H2S production. Significant problems with H2S donors are that the chemistry of H2S release is often unclear (for some, their pharmacologically similar effects to H2S are used as an indication of their H2S donor ability) and that the decomposition products could be reactive. The chemistry and biological applications of H2S releasing agents has been extensively covered.104–110 In this section, we summarize the main classes of H2S donors, which are grouped based on the mechanism of their H2S release: (i) donors that require thiols to release H2S, (ii) donors that release H2S by hydrolysis (with or without light), (iii) donors that release H2S in reaction with bicarbonate, and (iv) COS-releasing donors that yield H2S in the presence of carbonic anhydrase (Figure 1).
Figure 1.
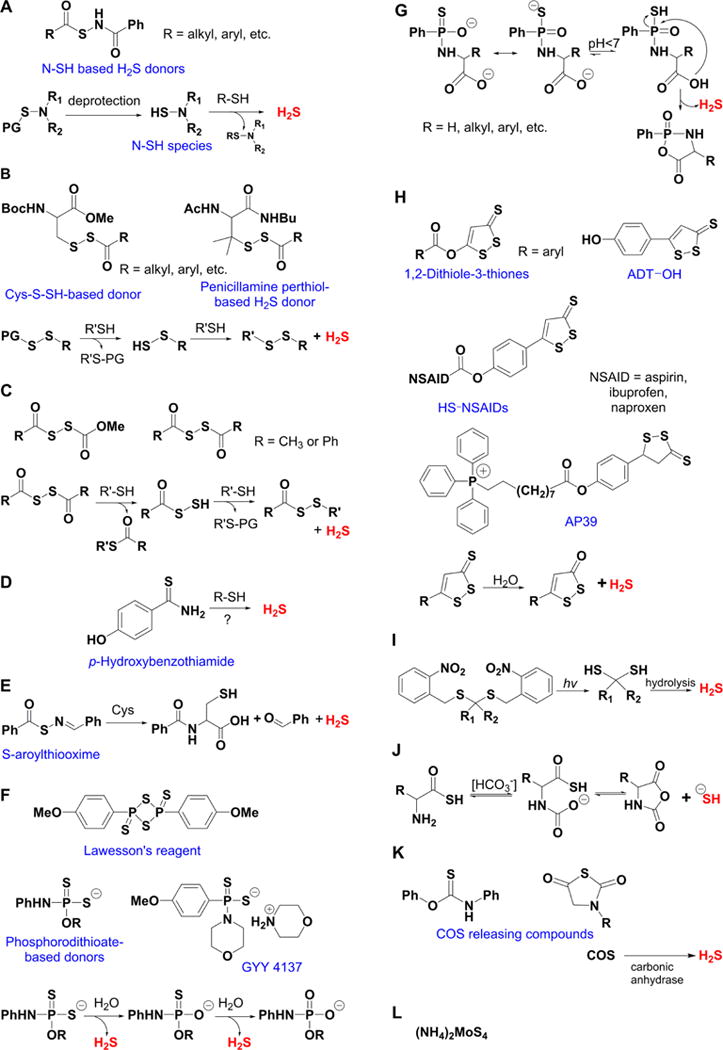
Overview of different classes of H2S donors. (A) Compounds based on the N-mercapto template (N-SH species) and the proposed mechanism for H2S release. PG, protective group. (B) Perthiol-based compounds and proposed thiol-dependent mechanism of H2S release. (C) Dithioperoxyanhydrides can also serve as H2S donors upon reaction with thiols. (D) Arylthioamides release H2S in the presence of thiols via an uncharacterized mechanism. (E) S-Aroylthiooximes release H2S in the presence of aminothiols. (F) Chemical structures of Lawesson’s reagent and its derivative, GYY4137, the most widely used H2S donor, and the proposed mechanism for H2S release from GYY4137. (G) Phosphorothioate-based H2S donors that release H2S in a pH-dependent manner. (H) Another widely used class of molecules is 1,2-dithiole-3-thiones. They can be coupled to nonsteroid antiinflammatory drugs (NSAID) such as aspirin, ibuprofen, or naproxen, or to triphenylphsophonium group (AP39) which directs them to mitochondria. This class of molecules is believed to release H2S via hydrolysis. (I) Example of photo cleavable gem-dithiol based H2S donors, which undergo hydrolysis to release H2S. (J) Thioamino acids release H2S in reactions with bicarbonate. (K) COS, released by COS donors, forms H2S in the presence of carbonic anhydrase. (L) Ammonium tetrathiomolibdate is shown to act as H2S donor in vivo.
The simplest and oldest known thiol-activated H2S donors are active principles of garlic.111 Their chemistry is covered in section 8.6 as these compounds also release low molecular weight persulfides. The first synthetic thiol-activated donors were compounds based on the N-mercapto template (Figure 1A).112 Since N-SH species are unstable, acyl groups were introduced to protect the mercapto group and enhance stability. In the presence of thiols such as GSH or cysteine, these compounds decompose to give H2S. Similarly to N-SH donors, tertiary perthiol-based compounds were reported as H2S donors, e.g., pencillamine-perthiol (Figure 1B).113 It is important to note, however, that H2S release from these donors also results in the formation of mixed disulfides, which could introduce other modifications on proteins and initiate signaling. Nonetheless, the protective effects of pencillamine-perthiol based H2S donors in myocardial ischemia/reperfusion injury have been reported. Dithioperoxyanhydrides were also recently reported as potential thiol-activated H2S donors.114 Again, these compounds require 2 mol of thiol and release 1 mol of H2S and a mixed disulfide (Figure 1C). Arylthioamides represent the fourth class of thiol-activated H2S donors (Figure 1D). However, these compounds show very weak H2S formation even in the presence of high concentrations of glutathione or cysteine.115 Nonetheless, when administered orally to rats, they induced a drop in blood pressure, reminiscent of the effect of H2S. S-Aroylthiooximes have also been proposed as H2S donors in the presence of aminothiols and show potential for the preparation of H2S releasing materials (Figure 1E).116,117
Some compounds, such as Lawesson’s reagent (2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane-2,4-disulfide) are known to release H2S by spontaneous hydrolysis in aqueous solution.118 In fact, this compound was shown to be beneficial in regulating colon ulceration in a rat colitis model.119 A derivative of Lawesson’s reagent, GYY4137 (morpholin-4-ium metoxyphenyl(morpholino) phosphino-dithioate) is one of the most widely used H2S donors (Figure 1F).120–124 The pH-sensitive H2S release (the lower the pH the greater the release) has been confirmed both colorimetrically and amperometrically, and the mechanism of H2S generation recently elucidated.125 This is a two-step process; the first, faster step involves straightforward sulfur–oxygen exchange with water, while the second, slower step, results in complete hydrolysis to an arylphosphonate (Figure 1F). GYY4137 has vasodilatory and anti-inflammatory effects akin to H2S, and considering the time scale for most of its reported biological effects, the probable source of H2S is the first reaction step (Figure 1F).125 Using a core structure of GYY4137 or the phosphorothioate as a template, new compounds that undergo pH-dependent cyclization and subseqent H2S release have been reported recently. Such donors could have particular application in ischemia-reperfusion injury where a pH drop is expected in ischemic tissues (Figure 1G).126
1,2-Dithiole-3-thiones represent another class of H2S-releasing molecules127 that is widely used in the design of H2S donors and is often coupled to some other pharmacologically active moiety, e.g., nonsteroidal anti-inflammatory drugs,128–130 adenosine,131 or targeted to mitochondria with the lipophilic triphenylphosphonium cation (Figure 1H).132–134 Hydrolysis is proposed to be involved in the mechanism of H2S release, and the hydrolysis products were recently identified by mass spectrometry. Furthermore, this class of molecules was shown to directly persulfidate glutathione,131 while mitochondrially targeted AP39 (Figure 1H), even at nanomolar concentrations, increased intracellular persulfide levels more strongly than any H2S donor.135 Some NSAID conjugated hybrids have entered phase I clinical trial.136 Some effort has been made in preparing photocleavable gemdithiol based H2S donors, which then undergo hydrolysis to release H2S (Figure 1I).137
Thio-amino acids (thioglycine and thiovaline) reportedly react with bicarbonate and are converted to the corresponding N-carboxyanhydrides with concomitant release of H2S (Figure 1J).138 Considering the high concentration of bicarbonate in the biological milieu and its common use as a buffer in cell culture media, these compounds could potentially be useful as H2S donors. Unlike other reported classes, these donors reach their plateau after 1 h and could be classified as donors with moderate-to-fast H2S release.
Some compounds are able to release carbonyl sulfide (COS; e.g., thiocarbamates and N-thiocarboxyanhydrides). COS is converted into H2S by the action of the ubiquitous enzyme carbonic anhydrase (Figure 1K).139,140 This chemistry has been explored to create molecules that release COS (and subsequently H2S) upon light activation or intracellular reaction with reactive oxygen and nitrogen species.141,142
The first metal complex-based H2S donor has been reported recently, ammonium tetrathiomolybdate (Figure 1L).143 (NH4)2MoS4 was shown to slowly release H2S in buffers and cell culture143 and exhibited cytoprotective effects in a rat model of ischemia-reperfusion injury.144
3.4. Methods for H2S Measurement
Unlike H2S, the deprotonated species, HS− and S2−, absorb in the ultraviolet region with absorption coefficients at 230 nm of 8.0 × 103 and 4.6 × 103 M−1 cm−1, respectively, at 25 °C.145 In principle, H2S solutions can be diluted in buffer at pH ∼9.6 (e.g., carbonate buffer), where H2S is predominantly in the HS− form, and the concentration can be determined from the absorbance at 230 nm.86 This approximation is, however, useful only for very pure solutions since the presence of H2S oxidation products as well as other components in the case of complex mixtures can cause interference.
To standardize stock solutions, classical iodometric titrations can be performed. For this, H2S is trapped in zinc acetate to minimize its diffusion and then reacted with excess iodine in acidic medium. The remaining iodine is titrated with sodium thiosulfate, using starch as an indicator (eqs 18 and 19). However, the presence of other reductants can lead to error.
| (18) |
| (19) |
The development of H2S detection tools has been rapidly expanding and has been covered in several review articles. In the following sections, an overview of the most widely used methods for H2S detection is provided.
3.4.1. Methylene Blue Method
This method is based on the synthesis of methylene blue from H2S and N,N-dimethyl-p-phenylenediamine in the presence of acid and ferric chloride (Chart 1). The oxidative coupling of two molecules of the diamine with H2S involves the initial one- or two-electron oxidation of the diamine to the cation radical or diimine intermediates, respectively, followed by nucleophilic attack of H2S to form a thiophenol derivative that reacts with a second molecule of the oxidized intermediate.146,147 Zinc chloride is also included in the assays to prevent H2S volatilization. The concentration of methylene blue is measured at 670 nm and compared to calibration curves obtained with samples of known concentrations of H2S that were similarly processed.148–150 Variations of this method include chromatographic separation of methylene blue151 and mass spectrometric detection.152 The methodological details and potential pitfalls of this method have been reviewed.32,101,153 Some of the key drawbacks are its low sensitivity (in the μM range), the release of sulfides from acid-labile stores (like iron sulfur clusters) that can lead to a gross overestimation of H2S, limited linear range for absorbance of methylene blue on concentration, and the potential for interference due to the presence of thiols or due to the turbidity of biological samples.
Chart 1.

Measurement of H2S through Formation of Methylene Blue
3.4.2. Lead Acetate
A simple way to follow the enzymatic synthesis of H2S is to use lead acetate and measure the formation of insoluble lead sulfide by the increase in turbidity at 390 nm. H2S concentrations are determined by comparison to a calibration curve generated with known concentrations of lead sulfide. Lead acetate is also useful for activity staining in gels; enzymes that produce H2S are identified as dark spots by soaking native gels in solutions containing the appropriate H2S-generating substrate(s) plus lead acetate.154 An alternative approach is to use lead acetate-soaked filter paper and to measure the appearance of black spots using densitometry.155,156 However, this method provides only semiquantitive data, and the sensitivity is quite low.
3.4.3. Electrochemical Sensors
Two types of sensors have been used: ion-selective electrodes and polarographic sensors. The H2S ion-selective electrodes use a silver sulfide membrane that specifically interacts with S2− creating a change in potential across the membrane. The electrodes are inexpensive, easy to operate, and highly selective. However, they require a long equilibration time and frequent reconditioning to remove interfering materials. Furthermore, they require a high pH that could interfere by releasing H2S from proteins. Second, the polarographic H2S sensors are based on measurement of the current produced from the oxidation of H2S by ferricyanide.157 These sensors contain a membrane through which H2S permeates into an internal solution of alkaline potassium ferricyanide, where chemical oxidation of H2S occurs concomitant with the reduction of ferricyanide to ferrocyanide. The latter is then reoxidized electrochemically. The polarographic sensors have a shorter response time and higher sensitivity, allowing for real-time monitoring of H2S down to ∼10 nM. However, they have the tendency to leak easily and have large residual currents due to impurities. Enzyme-based electrochemical H2S sensors have also been proposed and reviewed recently.158
3.4.4. Gas Chromatography
The determination of H2S in aqueous samples can be carried out following derivatization (e.g., to bis(pentafluorobenzyl)sulfide), extraction into organic solvents, and gas chromatography analysis with different detection systems.32,101 Alternatively, gas chromatography can be used for the direct determination of H2S gas removed from the headspace of reaction vessels and analyzed using a flame photometric detector or a sulfur chemiluminiscence detector that has high sensitivity.32,159,160 The concentration of H2S in the aqueous phase of the assay mixture at a given pH is then calculated based on mass conservation considerations knowing the pH of the solution, pK1 for H2S, and its solubility at the assay temperature. A big advantage of this method is that, when coupled to a sulfur chemiluminiscence detector, very low amounts of H2S (0.5 pmol) can be measured. Handling of the samples, however, requires particular care and gastight equipment.
3.4.5. Monobromobimane Derivatization
The fluorogenic reagent monobromobimane, which was originally introduced to label thiols (RSH),161 can also be used to measure H2S in the namolar range.162–164 Both thiols and H2S react with monobromobimane via a nucleophilic substitution process. In the case of thiols, a thioether is formed, while in the case of H2S, a bimane-substituted thiol is formed, which can react with a second monobromobimane forming dibimane sulfide (Chart 2). The latter can be detected by its fluorescence during HPLC separation or by mass spectrometry. This method has found a broad use lately; however, an important consideration with monobromobimane-based detection is the relatively slow reaction rate (k ≈ 10 M−1 s−1 at pH 8).162 Monobromobimane has also been used to quantify polysulfides and persulfides by mass spectrometry.165 The preparation of the corresponding standards is, however, challenging due to their instability; bimane polysulfides may be unstable too.
Chart 2.
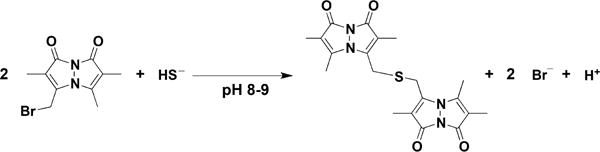
Reaction of Monobromobimane with H2S to form Dibimane Sulfide
3.4.6. Other Fluorescent Probes
The growing interest in detecting H2S in biological samples is spurring the development of H2S probes for tissue, cellular, and subcellular imaging. Such probes usually contain a fluorescence quencher that can be modified or removed by H2S. Various reactions and molecular scaffolds have been used, with different reaction rates, selectivity, and potential limitations. Since these probes have been recently reviewed,98,166–168 only selected examples are provided in Charts 3–5. One strategy is to detect the reduction of azide groups by H2S to amines in a variety of scaffolds including rhodamine, dansyl, and naphthalimide scaffolds (Chart 3A).169–171 Alternatively, the reduction of nitro groups to amines has been used171 (Chart 3B). A limitation of these probes is their slow reaction rates and the possible interference of other species, particularly thiols. Furthermore, the quantification of H2S from biological samples is not really possible. Considering that most of the probes react with H2S irreversibly, they actually remove sulfide from the intracellular pool and reach saturation rapidly giving a potentially incorrect impression of high intracellular steady state levels of H2S. Future development of reversible probes would permit measurements of actual changes in H2S levels.
Chart 3. Examples of Fluorescent Probes for H2S Detection Based on the Reduction of Azide or Nitro Groupsa.
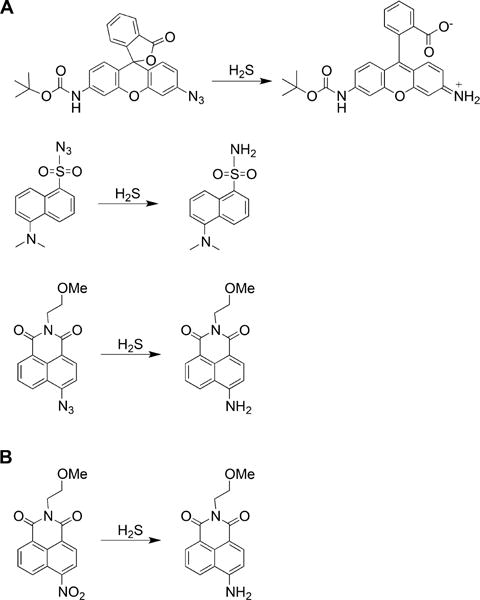
a(A) Reduction of azide groups in rhodamine (top), dansyl (middle), and naphthalimide (bottom) scaffolds. (B) Reduction of nitro group in the naphthalimide scaffold.
Chart 4. Examples of Fluorescent Probes for H2S Detection Containing Two Electrophilic Centersa.
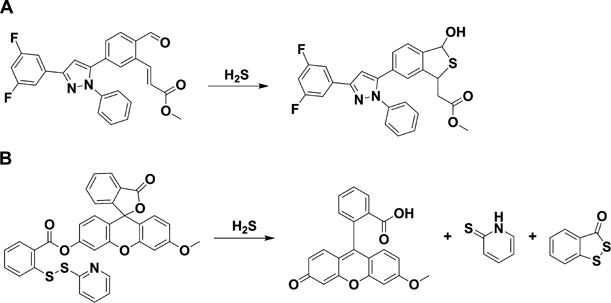
a(A) Probe containing an aldehyde and an acrylate group on a triaryl pyrazoline scaffold. (B) Probe containing an activated disulfide on a fluorescein scaffold.
To improve the selectivity over thiols, the double nucleophilicity of H2S can be exploited, as in the case of the reaction with monobromobimane (Chart 2). Probes have been developed that contain two electrophilic centers; for example, an aldehyde and an acrylate. H2S first reacts with the aldehyde forming a thiohemiacetal group that then undergoes a Michael addition reaction with the acrylate group (Chart 4A).172 Alternatively, the two electrophilic centers can be a disulfide and an ester. In the example shown in Chart 4B, H2S reacts with an activated disulfide forming a persulfide and a thiol. The persulfide then attacks the ester liberating a fluorophore and benzodithiolone.173
Another property that can be exploited for H2S detection is its high affinity for metals. Probes have been developed that contain a fluorophore bound to Cu2+, which quenches fluorescence. Binding of H2S to the metal ion results in CuS precipitation and an increase in fluorescence (Chart 5).174
Chart 5.
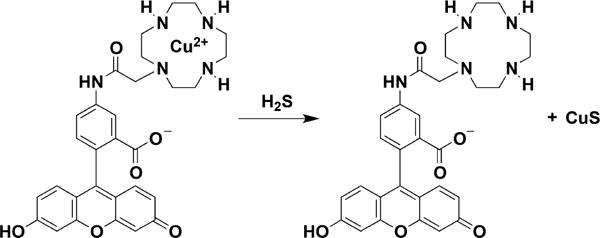
Example of a Probe for H2S Detection Based on the Release of Copper Sulfide from a Cyclen and Fluorescein Derivative
3.5. Endogenous Concentration of H2S
The methods described above can be used with appropriate precautions to detect and quantify H2S in simple solutions. However, their use with biological samples can be confounded by side reactivity, leading to highly variable estimates of H2S concentration. These differences can arise from the nature of the standards used, from sample loss due to the volatility and oxidation lability of H2S, from interference from species with similar reactivity (e.g., thiols and persulfides), and from the release of H2S from labile pools during sample handling (e.g., acidification, alkalinization, reduction). Biological samples contain labile sulfur compounds that release H2S upon certain chemical treatments.32,101 For example, exposure to acidic pH, which is associated with some analytical methods, liberates H2S from iron sulfur clusters. Addition of reductants such as dithiothreitol liberates H2S from sulfane sulfur compounds, particularly from persulfides, polysulfides, and elemental sulfur. Alkaline conditions result in H2S release from various sulfur-containing species, particularly thiols and disulfides. This potential for introducing artifacts has contributed to the estimates of H2S concentration in biological samples varying over 5 orders of magnitude.
As pointed out by Olson,153,175 it is important to critically evaluate the reliability of the reported values for H2S in biological samples. The concentration of H2S in tissues is often expressed as moles of H2S per gram protein or as moles of H2S per gram of wet weight. Cells are typically 10–20% protein and 60–75% water. Thus, 1 nmol of H2S (mg protein)−1 is equivalent to ∼200 μM H2S, and 1 nmol of H2S (mg wet weight)−1 is equivalent to ∼1500 μM. In cultured cells, H2S concentration is sometimes expressed as moles per cell. Considering that a human cell has a volume of the order of 10−12 L (i.e., 103 fL or 103 μm3), then 1 nmol of H2S (106 cells)−1 is equivalent to ∼1000 μM. Since, the human nose can detect ∼1 μM H2S in solutions,159,176 many reports of H2S concentrations in biological samples are undoubtedly in error.
Before 1996, when H2S was recognized as a physiological mediator, essentially all measurements of blood H2S had either failed to detect it or yielded extremely low values, consistent with the fact that H2S cannot be smelled in blood. Since 1996, reports of blood H2S concentrations had risen to an average of ∼50 μM.175,176 However, the use of more sensitive methods, e.g., polarographic sensor or gas chromatography coupled to a sulfur chemiluminiscence detector, together with greater rigor in sample preparation, are revealing that the concentration of H2S in blood is <100 nM and may be as low as ∼100 pM.159,177
Gas chromatography coupled to chemiluminiscence detection has revealed that the basal tissue H2S levels are quite low. According to one study, the basal H2S level is ∼10−15 nM in murine liver and brain.159 Another study reported levels of 0.004–0.055 μmol HsS kg−1 or 0.03–0.39 μmol (kg protein)−1, corresponding to 6–80 nM, in murine liver, brain, heart, muscle, esophagus, and kidney.178 In agreement with these low estimates, the steady-state concentration extrapolated from measurements of H2S production and consumption rates in murine liver, kidney, and brain were calculated to be 9–29 nM.179 Curiously, H2S levels in aorta are significantly higher (∼1.5 μM).178
The steady state concentration of H2S is the net result of its formation and decay rates. Production rates have been estimated to be 0.45, 0.3, and 0.1 pmol min−1 (mg tissue)−1 (i.e., ∼0.6, 0.4, and 0.2 μM min−1) in intact colon muscle, brain, and liver of mice in the presence of 10 mM cysteine; the production rate increased to 8, 7, and 20 pmol min−1 (mg tissue)−1 (i.e., ∼11, 10, and 28 μM min−1) respectively, in homogenized tissue.180 Another study reported H2S production rates by murine liver and brain homogenates of 106 and 1.2 nmol min−1 (g tissue)−1 (i.e., ∼151 and 1.7 μM min−1), respectively, in the presence of 10 mM cysteine.159 At a more physiologically relevant concentration of 0.1 mM cysteine, H2S production rates of 48 μmol h−1 (kg tissue)−1 (i.e., ∼ 12 μM min−1) and 29 μmol h−1 (kg tissue)−1 (i.e., 0.7 μM min−1) by murine liver and brain homogenates, respectively, were reported.179 The decay rates of H2S are high, and as expected, they decrease dramatically under hypoxic conditions.179 The apparent first order rate constant of H2S decay in murine liver under aerobic conditions was reported to be 277 min−1 at 37 °C.179 Thus, the very low steady-state tissue concentrations of H2S are primarily due to the high rate of its oxidation.179
4. ENZYMES INVOLVED IN H2S BIOGENESIS
At least three enzymes in the mammalian sulfur metabolic network have the potential to synthesize H2S.181–185 Two of these enzymes, cystathionine β-synthase (CBS) and CSE, comprise the transsulfuration pathway. The latter provides an avenue for synthesizing cysteine from the essential amino acid methionine, via the metabolic intermediate, homocysteine. The transsulfuration pathway enzymes exist predominantly in the cytoplasm although, under some conditions, they are reportedly located in other compartments such as the nucleus186 or the mitochondrion.187,188 The third enzyme, mercaptopyruvate sulfurtransferase (MST), resides in both mitochondrial and cytoplasmic compartments.189 It converts 3-mercaptopyruvate, derived from cysteine transamination, to pyruvate and transfers sulfur to a thiophilic acceptor forming a persulfide from which H2S can be released. In this section, an overview of the recent literature on the structure, mechanism, and regulation of these three enzymes is discussed with an emphasis on the recent literature pertaining to H2S biogenesis by the human enzymes.
4.1. Reactions Catalyzed by CBS
Located at the crossroads of the methionine cycle and the transsulfuration pathway, CBS commits sulfur to cysteine synthesis and catabolism, which in turn influences H2S biogenesis. CBS exhibits substrate promiscuity and catalyzes a multitude of reactions at the β-carbon of the substrates, serine and cysteine (Chart 6).154,185,190–192 In its role in the transsulfuration pathway, CBS catalyzes the β-replacement of serine by homocysteine forming cystathioninine and eliminating H2O (Chart 6, [1]). It can also catalyze the β-replacement of cysteine by homocysteine [2], by a second mole of cysteine [3], or by water [4], forming cystathionine, lanthionine, or serine, respectively, and eliminating H2S in the process. Finally, CBS can catalyze the β-elimination of cystine forming cysteine persulfide (Cys-SSH) [5]. Mutations in CBS are the most common cause of hereditary homocystinuria, an autosomal recessive disorder.193
Chart 6. Reactions Catalyzed by CBSa.
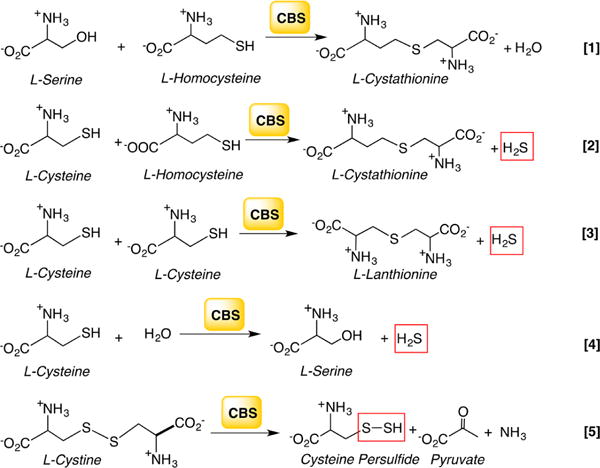
aReaction 1 generates cystathionine in the canonical transsulfuration pathway. Reactions 2–4 generate H2S from cysteine and/or homocysteine, and reaction 5 produces Cys-SSH from cystine.
4.1.1. Organization of CBS and Properties of the Heme Cofactor
Human CBS is a homodimer with a subunit molecular weight of ∼63 kDa. Its propensity for aggregation leads to its isolation as higher order oligomers ranging from 4-to 16-mers. The crystal structures of full-length human194,195 and Drosophila196 CBS have been obtained for the dimers. CBS is unique in being the only known PLP enzyme that is also a hemeprotein.197 It is a modular protein with an N-terminal domain spanning ∼70 residues, which binds a regulatory heme b cofactor (Figure 2A). This is followed by a middle catalytic core (spanning residues 71–411, human numbering), which houses the PLP cofactor and resembles the fold II or β-class of PLP enzymes.198 The catalytic core is conserved across organisms regardless of whether CBS contains or lacks the heme domain. A Cys272-X-X-Cys275 motif present in the catalytic core is seen in the reduced dithiol and oxidized disulfide state in two structures199,200 and could potentially render CBS sensitive to regulation by metal ions or to oxidation. CBS is reportedly inhibited by free copper (10–25 μM), although a connection between this observation and chelation by the CXXC motif has not been made.201 The C-terminal domain (spanning residues 412–551) comprises a tandem repeat of two “CBS domains”, which is a β–α–β–β–α secondary structure motif found in diverse proteins that often binds adenosine derivatives and is associated with energy sensing.202 In CBS, the C-terminal domain binds S-adenosylmethionine (AdoMet),203 an allosteric activator.204 Hence, ∼40% of the protein is involved in the N- and C-terminal regulatory domains, which exert allosteric control over the CBS-catalyzed reaction.
Figure 2.
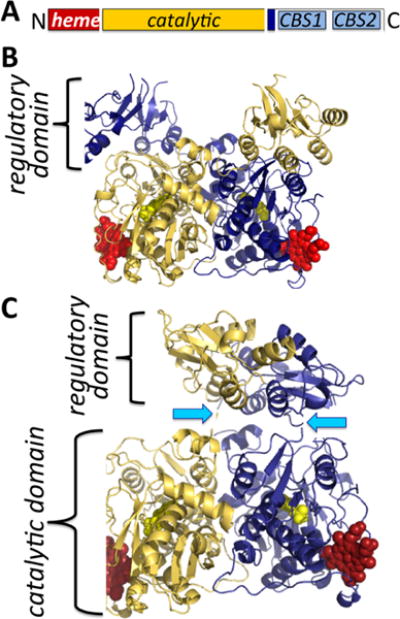
Organization and structure of human CBS. (A) CBS is a modular protein with regulatory domains at its N- and C-termini. The C-terminal domain comprises a tandem repeat of two CBS domains, CBS1 and CBS2. The structures of human CBS in the absence (PDB: 4L27) (B) and presence (PDB: 4PCU) (C) of AdoMet show that a large conformational rearrangement accompanies the transition from the basal to the activated state. The protomers are shown in blue and yellow, respectively, the heme (red) and PLP (yellow) are in sphere representation, and the blue arrows point to the intervening linker region between the catalytic core and the C-terminal domain.
Three structures of full-length CBS have been reported: two of human CBS in the presence and absence of AdoMet194,195 and a third of Drosophila CBS,196 which does not bind AdoMet and exists in a hyperactivated state.205 These structures reveal that AdoMet binding elicits a remarkable conformational rearrangement. In the absence of AdoMet, an intersubunit crossover of the C-terminal domains places each by the active site entrance of the other subunit, impeding substrate access (Figure 2B). In the presence of AdoMet, the C-terminal domain dimerizes atop the catalytic domains (Figure 2C). This structural rearrangement explains why AdoMet binding204 or truncation of the C-terminal domain entirely,206 activates CBS, i.e., by facilitating substrate access to the active site.
Although held by an unstructured N-terminal loop and relatively exposed, the heme in CBS is tightly bound. The first structures of truncated CBS lacking the C-terminal regulatory domain revealed that Cys52 and His65 coordinate the heme iron (Figure 3).199,200 The low-spin heme iron retains these ligands in both the ferric and ferrous oxidation states. Upon reduction, the Soret peak shifts from 428 to 448 nm while the α/β absorption bands shift from a broad feature centered at ∼550 nm (in ferric CBS) to 571 and 540 nm (in ferrous CBS).207 Early NMR, pulsed EPR,208 and resonance Raman209,210 studies had ruled out a catalytic role for the heme. 31P NMR studies demonstrated that the spin–lattice relaxation rates in the paramagnetic ferric (6.34 ± 0.01 s) and diamagnetic ferrous (5.04 ± 0.06 s) states were similar, indicating that the PLP and heme cofactors are not proximal to each other.208 The crystal structures revealed that an ∼20 Å distance separates the heme and PLP sites (Figure 3)199,200 and that this distance is not modulated by the presence or absence of the C-terminal regulatory domain or its allosteric effector, AdoMet.194,195 Yeast CBS lacks the heme cofactor but is highly active,211 further arguing against a catalytic role for this cofactor. In fact, deletion of the N-terminal 69 residues in human CBS results in a heme-less variant, which albeit less stable, retains ∼40% of wild-type activity.212
Figure 3.
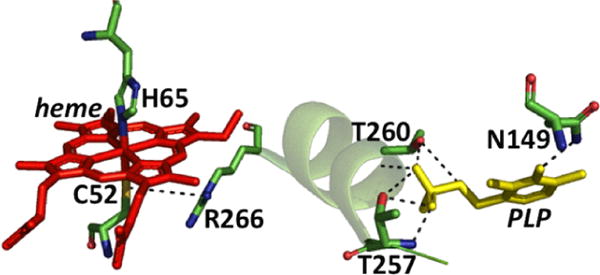
Close up of the CBS structure. The interactions between the Cys52 heme ligand and Arg266 at one end of the α-helix and between Thr257 and Thr260 and the phosphate group of PLP at the other are shown. Asn149 hydrogen bonds with the C4 oxygen in PLP.
A rhombic EPR signal is associated with ferric CBS with g values of 2.5, 2.3, and 1.86, which is similar to that of model heme complexes and heme proteins with imidazole/thiolate ligands.213 Resonance Raman studies using 34S-labeled CBS identified the ν(Fe–S) vibration at 312 cm−1.209 Exposure to mercuric chloride, a thiol chelator, resulted in CBS converting from a 6-coordinate low-spin to 5-coordinate high-spin state with a Soret maximum at 395 nm and a rhombic g = 6 EPR signal.213 The spin-state change induced by mercuric chloride was correlated with a loss of CBS activity,210 consistent with long-range communication between the heme and PLP sites. 31P NMR studies also provided evidence for long-range signal transmission by revealing that the chemical shift of the phosphorus nucleus in PLP shifted from 5.4 to 2.2 ppm upon reduction of the heme iron.208
The ferric heme in CBS, which is coordinately saturated, is relatively inert to exchange by exogenous ligands.214 The reduction potential of the Fe3+/Fe2+ couple is −350 ± 4 mV for full-length CBS215 and −291 ± 5 mV for truncated CBS216 lacking the C-terminal regulatory domain. Following reduction, ferrous CBS can bind exogenous ligands such as CO, NO•, cyanide, and various isonitriles.217–219 The heme cofactor reduces nitrite to ferrous heme-bound NO•.220 Binding of these ligands is associated with loss of activity, where characterized. Despite the low reduction potential for the heme iron in full-length CBS, it can be reduced by an NADPH-driven flavin oxidoreductase when coupled to carbonylation by CO, establishing the potential physiological relevance of this reaction.221 Ferrous CBS does not bind O2; instead it undergoes rapid oxidation (1.1 × 105 M−1 s−1) apparently by an outer sphere mechanism generating superoxide radical and ferric heme.216 Reoxidation of the ferrous-nitrosyl heme on CBS leads to peroxynitrite formation.222 Thus, the heme redox activity makes CBS a potential source of both reactive oxygen (O2•−) and nitrogen (ONOO−) species.
AdoMet increases the affinity of the CBS heme for NO• (2-fold) and CO (5-fold) and thereby sensitizes the enzyme to inhibition.223 Hence, in the interplay between the N- and C-terminal regulatory domains, heme supersedes AdoMet as an allosteric regulator. AdoMet activates ferric CBS but potentiates the inhibition of ferrous CBS by CO or NO•. Together with the effect of the C-terminal domain on the heme redox potential discussed above, the effect of AdoMet on the affinity of the heme ligands, CO and NO•, hints at long-range communication between the N- and C-terminal domains, which are >50 Å apart in the structure of AdoMet-bound CBS.195
4.1.2. Catalytic Mechanism of CBS
The ping pong reaction cycle of CBS involves the following steps: (i) binding of the first substrate (serine or cysteine), which results in displacement of Lys119 and formation of the corresponding external aldimine, (ii) abstraction of the α-proton by Lys119 leading to a resonance stabilized carbanion, (iii) elimination of water or H2S leading to aminoacrylate formation, (iv) addition of the second substrate (homocysteine, cysteine, or water) to give the corresponding product external aldimine, and (v) reformation of the Schiff base with Lys119 leading to product release (Chart 7). Support for this reaction mechanism has been obtained by stopped-flow kinetic studies on human224 and yeast225 enzymes as well as from kinetic studies on a heme-less variant of human CBS.212 Since the absorbance of the heme cofactor obscures the PLP cofactor, difference stopped flow spectrometry had to be used to monitor PLP-bound reaction intermediates in the human enzyme.224 Mutation of Lys119 to alanine reduces CBS activity ∼1000-fold and the exogenous base, ethylamine, leads to a 2-fold higher activity, consistent with the role of Lys119 as a general base in addition to its involvement in Schiff base formation.212
Chart 7. Reaction Mechanism of CBS and Structures of Key Intermediatesa.
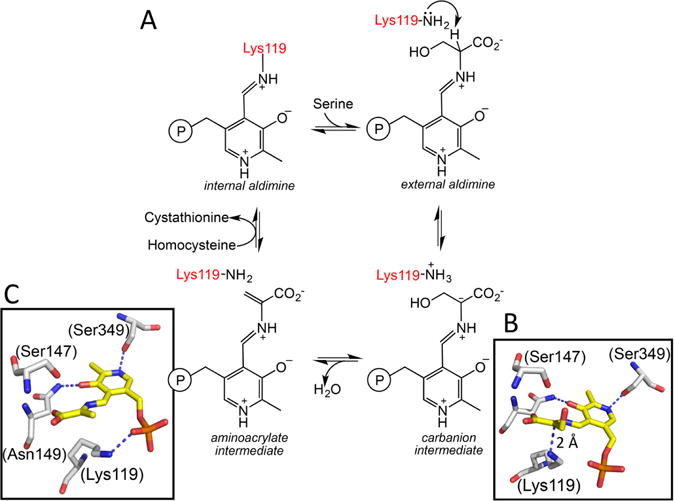
a(A) A minimal mechanism is shown for the β-replacement of serine by homocysteine to generate cystathionine and water. Structures of the carbanion (B) and aminoacrylate (C) intermediates trapped in Drosophila CBS (PDB: 2PC4 and 2PC3) are shown. The numbering of residues shown in parentheses in B and C are for human CBS. The corresponding residues in the fly protein are Lys88, Ser116, Asn118, and Ser318, respectively. An sp2 hybridized α carbon and sp2 hybridized α and β carbons are seen in the carbanion and aminoacrylate intermediates, respectively. Lys119 undergoes a major positional shift in the aminoacrylate intermediate where it is no longer required to stabilize the carbanion.
The active site of CBS displays a constellation of conserved interactions common to members of the fold II family of PLP enzymes. PLP is tethered via Lys119 forming an internal aldimine in the resting human enzyme. At the other end of the PLP ring, the side chain of Ser349 is positioned to interact with the pyridinium nitrogen while Asn149 hydrogen bonds with the oxygen. Electrostatic contacts made between conserved threonine residues (Thr257 and Thr260) in a glycine-rich loop and the phosphate group of PLP further lock the cofactor in place. High resolution structures of Drosophila CBS have captured two reaction intermediates, the carbanion and the aminoacrylate species, providing detailed insights into the mechanism of their stabilization.196 A zwitterionic interaction between the ε-NH3+ group of the lysine, which forms a Schiff base with PLP in the resting enzyme, and the Cα (at 2.0 Å distance) and C4A (at 3.0 Å distance) stabilizes the carbanion intermediate (Chart 7B). In the aminoacrylate intermediate, the ε-NH3+ of the lysine, which is no longer needed to stabilize charge at Cα/C4A, is instead, parked near the phosphate group of PLP, with which it interacts (Chart 7C).
Synthesis of Cys-SSH from cystine represents a β-elimination reaction. It is expected to proceed via a similar reaction sequence up to the formation of the aminoacrylate intermediate, which is accompanied by elimination of the Cys-SSH product. After this point, a transschiffization (rather than a β-replacement) reaction regenerates the resting internal adimine form of the enzyme and releases the eneamine product, which is hydrolyzed in solution to the α-keto acid, pyruvate, and ammonia.
4.1.3. Relative Efficacy of H2S versus Cys-SSH Synthesis by CBS
Rat165 and human226 CBS catalyze the β-elimination of cystine, the oxidized form of cysteine, to form Cys-SSH. The kinetic parameters for human CBS catalyzed Cys-SSH formation are kcat = 0.11 s−1 and kcat/Km = 85 M−1 s−1 at pH 7.4 and 37 °C. Under Vmax conditions, the most efficient reaction for H2S synthesis by human CBS is the β-replacement of cysteine by homocysteine with the following kinetic parameters: kcat = 19.6 s−1 and kcat/Km(Cys) = 2882 M−1 s−1 at pH 7.4 and 37 °C. Since the intracellular milieu is reducing and the concentration of cystine is significantly lower than of cysteine, substrate levels regulate H2S synthesis from cysteine versus Cys-SSH synthesis from cystine. Kinetic simulations of reaction rates at physiologically relevant concentrations of cysteine, cystine, and homocysteine revealed that the contribution of CBS to Cys-SSH synthesis is negligible under these conditions being ∼30 000-fold lower than H2S synthesis.226 This analysis suggests that CBS is unlikely to be a significant source of Cys-SSH in cells.
4.2. Regulation of CBS
CBS is a busy hub of regulation, which is fitting since it directs sulfur away from the cycle of an essential amino acid, methionine, to other sulfur metabolites such as cysteine, glutathione (GSH), taurine, and H2S. Embedded in the CBS structure itself are two domains at the N- and C-termini, which exert their regulation in distinct ways and also appear to impact each other in ways that are poorly understood. In the following section, modulation of CBS activity by its regulatory domains and by posttranslational modifications is discussed.
4.2.1. Heme-Dependent Allosteric Regulation of CBS
The ferric heme in CBS, which is coordinately saturated, is relatively inert to exchange by exogenous ligands.214 On the other hand, ferrous CBS binds exogenous ligands such as CO and NO• with concomitant loss of activity.217,218 Binding of NO• to ferrous CBS is accompanied by a shift in the Soret peak from 448 to 390 nm218 and leads to a 5-coordinate heme from which both Cys52 and His65 are dissociated (Chart 9). Wild-type CBS exhibits a monophasic binding isotherm for NO• and a KD ≤ 0.23 μM.227 The rate constant for NO• binding to CBS exhibits a linear dependence on NO• concentration (8 × 103 M−1 s−1, pH 7.0 and 25 °C) and is enhanced ∼2-fold in the presence of AdoMet together with a 1.3-fold decrease in koff. CO displaces the Cys52 ligand and forms a 6-coordinate low-spin ferrous-CO species with a maximum at 420 nm (Chart 8). Binding of NO• is ∼100-fold faster than of CO, which is limited by the dissociation of Cys52 from the heme iron.228 Thus, NO• is presumed to bind by initial displacement of the His65 ligand (Chart 8).227
Chart 8. Binding of NO• and CO to Ferrous CBSa.
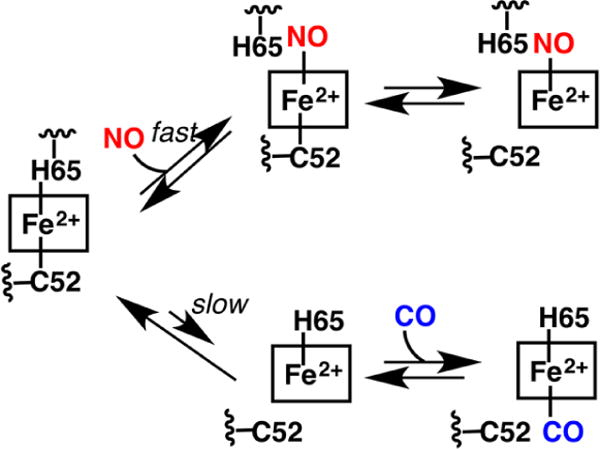
aBinding of NO• is fast and predicted to occur via displacement of the His65 ligand. Binding of CO is slow and limited by the slow dissociation of the Cys52 ligand.
The affinity of the CBS heme for CO (5-fold) and NO• (2-fold) is increased in the presence of AdoMet.223 Interestingly, deletion of the heme domain reverses the sensitivity of CBS to AdoMet, leading to a 1.5-fold decrease in activity.212 The heme ligand mutants, C52A/S, exhibit a similar magnitude of inhibition in the presence of AdoMet.229 The influence of the C-terminal domain on the heme redox potential (discussed above) and the effect of AdoMet on the affinity of the heme ligands, CO, and NO• hint at very long-range communication between the N- and C-terminal domains, which are >50 Å apart in the structure of AdoMet-bound CBS.195
Insights into how changes in the heme domain are communicated over an ∼20 Å distance to the active site have emerged from fluorescence and resonance Raman studies.230 The ketoenamine tautomer of PLP is key to reactivity since it facilitates the nucleophilic attack by the substrate amino group to form the external aldimine and subsequently stabilizes the carbanion following α-proton abstraction. Changes in the heme ligand environment (e.g., CO binding or heat treatment which displaces the Cys52 ligand)231 shift the PLP equilibrium from the ketoenamine to the enolimine tautomer, in which the proton relocates to the exocyclic oxygen at the C3 atom on the PLP ring. The salt bridge between Cys52 and Arg266 is postulated to be critical for stabilizing the active ketoenamine tautomer.230 Arg266 resides at one end of an α-helix. At the other end of the same α-helix are two conserved electrostatic interactions between Thr257 and Thr260 and the phosphate group of PLP (Figure 3). Loss of the Cys52-Arg266 salt bridge either via ligand exchange or in the pathogenic R266M mutant, stabilizes the inactive enolimine tautomer.230 The conservative R226K mutation leads to lengthening of the ferric Fe–S bond and perturbations in the PLP electronic spectrum.232 The allosteric communication between the heme and PLP sites is bidirectional since the pathogenic T257M mutation promotes loss of the Cys52 ligand in the ferrous state and a concomitant shift to the inactive enolimine tautomer.233
4.2.2. AdoMet-Dependent Allosteric Regulation of CBS
The C-terminal regulatory domain imparts both intrasteric and allosteric effects and is responsible for the propensity of the full-length protein to aggregate. Binding of AdoMet increases kcat ∼2-fold from 2.8 to 5.2 s−1, while deletion of the entire domain increases kcat 5-fold to 10 s−1 (all values calculated per monomer at 37 °C). The structures of human CBS with and without AdoMet provide molecular insights into the autoinhibitory effect of the C-terminal domain and its alleviation by AdoMet (Figure 2).194,195 While the catalytic cores in the two structures are virtually identical, the C-terminal domain undergoes a substantial rearrangement. In the absence of AdoMet, the C-terminal domain of each subunit sits on the catalytic core of the adjacent subunit, impeding access to the active site. A combination of hydrophobic interactions between residues in the CBS2 motif (Ile537, Leu540, and Ala544) and the catalytic core (Ile166, Val189, Val206, Leu210, and Ile214) and hydrogen bonding interactions between residues in the CBS1 motif (Thr460, Asn463, Ser466, and Tyr484) and a loop at the active site entrance (Glu201, Asn194, Arg196, and Asp198) lock in this conformation. AdoMet binds in a cleft between the CBS1 and CBS2 domains, which is solvent exposed and is stabilized via hydrophobic interactions and a network of hydrogen bonds. Binding of AdoMet leads to a major structural rearrangement in which the C-terminal domains uncross and dimerize in a head-to-tail fashion on top of the catalytic domain with which all interactions are broken. A flexible linker between the catalytic core and the C-terminal domain (spanning residues 381–411) is critical for mediating the AdoMet-induced conformational change, which leads to unobstructed access to the active site. The structure of human CBS with AdoMet is very similar to that of full-length Drosophila CBS, which is hyperactive in its basal state but does not bind AdoMet.196
A number of pathogenic mutations in the C-terminal domain (P422L, P427L, I435T, D444N, S466L, and L540Q) render the protein more active than wild-type CBS but insensitive to further activation by AdoMet,234–236 begging the question as to why they are disease causing. Curiously, a subset of pathogenic mutations in the catalytic core of CBS (A114V, A158V, V168M, A226T, R224H, T262M, I278T, A331V, and T353M) are functionally suppressed by deleting the C-terminal domain (i.e., the last 145 residues)237 or selecting for mutations in this domain that suppress the most common patient mutation in CBS, I278T.238 The suppressor mutants are unresponsive to AdoMet suggesting that the mutations stabilize the activated conformation even in the absence of the allosteric ligand.
4.2.3. Regulation of CBS by Covalent Modifications
CBS is regulated by at least three types of covalent modifications: (i) sumoylation, (ii) glutathionylation, and (iii) phosphorylation. CBS is a target of modification by the small ubiquitin-like modifier-1 protein (SUMO-I). A number of proteins belonging to the sumoylation machinery were identified as potential interacting partners of human CBS from a yeast two-hybrid screen and included Pc2, PIAS1, PIAS3, Ubc9, and RanBPM.186 Of these, Ubc9 is an E2 conjugating enzyme, while PIAS1, PIAS3, and Pc2 are E3 SUMO ligases, which confer target specificity and reaction efficiency. Under in vitro conditions, Pc2 enhances CBS sumoylation, which is correlated with a 70% decrease in activity.239
The C-terminal regulatory domain is required for the interaction between CBS and the other sumoylation machinery proteins noted above although the modification itself appears to occur in the catalytic core. Mutation of Lys211, embedded in a canonical ΨKXE sumoylation motif and exposed to solvent, leads to loss of sumoylation in vitro, suggesting that this lysine might be tagged by SUMO1. Sumoylation of CBS is correlated with its nuclear localization and can be visualized in cells and in tissue when care is taken to deactivate desumoylases.186 Interestingly, in porcine brain, sumoylated CBS appears to be the dominant form of the protein. The physiological significance of CBS sumoylation is presently unknown. It could serve to translocate CBS to the nucleus under conditions of stress (e.g., hydrogen peroxide, heat shock, heavy metals, or ethanol treatment) that result in a global increase in sumoylation,240–242 leading to a local increase in H2S and/or GSH synthesis assuming that CSE, which is sumoylated in vitro,239 also relocates under these conditions.
Glutathionylation of CBS is observed under both in vitro conditions and in cultured cells challenged with H2O2.243 This modification leads to a 2–3-fold increase in CBS activity and occurs at Cys346, which resides in the catalytic core near the dimer interface and is not particularly surface exposed. Glutathionylation of CBS renders the protein insensitive to further activation by AdoMet. Cys346 resides in a loop between two α-helices, which are involved in the interface between the catalytic core and the linker region in the (AdoMet-induced) activated conformation of CBS. Hence, modification at Cys346 might stabilize the activated conformation of CBS even in the absence of AdoMet. The functional significance of glutathionylation appears to be to up-regulate transsulfuration flux under oxidizing conditions, which deplete GSH pools, leading to greater synthesis of cysteine. The transsulfuration pathway is known to be an important feeder for cysteine, the limiting reagent for GSH synthesis.244,245 Glutathionylation of CBS, which is transiently increased in cells in response to oxidative stress, accounts for the increased flux of sulfur through the transsulfuration pathway under these conditions.
In the urothelium, CBS is reportedly phosphorylated at Ser227 and Ser525 in a cGMP/protein kinase G-dependent reaction.246 Phosphorylation was triggered with 8-bromo-cGMP, a stable analogue of cGMP, which caused an ∼2-fold increase in H2S production in urothelial cell lysates. H2S production was, however, tested in the presence of a high concentration of cysteine, which is a more effective substrate for CSE than for CBS, both of which are present in the urothelial cells. It was concluded that Ser227 rather than Ser525 is important for the phosphorylation-induced increase in CBS activity based on the responses of transfected cell lines harboring the S227A or S525A mutations.246 While S525A CBS expressing cells exhibited increased H2S production when treated with 8-bromo cGMP, S227A CBS expressing cells showed very low H2S synthesis, which was not responsive to 8-bromo cGMP. It should be noted, however, that the S227A mutant itself showed very low activity indicating that the mutant is catalytically compromised even in the absence of phosphorylation. A physiological role for H2S synthesis in bladder relaxation has been proposed.247
4.3. γ-Cystathionase (CSE)
CSE is the second enzyme in the transsulfuration pathway and is also dependent on the PLP cofactor, for catalysis.181,182 It catalyzes the γ-elimination of cystathionine to give cysteine, α-ketobutyrate, and ammonia (Chart 9).248 Cysteine synthesis via the transsulfuration pathway is a quantitatively significant source of this amino acid, supplying ∼50% of the cysteine present in the hepatic GSH pool.245 Like CBS, CSE exhibits substantial substrate promiscuity and catalyzes a complex array of H2S generating reactions involving chemistry at both the β-and γ-carbons of the substrate.249 In addition, CSE catalyzes both Cys-SSH and homocysteine persulfide (Hcy-SSH) synthesis from cystine and homocystine, respectively.165,226 Mutations in CSE lead to cystathionuria, an autosomal recessive disorder that is generally benign.250,251 In contrast to CBS, very few pathogenic mutations have been reported in CSE.251,252
Chart 9. Reactions Catalyzed by CSEa.
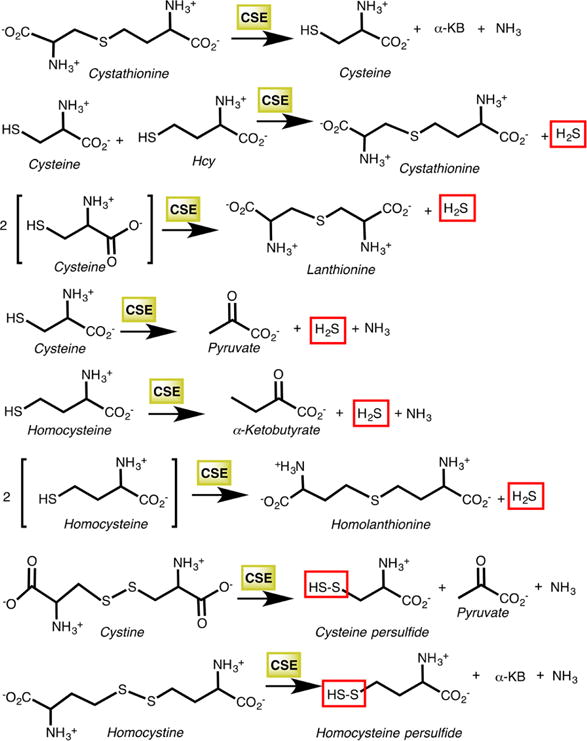
aThe first reaction is the cleavage of cystathionine to cysteine, α-ketobutyrate (α-KB), and ammonia in the canonical transsulfuration pathway. The next five reactions produce H2S, while the last two generate the corresponding persulfides from cystine and homocystine.
A significant difference between CBS and CSE is the ability of the latter to form a Schiff base with either cysteine or homocysteine bound to PLP, leading to chemical transformations at either the β- or γ-carbon of the substrate. This renders CSE-catalyzed H2S synthesis responsive to homocysteine concentrations and H2S production is predicted to increase between 20- and 200-fold in homocystinuria.249 This explains the clinical observation that homolanthionine (the condensation product of 2 mol of homocysteine) is present in urine of homocystinuric patients with CBS deficiency253 and emphasizes an underappreciated role for CSE in homocysteine management. Under Vmax conditions, the highest kcat for H2S generation is for the γ-replacement reaction of homocysteine by a second mole of the same substrate. This is followed by the γ-elimination of H2S from homocysteine or by the β-replacement of 1 mol of cysteine by another. In addition to H2S, these reactions produce the novel sulfur metabolites, homolanthionine and lanthionine (Chart 9). At physiologically relevant concentrations of substrates, the β-elimination of cysteine is predicted to be the major H2S-producing reaction catalyzed by CSE.249
The early part of the CSE-catalyzed reactions proceeds through the same steps as the CBS reaction (Chart 9) leading up to the carbanion intermediate. Thereafter, a second deprotonation at the β-carbon sets up the subsequent γ-elimination of H2S and formation of the β–γ unsaturated imine intermediate I, which is fully conjugated and can suffer one of two fates (Chart 10). Intermediate I can continue down the γ-elimination path, which involves protonation at the γ-carbon followed by imine hydrolysis to give an eneamino acid. Tautomerization of the eneamino acid to the keto acid (α-ketobutyrate) appears to be enzyme catalyzed as revealed by the stereochemical analysis of the α-ketobutyrate formed by γ-elimination of homoserine. In the γ-replacement path, a second amino acid (e.g., homocysteine or cysteine) adds to the electrophilic γ-carbon in intermediate I forming a condensation product. Following α-carbon protonation and Schiff base exchange with the active site lysine (Lys212 in human CSE), the resting internal aldimine is reformed.
Chart 10. Outline of CSE Mechanism Illustrated for the γ-Elimination of Homocysteine (Red Box) or the γ-Replacement of Homocysteine by a Second mole of the Same Substrate (Blue Box)a.
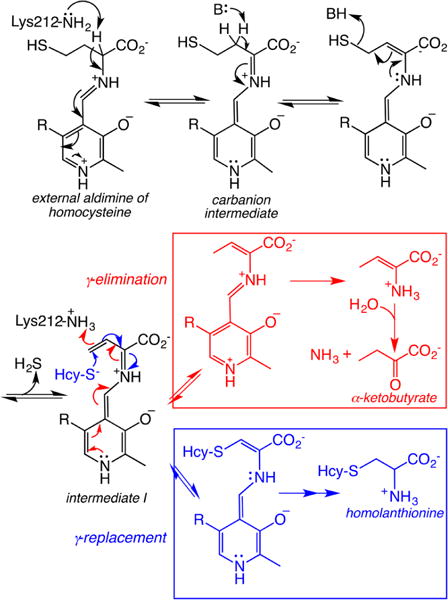
aHcy-S− denotes homocysteine. The first few steps until intermediate I are common to both pathways.
4.3.1. Relative Efficacy of H2S versus Cys-SSH or Hcy-SSH Synthesis by CSE
Cys-SSH and homocysteine-persulfide (Hcy-SSH) are formed by the CSE-catalyzed β-elimination of cystine and γ-elimination of homocystine, respectively (Chart 9).165,226 The kinetic parameters for CSE-catalyzed Cys-SSH formation are kcat = 0.21 s−1 and kcat/Km = 1.75 × 103 M−1 s−1 at pH 7.4 and 37 °C.226 Due to the insolubility of homocystine, the kinetic parameters for Hcy-SSH synthesis were determined at pH 8.5 and 37 °C and reported to be kcat = 1.5 s−1 and kcat/Km = 221 M−1 s−1. The activity of CSE at pH 7.4 is ∼11-fold lower than at pH 8.5. At physiologically relevant concentrations of substrate, Hcy-SSH synthesis by CSE is predicted to be negligible. H2S synthesis is estimated to represent ∼98.7% and Cys-SSH only 1.3% of CSE activity. However, a note of caution here is important. The intracellular concentration of cystine is low and not well determined. Allowing for a 25-fold higher hepatic cystine concentration (i.e., 5 μM) than previously reported (i.e., 0.2 μM),165 Cys-SSH and H2S synthesis are estimated to represent 33% and 66%, respectively of total CSE activity. CSE rather than CBS is predicted to be the major contributor of hepatic Cys-SSH synthesis at physiological concentrations of cystine, due to the higher protein levels of CSE versus CBS in this tissue.254 The kinetic analysis of H2S versus Cys-SSH synthesis suggests that, under conditions that lead to increased intracellular cystine levels (e.g., oxidizing conditions), Cys-SSH formation could be elevated.
4.3.2. Structural Organization of CSE
The structures of human CSE with (Figure 4) and without the PLP cofactor and with the suicide inactivator, propargylglycine (PPG) have been reported.255 The protein is a homotetramer (45 kDa monomers) and comprises two domains: a larger PLP domain (residues 9–263) and smaller C-terminal domain (264–401). The PLP is anchored via a Schiff base linkage to Lys212 and Asp187 is involved in an electrostatic interaction with the pyridinium nitrogen (Figure 5A). Tyr114 engages in a π-stacking interaction with the pyridinium ring. Residues contributed from the adjacent subunit also interact with the PLP. Reactions at the γ-carbon require a two-base mechanism (Chart 10); Tyr114 and Lys212 could serve this role.256 On the other hand, reactions at the β-carbon as in the synthesis of H2S from cysteine, involve a single base, i.e., Lys212. Consistent with this model, mutation of Tyr114 to phenylalanine not only fails to inhibit but, in fact, enhances H2S synthesis from cysteine 3.6-fold compared to wild-type CSE.257 Unfortunately the canonical cystathionine cleavage activity of this mutant was not reported.
Figure 4.
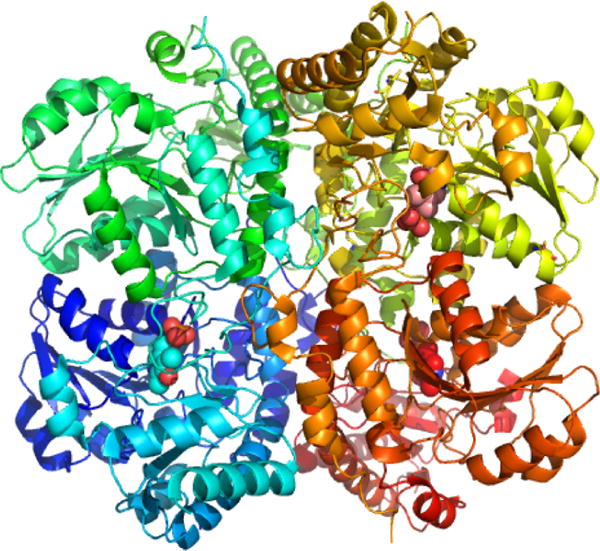
Structure of human CSE (PDB: 2NMP). Each of the four monomers is shown in a different color, and the three PLPs visible in the structure are in sphere representation.
Figure 5.
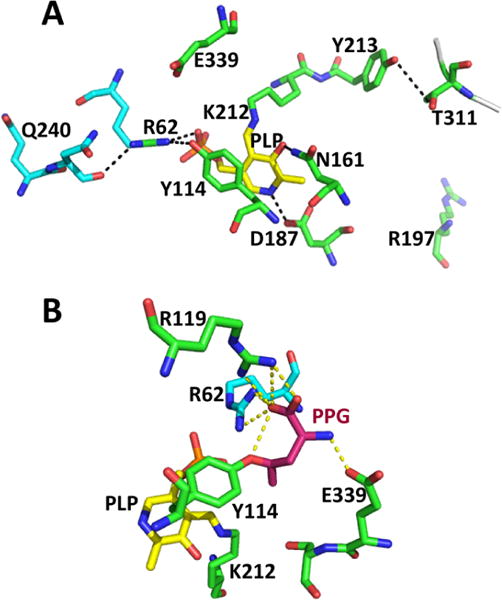
Close-up of the active site structure of human CSE. (A). Interactions between PLP (yellow) and residues donated by the two subunits (in green and cyan) are highlighted (PDB: 2NMP). (B). Structure of PPG-inactivated CSE (PDB: 3COG). The coloring is the same is in panel A. PPG is covalently linked to Tyr114 and is shown in pink.
A number of other active site residues are also highly conserved in CSE including Tyr60, Arg62, Thr189, and Arg375. Replacement of any of these residues with alanine leads to loss of H2S synthesis activity with the exception of the E339A mutant, which exhibits a 3-fold enhancement in the catalytic efficiency of H2S synthesis from cysteine.257 Based on a structure-based sequence alignment analysis, the residue corresponding to Glu339 in human CSE was predicted to be an important determinant of reaction specificity, i.e., for β- versus γ-elimination.256 Hydrophobic residues at this position are found in enzymes that catalyze β-elimination reactions as in plant and bacterial cystathionine β-lyases. In human CSE the hydrophobic E339Y substitution increased the catalytic efficiency of H2S synthesis 7-fold.257
Remarkably, the structure of inactivated CSE revealed that the PPG is not bound to PLP; instead, its Cγ is covalently linked to Tyr114 through a vinyl ether linkage (Figure 5B). In fact, the PPG is rotated 180° away from the PLP site, and its amino and carboxyl groups are involved in hydrogen bonding interactions with Arg62 (donated by an adjacent monomer), Glu339, and Arg119. In contrast, the PPG-bound structures of the Trichomonas vaginalis methionine γ-lyase (PDB: 1E5E) and the E. coli CsdB (PDB: 1I29),258 which catalyzes cysteine desulfuration and selenocysteine deselenation, respectively, show that the inhibitor is linked via a Schiff base to the PLP. In methionine γ-lyase, the Cγ of PPG is covalently linked to a tyrosine residue that is homologous to Tyr114 in human CSE. No additional covalent linkages are seen between PPG and CsdB, which is reversibly inhibited by PPG in contrast to irreversible inhibition of CSE and methionine γ-lyase.
4.3.3. Regulation of CSE
CSE levels are markedly reduced in malignant lymphoid cells, which exhibit cysteine auxotrophy.259 Since CSE deficiency can be clinically benign, it has been the subject of limited mechanistic and epidemiological scrutiny, and we understand little about how this enzyme is regulated in comparison to CBS. CSE has two CXXC motifs of which one, Cys307-X-X-Cys310, is relatively surface exposed. The second motif, Cys252-X-X-Cys255, is more buried and proximal to the dimer–dimer interface. The potential role of these CXXC motifs in allosteric regulation via redox changes or metal coordination is not known.
Protein kinase G-dependent phosphorylation of CSE at Ser377 has been reported to inhibit H2S production in the carotid body.260 However, Ser377 is completely buried and it is unclear how this residue can be phosphorylated or how the insertion of a phosphate group in the protein’s interior is stabilized. H2S synthesis by CSE reportedly increased in the presence of calcium/calmodulin,12 although other laboratories have not been able to reproduce this observation.261 Sumoylation of human CSE has been reported in vitro239 but the physiological relevance of this modification remains to be assessed.
4.4. Regulation of H2S Synthesis by the Transsulfuration Pathway
The multitude of reactions catalyzed by CBS and CSE165,190,226,249 begs the question as to how the trans-sulfuration pathway responds to cellular demands for cysteine versus H2S synthesis. At one level, this pathway might be controlled by gene expression, i.e., by the predominance or complete absence of one of the transsulfuration enzymes in some cells. For instance, if CBS expression is turned off, then cystathionine will not be synthesized in that cell type and therefore unavailable as a substrate for CSE (extracellular cystathionine levels are very low). Under these conditions, only the H2S synthesis reactions of CSE will be catalyzed. On another level, substrate levels control the dominant flux (i.e., serine versus cysteine for CBS and cysteine/homocysteine versus cystathionine for CSE). Finally, allosteric regulation by ligands whose concentrations change transiently in response to stimuli could regulate the dominant metabolic track that CBS and CSE operate on.
All three strategies are involved in regulating flux through the transsulfuration pathway and in triggering metabolic track switching in response to cellular needs.262 Binding of ligands, e.g., CO or NO• to the heme sensor in CBS, can flip the operating preference of the transsulfuration pathway from cysteine to H2S synthesis (Figure 6).262 Thus, despite the similar catalytic efficiencies for serine (2650 M−1 s−1) and cysteine (2882 M−1 s−1), CBS preferentially synthesizes cystathionine (from serine) over H2S (from cysteine) under basal conditions due to the higher cellular concentration of serine (∼1–2 mM) than cysteine (∼50–100 μM). The major product of CBS, i.e., cystathionine, is a more efficient substrate for CSE (8200 M−1 s−1) than is cysteine (270 M−1 s−1) or homocysteine (350 M−1 s−1).249 Hence, under basal conditions or upon AdoMet activation of CBS, the predominant flux in the transsulfuration pathway is toward cysteine synthesis (Figure 6, left). In contrast, under conditions that trigger H2S-signaling such as endoplasmic reticulum stress,263 CBS is inhibited by CO,217 a product of heme oxygenase-1 that is induced under these conditions.264 Furthermore, AdoMet exacerbates CO inhibition of CBS.223 In the absence of competition from cystathionine, CSE preferentially uses cysteine (which is ∼100-fold more abundant than homocysteine) to produce H2S (Figure 6, right). The track-switching model has important implications for sulfur metabolism. For instance, it predicts that H2S homeostasis will be dysregulated in CBS deficiency-induced hyperhomocysteinemia and suggests that H2S-dependent signaling cascades are perturbed in complex diseases like cardiovascular, neurodegenerative, neoplastic, and metabolic diseases where compromised endoplasmic reticulum function is a significant factor.265 Finally, there is growing evidence for crosstalk between NO•, CO, and H2S signaling pathways,262,266–268 but the molecular mechanisms underlying this interconnectedness are far from understood. The transsulfuration pathway represents a platform for the interplay between them via a heme sensor embedded in CBS.
Figure 6.
Heme-regulated switching in the transsulfuration pathway from cysteine (left) to H2S (right) synthesis.
4.5. Mercaptopyruvate Sulfur Transferase
MST is a sulfurtransferase that is found in the cytoplasm and in the mitochondrion.189,269 The specific activity of MST is reported to be 3-fold higher in mitochondria than in the cytosol in rat liver.189 MST catalyzes the transfer of the sulfur atom from 3-mercaptopyruvate, which is derived from cysteine via the action of cysteine aminotransferase (CAT, identical to aspartate aminotransferase), a PLP enzyme that utilizes α-ketoglutarate as a cosubstrate (Chart 11). In the first half reaction, sulfur transfer from mercaptopyruvate to an active site cysteine residue (Cys248 in the human MST sequence) results in a stable Cys-SSH intermediate and formation of pyruvate. In the second half reaction, the outer sulfur from the Cys-SSH intermediate is transferred to a nucleophilic acceptor, which can be a small molecule thiol or the protein, thioredoxin. H2S is subsequently liberated from the sulfur acceptor (Chart 11B).270,271
Chart 11. Reaction Catalyzed by MSTa.
a(A) 3-Mercatopyruvate is synthesized by L-cysteine aminotransferase (CAT), which requires α-ketoglutarate as a cosubstrate. In the first half reaction, MST transfers the sulfur atom from 3-mercaptopyruvate to an active site cysteine forming a Cys-SSH intermediate. (B) In the second half reaction, the outer sulfur from Cys-SSH is transferred to a small molecule thiol acceptor (RSH) or to thioredoxin (Trx) and subsequently released as H2S. (C) 3-Mercatopyruvate can be synthesized from D-cysteine via the action of D-amino acid oxidase (DAO).
Alternatively, the sulfur transfer can occur to cyanide, generating thiocyanate.272 Based on the kcat/Km parameters for sulfur transfer from mercaptopyruvate, the following order for decreasing catalytic efficiency has been reported for various biologically relevant acceptors: thioredoxin ≫ cyanide ≈ dihydrolipoic acid > cysteine > homocysteine > GSH.271 Dithiothreitol and mercaptoethanol can serve as surrogate acceptors in vitro. In nature, MST variants fused to thioredoxin are found273,274 suggesting that thioredoxin is the physiological sulfur acceptor for MST.275 In principle, other proteins could serve as sulfur acceptors by interacting directly with MST; however, the identities of such protein acceptors if they exist, are not known.
An alternative route for 3-mercaptopyruvate synthesis is via the oxidation of D-cysteine by the FAD-dependent peroxisomal enzyme, D-amino acid oxidase (Chart 11C), which was first inferred from studies on cyanide detoxification by rat hepatocytes.276 H2S production from D-cysteine is repressed by indole 2-carboxylate, an inhibitor of D-amino acid oxidase.277 While the highest MST levels are seen in liver, large intestine and kidney, the highest D-amino acid oxidase levels are seen in kidney and cerebellum. H2S production from D-cysteine is apparently significantly higher than from L-cysteine in kidney and in cerebellum.277 However, a cellular source for D-cysteine is not known.
MST belongs to the rhodanese superfamily and comprises two domains: an N-terminal domain extending from residues 1–138 and a C-terminal domain extending from residues 165–285 (Figure 7A). A long linker (residues 139–164) connects the two domains. The active site is located in a cleft between the two domains with each contributing residues to it. The structure of human MST has been captured with the persulfide intermediate (Cys248-SSH) and pyruvate (Figure 7B).271 A second structure was solved with an apparently unproductive complex in which the 3-mercaptopyruvate substrate forms a mixed disulfide with Cys248.271 Mutation of the active site cysteine to serine in rat MST leads to complete loss of activity.278
Figure 7.
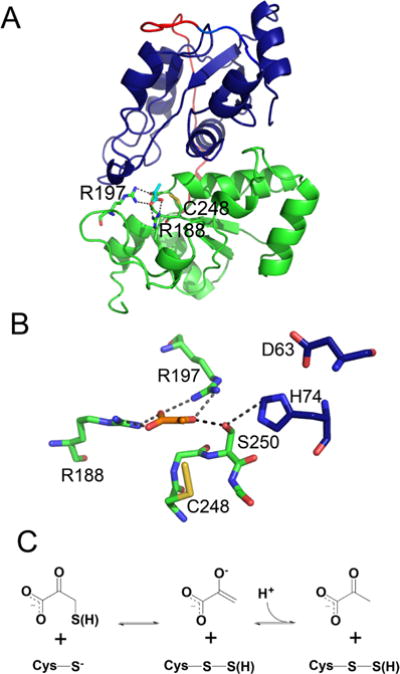
Structure and mechanism of human MST. (A) The N and C-terminal domains of human MST (PDB: 4JGT) are shown in blue and green, respectively, and the linker is in red. Pyruvate (cyan) and key active site residues including Cys248 in the Cys-SSH state are shown in stick representation. (B) Close up of the active site captured in a product complex with pyruvate (orange) and Cys-SSH (PDB: 4JGT). Two residues in the catalytic triad (D63 and H74) are donated by the N-terminal domain and are shown in blue. (C) Mechanism of the reaction between the 3-mercaptopyruvate substrate and the Cys248 thiolate.
Two conserved arginine residues, Arg188 and Arg197, which were known from mutagenesis studies to be important for substrate orientation,278 form ionic and hydrogen-bonding interactions with the carboxylate group of the substrate and product (Figure 7B). Arg197 additionally forms a hydrogen bond with the carbonyl group of the substrate/product. A Ser-His-Asp catalytic triad, first noted in the Leishmania MST crystal structure,279 is also present in human MST (Ser250-His74-Asp63). The N-terminal domain contributes Asp63 and His74 to the triad. The Cys248 residue is predominantly deprotonated, acoording to a pKa of 7.2.271 The reaction catalyzed by MST is proposed to involve attack by the thiolate of Cys248 on the sulfur of 3-mercaptopyruvate resulting in the formation of pyruvate and a persulfide (Cys-SSH). This product complex has been captured crystallographically (Figure 7B,C).271 The outer sulfur of the persulfide, most likely in the anionic Cys-SS− state, is surrounded by an hexapeptide loop and forms hydrogen bonds or other types of polar interactions with the four backbone amides of Gly249, Ser250, Val252 and Thr253 and with the hydroxyl of the latter. The role of the Ser250 hydroxyl in the catalytic triad might be to interact with the carbonyl group of mercaptopyruvate and facilitate catalysis by polarizing the C═O bond.279 An additional proposal, based on a QM/MM study, suggests that Ser250 is involved in the deprotonation of the 3-mercaptopyruvate substrate thiol and in the stabilization of the transient enolate oxyanion.280 However, mutation of the corresponding serine to alanine or lysine in rat MST decreases kcat/Km by a modest ∼4–10-fold, suggesting a relatively minor contribution of this residue to catalysis.278 The mechanism of sulfur transfer is proposed to consist of a stepwise process consisting of deprotonation of the 3-mercaptopyruvate thiol, sulfur atom transfer to the Cys248 thiolate to form nascent persulfenate and pyruvate enolate anions, and protonation of the enolate to the corresponding pyruvate enol, which tautomerizes to the keto form.280 The calculated activation barrier for this process is ∼67 kJ mol−1. In contrast, an alternative process of SH transfer that does not require the initial deprotonation of the 3-mercaptopyruvate thiol has a much higher calculated activation energy barrier, 180 kJ mol−1.280 The electrostatic repulsion between the thiolates of the substrate and of Cys248 during the sulfur atom transfer is proposed to be reduced by interactions of the sulfur with the surrounding amide and hydroxyl groups. Release of pyruvate followed by attack of the thiol acceptor on Cys-SSH moves the sulfane sulfur out of the MST active site, regenerating the resting enzyme (Chart 11). The attack by the thiolate acceptor on the outer sulfur of the persulfide would be favored by the specific geometry of the active site and by the increase in electrophilicity of the outer sulfur caused by hydrogen bonding to the amide and hydroxyl groups of the surrounding loop.280
4.5.1. Regulation of MST
Our understanding of how CAT/MST-dependent H2S synthesis is regulated is very limited. The active site cysteine in MST is sensitive to oxidation and could potentially be involved in redox regulation of the enzyme.275 Reversible inhibition of rat MST by treatment with stoichiometric quantities of hydrogen peroxide or tetrathionate and rescue of the resulting cysteine sulfenate by reductants such as dithiothreitol or thioredoxin has been reported.275 It is not known whether the active site cysteine residue is also susceptible to overoxidation, which would lead to irreversible inactivation.
A role for redox regulation of rat MST via formation of an intersubunit disulfide bond has been reported. Of the three surface-exposed cysteines in rat CST, two (Cys154 and Cys263) form an intersubunit linkage under oxidizing conditions.275,281 Reduction of the disulfide bond by thioredoxin increases MST activity 4.6-fold. These cysteines are not conserved in the human protein, which is only observed to exist as a monomer.271
Calcium (0–2.9 μM) reportedly inhibits CAT/MST-dependent H2S production in mouse retinal lysate.282 The inhibitory effect of calcium appears to be on CAT rather than MST since H2S production from 3-mercaptopyruvate was unaffected by the presence of calcium. The mechanisms by which calcium regulates CAT remain to be elucidated.
4.6. Inhibitors of H2S Biogenesis
The paucity of specific inhibitors of H2S-producing enzymes has limited advancements in the field and led to the indiscriminate use of nonspecific PLP enzyme inhibitors such as aminooxyacetic acid and hydroxylamine to “target” H2S production.181 Alternatively, aspartate, the preferred substrate for CAT/AAT, has been used to indirectly inhibit MST-dependent H2S production. Historically, the acetylenic substrate analog, propargylglycine, was the first mechanism-based inactivator designed to target CSE.283 The structure of human CSE revealed that propargylglycine is covalently linked to Tyr114 in the active site but is not attached to the PLP via a Schiff base.255 Propargylglycine exhibits low bioavailability and is typically used at high (1–10 mM) concentrations in cell culture experiments. It also exhibits off-target activity with alanine aminotransferase,284 limiting its utility.
An assessment of the commonly used generic inhibitors confirmed a lack of selectivity for CBS versus CSE with three compounds: hydroxylamine, aminooxyacetic acid, and trifluoroalanine.285 In contrast, two compounds showed selectivity for CSE. Of these, β-cyano-L-alanine (IC50 = 14 μM for human CSE285), a plant-derived neurotoxin, is known to act as a suicide inhibitor of PLP enzymes, which abstract a proton from the β-carbon of substrates, e.g., alanine aminotransferase.286,287 Injection of β-cyano-L-alanine induces convulsions and rigidity and leads to cystathionuria in rat, indicating CSE inhibition in vivo.288 Aminoethoyxvinylglycine (reported IC50 = 1 μM285 and Ki = 10.5 μM289 for human CSE) is a known inhibitor of ethylene synthesis by the plant enzyme, 1-aminocyclopropane-1-carboxylate synthase290 and of bacterial cystathionine β-lyase.291 It is likely to be a more useful reagent in cell culture experiments although information on its bioavailability and toxicity in cell culture and in whole animals is limited. Aminoethoxyvinyl glycine is a slow-binding but reversible inhibitor of cystathionine β-lyase.291
More recent efforts to address the gap in selective targeting of H2S-generating enzymes have involved high throughput screening292–294 and rational mechanism-based inhibitor design.295 In one study, CBS inhibitors were designed to mimic the product, cystathionine but the α-amino group that forms a Schiff base, was substituted by the heteroatomic functional groups: –NHNH2, –ONH2, and –NHOH to form a hydrazone, oxime, and nitrone linkage, respectively, with the PLP.295 The inhibitors showed modest potency against CBS, but their activity against CSE, which also binds cystathionine, was not tested. A high throughput assay of a library of 1900 compounds yielded a O-polymethoxylated flavone, tangeritin, and 1,4-napthaquinone, exhibiting modest IC50 values and are unlikely to be useful inhibitors for cell culture studies.293 A second high throughput screen of 6491 compounds yielded mostly large flavonoids as CBS inhibitors with a subset showing selectivity against CSE although selectivity against MST was not evaluated. These compounds are unlikely to selectively inhibit CBS in the cell.294 A third screen identified benserazide, a known DOPA decarboxylase inhibitor used for managing Parkinson’s disease, as a CBS inhibitor with modest selectivity against CSE and MST.292
5. ENZYMATIC H2S OXIDATION
The toxicity of H2S is associated with its inhibition of cytochrome c oxidase (half maximal inhibition occurs at ∼0.3 μM in cell extracts and ∼20 μM in intact cells).296 Steady-state H2S concentrations are maintained at low levels (6–80 nM)159,179 except in aorta, where the concentration is reportedly ∼20–200-fold higher.178 Hence, cells have strategies for avoiding H2S build-up. One such strategy involves the canonical H2S oxidation pathway, which exists in the mitochondrion and converts H2S to thiosulfate and sulfate,297 with the product distribution being tissue specific.298–300 Alternatively, H2S can be oxidized by globins to thiosulfate and protein-bound hydropolysulfides.301,302 Much less is known about the enzymology of H2S oxidation compared to its biogenesis, and in this section, the structures and functions of the human proteins in the mitochondrial H2S oxidation pathway are discussed, while globins that have H2S oxidation capacity are covered in section 6.
5.1. Mitochondrial H2S Oxidation Pathway
The eight-electron oxidation of H2S to sulfate starts in the mitochondrial matrix and is completed in the intermitochondrial membrane space where sulfite oxidase resides (Figure 8).
Figure 8.
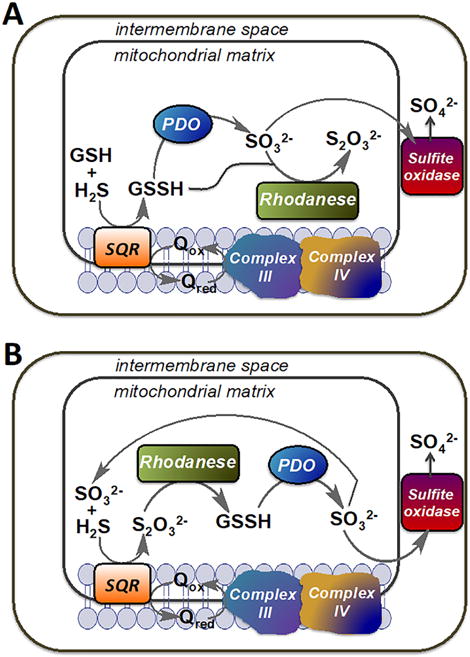
Alternative models describing the organization of the mitochondrial sulfide oxidation pathway. (A) In this model, GSH is the sulfur acceptor from SQR and the product, GSSH, is utilized by either PDO or by rhodanese generating sulfite and thiosulfate, respectively. (B) In this model, sulfite is the sulfur acceptor from SQR and the product, thiosulfate, is utilized by rhodanese to generate GSSH, which is subsequently oxidized by PDO to sulfite. Sulfite is eventually oxidized to sulfate by sulfite oxidase. Q represents coenzyme Q.
The first enzyme in the pathway is sulfide quinone oxidoreductase (SQR), which catalyzes a two-electron oxidation of H2S to persulfide and reduces coenzyme Q (CoQ). The latter enters the electron transfer chain at the level of complex III, thus connecting H2S oxidation to ATP and reactive oxygen species formation.303,304 Beyond this step, there is controversy regarding how the pathway is wired. Depending on whether the persulfide acceptor from SQR is GSH or sulfite, glutathione persulfide (GSSH) or thiosulfate results. GSSH synthesis by SQR would set up a competition between its utilization by persulfide dioxygenase (PDO also referred to as ETHE1), the product of the ethe1 gene, and by rhodanese (Figure 8A). Alternatively, if thiosulfate is the product of SQR, it would first have to undergo a sulfur transfer reaction catalyzed by rhodanese to form GSSH, which would then be oxidized to sulfite by PDO (Figure 8B). Sulfite is eventually oxidized to sulfate via sulfite oxidase.
5.2. Sulfide Quinone Oxidoreductase
SQR is a member of the flavin disulfide reductase superfamily, which catalyzes pyrimidine nucleotide-dependent reduction of substrates.305 Like other family members, SQR houses two redox centers, an active site disulfide and a noncovalent FAD cofactor that relays electrons from H2S to CoQ. Human SQR is a dimeric306 membrane-anchored protein with a globular domain that faces the mitochondrial matrix. The structure of a mammalian SQR is not yet available, but structures of SQR from Acidianus ambivalens307 (Figure 9) and other bacteria308,309 provide insights into its likely active site architecture.
Figure 9.
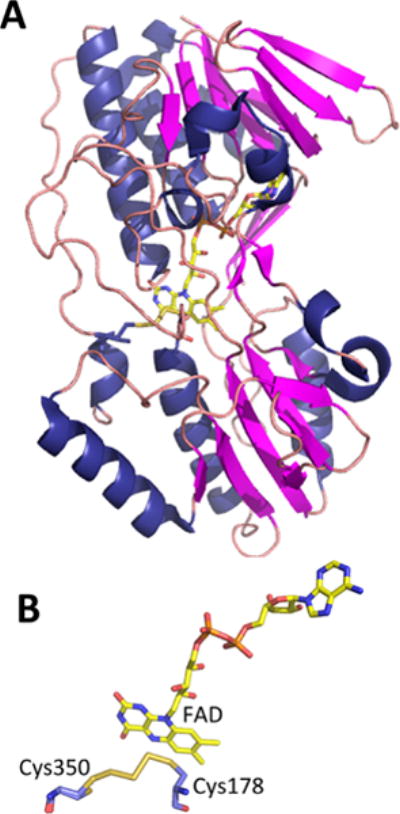
Structure of SQR from A. ambivalens. (A). Structure of an SQR subunit (PDB: 3H8L) in which the covalently bound FAD is shown in stick representation. (B) Close-up of the active site showing FAD and a bridging trisulfide intermediate between Cys350 and Cys178.
SQR is a combination of a sulfurtransferase that generates an active site Cys-SSH intermediate and an oxidoreductase, which oxidizes H2S as it reduces CoQ with FAD serving as an intermediate electron carrier (Chart 12). The sulfane sulfur from Cys-SSH is transferred to an acceptor, which can be GSH, sulfite, cyanide, or a second equivalent of H2S.297,310,311 As discussed in more detail below, the identity of the acceptor under physiological conditions is a subject of controversy. It is noteworthy that the bacterial SQRs do not require sulfur acceptors; instead, they form polysulfide or cyclooctasulfur products. In fact, a trisulfide intermediate is seen in the A. ambivalens SQR structure (Figure 9B).307 Also unlike some bacterial SQRs, the FAD in human SQR is bound noncovalently.310 It exhibits maxima at 385 and 450 nm and a promiment shoulder at 473 nm. The two-electron redox potential of the bound FAD is −123 ± 7 mV.306
Chart 12. Overview of the Reaction Catalyzed by SQRa.
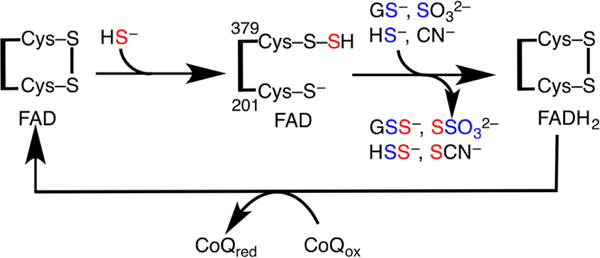
aIn the sulfurtransferase steps, HS− attacks the disulfide bond in SQR forming a persulfide at Cys379 and the sulfane sulfur (in red) is transferred to an acceptor (GSH, sulfite, sulfide, or cyanide). In the electron transfer steps, two electrons are transferred from HS− through the disulfide to FAD and then to CoQ.
The detailed mechanism of SQR is complex and involves a two-step sulfur transfer and a multistep electron transfer through the protein (Chart 13).306,310,311 The initial step involves nucleophilic attack of H2S on the active site disulfide (presumably formed between Cys301 and Cys379 in human SQR) and leads to the formation of Cys379-SSH and Cys201-S− on the re face of FAD. At this step, the formation of an unusually strong charge transfer complex is observed, which exhibits a maximum at 673 nm and extends out to 900 nm.306 The bimolecular rate constant for the formation of the H2S-dependent charge transfer species is 3.4 × 105 M−1 s−1 at pH 7.4 and 4 °C.306 In the presence of CoQ, the rate constant for the formation of the charge transfer complex increases 2.9-fold indicating that the reaction occurs more efficiently in a ternary complex.
Chart 13. Postulated Reaction Mechanism of SQRa.
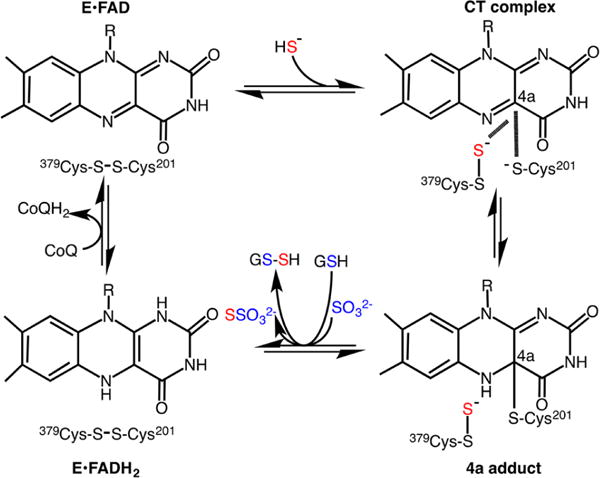
aNucleophilic attack of HS− on the active site disulfide results in the formation of a Cys-SSH intermediate at Cys379 and a charge transfer (CT) complex, which collapses to a postulated 4a adduct. A sulfur acceptor viz. GSH or sulfite moves the sulfane sulfur from the active site, forming GSSH or thiosulfate (S2O32−), respectively, and restoring the active site disulfide. Electron transfer from the reduced flavin, FADH2, to CoQ completes the catalytic cycle.
A charge transfer complex is also formed in the presence of sulfite, which presumably adds to the disulfide bond forming a sulfocysteine intermediate (Cys379-SSO32−). However, the bimolecular rate constant for sulfite addition is only 1 × 102 M−1 s−1 at pH 7.4 and 4 °C, which is 3000-fold lower than the kon for H2S and is unlikely to be significant except perhaps under pathological conditions when sulfite concentrations are elevated.
A variety of small molecules can accept the sulfane sulfur from the Cys-SSH intermediate in SQR, exhibiting a range of catalytic efficiencies (kcat/KM): sulfite (1.7 × 106 M−1 s−1), cyanide (5.1 × 105 M−1 s−1, pH 8.5), H2S (2.3 × 105 M−1 s−1), and GSH (5.1 × 103 M−1 s−1) at pH 7.4 and 25 °C unless noted otherwise.310,311 Cysteine and homocysteine can also serve as sulfane sulfur acceptors and exhibit catalytic efficiencies that are similar to GSH.311
The controversy regarding the physiological sulfur acceptor (Figure 8) originated from the reported inability of GSH to support SQR activity,310 which has since been shown to be incorrect.311 While GSH is abundant (1–10 mM depending on the cell type), sulfite, which is reactive, is present at low steady-state concentrations. A recent study reported that the intracellular sulfite concentration in rat liver is 9.2 μM based on HPLC analysis of tissue lysates, albeit without mass spectrometric or any other experimental validation of the identity of the compound that comigrated with authentic sulfite.312 Based on the kcat/KM values discussed above, the estimated apparent kcat at 10 μM sulfite is 16 versus 36 s−1 at 7 mM GSH, as previously predicted by kinetic simulations.311 In addition to the high probability that tissue sulfite concentrations were not determined accurately,312 the use of sulfite as the primary sulfane sulfur acceptor by SQR runs counter to logic for the following reasons. First, it would appear unlikely that if the SQR active site evolved to use a small molecule like sulfite as a substrate, that it could just coincidentally bind the tripeptide substrate, GSH, too. The crystal structure of human SQR should provide needed insights into this debate. Second, the dependence of the first step in a H2S oxidation pathway on sulfite, a six-electron oxidized product of the pathway (Figure 8B), would appear to be organizationally illogical and was described as “convoluted” even by its proponents.310 Third, thiosulfate is a poor substrate for rhodanese for GSSH synthesis,311 which is required for continued oxidation since GSSH is the only known substrate for PDO. The only other known thiosulfate sulfurtransferase that catalyzes the conversion of thiosulfate to GSSH is located in the cytoplasm313 and its involvement in the mitochondrial H2S oxidation pathway would require the unlikely translocation of the reactive GSSH intermediate across compartments. For these reasons, it appears likely that the flow of sulfur through the H2S oxidation pathway is HS− → GSSH → SO32− → SO42− + S2O32− as shown in Figure 8A.
5.3. Persulfide Dioxygenase
PDO is a nonheme mononuclear iron-containing mitochondrial matrix protein, which belongs to the metallo β-lactamase superfamily family and has a subunit molecular mass of 28 kDa.314 It catalyzes an oxygen-dependent oxidation of GSSH giving sulfite and GSH (eq 20).297,315,316
| (20) |
As isolated, the iron is predominantly in the ferrous oxidation state317 and is coordinated by a 2His-1Asp facial triad.318,319 Mutations in PDO lead to ethylmalonic encephalopathy, which is inherited as an autosomal recessive disease and is associated with severe neurological and gastrointestinal clinical presentations.315,320,321 Thiosulfate and H2S accumulate in PDO deficiency, and intriguingly, a tissue-specific reduction in cytochrome c levels is seen in muscle and brain.315,322,323
In solution, human PDO behaves as a mixture of monomer and dimer316,319 and, like the Arabidopsis thaliana and bacterial enzymes,318,324 crystallizes as a dimer.319 The structure of human PDO reveals an αββα fold typical of metallo β-lactamase family members (Figure 10A).
Figure 10.
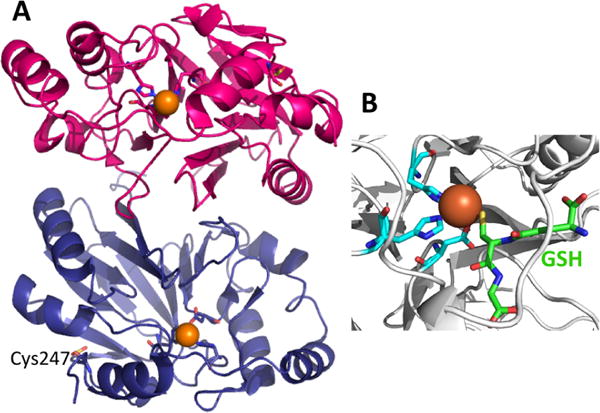
Crystal structure of PDO. (A) Structure of human PDO (PDB: 4CHL) in which the protomers are shown in pink and blue, the iron ion as an orange sphere, and the coordinating histidines and aspartate in stick representation. Cys247 is oxidized as cysteine sulfinate and is also shown in stick representation. (B). Close up of the P. putida PDO (PDB: 4YSL) with bound GSH (green). The cysteine sulfur of GSH is proximal to the iron ion, which is coordinated by a 2His-1Asp facial triad.
His79, His135, and Asp154 coordinate iron together with three water molecules, completing an octahedral coordination. A deep channel exists that leads to the active site and is predicted to be where the GSSH substrate binds. In fact, the interactions between the product, GSH, and the active site residues are visible in the Pseudomonas putida PDO structure where the sulfur of GSH is within 2.5 Å of the iron (Figure 10B).324 GSH binding displaces a single water ligand. Interestingly, Cys247, located near the surface, is oxidized to cysteine sulfinate in the structures of human and A. thaliana PDO.318,319 It is not known whether this oxidative modification is autocatalytic and whether it has mechanistic/regulatory import or is silent.319
Both GSSH and coenzyme A persulfide serve as substrates; the specific activity of PDO is, however, ∼50-fold higher with GSSH than with coenzyme A persulfide. The kcat/KM with GSSH is 1.4 × 105 M−1 s−1 at pH 7.4, 22 °C. There has been limited interrogation of the reaction mechanism of PDO. In analogy with related dioxygenases such as cysteine dioxygenase, a mechanism has been proposed in which binding of GSSH displaces one or more water molecules, simultaneously creating the binding site for O2 (Chart 14, [1–2]).316,319 The crystal structure of the P. putida PDO with bound GSH324 and the structure of human PDO in which GSSH has been modeled,319 show monodentate coordination by the sulfur atom. This is distinct from the bidentate coordination seen in cysteine dioxygenase in which the amine and sulfhydryl groups of cysteine serve as ligands to iron.325 In the mechanism proposed for PDO,316,319 binding of GSSH and of O2 leads to an FeIII-superoxo intermediate (Chart 14, [3]) that is in resonance with an FeII-superoxo species in which the sulfane sulfur ligand has a partial cation character [4]. Recombination of the sulfane sulfur radical cation and the FeII-superoxo species leads to a cyclic peroxo intermediate [5]. Following O–O bond cleavage, a reactive metal bound oxygen and a sulfoxy cation [6] are formed. Alternatively, cleavage of the Fe–O bond and transfer of the activated oxygen to the sulfoxy sulfur cation gives [7]. Rearrangement of either [6] or [7] gives [8] (Chart 14), which following hydrolysis, yields sulfite.
Chart 14. Postulated Reaction Mechanism of PDOa.
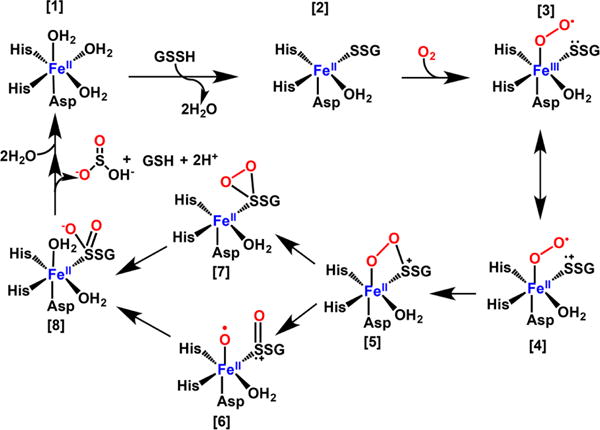
aBinding of GSSH to the resting enzyme [1] creates a binding site for O2 [2]. Formation of a superoxo-FeIII [3] in resonance with a biradical FeII species [4] leads to formation of a cyclic peroxo-FeII species [5]. Cleavage of the O–O bond gives [6]. Alternatively, cleavage of the Fe–O bond gives [7]. Binding of H2O [8], sets up hydrolysis and formation of the product, sulfite. Alternatively, the water that remained coordinated to the metal center could be used for the final hydrolysis step.
Two patient mutations in PDO, T1521 and D196N, have been characterized biochemically.316 Both mutations affect the iron content of PDO, decrease its thermal stability, and have smaller effects on either the Km for GSSH and/or on kcat. The mutated residues are distal from the active site and the decrease in thermal stability (by 10–15 °C) is likely to be the major biochemical penalty leading to disease.
5.4. Rhodanese
Rhodanese is a sulfurtransferase found in the mitochondrial matrix. Historically, rhodanese was thought to have a role in cyanide detoxification since it can convert thiosulfate and cyanide to thiocyanate (Chart 15A).326 More recently, its role in the mitochondrial H2S oxidation pathway has been demonstrated where it preferentially catalyzes thiosulfate synthesis versus utilization (Chart 15, B vs C).297,311 Elevated rhodanese expression is correlated with lower adiposity and knockout of the rhodanese gene in mouse leads to markedly increased diabetes.327 Other phenotypic and metabolic expressions of rhodanese deficiency have not yet been described.
Chart 15. Reactions Catalyzed by Rhodanesea.
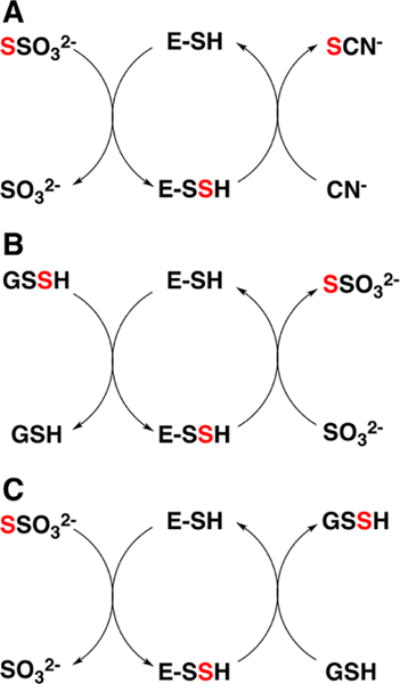
aRhodanese exhibits varied sulfur transferase activities including: (A) thiosulfate:cyanide sulfurtransferase, (B) GSSH:sulfite sulfurtransferase, and (C) thiosulfate:GSH sulfurtransferase. E-SSH denotes the enzyme-bound Cys-SSH intermediate. The red color traces the fate of the sulfur from the donor to the acceptor.
Rhodanese, like MST, belongs to the sulfurtransferase superfamily, characterized by a “rhodanese” domain fold with an α/β topology named after this protein, which is present in a single copy, in tandem repeats or fused with other proteins in members of this family.328,329 Human rhodanese is a monomeric protein with a molecular mass of 33 kDa.330 Two polymorphic variants of rhodanese have been described, which lead to E102D and P285A substitutions.331 The Glu102 residue is located at the entrance to the active site pocket and is ∼19 Å away from the catalytic cysteine, Cys257. Pro285 is surface exposed and ∼17 Å away from the active site. Interestingly, both variants exhibit greater thermal stability than wild-type rhodanese.330
The structure of bovine liver rhodanese332 serves as a useful model for the human protein with which it shares 89% sequence identity (Figure 11). The structure comprises two globular domains of approximately equal length and an active site that is housed in a cleft between the two domains. The C-terminal domain provides the catalytic cysteine and a mixture of hydrophilic and hydrophobic residues wall in the active site. The reaction mechanism of rhodanese involves an active site Cys-SSH intermediate from which the sulfane sulfur is transferred to an acceptor. In the bovine rhodanese structure, the Cys-SSH intermediate is stabilized by hydrogen bonds from the hydroxyl group of Thr252 and the backbone amides of Arg248, Lys249, and Val251.332 The catalytic triad present in MST is absent in rhodanese. This coincides with its preferential substrates being predominantly deprotonated at physiological pH.280
Figure 11.
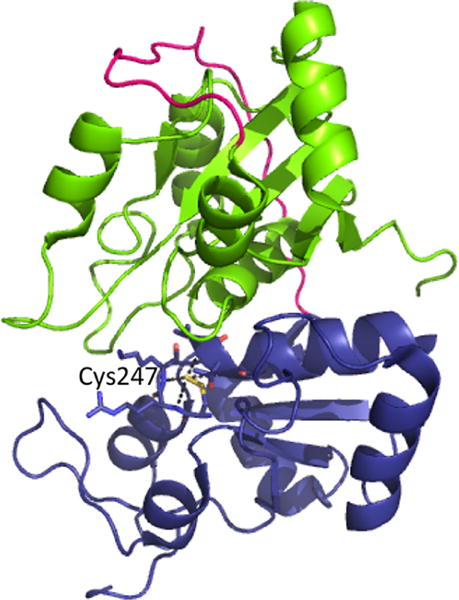
Structure of bovine rhodanese (PDB: 1RHD). The N- and C-terminal domains are shown in green and blue with the intervening linker in pink. A Cys-SSH intermediate is stabilized at Cys247 in the active site, via hydrogen bonding interactions with neighboring residues.
Like MST, the reaction catalyzed by rhodanese involves two sulfur transfer reactions: from the sulfur donor to the active site cysteine and from Cys-SSH to the sulfur acceptor (Chart 15). The various sulfur transfer reactions catalyzed by wild-type rhodanese and its polymorphic variants have been characterized.311,330 Human rhodanese preferentially catalyzes sulfur transfer in the direction of GSSH → S2O32− (kcat/Km(sulfite) = 6.5 × 106 M−1 s−1 at pH 7.4, 37 °C) versus in the reverse direction, S2O32− → GSSH (kcat/Km(GSH) = 0.03 × 103 M−1 s−1 at pH 7.4, 37 °C). Based on these values, rhodanese exhibits an estimated 217 000-fold discrimination against utilization of thiosulfate versus GSSH as a sulfur donor. The Km values for GSSH (450 μM) and sulfite (60 μM) in the GSSH → S2O32− sulfurtransfer reaction are lower than the Km values for the substrates, thiosulfate (340 μM) and GSH (21 mM), in the S2O32− → GSSH direction. Like wild-type rhodanese, the polymorphic variants also exhibit a marked preference for making rather than utilizing thiosulfate and exhibit comparable catalytic efficiencies for this reaction (kcat/Km(sulfite) = 1.5–4.2 × 103 M−1 s−1 at pH 7.4, 37 °C).330
Interestingly, cysteine and homocysteine can replace GSH as sulfur acceptors in the S2O32− → GSSH sulfur transfer reaction with catalytic efficiencies (kcat/Km(Hcy or Cys) ≈ 0.4 × 103 M−1 s−1 at pH 7.4, 37 °C) that are ∼13-fold higher than with GSH.311 However, the concentration of these amino acids is low in most tissues, and their high Km values (∼21 mM each) make them unlikely substrates for the reverse reaction under physiological conditions compared to GSH. In some tissues like kidney, which has high cysteine,333 or under pathological conditions like homocystinuria, which is characterized by elevated homocysteine,193 these sulfur containing amino acids might become relevant substrates, albeit in the less favorable reverse sulfur transfer reaction from thiosulfate.
The thiosulfate:cyanide transfer kinetics of wild-type rhodanese are characterized by relatively low catalytic efficiency (kcat/Km(KCN) = 31 × 103 M−1 s−1 at pH 7.4, 25 °C) and high Km for cyanide (29 mM) making a role for rhodanese in cyanide detoxification unlikely. Interestingly, the E102D mutant shows higher efficiency (kcat/Km(KCN) = 534 × 103 M−1 s−1) while the P285A mutant shows similar efficiency (48 × 103 M−1 s−1) as the wild-type protein in the cyanide detoxification assay.
5.5. Sulfite Oxidase
The nearly ubiquitous presence and conserved architecture of sulfite oxidases is consistent with the evolutionarily ancient role of this protein in protecting against sulfite-induced damage.334 Sulfite oxidase is a molybdenum containing protein, which catalyzes the two-electron oxidation of sulfite to sulfate in which water serves as the oxygen source (eq 21).
| (21) |
In humans, sulfite oxidase is a soluble enzyme found in the mitochondrial intermembrane space. Electrons from the sulfite oxidation reaction are passed via a heme cofactor found in vertebrate sulfite oxidases to cytochrome c and from there to complex IV. The important role of sulfite oxidase in detoxifying sulfite is borne out by its presence in the peroxisomal compartment in plant cells where it functions to remove toxic sulfite derived from atmospheric sulfur dioxide or from catabolism of sulfur containing amino acids, rather than a role in sulfur assimilation in the chloroplast.335 Sulfite oxidase deficiency is an autosomal recessive disorder that presents with severe neonatal neurological problems.336 It can result from defects in the synthesis of the molybdopterin cofactor or from mutations in the gene encoding sulfite oxidase itself.
Sulfite oxidase is a homodimer with a subunit molecular mass of 52 kDa and contains a heme b cofactor housed in the N-terminal domain that is connected via a flexible linker to the central molybdopterin-binding domain, which in turn is followed by the C-terminal dimerization domain. The structure of chicken sulfite oxidase337 (Figure 12) serves as a useful model for the human protein with which it shares 68% sequence identity. The 5-coordinate molybdenum center has square pyramidal geometry (Chart 16A). Of the three sulfur ligands, two are derived from the dithiolene group of the molybdopterin cofactor, while the third is donated by Cys207 (human sequence numbering). The remaining coordination sites are occupied by equatorial and apical oxo ligands. The substrate-binding pocket in chicken sulfite oxidase comprises Arg138, Arg190, and Arg450 in addition to Tyr322 and Trp204. A pathogenic mutation in a patient with severe sulfite oxidase deficiency has been mapped to Arg160, which corresponds to Arg138 in the chicken sequence.338
Figure 12.
Structure of dimeric chicken sulfite oxidase (PDB: 1SOX). The two subunits are shown in blue and magenta, respectively, and the heme (orange) and molybdopterin (MPT, cyan) cofactors are in sphere representation.
Chart 16. Molybdopterin Cofactor in Sulfite Oxidase and Redox Changes during the Catalytic Cyclea.
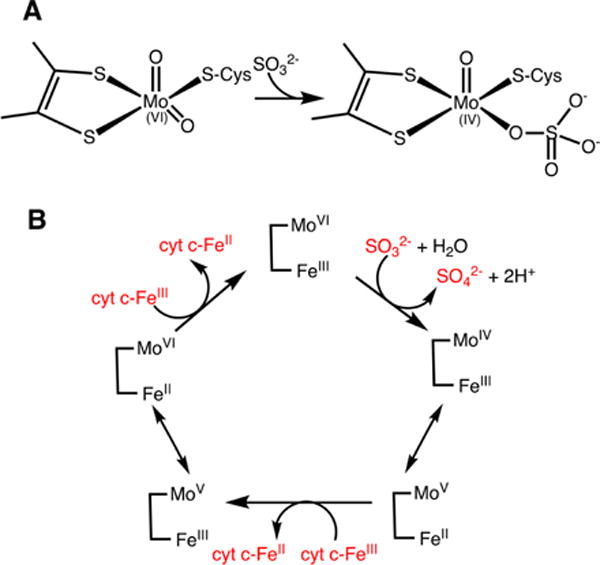
a(A) Attack of sulfite on an oxo/hydroxyl ligand reduces the molybdenum ion. (B) The reaction cycle of sulfite oxidase involves an initial two-electron reduction of the molybdenum center, which is subsequently oxidized in two one-electron steps via intramolecular electron transfer to the heme. The latter in turn, transfers electrons to the heme in cytochrome c (cyt c) in an intermolecular process.
The catalytic cycle of sulfite oxidase comprises reductive and oxidative half reactions. Sulfite binds transiently to an equatorial oxo/hydroxyl ligand and reduces the molybdenum center to MoIV (Chart 16A). Sulfite is released as sulfate following hydrolysis. The kcat/Km(sulfite) is 4.7 × 106 M−1 s−1 at pH 7.5 and 25 °C.339 In the oxidative cycle, sequential one-electron transfers occur from MoIV to the exogenous electron acceptor, i.e. heme iron in cytochrome c, via the heme b cofactor in sulfite oxidase (Chart 16B). The catalytic mechanism of sulfite oxidase has been extensively characterized by spectroscopic and rapid reaction kinetic methods combined with mutagenesis studies and has been reviewed recently.334
6. CHEMICAL BIOLOGY OF H2S
Notwithstanding the growing body of evidence for the biological roles of H2S, the gap between the physiological effects of H2S and its mechanism of action remains large. Based on chemical principles, H2S reactivity can be categorized into three reaction groups: (i) binding to and/or redox reactions with metal centers, (ii) cross-talk with and scavenging of reactive oxygen (ROS) and reactive nitrogen species (RNS), and (iii) oxidative modification of protein cysteines to form the corresponding persulfides (Figure 13).340–342 In this section, we provide an overview of the reactions grouped in (i) and (ii) while protein persulfidation is discussed in section 7.
Figure 13.
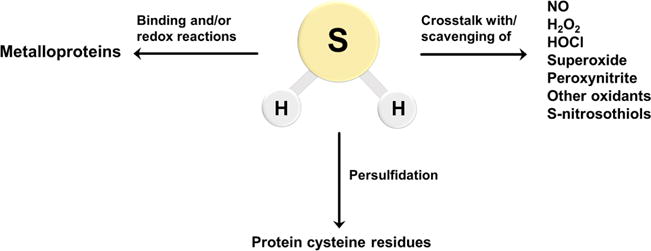
Biological reactivity of H2S. H2S can react directly with oxidants such as superoxide, HOCl, and ONOO−. It can also react with NO• and S-nitrosothiols leading to the formation of other signaling species (right arrow). Metal centers in proteins can bind H2S for delivery to specific targets, be reduced by H2S, or catalyze sulfide oxidation chemistry (left arrow). H2S is also involved in the modification of protein cysteine residues leading to persulfide (Cys-SSH) formation (central arrow).
6.1. Interaction of H2S with Metal Centers
Different possilbilites exist for the interaction between H2S and a metal center. First, H2S (or HS−) can coordinate the metal ion. Second, H2S can reduce the metal center, concomitantly forming HS• and other downstream sulfur oxidation products. Third, H2S can modify heme porphyrins covalently.
The first described biological effect of H2S identified in 1929 by Keilin was its toxicity, which was ascribed to inhibition of respiration by targeting cytochrome c oxidase (CcO).343 The reaction with H2S was later used to stabilize cytochrome c oxidase for its spectral characterization.344,345 CcO is the final acceptor in the mitochondrial electron transport chain, which uses electrons delivered by cytochrome c to reduce oxygen to water.346 It contains two copper centers (CuA and CuB) and two heme iron centers (a and a3).347–349 Oxygen binds to ferrous heme a3. NO• and CO also inhibit the enzyme reversibly.350–352 Inhibition of CcO by H2S is almost as strong as with CN−, with Ki of ∼0.2 μM,353 and it is noncompetitive with respect to both cytochrome c and oxygen. The work of Nicholls and colleagues was instrumental in pointing out that, in addition to inhibiting CcO, H2S might also serve as a substrate/electron donor.353–356 They observed that the initial product of H2S/CcO (aa3) interaction is not an inhibited form of the enzyme and that >1 mol of sulfide/mol CcO was required for full inhibition.355 Binding of H2S to catalytically active CcO (kon = 1.5 × 104 M−1 s−1, koff = 6 × 10−4 s−1, and KD = 4 × 10−8 M−1) is much tighter than the binding of ligands such as azide or fluoride (Chart 17).
Chart 17. Interaction of H2S with Cytochrome c Oxidasea.
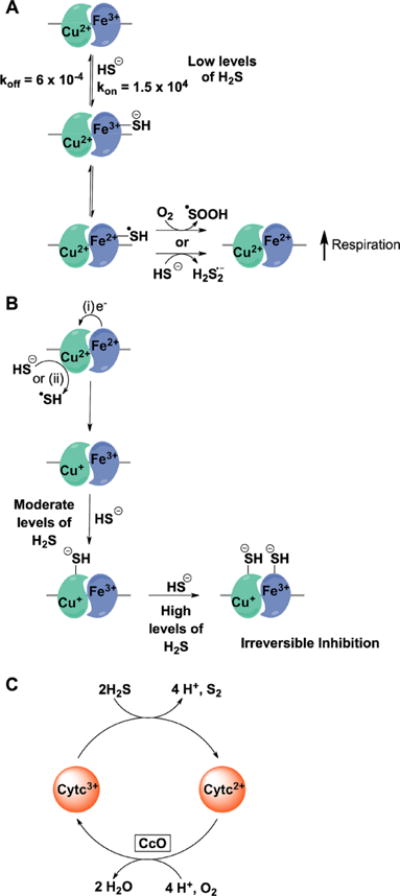
a(A) At low concentrations, H2S binds to ferric heme a3 and reduces it with concomitant formation of HS• which can react with either another molecule of H2S or with oxygen. Reduction of heme a3 leads to an increase in oxygen consumption. (B) At moderate levels, H2S interacts with the CuB+ center forming a stable CuB–SH− complex, which is difficult to oxidize. CuB+ is formed by electron transfer from ferrous heme a3 or by a direct reduction by H2S. At higher levels of H2S, the CuB–SH− complex induces a conformational change and causes further binding of H2S to ferric heme a3. (C) Alternatively, H2S can reduce cytochrome c thereby increasing CcO reduction and respiration.
A generalized mechanism has been proposed to explain the interaction of H2S with CcO.356,357 At low levels (1:1 ratio of H2S:CcO), H2S reduces ferric heme a3 with concomitant oxidation to HS•. The reduction of this heme iron by H2S is thermodynamically unfavorable, and it is likely that HS• removal, e.g., by reaction with HS− to form H2S2•− (eq 9), pulls the reduction in the forward direction. Heme iron reduction promotes oxygen binding and reduction, explaining why low concentrations of H2S stimulate respiration.355,358 Alternatively, HS• can react with oxygen to form HSOO• (eq 10) further contributing to oxygen consumption. At moderate concentrations (2–3 fold excess of H2S), H2S coordinates to the CuB center forming a stable Cu–SH2 complex as documented by EPR. It is likely that HS− reduces CuBII first and then coordinates to CuBI forming a stable complex that is difficult to reoxidize. In the presence of a large excess of H2S, HS− binds to ferric heme a3, in a process that is likely aided by a conformational change caused by HS− binding to CuBI (Chart 17).
Using a synthetic CcO model system, a similar behavior was observed, i.e., that at low H2S concentration, the ferric iron center was reduced but stable H2S binding was not observed.359 They also noted that cytochrome c can be reduced at low H2S concentrations, thus injecting more reducing equivalents into the electron transfer chain and stimulating oxygen consumption (Chart 17).359 Second order rate constants for H2S-induced cytochrome c reduction observed under aerobic (81 ± 5 M−1 s−1) and anaerobic (480 ± 2 M−1 s−1) conditions differed ∼6-fold at 25 °C.97
Another well-documented reaction of H2S is its reaction with hemoglobin (Hb) and myoglobin (Mb), known since the 19th Century,360–362 when a green compound was reported to form upon treatment of oxy-Hb or oxy-Mb with H2S.363 Although H2S poisoning resulting in sulfhemoglobinemia is rare, misuse of sulfadrugs (sulfonamides) can lead to “green blood”.364 Sulfhemoglobin (λmax ∼ 618 nm), results from covalent addition of sulfur to a double bond in one of the pyrrole rings365–369 leading to the formation of a chlorin type heme (Chart 18A).370 This covalent modification results in significant delocalization of π electron density away from the iron, reducing its affinity for O2 (∼2500-fold in Mb and ∼135-fold in Hb).369 Mb can be recovered by treating sulfmyoglobin with azide or cyanide, which probably react with the sulfur inserted in the pyrrole ring.371
Chart 18. Sulfhemoglobin Formationa.
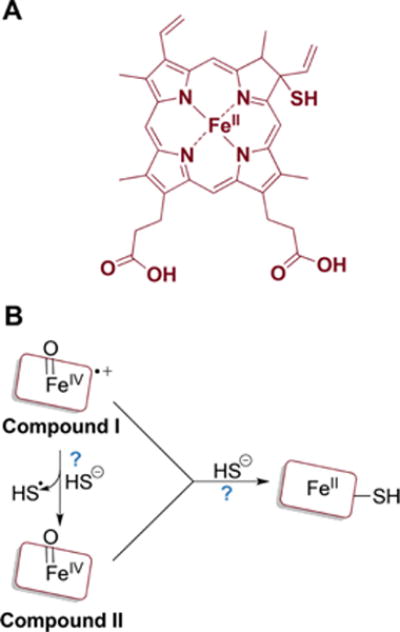
a(A) One of the proposed structures of sulfheme. (B) The mechanism of sulfheme formation is not fully understood (as denoted by the question marks) but starts with compound I or II reacting with H2S and results in sulfur being incorporated into the porphyrin ring.
Despite extensive studies, the actual mechanism of sulfheme formation is still unclear. It is postulated to involve the formation of an oxoferryl [FeIV═O Por•+] or [FeIV═O] intermediate (Chart 18B).372 Sulfmyoglobin is formed stoichiometrically in the reaction between H2S and metmyoglobin peroxide.371,373 Sulfcatalase is formed in the reaction between compound II catalase and H2S.371 H2S inhibits the heme in catalase in two ways: by irreversibly modifying the porphyrin and by reversibly ligating to the iron.371 Like catalase, lactoperoxidase compound II reacts with H2S to form sulflactoperoxidase.374 Studies on myeloperoxidase, a hemeprotein that produces hypochlorous acid and other oxidants for killing pathogens,375 indicate that H2S is a potent inhibitor (IC50 = 1 μM). H2S exhibits high bimolecular rate constants for reactions with compound I (1.1 × 106 M−1 s−1) and compound II (2 × 105 M−1 s−1).376 Surprisingly, the reaction of H2S with FeIII, FeII, compound I, or compound II resulted in the formation of a ferrous–H2S complex.
The activity of soluble guanylate cyclase (sGC) can also be modulated by H2S. Essential for NO• sensing, ferric sGC is reduced by HS−, which in turn facilitates NO• binding and activation of cyclic guanosine monophosphate synthesis.377
6.1.1. Catalytic H2S Oxidation by Methemoglobin, Myoglobin, and Neuroglobin
The ability of free hemin to oxidize H2S to thiosulfate was also known for a long time.378 In fact, the design of a H2S sensor is based on its affinity for ferricmyoglobin (FeIII–Mb or metmyoglobin).379 These observations presaged the discovery of catalytic H2S oxidation by methemoglobin (FeIII–Hb)301 and FeIII–Mb302 to a mixture of thiosulfate and iron-bound hydropolysulfides (Chart 19). Red blood cells lack mitochondria and, therefore, do not have the canonical H2S oxidation pathway. Yet, these cells have MST and, therefore, the capacity to make H2S301,380 raising the question as to whether alternative mechanisms exist for disposing H2S in these and other cells. The search for an answer to this question resulted in the discovery of catalytic H2S oxidation by globins containing iron in the ferric oxidation state, as described below.
Chart 19. Minimal Reaction Mechanism for Ferric Globin-Dependent Sulfide Oxidation to Thiosulfate and Iron-Bound Hydropolysulfidesa.
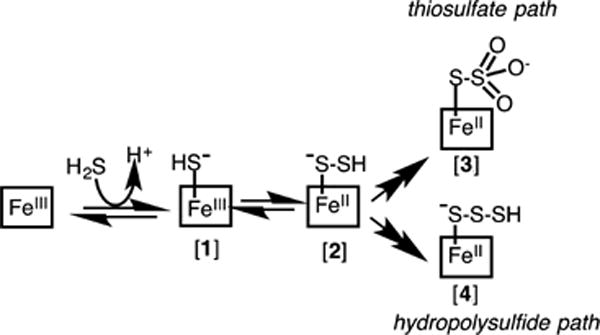
aDetails of the oxidation chemistry that lead to the products have been omitted for clarity.
Binding of H2S to FeIII–Hb or FeIII-Mb to give the corresponding HS−–FeIII species (Chart 19, [1]) is readily monitored by a shift in the Soret maximum from 405 → 423 nm in Hb301 and from 409 →428 nm in Mb.302 A concomitant resolution of the α/β bands at 577 and 541 nm in Hb and 578 and 545 nm in Mb is observed. The bimolecular rate constant for H2S binding to FeIII–Hb is 3.2 × 103 M−1 s−1, the koff is 0.05 s−1 and KD is 17 μM at pH 7.4, 37 °C. The corresponding values for Mb are kon = 1.6 × 104 M−1 s−1, koff = 1.6 s−1, and KD = 96 μM. The KD values represent upper limits since the rate constant for H2S binding to FeIII–Hb and FeIII–Mb increases with decreasing pH and only ∼20% of the dissolved sulfide exists as H2S at pH 7.4, where the measurements were made. Binding of H2S to sperm whale FeIII–Mb has also been reported (KD = 18.5 μM at pH 7.5 and 20 °C).381
Binding of H2S to FeIII–Hb results in the conversion of a high-spin g = 5.83 signal to a low-spin rhombic signal with g values of 2.51, 2.25, and 1.86.301 Similar changes are observed with FeIII–Mb, which converts from a high-spin g = 5.92 signal to a low-spin g = 2.57, 2.27, and 1.85 signal.302 Signal integration reveals less than stoichiometric spin concentration associated with the low-spin species indicating the presence of spin silent (diamagnetic and/or an integer spin species) intermediate(s) even at the earliest time point at which the spectra were recorded following H2S addition. Computational modeling suggested that the electronic structure of the S = 5/2 species can be described as a resonance hybrid of high-spin FeIII-SH− and high-spin FeII–•SH. However, this model is 116 kJ/mol higher in energy than the S = 1/2 model. Therefore, the initial intermediate is best described as FeIII–SH− with probably a small contribution of the HS• coordinated structure.
The HS−–FeIII species has been characterized by multiple approaches. These include resonance Raman spectroscopy and X-ray absorption spectroscopy, which reveal the initial formation of a 6-coordinate low-spin ferric species. The resonance Raman spectrum reveals the subsequent formation of high-spin ferrous species, albeit it is unclear whether the signal represents one or more likely, multiple intermediates. Coordination of H2S to Mb was observed by ultrahigh resolution ESI time-of-flight cryo-MS under anaerobic conditions even when H2S was in excess. Additional evidence for the initially formed ferric sulfide species comes from the X-ray structure of H2S treated with hemoglobin solved at 1.8 Å resolution (Figure 14A).382 The structure shows extra density above the iron on the distal side of the heme, which was assigned as sulfide based on the sulfur anomalous difference map. The Fe–S distance is 2.2 Å in both the α and β-subunits of hemoglobin and the HS−–FeIII intermediate is stabilized via hydrogen bonding to a histidine. Interestingly, a second sulfide was captured at the surface of the α-subunit, at the mouth of the PHE path, previously proposed to serve as an entry/exit channel for iron ligands.
Figure 14.
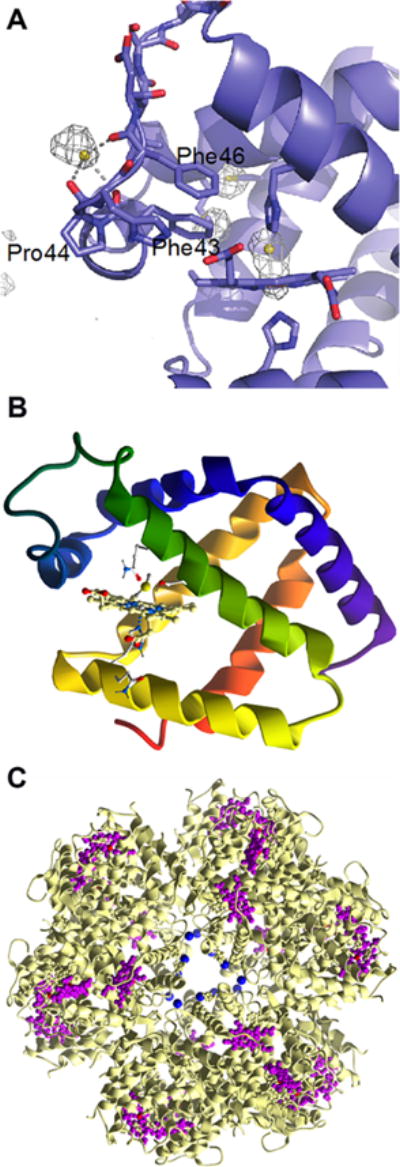
Binding of H2S to protein metal centers. (A) Structure of the α subunit of human hemoglobin showing HS− bound at the entry/exit point of the so-called Phe path that leads to the distal face of the heme. A second sulfide is bound to the heme iron (PDB: 5UCU). (B) Close up of the heme in hemoglobin I from Lucina pectinata with HS− coordinated to the iron ion (PDB: 1MOH). (C) Structure of hemoglobin-like protein C1 from Riftia pachyptila (PDB: 1YHU) with Zn2+ shown in blue and iron hemes in purple.
Bound, H2S probably exists in equilibrium with [FeII–HS•] which could react with another HS− to form H2S2•−. Coordinated H2S2•− could react further with HS− and oxygen leading to the propagation of hydropolysulfide chain coordinated to Fe2+ or to formation of thiosulfate (Chart 19). In principle, the ferrous heme-bound HS• radical could also react with O2 to form HSO2• (see section 2).
Under anaerobic conditions, binding of 1 equivalent of H2S to ferric iron is observed. However, under aerobic conditions, net consumption of H2S is seen with formation of thiosulfate and hydropolysulfides, which remain iron bound. In the proposed mechanism, the second intermediate is an iron-bound hydrodisulfide (Chart 19, [2]), which has been observed by cryo-MS on FeIII–Mb samples treated with Na2S. Exposure of FeIII–Hb to Na2S2 results in a shift in the Soret peak from 405 to 421 nm and in the appearance of α/β peaks at 575 and 543 nm, which is similar to the spectrum of HS−–FeIII.382 Under aerobic conditions, thiosulfate is formed from Na2S2 in the presence of FeIII–Hb.
Since the intracellular milieu is reducing, the fate of the iron-bound hydropolysulfides in the presence of physiologically relevant reductants is a pertinent issue. In the presence of GSH, the iron-bound hydropolysulfides are unstable, and GSSH, GSSG, and H2S products are observed.382 If formed, hydropolysulfides generated via globin-dependent oxidation are unlikely to be stable in the cell and would be converted to GSSH or other persulfides.
The catalytic nature of H2S oxidation by Hb and Mb at the expense of oxygen is evident from the stoichiometric excess of products formed over heme iron concentration. Furthermore, exposure of sulfide-treated FeIII–Hb to NADPH/flavin oxidoreductases led to the formation of O2–FeII-Hb with a shift in the Soret peak from 423 to 415 nm. Collectively, these results establish that (i) FeIII–Hb and FeIII–Mb can catalyze multiple rounds of sulfide oxidation, and (ii) the O2-liganded globin can be reformed in the presence of reductases like methemoglobin reductase.
Unlike Hb and Mb in which the distal heme site is available for binding exogenous ligands, the heme in neuroglobin has bis-histidine coordination.383 The function of neuroglobin, which is highly expressed in neuronal tissues and in some metabolically active tissues, is not known.384 Despite the coordinately saturated iron site, ferrous neuroglobin can bind O2, CO and NO•.385–388 The presence of the distal histidine ligand does in fact mute the reactivity of ferric neuroglobin (FeIII–Nb) toward H2S and leads to slow reduction to the ferrous state and to inefficient formation of thiosulfate and hydropolysulfides.389 In the presence of sulfide, the Soret peak of FeIII–Nb shifts from 412 to 415 nm and the α/β bands are broad and centered at 540 nm with a 575 nm shoulder. It is unclear what this spectral change represents, but it is likely to be a mixture of species as also suggested by EPR and resonance Raman spectroscopy. The EXAFS data do not show evidence for an iron–sulfur bond in sulfide-treated neuroglobin, indicating that the sulfide oxidation products are formed even in the absence of direct coordination to iron. The kon for the interaction of sulfide with FeIII–Nb is 13.8 M−1 s−1 at pH 7.4 and 25 °C, which is significantly smaller than the values for FeIII–Hb and FeIII–Mb. The koff and KD for the interaction of sulfide with FeIII–Nb are 5 × 10−3 s−1 and 370 μM, respectively. As expected, the H64A mutation of the distal histidine residue allows direct binding of sulfide as confirmed by EXAFS analysis, increases the rate constant for sulfide binding 4000-fold, and supports active oxidation of sulfide to thiosulfate and protein bound hydropolysulfides. A rich array of oxidation products were identified with the H64A mutant using cryo-MS including hydropolysulfides with 2–6 sulfur atoms and variously oxygenated derivatives in addition to thiosulfate and sulfate.389
Collectively, the studies on the globins reveal the potential for ferric-iron dependent sulfide oxidation chemistry, whose relative importance in the cell awaits evaluation. An open ligation site promotes sulfide coordination and oxidation chemistry, and in its absence, iron reduction is supported. The relatively low steady-state concentration of H2S likely reduces the prevalence of reactions between sulfide and heme or nonheme iron (or other metalloproteins) except in special cases like red blood cells where FeIII–Hb represents 1–3% of total hemoglobin, whose concentration is high (∼5 mM).
6.1.2. Binding and Transport of H2S by Globins
Specialized hemoglobins that transport H2S are found in organisms that are adapted to life in sulfide-rich environments. The best-studied example of such a Hb is from the clam, Lucina pectinata (Figure 14B),381,390–403 which lives in H2S-rich waters. The monomeric L. pectinata Hb hemoglobin I (HbI) transports H2S to symbiotic bacteria, which assimilate it and provide the host with a source of organic sulfur. HbI exhibits a high association constant (kon = 2.3 × 105 M−1 s−1) and an unusually low dissociation constant (koff = 0.22 × 10−3 s−1) for H2S,390 which suggests the stabilization of distal sulfide ligand by the active site.391,395,396,398,400,401 In human Hb, a histidine residue hydrogen bonds with the iron-bound sulfide. The corresponding residue in HbI is a glutamine, which has a flexible side chain. Mutation of the glutamine residue in HbI to valine precludes heme reduction, while mutation to histidine promotes formation of sulfhemoglobin. Another difference from human Hb, is the presence of phenylalanines in HbI that form a hydrophobic pocket around the sulfide.401 It is unclear whether H2S release occurs via slow dissociation or by heme iron reduction. Introduction of positively charged substituents on the porphyrin ring changes the reactivity of metal porphyrins from simple binding of H2S to catalytic oxidation of H2S.404 Hence, at low concentrations, H2S release could be due to its dissociation from the heme iron, while at high concentrations, heme reduction and H2S/hydropolysulfide delivery might predominate.405
Another sulfide-adapted organism, the giant tubeworm, Riftia pachyptila, lives in deep-sea hydrothermal vents in symbiotic relationship with sulfide-oxidizing bacteria that need both H2S and O2.406–408 The Riftia hemoglobins are large proteins with a molecular mass of ∼3500 kDa (Figure 14C). Binding of H2S and O2 occurs at separate sites. While O2 binds at the heme iron site, it is unclear where H2S binds. The protein contains 12 Zn2+ ions, which have been suggested as potential sites for H2S binding.408
6.1.3. Interaction of H2S with ZnII-Containing Proteins
The interaction of H2S with ZnII-containing proteins is poorly studied. It is reported that H2S represses androgen receptor transactivation by targeting the second zinc-finger module.409 Phosphodiesterase 5, a ZnII-containing enzyme, is inhibited by nanomolar H2S concentrations.410 Zinc–hydrogensulfido complexes are not easy to prepare and isolate and require bulky apolar ligands.411 The synthesis of a stable zinc hydrogensulfido complex with the tris(2-pyridylmethyl)amine ligand has been reported.412 H2S was released from the complex in acidic medium or transferred to a zinc center with higher affinity via intermediate formation of a μ-sulfido dinuclear species. The ability of ZnII to coordinate HS− was reported to depend on the ability of the HS− ligand to form hydrogen bonds.413 Chemical modifications on the ligand that precluded hydrogen bonding with HS− resulted in decomposition of the complex and ZnS precipitation. This study highlighted the importance of the second coordination sphere in stabilizing the Zn-HS− adduct, suggesting that the protein environment could do the same.
6.2. Interaction of H2S with ROS and Other Biologically Relevant Oxidants
Being at the lowest oxidation state of −2, the sulfur in H2S can only undergo oxidation. Oxidation leads to sulfate (SO42−), sulfite (SO32−), thiosulfate (S2O32−), persulfides (RSS−), organic (RSSnSR) and inorganic (HSSnSR) polysulfides, and elemental sulfur (Sn). The direct reaction of H2S with O2 is thermodynamically disfavored (see section 2).63,414 Given the high one-electron reduction potential (E°′(HS•, 2H+/H2S) = +0.91–0.94),64,65 only relatively strong one-electron oxidants can oxidize H2S to HS•, with further reaction of HS• providing an additional driving force. Indeed, several biologically relevant oxidants can support the one-electron oxidation of H2S, such as hydroxyl radical,415,416 carbonate radical,65 nitrogen dioxide,61 and myeloperoxidase oxoferryl compounds I and II;376 the rate constants of these reactions are shown in Table 2. The list of one-electron oxidants that can oxidize H2S can probably be extended to peroxyl and phenoxyl radicals as well as to other metal centers (see section 6.1). The superoxide radical can also oxidize H2S.97 The apparent rate constants at pH 7.4 vary depending on the oxidant and are similar to those reported for cysteine and GSH.61 Mixtures of polysulfides and polysulfide radical anions (S2•− and S3•−) are observed in reaction mixture containing superoxide and H2S in DMSO.97
Table 2.
Rate Constants for the Reaction of H2S with Biologically-Relevant Oxidants
| reduction potential
|
kinetics of reaction with H2S
|
||||
|---|---|---|---|---|---|
| oxidant | couple | E°′ (V) | ref | k(M−1 s−1) | ref |
| One-Electron Oxidant | |||||
| hydroxyl radical | HO•, H+/H2O | +2.31 | 414 | 1.1 × 1010 (pH 7) | 415,416 |
| oxygen | O2(g)/O2•− | −0.35a | 414 | very slow | |
| carbonate radical | CO3•−, H+/HCO3− | +1.77b | 63 | 2.0 × 108 (pH 7, 20 °C) | 65 |
| nitrogen dioxide | NO2•/NO2− | +1.04 | 63 | 1.2 × 107 (pH 7.5, 25 °C) | 61 |
| superoxide radical | O2•−, 2H+/H2O2 | +0.91 | 414 | ~208 (DMSO) | 97 |
| myeloperoxydase compound I | CI/Fe3+ | +1.35 | 421 | 1.1 × 106 (pH 7.4, 25 °C) | 376 |
| myeloperoxydase compound II | CII/Fe3+ | +0.97 | 421 | 2.0 × 105 (pH 7.4, 25 °C) | 376 |
| Two-Electron Oxidant | |||||
| hydrogen peroxide | H2O2, 2H+/2H2O | +1.35 | 414 | 0.48–0.73 (pH 7.4, 37 °C) | 61,73 |
| peroxynitrite | ONOOH, H+/NO2−, H2O | +1/30 | 422 | 6.7 × 103 (pH 7.4, 37 °C) | 420 |
| hypochlorite | HOCl, H+/Cl−, H2O | +1.28 | 421 | 0.8–20 × 108 (pH 7.4, 37 °C) | 61,418 |
| tauramine-chloramine | – | 303 (pH 7.4, 37 °C) | 61 | ||
The reduction potential based on a standard state of 1 M O2 is −0.18.414
Extrapolated to pH 7 from E°(CO3•−/CO32−) = 1.57 V assuming a pKa of 10.32 for HCO3−.
The initial oxidation product of H2S is the sulfyil radical (HS•). HS• is an oxidizing free radical capable of reacting with electron donors including ascorbate and GSH. Importantly, the one-electron oxidation of H2S can unleash oxygen-dependent free radical chain reactions amplifying the initial oxidative event.61,65 Although the reaction of HS• with a second HS• to form HSSH has a high rate constant ((6–9) × 109 M−1 s−1, eq 7),65 this reaction is unlikely to occur in most contexts because of its dependence on the square of HS• concentration. Alternatively, HS• can react with O2 to form SO2•− ((5–7) × 109 M−1 s−1, eq 10),65 a reducing radical which in turn can react with O2 forming O2•−. HS• can also react reversibly with HS− forming HSSH•− (forward and reverse rate constants, 5.4 × 109 M−1 s−1 and 5.3 × 105 s−1);65 the latter can also react with O2 forming O2•− (eq 11 and 12).65,97,415 Superoxide radical (O2•−) can dismutate, spontaneously or enzymatically, to O2 and H2O2414
The rate constants for reaction of H2S with two-electron oxidants (Table 2) are also comparable to those of low molecular weight thiols.61 The reaction with hydroperoxides (ROOH) initially forms HSOH, which can react with a second HS− to form HSSH.61,73 In the case of hydrogen peroxide, the final products depend on the initial ratio of hydrogen peroxide to H2S and consist mainly of polysulfides, elemental sulfur, and, in the presence of excess oxidant, sulfate.73,417 By analogy to thiols, the reaction with hypochlorous acid is likely to form HSCl that quickly hydrolyzes to HSOH.418
The reaction of peroxynitrite with H2S is more complex than its reaction with thiols and generates novel products.61,419,420 The decay of peroxynitrite in the presence of H2S is first order in peroxynitrite and first order in H2S; the second order rate constant is 6.7 × 103 M−1 s−1 (pH 7.4, 37 °C).420 The pH-dependence is bell-shaped, consistent with HS− and ONOOH being the reacting species. Computational modeling suggests that the reaction starts with the nucleophilic substitution of HS− on ONOOH to give HSOH and NO2− as initial products. The reaction then proceeds to the formation of “yellow” products that absorb at 408 nm.419 The increase in absorbance at 408 nm occurs with a lag phase, consistent with the formation of intermediates that precede formation of the yellow products.420 Free radical scavengers or nitrite had no effect on the amount of yellow product formed, but the yield increased when peroxynitrite was in excess. Thus, it was proposed that the reaction of HSSH with peroxynitrite leads to formation of the yellow products, and indeed, mixtures of HSSH and peroxynitrite in acetonitrile yielded products with similar absorbance spectra.420 Based on mass spectrometric and computational studies, it was proposed that at least one of the yellow products is HSNO2 or its isomer HSONO. In addition to the direct reaction of peroxynitrous acid with HS−, the free radicals derived from peroxynitrite (nitrogen dioxide, hydroxyl and carbonate radical) can also react with H2S.61
The probability of H2S acting as a direct scavenger of oxidants in biological systems depends on kinetic factors, i.e., on the products of the rate constants times H2S concentration. While the reactions of H2S with some oxidants display relatively high rate constants, comparable to those of LMW thiols, the tissue concentrations of H2S (submicromolar, see section 3.5) are very low, i.e., several orders of magnitude lower than those of other reductants (e.g., millimolar for some thiols). Thus, it can be concluded that the direct reaction of H2S with oxidants would not be fast enough in biological contexts to support a significant scavenging role. Furthermore, H2S would not be able to compete with thiols for one- and two-electron oxidants, unless high local concentrations H2S were reached as, for example, with bolus administration of exogenous H2S. In conclusion the biological “antioxidant” effects ascribed to H2S are unlikely to be due to direct scavenging of oxidants by H2S but rather to indirect effects on enzymes, transporters, and/or other targets in signaling pathways.
6.3. Reaction of H2S with NO• and Its Metabolites
NO• has important signaling roles in mammals including blood pressure regulation,423,424 immune defense,425,426 and neurotransmission.427–429 Most of the “classical” effects of NO•, such as vasodilation or neuro-modulation, are mediated by coordination of NO• to the heme iron in sGC, which activates the enzyme to generate cyclic guanosine monophosphate (cGMP), a powerful second messenger.430,431 But not all of the actions of NO• proceed via cGMP signaling (Chart 20). NO• can also lead to an oxidative posttranslational modification of cysteine called S-nitrosation.432–436 How S-nitrosothiols are formed in the cells is still a matter of debate.436–438 NO• can also undergo one-electron reduction to form nitroxyl (HNO, IUPAC name: hydridooxidonitrogen, azanone, nitrosylhydride),439–442 a powerful vasodilator.267,439,442 NO• is oxidized to nitrite and nitrate. Nitrite is now recognized as an important metabolite that can be reduced to NO•.443–445 Finally, peroxynitrite and its protonated form (ONOOH) can be generated in a diffusion controlled reaction between NO• and O2•− (∼1 × 1010 M−1 s−1).446–448
Chart 20. Interaction of H2S with NO• and Its Metabolitesa.
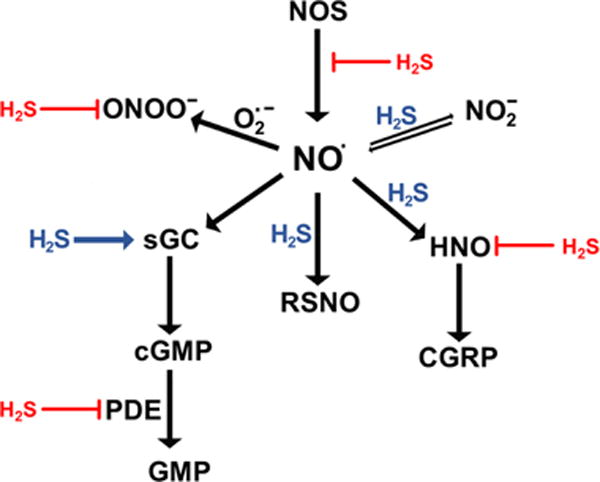
aNO• signals via the classical soluble guanylate cyclase (sGC)/cyclic GMP (cGMP) cascade. H2S can reduce sGC to increase NO• binding and cGMP production. cGMP is deactivated by phosphodiesterase 5 (PDE), an enzyme that is inhibited by H2S. Oxidation of NO• to nitrosonium (NO+) ion leads to modification of cysteine residues and formation of S-nitrosothiols (RSNO), a process that H2S can facilitate. NO• can be reduced by H2S to form HNO. HNO activates the release of calcitonin gene-related peptide (CGRP), a vasodilator, but HNO can also be trapped by H2S. NO• is oxidized to nitrite, which can be reduced back to NO•, a process that H2S can facilitate. NO• reacts with superoxide to form peroxynitrite (ONOO−). H2S can scavenge ONOO−.
H2S interferes with NO• signaling, either by reacting with NO• or its downstream metabolites267,419,420,449–453 or by modulating NO• production268,454,455 and cGMP levels.266,377,410 The first report on H2S-induced vasodilatory effects demonstrated its synergy with NO•.7 Inhibition of endothelial NO synthase (eNOS) leads to abrogation of H2S-induced vasodilation,7,266,267 while deletion of CSE prevented the vasodilatory effects of acetylcholine and NO•.266 In addition, the cardioprotective effects of H2S were abolished in eNOS−/− mice.268 Different mechanisms have been proposed for this crosstalk that are covered in section 10. In this section, we focus on the chemical aspects of the direct reactions between NO• (and its metabolites) and H2S.
6.3.1. Direct Reaction between NO• and H2S
Studies on the direct reaction between NO• and H2S date back to the 19th and early 20th century. The reaction of gaseous NO• and H2S was reported to form, among other products, nitrous oxide (N2O) and elemental sulfur.456–461 Formation of N2O was difficult to explain as a single step process. N2O is a product of HNO dimerization439–442 so HNO formation could be the actual intermediate step.461
It has been suggested that NO• and H2S can form HNO in vivo.451,452 The combination of H2S and NO• donors was observed to have the same effects in murine heart as the application of the HNO donor, Angeli’s salt.452 Indeed, when the reaction between H2S and NO• was studied at pH 7.4 under anaerobic conditions, the rate of HNO formation was first order on both NO• and H2S 267. The combination of NO• and H2S (2 μM each) yielded a peak HNO concentration of ∼0.5 μM, similar to the effects of 1 mM Angeli’s salt. Intracellular HNO production, detected by an HNO fluorescence sensor was also shown to depend on both NO• and H2S.267
Direct one-electron transfer from HS− to NO• to give HNO and S•− (eq 22) is thermodynamically unfavorable (ΔG0′ = +102 kJ/mol).64 An alternative mechanism is
| (22) |
the formation of HSNO•− (eq 23), which is similar to the reaction
| (23) |
reported between NO• and aromatic and “pseudoaromatic” alcohols such as tyrosine, hydroquinone, and ascorbic acid.462 HSNO•− is a powerful reducing agent (RSNO•−/RSNO, E < −1 V)463 that can initiate a cascade of reactions, leading to N2O and Sn formation (eqs 24–28).
| (24) |
| (25) |
| (26) |
| (27) |
| (28) |
Endogenous HNO production would be critically dependent on NO• and H2S being produced in close proximity since both can engage in competing reactions with other molecules (Figure 15A). The transient receptor potential channel A1 (TRPA1), a biological sensor that regulates HNO-induced release of the powerful vasodilator calcitonin gene related peptide, colocalizes with CBS267 and nNOS,464 potentially forming a functional unit for HNO formation and its action.267,465 Studies with an HNO-responsive two-photon ratiometric fluorescence imaging probe confirmed that endogenous HNO generation is dependent on endogenous H2S and NO• formation in cells and brain tissues.466
Figure 15.
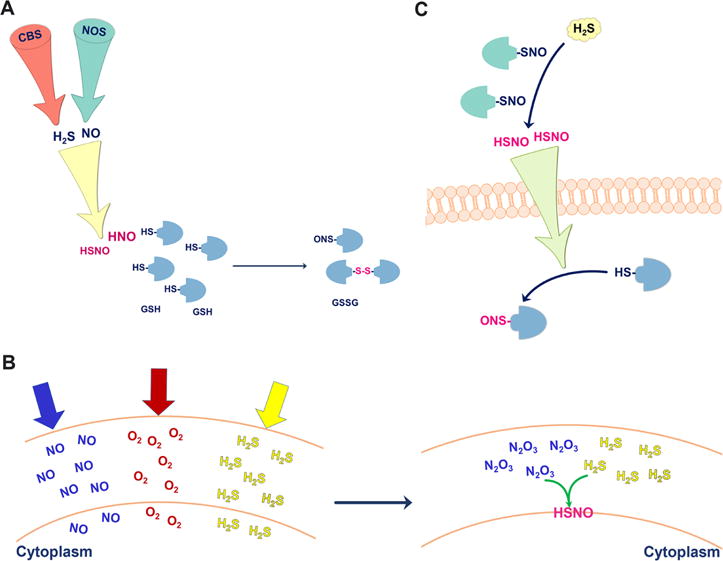
Signaling aspects of NO•/H2S cross-talk. (A) To form HNO and minimize side reactions, H2S and NO2• have to be produced in proximity. HNO (and possibly HSNO) reacts with protein thiols and glutathione. (B) All three gases, NO•, O2, and H2S tend to accumulate in membranes. NO• and O2 form N2O3, which readily reacts with H2S to form HSNO. (C) HSNO formed in the reaction of protein S-nitrosothiols with H2S can diffuse through the cell membrane and transfer the “NO+” group to another protein target.
6.3.2. Reaction of H2S with S-Nitrosothiols and Metal-Nitrosyls
Formation of a new S-nitrosothiol, HSNO (IUPAC name: nitrososulfane or (hydridosulfanido)oxidonitrogen), in the reaction of H2S with S-nitrosothiols (or other nitrosocontaining species) was first proposed in 2006.467,468 HSNO was known as a product of cis-HNSO photolysis in argon matrices, where it has been studied computationally and by IR spectroscopy.469–472 The crystal structure of the bis-(triphenylphosphine)iminium SNO salt (PNP+SNO−) was reported,473 but a detailed study in aqueous solution was missing.
Using pulse radiolysis to generate HS• and NO•, formation of a species with the spectral characteristics (λmax ≈ 330 nm) of an S-nitrosothiol was observed (Chart 21).449 The species was short-lived with a half-life of ∼12 μs449 yielding an estimate of ∼107 M−1 s−1 for the rate constant for the reaction of HSNO with sulfide anion (eq 26).64
Chart 21.
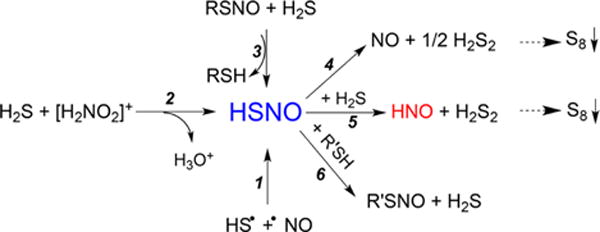
Reaction Paths for HSNO Generation (1–3) and Its Biologically Relevant Reactions (4–6)
HSNO was also detected in reactions of H2S with “NO+” carriers: acidified nitrite,449 N2O3,474 metal nitrosyls,450,475–477 and S-nitrosothiols.449,453 For example, in the reaction with acidified nitrite, a brown-red intermediate was formed prior to the solution turning milky white. ESI-TOF MS analysis of the acidic and neutralized solution of the brown-red intermediate revealed the parent ion mass and isotopic pattern expected for HSNO.449
Reaction of thiosemicarbazide with NO• results in HSNO formation under physiological conditions; thiosemicarbazides are therefore proposed to serve as a tool capable of transforming intracellular NO• into HSNO.478
In the reaction of H2S with N2O3 (eq 29), the facile formation of stable HSNO was demonstrated by Fourier-transform microwave spectroscopy.474
| (29) |
Generation of HNO and N2O (eq 26 and 28) was confirmed when 18O labeled N2O3 was reacted with excess of H2S.474 HSNO formation from N2O3 and H2S could be important for intracellular RSNO generation. Formation of N2O3 is deemed to be kinetically improbable due to the low intracellular concentration of NO• compared to O2.437 However, N2O3 could be formed in the lipid bilayers where NO• and O2 accumulate.437 H2S can also accumulate in lipid bilayers based on its partition coefficient57 creating conditions that might be conducive for HSNO formation (Figure 15B). Reaction of HSNO with thiols (Chart 21 and Figure 15C) can result in transnitrosation. HSNO can act as an “NO+” carrier from one protein to another and across the cell membrane (Figure 15C). This idea is supported by the ability of H2S to promote nitrosation of BSA outside a dialysis bag containing nitrosated BSA but also nitrosation of hemoglobin in the red blood cells from extracellular nitrosated BSA.449 NaHS treatment during cardiac ischemia was reported to increase tissue S-nitrosation479 possibly via HSNO formation which would then act as transnitrosating agent.
Using a combination of spectroscopic approaches and ESI-TOF MS to study the trans-nitrosation reaction between S-nitrosoglutathione and H2S, HSNO was detected within 30 min.449 Greater than equimolar H2S concentration promoted N2O and hydroxylamine formation via intermediate HNO generation (eqs 26, 28, and 30).
| (30) |
An unexplained observation made during the transnitrosation reaction between GSNO and H2S was that the solution turned yellow (λmax = 412 nm).449,480 MS analysis of the RSNO/H2S reaction mixture identified, among other products, SSNO− which was suggested to be a stable yellow product.481 SSNO− formation from RSNO and H2S was proposed to occur via eqs 31–33.453,480,481
| (31) |
| (32) |
| (33) |
Whether SSNO− is stable enough to mediate biological effects has been the subject of debate. Crystalline PNP+SSNO− synthesized by a published method473 has been used to chemically characterize SSNO−.69 Crystalline SSNO− and SSNO− dissolved in organic solvent are air and water sensitive.69,473 The reduction potential of SSNO− was determined to be −0.21 V versus NHE, which is within the range of physiological reductants like glutathione with which it reacted readily.69 Furthermore, SSNO− decomposed rapidly in the presence of H2S and cyanide forming SNO−.69,482 15N/14N NMR and cryo-ESI TOF MS analyses confirmed the formation of SNO−/HSNO in the reaction of SSNO− with HS− and CN− (eqs 34 and 35).482
| (34) |
| (35) |
A role for SSNO− in signaling is doubtful based on kinetic grounds (the apparent rate constant for its formation estimated from published data481 is 10−14 M−1 s−1) as well.64 Considering the very low concentrations of HS− (section 3.5) and RSNO436,437 and the very high concentrations of thiol, the reaction of SNO− with HS− (eq 32) in a cellular millieu seems unlikely. Furthermore, HS2−, which is intrinsically unstable and readily reduced by thiols, is unlikely to persist long enough or to react specifically with “NO+” to form SSNO− (eq 33). Even if formed, SSNO− would readily react with thiols to form HSNO.482 In summary, HSNO remains the chemically most plausible nitrosating agent that can react with cysteines (Chart 21 and Figure 15) and engage in transnitrosation reactions.482,483
6.3.3. Metal-Catalyzed Reaction between Nitrite and Sulfide
Although nitrite and H2S do not react directly at pH 7.4,97 NO• generation has been reported in cells treated with these two reagents.450 Intracellular HNO generation which was localized to mitochondria was observed when cells were treated with 100 μM nitrite and sulfide but not in cells depleted of mitochondria.450 This result implicated a role for mitochondrial proteins in catalyzing the reaction between nitrite and H2S. To understand the possible role of heme iron in the reaction mechanism for HNO generation from nitrite and sulfide, a water-soluble iron–porphyrin was used as a model system.450 Initial binding of nitrite to ferric heme and subsequent oxygen atom transfer484,485 to H2S to give HSOH was the predominant reaction observed when nitrite was in excess. The [FeII(NO)] ↔ [FeIII(NO−)] complex then slowly released HNO.450,485 When sulfide was in excess, it reduced ferric to ferrous heme so that the classic nitrite reductase activity of FeII heme was observed. The formed [FeIII(NO)]↔[FeII(NO+)] reacted with HS− to form an [FeII(HSNO)] complex (Chart 22).450 These results support the feasibility of H2S reacting with metal-nitrosyls to form S-nitrosothiols via HSNO, which represents an alternative mechanism for the physiological generation of HNO.
Chart 22. Proposed Reaction Mechanism for H2S-Assisted Nitrite Reduction Catalyzed by an Iron Porphyrin Compounda.
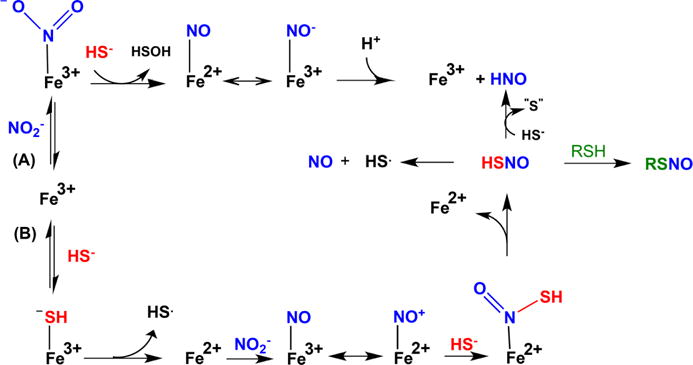
aPathway A, which predominates when nitrite is in excess over H2S, represents a classical oxygen atom transfer; nitrite coordination to ferric heme leads to HSOH and [Fe2+(NO)] ↔ [Fe3+(NO−)], which releases HNO slowly. Pathway B predominates when H2S is in excess over nitrite; reduction of ferric heme by H2S is followed by nitrite reduction to [Fe3+(NO)] ↔ [Fe2+(NO+)] species, which is scavenged by HS− giving HSNO. Either free or coordinated HSNO causes transnitrosation of protein thiols or, in the reaction with H2S, generates HNO.
Another metalloprotein, a molibdopterin-containing xanthine oxidase, was reported to catalyze H2S-stimulated nitrite reduction in endothelial cells and in mice injected with Na2S. However, the mechanism of this reaction was not elucidated.486
7. PROTEIN PERSULFIDATION
Protein persulfidation, an oxidative posttranslation modification of cysteines, represents a mechanism by which H2S signals. This modification is also referred to in the literature as “sulfhydration”,19 which implies “hydration” and is inaccurate. Instead, the process involves “sulfuration”, i.e., the addition of a sulfur atom.64,487 The term “persulfuration” has been also used, but the term “persulfidation” has been gaining wide acceptance and is used here. Other ways to describe RSSH are hydropersulfide, or hydrodisulfide, or as a disulfane derivative (e.g., CH3SSH is methyldisulfane64). A less ambiguous name for RSSH is hydridodisulfide. In this review, the term “persulfide” is used to designate RSSH/RSS−.
Contrary to the chemically incorrect claim that H2S can directly modify cysteine residues to form persulfides, the reaction between H2S and thiols (eq 36) requires an oxidant.64,181
| (36) |
Due to their instability and greater reactivity than thiols, working with persulfides is challenging. In the following subsections, progress on developing methods to study persulfides and our current understanding of their reactivity are discussed.
7.1. Model Systems to Study Protein Persulfidation
Persulfides are relatively unstable in aqueous solution and are typically synthesized immediately before use. Several model systems have been used to study persulfide chemistry. These models are grouped in two categories: (i) low molecular weight (LMW) persulfides and (ii) protein persulfide models.
7.1.1. LMW Persulfide Models
Several LMW persulfides have been reported that are either synthesized in situ or have been characterized following purification. A synthetic method for persulfides dates back to 1954 when alkyl- and arylpersulfides were prepared from sulfenyl chloride and thiols.488 The resulting acyldisulfide was hydrolyzed by HCl to give persulfide (Chart 23A, [I]). Persulfides can also be prepared from methoxycarbonyl disulfides, which would undergo alkoxide-induced displacement of the RSS− anion (Chart 23A, [II]).489 Alternatively acyl disulfides can be synthesized in the reaction of dialkyl thiosulfones with thioacid (Chart 23A, [III]).490 In fact, the acidic hydrolysis of acyl disulfides has become a general synthetic strategy for the preparation of small molecule persulfides like ethyl-, t-butyl-, benzyl-, diphenylmethyl-, trityl-, adamantyl-, and penicillamine-derived persulfide (Chart 23B).488,491–500 The hydrophobic LMW persulfides need to be handled in organic solvents and are consequently protonated,494,495 which reduces their nucleophilicity. In contrast, persulfides are deprotonated and more reactive at physiological pH (see section 6.2).
Chart 23.
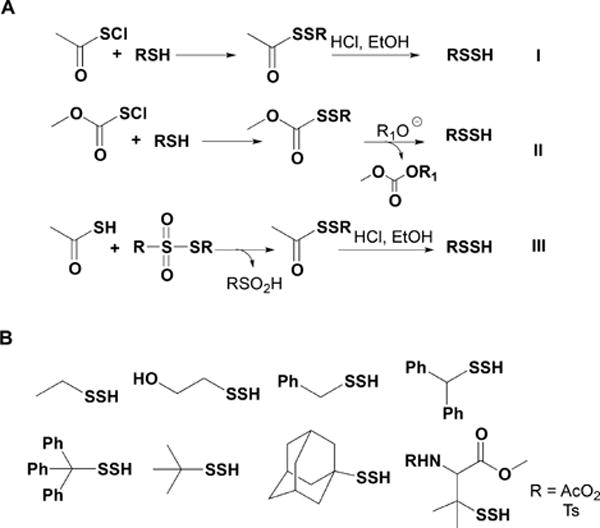
Synthetic Strategies for the Preparation of Low Molecular Weight Persulfides (A) and Structures of Some of the Persulfides Prepared following These Synthetic Routes (B)
A water-soluble penicillamine-derived LMW persulfide has been prepared.501 The synthetic protocol involved acylprotected disulfides and gave high yields (Chart 24). At pH 2.7, <5% degradation of the acyl-protected disulfide of penicillamine was observed after 120 min at room temperature, allowing relatively stable stock solutions to be prepared. When placed in buffers with pH >6 the disulfide underwent S- to N-methoxycarbonyl transfer, generating N-methoxycarbonyl penicillamine persulfide, a persulfide related to a commonly used S-nitrosothiol (Chart 24).
Chart 24.
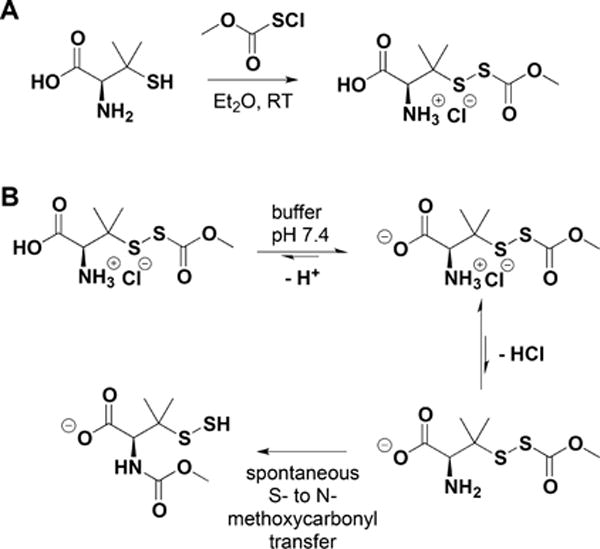
Synthesis of an Acyl-Protected Disulfide of Penicillamine (A) and Its Rearrangement to N-Methoxycarbonyl Penicillamine Persulfide via S- to N-Methoxycarbonyl Transfer at pH 7.4 (B)
LMW persulfides can be generated in situ in aqueous solution by mixing disulfides (such as cystine or GSSG) with H2S in equimolar ratio (Chart 25A).60,502–505 As this is an equilibrium process, the reaction mixture contains unreacted disulfides and H2S in addition to the persulfide. Alternatively, persulfides can be prepared in situ by CBS- or CSE-catalyzed conversion of cystine or homocystine to the corresponding persulfides.165,226 Cysteine persulfide and homocysteine persulfide are formed via α,β or α,γ elimination reactions, respectively (Chart 25B; for details see section 3). The LMW persulfide, GSSH, can also be formed in situ. Rhodanese in the presence of thiosulfate313,330 (or p-toluenethiosulfonate)506,507 and glutathione forms GSSH (Chart 25C) while SQR forms GSSH via sulfurtransfer from H2S to GSH (Figure 8).311 Another route for preparing GSSH is to reduce the trisulfide (GSSSG) with glutathione reductase and NADPH (Chart 25D).165,508
Chart 25. Strategies for the in Situ Preparation of LMW Persulfidesa.
a(A) Cysteine and glutathione persulfides can be prepared by the reaction of H2S with cystine and glutathione disulfide, respectively. (B) Cysteine persulfide can be generated from cystine and CSE or CBS. (C) Rhodanese can be used to transfer sulfur to glutathione. (D) Glutathione reductase (GR) uses electrons from NADPH to reduce glutathione trisulfide to persulfide and GSH.
7.1.2. Protein Persulfide Models
A commonly used strategy for preparing protein persulfides is the reaction of activated disulfides with equimolar H2S. For example, a protein with one reactive cysteine is first treated with Ellman’s reagent, 5,5′-dithiobis(2-nitrobenzoate) (DTNB), to form a mixed disulfide.60,505,509 The thionitrobenzoate anion (TNB) is a good leaving group, and in the next step the protein–TNB mixed disulfide is reacted with an equimolar concentration of H2S to generate the protein persulfide (Figure 16A).60,509 The concomitant release of TNB, which has a strong absorbance at 412 nm,510 provides a simple method for quantifying the reaction yield. Persulfides of papain,505,509 glutathione peroxidase 3,509 and human serum albumin60 have been prepared using this approach.
Figure 16.
Strategies for the preparation of protein persulfides. (A) Protein thiols react first with DTNB forming mixed disulfides that react with H2S forming persulfides. Thionitrobenzoate is a good leaving group and its UV–visible absorbance can be used to estimate the yield of persulfides. (B) Protein sulfenic acids, when stable, can be used as precursors for persulfide preparation. (C) Protein thiols can be mixed with inorganic polysulfides or with a mixture of HOCl and H2S. Besides persulfides, polythiolated products are also formed. (D) Protein thiols can react with 9-fluorenylmethyl disulfide. The products undergo alkaline hydrolysis forming persulfides.
The reaction of sulfenic acid with H2S (for details see section 8.2) can also be used to prepare protein persulfides.511 The primary obstacle with this approach is that sulfenic acid modifications on proteins are generally unstable. An exception is the sulfenic acid derivative of serum albumin, which is relatively stable512,513 and has been exploited to generate the corresponding persulfide (Figure 16B).60,511
A less specific approach for protein persulfidation involves mixing the protein with H2S and an oxidant, such as HOCl, or mixing the protein with polysulfide salts (Figure 16C).514 Uncontrolled protein poly thiolation is an inevitable outcome of this approach (see section 8.3), and the use of these methods is discouraged.
Alternatively, a protein thiolate can be reacted with 9-fluorenylmethyl disulfide to form a mixed disulfide which is then exposed to alkaline pH to promote hydrolysis generating the protein persulfide (Figure 16D).515 However, the alkaline conditions could lead to protein denaturation.
7.2. Persulfide Reactivity
Persulfides have characteristics in common with thiols, disulfides, polysulfides, hydroperoxides, and sulfenic acids. The chemistry of persulfides is very rich, and persulfides are very versatile molecules that are being assigned roles of increasing importance in biology.
The crystal structure of tritylpersulfide shows an S–S bond length of 2.0396 Å, which fits well with the S–S bond length observed in crystals of inorganic polysulfides. The CSSH dihedral angle is 82.2°,494 which is close to the CSSC dihedral angle of 83° seen in unstrained disulfides.516 The crystal structures of some proteins involved in sulfur metabolism have been obtained with cysteine residues modified to persulfides, e.g., the structures of bovine rhodanese332,517 and of human MST.271 In addition, the crystal structures of some proteins that contain free cysteine persulfide bound as a ligand have been obtained.518,519
In alkaline solutions, persulfides show an absorption maximum at 335–340 nm494,495,501,502 and a relatively low absorption coefficient (∼310 M−1 cm−1).31 The IR spectra of alkyl and aryl persulfides show a weak S–H stretch at ∼2500 cm−1 (Table 3), which is shifted to lower wavenumbers than thiols (∼2570 cm−1), consistent with the presence of a stronger S–H bond in thiols.494,495,520 Similar shifts are observed in the Raman spectra, with the additional presence of a band in the 200–500 cm−1 region due to the S–S bond. 1H NMR spectra of persulfides in organic solvents show shifts in the S–H proton relative to the corresponding thiols. For example, the S–H proton shows an ∼0.4 ppm upfield shift in tritylpersulfide and an ∼1.2 ppm downfield shift in benzenepersulfide and adamantylpersulfide (Table 3).494,495
Table 3.
Basic Physicochemical and Thermodynamic Properties of LMW Persulfides
| values | refs | |
|---|---|---|
| λmax | 335–340 nma | 31,494,495,501,502 |
| IR (S–H stretch) (cm−1) | 2490–2510 | 488–500 |
| (S–S stretch) (cm−1) | 200–500 | |
| 1H NMR | 2.7–3 ppm | 494,495,501,502 |
| S–S bond length | 2.04 Å | 495 |
| E°′(RSS−, 2H+/RSH, HS−) | −0.18 Vb | 64 |
| E°′(RSS•/RSS−) | +0.68 Vb | 64 |
Alkaline pH, but organic solvents as well.
Versus SHE.
7.2.1. Persulfide Acidity
Protonated persulfides can ionize to form the corresponding anionic persulfides (eq 37).
| (37) |
In agreement with the weaker S–H bond in persulfides than in thiols, the acidity of persulfides is predicted to be higher. There are very few reported experimental measurements of persulfide pKa. From the pH dependence of the rate of hydrogen atom transfer from 2-[(3-aminopropyl)amino]ethane persulfide to a carbon-centered free radical, the pKa of the persulfide was estimtated to be 6.2 ± 0.1 in comparison to a pKa of 7.6 ± 0.1 for the corresponding thiol.489 A computational study estimated that the pKa of cysteine persulfide (4.3) is ∼4 units lower than of cysteine thiol (8.29).60 The available data suggests that at physiological pH, RSS− will predominate over RSSH and that the [RSS−]/[RSSH] ratio can be ∼104-fold higher than the corresponding [RS−]/[RSH]. The acidity of persulfides and thiols on proteins will be modulated by their microenvironment, i.e., by the presence of functional groups.
7.2.2. Persulfide Nucleophilicity
Ionized persulfides (RSS−) are nucleophilic. Although both sulfurs in the ionized and in the protonated species have lone pairs of electrons, the outer or terminal sulfur in RSS− is the more nucleophilic center (eq 38).
| (38) |
Basicity and nucleophilicity are generally correlated, and the stronger the base, the greater the nucleophilicity.521 Since the pKa of persulfides is significantly lower than that of the corresponding thiol,60 persulfide would be expected to be less nucleophilic. However, the presence of a vicinal sulfur atom with lone electron pairs increases the nucleophilicity of the terminal sulfur atom via the alpha effect.522,523 Examples of the alpha effect enhancing nucleophilicity include HOO− relative to HO− and NH2NH2 and NH2OH relative to NH3.521 As noted previously, nucleophilicity is a kinetic concept and needs to be evaluated from the rate constants for the relevant reactions. A comparison between the reactivity of the persulfide versus thiol in human serum albumin toward 4,4′-dithiodipyridine provided a quantitative estimate of the magnitude of the alpha effect.60 The pH-independent rate constant for the reaction of the albumin persulfide with 4,4′-dithiodipyridine was 3-fold greater than for the thiolate. At pH 7.4, the persulfide is estimated to be fully ionized while the thiol is only partially ionized (the pKa of the single thiol is 8.1).60 Thus, the observed rate constant at pH 7.4 was 20-fold greater for persulfide than for the thiol.60
Computational evaluations of the HOMO energies are consistent with a higher nucleophilicity of persulfides than thiols. In one estimate, the energy of the HOMO of methylpersulfide was ∼29 kJ mol−1 higher than that of methylthiolate.103 In another estimate, the HOMO of cysteine persulfide was 51 kJ mol−1 higher than that of cysteine thiolate and, in addition, cysteine persulfide had lower chemical hardness than cysteine thiolate.60
The nucleophilicity of persulfides is evident from their reactivity toward thiol alkylating agents such as 1-chloro-2,4-dinitrobenzene,499 iodoacetamide,505,509,515 N-ethylmaleimide,505,509,515 methyl acrylate,515 monobromobimane,165,515 benzyl bromide,515 2-methylsulfonyl benzothiazol,135,511,515,524 and methylmethanethiosulfonate509 (Chart 26A). In contrast to thiols, which form thioeters with these reagents, persulfides form disulfides, which can be reduced to the corresponding thiols with reductants such as dithiothreitol. Interestingly, 9-fluorenylmethyl-based thioester models of LMW persulfide reacted with methylsulfonyl benzothiazol to give a trisulfide due to the reactivity of the initial benzothiazol disulfide derivative,515 which was not the case for other LMW persulfides or for protein (bovine serum albumin and glutathione peroxidase-3) persulfides.135,511,524 The nucleophilicity of persulfides is also revealed by their reaction with disulfides such as 5,5′-dithiobis(2-nitrobenzoic acid),60,509 N-acetylcysteine piridyldisulfide,509 and 4,4′-dithiodipyridine,60 to form trisulfides and other products (Chart 26B).
Chart 26. Reactions of Persulfides with Electrophilesa.
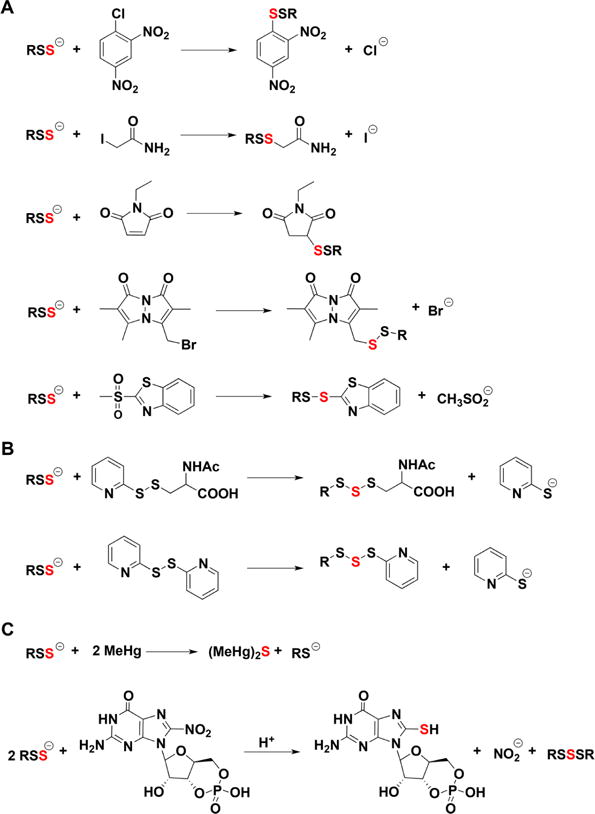
a(A) Reactions with thiol alkylating agents. (B) Reactions with disulfides. (C) Reactions with methylmercury and nitroguanosine 3′,5′-cyclic monophosphate.
Persulfides also react with electrophiles such as 8-nitroguanosine 3′,5′-cyclic monophosphate (8-nitro-cGMP) giving HS-cGMP,165,501 with methylmercury525 (Chart 26C) and, as described in the following sections, with one- and two-electron oxidants. In summary, persulfides are better nucleophiles than thiols because of the greater availability of RSS− versus RS− at neutral pH and higher intrinsic reactivity due to the alpha effect.
7.2.3. Reaction of Persulfides with Two-Electron Oxidants
Another manifestation of the nucleophilicity of the persulfides is their reactivity with two-electron oxidants. For example, the apparent rate constant of the reaction of albumin persulfide with peroxynitrite (1.2 × 104 M−1 s−1 at 20 °C) is 4-fold higher than of the corresponding thiol (2.7 × 103 M−1 s−1).60 By analogy with thiols, the immediate product of the reaction between a persulfide and a hydroperoxide is likely to be an unstable perthiosulfenic acid (RSSOH), which undergoes further reactions forming polysulfides (RSnR and RSn−) and, in the presence of excess oxidant, perthiosulfinic and perthiosulfonic acids (RSSO2H and RSSO3H). The latter have been detected as oxidation products of papain, albumin, and glutathione peroxidase.60,509,511 Importantly, in contrast to the thiol-derived sulfinic and sulfonic acids (RSO2H and RSO3H), which are generally considered to be irreversible oxidation products, the corresponding persulfide derivatives RSSO2H and RSSO3H can be reduced back to thiol by common reductants.135,340 Higher recovery of thiols after exposure of persulfides versus thiols to hydrogen peroxide (or HNO) followed by dithiothreitol treatment was demonstrated.526
The higher reactivity of persulfides to oxidants and the recovery of thiols following reduction of the resulting oxidation products, support the proposal that persulfides can serve a protective functions for protein thiols (Chart 27).135,340,341,527
Chart 27. Protein Persulfidation Can Protect Proteins from Overoxidationa.
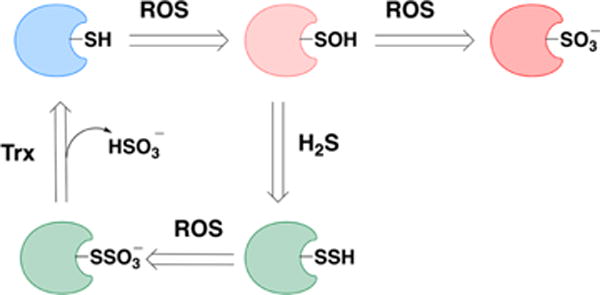
aA thiol can be oxidized to a sulfenic acid. The latter can be reduced back to thiol or be further oxidized to sulfonate, an irreversible modification. Persulfides, if exposed further to oxidants, will form S-sulfocysteines (−SSO3−). Enzymes such as thioredoxin can reduce the S–S bonds and restore the native thiol.
7.2.4. Reaction or Persulfides with One-Electron Oxidants
Persulfides are excellent one-electron reductants and are in fact better than thiols or H2S.528 This is consistent with the lower energy of dissociation of the S–H bond (293 kJ mol−1) in comparison to thiols and H2S (385 kJ mol−1).529 It is also consistent with the one-electron reduction potential of persulfides (E°′(RSS•/RSS−) = 0.68 V), which is lower than those of the corresponding thiol, (E°′(RS•, H+/RSH) = 0.96 V) and of H2S (E°′(S•−, H+/HS−) = 0.91 V),64 respectively. Thus, persulfides can be oxidized by weaker oxidants than thiols or H2S, and RSS• is less oxidizing than RS• or HS•.
Depending on the nature of the one-electron oxidant (A1• or A2• in eqs 39 and 40, e.g., carbon centered radical or peroxyl CCl3OO• radicals, respectively) the persulfide can react through a hydrogen atom or electron transfer mechanism, which affects the pH dependence of the process.528
| (39) |
| (40) |
Exposure of aralkyl persulfides to ferric salts under organic solvents led to the formation of tetrasulfide and ferrous ion, showing that persulfides can be oxidized by ferric ions (eqs 41 and 42).530
| (41) |
| (42) |
Importantly, persulfides were unable to oxidize ferrous salts.530 This contrasts with the behavior of hydrogen peroxide and alkyl hydroperoxides, which oxidize ferrous to ferric ion with concomitant formation of a hydroxyl radical via the Fenton reaction (eq 43).
| (43) |
Persulfides can reduce ferricyanide,531 ferric cytochrome c,501,505,532 and metmyoglobin.531 In addition, persulfides have been proposed to react with carbon centered radicals,489 peroxyl radicals,528,533 and the nitroxide TEMPOL,531 yielding the perthiyl radical. All of these reactions appear to be faster with persulfides than with thiols. For example, the rate constant for the reaction of 2-[(3-aminopropyl)amino]ethane persulfide and the carbon-centered α-hydroxyalkyl radical derived from isopropanol is 2.4 × 109 M−1 s−1, which is 1 order of magnitude higher than the corresponding reaction of the thiol.489
Penicillamine-derived persulfide reduces ferric cytochrome c quantitatively in contrast to penicillamine and glutathione, which do not show significant reduction.501 However, this reaction is thermodynamically uphill given the mismatch in redox potentials between the persulfide (+0.68 V) and cytochrome c (E°′(cyt c3+/cyt c2+) = +0.26 V).64 Persulfides react directly with oxygen, albeit slowly (k < 0.3–0.4 M−1 s−1).103 This reaction also faces thermodynamic and kinetic barriers due to a mismatch in the redox potentials and the triplet state of oxygen and reveals the intrinsically higher reactivity of persulfides with respect to thiols and H2S. A likely explanation for why each of these unfavorable reactions occurs is that the perthiyl radical product is efficiently removed via recombination forming tetrasulfide (eq 44). Perthiyl radicals decay predominantly through second order processes with rate constants of 1–6 × 109 M−1 s−1.528,533,534 The resulting RSSSSR can decompose to give RSSR and S2 (eq 45).
| (44) |
| (45) |
Perthiyl radicals have been characterized by pulse radiolysis, flash photolysis, and EPR spectroscopy.528 The unpaired electron in perthiyl radicals is delocalized between the two sulfur atoms. The resonance stabilization energy of perthiyl radicals relative to thiyl radicals is estimated in 8.8 kJ mol−1.489,528 This inherent stability of perthiyl radicals contributes to the efficiency of persulfides as reductants.
In addition to the radical recombination reaction to form tetrasulfides, perthiyl radicals can also act as oxidants, but the rate constants of these reactions are smaller than those of thiyl radicals.528 For example, the rate constant for hydrogen atom abstraction by the perthiyl radical from a polyunsaturated fatty acid is 1.2 × 106 M−1 s−1, 1 order of magnitude lower than the corresponding reaction of a thiyl radical, 1.4 × 107 M−1 s−1. The rate constant for the reaction of a perthiyl radical with ascorbate is (1–6) × 106 M−1 s−1, which is 1 order of magnitude smaller than that of the thiyl radical.528
Perthiyl radicals reportedly react with oxygen with a second order rate constant of 5 × 106 M−1 s−1 initially forming an RSSOO• species and ultimately forming inorganic sulfate.534 However, recent studies have not confirmed this experimentally, and computational studies suggest that the reaction of perthiyl radicals with oxygen is thermodynamically uphill.531,533 In contrast, thiyl radical reacts reversibly with oxygen forming thioperoxyl radicals (RSOO•) with forward and reverse rate constants of ∼109 M−1 s−1 and ∼105 s−1, respectively,535,536 a reaction that contributes to oxidative damage via chain propagation.
A perthiyl radical can react with RSS− to form RSSSSR•−, which is unstable,537 has not been detected directly, and reacts with oxygen to form O2•− and the more stable RSSSSR (eqs 46 and 47). Analogously, RSS• can react with RS− to form RSSSR•−.528
| (46) |
| (47) |
In contrast to thiyl radicals that react with NO• very rapidly forming nitrosothiols,538 perthiyl radicals do not appear to react with NO•. The apparent lack of reactivity has been attributed to the relatively high stability of the radicals and to the weakness of the N–S bond in RSSNO. Accordingly, attempts to prepare RSSNO failed with immediate NO• generation from the reaction mixture.531,539
In summary, persulfides are excellent one-electron reductants, which can be explained by their ease of oxidation, by the high relative stability of the perthiyl radical, and by the rapid radical recombination, which facilitates product removal.
7.2.5. Electrophilicity
Persulfides are relatively weak electrophiles. The reactions of persulfides in the protonated state with a general nucleophile Nu−, are shown in eqs 48 and 49. When the inner sulfur is the site of nucleophilic attack, H2S is released. When the outer sulfur is the site of attack sulfur transfer to the nucleophile occurs with elimination of the thiol.
| (48) |
| (49) |
Persulfides can react with cyanide,540 thiolates,501,509,540 sulfite,540 phosphines,496 and amines.541 Thiols and H2S are formed as reaction products, together with Sn and polysulfides. In organic solvents, cyanide, amines, hydroxide, and halides react as bases rather than as nucleophiles, abstracting a proton from RSSH and promoting its decay.103,495,541,542
Computational modeling of the lowest unoccupied molecular orbital (LUMO) of methylpersulfide (CH3SSH) shows that attack at either sulfur atom is possible, while the electrostatic potential surface analysis shows a slight preference for attack on the outer sulfur.103
Steric hindrance is a critical factor that can bias nucleophilic attack toward the outer sulfur, while unhindered persulfides can be attacked on the inner sulfur releasing H2S.495,543 The importance of steric hindrance has been documented by comparing the base-promoted decay of trityl- and adamantylpersulfides versus benzylpersulfide. In the former cases, thiol and Sn were the reaction products, while polysulfides (RSSnSR) and H2S were formed with the more accessible benzylpersulfide.494,495
Another factor that critically influences the site of attack is the acidity of the leaving group. H2S has a pKa of 6.98, while thiols have higher pKa values (8.29 and 8.94 for cysteine and glutathione, respectively, 25 °C).544 It is therefore expected that nucleophiles will attack cysteine persulfides at the inner sulfur releasing the sulfide anion (HS−), which is the better leaving group. With protein persulfides, geometric considerations around the persulfide also determine the electrophilicity of the two sulfur atoms. For example, in MST, attack on the outer sulfur is favored by steric and inductive effects, which are governed by active site residues that also render the persulfide highly electrophilic.280
Persulfides can also react with substituted phosphines (R3P) in a process that is reminiscent of hydroperoxides. The major products formed are phosphine sulfide (R3P═S) and thiol.496 Analysis of the reaction products provided evidence that, while the attack can occur at either sulfur atom, attack at the outer sulfur predominates, particularly when the persulfide is sterically hindered.545 Phosphines have been used to detect persulfides in biological samples (see section 7.3.2).
The reaction of persulfides with cyanide to give thiols and thiocyanate (eq 50) deserves mention.
| (50) |
This reaction can be used for the detection of persulfides (see section 7.3.2) since thiocyanate can react with ferric ions forming a red complex that absorbs at 460 nm and can be quantified spectrophotometrically (eq 51).31
| (51) |
The reaction of persulfides with cyanide is favored at pH 7.4 relative to pH 10, suggesting that RSSH is the reactive species.103 From a mechanistic standpoint, cyanide has been postulated to react with the RS(S)H tautomer of persulfide.546 However, computational modeling of the reaction of methylpersulfide (CH3SSH) with cyanide in a polar medium predicted a linear transition state with an ∼50 kJ mol−1 activation free energy barrier, which is compatible with a nucleophilic displacement mechanism. While the activation free energy barrier for the nucleophilic attack on the inner sulfur was 3 kJ mol−1 lower than the attack on the outer sulfur, the free energy change for the reaction was higher by 96 kJ mol−1, indicating that the reaction on the outer sulfur is thermodynamically favored.103 MST and rhodanese catalyze transfer of the outer sulfur from their active site cysteine persulfides to cyanide and to other acceptors (thiols, sulfite). In addition, thiols, which are present at millimolar concentrations inside cells,547 are likely to react with LMW persulfides.
Reaction with a thiol at the inner sulfur can give rise to disulfide and H2S (eq 52).
| (52) |
Penicillamine-derived persulfide reacts with glutathione to generate H2S,501 while glutathione peroxidase-3 persulfide formed a mixed disulfide between the protein and the thiol in the reactions with glutathione and N-acetyl cysteine.509 The general reaction described in eq 52 provides a mechanism for H2S generation from persulfide (see section 8.9).
Reaction at the outer sulfur with a LMW or protein thiol results in trans-persulfidation (eq 53). This reaction is relevant for some proteins involved in iron–sulfur cluster formation and H2S biosynthesis and oxidation. The role of trans-persulfidation in protein persulfide formation and signaling, and in enzyme-catalyzed depersulfidation is discussed in section 8.
| (53) |
7.2.6. Spontaneous Decay of Persulfides
Persulfides are unstable in aqueous solution, which poses challenges for their characterization. For example, real-time MS analysis of penicillamine-derived persulfide showed that it decays with a half-life of 2.7 min at 23 °C;135 somewhat higher values have been reported for the decay of CysSSH (35 min at 37 °C).226 The decay represents a disproportionation reaction involving two molecules of persulfide (eqs 54–56),542 which is consistent with the sulfur atoms possessing both electrophilic and nucleophilic character. The importance of RSSH ionization is evidenced by the dependence of the decay rate of persulfides in organic solvents on the strength of the added base.494 Acidic conditions also appear to favor decay.495,499
| (54) |
| (55) |
| (56) |
The decay products vary depending on the site of the original attack. Persulfides with bulky substituents react preferentially at the outer sulfur yielding thiol and elemental sulfur (eqs 54 and 55), while those with small substitutents react at the inner sulfur yielding polysulfanes and H2S (eq 56).226,494,495,501 Attack at the inner sulfur is also favored by the release of HS−, which is a better leaving group than RS− as discussed previously. Cysteine persulfide predominantly decays via the reaction shown in eq 56,226 and a similar behavior has been reported for the penicillamine-derived persulfide.501
In addition to disproportionation, persulfides can undergo thermal or light-induced homolysis of the S–S bond giving the corresponding RS• and HS• radicals.548 This behavior is expected from the S–S bond energies of HS-SH (276 kJ mol−1) and CH3S-SCH3 (309 kJ mol−1).529 Homolysis of persulfides is very slow and is unlikely to contribute to their decay at room temperature and under moderate light.548
7.3. Methods for Detecting Persulfidated Cysteines
Spectrophotometric analysis of protein persulfides is of limited utility as they absorb between 335 and 340 nm and have a weak extinction coefficient (∼300 M−1 s−1).31,549 The IR spectra of thiols and persulfides are similar. However, a band in the 200–500 cm−1 region due to the S–S bond is characteristic of persulfides (Table 3).491–494 With a few exceptions (Figure 17A) these spectroscopic methods have limited applicability for detecting protein persulfides in complex mixtures.
Figure 17.
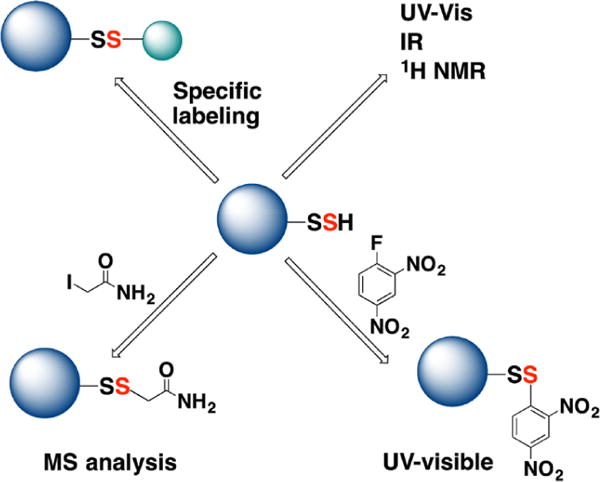
Methodological approaches for the characterization of protein persulfides. (A) When pure, protein persulfides can be characterized by UV–visible, IR, and 1H NMR spectroscopy. (B) Protein persulfides react with 1-fluoro-2,4-dinitrobenzene to form mixed disulfides. DTT releases 2,4-dinitrobenzenethiol, which absorbs at 408 nm under alkaline conditions. (C) Persulfides can be labeled with thiol blocking reagents such as iodoacetamide and analyzed by MS. (D) Protein persulfides can be tagged through different strategies that rely on either their electrophilic or their nucleophilic character.
The major challenge in developing labeling methods is to discriminate between the reactivity of persulfides and thiols, disulfides, sulfenic acids, and polysulfides. Consequently, not too many reliable methods are currently available. The available methods for intracellular persulfide detection can be grouped into two categories: (i) methods for protein persulfide labeling (which rely on the nucleophilic nature of the outer sulfur) and (ii) methods for sulfane sulfur detection (which rely on the electrophilic nature of the persulfide).
7.3.1. Methods for Protein Persulfide Labeling
Due to their greater nucleophilicity, persulfides react faster with commonly used thiol blocking electrophiles than the corresponding thiols60 and yield distinct products. Thus, alkylation of thiols yields thioethers while disulfides are formed from persulfides.509 Several methods exploit these characteristics of persulfides for their detection.
A spectroscopic method for the indirect detection of protein persulfides relies on the reaction of protein persulfides with 1-fluoro-2,4-dinitrobenzene to form mixed disulfides.550 Subsequent treatment with 1,4-dithiothreitol (DTT) releases 2,4-dinitrobenzenethiol with a characteristic absorbance at 408 nm under alkaline (1 M NaOH) conditions. Using an extinction coefficient of 13 800 M−1 cm−1 for 2,4-dinitrobenzenethiol, the protein persulfide concentration can be estimated (Figure 17B).550
MS can be used to directly detect the presence of persulfides in proteins.19 However, the mass increase due to the addition of one sulfur atom (Δm/z = 31.97207) is very similar to that caused by the addition of two oxygen atoms (Δm/z = 31.98984) and can only be distinguished in small peptides but not whole proteins.19 The relative instability of persulfides further limits their direct detection by MS analysis. To circumvent these problems, persulfidated proteins can be blocked with agents such as N-ethylmaleimide or iodoacetamide (Figure 17C) and then analyzed by MS.505,509 This treatment stabilizes the persulfide modification, and the detection of a Δm/z of 32 with respect to the nonpersulfidated peptide, confirmed by daughter ions, provides strong evidence for the presence of persulfide.
In principle, treatment of cells with H235S followed by Western blot analysis and radioactivity measurement can provide a semiquantitive estimate of protein persulfidation levels. However, the inavailability of H235S and the instability of persulfides limit this approach.
The modified biotin switch method first used for proteomic analysis of persulfides19 was based on the premise that, unlike thiols, persulfides would not react with the electrophilic thiol-blocking reagent, S-methylmethanethiosulfonate (MMTS). The strategy involved initial blocking of thiols with MMTS followed by persulfides labeling with N-[6-(biotinamido)hexyl]–3′-(2′-pyridyldithio)propionamide (biotin-HPDP; Figure 18A). One problem with this method is that MMTS treatment induced intra- and intermolecular protein disulfides.551 However, the more important flaw with the modified biotin swtich approach for persulfide proteome mapping is that MMTS and its analogue S-4-bromobenzyl methanethiosulfonate react readily with protein persulfides.509 Unfortunately, despite these limitations the modified biotin switch method continues to be used and has been shown to illustrate decreased labeling in CSE19,552–554 and CBS555,556 knockout cell lines and tissues and increased labeling in response to H2S treatment. In some cases, cysteines identified as persulfidated targets have been validated by mutagenesis.552,554,557 A plausible explanation for how some persulfides might become labeled by the modified biotin-switch assay is that persulfides react with MMTS faster than free thiols509 and the resulting RS–S–S–Me reacts with residual free thiols regenerating free thiols at sites that carried the persulfide modification originally. The newly formed thiol is subsequently labeled by biotin-HPDP or by fluorescently tagged maleimides (Figure 18B). Given the chemical issues in its design and the resulting misrepresentation of the persulfide proteome, the use of the modified biotin switch method is discouraged.
Figure 18.
Modified biotin switch assay for persulfide labeling. (A) MMTS was proposed to selectively block free thiols leaving persulfides unmodified and ready for reaction with a biotin-derivatized reactive disulfide (biotin-HPDP). (B) Mechanistic explanation for persulfide labeling with the biotin switch assay. MMTS reacts with persulfides more readily than with thiols forming a trisulfide product. This trisulfide is attacked by unreacted thiols leaving a free cysteine that can react with biotin-HPDP.
A second method for persulfide detection involves blocking free thiols and persulfides with Cy5-maleimide and subsequently reducing the R–S–S–maleimideCy5 adduct (Figure 19),552 which results in the loss fluorescence. The loss of red fluoresecence is detected following separation of proteins by gel electrophoresis. While relatively simple, the limitation of this method is that it is based on the absence of a signal associated with persulfides rather than on a positive signal, which can be coupled to MS for proteomic analysis. Furthermore, since maleimides are known to react with amines, extensive labeling can give high backgrounds obscuring changes in signal intensity when persulfides are present at low concentrations.
Figure 19.
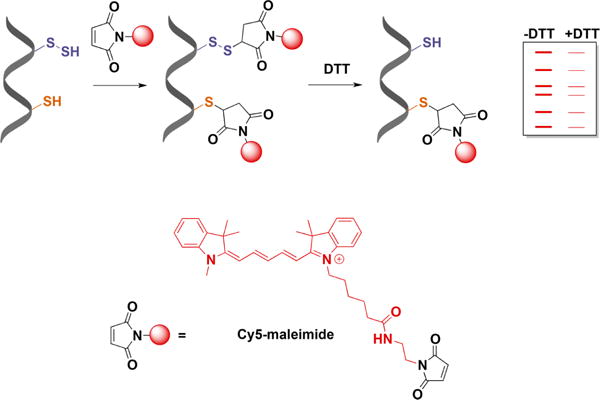
Persulfide detection by differential fluorescence tagging. Both thiols and persulfides are initially blocked with Cy5-maleimide. DTT treatment then removes the fluorescent tag from the persulfides. Proteins are separated by electrophoresis, and the loss of fluorescence caused by DTT is used as a measure of persulfidation.
A method that allows trapping of persulfidated proteins and subsequent MS analysis60 is the basis of the biotin thiol assay (BTA).558 Biotin maleimide60 (or maleimide-PEG2-biotin)558 is initially used to react with thiols and persulfides (Figure 20). Proteins are then trypsinized, and thiol- and persulfide-tagged biotinylated peptides are immobilized on streptavidin beads. Persulfidated peptides are reduced and peptides are eluted from the beads and derivatized with iodoacetamide for subsequent MS analysis. The use of heavy (deuterated) and light iodoacetamide allows for quantitative analysis.558 Using the BTA method >800 proteins have been identified as persulfidation targets.558 Modifications of this method referred to as qPerS-SID559 and ProPerDP514 have been reported in which biotinylated iodoacetamide is used instead of biotin maleimide and (tris(2-carboxyethyl)phosphine) instead of DTT.
Figure 20.
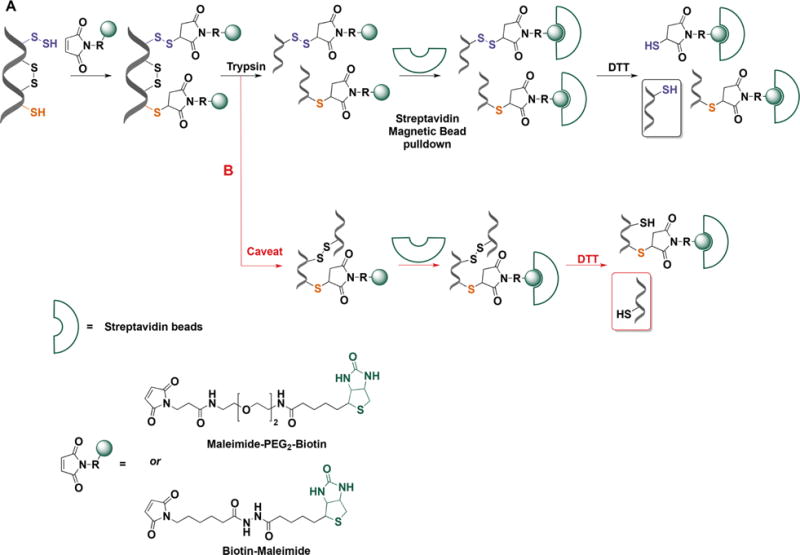
Persulfide labeling with biotin-tagged alkylating reagents. (A) In the first step proteins are mixed with maleimide-biotin (or maleimide-PEG2-biotin) to tag both thiols and persulfides. Proteins are trypsinized and the biotinylated peptides are bound to streptavidin beads. Persulfidated peptides attach to streptavidin beads via disulfide bonds. DTT treatment facilitates elution from the beads and subsequent MS analysis. (B) A possible caveat is that peptides connected by disulfide bonds and containing a thiol or persulfide could be released from the beads with DTT. However, the concentration of disulfide bonds in intracellular proteins is low and this not expected to be a quantitiatvely major drawback of the method.
One difference between the ProPerDP and BTA method is that in the former intact proteins rather than peptides are eluted from the streptavidin beads (Figure 21). Immobilization of intact proteins on streptavidin beads is, however, not recommended since it leads to an underestimation of persulfide targets. For example, a protein containing three cysteine residues (e.g., GAPDH) of which only one is persulfidated will be eluted with a lower yield as binding can occur via any of the cysteine sites. This could explain the relatively low number of protein persulfidation targets identified by the ProPerDP method (Figure 21).514
Figure 21.
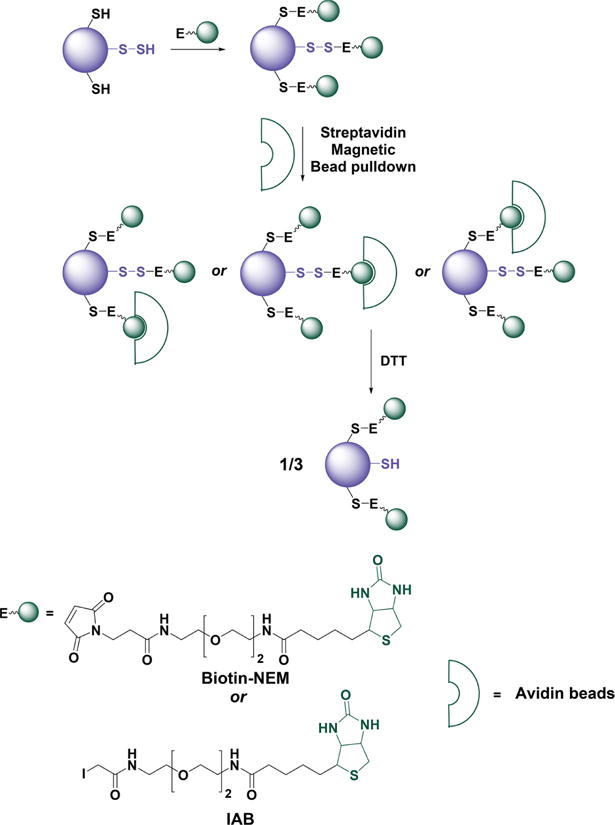
Caveats of whole protein labeling with biotin-tagged alkylating reagents. Proteins containing several cysteine residues, of which only one is persulfidated, are labeled with biotin-maleimide (biotin-NEM) or biotin iodoacetamide (IAB). Since all labels have equal chances of binding to streptavidin beads, the elution with DTT will result in lower than expected yield because the protein will remain bound through the tagged thiols.
Combining the persulfide labeling qPerS-SID approach with the SILAC (stable isotope labeling with amino acid in culture) method allows for quanititative persulfide proteomics analysis559 (Figure 22). However, the selectivity of the qPerS-SID and similar methods would be improved by optimizing the initial blocking conditions. As described for the BTA method,558 limiting the concentration of the blocking reagent and the duration of labeling, decreases background labeling and identification of false positives (e.g., due to the reactivity of sulfenic acids with IA-biotin560). A potential limitation of persulfide tagging methods is that the reduction step also reduces disulfide bonds that were originally present within and across proteins. These disulfide-containing peptides/proteins will, however, only be released from streptavidin beads if they contain a biotin-tagged persulfide but not a biotin-tagged thiol (Figure 20B). In the former case, the identity of the cysteine that was the site of persulfidation will not be revealed by the MS/MS analysis. However, disulfides are not common in intracellular proteins, and the interference by disulfide-containing peptides/proteins in persulfide identification is expected to be low.
Figure 22.
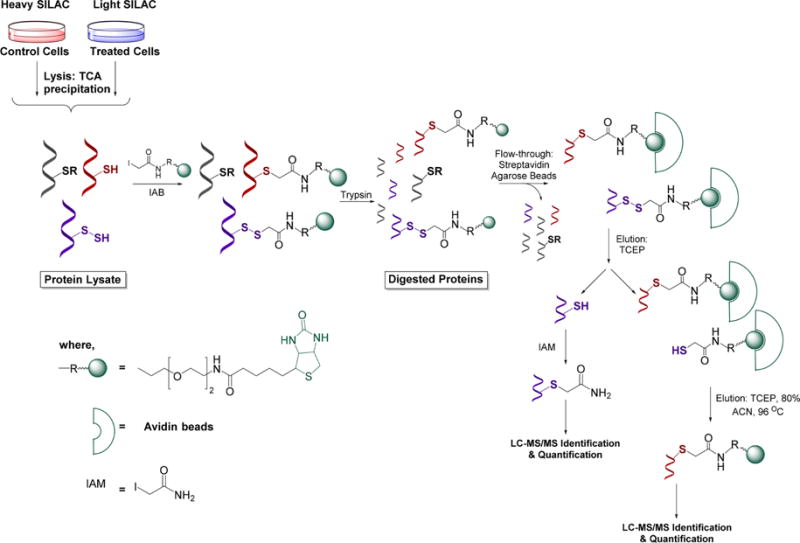
qPerS-SID (quantitative persulfide site identification) approach. Control cells and H2S-treated cells are grown in heavy and light SILAC (stable isotope labeling with amino acids in cell culture) media, respectively. After cell lysis protein extracts are mixed 1:1 and exposed to iodoacetamide-PEG-biotin to labelthiols and persulfides. Proteins are then trypsinized and labeled peptides bound to streptavidinbeads. Since persulfidated peptides attach to streptavidin beads via disulfide bonds they are eluted with TCEP (tris(2-carboxyethyl)phosphine). Released peptides are subjected to LC-MS/MS identification and quantification (enabled by the SILAC approach).
A related approach that was used to detect tyrosine phosphatase 1B (PTP1B) persulfidation561 used iodoacetamide to initially block free thiols and persulfidated cysteines, followed by reduction and capture of the newly exposed cysteine thiol with biotinylated iodoacetamide. Although potentially useful to identify potential persulfidation sites on purified proteins, DTT treatment also reduces other oxidative cysteine modifications that might exist on the protein (e.g., nitrosothiol and sulfenic acid) confounding the result.
In another approach, thiols and persulfides are blocked with a maleimide derivative with a peptide arm (MalP).562 The persulfidated protein is then detected directly in gel upon loss of the succinimide-peptide moiety (the product of maleimide reaction with a sulfhydryl) upon cleavage of the S–S bond by DTT. The size of the MalP derivative was optimized to cause a detectable shift in protein migration by denaturing polyacrylamide gel electrophoresis.562 The best results were obtained with a 16-mer of MalP (MalP16:1.95 kDa; Figure 23). This method is useful for monitoring persulfidation changes in a known target but not for proteome wide analysis.
Figure 23.
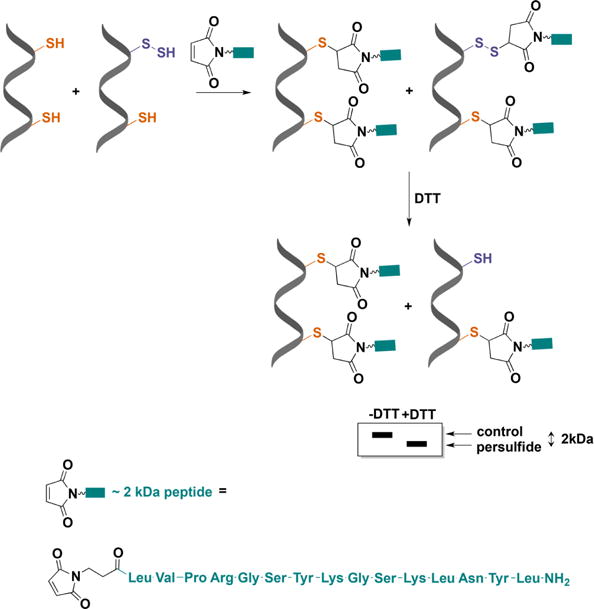
Detection of protein persulfidation by differential peptide tagging. An engineered maleimide with a peptide arm and a molecular mass of ∼2 kDa (MalP) reacts with both thiols and persulfides. DTT removes the mixed disulfides formed with persulfides and increases the electrophoretic mobility of the protein.
An alternative tag-switch strategy for identifying protein persulfides is based on the premise that the disulfide bonds resulting from alkylation of protein persulfides with an appropriate thiol blocking reagent show enhanced reactivity to nucleophiles than protein disulfides in which the electrophilicity of the two sulfur atoms is similar (Figure 24).135,511,524 Therefore, it is possible to introduce a tag-switching reagent (containing both the nucleophile and a reporting molecule) to label only the persulfide products. It should be noted that thiol products are thioethers, which are not expected to react with the nucleophile. Typical thiol-blocking reagents such as maleimides and iodoacetamides are not suited for the tagswitch technology. However, a reagent that gives a mixed aromatic disulfide linkage when reacting with persulfides provides the differential reactivity criteria for the tag-switch technology. In the first step, MSBT or its more water-soluble analogue (benzothiazole-2-sulfonyl)-acetic acid (MSBT-A)563 is used to block thiols and persulfides. In the next step, a biotinylated derivative of methyl cyanoacetate serves as a nucleophile to label the inner sulfur atom of the benzothiazole-blocked persulfide. The selectivity of this method was demonstrated by the reactivity of persulfidated but not glutathionylated, sulfenylated or unmodified bovine serum albumin, which contains intramolecular disulfides in addition to a reactive cysteine.511,524 The sensitivity of this method has been increased with two new cyanoacetic acid derivatives containing the fluorescent BODIPY moiety (CN-BOT) or the Cy3-dye (CN-Cy3; Figure 24B). These probes allow detection of persulfidated proteins by fluorescence confocal microscopy or in gels.135 The reactivity of sulfenic acids with cyanoacetic acid-derivatives is a potential concern and this can be prevented by blocking sulfenic acids with dimedone prior to the reaction with MSBT,511,524 although no difference in detected persulfide levels was observed between dimedone pretreated and untreated samples.511,524 The concern that highly reactive protein disulfides would cross-react with cyanoacetic acid-derivatives is overcome by the denaturing conditions of the assay in which the different protein benzothiazole derivatives would be expected to show similar reactivity. The method has been successfully used for mining of the persulfidation proteome in Arabidopsis thaliana, identifying >2000 persulfidated protein targets (5% of the entire Arabidopsis proteome).564
Figure 24.
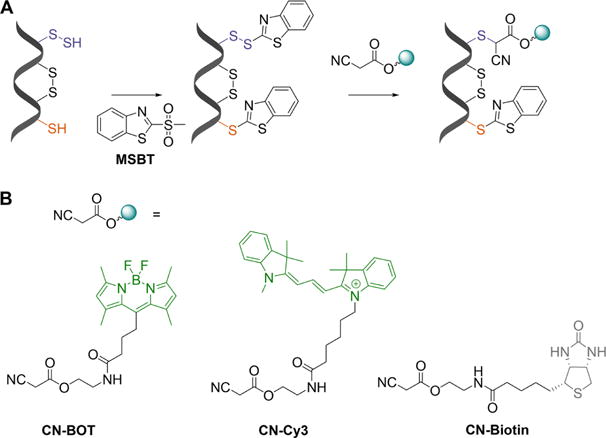
Cyanoacetic acid-based tag-switch method for persulfide labeling. (A) Both persulfides and thiols are initially blocked with MSBT. The product with a persulfidated cysteine is a mixed aromatic disulfide that can be nucleophilically attacked by a cyanoacetic acid–based probe causing a tag-switch. (B) Three different tags are attached to cyanoacetic acid: BODIPY (CN-BOT), Cy3 (CN-Cy3), and biotin (CN-biotin).
7.3.2. Methods for Sulfane Sulfur Detection
The simplest way to detect the total sulfane sulfur pool is by reducing the sample with DTT (Figure 25),100,101 which releases H2S. The latter can then be detected by one of several methods as discussed in section 3. However, this method can detect sulfurs from iron sulfur clusters, in addition to inorganic polysulfides and thiosulfates.565
Figure 25.
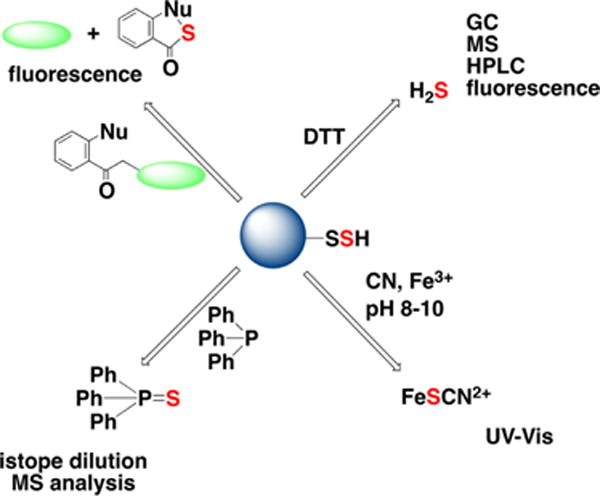
Overview of methodological approaches for total sulfane sulfur detection. Sulfane sulfur compounds release H2S upon DTT treatment. Sulfane sulfur can be detected by the cold cyanolysis method; samples are incubated in alkaline (pH 8–10) cyanide solutions to release SCN−, which is then quantified spectrophotometrically as a complex with Fe3+. Sulfane sulfur can be extracted by triarylphosphines in the form of triarylphosphine sulfide, which can be quantified by isotope dilution MS analysis. Finally, different fluorescence probes (e.g., the SSP series) can detect sulfane sulfur in protein persulfides and low molecultar weight hydropolysulfides in cells (described in greater detail in Chart 28).
Cold cyanolysis is widely used to detect sulfane sulfur (Figure 25).31,60,566,567 In this method, the cyanide anion attacks the sulfur–sulfur bond103,546 in persulfides, polysulfides (RSSnR), and aryl thiosulfonates (pH 8.5–10, 10 °C to room temperature). The resulting thiocyanate is converted to ferric thiocyanate and measured by its characteristic absorbance at 460 nm. When the reaction is performed at higher temperatures (referred to as “hot cyanolysis”), sulfane sufur from thrithionate (−O3SSnSO3−) and thiosulfate (−SSO3−) can be detected as well.31 The reactivity of sulfane sufur toward cyanolysis decreases in the following order: persulfide > polysulfide > thiosulfonate > polythionate = thiosulfate > elemental sulfur.
Fluorescent probes have been developed to visualize and quantify sulfane sufur (Figure 25) in cells. The first such probes described were SSP1 and SSP2 (Chart 28A).568 Sulfane sufur reacts with a nucleophilic thiol in the nonfluorescent SSP1/SSP2 probe to form a persulfide intermediate, which in turn reacts with an electrophilic ester group leading to spontaneous cyclization and release of the fluorophore (Chart 28B). Negligible basal fluorescence is seen in cells treated with SSP1 or SSP2 indicating that they are either inefficient at detecting protein and/or LMW persulfides or that the concentrations of these compounds are very low.
Chart 28. Fluorescent Probes for Sulfane Sulfura.
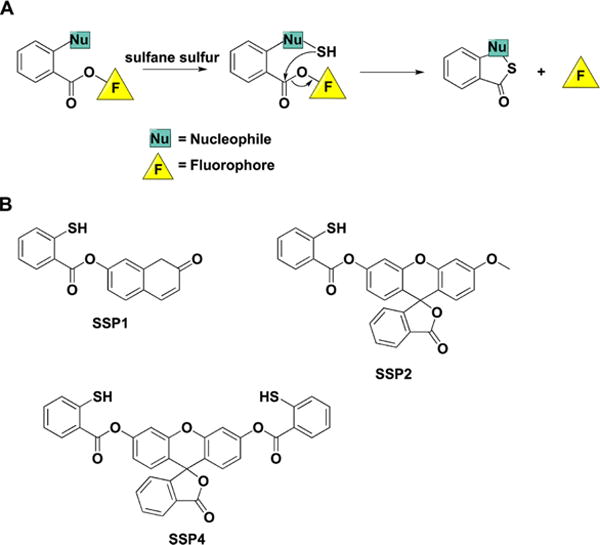
a(A) Internal cyclization and fluorophore release following the initial reaction of the probe with sulfane sulfur compounds. (B) Structures of the synthetic probes that exhibit this type of chemical reactivity.
Electrophilic probes like DSPI-3, which initially reacts with inorganic polysulfides and releases the fluorophore upon internal cyclization, have been reported.569–571 (Chart 29). While DSPI-3 reacts readily with thiols as well, the subsequent intramolecular cyclization leading to generation of a fluorescent signal does not occur (Chart 29A). However, if the thiol adduct were to react further with polysulfides, fluorophore release would occur.569 The reaction of protein persulfides or LMW persulfides with DSP probes has not been tested. Optimization of this class of probes for greater selectivity for polysulfide has been reported together with the development of a FRET probe, which detects both H2S and polysulfides (Chart 29C).571
Chart 29. Electrophilic Probes that Release a Fluorophore upon Reaction with Inorganic Polysulfidesa.
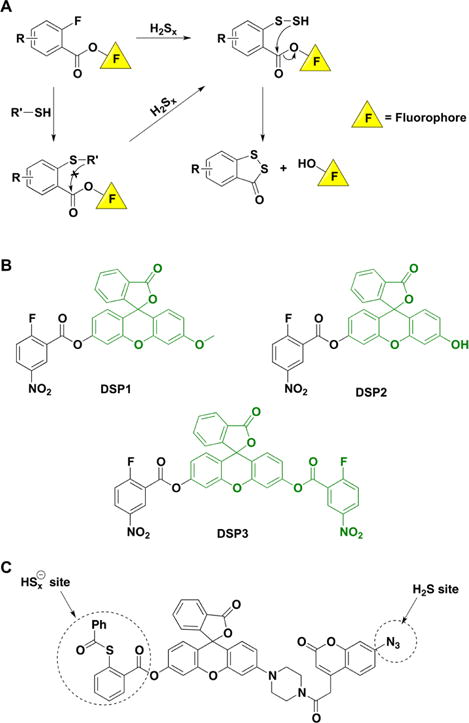
a(A) Reactions leading to fluorophore release. (B) Structures of the synthetic probes that exhibit this type of reactivity. (C) FRET-based probe for the simultaneous detection of H2S and sulfane sulfur-containing species.
A ratiometric near-IR fluorescence probe (Cy-Dise) for cysteine persulfide based on a selenium–sulfur exchange reaction has been reported (Chart 30A).572 The method exploits the lower pKa and greater nucleophilicity of cysteine persulfide compared to cysteine thiol. It is unclear however how this probe is selective for cysteine persulfide versus cysteine thiols with low pKas, other persulfides, or inorganic polysulfides. The same limitation is also true with a persulfide probe that combines nucleophile-induced xanthene fluorescence quenching with coumarin as a FRET donor (Chart 30B).573
Chart 30.
Ratiometric Near-IR Fluorescence Probe for Cysteine Persulfide Detection (A) and FRET Probe Designed for Persulfide Detection (B)
Isotope dilution MS is a reliable method for quantifying total sulfane sulfur pool574 and is based on their known reactivity with triphenylphosphines (Figure 25). This method relies on the use of 13C isotope-labeled triarylphosphine sulfide as an internal standard spiked into the biological sample treated with triarylphosphine. From the ratio of the MS signal intensities of the internal standard and triarylphosphine sulfide, an estimate of the total sulfane concentration is obtained (Chart 31).574
Chart 31. Isotope Dilution Mass Spectrometry Approach for the Detection of Sulfane Sulfur Using Substituted Phosphinesa.
aThe reaction of triarylphosphine (P2) with sulfane sulfur compounds. After incubation of P2 with cell or tissue samples to form PS2, PS1 (13C-labeled triarylphosphine sulfide) is added as internal standard and the samples analyzed by MS.
8. CELLULAR MECHANISMS OF PERSULFIDE FORMATION AND REMOVAL
An initial estimate based on the modified biotin tag switch assay was that up to 25% of all proteins are persulfidated.19 The BTA method identified 834 persulfidated proteins in a pancreatic beta cell line,558 representing ~5% of the proteome. The cyanoacetate-based tag-switch method confirmed that ~5% of the entire plant proteome is persulfidated.564 Significantly lower steady-state protein persulfide levels were reported in HEK293 cells (0.15% of the proteome) and in murine liver (1.2% of the proteome) using the ProPerDP method.514 Unexpectedly high concentrations of LMW persulfides (150 μM GSSH in brain and 1–4 μM cysteine persulfide in different mouse tissues) were reported in an MS study.165 The isotope-dilution MS method yielded estimates of sulfane sulfur levels in murine plasma and erythrocytes of 4.7–13.1 and 2.3–3.7 nmol/g protein, respectively, and higher values in other organs: 57.0 (liver), 150.9 (kidney), 46.0 (brain), 61.8 (heart), 56.1 (spleen), and 20.8 (lung) nmol/g tissue.574 Extrapolating from these values, this study suggests that the sulfane sulfur concentration in plasma and tissues is in the low micromolar range. In the next section, routes for persulfide formation and removal are described.
8.1. Reaction between H2S and Disulfides
Early reports indicated that cystine and other LMW and protein disulfides react at alkaline pH with sodium sulfide. The product absorbed at 320–350 nm, was unstable, reacted with cyanide, and was assigned as persulfide (eq 57).502,575,576
| (57) |
Thermochemical calculations and computational modeling of the reaction with cystine suggest that the formation of the RSSH and RS− products is uphill by +56.1 kJ mol−1, but that fast equilibration to RSS− and RSH due to the lower pKa of the persulfide drives the reaction in the forward direction. The reaction is also driven by further reactions of the unstable persulfide product.60,64
The reactions of H2S with typical LMW disulfides are slow.60,179,503–505 For example, the rate constant for the reaction of H2S with cystine at pH 7.4 and 25 °C is 0.6 M−1 s−1.60 The reaction of H2S with GSSG is also slow and reversible and leads to GSSH and a mixture of products.504,505 Nevertheless, the protein environment can accelerate the reaction. For example, in SQR, the reaction of H2S with an active site disulfide is accelerated by ~106-fold with respect to free cystine.306,310,311
Reaction of H2S with typical disulfides is slower than the analogous reaction of RSH (the thiol–disulfide exchange reactions), probably due to the lack of inductive/field or solvation effects attributed to the adjacent methylene group in thiolates.60 The logarithm of the pH-independent rate constant for the reaction of HS− with disulfides decreases linearly with a slope of −0.75 as the pKa of the thiol that constitutes the disulfide increases, consistent with acidic thiols being better leaving groups. Computational modeling predicts a linear transition state as expected for a concerted SN2 mechanism.60 The influence of the thiol pKa on kinetics supports the prediction that proteins that contain disulfides formed with acidic thiols are better targets for H2S. In asymmetrical disulfides (RSSR′) as found in proteins the more acidic thiol can be expected to be the leaving group although steric constraints would also influence which sulfur was attacked.
In the cytosol, the concentration of LMW and protein disulfides is very low.547,577,578 Hence, the direct reaction between H2S and disulfides could be more relevant in compartments such as the endoplasmic reticulum and under oxidizing conditions.511
8.2. Reaction between H2S and Sulfenic Acids
Sulfenic acids (RSOH) are usually formed by reaction of a thiol with a hydroperoxide or with hypochlorous acid.579 The sulfur in sulfenic acid is a weak nucleophile and also a soft electrophile.580–582 Sulfenic acids are typically unstable and decay mainly by reaction with thiols forming disulfides.581,582 In addition sulfenic acids can react with H2S (eq 58).
| (58) |
The formation of persulfide in this reaction was confirmed using the sulfenic acid formed on Cys34 of human albumin.60,511 This sulfenic acid is located in a cleft with no neighboring thiols. The second-order rate constant for this reaction is 270 M−1 s−1 at pH 7.4 and 25 °C.60 The pH-independent rate constant with H2S is ~4-fold higher than with the corresponding thiol60 and suggests that steric constraints influence the effective access of the nucleophile.
Intracellular persulfide levels increase when cells are treated with H2O2, suggesting the relevance of sulfenic acid for priming the persulfide modification on proteins. Persulfide levels in H2O2-treated cells were decreased by inhibiting CBS and CSE.60 Since H2O2 can also promote disulfide formation in cells, the effect of diamide, which only supports disufide formation, was compared to the effect of H2O2. However, diamide had the opposite effect, leading to lower persulfide levels, which is consistent with the kinetic data that the reaction between disulfides and H2S is slow and therefore unlikely to be physiologically significant except in special cases.135 Under conditions of endoplasmic reticulum stress,558 which leads to enhanced ROS production,583 increased persulfidation was observed.
Sulfenic acid can be further oxidized to sulfinic acid (RSO2H) and sulfonic acid (RSO3H)580 which are typically irreversible modifications.584–587 As discussed previously in section 7.2.4, reaction of H2S with protein sulfenic acids to form persulfides could protect against overoxidation and irreversible protein damage. Namely, oxidation of persulfidated residues would result in the formation of RSSO2H and RSSO3H, which could be reduced back to thiols in the cell.
8.3. Reaction between H2S and S-Nitrosothiols as a Source of Persulfides
S-Nitrosothiols represent another post-translational modification of cysteine residues important for the regulation of protein function,432–437 as discussed in section 6.3. When reacting with thiols, S-nitrosothiols usually undergo trans-nitrosation, a reaction that is largely thermoneutral (eq 59).437
| (59) |
An alternative thiol-assisted decomposition of S-nitrosothiols has been proposed in which disulfide and HNO are formed as products.588,589
| (60) |
Consistent with this mechanism, an increase in protein S-glutathionylation is seen in cells treated with GSNO.590 However, since this reaction is thermodynamically unfavorable (~+30 kJ/mol), it would be relevant only if it were coupled to product removal.64,449
The expected product for the reaction of H2S with RSNO is HSNO.449,453 Formation of HNO and a protein persulfide (eq 61) is thermodynamically unfavored (+26 kJ/mol),64 although the protein microenvironment could facilitate this reaction.
| (61) |
The dichotomous reactivity of RSNO with nucleophiles can be explained by the unusual electronic structure of the —SNO group.591 The resonance representations of RSNO include the standard Lewis structure with a single S—N bond, a zwitterionic structure with an S═N double bond (RS+═N—O−) and an ionic structure I (RS−…NO+) (Figure 26A). Direct interaction with charged and polar residues in the protein microenvironment could affect the electronic structure of the —SNO group by increasing the electrophilicity of the S atom and therefore its susceptibility to nucleophilic attack (eq 61) or by weakening the S—N bond and increasing the electrophilicity of the N atom thereby promoting transnitrosation (eq 59).592 In addition, external electric fields created by the protein environment could influence the electronic structure of RSNOs so as to favor thiolation (or persulfidation in case of the reaction with HS−) over trans-nitrosation reaction (Figure 26B).592,593
Figure 26.
Factors that might favor protein persulfidation over trans-nitrosation in the reaction of an S-nitrosothiol (RSNO) with H2S. (A) Resonance structures of RSNO. Nucleophilic attack on the sulfur is favored in Structure D. (B) Interactions with the protein environment could stabilize the resonance structure D. Positively charged Arg or Lys residues could stabilize the NO moiety, while negatively charged Glu or Asp residues could stabilize the sulfur. In addition electric fields (EF) created by the protein environment could selectively stabilize a resonsance structure, e.g., D, promoting persulfidation and HNO release.
S-Nitrosation and persulfidation might differentially regulate protein function.594 In one study, the persulfide and S-nitrosothiol proteomes reportedly exhibited a 36% overlap.558 Further work is needed to delineate the effects of these modifications on proteins function.
8.4. Role of Polysulfides in Protein Persulfidation
Inorganic polysulfides (HSSnS−) and polysulfanes (HSSnSH) are sulfane sulfur compounds and prone to nucleophilic attack by thiolates. The high abundance of contaminating inorganic polysulfides in H2S solutions (particularly when NaHS is the source) and their chemical reactivity have led to their contributing to the numerous effects originally ascribed to H2S. For instance, at nanomolar concentrations, polysulfides activate TRPA1595 while H2S at micromolar concentrations fails to do so.267,595 Similarly, polysulfide contaminants in an NaHS-derived solution led to disulfide bond formation in the lipid phosphatase and tensin homologue (PTEN),596 by attack of a proximal cysteine on the initially polythiolated cysteine (Chart 32A). Inorganic polysulfides also contain negatively charged terminal sulfurs that could react with electrophiles. Polysulfides or H2S solutions containing traces of Fe3+ or Cu2+, which stimulate polysulfide formation, were shown to reduce disulfide bonds in human immunoglobulins.97 Hence, an additional caution with the use of H2S solutions containing polysulfide contaminants is that they can react with disulfide bonds forming RSH and RSnS− (Chart 32B).
Chart 32. Reactions of Inorganic Polysulfides with Proteinsa.
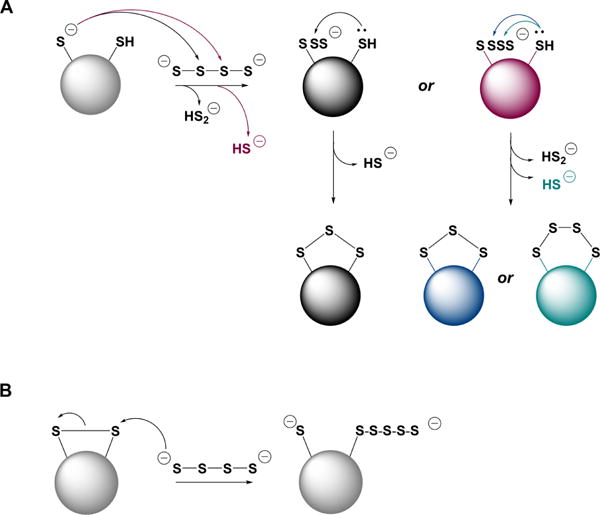
a(A) Inorganic polysulfides can react as electrophiles with a protein thiolate. The resulting polythiolated cysteine can have different numbers of S atoms and can in turn, be attacked by a proximal cysteine forming a family of products, e.g., intramolecular disulfides, trisulfides, etc. (B) Inorganic polysulfides can react as nucleophiles with an intramolecular protein disulfide forming thiol and polythiolated cysteine.
While much emphasis has recently been placed on the potential role of inorganic polysulfides as signaling molecules that are responsible for the effects of H2S,597,598 it is unclear and unlikely that they serve this role under physiological conditions. Polysulfides are charged and full protonation is almost impossible under physiological conditions, making diffusion across membranes very slow. Considering the very low steady state levels of H2S and its tightly regulated oxidation (see sections 3.5 and 5), it is difficult to envision that stochastic oxidation could result in significant amounts of inorganic polysulfides. Polysulfides are unstable and can be readily reduced so it is additionally unclear how they endure reducing intracellular environement. Finally, to serve in signaling, synthesis of polysulfide (which represents a family of catenated sulfur compounds of variable length) should be tightly regulated and their action on targets should be exerted with specificity. To date, none of these criteria are met by polysulfides. An enzymatic reaction in mammals that leads to regulated polysulfide synthesis is not known. Instead, polysulfide synthesis appears to be stochasticity controlled by the concentration of oxygen and metals on the one hand and protein thiols/disulfides on the other.
The reported production of polysulfides (H2S2 and H2S3) from 3-mercaptopyruvate by MST599 was problematic for the following reasons. Polysulfide formation from 3-mercaptopyruvate was observed when the reaction was run in the absence of a sulfur acceptor. Under these conditions, the KM for 3-mercaptopyruvate was reportedly 4.5 mM, although substrate inhibition led to complete loss of enzyme activity above 2 mM concentration.271 Furthermore, the KM value obtained in the H2S3 synthesis assay is at least 10-fold higher than the KM for 3-mercaptopyruvate (20—350 μM) in the presence of acceptors, which leads to H2S synthesis. Therefore, conditions supporting polysulfide synthesis by MST are unlikely to exist in the cell since MST exhibits a low KM (2.5 μM) for its physiological acceptor, thioredoxin.271
The chemical characteristics of inorganic polysulfides and polysulfanes make them unlikely to be signaling molecules as well.600,601 The equilibrium between elemental sulfur and aqueous polysulfide at 25 °C was studied either by adding acid to polysulfide solutions until the sulfur precipitated, or by dissolving elemental sulfur in aqueous polysulfide solution until an equilibrium was established. The ratio between the Sn and HS− species was strongly dependent on the alkalinity of the solution.
| (62) |
Since the polysulfide dianions of different chain-lengths are in an equilibrium (eq 63) that is rapidly established, it is not possible to reliably separate polysulfide species by ion chromatography.603
| (63) |
The pKas were studied using solutions of pure salts of S22− to S52− and a special streaming apparatus, which mixed polysulfides with HCl and allowed determination of the pH within 10−2 s.604 The short mixing time averted decomposition of the protonated polysulfide into sulfur and monosulfide. Only the pentasulfide did not equilibrate as multiple species upon acidification, which allowed precise determination of its pKa. The shorter polysulfides disproportionated within the mixing time of the experiment.604 S22− and S32− could not be detected in an aqueous solution of potassium trisulfide which equilibrated to 18% HS−, 62% S42−, and 20% S52−.604 In fact, other groups have confirmed that HS2− and S32− do not exist at detectable concentrations in neutral solutions.49,605–609 Tetra-sulfide dianion is the predominant species until a pH of 10–11. However, even tetrasulfides disproportionate to give the pentasulfide (eq 64),605,606,608,609 although this reaction is slow.
| (64) |
Polysulfanes can be prepared by rapid acidification of crude sodium sulfane (Na2Sn) to produce raw sulfane (H2Sn). H2S3 (as a side product) and H2S2 can be collected by fractional distillation of raw sulfane at room temperature and at −80 °C, respectively.69,70 Sulfanes are liquids that are miscible with carbon disulfide, benzene, tetrachloromethane, and dry diethyl ether. H2S2 and H2S3 are colorless/pale yellow, but the higher sulfanes are more yellow.69,70 Using 1H NMR, the existence of sulfanes with up to 35 sulfur atoms was demonstrated.610 Exposure of pure sulfanes to air/humidity caused immediate decomposition. In the cases of H2S2 and H2S3 the decomposition is explosive.69
| (65) |
The pKa values of H2S2 are pK1 5.0 and pK2 9.7. The pKa values of H2S3 are pK1 4.2 and pK2 7.5. In fact, ab initio MO calculations confirm that the acidity of sulfanes increases with the number of sulfur atoms in the molecule.611
Instability of aqueous polysulfide solutions (particularly S22− to S42−) is also due to their rapid autoxidation in air at temperatures between 23 and 40 °C forming thiosulfate and elemental sulfur (eq 66).612 No other sulfur containing species are detected in these reactions.
| (66) |
Polysulfides can also react with sulfite in neutral solution to give thiosulfate and HS− (eq 67).613
| (67) |
Studies in which inorganic polysulfides are trapped as organic polysulfanes (e.g., with monobromobimane) in order to establish their intracellular formation need to be viewed with caution. Organic polysulfanes (RSSnSR) are also unstable; even pure substances tend to decompose by equilibration with other chain lengths and by formation of elemental sulfur. These reactions are accelerated by light and heat, and by the presence of nucleophiles.614 Due to the intrinsic instability of inorganic and organic polysulfanes and polysulfides neither standards nor products used in this methodological approach are stable.
Based on all these chemical characteristics, it is very difficult to envision a biological setting that is conducive to the regulated production of polysullfides or their utilization in signaling in mammalian systems. However, polysulfides with the caveats of their instability noted above might have some pharmacological potential.
8.5. Persulfide Formation via Radical Reactions
Strong one-electron oxidants can react with H2S and RSH forming HS• and RS•, respectively. Rapid free radical recombination between HS• and RS• would lead to persulfide formation (eq 68), although this is highly likely to be an insignificant source of persulfides in cells due to the low concentration of the free radicals.
| (68) |
An alternative and more likely, radical pathway for RSSH formation is via the reaction of HS• with RS− or, conversely, RS• with HS−, to form the radical anion, RSSH•−. The latter can react with oxygen forming RSSH and O2•− (eqs 69–71).
| (69) |
| (70) |
| (71) |
The potential importance of free radical reactions in persulfide synthesis was demonstrated by increased protein persufidation in cell lysates treated with metal ions (Fe3+ or Cu2+) and H2S.511 Similarly, persulfidation of BSA was strongly induced by treatment with H2S and a water-soluble heme iron.511 The reactivity of H2S and RSH with metal ions under aerobic conditions should serve as a caution for handling cell lysates, which could contain higher free metal ion concentrations and in the presence of H2S could lead to an artifactual increases in persulfidation.
H2S can react with the oxidized form of several metalloproteins like cytochrome c97,505 (see section 5.1) leading to their reduction and formation of HS•. It is possible that, under conditions of increased H2S production or decreased oxidation (e.g., during hypoxia),615 ferric cytochrome c is reduced while H2S is oxidized to HS• leading to increased protein persulfidation in mitochondria. The role of metalloprotein-assisted HS• generation in protein persulfidation remains to be investigated.
The chemistry of polysulfides is closely related to that of the radical monoanions Sx•− (eqs 72 and 73) in that they are in equilibrium with each other.
| (72) |
| (73) |
In solution these radicals are formed by homolytic dissociation of the polysulfide anions, a process that is enhanced in solvents of lower polarity than water and/or by higher temperatures.600,616 In aqueous solutions at room temperature, the reactions given in eqs 72 and 73 are shifted to the left. However, if the solutions are very dilute, then radical anions could be more abundant. The radical anions would be reactive toward thiols leading to protein polythiolation and O2•− formation (eqs 74 and 75).
| (74) |
| (75) |
8.6. Reaction between Thiols and Activated Organic Disulfides and Polysulfides
Organosulfur compounds in garlic can react with thiols and serve as a source of persulfides and H2S. Allicin (diallyl thiosulfinate) is synthesized from alliin after release of alliinase when garlic is crushed. Allicin is rapidly metabolized in aqueous solution to diallyl sulfide, diallyl disulfide, diallyl trisulfide, and ajoene (Chart 33A).105,110,111,617
Chart 33. Active Principles of Garlic and the Mechanisms for LMW Persulfide Generation from Thema.
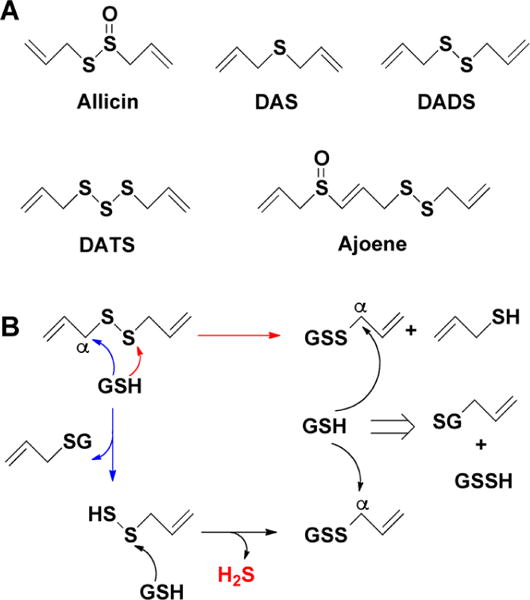
a(A) Allicin (diallyl thiosulfinate) is rapidly metabolized in aqueous solutions into diallyl sulfide (DAS), diallyl disulfide (DADS), diallyl trisulfide (DATS), and ajoene. (B) Glutathione-promoted decomposition of DADS and generation of allylpersulfide, glutathione persulfide (GSSH), and H2S.
GSH reacts with the active principles in garlic leading to H2S release.618 These reactions are facilitated by allyl substituents and by increasing numbers of sulfur atoms. A similar structure–activity correlation has been reported for the cancer-preventative effects of garlic-derived organic polysulfides.619 In addition to H2S, LMW persulfides are also formed in the reaction of GSH with organic disulfides and polysulfides resulting from nucleophilic substitution at the α-carbon, yielding S-allyl-glutathione and allyl persulfide (Chart 33B).618 The latter reacts with GSH releasing H2S and allyl-glutathione disulfide, which in turn is an additional target for nucleophilic substitution leading to GSSH formation. Diallyltrisulfide and higher order diallylpolysulfides react in a similar manner and have the additional possibility of undergoing a direct nucleophilic attack by GSH at the sulfane sulfur.618 In addition to GSH, other thiolates in cells can engage in similar chemical reactions with garlic-derived allyl sulfides.
8.7. Elimination Reactions of Disulfides
Disulfides degrade under alkaline conditions giving rise to a variety of products. Base-promoted elimination of disulfides leads to the formation of the corresponding persulfide and dehydroalanine derivative (Chart 34).576,620,621
Chart 34.
Base-Promoted Persulfide Generation from Cystine
Elimination reactions of disufide substrates (cysteine and homocystine) are catalyzed by CBS and CSE forming the corresponding persulfides165,226,622–624 At physiologically relevant substrate concentrations, the contributions of CBS and CSE to Cys-SSH generation is low and homocysteine persulfide synthesis by CSE is negligible (see sections 4.1.3 and 4.3.1).226 However, under pathological conditions that lead to cystine accumulation, Cys-SSH synthesis by CSE might become a contributing factor to persulfide synthesis.
8.8. Sulfur Transfer Reactions
Sulfurtransferases react with their substrates forming a persulfide intermediate from which the sulfane sulfur is transferred to an acceptor. In addition to MST, rhodanese and SQR that are directly involved in H2S synthesis or clearance (sections 4 and 5), the cysteine desulfurases catalyze sulfur transfer reactions needed in biosynthetic pathways e.g. synthesis of iron sulfur clusters625–636 and thionucleosides.629,633,634,636 While some sulfurtransferases (e.g cysteine desulfurase) utilize the PLP cofactor, others do not (e.g., MST). These enzyme promote the formation of a Cys-SSH in the active site and can, in principle, transfer the sulfane sulfur to a thiol on a target protein or to a LMW thiol. The role of these enzymes in catalyzing protein persulfidation remains to be determined.
8.9. Reaction between Thiols and Persulfides (Transpersulfidation)
As discussed in section 7.2, nucleophilic attack by thiolates on persulfides occurs predominantly on the inner sulfur with formation of disulfide and release of H2S (eq 52). This chemistry has been documented with LMW persulfides and with protein persulfide models.494,501,509 However, proteins such as the sulfurtransferases modulate the reactivity of persulfides such that the outer sulfur is transferred and H2S is not released (see section 5.2). Very high levels of LMW persulfides in cells, tissue and circulation (50–100 μM) have been reported165 leading to the suggestion that Cys-SSH and/or GSSH are major biologically relevant transpersulfidating reagents.527 However, high concentrations of reactive persulfides and especially in an oxidizing compartment like blood are unlikely. Cys-SSH and GSSH have been proposed to be major biologically relevant transpersulfidating reagents that can even be transported across the membrane.165,637 However, the low tissue levels of sulfane sulfur compounds32,574 and the fact that their reaction with thiolates, which are abundant in cells, favor H2S release, argue against this possibility.
The transfer of the terminal sulfur from a persulfide to a thiolate, constitutes a transpersulfidation reaction, and has been documented in several proteins. Sulfur-containing cofactors and modified thionucleosides, as well as iron–sulfur clusters obtain their sulfur atom via transpersulfidation reactions.636,638 It is likely that steric factors, the acidity of the parent thiol and the protein microenvironment determine the predominance of thiolate attack on the outer versus the inner sulfur of the persulfide.
The reaction mechanism of transpersulfidation by the LMW persulfides (eq 53) has been proposed to involve the tautomeric thiosulfoxide species (Chart 35) as the sulfur donor.33,34,546 The S═S bond in thiosulfoxides can be considered to be either a double639 or “semipolar”640 bond depending on the electronegativity of substituents. Nevertheless, computational studies reveal that, although the thiosulfoxide is only 5 kJ/mol less stable than the corresponding persulfide, the energy barrier for tautomerization is very high, i.e., >100 kJ/mol.641 Hence, LMW thiosulfoxides appear to be unlikely donors in uncatalyzed cellular transpersulfidation reactions.
Chart 35.
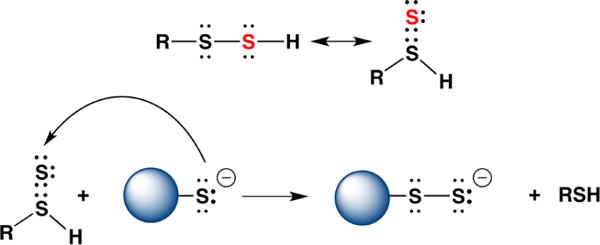
Tautomerization of a Persulfide to a Thiosulfoxide as a Postulated Mechanism for Transpersulfidation
Transpersulfidation can, however, occur in the active site of certain enzymes. A computational analysis of the sulfurtransfer reaction280 based on cystal structure of human MST271 suggests that the persulfide anion is the sulfur donor to the thiolate acceptor, in a reaction that is facilitated by the increase in electrophilicity of the outer sulfur through multiple hydrogen bonding interactions. Therefore, the active site geometry and electronics favor transfer of the terminal sulfur, i.e. transpersulfidation to either LMW or a protein thiol (Figure 7).
MST transfers the sulfane sulfur to sulfite forming thiosulfate, albeit very inefficiently.642 Treatement of red blood cells with the MST substrate, 3-mercaptopyruvate, reportedly inhibited many glycolytic enzymes including hexokinase, phosphofructokinase, pyruvate kinase, glucose-6-phosphate dehydrogenase and 6 phosphogluconate dehydrogenase.643 Transpersulfidation of the sulfane sulfur from MST to those enzymes was suggested but not established as the mechanism of inhibition.643 An increase of persulfidation was reported in cells treated with D-cysteine,135 a source of 3-mercaptopyruvate via D-amino acid oxidase.277 The cryo-EM structure of complex I isolated from Yarrowia lipolytica indicates the presence of an additional subunit, which was identified to be an accessory sulfurtransferase subunit. This rhodanese/MST-like subunit was capable of using 3-mercaptopyruvate as a substrate and, in the presence of thiols, released H2S. The role of this sulfurtransferase subunit on the structure and function of complex I remains to be elucidated.644
There are limited examples of rhodanese-catalyzed sulfur transfer to protein acceptors. Although a role for rhodanese in iron sulfur biogenesis had been previously proposed,645–649 it has since been shown to not be involved in this process. Rhodanese catalyzed sulfur transfer from thiosulfate to malate dehydrogenase as monitored by 35S radiolabel transfer and led to an almost 2-fold increase in activity. Hence, persulfidation could regulate energy metabolism via the citric acid cycle.650 In the presence of thiosulfate, bovine rhodanese restored the activity of partially inactivated NADH dehydrogenase, a subunit of complex I.651
Rhodanese has been identified as a candidate obesity-resistance gene with increased expression in adipocytes being correlated with leaness.327 Overexpression of rhodanese in adipocytes protected mice from diet-induced obesity and insulin-resistant diabetes and rhodanese-deficient mice showed aggravated development of diabetes.327 An earlier study had correlated low rhodanese expression with increased whole cell ROS and mitochondrial O2•− levels and higher mortality in hemodialysis patients.652 The link between these observations and changes in intracellular persulfide levels and whether rhodanese in fact catalyzes protein persulfidation, remains to be elucidated.
The rhodanese homology domain has been identified in ∼500 proteins in the three major evolutionary phyla. In the human genome, there are 47 examples of rhodanese-domain containing proteins.328 Cdc25 phosphatase, an activator of cell division kinases during the cell cycle, is an example of rhodanese domain-containing protein. The crystal structure of the catalytic domain of human Cdc25A reveals a small α/β domain with a rhodanese domain fold.653 It is not known, however, whether the signaling role of Cdc25 involves transpersulfidation. Other proteins that have a rhodanese domain and form an active site Cys-SSH are adenylyltransferase and the MOCS3 sulfurtransferase.654,655 The roles of these proteins in catalyzing transpersulfidation chemistry are, however, likely to be restricted to the specific pathways in which they are involved e.g. molybdopterin biosynthesis.
8.10. Depersulfidation
Signaling via protein persulfidation requires the existence of cellular mechanisms for removal of the persulfide modification and inactivation of the signal. Thioredoxin (Trx) is a 12 kDa disulfide oxidoreductase, which serves as a redox partner for a wide variety of client proteins.656–659 In humans, Trx1 is present in the cytosol and the nucleus while Trx2 is present in mitochondria. Trx contain two cysteines (Figure 27A) in an active site CXXC motif.656 The pKa values of the nucleophilic cysteine is between 6 and 7 and for the resolving cysteine is between 8 and 9.660–662 Reduction of a client protein disulfide starts with the attack of the nucleophilic cysteine to form an intermolecular mixed disulfide, followed by subsequent attack of the resolving cysteine on the mixed disulfide to form the fully oxidized Trx. The two-electron reduction potential of the disulfide/dithiol couple in thioredoxin is −284 mV at pH 7.0 and 25 °C.656,663 The recognition between Trx and its client proteins is postulated to be entropically driven.664 Oxidized Trx is reduced by the FAD-containing selenoprotein, thioredoxin reductase (TrxR1). Three isoforms of this protein exist in mammals: cytosolic TrxR1, mitochondrial TrxR2 and TrxR3 present only in testis. TrxR uses NADPH as a source of electrons (Figure 27B). Excellent reviews on the Trx/TrxR system have been published.656–659
Figure 27.
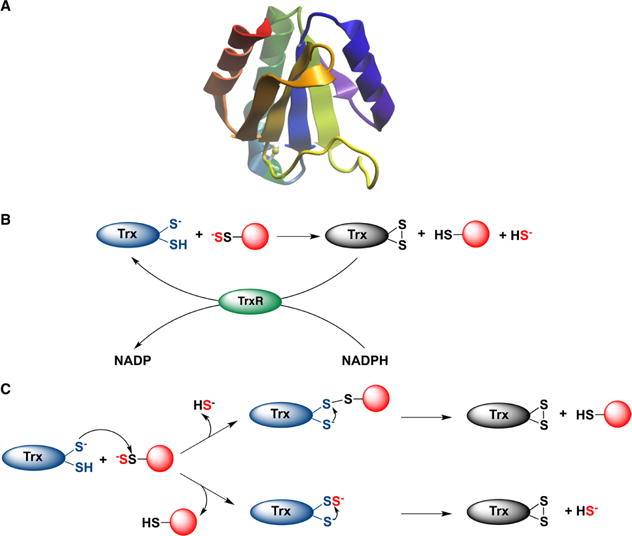
Depersulfidation by thioredoxin (Trx). (A) The structure of human thioredoxin (PDB: 5DQY). (B) Thioredoxin reduces protein persulfides and releases H2S. Oxidized Trx is reduced by thioredoxin reductase (TrxR) at the expense of NADPH. (C) Two possible mechanisms for protein depersulfidation. Top: The nucleophilic thiol attacks the inner sulfur of the protein persulfide forming a mixed protein-Trx disulfide and releasing H2S. In the next step the resolving cysteine reduces the mixed disulfide forming fully oxidized Trx. Bottom: Trx undergoes persulfidation, forming Trx persulfide, which is reduced by the resolving cysteine with concomitant release of H2S.
As discussed in Section 4.5, the Trx/TrxR is involved in the sulfur transfer reaction catalyzed by MST,270,271,273 and formation of thioredoxin persulfide was demonstrated with the Trichomonas vaginalis MST.274 Trx is ∼200-fold more efficient at reducing the Cys-SSH in PTP1B than DTT.561 Addition of Trx to cell lysate resulted in H2S generation, while the treatment of cells with auranofin, a TrxR inhibitor, increased total intracellular persulfidation levels.135 Trx reduced penicillamine-derived HSA persulfide and Cys-SSH.135 The first order rate constant for the reaction of Trx with Cys-SSH was estimated to be 4.5 × 103 M−1 s−1 at 23 °C and pH 7.4, which is almost 10-fold higher than with Cys-SS-Cys. A similar rate constant was observed with HSA-SSH (4.1 × 103 M−1 s−1).135 The Trx/TrxR/NADPH system exhibited Michaelis–Menten-like kinetic behavior. These results are consistent with an important role for the Trx/TrxR system in protein depersulfidation.135,514 The involvement of a related protein, TRP14 (thioredoxin-related protein of 14 kDa),665 was demonstrated by its silencing, which resulted in increased persulfide levels.514 TRP14 might be important as a depersulfidase particularly under conditions of oxidative stress when Trx is tied up with the peroxiredoxin system.514
Depersulfidation by Trx can occur via one of two mechanisms: (i) transfer of the outer sulfur from the persulfide to the nucleophilic cysteine of Trx leading to the transient formation of Trx-SSH which is subsequently resolved forming H2S and oxidized Trx and (ii) a nucleophilic attack on the inner sulfur of the persulfide with elimination of H2S and formation of a mixed Trx-client disulfide complex, which is resolved (Figure 27C).135 While the formation of mixed disulfides is part of the disulfide reductase activity of Trx, the mechanism of the depersulfidase activity remains to be established.
Increased Trx levels are associated with diseases such as rheumatoid arthritis, hepatitis C and HIV-1 infections.666–668 HIV-1 patients with high viral load have increased levels of circulating Trx.666,667 An inverse correlation was seen between total plasma sulfane sulfur levels and viral load in HIV-1 patients. Indirectly, this result is consistent with a role for the Trx system in depersulfidation in vivo.135
The glutaredoxin system (GSH/glutathione reductase (GR)/glutaredoxin (Grx)) could also be involved in catalyzing depersulfidation in vivo (Chart 36).514 GR activity has been reported in cytoplasm and in organelles (ER, lysosome, mitochondria and nucleus). GR regulates cellular redox status by maintaining low levels of GSSG and is important in protecting cells from oxidative stress.669–671
Chart 36. Glutaredoxin-Catalyzed Protein Depersulfidationa.
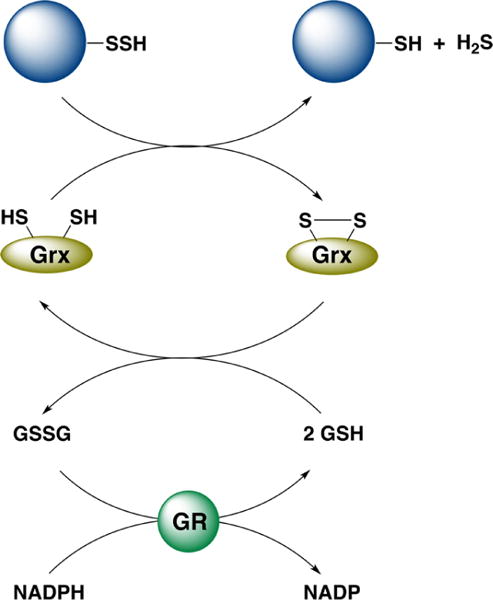
aGlutaredoxin (Grx) reduces a protein persulfide, oxidized Grx is reduced by glutathione (GSH), and GSSG is reduced by glutathione reductase (GR) at the expense of NADPH.
The Grx system efficiently reduced polysulfides and BSA-SSH in vitro. In murine hepatocytes with the double knockout (TrxR1/GR null), increased persulfidation was observed.514 GR null cells, however, showed no difference in persulfidation levels compared to controls.514 Since the in vitro assay requires addition of GSH, the observed polysulfide and protein persulfide reduction could have resulted from their direct reduction by GSH.
Beside Grx, the Trx fold is also found in several other classes of enzymes, such as Dsb (disulfide bond formation protein) proteins, glutathione S-transferase, and protein disulfide isomerase (PDI) families.656 Further systematic studies should unravel the potential role, if any, of these enzymes in protein depersulfidation.
9. PERSULFIDATION IN ACTION
As a growing list of persulfidated proteins is being identified, the stage is being set for making the connection between these targets and the molecular mechanisms of H2S action. In this section, we provide an overview of protein targets and downstream signaling pathways that are affected by H2S-induced persulfidation with the caution that, in many systems, the persulfidation target has not been rigorously established and correlated with functional effects.
9.1. Persulfidation of KATP Channels
Although CSE knockout mice exhibit hypertension,12 which is consistent with a role for H2S as an endogenous regulator of blood pressure,7,672 subsequent studies have revealed that this effect is mediated via cross-talk with NO•,266,267,451,673 an established vasodilator. Glibenclamide, a selective potassium ATP channel (KATP) blocker, partially inhibits the vasodilatory effects of H2S.672 Persulfidation of Cys43 on the Kir6.1 subunit of the KATP channel in smooth muscle cells prevents its association with ATP and promotes binding to phosphathidylinositol-4,5-bisphosphate,553 which leads to channel opening, to K+ influx, and, subsequently, to smooth muscle cell relaxation.
9.2. Persulfidation of Keap-1, p66Shc, and RAGE
A major mechanism for upregulating antioxidant enzymes involves activation of the antioxidant response element (ARE) by the oxidative-stress sensor protein Kelch-like ECH-associated protein 1 (Keap1) and the transcription factor nuclear factor (erythroid-derived 2)-like 2 (Nrf-2).674–676 Normally, Keap1 binds to the Neh2 domain of Nrf-2 and sequesters it in the cytoplasm, where it is targeted for proteosomal degradation. Electrophilic agents such as sulforaphane can modify Keap1, promoting Nrf-2 nuclear accumulation and ARE activation.674,676 A widely accepted model for the nuclear accumulation of Nrf-2 invokes modification of critical cysteines on Keap-1 resulting in a conformational change, which induces dissociation of the Keap1–Nrf-2 complex leading to nuclear translocation of Nrf-2 (Figure 28).674–676
Figure 28.
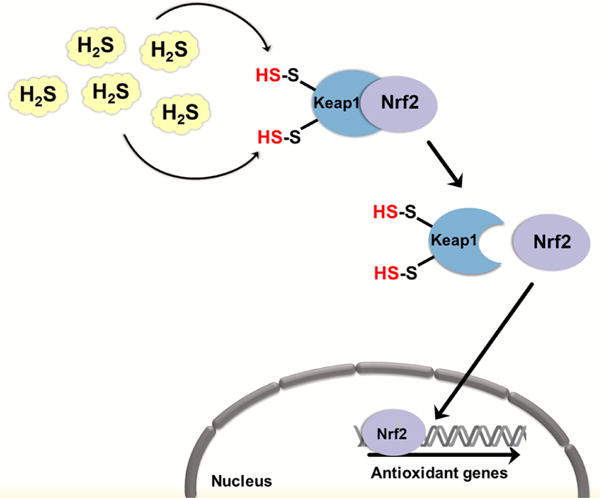
H2S may regulate cellular antioxidant defenses and prevent senescence by persulfidation of Keap1. In the cytosol, Keap1 represses Nrf-2 signaling by binding to it. Bound Nrf-2 is subjected to polyubiquitination and proteasomal degradation. Persulfidation of cysteine residues in Keap1 induces a conformational change, which results in Nrf-2 release. Nrf-2 translocates to the nucleus where it upregulates the expression of various antioxidant defense genes.
The cardioprotective effects of H2S, particularly in ischemia-reperfusion injury, depend on the nuclear translocation of Nrf-2 and activation of antioxidant defense enzymes.677,678 One group has reported that Keap-1 is persulfidated at Cys151 (identified using the modified biotin-switch assay) when cells are exposed to H2S,554 (Figure 28). In contrast, a second study has reported the formation of a disulfide bond between Cys226 and Cys613 in Keap1 in H2S-treated cells.679 Surprisingly, the same study reported that Cys226 and Cys613 were also persulfidated, which is not expected if persulfidation involves attack of the initially formed disulfide by H2S.679 Alternatively, persulfidation could occur by reaction of H2S with the sulfenic acid derivative of each cysteine. Activation of the Keap1-Nrf-2 signaling cascade resulted in the upregulation of enzymes involved in H2S metabolism, CBS, CSE, and SQR.679
An alternative mechanism proposed for regulating intracellular ROS production involves persulfidation of the p66Shc protein.680 The p66Shc protein is a member of the ShcA family with which it shares three functionally identical domains: the C-terminal Src homology 2 domain (SH2), the central collagen homology domain (CH1), and the N-terminal phosphotyrosine-binding domain (PTB).681 When cells are exposed to oxidative stress caused by exposure to UV light or to H2O2, p66Shc is activated by phosphorylation at Ser36. Activated p66Shc is then dephosphorylated and translocated to the mitochondrion, where it binds to cytochrome c and assists in the electron transport process.681 p66Shc−/− mice show a 30% increase in lifespan.681 Persulfidation of p66Shc at Cys59 inhibits its interaction with PKCβII and attenuates H2O2-induced p66Shc phosphorylation, a critical step in p66Shc-mediated mitochondrial ROS generation.680 H2S is known to protect against oxidative stress, which cannot readily be explained by a direct antioxidant role (see section 2). Inhibition of mitochondrial ROS production via persulfidation of p66Shc680 and upregulation of antioxidant defense enzymes via Keap1-Nrf-2 signaling554,679 provide a mechanistic basis for the protective effects of H2S.
Persulfidation of RAGE (receptor for AGE: advanced glycation end products) at Cys259 and Cys301 by H2S treatment or CBS overexpression attenuates cell death and senescence caused by both AGE and β-amyloid682 AGE are glycated proteins and lipids observed in diabetic patients.683 Dimeric RAGE is processed in the ER and delivered to the cell membrane. Persulfidated RAGE monomers are less stable, which disrupts their translocation from the ER to the plasma membrane and leads to increased protection against cell death and senescence.682
9.3. Persulfidation and ER Stress
ER stress induces a major transcriptional, translational, and metabolic reprogramming in cells and is associated with the development of many diseases, ranging from metabolic dysfunction to neurodegeneration.684 ATF4, a master transcriptional regulator, is induced during the ER stress response and upregulates CSE263 and the cysteine transporter, Slc7a11.558 Persuflidation of a number of protein targets is increased during ER stress and is correlated with reprogramming of energy metabolism toward increased glycolytic flux in pancreatic beta cells (Figure 29).558
Figure 29.
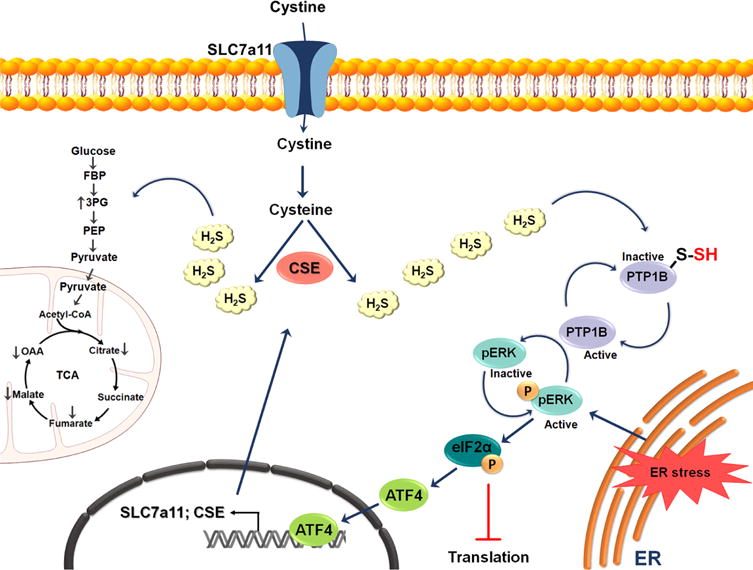
Possible role of H2S in endoplasmic reticulum (ER) stress. Under ER stress, the activity of the transcription factor ATF4 is increased, resulting in the upregulation of CSE and the cystine transporter Slc7a11. The subsequent increased production of H2S leads to the persulfidation of protein tyrosine phosphatase 1B (PTP1B) and consequently to an increase in pERK phosphorylation. pERK activation results in global inhibition of protein translation by activation of eukaryotic translation initiation factor 2α (eIF2α). eIF2α induces ATF4 nuclear translocation. The increased production of H2S during ER stress also results in persulfidation of glycolytic and tricarboxylic acid (TCA) cycle enzymes.
The ER-stress response also leads to persulfidation of the protein tyrosine phosphatase (PTP) family of enzymes.561 PTPs are cysteine hydrolases and are sensitive to oxidation.685 PTP-1B is a members of this class of enzymes that is located on the cytoplasmic face of the ER and plays an important role in ER stress signaling.686 Persulfidation of Cys215 in PTP-1B leads to the loss of enzymatic activity and, consequently, to increased phosphorylation and activation of PERK,561 which results in global inhibition of protein translation (Figure 29). ER stress conditions induce ROS production, which could promote protein sulfenylation and potentiate subsequent persulfidation.
9.4. H2S Effects on GAPDH
GAPDH is an important glycolytic enzyme and exhibits high reactivity toward H2O2, which oxidizes the nucleophilic Cys152 residue in the active site, inactivating the enzyme. The redox sensitivity of GAPDH is important for metablic adaptation to increased intracellular H2O2687 levels. S-Nitrosation of the catalytic Cys152 abolishes GAPDH activity and promotes its binding to the E3-ubiquitin-ligase, Siah1.688 The latter possesses a nuclear localization tag and leads to nuclear accumulation of GAPDH. Stabilization of Siah1 by GAPDH promotes degradation of nuclear proteins and leads to apoptosis.688 Persulfidation of GAPDH reportedly also promotes Siah1 binding although the modification site was not rigorously determined.556
Besides the active site Cys152 residue, human GAPDH has two other cysteines, Cys156 and Cys247. Due to its high abundance, GAPDH is a commonly identified target in proteomic searches including persulfide proteomic data sets.19,558 While some studies have reported activation of GAPDH in response to persulfidation,19,558 the connection between persulfidation of Cys152 and GAPDH activity has not been rigorously established. Cys152 functions as a nucleophile in the reaction cycle, and given the inhibitory effect of S-nitrosation of Cys152 on activity, it would appear a priori that persulfidation of GAPDH would also be inhibitory. Consistent with this prediction, persulfidation of purified GAPDH at Cys152 was shown to inhibit activity.689 A caveat of this study is that persulfidation induced by NaHS or polysulfides was observed at Cys156 and Cys247 in wild-type enzyme but not at Cys152 although modification at these other two sites did not affect enzyme activity. Cys152 was persulfidated only when Cys156 was mutated to serine, and while the mutation did not affect the activity of unmodified enzyme, activity was inhibited upon persulfidation at Cys152.689
9.5. Persulfidation of NFkB
Nuclear factor κB (NF-κB) is an antiapoptotic transcription factor. Under basal conditions, it is sequestered in the cytosol via interaction with the inhibitor, IκBα.690 During inflammation, cells produce tumor necrosis factor α (TNF-α), which can potentially lead to cell death.691 H2S is known to protect against inflammation, but the underlying mechanism is not known.692 The discovery that NF-κB is persulfdiated at Cys38 in the p65 subunit suggests a potential mechanism of H2S action.552 Persulfidation was suggested to promote binding of NF-κB to the coactivator ribosomal protein S3, increasing its binding to promoters of antiapoptotic genes, including CSE (Figure 30). An opposing mechanism was however suggested by the report that H2S suppresses oxidized low-density lipoprotein-induced macrophage inflammation by inhibiting NF-κB.693 Persulfidation of Cys38 was suggested to lead to cytoplasmic retention of NF-κB, inhibiting its DNA binding activity.
Figure 30.
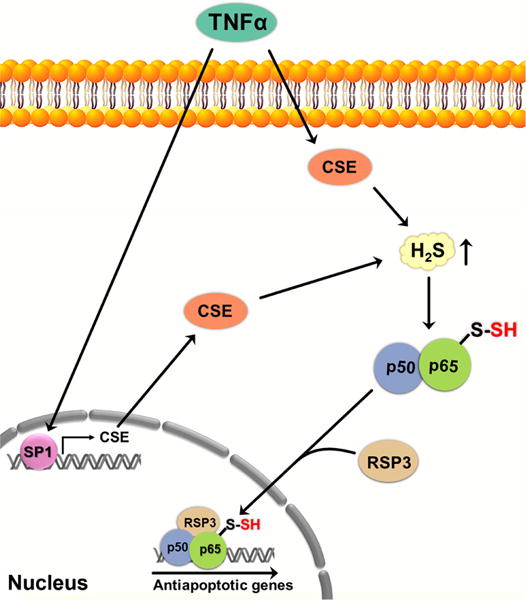
Persulfidation of NF-κB may regulate apoptosis. The proinflammatory cytokine TNFα, involved in the control of inflammatory reactions, stimulates CSE transcription by activating the SP1 transcription factor, resulting in increased H2S levels. H2S induces persulfidation of Cys38 in the p65 subunit of NF-κB, enhancing the binding of NF-κB subunits to the coactivator RPS3. The activator complex then migrates to the nucleus where it upregulates the expression of several antiapoptotic genes. TNFα, tumor necrosis factor α; CSE, cystathionine γ lyase; p50 and p65 subunits of NF-κB; RPS3, ribosomal protein S3; SP1, specificity protein-1.
9.6. MEK1/PARP-1 Activation and DNA Damage Repair
DNA damage stimulates a complex and highly concerted DNA damage repair response, which includes binding of poly(ADP- ribose)ation polymerases (PARPs) to DNA strand breaks and catalysis of poly(ADP-ribose)ation. Poly(ADP-ribose)ation attracts other DNA damage repair proteins.694 The MEK/ERK signaling cascade plays an important role in activating PARPs.695 Persulfidation of Cys341 in MEK1 reportedly facilitates the translocation of phosphorylated ERK1/2 into the nucleus, where it activates PARP-1 and increases the DNA damage repair yield.557
9.7. Persulfidation of Parkin
Mutations in parkin, an E3 ubiquitin ligase, are associated with the etiology of Parkinson’s disease, which is caused by the death of dopamine-generating cells in the substantia nigra.696,697 Parkin contains reactive cysteine residues that are susceptible to oxidative modifications. For example, S-nitrosation of parkin inhibits its activity.698 Parkin can be persulfidated at Cys59, Cys95, and Cys182.594 The activity of parkin is reportedly increased upon persulfidation and is correlated with the rescue of neurons from cell death by removal of damaged proteins (Figure 31). A decrease in parkin persulfidation has been reported in brain from Parkinson’s disease patients, while S-nitrosation is increased in the same samples.594 If substantiated, the activity of parkin would appear to be differentially regulated by modifications at the same cysteine residues and H2S donors could have therapeutic potential in the early treatment of Parkinson’s disease.
Figure 31.
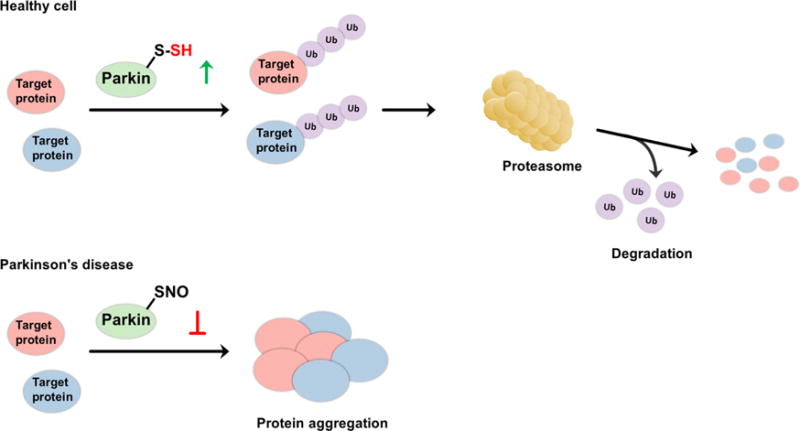
Possible regulatory role of H2S on the catalytic activity of parkin. (A) In healthy subjects, parkin, a E3 ubiquitin ligase, is persulfidated, which increases its enzymatic activity. This leads to ubiquitination of diverse substrates and their subsequent proteasomal degradation. (B) In patients with Parkinson’s disease, parkin is S-nitrosated. The decreased catalytic activity results in protein aggregation, accumulation of toxic proteins, and cell death.
Parkin is also a regulator of mitophagy, which leads to the removal of damaged mitochondria, particularly during ischemia-reperfusion injury.699 The pharmacological potential of H2S in preventing ischemia-reperfusion injury,677,678 might be mediated in part by the persulfidation of parkin.
9.8. Persulfidation of the TRP Channels
CBS-deficient patients exhibit a variety of phenotypes, including osteoporosis,700 which is characterized by a low bone density and increased risk of fracture. Bone marrow mesenchymal stem cells are nonhematopoietic multipotent stem cells responsible for bone formation and balancing osteoclast-mediated bone resorption to maintain bone mineral density. H2S donors reportedly protect MC3T3-E1 osteoblasts against H2O2-induced oxidative damage, although the mechanism of this effect is not known.701 CBS deficiency reportedly resulted in aberrant intracellular Ca2+ influx due to reduced persulfidation of multiple TRP channels.555 Decreased Ca2+ influx downregulates PKC/Erk-mediated Wnt/β-catenin signaling, which is important for controlling osteogenic differentiation of bone marrow mesenchymal stem cells that are postulated to produce H2S to regulate their self-renewal and osteogenic differentiation.555
9.9. Persulfidation Targets Revealed by Proteomic Approaches
Early studies on xanthine oxidase and aldehyde oxidase, which are molybdopterin containing enzymes, reported that they are regulated by persulfidation. However, the location of the persulfide was not established, and based on later structural and spectroscopic studies, the labile sulfur was found to be a sulfide ligand to molybdenum rather than persulfide.702 Early studies on Cu,ZnSOD reported the presence of an absorbance peak at 325 nm703 that was assigned to persulfidation at Cys111.704 Persulfidation blocked copper-induced protein aggregation but did not affect SOD activity.705 In addition to being modified by persulfidation at Cys111, two SOD monomers can be covalently linked via a polysulfane bridge (up to 5 sulfur atoms) between their Cys111 residues.706
Persulfide proteome analysis in response to increased endogenous H2S levels due to ER stress558 or exogenous treatment with GYY4137 and polysulfides559 has identified many persulfidated proteins. Under ER stress conditions, a total of 827 proteins were identified558 of which 178 overlapped with the much smaller 208 persulfidated protein data set reported in the GYY4137 treatment study.559 Enrichment of persulfidated proteins involved in translation, glycolysis, and the TCA cycle was reported in both studies in addition to the heat shock proteins Hsp70 and Hsp90 proteins and proteins involved in actin remodeling (actin, actinin, cofilin, and actin-related proteins),558,559 Actin and Hsp70 and Hsp90 were also identified previously in studies that yielded very limited persulfide target identification.19,165,511 Significant overlap was observed between persulfidation, sulfenylation, S-nitrosation and glutathionylation targets.558,559 Although no consensus sequence motif could be identified around persulfidated cysteines, there was some enrichment of the location of target cysteines at the N-termini of alpha helices.558
10. PHYSIOLOGICAL AND PHARMACOLOGICAL EFFECTS OF H2S
The past two decades have witnessed an increasing interest in understanding the physiological and pharmacological effects of H2S and its donors. However, with the recent development of analytical tools for H2S detection (see section 3.4), it has become clear that the actual endogenous values of H2S are quite low (see section 3.5), while the vast majority of physiological experiments were performed with supra-physiological amounts of H2S. Therefore, in this section, we provide an overview of processes relevant to human biology that are regulated by endogenous H2S or are responsive to the pharmacological treatment of H2S or its donors. Microbial707–713 and plant714–722 sulfur metabolism and the physiological roles of H2S in these systems are not covered.
10.1. H2S and the Nervous System
H2S affects hippocampal long-term potentiation by acting on N-methyl-D-aspartate (NMDA)-type glutamate receptors albeit only when applied together with weak tetanic stimulation at active but not quiescent synapses.6 Under these conditions, H2S facilitated NMDA receptor-mediated currents by activating adenylyl cyclase and the downstream cyclic adenosine mono-phosphate (cAMP)/protein kinase A (PKA) cascades.723 Although persulfidation of the NMDA receptor has been implicated, it has not been demonstrated.
Several ion channels have been identified as potential targets of H2S.724–736 H2S increases intracellular Ca2+ levels and subsequent Ca2+ waves in primary astrocyte cultures and hippocampal slices from rats.724 Similar results were obtained with microglial cells and with a neuroblastoma cell line.725,726 Furthermore, H2S affected intracellular acidification in a concentration-dependent manner in primary cultured microglia and astrocytes by regulating the activities of the Cl−/HCO3− exchanger and Na+/H+ exchanger.727
H2S is postulated to have both pro- and antinociceptive effects in the peripheral nervous system.728–736 Colonic luminal administration of NaHS caused nociceptive behavior manifested as abdominal allodynia/hyperalgesia in mice, which was abolished by a T-type channel blocker.728 NaHS-induced nociception also caused reversible T-type Ca2+ channel-dependent hyperalgesia in the rat spinal cord and peripheral tisssues.730,731 These results suggest that sensitization/activation of T-type Ca2+ channels might be involved728,731 although TRP channels have also been suggested to mediate pronociceptive effects of H2S in rodent models.732–734 In contrast, subcutaneous injection of millimolar H2S solutions did not cause pain in humans.267 The antinociceptive effects of H2S have been linked to activation of KATP channels.129,735,737 However, most of these effects were caused at high, rather than pharmacological, doses of H2S.
As a pharmacological agent, H2S can improve disease outcomes in different pathological settings (Figure 32). For example, neuronal cell death caused by peroxynitrite was significantly attenuated by H2S treatment.738 Neuronal injury induced by H2O2 in primary cultured rat astrocytes impaired glutamate uptake, whereas treatment with H2S exerted a neuroprotective effect by increasing glutamate uptake.739 Pharmacological H2S treatment has also shown some promise for treating Parkinson’s disease as discussed in section 9.7.594
Figure 32.
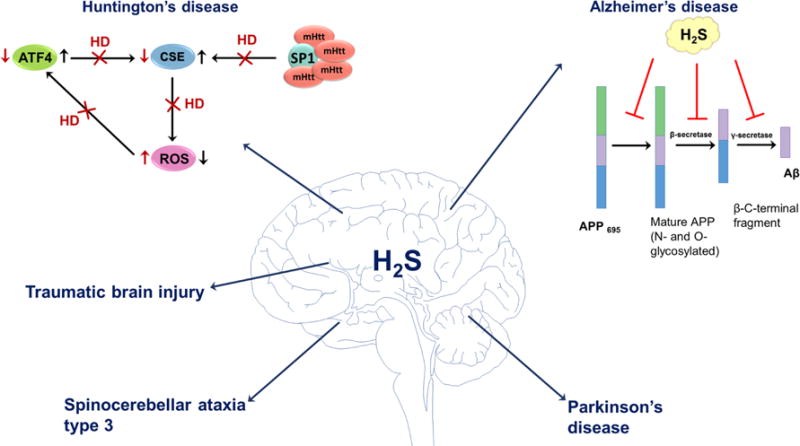
Some possible physiological roles of H2S in neurodegenerative disorders. H2S has been shown to be involved in pathogenesis of Huntington’s, Alzheimer’s, and Parkinson’s diseases, spinocerebellar ataxia, and traumatic brain injury. In healthy subjects, the expression of CSE is regulated by SP1 and ATF4 transcription factors. In Huntington’s disease, abnormal mutated huntingtin (mHtt) protein binds to SP1 and inhibits its activity. Reduced CSE expression results in oxidative stress that subsequently affects ATF4 expression. H2S also inhibits the production of amyloid beta (Aβ) at different catalytic steps of Aβ. The mature isoform of APP is cleaved by β- and γ-secretases forming Aβ. H2S interferes with APP maturation and inhibits the activity of β- and γ-secretases leading to the decreased production of Aβ. In Parkinson’s disease, H2S induces persulfidation of parkin and increases E3 ubiquitin ligase activity. H2S has beneficial effects in spinocerebellar ataxia type 3, where it regulates protein persulfidation and improves SCA3-associated tissue degeneration. In traumatic brain injury H2S exerts antiapoptotic effects and down-regulates the expression of autophagy-related proteins, reduces brain edema and improves the recovery of motor and cognitive dysfunction.
H2S levels are reported to be substantially reduced in brain of Alzheimer’s disease patients as compared to healthy individuals.740,741 In a rat model of Alzheimer’s disease, pretreatment with NaHS improved learning and memory deficits.742 Treatment of PC12 cell line with H2S inhibited expression of BACE-1 (beta-site amyloid precursor protein cleaving enzyme-1) mRNA and protein, a major β-secretase involved in amyloid beta (Aβ) production.743 The PI3K/Akt signaling pathway is reportedly involved in the H2S-induced decrease in BACE-1 expression and Aβ release. H2S treatment of SH-SY5Y cells suppressed Aβ formation probably by inhibition of amyloid precursor protein glycosylation and γ-secretase activities.744 These studies suggest that the H2S might exerts its effects on different steps involved in Aβ generation (Figure 32).743,744
H2S production is reported to be markedly decreased in Huntington’s disease.745,746 Mutant huntingtin protein inhibits Sp1, a transcriptional activator of CSE, leading to decreased protein expression and consequently, H2S production (Figure 32).746 H2S showed beneficial effects for spinocerebellar ataxia type 3 (SCA3), a neurodegenerative disease caused by polyQ repeats in ataxin-3, which leads to protein aggregation and subsequent neuronal dysfunction and death.747 In a Drosophila model of SCA3, CSE overexpression or treatment with thiosulfate reduced levels of oxidized proteins, inhibited the immune response and prevented SCA3-associated tissue degeneration. These beneficial effects were correlated with an increase in persulfidation levels.747
10.2. H2S and the Cardiovascular System
H2S exerts multiple effects in the cardiovascular system including attenuating myocardial ischemia reperfusion injury, promoting angiogenesis, relaxing smooth muscle cells, and regulating blood pressure.748–752 CSE is believed to be the primary H2S producing enzyme in the cardiovascular system. It is expressed in vascular endothelial cells, smooth muscle cells, and cardiomyocytes.12,672,753
Relaxation of rat aortic tissue in vitro was one of the first described effects of H2S in the cardiovascular system and occurred in synergy with NO•.7 Intravenous application of H2S decreased blood pressure in rats, which was suppressed by glibenclamide, a KATP channel blocker.672,754–756 Exposure of isolated vascular smooth muscle cells to H2S increased KATP currents. A vasodilatory role for endogenously synthesized H2S appeared to be supported by the observation that CSE knockout mice develop age-related hypertension.12 However, a contradictory result was reported by a second group, which found no changes in blood pressure in CSE knockout mice.757 H2S has been described as an endothelium-derived hyperpolarizing factor (Figure 33),756 acting primarily via activation of the KATP channel, which can be persulfidated at Cys43 in the pore-forming Kir 6.1 subunit553 (see section 9.1). However, the mechanism by which Cys43 is persulfidated and whether H2S acts alone as an endothelium-derived hyperpolarizing factor are unclear.
Figure 33.
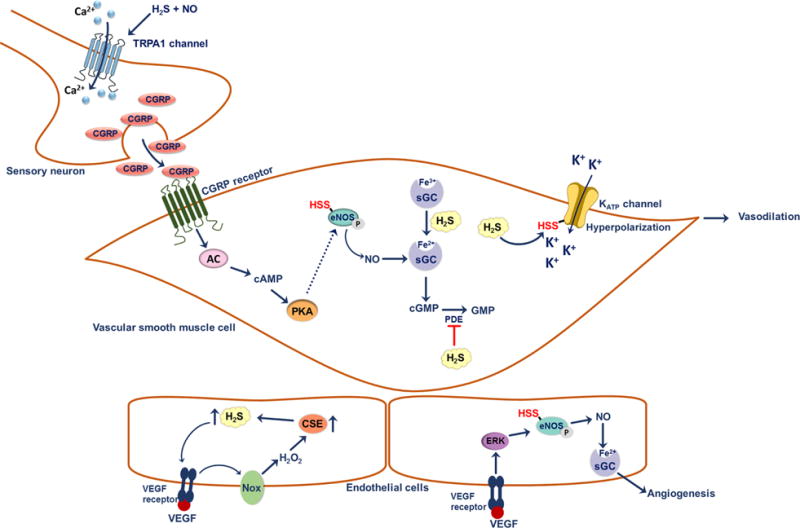
Possible signaling roles of H2S in the vascular system. At a sensory nerve ending, H2S interacts with NO• to give HNO. HNO activates TRPA1 channels; this results in Ca2+ influx and subsequent release of calcitonin gene-related peptide (CGRP). Binding of CGRP to its receptor on vascular smooth muscle cells activates the adenylate cyclase and the cyclic adenosine monophosphate (cAMP)-controlled downstream signaling pathways. As a result of elevated cAMP, protein kinase A (PKA) is activated and could potentially increase the activity of eNOS. Persulfidation of Cys443 on eNOS increases the activity of the enzyme as well as its ability to be phosphorylated, which results in increased production of NO• and activation of soluble guanylate cyclase (sGC). H2S potentiates the binding of NO• to sGC by reducing ferric heme to the ferrous state. Degradation of cGMP by phosphodiesterase (PDE) is prevented by H2S. These effects result in vasodilation of smooth muscle cells. Persulfidation of Cys43 on KATP channel enhances its activity, resulting in the influx of K+ and hyperpolarization of vascular smooth muscle cells. H2S also plays a role in angiogenesis. Binding of VEGF to its receptor on endothelial cells induces the production of H2O2 by activating NADPH oxidase (Nox). H2O2 supposedly increases CSE expression leading in turn, to increased H2S production. H2S stimulates activation of the Akt signaling cascade, which results in the phosphorylation of eNOS, increasing its activity. NO• acts as a pro-angiogenic factor.
There is growing evidence that the vasodilatory effects of H2S are intricately linked to NO• signaling pathways.7,266–268,465,758 For instance, the KATP channel-based vasodilatory effect of H2S is attenuated in the absence of NO•,672,754 and inhibition of eNOS abrogated H2S-induced vasorelaxation.7,266,267 Furthermore, HNO, a product of the reaction between H2S and NO•, activates TRPA1.267 Stimulation of the calcitonin gene-related receptor on smooth muscle cells activates adenylate cyclase, which then generates cAMP, a powerful secondary messenger responsible for vasodilation (Figure 33).267
Regulation of cGMP levels appears to be an important mechanism by which H2S potentiates the effects of NO• particularly in the context of angiogenesis.266,410,758,759 H2S inhibits the phosphodiesterase410,758,759 slowing cGMP degradation and increasing its half-life. Furthermore, binding of H2S to soluble guanylate cyclase leads to reduction of the heme iron to the ferrous state, which binds NO•377 (Figure 33). H2S also regulates eNOS activity and expression. Phosphorylation of eNOS increases its enzymatic activity and is enhanced in cells and blood vessels treated with H2S apparently via persulfidation at Cys443266,268,454 (Figure 33). Persulfidation increases eNOS activity and its ability to be phosphorylated, while also preventing the inhibitory S-nitrosation modification at the same cysteine residue.454 Surprisingly, H2S inhibits neuronal NOS and inducible NOS by directly binding to the iron heme center but does not inhibit purified eNOS.
Circulating H2S levels in patients with chronic heart disease or heart failure are significantly reduced compared to age-matched controls.760 The pharmacological potential of H2S has been demonstrated in a myocardial ischemia/reperfusion injury model677,678,750 where H2S administration at the time of reperfusion reduced infarct size by 72%.750 The protective effect of H2S was linked to preservation of mitochondrial function, reduction of cardiomyocyte apoptosis, anti-inflammatory responses, and antioxidant effects.750 Heart Nrf-2 and phosphorylated Akt levels were significantly higher in H2S treated mice suggesting that antioxidant gene expression was increased.677 Upregulation of the antiapoptotic Blc-2 protein and down-regulation of the pro-apoptotic factors Bax and caspase 2 were seen after H2S treatment.761 These effects could be regulated by persulfidation of Keap1 as described in section 9.2.
Binding of vascular endothelial growth factor (VEGF) to its receptor increases CSE expression via intermediate production of H2O2 as a signaling molecule.762 H2S production stimulates the Akt pathway, resulting in eNOS phosphorylation and higher NO• levels. The VEGF receptor might be a direct target of H2S, which reportedly reduces the Cys1045−Cys1024 disulfide bond and disrupts the active conformation of the receptor.763
Antiatherosclerotic properties of H2S have been reported.764,765 CSE knockout mice fed an atherogenic diet developed atherosclerotic lesions and exhibited a different plasma lipid profile than wild-type mice.765 Expression of the adhesion molecule ICAM-1, which is important for the development of atherosclerotic lesions, was significantly elevated in aorta of CSE knockout mice on an atherogenic diet.764 Additionally, NFκB expression was elevated in smooth muscle cells isolated from CSE knockout mice.764
10.3. H2S and Inflammation
H2S reportedly elicits proinflammatory and anti-inflammatory effects in various models of inflammation.766 H2S had a proinflammatory effect on acute pancreatitis and associated lung injury and treatment with the CSE inhibitor, D,L-propargylglycine, significantly decreased pancreatic and lung injury.767 The proinflammatory effect of H2S was diminished in CSE knockout mice in which acute pancreatitis and associated lung injury was induced.768 Furthermore, in an ischemia reperfusion injury model, a reduced inflammatory response was observed in kidneys of CSE knockout mice.769 A proinflammatory response to H2S has also been reported in several models of sepsis,770–772 and H2S levels are increased in patients with septic shock.692
An anti-inflammatory effect of H2S has been reported in subjects with intestinal ischemic damage and ethanol-induced gastritis.773–775 CSE expression is decreased in the gastric mucosa by nonsteroidal anti-inflammatory drugs776–778 and NaHS treatment decreased expression of TNF-α, intercellular adhesion molecule 1 (ICAM-1), and lymphocyte-associated antigen-1.776,777 H2S synthesis is markedly increased in colon ulcers, and it promotes the rapid restoration of the epithelial barrier integrity and the repair of the damaged tissue.779 The slow releasing H2S donor GYY4137 also exerts an anti-inflammatory effect121,780 by inhibiting NFkB activity. Persulfidation of the p65 subunit of NFkB is proposed to increase its interaction with the ribosomal protein S3 and to upregulate several antiapoptotic genes (see section 9.5).552
10.4. H2S and the Respiratory System
H2S treatment reportedly attenuates bleomycin-induced pulmonary fibrosis in rats.781 H2S treatment suppressed the migration, proliferation, and myofibroblast trans-differentiation of a human lung fibroblast cell line. The inhibitory effects of H2S were correlated with a decrease in ERK phosphorylation.782,783 Transforming growth factor β1 (TGF-β1) is a master regulator of fibrosis, and its inhibition by H2S resulted in decreased vimentin expression and increased E-cadherin levels.784 A bronchodilatory effect of H2S was ascribed to inhibition of Ca2+ release from the ER.785 H2S treatment induced relaxation of mouse tracheal smooth muscle cells by activating the calcium-activated potassium channel.786 Furthermore, H2S reportedly plays a role in the pathology and treatment of chronic obstructive pulmonary disease.787
10.5. H2S and the Renal System
H2S appears to play an important role in the onset and progression of renal diseases. Plasma H2S levels are lower in patients with diabetic versus nondiabetic nephropathy undergoing chronic hemodialysis.788 CBS and CSE expression is downregulated in experimental models of diabetes.789,790
An intrarenal infusion of NaHS increased blood flow, glomerular filtration rate, and urinary sodium and potassium excretion in rats.791,792 H2S inhibited the Na+/K+/2Cl− cotransporter and Na+/K+/ATPase activity in proximal kidney tubule epithelial cells792 and decreased cAMP levels in different renal cell types.793,794 H2S also decreased renin production in rat kidney.794 CSE overexpression increased endogenous H2S production and suppressed isoproterenol-induced renin release.794 However, the underlying mechanism by which H2S regulates renin release is unclear.
Conflicting results on the effects of H2S on kidney ischemia/reperfusion injury have been reported.769,795 Increased damage and mortality after renal ischemia/reperfusion was reported in CSE knockout mice, and H2S treatment protected mice from ischemia-induced renal injury and decreased mortality.795 A second study using a different strain of CSE knockout mice failed to observe a significant difference between wild-type and knockout animals exposed to kidney ischemia/reperfusion.769
10.6. H2S and the Liver
All H2S-producing enzymes are expressed in the liver, which plays an important role in glucose and lipid homeostasis, xenobiotic metabolism, and antioxidant defense.796–799 H2S is suggested to be an important modulator of hepatic micro-circulation.800 H2S donors such as diallyl trisulfide attenuate ethanol-induced liver injury in mice and increase the activity of mitochondrial antioxidant enzymes.801 The hepatoprotective effects of H2S donors were correlated with Nrf2 translocation and increased expression of antioxidant genes, which is regulated by Keap1 persulfidation (see section 9.2).554,679
10.7. H2S and the Gut
Some gut microbes use sulfate as a terminal electron acceptor for respiration and produce H2S using the dissimilatory sulfite reductase enzyme complex.710,712 Desulfovibrio is the predominant sulfate reducing bacterium in the human intestine, while Desulfobacter, Desulfomonas, Desulfobulbus, and Desulfotomaculum are found at lower levels.802 Very high H2S (up to 1000 ppm) has been reported in the rat cecum,803 and 0.2−30 ppm of H2S has been reported in human flatus.804 It is estimated that approximately half of the fecal H2S is produced by gut microbes with the remainder being derived from host metabolism.805 An imbalance in the number or composition of gut microbes is associated with various diseases.712 Sulfate reducing bacteria are resistant to a broad spectrum of antibiotics, and repeated use of these drugs might favor a bloom of these bacteria.806 An increase in the number of sulfate reducers has been observed in patients with ulcerative colitis, inflammatory bowel disease and Crohn’s disease,807 periodontitis,808 pouchitis,804 and irritable bowel syndrome.809 Probiotic and prebiotic treatments reduce the numbers of Desulfovibrio bacteria810,811 and the levels of proinflammatory cytokines IFN-γ, TNF-α, and IL-1β.811
10.8. H2S and the Reproductive System
CBS and CSE are expressed in the human corpus cavernosum, the muscular trabeculae, and smooth muscle components of the penile artery.812,813 The administration of CSE inhibitors into corpus cavernosum impaired the normal intracavernosal pressure response to cavernous nerve electrostimulation suggesting a possible role for H2S in penile tissue smooth muscle relaxation.814,815 NaHS treatment induced relaxation of rabbit815 and human812 corpus cavernosum strips in a concentration-dependent manner. An H2S-donating derivative of sildenafil has been developed for potential use in treating erectile dysfunction.816
H2S also exerts effects on male and female fertility.817–823 Expression of H2S-producing enzymes and H2S synthesis have been reported in uterus, vagina, and placenta.818–821 NaHS was shown to reversibly attenuate the contractile response of isolated rat uterus and delay parturition824–827 and mediate spontaneous contractions of the human oviduct.823
10.9. H2S and Oxygen Sensing and Hibernation
Exposure to low concentrations of H2S (80 ppm) induced a suspended animation-like state in mice, by decreasing metabolic rate and core body temperature.13 The H2S-induced depression of the metabolic rate observed in mice could be beneficial in patients with major trauma or cardiac arrest.828
Hypoxia increases H2S production in carotid bodies of rat and mice.829 In CSE knockout mice and wild-type rats treated with a CSE inhibitor, the hypoxic sensitivity of carotid bodies is markedly impaired. CO reportedly abolished CSE activity and reduced H2S generation in rat carotid bodies via protein kinase G-dependent phosphorylation of CSE.829,830 In hypoxia, CO levels and CSE phosphorylation decrease, leading to increased H2S production (Figure 34).260
Figure 34.
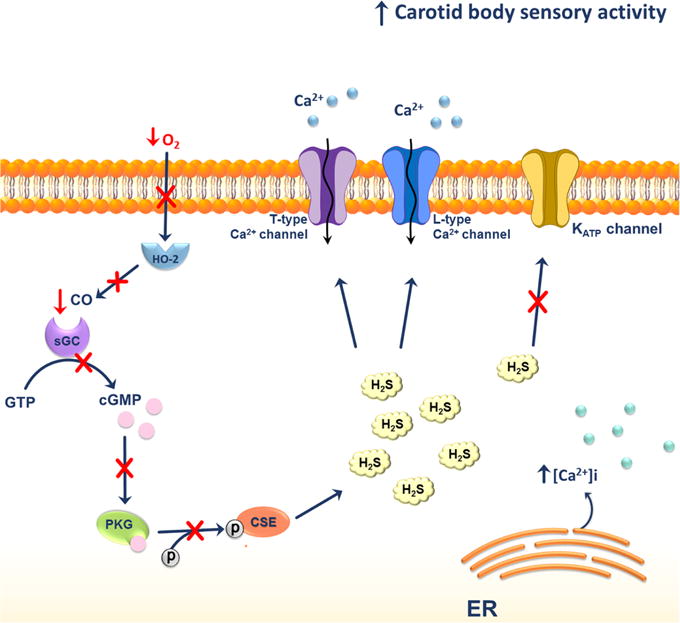
Possible H2S effects on glomus cells of carotid bodies under hypoxic conditions. Under hypoxic conditions the levels of H2S in glomus cells of carotid bodies are elevated. This could be a result of decreased phosphorylation of CSE due to the lack of CO produced by heme oxygenase-2 (HO-2). The lack of CO results in the inhibition of cyclic guanosine monophosphate (cGMP)-stimulated activation of phosphokinase G. The overall increase in H2S levels activates the L-type Ca2+ and T-type voltage-gated Ca2+ channels and mobilizes Ca2+ from the endoplasmic reticulum (ER).
The stimulatory effect of H2S on carotid body sensory activity is completely abolished by cadmium chloride, a nonselective inhibitor of voltage-activated Ca2+ channels.829,831 H2S treatment and hypoxia induced an increase in intracellular Ca2+ concentrations in rat glomus cells. Inhibition of H2S production prevented carotid body activation and hypertension in rodents exposed to intermittent hypoxia, which is a model for obstructive and central sleep apnea.
Treatment of Caenorhabditis elegans with H2S increased thermotolerance and longevity at higher temperatures.832 Several proteins were associated with these effects including hypoxia-inducible factor-1 and SKN-1, a homologue of mammalian Nrf-2. Furthermore, knockout of the MST ortholog reduced lifespan in C. elegans,833 and the effect was rescued by GYY4137. H2S and ROS are postulated to play important roles in extending lifespan in C. elegans.156 Decreased expression of CBS significantly reduced the lifespan of germline-deficient C. elegans mutants compared to the wild type strain.156 The role of protein persulfidation in lifespan extension has not been examined.
10.10. H2S and Cancer
The roles of H2S in cancer development and progression are still controversial. Some of the biological effects of H2S that might be relevant to cancer biology include stimulation of angiogenesis, regulation of intracellular signaling and cell death, and cellular bioenergetics.834–836 Many of the effects exhibit a biphasic dose−response curve; low concentrations of H2S are cytoprotective and high concentrations are cytotoxic.835–839
Overexpression of CBS at protein and mRNA levels has been reported in primary epithelial ovarian cancer tissue840–842 and in several breast cancer cell lines.843–845 However, CBS expression is reportedly suppressed in gastric and colorectal cancers, glioma, and hepatocellular carcinoma.846–848 Additional studies are needed to elucidate the roles of CBS in cancer development and progression. Similar contradictory observations have been reported for CSE, which was shown to be both up- and down-regulated in several cancer types.849–853 The pro-carcinogenic effects of H2S also contradict studies using H2S donors, which show anticarcinogenic effects.837–839,854 A deeper understanding of whether and how H2S plays a role in cancer etiology and progression is needed.
11. CONCLUSIONS
While an increasing number of signaling roles and physiological effects are being attributed to H2S, our understanding of the underlying mechanisms lags far behind. Significant barriers that have thwarted progress in the field include the technical challenges of working with a redox active and volatile molecule whose salts are often contaminated with polysulfides, which can be more reactive than H2S itself. The scarcity of sensitive, rigorous, and readily available methods for quantifying H2S or persulfides poses additional challenges. The identities of the preferred targets for H2S and of its downstream signaling intermediates are not yet completely understood. While persulfide proteomic analyses are beginning to reveal a rich trove of protein targets, the functional validation of how persulfidation affects individual proteins and metabolic flux has been challenging, since quantitative methods for introducing the persulfidation modification and stabilizing it under assay conditions are not readily available. Solving this methodological problem would lead to insights into how H2S signals and could identify important therapeutic targets. It would also lead to the resolution of the many contradictory effects of H2S that have been reported with purified proteins, at the cellular and organismal levels.
The contributions of the enzymes in H2S-producing and H2S-oxidizing pathways in controlling H2S levels and the conditions that permit spiking of H2S from low nanomolar steady-state concentrations to levels which trigger signaling are other important gaps in the field. Furthermore, the relative contributions of the three H2S-producing enzymes in different tissues are largely unknown. The development of selective inhibitors for CBS, CSE, MST, and SQR in particular will be very useful as molecular reagents together with gene editing technology for dissecting the roles of these enzymes in H2S homeostasis. Expanding the tool set for selective and ratiometric fluorescence visualization of protein persulfidation will facilitate elucidation of the spatiotemporal changes that occur during H2S signaling.
The field of H2S biology would benefit greatly by being strongly grounded in chemical studies on H2S and persulfide formation and decay kinetics and reactivity, to provide a more rigorours framework for understanding the potential roles of persulfidation in cell signaling. Proteomic studies suggest significant overlap between persulfidation and other oxidative cysteine modifications. Teasing apart why modifications like S-nitrosation, glutathionylation, and sulfenylation or persulfidation target the same cysteines in some proteins will provide important insights into cross-talk between oxidative cysteine-based signaling pathways.
Regulation of persulfidation is another area that is poorly understood. Are catalysts involved, and if so, what are their identities? Enzyme-catalyzed trans-persulfidation reactions are expected to be important both for adding and removing the persulfide modification, and an understanding of how each process is regulated and how target specificity is achieved is critical for elucidating H2S-based signaling. The relative newness of the H2S chemical biology field and the large fundamental gaps in our understanding combine to portend an exciting future.
Acknowledgments
M.R.F. and J.Z. acknowledge support from ATIP Avenir grant and Investments for the future “Programme IdEx Bordeaux” (ANR-10-IDEX-03-02). B.A. is supported by grants from Comisión Sectorial de Investigación Cientifíca (CSIC), Universidad de la República, Uruguay. R.B. acknowledges support from NIH (HL58984 and GM112455). We are grateful to Dr. Ernesto Cuevasanta and Emilia Kouroussis for their assistance.
Biographies
Milos R. Filipovic received his undergraduate and postgraduate training in biochemistry (University of Belgrade). After his postdoc at Friedrich-Alexander University Erlangen-Nuremberg, where he also worked as a group leader for a few years, he moved to Institut de Biochimie et Génétique Cellulaires (IBGC, CNRS UMR5095) at the University of Bordeaux, as Idex Junior Chair and ATIP-Avenir awardee to lead the “Signaling by gasotransmitters” group. He is interested in redox signaling, in general, and in developing new tools to dissect molecular mechanisms by which different gasotransmitters signal in the human body, in particular.
Jasmina Zivanovic studied biology at the University of Belgrade where she also obtained her Ph.D. (in endocrinology). She is a postdoc researcher at IBGC CNRS UMR5095 in the “Signaling by gasotransmitters” group. Her research focus is on developing new tools for persulfide labelling to address the changes of persulfidation in ageing and ageing-related diseases.
Beatriz Alvarez is Associate Professor of Enzymology at the School of Sciences, University of the Republic, Montevideo, Uruguay. She received her M.Sc. and Ph.D. in Chemistry degrees from the University of the Republic. Her interests span the areas of redox biochemistry, kinetics and enzymology, especially in relation to biological thiols and hydrogen sulfide.
Ruma Banerjee was raised as a peripatetic army brat changing ten schools before graduating. She received her formal training in plant science (B.S. and M.S., University of Delhi), biochemistry (Ph.D., Rensselaer Polytechnic Institute, NY), and biophysics (postdoc, University of Michigan). She is currently the Vincent Massey Collegiate Professor and Associate Chair of Biological Chemistry at the University of Michigan Medical School. Her interests range from B12 trafficking and metalloenzymology to hydrogen sulfide homeo-stasis and signaling. She is an Associate Editor for Chemical Reviews and for the Journal of Biological Chemistry.
Footnotes
ORCID
Milos R. Filipovic: 0000-0003-0060-0041
Notes
The authors declare no competing financial interest.
References
- 1.Parker ET, Cleaves HJ, Dworkin JP, Glavin DP, Callahan M, Aubrey A, Lazcano A, Bada JL. Primordial Synthesis of Amines and Amino Acids in a 1958 Miller H2S-Rich Spark Discharge Experiment. Proc Natl Acad Sci U S A. 2011;108:5526–5531. doi: 10.1073/pnas.1019191108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patel BH, Percivalle C, Ritson DJ, Duffy CD, Sutherland JD. Common Origins of RNA, Protein and Lipid Precursors in a Cyanosulfidic Protometabolism. Nat Chem. 2015;7:301–307. doi: 10.1038/nchem.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Winogradsky S. Ueber Schwefelbakterien. Bot Zeitung. 1887;45:489–507. 513–523, 529–539, 545–559, 569–576, 585–594. [Google Scholar]
- 4.Ramazzini B. Baptiftam Conzattum; Padua: 1713. De Morbis Artificum Diatriba. [Google Scholar]
- 5.Scheele CW. Chemische Abhandlung von der Luft und dem Feuer (Chemical treatise on air and fire) Magnus Swederus; Upsala, Sweden: 1777. [Google Scholar]
- 6.Abe K, Kimura H. The Possible Role of Hydrogen Sulfide as an Endogenous Neuromodulator. J Neurosci. 1996;16:1066–1071. doi: 10.1523/JNEUROSCI.16-03-01066.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hosoki R, Matsuki N, Kimura H. The Possible Role of Hydrogen Sulfide as an Endogenous Smooth Muscle Relaxant in Synergy with Nitric Oxide. Biochem Biophys Res Commun. 1997;237:527–531. doi: 10.1006/bbrc.1997.6878. [DOI] [PubMed] [Google Scholar]
- 8.Grigorian RM, Vasilenko FD, Akulova RF, Timoshenko MA. The Effect of Hydrogen Sulfide Baths on the Peripheral Blood Circulation after Reconstructive Operations on the Major Arteries on the Extremities. Sov Med. 1963;26:46–51. [PubMed] [Google Scholar]
- 9.Lavrov VP. The Effect of Matsesta Hydrogen Sulfide Baths on the Status of the Myocardium and Coronary Arteries in Experimental Atherosclerosis. Vopr Kurortol Fizioter Lech Fiz Kult. 1968;33:313–316. [PubMed] [Google Scholar]
- 10.Bocconi G. The Biological Actions of Sulfurated Waters. Minerva Ecol Idroclimatol Fis Sanit. 1976;16:105–116. [PubMed] [Google Scholar]
- 11.Kruszyna H, Kruszyna R, Smith RP. Cyanide and Sulfide Interact with Nitrogenous Compounds to Influence the Relaxation of Various Smooth Muscles. Exp Biol Med. 1985;179:44–49. doi: 10.3181/00379727-179-42062. [DOI] [PubMed] [Google Scholar]
- 12.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, et al. H2S as a Physiologic Vasorelaxant: Hypertension in Mice with Deletion of Cystathionine -Lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blackstone E. H2S Induces a Suspended Animation-Like State in Mice. Science. 2005;308:518–518. doi: 10.1126/science.1108581. [DOI] [PubMed] [Google Scholar]
- 14.Asfar P, Calzia E, Radermacher P. Is Pharmacological, H2S-Induced “Suspended Animation” Feasible in the ICU? Crit Care. 2014;18:215. doi: 10.1186/cc13782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szabó C. Hydrogen Sulphide and Its Therapeutic Potential. Nat Rev Drug Discovery. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 16.Li L, Rose P, Moore PK. Hydrogen Sulfide and Cell Signaling. Annu Rev Pharmacol Toxicol. 2011;51:169–187. doi: 10.1146/annurev-pharmtox-010510-100505. [DOI] [PubMed] [Google Scholar]
- 17.Wang R. Physiological Implications of Hydrogen Sulfide: A Whiff Exploration That Blossomed. Physiol Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 18.Mustafa AK, Gadalla MM, Snyder SH. Signaling by Gasotransmitters. Sci Signaling. 2009;2:re2–re2. doi: 10.1126/scisignal.268re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, Snyder SH. H2S Signals Through Protein S-Sulfhydration. Sci Signaling. 2009;2:ra72–ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paul BD, Snyder SH. H2S Signalling through Protein Sulfhydration and beyond. Nat Rev Mol Cell Biol. 2012;13:499–507. doi: 10.1038/nrm3391. [DOI] [PubMed] [Google Scholar]
- 21.Carroll JJ, Mather AE. The Solubility of Hydrogen Sulphide in Water from 0 to 90°C and Pressures to 1 MPa. Geochim Cosmochim Acta. 1989;53:1163–1170. [Google Scholar]
- 22.Iliuta MC, Larachi F. Solubility of Total Reduced Sulfurs (Hydrogen Sulfide, Methyl Mercaptan, Dimethyl Sulfide, and Dimethyl Disulfide) in Liquids. J Chem Eng Data. 2007;52:2–19. [Google Scholar]
- 23.De Bruyn WJ, Swartz E, Hu JH, Shorter JA, Davidovits P, Worsnop DR, Zahniser MS, Kolb CE. Henry’s Law Solubilities and Śetchenow Coefficients for Biogenic Reduced Sulfur Species Obtained from Gas-Liquid Uptake Measurements. J Geophys Res. 1995;100:7245–7251. [Google Scholar]
- 24.Evans CL. The Toxicity of Hydrogen Sulphide and Other Sulphides. Q J Exp Physiol Cogn Med Sci. 1967;52:231–248. doi: 10.1113/expphysiol.1967.sp001909. [DOI] [PubMed] [Google Scholar]
- 25.Yant WP. Hydrogen Sulphide in Industry-Occurrence, Effects, and Treatment. Am J Public Health Nations Health. 1930;20:598–608. doi: 10.2105/ajph.20.6.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elkins HB. The Chemistry of Industrial Toxicology. John Wiley & Sons; New York: 1950. [Google Scholar]
- 27.Beauchamp RO, Bus JS, Popp JA, Boreiko CJ, Andjelkovich DA, Leber P. A Critical Review of the Literature on Hydrogen Sulfide Toxicity. CRC Crit Rev Toxicol. 1984;13:25–97. doi: 10.3109/10408448409029321. [DOI] [PubMed] [Google Scholar]
- 28.Ahlborg G. Hydrogen Sulfide Poisoning in Shale Oil Industry. AMA Arch Ind Hyg Occup Med. 1951;3:247–266. [PubMed] [Google Scholar]
- 29.Haggard HW. The Toxicity of Hydrogen Sulphide. J Ind Hyg Toxicol. 1925;7:113–121. [Google Scholar]
- 30.Poda GA. Hydrogen Sulfide Can Be Handled Safely. Arch Environ Health. 1966;12:795–800. doi: 10.1080/00039896.1966.10664486. [DOI] [PubMed] [Google Scholar]
- 31.Wood JL. Sulfane Sulfur. Methods Enzymol. 1987;143:25–29. doi: 10.1016/0076-6879(87)43009-7. [DOI] [PubMed] [Google Scholar]
- 32.Ubuka T. Assay Methods and Biological Roles of Labile Sulfur in Animal Tissues. J Chromatogr B: Anal Technol Biomed Life Sci. 2002;781:227–249. doi: 10.1016/s1570-0232(02)00623-2. [DOI] [PubMed] [Google Scholar]
- 33.Toohey JI. Sulfur Signaling: Is the Agent Sulfide or Sulfane? Anal Biochem. 2011;413:1–7. doi: 10.1016/j.ab.2011.01.044. [DOI] [PubMed] [Google Scholar]
- 34.Toohey J, Cooper A. Thiosulfoxide (Sulfane) Sulfur: New Chemistry and New Regulatory Roles in Biology. Molecules. 2014;19:12789–12813. doi: 10.3390/molecules190812789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rickard D, Luther GW. Chemistry of Iron Sulfides. Chem Rev. 2007;107:514–562. doi: 10.1021/cr0503658. [DOI] [PubMed] [Google Scholar]
- 36.Trindle C. Molecular Structure and Bonding, the Qualitative Molecular Orbital Approach (Gimarc, M.) J Chem Educ. 1979;56:A352. [Google Scholar]
- 37.Rickard D, Luther GW. Kinetics of Pyrite Formation by the H2S Oxidation of Iron (II) Monosulfide in Aqueous Solutions between 25 and 125°C: The Mechanism. Geochim Cosmochim Acta. 1997;61:135–147. [Google Scholar]
- 38.Drzaic PS, Marks J, Brauman JI. In: Gas Phase Ion Chemistry. Bowers MT, editor. Vol. 3 Elsevier; Amsterdam, The Netherlands: 1984. [Google Scholar]
- 39.Radzig AA, Smirnov BM. Reference Data on Atoms, Molecules, and Ions. Vol. 31 Springer; Berlin: 1985. (Springer Series in Chemical Physics). [Google Scholar]
- 40.McDaniel DH, Evans WG. Strong Hydrogen Bonds. III. Hydrogen Sulfide-Hydrosulfide Anion Interactions. Inorg Chem. 1966;5:2180–2181. [Google Scholar]
- 41.Drozdov AP, Eremets MI, Troyan IA, Ksenofontov V, Shylin SI. Conventional Superconductivity at 203 K at High Pressures in the Sulfur Hydride System. Nature. 2015;525:73–76. doi: 10.1038/nature14964. [DOI] [PubMed] [Google Scholar]
- 42.Fathe K, Holt JS, Oxley SP, Pursell CJ. Infrared Spectroscopy of Solid Hydrogen Sulfide and Deuterium Sulfide. J Phys Chem A. 2006;110:10793–10798. doi: 10.1021/jp0634104. [DOI] [PubMed] [Google Scholar]
- 43.Millero FJ. The Thermodynamics and Kinetics of the Hydrogen Sulfide System in Natural Waters. Mar Chem. 1986;18:121–147. [Google Scholar]
- 44.Fogg PGT, Young CL, Clever HL, Boozer EL, Hayduk W. Hydrogen Sulfide, Deuterium Sulfide and Hydrogen Selenide. Pergamon Press; Oxford, U.K: 1988. [Google Scholar]
- 45.Konopik N, Leberl O. Colorimetric pH Determination in the Range 10 to 15. IV. The Second Dissociation Constant of Hydrogen Sulphide. Monatsh Chem. 1949;80:781–787. [Google Scholar]
- 46.Kubli H. Die Dissoziation von Schwefelwas-Serstoff. Helv Chim Acta. 1946;29:1962–1973. [Google Scholar]
- 47.Ellis AJ, Golding RM. Spectrophotometric Determination of the Acid Dissociation Constants of Hydrogen Sulphide. J Chem Soc. 1959;127 [Google Scholar]
- 48.Knox J. Zur Kenntnis Der Ionengbildung Des Schwefels Und Der Komplexionen Des Quecksilber. Z Elektrochem Angew Phys Chem. 1906;12:477–481. [Google Scholar]
- 49.Maronny G. Constantes de Dissociation de L’hydrogène Sulfuré. Electrochim Acta. 1959;1:58–69. [Google Scholar]
- 50.Ellis AJ, Giggenbach W. Hydrogen Sulphide Ionization and Sulphur Hydrolysis in High Temperature Solution. Geochim Cosmochim Acta. 1971;35:247–260. [Google Scholar]
- 51.Giggenbach W. Optical Spectra of Highly Alkaline Sulfide Solutions and the Second Dissociation Constant of Hydrogen Sulfide. Inorg Chem. 1971;10:1333–1338. [Google Scholar]
- 52.Meyer B, Ward K, Koshlap K, Peter L. Second Dissociation Constant of Hydrogen Sulfide. Inorg Chem. 1983;22:2345–2346. [Google Scholar]
- 53.Licht S, Manassen J. The Second Dissociation Constant of Hydrogen Sulfide. J Electrochem Soc. 1987;134:918–921. [Google Scholar]
- 54.Myers RJ. The New Low Value for the Second Dissociation Constant for H2S: Its History, Its Best Value, and Its Impact on the Teaching of Sulfide Equilibria. J Chem Educ. 1986;63:687. [Google Scholar]
- 55.Schoonen MAA, Barnes HL. An Approximation of the Second Dissociation Constant for H2S. Geochim Cosmochim Acta. 1988;52:649–654. [Google Scholar]
- 56.Guenther EA, Johnson KS, Coale KH. Direct Ultraviolet Spectrophotometric Determination of Total Sulfide and Iodide in Natural Waters. Anal Chem. 2001;73:3481–3487. doi: 10.1021/ac0013812. [DOI] [PubMed] [Google Scholar]
- 57.Cuevasanta E, Denicola A, Alvarez B, Möller MN. Solubility and Permeation of Hydrogen Sulfide in Lipid Membranes. PLoS One. 2012;7:e34562. doi: 10.1371/journal.pone.0034562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mathai JC, Missner A, Kugler P, Saparov SM, Zeidel ML, Lee JK, Pohl P. No Facilitator Required for Membrane Transport of Hydrogen Sulfide. Proc Natl Acad Sci U S A. 2009;106:16633–16638. doi: 10.1073/pnas.0902952106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Riahi S, Rowley CN. Why Can Hydrogen Sulfide Permeate Cell Membranes? J Am Chem Soc. 2014;136:15111–15113. doi: 10.1021/ja508063s. [DOI] [PubMed] [Google Scholar]
- 60.Cuevasanta E, Lange M, Bonanata J, Coitiño EL, Ferrer-Sueta G, Filipovic MR, Alvarez B. Reaction of Hydrogen Sulfide with Disulfide and Sulfenic Acid to Form the Strongly Nucleophilic Persulfide. J Biol Chem. 2015;290:26866–26880. doi: 10.1074/jbc.M115.672816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carballal S, Trujillo M, Cuevasanta E, Bartesaghi S, Möller MN, Folkes LK, García-Bereguiaín MA, Gutiérrez-Merino C, Wardman P, Denicola A, et al. Reactivity of Hydrogen Sulfide with Peroxynitrite and Other Oxidants of Biological Interest. Free Radic Biol Med. 2011;50:196–205. doi: 10.1016/j.freeradbiomed.2010.10.705. [DOI] [PubMed] [Google Scholar]
- 62.Trujillo M, Alvarez B, Radi R. One- and Two-Electron Oxidation of Thiols: Mechanisms, Kinetics and Biological Fates. Free Radical Res. 2016;50:150–171. doi: 10.3109/10715762.2015.1089988. [DOI] [PubMed] [Google Scholar]
- 63.Armstrong DA, Huie RE, Koppenol WH, Lymar SV, Merényi G, Neta P, Ruscic B, Stanbury DM, Steenken S, Wardman P. Standard Electrode Potentials Involving Radicals in Aqueous Solution: Inorganic Radicals (IUPAC Technical Report) Pure Appl Chem. 2015;87:1139–1150. [Google Scholar]
- 64.Koppenol WH, Bounds PL. Signaling by Sulfur-Containing Molecules. Quantitative Aspects. Arch Biochem Biophys. 2017;617:3–8. doi: 10.1016/j.abb.2016.09.012. [DOI] [PubMed] [Google Scholar]
- 65.Das TN, Huie RE, Neta P, Padmaja S. Reduction Potential of the Sulfhydryl Radical: Pulse Radiolysis and Laser Flash Photolysis Studies of the Formation and Reactions of ·SH and HSSH· - in Aqueous Solutions. J Phys Chem A. 1999;103:5221–5226. [Google Scholar]
- 66.Darwent BD. Bond Dissociation Energies in Simple Molecules. National Standard Reference Data System, National Bureau of Standards. 1970:1–58. [Google Scholar]
- 67.Wood PM. The Potential Diagram for Oxygen at pH 7. Biochem J. 1988;253:287–289. doi: 10.1042/bj2530287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Karmann W, Meissner G, Henglein A. Pulsradiolyse Des Schwefelwasserstoffs in Wäßriger Lösung. Z Naturforsch, B: J Chem Sci. 1967;22:273–282. [Google Scholar]
- 69.Wedmann R, Zahl A, Shubina TE, Dürr M, Heinemann FW, Bugenhagen BEC, Burger P, Ivanovic-Burmazovic I, Filipovic MR. Does Perthionitrite (SSNO–) Account for Sustained Bioactivity of NO? A (Bio)chemical Characterization. Inorg Chem. 2015;54:9367–9380. doi: 10.1021/acs.inorgchem.5b00831. [DOI] [PubMed] [Google Scholar]
- 70.Feher F. Handbook of Preparative Inorganic Chemistry. Academic Press; New York: 1963. Sulfur, Selenium, Tellurium; pp. 341–456. [Google Scholar]
- 71.Zhu J, Petit K, Colson AO, DeBolt S, Sevilla MD. Reactions of Sulfhydryl and Sulfide Radicals with Oxygen, Hydrogen Sulfide, Hydrosulfide, and Sulfide: Formation of SO2-, HSSH-, HSS.2-and HSS. J Phys Chem. 1991;95:3676–3681. [Google Scholar]
- 72.Hoffmann MR, Lim BC. Kinetics and Mechanism of the Oxidation of Sulfide by Oxygen: Catalysis by Homogeneous Metal-Phthalocyanine Complexes. Environ Sci Technol. 1979;13:1406–1414. [Google Scholar]
- 73.Hoffmann MR. Kinetics and Mechanism of Oxidation of Hydrogen Sulfide by Hydrogen Peroxide in Acidic Solution. Environ Sci Technol. 1977;11:61–66. [Google Scholar]
- 74.O’Brien DJ, Birkner FB. Kinetics of Oxygenation of Reduced Sulfur Species in Aqueous Solution. Environ Sci Technol. 1977;11:1114–1120. [Google Scholar]
- 75.Kotronarou A, Hoffmann MR. Catalytic Autoxidation of Hydrogen Sulfide in Wastewater. Environ Sci Technol. 1991;25:1153–1160. [Google Scholar]
- 76.Zhang J-Z, Millero FJ. The Products from the Oxidation of H2S in Seawater. Geochim Cosmochim Acta. 1993;57:1705–1718. [Google Scholar]
- 77.Sun J, Stanbury DM. Trace Metal-Ion Catalysis of Oxidation of Aqueous Hydrogen Sulfide by Outer-Sphere Oxidants. Inorg Chim Acta. 2002;336:39–45. [Google Scholar]
- 78.Panda SK, Andersson JT, Schrader W. Characterization of Supercomplex Crude Oil Mixtures: What Is Really in There? Angew Chem, Int Ed. 2009;48:1788–1791. doi: 10.1002/anie.200803403. [DOI] [PubMed] [Google Scholar]
- 79.Holleman AF, Wiberg E. Lehrbuch Der Anorganischen Chemie. Walter de Gruyter & Co; Berlin: 1995. [Google Scholar]
- 80.Kotronarou A, Mills G, Hoffmann MR. Oxidation of Hydrogen Sulfide in Aqueous Solution by Ultrasonic Irradiation. Environ Sci Technol. 1992;26:2420–2428. [Google Scholar]
- 81.Poulton SW, Krom MD, Rijn JV, Raiswell R. The Use of Hydrous Iron (III) Oxides for the Removal of Hydrogen Sulphide in Aqueous Systems. Water Res. 2002;36:825–834. doi: 10.1016/s0043-1354(01)00314-1. [DOI] [PubMed] [Google Scholar]
- 82.Ruhland B, Helmut F. Stripping of Sulfide-Containing Liquids, Especially from Leather or Petrochemical Industries, with Carbon Dioxide-Containing Gas Involves Recycling of Hydrogen Sulfide-Containing Off-Gas. DE 10204654A1. 2003 [Google Scholar]
- 83.Hardison LC. Removal of Hydrogen Sulfide from Sour Water without Loss of Heavy Metal. US 5292440A. 1992 [Google Scholar]
- 84.Piché S, Larachi F. Hydrosulfide Oxidation Pathways in Oxic Solutions Containing iron(III) Chelates. Environ Sci Technol. 2007;41:1206–1211. doi: 10.1021/es061752h. [DOI] [PubMed] [Google Scholar]
- 85.Martell AE, Motekaitis RJ, Chen D, Hancock RD, McManus D. Selection of New Fe(III)/Fe(II) Chelating Agents as Catalysts for the Oxidation of Hydrogen Sulfide to Sulfur by Air. Can J Chem. 1996;74:1872–1879. [Google Scholar]
- 86.Hughes MN, Centelles MN, Moore KP. Making and Working with Hydrogen Sulfide. Free Radic Biol Med. 2009;47:1346–1353. doi: 10.1016/j.freeradbiomed.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 87.Steudel R, Eckert B. Solid Sulfur Allotropes Sulfur Allotropes. Top Curr Chem. 2003;230:1–80. [Google Scholar]
- 88.Steudel R. Liquid Sulfur Liquid Sulfur. Top Curr Chem. 2003;230:81–116. [Google Scholar]
- 89.Steudel R. Aqueous Sulfur Sols. Top Curr Chem. 2003;230:153–166. [Google Scholar]
- 90.Tebbe FN, Wasserman E, Peet WG, Vatvars A, Hayman AC. Composition of Elemental Sulfur in Solution: Equilibrium of S6, S7 and S8 at Ambient Temperatures. J Am Chem Soc. 1982;104:4971–4972. [Google Scholar]
- 91.Hojo M, Sawyer DT. Hydroxide-Induced Reduction of Elemental Sulfur (S8) to Trisulfur Anion Radical (S3•−) Inorg Chem. 1989;28:1201–1202. [Google Scholar]
- 92.Bartlett PD, Meguerian G. Reactions of Elemental Sulfur. I. The Uncatalyzed Reaction of Sulfur with Triarylphosphines 1. J Am Chem Soc. 1956;78:3710–3715. [Google Scholar]
- 93.Piery M, Bonnamour S, De Milhaud M. Dégagement d’Hydrogène Sulfure Par Le Voies Respiratoires Après Injections Intraveineuse De Soufre Colloïdal. Compt Rend Soc Biol. 1924;(91):679–720. [Google Scholar]
- 94.Bernheim F, Bernheim MLC. The Action of Colloidal Sulfur on Liver Oxidations. J Biol Chem. 1932;96:331–339. [Google Scholar]
- 95.Searcy DG, Lee SH. Sulfur Reduction by Human Erythrocytes. J Exp Zool. 1998;282:310–322. doi: 10.1002/(sici)1097-010x(19981015)282:3<310::aid-jez4>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 96.Clinical Trials. Gov Identifier: NCT01989208.
- 97.Wedmann R, Bertlein S, Macinkovic I, Böltz S, Miljkovic JL, Muñoz LE, Herrmann M, Filipovic MR. Working with “H2S”: Facts and Apparent Artifacts. Nitric Oxide. 2014;41:85–96. doi: 10.1016/j.niox.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 98.Hartle MD, Pluth MD. A Practical Guide to Working with H2S at the Interface of Chemistry and Biology. Chem Soc Rev. 2016;45:6108–6117. doi: 10.1039/c6cs00212a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Brauer G. Handbook of Preparative Inorganic Chemistry. 2nd. Academic Press; New York: p. 1963. [Google Scholar]
- 100.Greiner R, Pálinkás Z, Bäsell K, Becher D, Antelmann H, Nagy P, Dick TP. Polysulfides Link H2S to Protein Thiol Oxidation. Antioxid Redox Signal. 2013;19:1749–1765. doi: 10.1089/ars.2012.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nagy P, Pálinkás Z, Nagy A, Budai B, Tóth I, Vasas A. Chemical Aspects of Hydrogen Sulfide Measurements in Physiological Samples. Biochim Biophys Acta, Gen Subj. 2014;1840:876–891. doi: 10.1016/j.bbagen.2013.05.037. [DOI] [PubMed] [Google Scholar]
- 102.Hartle MD, Meininger DJ, Zakharov LN, Tonzetich ZJ, Pluth MD. NBu4SH Provides a Convenient Source of HS–Soluble in Organic Solution for H2S and Anion-Binding Research. Dalt Trans. 2015;44:19782–19785. doi: 10.1039/c5dt03355a. [DOI] [PubMed] [Google Scholar]
- 103.Saund SS, Sosa V, Henriquez S, Nguyen QNN, Bianco CL, Soeda S, Millikin R, White C, Le H, Ono K, et al. The Chemical Biology of Hydropersulfides (RSSH): Chemical Stability, Reactivity and Redox Roles. Arch Biochem Biophys. 2015;588:15–24. doi: 10.1016/j.abb.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bełtowski J. Hydrogen Sulfide in Pharmacology and Medicine – An Update. Pharmacol Rep. 2015;67:647–658. doi: 10.1016/j.pharep.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 105.Kashfi K, Olson KR. Biology and Therapeutic Potential of Hydrogen Sulfide and Hydrogen Sulfide-Releasing Chimeras. Biochem Pharmacol. 2013;85:689–703. doi: 10.1016/j.bcp.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhao Y, Biggs TD, Xian M. Hydrogen Sulfide (H2S) Releasing Agents: Chemistry and Biological Applications. Chem Commun. 2014;50:11788–11805. doi: 10.1039/c4cc00968a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wallace JL, Wang R. Hydrogen Sulfide-Based Therapeutics: Exploiting a Unique but Ubiquitous Gasotransmitter. Nat Rev Drug Discovery. 2015;14:329–345. doi: 10.1038/nrd4433. [DOI] [PubMed] [Google Scholar]
- 108.Whiteman M, Perry A, Zhou Z, Bucci M, Papapetropoulos A, Cirino G, Wood ME. Phosphinodithioate and Phosphoramidodithioate Hydrogen Sulfide Donors. Handb Exp Pharmacol. 2015;230:337–363. doi: 10.1007/978-3-319-18144-8_17. [DOI] [PubMed] [Google Scholar]
- 109.Zhao Y, Pacheco A, Xian M. Medicinal Chemistry: Insights into the Development of Novel H2S Donors. Handb Exp Pharmacol. 2015;230:365–388. doi: 10.1007/978-3-319-18144-8_18. [DOI] [PubMed] [Google Scholar]
- 110.Pluth M, Bailey T, Hammers M, Hartle M, Henthorn H, Steiger A. Natural Products Containing Hydrogen Sulfide Releasing Moieties. Synlett. 2015;26:2633–2643. [Google Scholar]
- 111.Yagdi E, Cerella C, Dicato M, Diederich M. Garlic-Derived Natural Polysulfanes as Hydrogen Sulfide Donors: Friend or Foe? Food Chem Toxicol. 2016;95:219–233. doi: 10.1016/j.fct.2016.07.016. [DOI] [PubMed] [Google Scholar]
- 112.Zhao Y, Wang H, Xian M. Cysteine-Activated Hydrogen Sulfide (H2S) Donors. J Am Chem Soc. 2011;133:15–17. doi: 10.1021/ja1085723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhao Y, Bhushan S, Yang C, Otsuka H, Stein JD, Pacheco A, Peng B, Devarie-Baez NO, Aguilar HC, Lefer DJ, et al. Controllable Hydrogen Sulfide Donors and Their Activity against Myocardial Ischemia-Reperfusion Injury. ACS Chem Biol. 2013;8:1283–1290. doi: 10.1021/cb400090d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Roger T, Raynaud F, Bouillaud F, Ransy C, Simonet S, Crespo C, Bourguignon M-P, Villeneuve N, Vilaine J-P, Artaud I, et al. New Biologically Active Hydrogen Sulfide Donors. ChemBioChem. 2013;14:2268–2271. doi: 10.1002/cbic.201300552. [DOI] [PubMed] [Google Scholar]
- 115.Martelli A, Testai L, Citi V, Marino A, Pugliesi I, Barresi E, Nesi G, Rapposelli S, Taliani S, Da Settimo F, et al. Arylthioamides as H2S Donors: L -Cysteine-Activated Releasing Properties and Vascular Effects in Vitro and in Vivo. ACS Med Chem Lett. 2013;4:904–908. doi: 10.1021/ml400239a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Foster JC, Powell CR, Radzinski SC, Matson JB. S-Aroylthiooximes: A Facile Route to Hydrogen Sulfide Releasing Compounds with Structure-Dependent Release Kinetics. Org Lett. 2014;16:1558–1561. doi: 10.1021/ol500385a. [DOI] [PubMed] [Google Scholar]
- 117.Carter JM, Qian Y, Foster JC, Matson JB. Peptide-Based Hydrogen Sulphide-Releasing Gels. Chem Commun (Cambridge, U K) 2015;51:13131–13134. doi: 10.1039/c5cc04883d. [DOI] [PubMed] [Google Scholar]
- 118.Ozturk T, Ertas E, Mert O. Use of Lawesson’s Reagent in Organic Syntheses. Chem Rev. 2007;107:5210–5278. doi: 10.1021/cr040650b. [DOI] [PubMed] [Google Scholar]
- 119.Wallace JL, Vong L, McKnight W, Dicay M, Martin GR. Endogenous and Exogenous Hydrogen Sulfide Promotes Resolution of Colitis in Rats. Gastroenterology. 2009;137:569. doi: 10.1053/j.gastro.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 120.Li L, Whiteman M, Guan YY, Neo KL, Cheng Y, Lee SW, Zhao Y, Baskar R, Tan C-H, Moore PK. Characterization of a Novel, Water-Soluble Hydrogen Sulfide-Releasing Molecule (GYY4137): New Insights Into the Biology of Hydrogen Sulfide. Circulation. 2008;117:2351–2360. doi: 10.1161/CIRCULATIONAHA.107.753467. [DOI] [PubMed] [Google Scholar]
- 121.Li L, Salto-Tellez M, Tan C-H, Whiteman M, Moore PK. GYY4137, a Novel Hydrogen Sulfide-Releasing Molecule, Protects against Endotoxic Shock in the Rat. Free Radic Biol Med. 2009;47:103–113. doi: 10.1016/j.freeradbiomed.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 122.Li L, Fox B, Keeble J, Salto-Tellez M, Winyard PG, Wood ME, Moore PK, Whiteman M. The Complex Effects of the Slow-Releasing Hydrogen Sulfide Donor GYY4137 in a Model of Acute Joint Inflammation and in Human Cartilage Cells. J Cell Mol Med. 2013;17:365–376. doi: 10.1111/jcmm.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu Z, Han Y, Li L, Lu H, Meng G, Li X, Shirhan M, Peh MT, Xie L, Zhou S, et al. The Hydrogen Sulfide Donor, GYY4137, Exhibits Anti-Atherosclerotic Activity in High Fat Fed Apolipoprotein E −/− Mice. Br J Pharmacol. 2013;169:1795–1809. doi: 10.1111/bph.12246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Park C-M, Zhao Y, Zhu Z, Pacheco A, Peng B, Devarie-Baez NO, Bagdon P, Zhang H, Xian M. Synthesis and Evaluation of Phosphorodithioate-Based Hydrogen Sulfide Donors. Mol BioSyst. 2013;9:2430. doi: 10.1039/c3mb70145j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Alexander BE, Coles SJ, Fox BC, Khan TF, Maliszewski J, Perry A, Pitak MB, Whiteman M, Wood ME, et al. Investigating the Generation of Hydrogen Sulfide from the Phosphonamidodithioate Slow-Release Donor GYY4137. MedChem-Comm. 2015;6:1649–1655. [Google Scholar]
- 126.Kang J, Li Z, Organ CL, Park C-M, Yang C, Pacheco A, Wang D, Lefer DJ, Xian M. pH-Controlled Hydrogen Sulfide Release for Myocardial Ischemia-Reperfusion Injury. J Am Chem Soc. 2016;138:6336–6339. doi: 10.1021/jacs.6b01373. [DOI] [PubMed] [Google Scholar]
- 127.Caliendo G, Cirino G, Santagada V, Wallace JL. Synthesis and Biological Effects of Hydrogen Sulfide (H2S): Development of H2S-Releasing Drugs as Pharmaceuticals. J Med Chem. 2010;53:6275–6286. doi: 10.1021/jm901638j. [DOI] [PubMed] [Google Scholar]
- 128.Wallace JL, Caliendo G, Santagada V, Cirino G. Markedly Reduced Toxicity of a Hydrogen Sulphide-Releasing Derivative of Naproxen (ATB-346) Br J Pharmacol. 2010;159:1236–1246. doi: 10.1111/j.1476-5381.2009.00611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Distrutti E. 5-Amino-2-Hydroxybenzoic Acid 4-(5-Thioxo-5H-[1,2]dithiol-3yl)-Phenyl Ester (ATB-429), a Hydrogen Sulfide-Releasing Derivative of Mesalamine, Exerts Antinociceptive Effects in a Model of Postinflammatory Hypersensitivity. J Pharmacol Exp Ther. 2006;319:447–458. doi: 10.1124/jpet.106.106435. [DOI] [PubMed] [Google Scholar]
- 130.Vannini F, MacKessack-Leitch AC, Eschbach EK, Chattopadhyay M, Kodela R, Kashfi K. Synthesis and Anti-Cancer Potential of the Positional Isomers of NOSH-Aspirin (NBS-1120) a Dual Nitric Oxide and Hydrogen Sulfide Releasing Hybrid. Bioorg Med Chem Lett. 2015;25:4677–4682. doi: 10.1016/j.bmcl.2015.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lougiakis N, Papapetropoulos A, Gikas E, Toumpas S, Efentakis P, Wedmann R, Zoga A, Zhou Z, Iliodromitis EK, Skaltsounis A-L, et al. Synthesis and Pharmacological Evaluation of Novel Adenine–Hydrogen Sulfide Slow Release Hybrids Designed as Multitarget Cardioprotective Agents. J Med Chem. 2016;59:1776–1790. doi: 10.1021/acs.jmedchem.5b01223. [DOI] [PubMed] [Google Scholar]
- 132.Le Trionnaire S, Perry A, Szczesny B, Szabo C, Winyard PG, Whatmore JL, Wood ME, Whiteman M. The Synthesis and Functional Evaluation of a Mitochondria-Targeted Hydrogen Sulfide Donor, (10-Oxo-10-(4-(3-Thioxo-3H-1,2-Dithiol-5-Yl)phenoxy)-decyl)triphenylphosphonium Bromide (AP39) MedChemComm. 2014;5:728–736. [Google Scholar]
- 133.Gerő D, Torregrossa R, Perry A, Waters A, Le-Trionnaire S, Whatmore JL, Wood M, Whiteman M. The Novel Mitochondria-Targeted Hydrogen Sulfide (H2S) Donors AP123 and AP39 Protect against Hyperglycemic Injury in Microvascular Endothelial Cells in Vitro. Pharmacol Res. 2016;113:186–198. doi: 10.1016/j.phrs.2016.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ahmad A, Olah G, Szczesny B, Wood ME, Whiteman M, Szabo C. AP39, A Mitochondrially Targeted Hydrogen Sulfide Donor, Exerts Protective Effects in Renal Epithelial Cells Subjected to Oxidative Stress in Vitro and in Acute Renal Injury in Vivo. Shock. 2016;45:88–97. doi: 10.1097/SHK.0000000000000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wedmann R, Onderka C, Wei S, Szijártó IA, Miljkovic JL, Mitrovic A, Lange M, Savitsky S, Yadav PK, Torregrossa R, et al. Improved Tag-Switch Method Reveals That Thioredoxin Acts as Depersulfidase and Controls the Intracellular Levels of Protein Persulfidation. Chem Sci. 2016;7:3414–3426. doi: 10.1039/c5sc04818d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Clinical Trials. Gov Identifiers: NCT01926444.
- 137.Devarie-Baez NO, Bagdon PE, Peng B, Zhao Y, Park C-M, Xian M. Light-Induced Hydrogen Sulfide Release from “Caged” Gem -Dithiols. Org Lett. 2013;15:2786–2789. doi: 10.1021/ol401118k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhou Z, von Wantoch Rekowski M, Coletta C, Szabo C, Bucci M, Cirino G, Topouzis S, Papapetropoulos A, Giannis A. Thioglycine and L-Thiovaline: Biologically Active H2S-Donors. Bioorg Med Chem. 2012;20:2675–2678. doi: 10.1016/j.bmc.2012.02.028. [DOI] [PubMed] [Google Scholar]
- 139.Steiger AK, Pardue S, Kevil CG, Pluth MD. Self-Immolative Thiocarbamates Provide Access to Triggered H2S Donors and Analyte Replacement Fluorescent Probes. J Am Chem Soc. 2016;138:7256–7259. doi: 10.1021/jacs.6b03780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Powell CR, Foster JC, Okyere B, Theus MH, Matson JB. Therapeutic Delivery of H2S via COS: Small Molecule and Polymeric Donors with Benign Byproducts. J Am Chem Soc. 2016;138:13477–13480. doi: 10.1021/jacs.6b07204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhao Y, Bolton SG, Pluth MD. Light-Activated COS/H2S Donation from Photocaged Thiocarbamates. Org Lett. 2017;19:2278–2281. doi: 10.1021/acs.orglett.7b00808. [DOI] [PubMed] [Google Scholar]
- 142.Zhao Y, Pluth MD. Hydrogen Sulfide Donors Activated by Reactive Oxygen Species. Angew Chem, Int Ed. 2016;55:14638–14642. doi: 10.1002/anie.201608052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Xu S, Yang C-T, Meng F-H, Pacheco A, Chen L, Xian M. Ammonium Tetrathiomolybdate as a Water-Soluble and Slow-Release Hydrogen Sulfide Donor. Bioorg Med Chem Lett. 2016;26:1585–1588. doi: 10.1016/j.bmcl.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Dyson A, Dal-Pizzol F, Sabbatini G, Lach AB, Galfo F, dos Santos Cardoso J, Pescador Mendonça B, Hargreaves I, Bollen Pinto B, Bromage DI, et al. Ammonium Tetrathiomolybdate Following Ischemia/reperfusion Injury: Chemistry, Pharmacology, and Impact of a New Class of Sulfide Donor in Preclinical Injury Models. PLOS Med. 2017;14:e1002310. doi: 10.1371/journal.pmed.1002310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zuman P, Szafranski W. Ultraviolet Spectra of Hydroxide, Alkoxide, and Hydrogen Sulfide Anions. Anal Chem. 1976;48:2162–2163. [Google Scholar]
- 146.Kubán V, Dasgupta PK, Marx JN. Nitroprusside and Methylene Blue Methods for Silicone Membrane Differentiated Flow Injection Determination of Sulfide in Water and Wastewater. Anal Chem. 1992;64:36–43. doi: 10.1021/ac00025a008. [DOI] [PubMed] [Google Scholar]
- 147.Lawrence NS, Davis J, Jiang L, Jones TGJ, Davies SN, Compton RG. The Electrochemical Analog of the Methylene Blue Reaction: A Novel Amperometric Approach to the Detection of Hydrogen Sulfide. Electroanalysis. 2000;12:1453–1460. [Google Scholar]
- 148.Fischer E. Bildung von Methylenblau Als Reaktion Auf Schwefelwasserstoff. Ber Dtsch Chem Ges. 1883;16:2234–2236. [Google Scholar]
- 149.Fogo JK, Popowsky M. Spectrophotometric Determination of Hydrogen Sulfide. Anal Chem. 1949;21:732–734. [Google Scholar]
- 150.Siegel LM. A Direct Microdetermination for Sulfide. Anal Biochem. 1965;11:126–132. doi: 10.1016/0003-2697(65)90051-5. [DOI] [PubMed] [Google Scholar]
- 151.Haddad PR, Heckenberg AL. Trace Determination of Sulfide by Reversed-Phase Ion-Interaction Chromatography Using Pre-Column Derivatization. J Chromatogr. 1988;447:415–420. [PubMed] [Google Scholar]
- 152.Small JM, Hintelmann H. Methylene Blue Derivatization Then LC–MS Analysis for Measurement of Trace Levels of Sulfide in Aquatic Samples. Anal Bioanal Chem. 2007;387:2881–2886. doi: 10.1007/s00216-007-1140-3. [DOI] [PubMed] [Google Scholar]
- 153.Olson KR. A Practical Look at the Chemistry and Biology of Hydrogen Sulfide. Antioxid Redox Signal. 2012;17:32–44. doi: 10.1089/ars.2011.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chen X, Jhee K-H, Kruger WD. Production of the Neuromodulator H2S by Cystathionine β-Synthase via the Condensation of Cysteine and Homocysteine. J Biol Chem. 2004;279:52082–52086. doi: 10.1074/jbc.C400481200. [DOI] [PubMed] [Google Scholar]
- 155.Hine C, Harputlugil E, Zhang Y, Ruckenstuhl C, Lee BC, Brace L, Longchamp A, Treviño-Villarreal JH, Mejia P, Ozaki CK, et al. Endogenous Hydrogen Sulfide Production Is Essential for Dietary Restriction Benefits. Cell. 2015;160:132–144. doi: 10.1016/j.cell.2014.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wei Y, Kenyon C. Roles for ROS and Hydrogen Sulfide in the Longevity Response to Germline Loss in Caenorhabditis Elegans. Proc Natl Acad Sci U S A. 2016;113:E2832–E2841. doi: 10.1073/pnas.1524727113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Doeller JE, Isbell TS, Benavides G, Koenitzer J, Patel H, Patel RP, Lancaster JR, Darley-Usmar VM, Kraus DW. Polarographic Measurement of Hydrogen Sulfide Production and Consumption by Mammalian Tissues. Anal Biochem. 2005;341:40–51. doi: 10.1016/j.ab.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 158.Xu T, Scafa N, Xu L-P, Zhou S, Abdullah Al-Ghanem K, Mahboob S, Fugetsu B, Zhang X. Electrochemical Hydrogen Sulfide Biosensors. Analyst. 2016;141:1185–1195. doi: 10.1039/c5an02208h. [DOI] [PubMed] [Google Scholar]
- 159.Furne J, Saeed A, Levitt MD. Whole Tissue Hydrogen Sulfide Concentrations Are Orders of Magnitude Lower than Presently Accepted Values. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1479–R1485. doi: 10.1152/ajpregu.90566.2008. [DOI] [PubMed] [Google Scholar]
- 160.Vitvitsky V, Banerjee R. H2S Analysis in Biological Samples Using Gas Chromatography with Sulfur Chemiluminescence Detection. Methods Enzymol. 2015;554:111–123. doi: 10.1016/bs.mie.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kosower NS, Kosower EM, Newton GL, Ranney HM. Bimane Fluorescent Labels: Labeling of Normal Human Red Cells under Physiological Conditions. Proc Natl Acad Sci U S A. 1979;76:3382–3386. doi: 10.1073/pnas.76.7.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wintner EA, Deckwerth TL, Langston W, Bengtsson A, Leviten D, Hill P, Insko MA, Dumpit R, VandenEkart E, Toombs CF, et al. A Monobromobimane-Based Assay to Measure the Pharmacokinetic Profile of Reactive Sulphide Species in Blood. Br J Pharmacol. 2010;160:941–957. doi: 10.1111/j.1476-5381.2010.00704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Shen X, Pattillo CB, Pardue S, Bir SC, Wang R, Kevil CG. Measurement of Plasma Hydrogen Sulfide in Vivo and in Vitro. Free Radic Biol Med. 2011;50:1021–1031. doi: 10.1016/j.freeradbiomed.2011.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Shen X, Peter EA, Bir S, Wang R, Kevil CG. Analytical Measurement of Discrete Hydrogen Sulfide Pools in Biological Specimens. Free Radic Biol Med. 2012;52:2276–2283. doi: 10.1016/j.freeradbiomed.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ida T, Sawa T, Ihara H, Tsuchiya Y, Watanabe Y, Kumagai Y, Suematsu M, Motohashi H, Fujii S, Matsunaga T, et al. Reactive Cysteine Persulfides and S-Polythiolation Regulate Oxidative Stress and Redox Signaling. Proc Natl Acad Sci U S A. 2014;111:7606–7611. doi: 10.1073/pnas.1321232111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lin VS, Chen W, Xian M, Chang CJ. Chemical Probes for Molecular Imaging and Detection of Hydrogen Sulfide and Reactive Sulfur Species in Biological Systems. Chem Soc Rev. 2015;44:4596–4618. doi: 10.1039/c4cs00298a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Takano Y, Shimamoto K, Hanaoka K. Chemical Tools for the Study of Hydrogen Sulfide (H2S) and Sulfane Sulfur and Their Applications to Biological Studies. J Clin Biochem Nutr. 2016;58:7–15. doi: 10.3164/jcbn.15-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Feng W, Dymock BW. Fluorescent Probes for H2S Detection and Quantification. Handb Exp Pharmacol. 2015;230:291–323. doi: 10.1007/978-3-319-18144-8_15. [DOI] [PubMed] [Google Scholar]
- 169.Lippert AR, New EJ, Chang CJ. Reaction-Based Fluorescent Probes for Selective Imaging of Hydrogen Sulfide in Living Cells. J Am Chem Soc. 2011;133:10078–10080. doi: 10.1021/ja203661j. [DOI] [PubMed] [Google Scholar]
- 170.Peng H, Cheng Y, Dai C, King AL, Predmore BL, Lefer DJ, Wang B. A Fluorescent Probe for Fast and Quantitative Detection of Hydrogen Sulfide in Blood. Angew Chem, Int Ed. 2011;50:9672–9675. doi: 10.1002/anie.201104236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Montoya LA, Pluth MD. Selective Turn-on Fluorescent Probes for Imaging Hydrogen Sulfide in Living Cells. Chem Commun. 2012;48:4767. doi: 10.1039/c2cc30730h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Qian Y, Karpus J, Kabil O, Zhang S-Y, Zhu H-L, Banerjee R, Zhao J, He C. Selective Fluorescent Probes for Live-Cell Monitoring of Sulphide. Nat Commun. 2011;2:495. doi: 10.1038/ncomms1506. [DOI] [PubMed] [Google Scholar]
- 173.Liu C, Pan J, Li S, Zhao Y, Wu LY, Berkman CE, Whorton AR, Xian M. Capture and Visualization of Hydrogen Sulfide by a Fluorescent Probe. Angew Chem, Int Ed. 2011;50:10327–10329. doi: 10.1002/anie.201104305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sasakura K, Hanaoka K, Shibuya N, Mikami Y, Kimura Y, Komatsu T, Ueno T, Terai T, Kimura H, Nagano T. Development of a Highly Selective Fluorescence Probe for Hydrogen Sulfide. J Am Chem Soc. 2011;133:18003–18005. doi: 10.1021/ja207851s. [DOI] [PubMed] [Google Scholar]
- 175.Olson KR, DeLeon ER, Liu F. Controversies and Conundrums in Hydrogen Sulfide Biology. Nitric Oxide. 2014;41:11–26. doi: 10.1016/j.niox.2014.05.012. [DOI] [PubMed] [Google Scholar]
- 176.Olson KR. Is Hydrogen Sulfide a Circulating “gasotransmitter” in Vertebrate Blood? Biochim Biophys Acta, Bioenerg. 2009;1787:856–863. doi: 10.1016/j.bbabio.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 177.Whitfield NL, Kreimier EL, Verdial FC, Skovgaard N, Olson KR. Reappraisal of H2S/sulfide Concentration in Vertebrate Blood and Its Potential Significance in Ischemic Preconditioning and Vascular Signaling. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1930–R1937. doi: 10.1152/ajpregu.00025.2008. [DOI] [PubMed] [Google Scholar]
- 178.Levitt MD, Abdel-Rehim MS, Furne J. Free and Acid-Labile Hydrogen Sulfide Concentrations in Mouse Tissues: Anomalously High Free Hydrogen Sulfide in Aortic Tissue. Antioxid Redox Signal. 2011;15:373–378. doi: 10.1089/ars.2010.3525. [DOI] [PubMed] [Google Scholar]
- 179.Vitvitsky V, Kabil O, Banerjee R. High Turnover Rates for Hydrogen Sulfide Allow for Rapid Regulation of Its Tissue Concentrations. Antioxid Redox Signal. 2012;17:22–31. doi: 10.1089/ars.2011.4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Linden DR, Sha L, Mazzone A, Stoltz GJ, Bernard CE, Furne JK, Levitt MD, Farrugia G, Szurszewski JH. Production of the Gaseous Signal Molecule Hydrogen Sulfide in Mouse Tissues. J Neurochem. 2008;106:1577–1585. doi: 10.1111/j.1471-4159.2008.05502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kabil O, Banerjee R. Redox Biochemistry of Hydrogen Sulfide. J Biol Chem. 2010;285:21903–21907. doi: 10.1074/jbc.R110.128363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Singh S, Banerjee R. PLP-Dependent H2S Biogenesis. Biochim Biophys Acta, Proteins Proteomics. 2011;1814:1518–1527. doi: 10.1016/j.bbapap.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kabil O, Banerjee R. Enzymology of H2S Biogenesis, Decay and Signaling. Antioxid Redox Signal. 2014;20:770–782. doi: 10.1089/ars.2013.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kabil O, Motl N, Banerjee R. H2S and Its Role in Redox Signaling. Biochim Biophys Acta, Proteins Proteomics. 2014;1844:1355–1366. doi: 10.1016/j.bbapap.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Stipanuk MH, Beck PW. Characterization of the Enzymic Capacity for Cysteine Desulphhydration in Liver and Kidney of the Rat. Biochem J. 1982;206:267–277. doi: 10.1042/bj2060267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kabil O, Zhou Y, Banerjee R. Human Cystathionine β-Synthase Is a Target for Sumoylation. Biochemistry. 2006;45:13528–13536. doi: 10.1021/bi0615644. [DOI] [PubMed] [Google Scholar]
- 187.Teng H, Wu B, Zhao K, Yang G, Wu L, Wang R. Oxygen-Sensitive Mitochondrial Accumulation of Cystathionine -Synthase Mediated by Lon Protease. Proc Natl Acad Sci U S A. 2013;110:12679–12684. doi: 10.1073/pnas.1308487110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Fu M, Zhang W, Wu L, Yang G, Li H, Wang R. Hydrogen Sulfide (H2S) Metabolism in Mitochondria and Its Regulatory Role in Energy Production. Proc Natl Acad Sci U S A. 2012;109:2943–2948. doi: 10.1073/pnas.1115634109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Nagahara N, Ito T, Kitamura H, Nishino T. Tissue and Subcellular Distribution of Mercaptopyruvate Sulfurtransferase in the Rat: Confocal Laser Fluorescence and Immunoelectron Microscopic Studies Combined with Biochemical Analysis. Histochem Cell Biol. 1998;110:243–250. doi: 10.1007/s004180050286. [DOI] [PubMed] [Google Scholar]
- 190.Singh S, Padovani D, Leslie RA, Chiku T, Banerjee R. Relative Contributions of Cystathionine β-Synthase and γ-Cystathio-nase to H2S Biogenesis via Alternative Trans-Sulfuration Reactions. J Biol Chem. 2009;284:22457–22466. doi: 10.1074/jbc.M109.010868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Mishanina TV, Libiad M, Banerjee R. Biogenesis of Reactive Sulfur Species for Signaling by Hydrogen Sulfide Oxidation Pathways. Nat Chem Biol. 2015;11:457–464. doi: 10.1038/nchembio.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Braunstein AE, Goryachenkova EV, Lac ND. Reactions Catalysed by Serine Sulfhydrase from Chicken Liver. Biochim Biophys Acta. 1969;171:366–368. doi: 10.1016/0005-2744(69)90173-9. [DOI] [PubMed] [Google Scholar]
- 193.Mudd SH, Finkelstein JD, Irreverre F, Laster L. Homocystinuria: An Enzymatic Defect. Science. 1964;143:1443–1445. doi: 10.1126/science.143.3613.1443. [DOI] [PubMed] [Google Scholar]
- 194.Ereño-Orbea J, Majtan T, Oyenarte I, Kraus JP, Martínez-Cruz LA. Structural Basis of Regulation and Oligomerization of Human Cystathionine β-Synthase, the Central Enzyme of Transsulfuration. Proc Natl Acad Sci U S A. 2013;110:E3790–E3799. doi: 10.1073/pnas.1313683110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ereño-Orbea J, Majtan T, Oyenarte I, Kraus JP, Martínez-Cruz LA. Structural Insight into the Molecular Mechanism of Allosteric Activation of Human Cystathionine β-Synthase by S -Adenosylmethionine. Proc Natl Acad Sci U S A. 2014;111:E3845–E3852. doi: 10.1073/pnas.1414545111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Koutmos M, Kabil O, Smith JL, Banerjee R. Structural Basis for Substrate Activation and Regulation by Cystathionine Beta-Synthase (CBS) Domains in Cystathionine Beta-Synthase. Proc Natl Acad Sci U S A. 2010;107:20958–20963. doi: 10.1073/pnas.1011448107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kery V, Bukovska G, Kraus JP. Transsulfuration Depends on Heme in Addition to Pyridoxal 5′-phosphate. Cystathionine Beta-Synthase Is a Heme Protein. J Biol Chem. 1994;269:25283–25288. [PubMed] [Google Scholar]
- 198.Alexander FW, Sandmeier E, Mehta PK, Christen P. Evolutionary Relationships among Pyridoxal-5′-phosphate-Dependent Enzymes. Regio-Specific Alpha, Beta and Gamma Families. Eur J Biochem. 1994;219:953–960. doi: 10.1111/j.1432-1033.1994.tb18577.x. [DOI] [PubMed] [Google Scholar]
- 199.Meier M. Structure of Human Cystathionine Beta-Synthase: A Unique Pyridoxal 5′-phosphate-Dependent Heme Protein. EMBO J. 2001;20:3910–3916. doi: 10.1093/emboj/20.15.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Taoka S, Lepore BW, Kabil O, Ojha S, Ringe D, Banerjee R. Human Cystathionine Beta-Synthase Is a Heme Sensor Protein. Evidence That the Redox Sensor Is Heme and Not the Vicinal Cysteines in the CXXC Motif Seen in the Crystal Structure of the Truncated Enzyme. Biochemistry. 2002;41:10454–10461. doi: 10.1021/bi026052d. [DOI] [PubMed] [Google Scholar]
- 201.Bar-Or D, Rael LT, Thomas GW, Kraus JP. Inhibitory Effect of Copper on Cystathionine Beta-Synthase Activity: Protective Effect of an Analog of the Human Albumin N-Terminus. Protein Pept Lett. 2005;12:271–273. doi: 10.2174/0929866053587048. [DOI] [PubMed] [Google Scholar]
- 202.Bateman A. The Structure of a Domain Common to Archaebacteria and the Homocystinuria Disease Protein. Trends Biochem Sci. 1997;22:12–13. doi: 10.1016/s0968-0004(96)30046-7. [DOI] [PubMed] [Google Scholar]
- 203.Scott JW, Hawley SA, Green KA, Anis M, Stewart G, Scullion GA, Norman DG, Hardie DG. CBS Domains Form Energy-Sensing Modules Whose Binding of Adenosine Ligands Is Disrupted by Disease Mutations. J Clin Invest. 2004;113:274–284. doi: 10.1172/JCI19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Finkelstein JD, Kyle WE, Martin JJ, Pick A-M. Activation of Cystathionine Synthase by Adenosylmethionine and Adenosylethionine. Biochem Biophys Res Commun. 1975;66:81–87. doi: 10.1016/s0006-291x(75)80297-x. [DOI] [PubMed] [Google Scholar]
- 205.Kabil H, Kabil O, Banerjee R, Harshman LG, Pletcher SD. Increased Transsulfuration Mediates Longevity and Dietary Restriction in Drosophila. Proc Natl Acad Sci U S A. 2011;108:16831–16836. doi: 10.1073/pnas.1102008108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kery V, Poneleit L, Kraus JP. Trypsin Cleavage of Human Cystathionine β-Synthase into an Evolutionarily Conserved Active Core: Structural and Functional Consequences. Arch Biochem Biophys. 1998;355:222–232. doi: 10.1006/abbi.1998.0723. [DOI] [PubMed] [Google Scholar]
- 207.Taoka S, Ohja S, Shan X, Kruger WD, Banerjee R. Evidence for Heme-Mediated Redox Regulation of Human Cystathionine Beta-Synthase Activity. J Biol Chem. 1998;273:25179–25184. doi: 10.1074/jbc.273.39.25179. [DOI] [PubMed] [Google Scholar]
- 208.Kabil Ö, Toaka S, LoBrutto R, Shoemaker R, Banerjee R. Pyridoxal Phosphate Binding Sites Are Similar in Human Heme-Dependent and Yeast Heme-Independent Cystathionine β-Synthases. J Biol Chem. 2001;276:19350–19355. doi: 10.1074/jbc.M100029200. [DOI] [PubMed] [Google Scholar]
- 209.Green EL, Taoka S, Banerjee R, Loehr TM. Resonance Raman Characterization of the Heme Cofactor in Cystathionine Beta-Synthase. Identification of the Fe-S(Cys) Vibration in the Six-Coordinate Low-Spin Heme. Biochemistry. 2001;40:459–463. doi: 10.1021/bi0010874. [DOI] [PubMed] [Google Scholar]
- 210.Taoka S, Green EL, Loehr TM, Banerjee R. Mercuric Chloride-Induced Spin or Ligation State Changes in Ferric or Ferrous Human Cystathionine Beta-Synthase Inhibit Enzyme Activity. J Inorg Biochem. 2001;87:253–259. doi: 10.1016/s0162-0134(01)00336-1. [DOI] [PubMed] [Google Scholar]
- 211.Jhee KH, McPhie P, Miles EW. Yeast Cystathionine Beta-Synthase Is a Pyridoxal Phosphate Enzyme But, Unlike the Human Enzyme, Is Not a Heme Protein. J Biol Chem. 2000;275:11541–11544. doi: 10.1074/jbc.c000056200. [DOI] [PubMed] [Google Scholar]
- 212.Evande R, Ojha S, Banerjee R. Visualization of PLP-Bound Intermediates in Hemeless Variants of Human Cystathionine β-Synthase: Evidence That Lysine 119 Is a General Base. Arch Biochem Biophys. 2004;427:188–196. doi: 10.1016/j.abb.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 213.Ojha S, Hwang J, Kabil O, Penner-Hahn JE, Banerjee R. Characterization of the Heme in Human Cystathionine Beta-Synthase by X-Ray Absorption and Electron Paramagnetic Resonance Spectros-copies. Biochemistry. 2000;39:10542–10547. doi: 10.1021/bi000831h. [DOI] [PubMed] [Google Scholar]
- 214.Singh S, Madzelan P, Banerjee R. Properties of an Unusual Heme Cofactor in PLP-Dependent Cystathionine β-Synthase. Nat Prod Rep. 2007;24:631–639. doi: 10.1039/b604182p. [DOI] [PubMed] [Google Scholar]
- 215.Singh S, Madzelan P, Stasser J, Weeks CL, Becker D, Spiro TG, Penner-Hahn J, Banerjee R. Modulation of the Heme Electronic Structure and Cystathionine β-Synthase Activity by Second Coordination Sphere Ligands: The Role of Heme Ligand Switching in Redox Regulation. J Inorg Biochem. 2009;103:689–697. doi: 10.1016/j.jinorgbio.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Carballal S, Madzelan P, Zinola CF, Graña M, Radi R, Banerjee R, Alvarez B. Dioxygen Reactivity and Heme Redox Potential of Truncated Human Cystathionine β-Synthase. Biochemistry. 2008;47:3194–3201. doi: 10.1021/bi700912k. [DOI] [PubMed] [Google Scholar]
- 217.Taoka S, West M, Banerjee R. Characterization of the Heme and Pyridoxal Phosphate Cofactors of Human Cystathionine β-Synthase Reveals Nonequivalent Active Sites. Biochemistry. 1999;38:2738–2744. doi: 10.1021/bi9826052. [DOI] [PubMed] [Google Scholar]
- 218.Taoka S, Banerjee R. Characterization of NO Binding to Human Cystathionine Beta-Synthase: Possible Implications of the Effects of CO and NO Binding to the Human Enzyme. J Inorg Biochem. 2001;87:245–251. doi: 10.1016/s0162-0134(01)00335-x. [DOI] [PubMed] [Google Scholar]
- 219.Vadon-Le Goff S, Delaforge M, Boucher J-L, Janosik M, Kraus JP, Mansuy D. Coordination Chemistry of the Heme in Cystathionine β-Synthase: Formation of Iron(II)–Isonitrile Complexes. Biochem Biophys Res Commun. 2001;283:487–492. doi: 10.1006/bbrc.2001.4807. [DOI] [PubMed] [Google Scholar]
- 220.Gherasim C, Yadav PK, Kabil O, Niu W-N, Banerjee R. Nitrite Reductase Activity and Inhibition of H2S Biogenesis by Human Cystathionine SS-Synthas. PLoS One. 2014;9:e85544. doi: 10.1371/journal.pone.0085544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Kabil O, Weeks CL, Carballal S, Gherasim C, Alvarez B, Spiro TG, Banerjee R. Reversible Heme-Dependent Regulation of Human Cystathionine β-Synthase by a Flavoprotein Oxidoreductase. Biochemistry. 2011;50:8261–8263. doi: 10.1021/bi201270q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Carballal S, Cuevasanta E, Yadav PK, Gherasim C, Ballou DP, Alvarez B, Banerjee R. Kinetics of Nitrite Reduction and Peroxynitrite Formation by Ferrous Heme in Human Cystathionine β-Synthase. J Biol Chem. 2016;291:8004–8013. doi: 10.1074/jbc.M116.718734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Vicente JB, Colaço HG, Sarti P, Leandro P, Giuffrè A. S-Adenosyl-L-Methionine Modulates CO and NO• Binding to the Human H2S-Generating Enzyme Cystathionine β-Synthase. J Biol Chem. 2016;291:572–581. doi: 10.1074/jbc.M115.681221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Yadav PK, Banerjee R. Detection of Reaction Intermediates during Human Cystathionine β-Synthase-Monitored Turnover and H2S Production. J Biol Chem. 2012;287:43464–43471. doi: 10.1074/jbc.M112.414722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Singh S, Ballou DP, Banerjee R. Pre-Steady-State Kinetic Analysis of Enzyme-Monitored Turnover during Cystathionine β-Synthase-Catalyzed H2S Generation. Biochemistry. 2011;50:419–425. doi: 10.1021/bi1010893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Yadav PK, Martinov M, Vitvitsky V, Seravalli J, Wedmann R, Filipovic MR, Banerjee R. Biosynthesis and Reactivity of Cysteine Persulfides in Signaling. J Am Chem Soc. 2016;138:289–299. doi: 10.1021/jacs.5b10494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Vicente JB, Colaço HG, Mendes MIS, Sarti P, Leandro P, Giuffrè A. NO• Binds Human Cystathionine β-Synthase Quickly and Tightly. J Biol Chem. 2014;289:8579–8587. doi: 10.1074/jbc.M113.507533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Puranik M, Nielsen SB, Youn H, Hvitved AN, Bourassa JL, Case MA, Tengroth C, Balakrishnan G, Thorsteinsson MV, Groves JT, et al. Dynamics of Carbon Monoxide Binding to CooA. J Biol Chem. 2004;279:21096–21108. doi: 10.1074/jbc.M400613200. [DOI] [PubMed] [Google Scholar]
- 229.Ojha S, Wu J, LoBrutto R, Banerjee R. Effects of Heme Ligand Mutations Including a Pathogenic Variant, H65R, on the Properties of Human Cystathionine Beta-Synthase. Biochemistry. 2002;41:4649–4654. doi: 10.1021/bi011827o. [DOI] [PubMed] [Google Scholar]
- 230.Weeks CL, Singh S, Madzelan P, Banerjee R, Spiro TG. Heme Regulation of Human Cystathionine β-Synthase Activity: Insights from Fluorescence and Raman Spectroscopy. J Am Chem Soc. 2009;131:12809–12816. doi: 10.1021/ja904468w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Pazicni S, Cherney MM, Lukat-Rodgers GS, Oliveriusová J, Rodgers KR, Kraus JP, Burstyn JN. The Heme of Cystathionine Beta-Synthase Likely Undergoes a Thermally Induced Redox-Mediated Ligand Switch. Biochemistry. 2005;44:16785–16795. doi: 10.1021/bi051305z. [DOI] [PubMed] [Google Scholar]
- 232.Smith AT, Su Y, Stevens DJ, Majtan T, Kraus JP, Burstyn JN. Effect of the Disease-Causing R266K Mutation on the Heme and PLP Environments of Human Cystathionine β-Synthase. Biochemistry. 2012;51:6360–6370. doi: 10.1021/bi300421z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Yadav PK, Xie P, Banerjee R. Allosteric Communication between the Pyridoxal 5′-Phosphate (PLP) and Heme Sites in the H2S Generator Human Cystathionine β-Synthase. J Biol Chem. 2012;287:37611–37620. doi: 10.1074/jbc.M112.414706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Evande R, Blom H, Boers GHJ, Banerjee R. Alleviation of Intrasteric Inhibition by the Pathogenic Activation Domain Mutation, D444N, in Human Cystathionine Beta-Synthase. Biochemistry. 2002;41:11832–11837. doi: 10.1021/bi026248d. [DOI] [PubMed] [Google Scholar]
- 235.Janosík M, Kery V, Gaustadnes M, Maclean KN, Kraus JP. Regulation of Human Cystathionine Beta-Synthase by S-Adenosyl-L-Methionine: Evidence for Two Catalytically Active Conformations Involving an Autoinhibitory Domain in the C-Terminal Region. Biochemistry. 2001;40:10625–10633. doi: 10.1021/bi010711p. [DOI] [PubMed] [Google Scholar]
- 236.Mendes MIS, Santos AS, Smith DEC, Lino PR, Colaço HG, de Almeida IT, Vicente JB, Salomons GS, Rivera I, Blom HJ, et al. Insights into the Regulatory Domain of Cystathionine Beta-Synthase: Characterization of Six Variant Proteins. Hum Mutat. 2014;35:1195–1202. doi: 10.1002/humu.22616. [DOI] [PubMed] [Google Scholar]
- 237.Shan X, Kruger WD. Correction of Disease-Causing CBS Mutations in Yeast. Nat Genet. 1998;19:91–93. doi: 10.1038/ng0598-91. [DOI] [PubMed] [Google Scholar]
- 238.Shan X, Dunbrack RL, Christopher SA, Kruger WD. Mutations in the Regulatory Domain of Cystathionine Beta Synthase Can Functionally Suppress Patient-Derived Mutations in Cis. Hum Mol Genet. 2001;10:635–643. doi: 10.1093/hmg/10.6.635. [DOI] [PubMed] [Google Scholar]
- 239.Agrawal N, Banerjee R. Human Polycomb 2 Protein Is a SUMO E3 Ligase and Alleviates Substrate-Induced Inhibition of Cystathionine β-Synthase Sumoylation. PLoS One. 2008;3:e4032. doi: 10.1371/journal.pone.0004032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Kurepa J, Walker JM, Smalle J, Gosink MM, Davis SJ, Durham TL, Sung D-Y, Vierstra RD. The Small Ubiquitin-like Modifier (SUMO) Protein Modification System in Arabidopsis. J Biol Chem. 2003;278:6862–6872. doi: 10.1074/jbc.M209694200. [DOI] [PubMed] [Google Scholar]
- 241.Hong Y, Rogers R, Matunis MJ, Mayhew CN, Goodson M, Park-Sarge O-K, Sarge KD. Regulation of Heat Shock Transcription Factor 1 by Stress-Induced SUMO-1 Modification. J Biol Chem. 2001;276:40263–40267. doi: 10.1074/jbc.M104714200. [DOI] [PubMed] [Google Scholar]
- 242.Zhou W, Ryan JJ, Zhou H. Global Analyses of Sumoylated Proteins in Saccharomyces Cerevisiae. J Biol Chem. 2004;279:32262–32268. doi: 10.1074/jbc.M404173200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Niu W-N, Yadav PK, Adamec J, Banerjee R. S-Glutathionylation Enhances Human Cystathionine β-Synthase Activity under Oxidative Stress Conditions. Antioxid Redox Signal. 2015;22:350–361. doi: 10.1089/ars.2014.5891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Vitvitsky V, Thomas M, Ghorpade A, Gendelman HE, Banerjee R. A Functional Transsulfuration Pathway in the Brain Links to Glutathione Homeostasis. J Biol Chem. 2006;281:35785–35793. doi: 10.1074/jbc.M602799200. [DOI] [PubMed] [Google Scholar]
- 245.Mosharov E, Cranford MR, Banerjee R. The Quantitatively Important Relationship between Homocysteine Metabolism and Glutathione Synthesis by the Transsulfuration Pathway and Its Regulation by Redox Changes. Biochemistry. 2000;39:13005–13011. doi: 10.1021/bi001088w. [DOI] [PubMed] [Google Scholar]
- 246.d’Emmanuele di Villa Bianca R, Mitidieri E, Esposito D, Donnarumm E, Russo A, Fusco F, Ianaro A, Mirone V, Cirino G, Russo G, et al. Human Cystathionine-β-Synthase Phosphor-ylation on Serine227 Modulates Hydrogen Sulfide Production in Human Urothelium. PLoS One. 2015;10:e0136859. doi: 10.1371/journal.pone.0136859. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 247.d’Emmanuele di Villa Bianca R, Mitidieri E, Fusco F, Russo A, Pagliara V, Tramontano T, Donnarumma E, Mirone V, Cirino G, Russo G, et al. Urothelium Muscarinic Activation Phosphorylates CBSSer227 via cGMP/PKG Pathway Causing Human Bladder Relaxation through H2S Production. Sci Rep. 2016;6:31491. doi: 10.1038/srep31491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Zhu W, Lin A, Banerjee R. Kinetic Properties of Polymorphic Variants and Pathogenic Mutants in Human Cystathio-nine γ-Lyase. Biochemistry. 2008;47:6226–6232. doi: 10.1021/bi800351a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Chiku T, Padovani D, Zhu W, Singh S, Vitvitsky V, Banerjee R. H2S Biogenesis by Human Cystathionine Gamma-Lyase Leads to the Novel Sulfur Metabolites Lanthionine and Homolan-thionine and Is Responsive to the Grade of Hyperhomocysteinemia. J Biol Chem. 2009;284:11601–11612. doi: 10.1074/jbc.M808026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Scott CR, Dassell SW, Clark SH, Chiang-Teng C, Swedberg KR. Cystathioninemia: A Benign Genetic Condition. J Pediatr. 1970;76:571–577. doi: 10.1016/s0022-3476(70)80407-3. [DOI] [PubMed] [Google Scholar]
- 251.Kraus JP, Hašek J, Kožich V, Collard R, Venezia S, Janošíková B, Wang J, Stabler SP, Allen RH, Jakobs C, et al. Cystathionine γ-Lyase: Clinical, Metabolic, Genetic, and Structural Studies. Mol Genet Metab. 2009;97:250–259. doi: 10.1016/j.ymgme.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Wang J, Hegele RA. Genomic Basis of Cystathioninuria (MIM 219500) Revealed by Multiple Mutations in Cystathionine Gamma-Lyase (CTH) Hum Genet. 2003;112:404–408. doi: 10.1007/s00439-003-0906-8. [DOI] [PubMed] [Google Scholar]
- 253.Perry TL, Hansen S, MacDougall L. Homolanthionine Excretion in Homocystinuria. Science. 1966;152:1750–1752. doi: 10.1126/science.152.3730.1750. [DOI] [PubMed] [Google Scholar]
- 254.Kabil O, Vitvitsky V, Xie P, Banerjee R. The Quantitative Significance of the Transsulfuration Enzymes for H2S Production in Murine Tissues. Antioxid Redox Signal. 2011;15:363–372. doi: 10.1089/ars.2010.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Sun Q, Collins R, Huang S, Holmberg-Schiavone L, Anand GS, Tan C-H, Van-den-Berg S, Deng L-W, Moore PK, Karlberg T, et al. Structural Basis for the Inhibition Mechanism of Human Cystathionine γ-Lyase, an Enzyme Responsible for the Production of H2S. J Biol Chem. 2009;284:3076–3085. doi: 10.1074/jbc.M805459200. [DOI] [PubMed] [Google Scholar]
- 256.Messerschmidt A, Worbs M, Steegborn C, Wahl MC, Huber R, Laber B, Clausen T. Determinants of Enzymatic Specificity in the Cys-Met-Metabolism PLP-Dependent Enzyme Family: Crystal Structure of Cystathionine γ-Lyase from Yeast and Intrafamiliar Structure Comparison. Biol Chem. 2003;384:373–386. doi: 10.1515/BC.2003.043. [DOI] [PubMed] [Google Scholar]
- 257.Huang S, Chua JH, Yew WS, Sivaraman J, Moore PK, Tan C-H, Deng L-W. Site-Directed Mutagenesis on Human Cystathionine-γ-Lyase Reveals Insights into the Modulation of H2S Production. J Mol Biol. 2010;396:708–718. doi: 10.1016/j.jmb.2009.11.058. [DOI] [PubMed] [Google Scholar]
- 258.Mihara H, Fujii T, Kato S-i, Kurihara T, Hata Y, Esaki N. Structure of External Aldimine of Escherichia Coli CsdB, an IscS/Nifs Homolog: Implications for Its Specificity toward Selenocysteine. J Biochem. 2002;131:679–685. doi: 10.1093/oxfordjournals.jbchem.a003151. [DOI] [PubMed] [Google Scholar]
- 259.Sturman JA, Beratis NG, Guarini L, Gaull GE. Transsulfuration by Human Long Term Lymphoid Lines. Normal and Cystathionase-Deficient Cells. J Biol Chem. 1980;255:4763–4765. [PubMed] [Google Scholar]
- 260.Yuan G, Vasavda C, Peng YJ, Makarenko VV, Raghuraman G, Nanduri J, Gadalla MM, Semenza GL, Kumar GK, Snyder SH, et al. Protein Kinase G-Regulated Production of H2S Governs Oxygen Sensing. Sci Signaling. 2015;8:ra37–ra37. doi: 10.1126/scisignal.2005846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Mikami Y, Shibuya N, Ogasawara Y, Kimura H. Hydrogen Sulfide Is Produced by Cystathionine γ-Lyase at the Steady-State Low Intracellular Ca2+ Concentrations. Biochem Biophys Res Commun. 2013;431:131–135. doi: 10.1016/j.bbrc.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 262.Kabil O, Yadav V, Banerjee R. Heme-Dependent Metabolite Switching Regulates H2S Synthesis in Response to Endoplasmic Reticulum (ER) Stress. J Biol Chem. 2016;291:16418–16423. doi: 10.1074/jbc.C116.742213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Dickhout JG, Carlisle RE, Jerome DE, Mohammed-Ali Z, Jiang H, Yang G, Mani S, Garg SK, Banerjee R, Kaufman RJ, et al. Integrated Stress Response Modulates Cellular Redox State via Induction of Cystathionine γ-Lyase. J Biol Chem. 2012;287:7603–7614. doi: 10.1074/jbc.M111.304576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Liu X-M, Peyton KJ, Ensenat D, Wang H, Schafer AI, Alam J, Durante W. Endoplasmic Reticulum Stress Stimulates Heme Oxygenase-1 Gene Expression in Vascular Smooth Muscle. J Biol Chem. 2005;280:872–877. doi: 10.1074/jbc.M410413200. [DOI] [PubMed] [Google Scholar]
- 265.Cao SS, Kaufman RJ. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxid Redox Signal. 2014;21:396–413. doi: 10.1089/ars.2014.5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Modis K, Panopoulos P, Asimakopoulou A, Gero D, Sharina I, Martin E, et al. Hydrogen Sulfide and Nitric Oxide Are Mutually Dependent in the Regulation of Angiogenesis and Endothelium-Dependent Vasorelaxation. Proc Natl Acad Sci U S A. 2012;109:9161–9166. doi: 10.1073/pnas.1202916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Eberhardt M, Dux M, Namer B, Miljkovic J, Cordasic N, Will C, Kichko TI, de la Roche J, Fischer M, Suárez SA, et al. H2S and NO Cooperatively Regulate Vascular Tone by Activating a Neuroendocrine HNO–TRPA1–CGRP Signalling Pathway. Nat Commun. 2014;5:4381. doi: 10.1038/ncomms5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.King AL, Polhemus DJ, Bhushan S, Otsuka H, Kondo K, Nicholson CK, Bradley JM, Islam KN, Calvert JW, Tao Y-X, et al. Hydrogen Sulfide Cytoprotective Signaling Is Endothelial Nitric Oxide Synthase-Nitric Oxide Dependent. Proc Natl Acad Sci U S A. 2014;111:3182–3187. doi: 10.1073/pnas.1321871111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K, Kimura H. 3-Mercaptopyruvate Sulfurtransferase Produces Hydrogen Sulfide and Bound Sulfane Sulfur in the Brain. Antioxid Redox Signal. 2009;11:703–714. doi: 10.1089/ars.2008.2253. [DOI] [PubMed] [Google Scholar]
- 270.Mikami Y, Shibuya N, Kimura Y, Nagahara N, Ogasawara Y, Kimura H. Thioredoxin and Dihydrolipoic Acid Are Required for 3-Mercaptopyruvate Sulfurtransferase to Produce Hydrogen Sulfide. Biochem J. 2011;439:479–485. doi: 10.1042/BJ20110841. [DOI] [PubMed] [Google Scholar]
- 271.Yadav PK, Yamada K, Chiku T, Koutmos M, Banerjee R. Structure and Kinetic Analysis of H2S Production by Human Mercaptopyruvate Sulfurtransferase. J Biol Chem. 2013;288:20002–20013. doi: 10.1074/jbc.M113.466177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Nagahara N, Ito T, Minami M. Mercaptopyruvate Sulfurtransferase as a Defense against Cyanide Toxication: Molecular Properties and Mode of Detoxification. Histol Histopathol. 1999;14:1277–1286. doi: 10.14670/HH-14.1277. [DOI] [PubMed] [Google Scholar]
- 273.Williams RAM, Kelly SM, Mottram JC, Coombs GH. 3-Mercaptopyruvate Sulfurtransferase of Leishmania Contains an Unusual C-Terminal Extension and Is Involved in Thioredoxin and Antioxidant Metabolism. J Biol Chem. 2003;278:1480–1486. doi: 10.1074/jbc.M209395200. [DOI] [PubMed] [Google Scholar]
- 274.Westrop GD, Georg I, Coombs GH. The Mercaptopyruvate Sulfurtransferase of Trichomonas Vaginalis Links Cysteine Catabolism to the Production of Thioredoxin Persulfide. J Biol Chem. 2009;284:33485–33494. doi: 10.1074/jbc.M109.054320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Nagahara N, Katayama A. Post-Translational Regulation of Mercaptopyruvate Sulfurtransferase via a Low Redox Potential Cysteine-Sulfenate in the Maintenance of Redox Homeostasis. J Biol Chem. 2005;280:34569–34576. doi: 10.1074/jbc.M505643200. [DOI] [PubMed] [Google Scholar]
- 276.Huang J, Niknahad H, Khan S, O’Brien PJ. Hepatocyte-Catalysed Detoxification of Cyanide by L- and D-Cysteine. Biochem Pharmacol. 1998;55:1983–1990. doi: 10.1016/s0006-2952(98)00072-0. [DOI] [PubMed] [Google Scholar]
- 277.Shibuya N, Koike S, Tanaka M, Ishigami-Yuasa M, Kimura Y, Ogasawara Y, Fukui K, Nagahara N, Kimura H. A Novel Pathway for the Production of Hydrogen Sulfide from D-Cysteine in Mammalian Cells. Nat Commun. 2013;4:1366. doi: 10.1038/ncomms2371. [DOI] [PubMed] [Google Scholar]
- 278.Nagahara N, Nishino T. Role of Amino Acid Residues in the Active Site of Rat Liver Mercaptopyruvate Sulfurtransferase. CDNA Cloning, Overexpression, and Site-Directed Mutagenesis. J Biol Chem. 1996;271:27395–27401. doi: 10.1074/jbc.271.44.27395. [DOI] [PubMed] [Google Scholar]
- 279.Alphey MS, Williams RAM, Mottram JC, Coombs GH, Hunter WN. The Crystal Structure of Leishmania Major 3-Mercaptopyruvate Sulfurtransferase. A Three-Domain Architecture with a Serine Protease-like Triad at the Active Site. J Biol Chem. 2003;278:48219–48227. doi: 10.1074/jbc.M307187200. [DOI] [PubMed] [Google Scholar]
- 280.Huang G-T, Yu J-SK. Enzyme Catalysis That Paves the Way for S-Sulfhydration via Sulfur Atom Transfer. J Phys Chem B. 2016;120:4608–4615. doi: 10.1021/acs.jpcb.6b03387. [DOI] [PubMed] [Google Scholar]
- 281.Nagahara N. Regulation of Mercaptopyruvate Sulfurtransfer-ase Activity Via Intrasubunit and Intersubunit Redox-Sensing Switches. Antioxid Redox Signal. 2013;19:1792–1802. doi: 10.1089/ars.2012.5031. [DOI] [PubMed] [Google Scholar]
- 282.Mikami Y, Shibuya N, Kimura Y, Nagahara N, Yamada M, Kimura H. Hydrogen Sulfide Protects the Retina from Light-Induced Degeneration by the Modulation of Ca2+ Influx. J Biol Chem. 2011;286:39379–39386. doi: 10.1074/jbc.M111.298208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Abeles RH, Walsh CT, Acetylenic Enzyme Inactivators Inactivation of Gamma-Cystathionase, in Vitro and in Vivo, by Propargylglycine. J Am Chem Soc. 1973;95:6124–6125. doi: 10.1021/ja00799a053. [DOI] [PubMed] [Google Scholar]
- 284.Shinozijka S, Tanase S, Morino Y. Metabolic Consequences of Affinity Labeling of Cystathionase and Alanine Aminotransferase by L-Propargylglycine in Vivo. Eur J Biochem. 1982;124:377–382. doi: 10.1111/j.1432-1033.1982.tb06603.x. [DOI] [PubMed] [Google Scholar]
- 285.Asimakopoulou A, Panopoulos P, Chasapis CT, Coletta C, Zhou Z, Cirino G, Giannis A, Szabo C, Spyroulias GA, Papapetropoulos A. Selectivity of Commonly Used Pharmacological Inhibitors for Cystathionine β Synthase (CBS) and Cystathionine γ Lyase (CSE) Br J Pharmacol. 2013;169:922–932. doi: 10.1111/bph.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Alston TA, Porter DJT, Mela L, Bright HJ. Inactivation of Alanine Aminotransferase by the Neurotoxin β-Cyano-L-Alanine. Biochem Biophys Res Commun. 1980;92:299–304. doi: 10.1016/0006-291x(80)91552-1. [DOI] [PubMed] [Google Scholar]
- 287.Pfeffer M, Ressler C. β-Cyanoalanine, an Inhibitor of Rat Liver Cystathionase. Biochem Pharmacol. 1967;16:2299–2308. doi: 10.1016/0006-2952(67)90217-1. [DOI] [PubMed] [Google Scholar]
- 288.Ressler C, Nelson J, Pfeffer M. Metabolism of Beta-Cyanoalanine. Biochem Pharmacol. 1967;16:2309–2319. doi: 10.1016/0006-2952(67)90218-3. [DOI] [PubMed] [Google Scholar]
- 289.Steegborn C, Clausen T, Sondermann P, Jacob U, Worbs M, Marinkovic S, Huber R, Wahl MC. Kinetics and Inhibition of Recombinant Human Cystathionine Gamma-Lyase. Toward the Rational Control of Transsulfuration. J Biol Chem. 1999;274:12675–12684. doi: 10.1074/jbc.274.18.12675. [DOI] [PubMed] [Google Scholar]
- 290.Capitani G, McCarthy DL, Gut H, Grütter MG, Kirsch JF. Apple 1-Aminocyclopropane-1-Carboxylate Synthase in Complex with the Inhibitor L-Aminoethoxyvinylglycine. Evidence for a Ketimine Intermediate. J Biol Chem. 2002;277:49735–49742. doi: 10.1074/jbc.M208427200. [DOI] [PubMed] [Google Scholar]
- 291.Clausen T, Huber R, Messerschmidt A, Pohlenz HD, Laber B. Slow-Binding Inhibition of Escherichia Coli Cystathionine Beta-Lyase by L-Aminoethoxyvinylglycine: A Kinetic and X-Ray Study. Biochemistry. 1997;36:12633–12643. doi: 10.1021/bi970630m. [DOI] [PubMed] [Google Scholar]
- 292.Druzhyna N, Szczesny B, Olah G, Mo´dis K, Asimakopoulou A, Pavlidou A, Szoleczky P, Gerö D, Yanagi K, Törö G, et al. Screening of a Composite Library of Clinically Used Drugs and Well-Characterized Pharmacological Compounds for Cystathionine β-Synthase Inhibition Identifies Benserazide as a Drug Potentially Suitable for Repurposing for the Experimental Therapy of Colon. Pharmacol Res. 2016;113:18–37. doi: 10.1016/j.phrs.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Thorson MK, Majtan T, Kraus JP, Barrios AM. Identification of Cystathionine β-Synthase Inhibitors Using a Hydrogen Sulfide Selective Probe. Angew Chem, Int Ed. 2013;52:4641–4644. doi: 10.1002/anie.201300841. [DOI] [PubMed] [Google Scholar]
- 294.Niu W, Wu P, Chen F, Wang J, Shang X, Xu C. Discovery of Selective Cystathionine β-Synthase Inhibitors by High-Throughput Screening with a Fluorescent Thiol Probe. MedChem-Comm. 2017;8:198–201. doi: 10.1039/c6md00493h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.McCune CD, Chan SJ, Beio ML, Shen W, Chung WJ, Szczesniak LM, Chai C, Koh SQ, Wong PT-H, Berkowitz DB. Zipped Synthesis” by Cross-Metathesis Provides a Cystathionine β-Synthase Inhibitor That Attenuates Cellular H2S Levels and Reduces Neuronal Infarction in a Rat Ischemic Stroke Model. ACS Cent Sci. 2016;2:242–252. doi: 10.1021/acscentsci.6b00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Bouillaud F, Blachier F. Mitochondria and Sulfide: A Very Old Story of Poisoning, Feeding, and Signaling? Antioxid Redox Signal. 2011;15:379–391. doi: 10.1089/ars.2010.3678. [DOI] [PubMed] [Google Scholar]
- 297.Hildebrandt TM, Grieshaber MK. Three Enzymatic Activities Catalyze the Oxidation of Sulfide to Thiosulfate in Mammalian and Invertebrate Mitochondria. FEBS J. 2008;275:3352–3361. doi: 10.1111/j.1742-4658.2008.06482.x. [DOI] [PubMed] [Google Scholar]
- 298.Bartholomew TC, Powell GM, Dodgson KS, Curtis CG. Oxidation of Sodium Sulphide by Rat Liver, Lungs and Kidney. Biochem Pharmacol. 1980;29:2431–2437. doi: 10.1016/0006-2952(80)90346-9. [DOI] [PubMed] [Google Scholar]
- 299.Curtis CG, Bartholomew TC, Rose FA, Dodgson KS. Detoxication of Sodium 35 S-Sulphide in the Rat. Biochem Pharmacol. 1972;21:2313–2321. doi: 10.1016/0006-2952(72)90382-6. [DOI] [PubMed] [Google Scholar]
- 300.Furne J, Springfield J, Koenig T, DeMaster E, Levitt MD. Oxidation of Hydrogen Sulfide and Methanethiol to Thiosulfate by Rat Tissues: A Specialized Function of the Colonic Mucosa. Biochem Pharmacol. 2001;62:255–259. doi: 10.1016/s0006-2952(01)00657-8. [DOI] [PubMed] [Google Scholar]
- 301.Vitvitsky V, Yadav PK, Kurthen A, Banerjee R. Sulfide Oxidation by a Noncanonical Pathway in Red Blood Cells Generates Thiosulfate and Polysulfides. J Biol Chem. 2015;290:8310–8320. doi: 10.1074/jbc.M115.639831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Bostelaar T, Vitvitsky V, Kumutima J, Lewis BE, Yadav PK, Brunold TC, Filipovic M, Lehnert N, Stemmler TL, Banerjee R. Hydrogen Sulfide Oxidation by Myoglobin. J Am Chem Soc. 2016;138:8476–8488. doi: 10.1021/jacs.6b03456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Powell MA, Somero GN. Hydrogen Sulfide Oxidation Is Coupled to Oxidative Phosphorylation in Mitochondria of Solemya Reidi. Science. 1986;233:563–566. doi: 10.1126/science.233.4763.563. [DOI] [PubMed] [Google Scholar]
- 304.Goubern M, Andriamihaja M, Nubel T, Blachier F, Bouillaud F. Sulfide, the First Inorganic Substrate for Human Cells. FASEB J. 2007;21:1699–1706. doi: 10.1096/fj.06-7407com. [DOI] [PubMed] [Google Scholar]
- 305.Argyrou A, Blanchard JS. Flavoprotein Disulfide Reductases: Advances in Chemistry and Function. Prog Nucleic Acid Res Mol Biol. 2004;78:89–142. doi: 10.1016/S0079-6603(04)78003-4. [DOI] [PubMed] [Google Scholar]
- 306.Mishanina TV, Yadav PK, Ballou DP, Banerjee R. Transient Kinetic Analysis of Hydrogen Sulfide Oxidation Catalyzed by Human Sulfide Quinone Oxidoreductase. J Biol Chem. 2015;290:25072–25080. doi: 10.1074/jbc.M115.682369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Brito JA, Sousa FL, Stelter M, Bandeiras TM, Vonrhein C, Teixeira M, Pereira MM, Archer M. Structural and Functional Insights into Sulfide:quinone Oxidoreductase. Biochemistry. 2009;48:5613–5622. doi: 10.1021/bi9003827. [DOI] [PubMed] [Google Scholar]
- 308.Marcia M, Ermler U, Peng G, Michel H. The Structure of Aquifex Aeolicus Sulfide:quinone Oxidoreductase, a Basis to Understand Sulfide Detoxification and Respiration. Proc Natl Acad Sci U S A. 2009;106:9625–9630. doi: 10.1073/pnas.0904165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Cherney MM, Zhang Y, Solomonson M, Weiner JH, James MNG. Crystal Structure of Sulfide:Quinone Oxidoreductase from Acidithiobacillus Ferrooxidans: Insights into Sulfidotrophic Respiration and Detoxification. J Mol Biol. 2010;398:292–305. doi: 10.1016/j.jmb.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 310.Jackson MR, Melideo SL, Jorns MS. Human Sulfide:Quinone Oxidoreductase Catalyzes the First Step in Hydrogen Sulfide Metabolism and Produces a Sulfane Sulfur Metabolite. Biochemistry. 2012;51:6804–6815. doi: 10.1021/bi300778t. [DOI] [PubMed] [Google Scholar]
- 311.Libiad M, Yadav PK, Vitvitsky V, Martinov M, Banerjee R. Organization of the Human Mitochondrial Hydrogen Sulfide Oxidation Pathway. J Biol Chem. 2014;289:30901–30910. doi: 10.1074/jbc.M114.602664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Augustyn KDC, Jackson MR, Jorns MS. Use of Tissue Metabolite Analysis and Enzyme Kinetics To Discriminate between Alternate Pathways for Hydrogen Sulfide Metabolism. Biochemistry. 2017;56:986–996. doi: 10.1021/acs.biochem.6b01093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Melideo SL, Jackson MR, Jorns MS. Biosynthesis of a Central Intermediate in Hydrogen Sulfide Metabolism by a Novel Human Sulfurtransferase and Its Yeast Ortholog. Biochemistry. 2014;53:4739–4753. doi: 10.1021/bi500650h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Pettinati I, Brem J, Lee SY, McHugh PJ, Schofield CJ. The Chemical Biology of Human Metallo-β-Lactamase Fold Proteins. Trends Biochem Sci. 2016;41:338–355. doi: 10.1016/j.tibs.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Tiranti V, Viscomi C, Hildebrandt T, Di Meo I, Mineri R, Tiveron C, D Levitt M, Prelle A, Fagiolari G, Rimoldi M, et al. Loss of ETHE1, a Mitochondrial Dioxygenase, Causes Fatal Sulfide Toxicity in Ethylmalonic Encephalopathy. Nat Med. 2009;15:200–205. doi: 10.1038/nm.1907. [DOI] [PubMed] [Google Scholar]
- 316.Kabil O, Banerjee R. Characterization of Patient Mutations in Human Persulfide Dioxygenase (ETHE1) Involved in H2S Catabo-lism. J Biol Chem. 2012;287:44561–44567. doi: 10.1074/jbc.M112.407411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Holdorf MM, Bennett B, Crowder MW, Makaroff CA. Spectroscopic Studies on Arabidopsis ETHE1, a Glyoxalase II-like Protein. J Inorg Biochem. 2008;102:1825–1830. doi: 10.1016/j.jinorgbio.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.McCoy JG, Bingman CA, Bitto E, Holdorf MM, Makaroff CA, Phillips GN. Structure of an ETHE1-like Protein from Arabidopsis Thaliana. Acta Crystallogr, Sect D: Biol Crystallogr. 2006;62:964–970. doi: 10.1107/S0907444906020592. [DOI] [PubMed] [Google Scholar]
- 319.Pettinati I, Brem J, McDonough MA, Schofield CJ. Crystal Structure of Human Persulfide Dioxygenase: Structural Basis of Ethylmalonic Encephalopathy. Hum Mol Genet. 2015;24:2458–2469. doi: 10.1093/hmg/ddv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Tiranti V, D’Adamo P, Briem E, Ferrari G, Mineri R, Lamantea E, Mandel H, Balestri P, Garcia-Silva M-T, Vollmer B, et al. Ethylmalonic Encephalopathy Is Caused by Mutations in ETHE1, a Gene Encoding a Mitochondrial Matrix Protein. Am J Hum Genet. 2004;74:239–252. doi: 10.1086/381653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Mineri R, Rimoldi M, Burlina AB, Koskull S, Perletti C, Heese B, von Dobeln U, Mereghetti P, Di Meo I, Invernizzi F, et al. Identification of New Mutations in the ETHE1 Gene in a Cohort of 14 Patients Presenting with Ethylmalonic Encephalopathy. J Med Genet. 2008;45:473–478. doi: 10.1136/jmg.2008.058271. [DOI] [PubMed] [Google Scholar]
- 322.García-Silva MT, Campos Y, Ribes A, Briones P, Cabello A, Borbujo JS, Arenas J, Garavaglia B. Encephalopathy, Petechiae, and Acrocyanosis with Ethylmalonic Aciduria Associated with Muscle Cytochrome c Oxidase Deficiency. J Pediatr. 1994;125:843–844. [PubMed] [Google Scholar]
- 323.Burlina A, Zacchello F, Dionisi-Vici C, Bertini E, Sabetta G, Bennet MJ, Hale DE, Schmidt-Sommerfeld E, Rinaldo P. New Clinical Phenotype of Branched-Chain Acyl-CoA Oxidation Defect. Lancet. 1991;338:1522–1523. doi: 10.1016/0140-6736(91)92338-3. [DOI] [PubMed] [Google Scholar]
- 324.Sattler SA, Wang X, Lewis KM, DeHan PJ, Park C-M, Xin Y, Liu H, Xian M, Xun L, Kang C. Characterizations of Two Bacterial Persulfide Dioxygenases of the Metallo-β-Lactamase Superfamily. J Biol Chem. 2015;290:18914–18923. doi: 10.1074/jbc.M115.652537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Driggers CM, Cooley RB, Sankaran B, Hirschberger LL, Stipanuk MH, Karplus PA. Cysteine Dioxygenase Structures from pH4 to 9: Consistent Cys-Persulfenate Formation at Intermediate pH and a Cys-Bound Enzyme at Higher pH. J Mol Biol. 2013;425:3121–3136. doi: 10.1016/j.jmb.2013.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Westley J. Rhodanese. Adv Enzymol Relat Areas Mol Biol. 2006;39:327–368. doi: 10.1002/9780470122846.ch5. [DOI] [PubMed] [Google Scholar]
- 327.Morton NM, Beltram J, Carter RN, Michailidou Z, Gorjanc G, McFadden C, Barrios-Llerena ME, Rodriguez-Cuenca S, Gibbins MTG, Aird RE, et al. Genetic Identification of Thiosulfate Sulfurtransferase as an Adipocyte-Expressed Antidiabetic Target in Mice Selected for Leanness. Nat Med. 2016;22:771–779. doi: 10.1038/nm.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Bordo D. The rhodanese/Cdc25 Phosphatase Superfamily: Sequence-Structure-Function Relations. EMBO Rep. 2002;3:741–746. doi: 10.1093/embo-reports/kvf150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Cipollone R, Ascenzi P, Visca P. Common Themes and Variations in the Rhodanese Superfamily. IUBMB Life. 2007;59:51–59. doi: 10.1080/15216540701206859. [DOI] [PubMed] [Google Scholar]
- 330.Libiad M, Sriraman A, Banerjee R. Polymorphic Variants of Human Rhodanese Exhibit Differences in Thermal Stability and Sulfur Transfer Kinetics. J Biol Chem. 2015;290:23579–23588. doi: 10.1074/jbc.M115.675694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Billaut-Laden I, Allorge D, Crunelle-Thibaut A, Rat E, Cauffiez C, Chevalier D, Houdret N, Lo-Guidice J-M, Broly F. Evidence for a Functional Genetic Polymorphism of the Human Thiosulfate Sulfurtransferase (Rhodanese), a Cyanide and H2S Detoxification Enzyme. Toxicology. 2006;225:1–11. doi: 10.1016/j.tox.2006.04.054. [DOI] [PubMed] [Google Scholar]
- 332.Ploegman JH, Drent G, Kalk KH, Hol WG. Structure of Bovine Liver Rhodanese. I. Structure Determination at 2.5 A Resolution and a Comparison of the Conformation and Sequence of Its Two Domains. J Mol Biol. 1978;123:557–594. [PubMed] [Google Scholar]
- 333.Vitvitsky V, Dayal S, Stabler S, Zhou Y, Wang H, Lentz SR, Banerjee R. Perturbations in Homocysteine-Linked Redox Homeostasis in a Murine Model for Hyperhomocysteinemia. Am J Physiol Regul Integr Comp Physiol. 2004;287:R39–R46. doi: 10.1152/ajpregu.00036.2004. [DOI] [PubMed] [Google Scholar]
- 334.Kappler U, Enemark JH. Sulfite-Oxidizing Enzymes. J Biol Inorg Chem. 2015;20:253–264. doi: 10.1007/s00775-014-1197-3. [DOI] [PubMed] [Google Scholar]
- 335.Nowak K, Luniak N, Witt C, Wüstefeld Y, Wachter A, Mendel RR, Hänsch R. Peroxisomal Localization of Sulfite Oxidase Separates It from Chloroplast-Based Sulfur Assimilation. Plant Cell Physiol. 2004;45:1889–1894. doi: 10.1093/pcp/pch212. [DOI] [PubMed] [Google Scholar]
- 336.Shih VE, Abroms IF, Johnson JL, Carney M, Mandell R, Robb RM, Cloherty JP, Rajagopalan KV. Sulfite Oxidase Deficiency. N Engl J Med. 1977;297:1022–1028. doi: 10.1056/NEJM197711102971902. [DOI] [PubMed] [Google Scholar]
- 337.Kisker C, Schindelin H, Pacheco A, Wehbi WA, Garrett RM, Rajagopalan KV, Enemark JH, Rees DC. Molecular Basis of Sulfite Oxidase Deficiency from the Structure of Sulfite Oxidase. Cell. 1997;91:973–983. doi: 10.1016/s0092-8674(00)80488-2. [DOI] [PubMed] [Google Scholar]
- 338.Garrett RM, Johnson JL, Graf TN, Feigenbaum A, Rajagopalan KV. Human Sulfite Oxidase R160Q: Identification of the Mutation in a Sulfite Oxidase-Deficient Patient and Expression and Characterization of the Mutant Enzyme. Proc Natl Acad Sci U S A. 1998;95:6394–6398. doi: 10.1073/pnas.95.11.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Wilson HL, Rajagopalan KV. The Role of Tyrosine 343 in Substrate Binding and Catalysis by Human Sulfite Oxidase. J Biol Chem. 2004;279:15105–15113. doi: 10.1074/jbc.M314288200. [DOI] [PubMed] [Google Scholar]
- 340.Filipovic MR. Persulfidation (S-Sulfhydration) and H2S. Handb Exp Pharmacol. 2015;230:29–59. doi: 10.1007/978-3-319-18144-8_2. [DOI] [PubMed] [Google Scholar]
- 341.Zivanovic J, Filipovic MR. Hydrogen Sulfide: Stench from the Past as a Mediator of the Future. Biochem (London) 2016;38:12–17. [Google Scholar]
- 342.Li Q, Lancaster JR. Chemical Foundations of Hydrogen Sulfide Biology. Nitric Oxide. 2013;35:21–34. doi: 10.1016/j.niox.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Keilin D. Cytochrome and Respiratory Enzymes. Proc R Soc London, Ser B. 1929;104:206–252. [Google Scholar]
- 344.Chance B, Schoener B. High and Low Energy States of Cytochromes. I. In Mitochondria. J Biol Chem. 1966;241:4567–4573. [PubMed] [Google Scholar]
- 345.Chance B, Lee CP, Schoener B. High and Low Energy States of Cytochromes. II. In Submitochondrial Particles. J Biol Chem. 1966;241:4574–4576. [PubMed] [Google Scholar]
- 346.Lehninger AL, Nelson DL, Cox MM. Lehninger Principles of Biochemistry. Worth Publishers; New York: 2000. [Google Scholar]
- 347.Yoshikawa S, Shimada A. Reaction Mechanism of Cytochrome c Oxidase. Chem Rev. 2015;115:1936–1989. doi: 10.1021/cr500266a. [DOI] [PubMed] [Google Scholar]
- 348.Capaldi RA. Structure and Function of Cytochrome c Oxidase. Annu Rev Biochem. 1990;59:569–596. doi: 10.1146/annurev.bi.59.070190.003033. [DOI] [PubMed] [Google Scholar]
- 349.Kaila VRI, Verkhovsky MI, Wikström M. Proton-Coupled Electron Transfer in Cytochrome Oxidase. Chem Rev. 2010;110:7062–7081. doi: 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]
- 350.Cooper CE, Brown GC. The Inhibition of Mitochondrial Cytochrome Oxidase by the Gases Carbon Monoxide, Nitric Oxide, Hydrogen Cyanide and Hydrogen Sulfide: Chemical Mechanism and Physiological Significance. J Bioenerg Biomembr. 2008;40:533–539. doi: 10.1007/s10863-008-9166-6. [DOI] [PubMed] [Google Scholar]
- 351.Sarti P, Forte E, Mastronicola D, Giuffrè A, Arese M. Cytochrome c Oxidase and Nitric Oxide in Action: Molecular Mechanisms and Pathophysiological Implications. Biochim Biophys Acta, Bioenerg. 2012;1817:610–619. doi: 10.1016/j.bbabio.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 352.Cooper CE. Nitric Oxide and Cytochrome Oxidase: Substrate, Inhibitor or Effector? Trends Biochem Sci. 2002;27:33–39. doi: 10.1016/s0968-0004(01)02035-7. [DOI] [PubMed] [Google Scholar]
- 353.Nicholls P. Inhibition of Cytochrome c Oxidase by Sulphide. Biochem Soc Trans. 1975;3:316–319. doi: 10.1042/bst0030316. [DOI] [PubMed] [Google Scholar]
- 354.Nicholls P. The Effect of Sulphide on Cytochrome aa3. Isosteric and Allosteric Shifts of the Reduced Alpha-Peak. Biochim Biophys Acta, Bioenerg. 1975;396:24–35. doi: 10.1016/0005-2728(75)90186-3. [DOI] [PubMed] [Google Scholar]
- 355.Nicholls P, Kim JK. Oxidation of Sulphide by Cytochrome aa3. Biochim Biophys Acta, Bioenerg. 1981;637:312–320. doi: 10.1016/0005-2728(81)90170-5. [DOI] [PubMed] [Google Scholar]
- 356.Nicholls P, Kim JK. Sulphide as an Inhibitor and Electron Donor for the Cytochrome c Oxidase System. Can J Biochem. 1982;60:613–623. doi: 10.1139/o82-076. [DOI] [PubMed] [Google Scholar]
- 357.Hill BC, Woon TC, Nicholls P, Peterson J, Greenwood C, Thomson AJ. Interactions of Sulphide and Other Ligands with Cytochrome c Oxidase. An Electron-Paramagnetic-Resonance Study. Biochem J. 1984;224:591–600. doi: 10.1042/bj2240591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Koenitzer JR, Isbell TS, Patel HD, Benavides GA, Dickinson DA, Patel RP, Darley-usmar VM, Lancaster JR, Doeller JE, Kraus DW. Hydrogen Sulfide Mediates Vasoactivity in an O2 -Dependent Manner. Am J Physiol Hear Circ Physiol. 2006;292:H1953–H1960. doi: 10.1152/ajpheart.01193.2006. [DOI] [PubMed] [Google Scholar]
- 359.Collman JP, Ghosh S, Dey A, Decreau RA. Using a Functional Enzyme Model to Understand the Chemistry behind Hydrogen Sulfide Induced Hibernation. Proc Natl Acad Sci U S A. 2009;106:22090–22095. doi: 10.1073/pnas.0904082106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Araki T. Ueber Den Blutfarbstoff Und Seine Niiheren Umwanidlungsproducte. Ztschr f Phlysiol Chem. 1890;14:405–416. [Google Scholar]
- 361.Lewisson Zur Frage Uiber Ozon Im Blute. Virchow’s Arch Bd. 1868;XXXVI:15. [Google Scholar]
- 362.Hoppe-Seyler F. Einwirkung Des Schwefelwasserstoffgases Auf Das Blut. Centr f d med Wissensch. 1863;433:28. [Google Scholar]
- 363.Keilin D. On the Combination of Methaemoglobin with H2S. Proc R Soc London, Ser B. 1933;113:393–404. [Google Scholar]
- 364.Flexman AM, Del Vicario G, Schwarz SKW. Dark Green Blood in the Operating Theatre. Lancet. 2007;369:1972. doi: 10.1016/S0140-6736(07)60918-0. [DOI] [PubMed] [Google Scholar]
- 365.Berzofsky JA, Peisach J, Alben JO. Sulfheme Proteins. 3. Carboxysulfmyoglobin: The Relation between Electron Withdrawal from Iron and Ligand Binding. J Biol Chem. 1972;247:3774–3782. [PubMed] [Google Scholar]
- 366.Berzofsky JA, Peisach J, Blumberg WE. Sulfheme Proteins. I. Optical and Magnetic Properties of Sulfmyoglobin and Its Derivatives. J Biol Chem. 1971;246:3367–3377. [PubMed] [Google Scholar]
- 367.Berzofsky JA, Peisach J, Blumberg WE. Sulfheme Proteins. II. The Reversible Oxygenation of Ferrous Sulfmyoglobin. J Biol Chem. 1971;246:7366–7372. [PubMed] [Google Scholar]
- 368.Berzofsky JA, Peisach J, Horecker BL. Sulfheme Proteins. IV. The Stoichiometry of Sulfur Incorporation and the Isolation of Sulfhemin, the Prosthetic Group of Sulfmyoglobin. J Biol Chem. 1972;247:3783–3791. [PubMed] [Google Scholar]
- 369.Carrico RJ, Blumberg WE, Peisach J. The Reversible Binding of Oxygen to Sulfhemoglobin. J Biol Chem. 1978;253:7212–7215. [PubMed] [Google Scholar]
- 370.Chatfield MJ, La Mar GN. 1H Nuclear Magnetic Resonance Study of the Prosthetic Group in Sulfhemoglobin. Arch Biochem Biophys. 1992;295:289–296. doi: 10.1016/0003-9861(92)90520-7. [DOI] [PubMed] [Google Scholar]
- 371.Nicholls P. The Formation and Properties of Sulphmyoglobin and Sulphcatalase. Biochem J. 1961;81:374–383. doi: 10.1042/bj0810374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Ríos-González BB, Román-Morales EM, Pietri R, López-Garriga J. Hydrogen Sulfide Activation in Hemeproteins: The Sulfheme Scenario. J Inorg Biochem. 2014;133:78–86. doi: 10.1016/j.jinorgbio.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Libardi SH, Pindstrup H, Cardoso DR, Skibsted LH. Reduction of Ferrylmyoglobin by Hydrogen Sulfide. Kinetics in Relation to Meat Greening. J Agric Food Chem. 2013;61:2883–2888. doi: 10.1021/jf305363e. [DOI] [PubMed] [Google Scholar]
- 374.Nakamura S, Nakamura M, Yamazaki I, Morrison M. Reactions of Ferryl Lactoperoxidase (Compound II) with Sulfide and Sulfhydryl Compounds. J Biol Chem. 1984;259:7080–7085. [PubMed] [Google Scholar]
- 375.Nussbaum C, Klinke A, Adam M, Baldus S, Sperandio M. yeloperoxidase: A Leukocyte-Derived Protagonist of Inflammation and Cardiovascular Disease. Antioxid Redox Signal. 2013;18:692–713. doi: 10.1089/ars.2012.4783. [DOI] [PubMed] [Google Scholar]
- 376.Pálinkás Z, Furtmüller PG, Nagy A, Jakopitsch C, Pirker KF, Magierowski M, Jasnos K, Wallace JL, Obinger C, Nagy P. Interactions of Hydrogen Sulfide with Myeloperoxidase. Br J Pharmacol. 2015;172:1516–1532. doi: 10.1111/bph.12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Zhou Z, Martin E, Sharina I, Esposito I, Szabo C, Bucci M, Cirino G, Papapetropoulos A. Regulation of Soluble Guanylyl Cyclase Redox State by Hydrogen Sulfide. Pharmacol Res. 2016;111:556–562. doi: 10.1016/j.phrs.2016.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Sorbo B. On the Formation of Thiosulfate from Inorganic Sulfide by Liver Tissue and Heme Compounds. Biochim Biophys Acta. 1958;27:324–329. doi: 10.1016/0006-3002(58)90339-1. [DOI] [PubMed] [Google Scholar]
- 379.Strianese M, De Martino F, Pellecchia C, Ruggiero G, D’Auria S. Myoglobin as a New Fluorescence Probe to Sense H2S. Protein Pept Lett. 2011;18:282–286. doi: 10.2174/092986611794578378. [DOI] [PubMed] [Google Scholar]
- 380.Valentine WN, Frankenfeld JK. 3-Mercaptopyruvate Sulfurtransferase (EC 2.8.1.2): A Simple Assay Adapted to Human Blood Cells. Clin Chim Acta. 1974;51:205–210. doi: 10.1016/0009-8981(74)90031-x. [DOI] [PubMed] [Google Scholar]
- 381.Kraus DW, Wittenberg JB, Lu JF, Peisach J. Hemoglobins of the Lucina Pectinata/bacteria Symbiosis. II. An Electron Paramagnetic Resonance and Optical Spectral Study of the Ferric Proteins. J Biol Chem. 1990;265:16054–16059. [PubMed] [Google Scholar]
- 382.Vitvitsky V, Yadav PK, An S, Seravalli J, Cho U-S, Banerjee R. Structural and Mechanistic Insights into Hemoglobin-Catalyzed Hydrogen Sulfide Oxidation and the Fate of Polysulfide Products. J Biol Chem. 2017;292:5584–5592. doi: 10.1074/jbc.M117.774943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Burmester T, Weich B, Reinhardt S, Hankeln T. A Vertebrate Globin Expressed in the Brain. Nature. 2000;407:520–523. doi: 10.1038/35035093. [DOI] [PubMed] [Google Scholar]
- 384.Burmester T, Hankeln T. What Is the Function of Neuroglobin? J Exp Biol. 2009;212:1423–1428. doi: 10.1242/jeb.000729. [DOI] [PubMed] [Google Scholar]
- 385.Trent JT, Watts RA, Hargrove MS. Human Neuroglobin, a Hexacoordinate Hemoglobin That Reversibly Binds Oxygen. J Biol Chem. 2001;276:30106–30110. doi: 10.1074/jbc.C100300200. [DOI] [PubMed] [Google Scholar]
- 386.Kriegl JM, Bhattacharyya AJ, Nienhaus K, Deng P, Minkow O, Nienhaus GU. Ligand Binding and Protein Dynamics in Neuroglobin. Proc Natl Acad Sci U S A. 2002;99:7992–7997. doi: 10.1073/pnas.082244399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Van Doorslaer S, Dewilde S, Kiger L, Nistor SV, Goovaerts E, Marden MC, Moens L. Nitric Oxide Binding Properties of Neuroglobin. J Biol Chem. 2003;278:4919–4925. doi: 10.1074/jbc.M210617200. [DOI] [PubMed] [Google Scholar]
- 388.Nienhaus K, Kriegl JM, Nienhaus GU. Structural Dynamics in the Active Site of Murine Neuroglobin and Its Effects on Ligand Binding. J Biol Chem. 2004;279:22944–22952. doi: 10.1074/jbc.M401561200. [DOI] [PubMed] [Google Scholar]
- 389.Ruetz M, Kumutima J, Lewis BE, Filipovic MR, Lehnert N, Stemmler TL, Banerjee R. A Distal Ligand Mutes the Interaction of Hydrogen Sulfide with Human Neuroglobin. J Biol Chem. 2017;292:6512–6528. doi: 10.1074/jbc.M116.770370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Kraus DW, Wittenberg JB. Hemoglobins of the Lucina Pectinata/bacteria Symbiosis. I. Molecular Properties, Kinetics and Equilibria of Reactions with Ligands. J Biol Chem. 1990;265:16043–16053. [PubMed] [Google Scholar]
- 391.Pietri R, Lewis A, León RG, Casabona G, Kiger L, Yeh S-R, Fernandez-Alberti S, Marden MC, Cadilla CL, López-Garriga J. Factors Controlling the Reactivity of Hydrogen Sulfide with Hemeproteins. Biochemistry. 2009;48:4881–4894. doi: 10.1021/bi801738j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Ramos-Alvarez C, Yoo B-K, Pietri R, Lamarre I, Martin J-L, López-Garriga J, Negrerie M. Reactivity and Dynamics of H2S, NO, and O2 Interacting with Hemoglobins from Lucina Pectinata. Biochemistry. 2013;52:7007–7021. doi: 10.1021/bi400745a. [DOI] [PubMed] [Google Scholar]
- 393.Díaz-Ayala R, Moya-Rodríguez A, Pietri R, Cadilla CL, López-Garriga J. Molecular Cloning and Characterization of a (Lys)6-Tagged Sulfide-Reactive Hemoglobin I from Lucina Pectinata. Mol Biotechnol. 2015;57:1050–1062. doi: 10.1007/s12033-015-9896-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Gavira JA, Camara-Artigas A, De Jesús-Bonilla W, López-Garriga J, Lewis A, Pietri R, Yeh S-R, Cadilla CL, García-Ruiz JM. Structure and Ligand Selection of Hemoglobin II from Lucina Pectinata. J Biol Chem. 2008;283:9414–9423. doi: 10.1074/jbc.M705026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Fernandez-Alberti S, Bacelo DE, Binning RC, Echave J, Chergui M, López-Garriga J. Sulfide-Binding Hemoglobins: Effects of Mutations on Active-Site Flexibility. Biophys J. 2006;91:1698–1709. doi: 10.1529/biophysj.106.081646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Silfa E, Almeida M, Cerda J, Wu S, López-Garriga J. Orientation of the Heme Vinyl Groups in the Hydrogen Sulfide-Binding Hemoglobin I from Lucina Pectinata. Biospectroscopy. 1998;4:311–326. doi: 10.1002/(sici)1520-6343(1998)4:5<311::aid-bspy3>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 397.Cerda J, Echevarria Y, Morales E, López-Garriga J. Resonance Raman Studies of the Heme-Ligand Active Site of Hemoglobin I fromLucina Pectinata. Biospectroscopy. 1999;5:289–301. [Google Scholar]
- 398.Nguyen BD, Zhao X, Vyas K, La Mar GN, Lile RA, Brucker EA, Phillips GN, Olson JS, Wittenberg JB. Solution and Crystal Structures of a Sperm Whale Myoglobin Triple Mutant That Mimics the Sulfide-Binding Hemoglobin from Lucina Pectinata. J Biol Chem. 1998;273:9517–9526. doi: 10.1074/jbc.273.16.9517. [DOI] [PubMed] [Google Scholar]
- 399.Pietri R, Granell L, Cruz A, De Jesús W, Lewis A, León R, Cadilla CL, Garriga JL. Tyrosine B10 and Heme-Ligand Interactions of Lucina Pectinata Hemoglobin II: Control of Heme Reactivity. Biochim Biophys Acta, Proteins Proteomics. 2005;1747:195–203. doi: 10.1016/j.bbapap.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 400.Pietri R, León RG, Kiger L, Marden MC, Granell LB, Cadilla CL, López-Garriga J. Hemoglobin I from Lucina Pectinata: A Model for Distal Heme-Ligand Control. Biochim Biophys Acta, Proteins Proteomics. 2006;1764:758–765. doi: 10.1016/j.bbapap.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 401.Ramirez E, Cruz A, Rodriguez D, Uchima L, Pietri R, Santana A, López-Garriga J, López GE. Effects of Active Site Mutations in Haemoglobin I from Lucina Pectinata: A Molecular Dynamic Study. Mol Simul. 2008;34:715–725. doi: 10.1080/08927020802144114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Román-Morales E, Pietri R, Ramos-Santana B, Vinogradov SN, Lewis-Ballester A, López-Garriga J. Structural Determinants for the Formation of Sulfhemeprotein Complexes. Biochem Biophys Res Commun. 2010;400:489–492. doi: 10.1016/j.bbrc.2010.08.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Pietri R, Román-Morales E, López-Garriga J. Hydrogen Sulfide and Hemeproteins: Knowledge and Mysteries. Antioxid Redox Signal. 2011;15:393–404. doi: 10.1089/ars.2010.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Filipovic MR, Ivanovic-Burmazovic I. Removal of Hydrogen Sulphide (H2S): Catalytic Oxidation of Sulphide Species. WO2012175630A1. 2012 [Google Scholar]
- 405.Hartle MD, Prell JS, Pluth MD. Spectroscopic Investigations into the Binding of Hydrogen Sulfide to Synthetic Picket-Fence Porphyrins. Dalt Trans. 2016;45:4843–4853. doi: 10.1039/c5dt04563k. [DOI] [PubMed] [Google Scholar]
- 406.Zal F, Leize E, Lallier FH, Toulmond A, Van Dorsselaer A, Childress JJ. S-Sulfohemoglobin and Disulfide Exchange: The Mechanisms of Sulfide Binding by Riftia Pachyptila Hemoglobins. Proc Natl Acad Sci U S A. 1998;95:8997–9002. doi: 10.1073/pnas.95.15.8997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Bailly X, Vinogradov S. The Sulfide Binding Function of Annelid Hemoglobins: Relic of an Old Biosystem? J Inorg Biochem. 2005;99:142–150. doi: 10.1016/j.jinorgbio.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 408.Flores JF, Fisher CR, Carney SL, Green BN, Freytag JK, Schaeffer SW, Royer WE. Sulfide Binding Is Mediated by Zinc Ions Discovered in the Crystal Structure of a Hydrothermal Vent Tubeworm Hemoglobin. Proc Natl Acad Sci U S A. 2005;102:2713–2718. doi: 10.1073/pnas.0407455102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Zhao K, Li S, Wu L, Lai C, Yang G. Hydrogen Sulfide Represses Androgen Receptor Transactivation by Targeting at the Second Zinc Finger Module. J Biol Chem. 2014;289:20824–20835. doi: 10.1074/jbc.M114.559518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Pyriochou A, Roussos C, Roviezzo F, Brancaleone V, Cirino G. Hydrogen Sulfide Is an Endogenous Inhibitor of Phosphodiesterase Activity. Arterioscler Thromb Vasc Biol. 2010;30:1998–2004. doi: 10.1161/ATVBAHA.110.209783. [DOI] [PubMed] [Google Scholar]
- 411.Galardon E, Tomas A, Selkti M, Roussel P, Artaud I. Synthesis, Characterization, and Reactivity of Alkyldisulfanido Zinc Complexes. Inorg Chem. 2009;48:5921–5927. doi: 10.1021/ic900238v. [DOI] [PubMed] [Google Scholar]
- 412.Galardon E, Tomas A, Roussel P, Artaud I. Synthesis, Stability, and Reactivity of [(TPA)Zn(SH)]+ in Aqueous and Organic Solutions. Eur J Inorg Chem. 2011;2011:3797–3801. [Google Scholar]
- 413.Hartle MD, Delgado M, Gilbertson JD, Pluth MD. Stabilization of a Zn(ii) Hydrosulfido Complex Utilizing a Hydrogen-Bond Accepting Ligand. Chem Commun (Cambridge, U K) 2016;52:7680–7682. doi: 10.1039/c6cc01373b. [DOI] [PubMed] [Google Scholar]
- 414.Koppenol WH, Stanbury DM, Bounds PL. Electrode Potentials of Partially Reduced Oxygen Species, from Dioxygen to Water. Free Radic Biol Med. 2010;49:317–322. doi: 10.1016/j.freeradbiomed.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 415.Mills G, Schmidt KH, Matheson MS, Meisel D. Thermal and Photochemical Reactions of Sulfhydryl Radicals. Implications for Colloid Photocorrosion. J Phys Chem. 1987;91:1590–1596. [Google Scholar]
- 416.Ross AB, Mallard WG, Helman WP, Buxton GV, Huie RT, Neta P. NDRL/NIST Solution Kinetics Database. 1998 version 3. [Google Scholar]
- 417.Rabai G, Orban M, Epstein IR. A Model for the pH-Regulated Oscillatory Reaction between Hydrogen Peroxide and Sulfide Ion. J Phys Chem. 1992;96:5414–5419. [Google Scholar]
- 418.Nagy P, Winterbourn CC. Rapid Reaction of Hydrogen Sulfide with the Neutrophil Oxidant Hypochlorous Acid to Generate Polysulfides. Chem Res Toxicol. 2010;23:1541–1543. doi: 10.1021/tx100266a. [DOI] [PubMed] [Google Scholar]
- 419.Filipovic MR, Miljkovic J, Allgäuer A, Chaurio R, Shubina T, Herrmann M, Ivanovic-Burmazovic I. Biochemical Insight into Physiological Effects of H2S: Reaction with Peroxynitrite and Formation of a New Nitric Oxide Donor, Sulfinyl Nitrite. Biochem J. 2012;441:609–621. doi: 10.1042/BJ20111389. [DOI] [PubMed] [Google Scholar]
- 420.Cuevasanta E, Zeida A, Carballal S, Wedmann R, Morzan UN, Trujillo M, Radi R, Estrin DA, Filipovic MR, Alvarez B. Insights into the Mechanism of the Reaction between Hydrogen Sulfide and Peroxynitrite. Free Radic Biol Med. 2015;80:93–100. doi: 10.1016/j.freeradbiomed.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 421.Davies MJ, Hawkins CL, Pattison DI, Rees MD. Mammalian Heme Peroxidases: From Molecular Mechanisms to Health Implications. Antioxid Redox Signal. 2008;10:1199–1234. doi: 10.1089/ars.2007.1927. [DOI] [PubMed] [Google Scholar]
- 422.Koppenol WH, Kissner R. Can O=NOOH Undergo Homolysis? Chem Res Toxicol. 1998;11:87–90. doi: 10.1021/tx970200x. [DOI] [PubMed] [Google Scholar]
- 423.Palmer RM, Ferrige AG, Moncada S. Nitric Oxide Release Accounts for the Biological Activity of Endothelium-Derived Relaxing Factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 424.Palmer RM, Ashton DS, Moncada S. Vascular Endothelial Cells Synthesize Nitric Oxide from L-Arginine. Nature. 1988;333:664–666. doi: 10.1038/333664a0. [DOI] [PubMed] [Google Scholar]
- 425.Bogdan C. Nitric Oxide and the Immune Response. Nat Immunol. 2001;2:907–916. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 426.Bogdan C, Röllinghoff M, Diefenbach A. The Role of Nitric Oxide in Innate Immunity. Immunol Rev. 2000;173:17–26. doi: 10.1034/j.1600-065x.2000.917307.x. [DOI] [PubMed] [Google Scholar]
- 427.Gamper N, Ooi L. Redox and Nitric Oxide-Mediated Regulation of Sensory Neuron Ion Channel Function. Antioxid Redox Signal. 2015;22:486–504. doi: 10.1089/ars.2014.5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Santos AI, Martínez-Ruiz A, Araujó IM. S-Nitrosation and Neuronal Plasticity. Br J Pharmacol. 2015;172:1468–1478. doi: 10.1111/bph.12827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Nott A, Riccio A. Nitric Oxide-Mediated Epigenetic Mechanisms in Developing Neurons. Cell Cycle. 2009;8:725–730. doi: 10.4161/cc.8.5.7805. [DOI] [PubMed] [Google Scholar]
- 430.Friebe A, Koesling D. Regulation of Nitric Oxide-Sensitive Guanylyl Cyclase. Circ Res. 2003;93:96–105. doi: 10.1161/01.RES.0000082524.34487.31. [DOI] [PubMed] [Google Scholar]
- 431.Moncada S, Palmer RM, Higgs EA. Nitric Oxide: Physiology, Pathophysiology, and Pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 432.Foster MW, Hess DT, Stamler JS. Protein S-Nitrosylation in Health and Disease: A Current Perspective. Trends Mol Med. 2009;15:391–404. doi: 10.1016/j.molmed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Hess DT, Stamler JS. Regulation by S-Nitrosylation of Protein Post-Translational Modification. J Biol Chem. 2012;287:4411–4418. doi: 10.1074/jbc.R111.285742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Seth D, Stamler JS. The SNO-Proteome: Causation and Classifications. Curr Opin Chem Biol. 2011;15:129–136. doi: 10.1016/j.cbpa.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Lima B, Forrester MT, Hess DT, Stamler JS. S-Nitrosylation in Cardiovascular Signaling. Circ Res. 2010;106:633–646. doi: 10.1161/CIRCRESAHA.109.207381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Broniowska KA, Hogg N. The Chemical Biology of S-Nitrosothiols. Antioxid Redox Signal. 2012;17:969–980. doi: 10.1089/ars.2012.4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Lancaster JR. How Are Nitrosothiols Formed de Novo in Vivo? Arch Biochem Biophys. 2017;617:137–144. doi: 10.1016/j.abb.2016.10.015. [DOI] [PubMed] [Google Scholar]
- 438.Zhang Y, Hogg N. The Mechanism of Transmembrane S-Nitrosothiol Transport. Proc Natl Acad Sci U S A. 2004;101:7891–7896. doi: 10.1073/pnas.0401167101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Miljkovic JL, Filipovic MR. HNO/Thiol Biology as a Therapeutic Target. Springer International Publishing; Berlin: 2016. pp. 335–375. [Google Scholar]
- 440.Shafirovich V, Lymar SV. Nitroxyl and Its Anion in Aqueous Solutions: Spin States, Protic Equilibria, and Reactivities toward Oxygen and Nitric Oxide. Proc Natl Acad Sci U S A. 2002;99:7340–7345. doi: 10.1073/pnas.112202099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Bartberger MD, Liu W, Ford E, Miranda KM, Switzer C, Fukuto JM, Farmer PJ, Wink DA, Houk KN. The Reduction Potential of Nitric Oxide (NO) and Its Importance to NO Biochemistry. Proc Natl Acad Sci U S A. 2002;99:10958–10963. doi: 10.1073/pnas.162095599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Fukuto JM, Carrington SJ. HNO Signaling Mechanisms. Antioxid Redox Signal. 2011;14:1649–1657. doi: 10.1089/ars.2010.3855. [DOI] [PubMed] [Google Scholar]
- 443.Gladwin MT, Schechter AN, Kim-Shapiro DB, Patel RP, Hogg N, Shiva S, Cannon RO, Kelm M, Wink DA, Espey MG, et al. The Emerging Biology of the Nitrite Anion. Nat Chem Biol. 2005;1:308–314. doi: 10.1038/nchembio1105-308. [DOI] [PubMed] [Google Scholar]
- 444.Lundberg JO, Weitzberg E, Shiva S, Gladwin MT. Nitrite and Nitrate in Human Health and Disease. Humana Press; Totowa, NJ: 2011. The Nitrate–Nitrite–Nitric Oxide Pathway in Mammals; pp. 21–48. [Google Scholar]
- 445.Lundberg JO, Weitzberg E, Gladwin MT. The Nitrate– nitrite–nitric Oxide Pathway in Physiology and Therapeutics. Nat Rev Drug Discovery. 2008;7:156–167. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- 446.Kissner R, Nauser T, Bugnon P, Lye PG, Koppenol WH. Formation and Properties of Peroxynitrite as Studied by Laser Flash Photolysis, High-Pressure Stopped-Flow Technique, and Pulse Radiolysis. Chem Res Toxicol. 1997;10:1285–1292. doi: 10.1021/tx970160x. [DOI] [PubMed] [Google Scholar]
- 447.Szabo´ C, Ischiropoulos H, Radi R. Peroxynitrite: Biochemistry, Pathophysiology and Development of Therapeutics. Nat Rev Drug Discovery. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 448.Pacher P, Beckman JS, Liaudet L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Filipovic MR, Miljkovic JL, Nauser T, Royzen M, Klos K, Shubina T, Koppenol WH, Lippard SJ, Ivanović–Burmazović I. Chemical Characterization of the Smallest S-Nitrosothiol, HSNO; Cellular Cross-Talk of H2S and S-Nitrosothiols. J Am Chem Soc. 2012;134:12016–12027. doi: 10.1021/ja3009693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Miljkovic JL, Kenkel I, Ivanović–Burmazović I, Filipovic MR. Generation of HNO and HSNO from Nitrite by Heme-Iron-Catalyzed Metabolism with H2S. Angew Chem, Int Ed. 2013;52:12061–12064. doi: 10.1002/anie.201305669. [DOI] [PubMed] [Google Scholar]
- 451.Yong Q-C, Hu L-F, Wang S, Huang D, Bian J-S. Hydrogen Sulfide Interacts with Nitric Oxide in the Heart: Possible Involvement of Nitroxyl. Cardiovasc Res. 2010;88:482–491. doi: 10.1093/cvr/cvq248. [DOI] [PubMed] [Google Scholar]
- 452.Yong Q-C, Cheong JL, Hua F, Deng L-W, Khoo YM, Lee H-S, Perry A, Wood M, Whiteman M, Bian J-S. Regulation of Heart Function by Endogenous Gaseous Mediators-Crosstalk between Nitric Oxide and Hydrogen Sulfide. Antioxid Redox Signal. 2011;14:2081–2091. doi: 10.1089/ars.2010.3572. [DOI] [PubMed] [Google Scholar]
- 453.Cortese-Krott MM, Fernandez BO, Santos JLT, Mergia E, Grman M, Nagy P, Kelm M, Butler A, Feelisch M. Nitrosopersulfide (SSNO(–)) Accounts for Sustained NO Bioactivity of S-Nitrosothiols Following Reaction with Sulfide. Redox Biol. 2014;2:234–244. doi: 10.1016/j.redox.2013.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Altaany Z, Ju Y, Yang G, Wang R. The Coordination of S-Sulfhydration, S-Nitrosylation, and Phosphorylation of Endothelial Nitric Oxide Synthase by Hydrogen Sulfide. Sci Signaling. 2014;7:ra87–ra87. doi: 10.1126/scisignal.2005478. [DOI] [PubMed] [Google Scholar]
- 455.Heine CL, Schmidt R, Geckl K, Schrammel A, Gesslbauer B, Schmidt K, Mayer B, Gorren ACF. Selective Irreversible Inhibition of Neuronal and Inducible Nitric-Oxide Synthase in the Combined Presence of Hydrogen Sulfide and Nitric Oxide. J Biol Chem. 2015;290:24932–24944. doi: 10.1074/jbc.M115.660316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Berzelius JJ. In: Traitè de Chimie. Oeuvres de Berzelius JJ, editor. Paris: F. Didot frèrès; 1831. [Google Scholar]
- 457.LeConte MC. Action Des Hydracides Sur Les Acides Oxygénés. Ann Chim Phys. 1847:180–183. [Google Scholar]
- 458.Pierce JA. A Study of the Reaction between Nitric Oxide and Hydrogen Sulphide. J Phys Chem. 1928;33:22–36. [Google Scholar]
- 459.Dunnicliff HB, Mohammad S, Kishen J. The Interaction between Nitric Oxide and Hydrogen Sulphide in the Presence of Water. J Phys Chem. 1930;35:1721–1734. [Google Scholar]
- 460.Kurtenacker A, Löschner H. Über Die Einwirkung von Stickoxyd Auf Thiosulfat Und Sulfid. Zeitschrift för. Anorg und Allg Chemie. 1938;238:335–349. [Google Scholar]
- 461.Kulik MD. Method for Removing Hydrogen Sulfide and Nitric Oxide from Gaseous Mixtures. US4314977A. 1982 [Google Scholar]
- 462.Suarez SA, Neuman NI, Muñoz M, Álvarez L, Bikiel DE, Brondino CD, Ivanović-Burmazović I, Miljkovic JL, Filipovic MR, Martí MA, et al. Nitric Oxide Is Reduced to HNO by Proton-Coupled Nucleophilic Attack by Ascorbate, Tyrosine, and Other Alcohols. A New Route to HNO in Biological Media? J Am Chem Soc. 2015;137:4720–4727. doi: 10.1021/ja512343w. [DOI] [PubMed] [Google Scholar]
- 463.Arulsamy N, Bohle DS, Butt JA, Irvine GJ, Jordan PA, Sagan E. Interrelationships between Conformational Dynamics and the Redox Chemistry of S-Nitrosothiols. J Am Chem Soc. 1999;121:7115–7123. [Google Scholar]
- 464.Poole DP, Pelayo JC, Cattaruzza F, Kuo Y, Gai G, Chiu JV, Bron R, Furness JB, Grady EF, Bunnett NW. Transient Receptor Potential Ankyrin 1 Is Expressed by Inhibitory Motoneurons of the Mouse Intestine. Gastroenterology. 2011;141:565–575. doi: 10.1053/j.gastro.2011.04.049. [DOI] [PubMed] [Google Scholar]
- 465.Dux M, Will C, Vogler B, Filipovic MR, Messlinger K. Meningeal Blood Flow Is Controlled by H2S-NO Crosstalk Activating a HNO-TRPA1-CGRP Signalling Pathway. Br J Pharmacol. 2016;173:431–445. doi: 10.1111/bph.13164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Zhou Y, Zhang X, Yang S, Li Y, Qing Z, Zheng J, Li J, Yang R. Ratiometric Visualization of NO/H2S Cross-Talk in Living Cells and Tissues Using a Nitroxyl-Responsive Two-Photon Fluorescence Probe. Anal Chem. 2017;89:4587–4594. doi: 10.1021/acs.analchem.7b00073. [DOI] [PubMed] [Google Scholar]
- 467.Whiteman M, Li L, Kostetski I, Chu SH, Siau JL, Bhatia M, Moore PK. Evidence for the Formation of a Novel Nitrosothiol from the Gaseous Mediators Nitric Oxide and Hydrogen Sulphide. Biochem Biophys Res Commun. 2006;343:303–310. doi: 10.1016/j.bbrc.2006.02.154. [DOI] [PubMed] [Google Scholar]
- 468.Ali MY, Ping CY, Mok Y-Y, Ling L, Whiteman M, Bhatia M, Moore PK. Regulation of Vascular Nitric Oxide in Vitro and in Vivo a New Role for Endogenous Hydrogen Sulphide? Br J Pharmacol. 2006;149:625–634. doi: 10.1038/sj.bjp.0706906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Müller RP, Nonella M, Russegger P, Huber JR. UV-, VIS- and IR-Light-Induced Isomerization of HSNO in a Low-Temperature Matrix. Chem Phys. 1984;87:351–361. [Google Scholar]
- 470.Tchir PO, Spratley RD. The Photolysis of Matrix Isolated Cis-Thinoylimide. 1. The Identification and Infrared Spectra of Cis-HOSN, HSNO and SNO. Can J Chem. 1975;53:2318–2330. [Google Scholar]
- 471.Ivanova LV, Anton BJ, Timerghazin QK. On the Possible Biological Relevance of HSNO Isomers: A Computational Investigation. Phys Chem Chem Phys. 2014;16:8476–8486. doi: 10.1039/c4cp00469h. [DOI] [PubMed] [Google Scholar]
- 472.Timerghazin QK, Peslherbe GH, English AM. Structure and Stability of HSNO, the Simplest S-Nitrosothiol. Phys Chem Chem Phys. 2008;10:1532–1539. doi: 10.1039/b715025c. [DOI] [PubMed] [Google Scholar]
- 473.Seel F, Kuhn R, Simon G, Wagner M. PNP-Perthionitrit Und PNP-Monothionitrit/PNP-Perthionitrite and PNP-M Onothionitrite. Z Naturforsch, B: J Chem Sci. 1985;40:1607–1617. [Google Scholar]
- 474.Nava M, Martin-Drumel M-A, López CA, Crabtree KN, Womack CC, Nguyen TL, Thorwirth S, Cummins CC, Stanton JF, McCarthy MC. Spontaneous and Selective Formation of HSNO, a Crucial Intermediate Linking H2S and Nitroso Chemistries. J Am Chem Soc. 2016;138:11441–11444. doi: 10.1021/jacs.6b05886. [DOI] [PubMed] [Google Scholar]
- 475.Chatterjee D, Sarkar P, Oszajca M, van Eldik R. Formation of [RuIII(edta)(SNO)]2− in RuIII(edta)-Mediated S-Nitrosylation of Bisulfide Ion. Inorg Chem. 2016;55:5037–5040. doi: 10.1021/acs.inorgchem.6b00615. [DOI] [PubMed] [Google Scholar]
- 476.Quiroga SL, Almaraz AE, Amorebieta VT, Perissinotti LL, Olabe JA. Addition and Redox Reactivity of Hydrogen Sulfides (H2S/HS−) with Nitroprusside: New Chemistry of Nitrososulfide Ligands. Chem Eur J. 2011;17:4145–4156. doi: 10.1002/chem.201002322. [DOI] [PubMed] [Google Scholar]
- 477.Filipovic MR, Eberhardt M, Prokopovic V, Mijuskovic A, Orescanin-Dusic Z, Reeh P, Ivanovic-Burmazovic I. Beyond H2S and NO Interplay: Hydrogen Sulfide and Nitroprusside React Directly to Give Nitroxyl (HNO). A New Pharmacological Source of HNO. J Med Chem. 2013;56:1499–1508. doi: 10.1021/jm3012036. [DOI] [PubMed] [Google Scholar]
- 478.Islam ASM, Bhowmick R, Pal K, Katarkar A, Chaudhuri K, Ali MA. Smart Molecule for Selective Sensing of Nitric Oxide: Conversion of NO to HSNO Relevance of Biological HSNO Formation. Inorg Chem. 2017;56:4324–4331. doi: 10.1021/acs.inorgchem.6b02787. [DOI] [PubMed] [Google Scholar]
- 479.Sun J, Aponte AM, Menazza S, Gucek M, Steenbergen C, Murphy E. Additive Cardioprotection by Pharmacological Postconditioning with Hydrogen Sulfide and Nitric Oxide Donors in Mouse Heart: S-Sulfhydration vs. S-Nitrosylation. Cardiovasc Res. 2016;110:96–106. doi: 10.1093/cvr/cvw037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Munro AP, Williams DLH. Reactivity of Sulfur Nucleophiles towards S-Nitrosothiols. J Chem Soc Perkin Trans2. 2000;24:1794–1797. [Google Scholar]
- 481.Cortese-Krott MM, Kuhnle GGC, Dyson A, Fernandez BO, Grman M, DuMond JF, Barrow MP, McLeod G, Nakagawa H, Ondrias K, et al. Key Bioactive Reaction Products of the NO/H2S Interaction Are S/N-Hybrid Species, Polysulfides, and Nitroxyl. Proc Natl Acad Sci U S A. 2015;112:E4651–E4660. doi: 10.1073/pnas.1509277112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Wedmann R, Ivanovic-Burmazovic I, Filipovic MR. Nitrosopersulfide (SSNO−) Decomposes in the Presence of Sulfide, Cyanide or Glutathione to Give HSNO/SNO−: Consequences for the Assumed Role in Cell Signalling. Interface Focus. 2017;7:20160139. doi: 10.1098/rsfs.2016.0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Ivanovic-Burmazovic I. HNO Generation From NOradical Dot, Nitrite, Inorganic or Organic Nitrosyls, and Crosstalk With H2S. Chemistry and Biology of Nitroxyl (HNO) Science direct. 2017:67–104. [Google Scholar]
- 484.Heinecke J, Ford PC. Formation of Cysteine Sulfenic Acid by Oxygen Atom Transfer from Nitrite. J Am Chem Soc. 2010;132:9240–9243. doi: 10.1021/ja102221e. [DOI] [PubMed] [Google Scholar]
- 485.Heinecke JL, Khin C, Pereira JCM, Suárez SA, Iretskii AV, Doctorovich F, Ford PC. Nitrite Reduction Mediated by Heme Models. Routes to NO and HNO? J Am Chem Soc. 2013;135:4007–4017. doi: 10.1021/ja312092x. [DOI] [PubMed] [Google Scholar]
- 486.Bir SC, Kolluru GK, McCarthy P, Shen X, Pardue S, Pattillo CB, Kevil CG. Hydrogen Sulfide Stimulates Ischemic Vascular Remodeling through Nitric Oxide Synthase and Nitrite Reduction Activity Regulating Hypoxia-Inducible Factor-1α and Vascular Endothelial Growth Factor-Dependent Angiogenesis. J Am Heart Assoc. 2012;1:e004093–e004093. doi: 10.1161/JAHA.112.004093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Toohey JI. The Conversion of H2S to Sulfane Sulfur. Nat Rev Mol Cell Biol. 2012;13:803–803. doi: 10.1038/nrm3391-c1. [DOI] [PubMed] [Google Scholar]
- 488.Böhme H, Zinner G. Über Darstellung Und Eigenschaften von Alkyl-Hydro-Polysulfiden. Justus Liebigs Ann Chem. 1954;585:142–149. [Google Scholar]
- 489.Everett SA, Folkes LK, Wardman P, Asmus KD. Free-Radical Repair by a Novel Perthiol: Reversible Hydrogen Transfer and Perthiyl Radical Formation. Free Radical Res. 1994;20:387–400. doi: 10.3109/10715769409145638. [DOI] [PubMed] [Google Scholar]
- 490.Park C-M, Weerasinghe L, Day JJ, Fukuto JM, Xian M. Persulfides: Current Knowledge and Challenges in Chemistry and Chemical Biology. Mol BioSyst. 2015;11:1775–1785. doi: 10.1039/c5mb00216h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Tsurugi J, Abe Y, Kawamura S. Synthesis of 2-Substituted Ethyl Hydrodisulfides. Bull Chem Soc Jpn. 1970;43:1890–1892. [Google Scholar]
- 492.Tsurugi J, Nakabayashi T. Synthesis of Dibenzhydryl and Dibenzyl Penta- and Hexasulfides. J Org Chem. 1959;24:807–810. [Google Scholar]
- 493.Nakabayashi T, Tsurugi J, Yabuta T. Organic Polysulfides. 1 IV. Synthesis of Bis(triphenylmethyl) Polysulfides. J Org Chem. 1964;29:1236–1238. [Google Scholar]
- 494.Bailey TS, Pluth MD. Reactions of Isolated Persulfides Provide Insights into the Interplay between H2S and Persulfide Reactivity. Free Radic Biol Med. 2015;89:662–667. doi: 10.1016/j.freeradbiomed.2015.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Bailey TS, Zakharov LN, Pluth MD. Understanding Hydrogen Sulfide Storage: Probing Conditions for Sulfide Release from Hydrodisulfides. J Am Chem Soc. 2014;136:10573–10576. doi: 10.1021/ja505371z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Tsurugi J, Nakabayashi T, Ishihara T. Aralkyl Hydro-disulfides. 1 III. The Reaction with Tertiary Phosphines. J Org Chem. 1965;30:2707–2710. [Google Scholar]
- 497.Aycock DF, Jurch GR. Synthesis of Alkyl Trithioperesters (Alkyl Thiocarbonyl Disulfides) J Org Chem. 1979;44:569–572. [Google Scholar]
- 498.Heimer NE, Field L. Biologically Oriented Organic Sulfur Chemistry. 23. A Hydrodisulfide from a Sulfonamide Derivative of Penicillamine. J Org Chem. 1984;49:1446–1449. [Google Scholar]
- 499.Heimer NE, Field L, Neal RA. Biologically Oriented Organic Sulfur Chemistry. 21. Hydrodisulfide of a Penicillamine Derivative and Related Compounds. J Org Chem. 1981;46:1374–1377. [Google Scholar]
- 500.Heimer NE, Field L, Waites JA. Organic Disulfides and Related Substances. 44. Preparation and Characterization of 1-Adamantyl Hydrodisulfide as a Stable Prototype of the Series. J Org Chem. 1985;50:4164–4166. [Google Scholar]
- 501.Artaud I, Galardon E. A Persulfide Analogue of the Nitrosothiol SNAP: Formation, Characterization and Reactivity. ChemBioChem. 2014;15:2361–2364. doi: 10.1002/cbic.201402312. [DOI] [PubMed] [Google Scholar]
- 502.Rao GS, Gorin G. Reaction of Cystine with Sodium Sulfide in Sodium Hydroxide Solution. J Org Chem. 1959;24:749–753. [Google Scholar]
- 503.Liu DK, Chang SG. Kinetic Study of the Reaction between Cystine and Sulfide in Alkaline Solutions. Can J Chem. 1987;65:770–774. [Google Scholar]
- 504.Vasas A, Dóka É, Fábián I, Nagy P. Kinetic and Thermodynamic Studies on the Disulfide-Bond Reducing Potential of Hydrogen Sulfide. Nitric Oxide. 2015;46:93–101. doi: 10.1016/j.niox.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 505.Francoleon NE, Carrington SJ, Fukuto JM. The Reaction of H(2)S with Oxidized Thiols: Generation of Persulfides and Implications to H(2)S Biology. Arch Biochem Biophys. 2011;516:146–153. doi: 10.1016/j.abb.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 506.Mintel R, Westley J. The Rhodanese Reaction. Mechanism of Sulfur-Sulfur Bond Cleavage. J Biol Chem. 1966;241:3381–3385. [PubMed] [Google Scholar]
- 507.Villarejo M, Westley J. Mechanism of Rhodanese Catalysis of Thiosulfate-Lipoate Oxidation-Reduction. J Biol Chem. 1963;238:4016–4020. [PubMed] [Google Scholar]
- 508.Moutiez M, Aumercier M, Teissier E, Parmentier B, Tartar A, Sergheraert C. Reduction of a Trisulfide Derivative of Glutathione by Glutathione Reductase. Biochem Biophys Res Commun. 1994;202:1380–1386. doi: 10.1006/bbrc.1994.2083. [DOI] [PubMed] [Google Scholar]
- 509.Pan J, Carroll KS. Persulfide Reactivity in the Detection of Protein S-Sulfhydration. ACS Chem Biol. 2013;8:1110–1116. doi: 10.1021/cb4001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Ellman GL. Tissue Sulfhydryl Groups. Arch Biochem Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 511.Zhang D, Macinkovic I, Devarie-Baez NO, Pan J, Park C-M, Carroll KS, Filipovic MR, Xian M. Detection of Protein S-Sulfhydration by a Tag-Switch Technique. Angew Chem, Int Ed. 2014;53:575–581. doi: 10.1002/anie.201305876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.Turell L, Botti H, Carballal S, Ferrer-Sueta G, Souza JM, Durán R, Freeman BA, Radi R, Alvarez B. Reactivity of Sulfenic Acid in Human Serum Albumin. Biochemistry. 2008;47:358–367. doi: 10.1021/bi701520y. [DOI] [PubMed] [Google Scholar]
- 513.Alvarez B, Carballal S, Turell L, Radi R. Formation and Reactions of Sulfenic Acid in Human Serum Albumin. Methods Enzymol. 2010;473:117–136. doi: 10.1016/S0076-6879(10)73005-6. [DOI] [PubMed] [Google Scholar]
- 514.Dóka É, Pader I, Bíró A, Johansson K, Cheng Q, Ballagó K, Prigge JR, Pastor-Flores D, Dick TP, Schmidt EE, et al. A Novel Persulfide Detection Method Reveals Protein Persulfide- and Polysulfide-Reducing Functions of Thioredoxin and Glutathione Systems. Sci Adv. 2016;2:e1500968. doi: 10.1126/sciadv.1500968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Park C-M, Johnson BA, Duan J, Park J-J, Day JJ, Gang D, Qian W-J, Xian M. 9-Fluorenylmethyl (Fm) Disulfides: Biomimetic Precursors for Persulfides. Org Lett. 2016;18:904–907. doi: 10.1021/acs.orglett.5b03557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Schmidt B, Ho L, Hogg PJ. Allosteric Disulfide Bonds. Biochemistry. 2006;45:7429–7433. doi: 10.1021/bi0603064. [DOI] [PubMed] [Google Scholar]
- 517.Ploegman JH, Drent G, Kalk KH, Hol WG. The Structure of Bovine Liver Rhodanese. II. The Active Site in the Sulfur-Substituted and the Sulfur-Free Enzyme. J Mol Biol. 1979;127:149–162. doi: 10.1016/0022-2836(79)90236-5. [DOI] [PubMed] [Google Scholar]
- 518.Souness RJ, Kleffmann T, Tchesnokov EP, Wilbanks SM, Jameson GB, Jameson GNL. Mechanistic Implications of Persulfenate and Persulfide Binding in the Active Site of Cysteine Dioxygenase. Biochemistry. 2013;52:7606–7617. doi: 10.1021/bi400661a. [DOI] [PubMed] [Google Scholar]
- 519.Clausen T, Kaiser JT, Steegborn C, Huber R, Kessler D. Crystal Structure of the Cystine C-S Lyase from Synechocystis: Stabilization of Cysteine Persulfide for FeS Cluster Biosynthesis. Proc Natl Acad Sci U S A. 2000;97:3856–3861. doi: 10.1073/pnas.97.8.3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Tsurugi J, Kawamura S, Horii T. Aryl Hydrodisulfides. J Org Chem. 1971;36:3677–3680. [Google Scholar]
- 521.Anslyn EV, Dougherty DA. Modern Physical Chemistry. University Science Books; South Orange, NJ: 2006. [Google Scholar]
- 522.Jencks WP, Carriuolo J. Reactivity of Nucleophilic Reagents toward Esters. J Am Chem Soc. 1960;82:1778–1786. [Google Scholar]
- 523.Edwards JO, Pearson RG. The Factors Determining Nucleophilic Reactivities. J Am Chem Soc. 1962;84:16–24. [Google Scholar]
- 524.Park C-M, Macinkovic I, Filipovic MR, Xian M. Use of The “tag-Switch” method for the Detection of Protein S-Sulfhydration. Methods Enzymol. 2015;555:39–56. doi: 10.1016/bs.mie.2014.11.033. [DOI] [PubMed] [Google Scholar]
- 525.Abiko Y, Yoshida E, Ishii I, Fukuto JM, Akaike T, Kumagai Y. Involvement of Reactive Persulfides in Biological Bismethylmercury Sulfide Formation. Chem Res Toxicol. 2015;28:1301–1306. doi: 10.1021/acs.chemrestox.5b00101. [DOI] [PubMed] [Google Scholar]
- 526.Millikin R, Bianco CL, White C, Saund SS, Henriquez S, Sosa V, Akaike T, Kumagai Y, Soeda S, Toscano JP, et al. The Chemical Biology of Protein Hydropersulfides: Studies of a Possible Protective Function of Biological Hydropersulfide Generation. Free Radic Biol Med. 2016;97:136–147. doi: 10.1016/j.freeradbiomed.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Ono K, Akaike T, Sawa T, Kumagai Y, Wink DA, Tantillo DJ, Hobbs AJ, Nagy P, Xian M, Lin J, et al. Redox Chemistry and Chemical Biology of H2S, Hydropersulfides, and Derived Species: Implications of Their Possible Biological Activity and Utility. Free Radic Biol Med. 2014;77:82–94. doi: 10.1016/j.freeradbiomed.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528.Everett SA, Wardman P. Perthiols as Antioxidants: Radical-Scavenging and Prooxidative Mechanisms. Methods Enzymol. 1995;251:55–69. doi: 10.1016/0076-6879(95)51110-5. [DOI] [PubMed] [Google Scholar]
- 529.Benson SW. Thermochemistry and Kinetics of Sulfur-Containing Molecules and Radicals. Chem Rev. 1978;78:23–35. [Google Scholar]
- 530.Kawamura S, Abe Y, Tsurugi J. Aralkyl Hydrodisulfides. X. Reactions with Iron Salts. J Org Chem. 1969;34:3633–3635. [Google Scholar]
- 531.Bianco CL, Chavez TA, Sosa V, Saund SS, Nguyen QNN, Tantillo DJ, Ichimura AS, Toscano JP, Fukuto JM. The Chemical Biology of the Persulfide (RSSH)/perthiyl (RSS•) Redox Couple and Possible Role in Biological Redox Signaling. Free Radic Biol Med. 2016;101:20–31. doi: 10.1016/j.freeradbiomed.2016.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Prütz WA. Catalytic Reduction of Fe(III)-Cytochrome-c Involving Stable Radiolysis Products Derived from Disulphides, Proteins and Thiols. Int J Radiat Biol. 1992;61:593–602. doi: 10.1080/09553009214551401. [DOI] [PubMed] [Google Scholar]
- 533.Chauvin J-PR, Haidasz EA, Griesser M, Pratt DA. Polysulfide-1-Oxides React with Peroxyl Radicals as Quickly as Hindered Phenolic Antioxidants and Do so by a Surprising Concerted Homolytic Substitution. Chem Sci. 2016;7:6347–6356. doi: 10.1039/c6sc01434h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Everett SA, Schoeneich C, Stewart JH, Asmus KD. Perthiyl Radicals, Trisulfide Radical Ions, and Sulfate Formation: A Combined Photolysis and Radiolysis Study on Redox Processes with Organic Di- and Trisulfides. J Phys Chem. 1992;96:306–314. [Google Scholar]
- 535.Quintiliani M, Badiello R, Tamba M, Esfandi A, Gorin G. Radiolysis of Glutathione in Oxygen-Containing Solutions of pH7. Int J Radiat Biol Relat Stud Phys, Chem Med. 1977;32:195–202. doi: 10.1080/09553007714550891. [DOI] [PubMed] [Google Scholar]
- 536.Tamba M, Simone G, Quintiliani M. Interactions of Thiyl Free Radicals with Oxygen: A Pulse Radiolysis Study. Int J Radiat Biol Relat Stud Phys, Chem Med. 1986;50:595–600. doi: 10.1080/09553008614550991. [DOI] [PubMed] [Google Scholar]
- 537.Kende I, Pickering TL, Tobolsky AV. The Dissociation Energy of the Tetrasulfide Linkage. J Am Chem Soc. 1965;87:5582–5586. [Google Scholar]
- 538.Madej E, Folkes LK, Wardman P, Czapski G, Goldstein S. Thiyl Radicals React with Nitric Oxide to Form S-Nitrosothiols with Rate Constants near the Diffusion-Controlled Limit. Free Radic Biol Med. 2008;44:2013–2018. doi: 10.1016/j.freeradbiomed.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 539.Bailey TS, Henthorn HA, Pluth MD. The Intersection of NO and H2S: Persulfides Generate NO from Nitrite through Polysulfide Formation. Inorg Chem. 2016;55:12618–12625. doi: 10.1021/acs.inorgchem.6b01660. [DOI] [PubMed] [Google Scholar]
- 540.Kawamura S, Otsuji Y, Nakabayashi T, Kitao T, Tsurugi J. Aralkyl Hydrodisulfides. IV. The Reaction of Benzyl Hydrodisulfide with Several Nucleophiles. J Org Chem. 1965;30:2711–2714. [Google Scholar]
- 541.Tsurugi J, Abe Y, Nakabayashi T, Kawamura S, Kitao T, Niwa M. Aralkyl Hydrodisulfides. XI. Reaction with Amines. J Org Chem. 1970;35:3263–3266. [Google Scholar]
- 542.Kawamura S, Kitao T, Nakabayashi T, Horii T, Tsurugi J. Aralkyl Hydrodisulfides. VIII. Alkaline Decomposition and Its Competition with Nucleophiles. J Org Chem. 1968;33:1179–1181. [Google Scholar]
- 543.Kawamura S, Nakabayashi T, Kitao T, Tsurugi J. Aralkyl Hydrodisulfides. VI. The Reaction of Benzhydryl Hydrosulfide with Several Neucleophiles. J Org Chem. 1966;31:1985–1987. [Google Scholar]
- 544.Portillo-Ledesma S, Sardi F, Manta B, Tourn MV, Clippe A, Knoops B, Alvarez B, Coitiño EL, Ferrer-Sueta G. Deconstructing the Catalytic Efficiency of Peroxiredoxin-5 Peroxidatic Cysteine. Biochemistry. 2014;53:6113–6125. doi: 10.1021/bi500389m. [DOI] [PubMed] [Google Scholar]
- 545.Nakabayashi T, Kawamura S, Kitao T, Tsurugi J. Aralkyl Hydrodisulfides. 1 V. The Reaction of 35 S-Labeled Aralkyl Hydrodisulfides with Triphenylphosphine. J Org Chem. 1966;31:861–864. [Google Scholar]
- 546.Toohey JI. Sulphane Sulphur in Biological Systems: A Possible Regulatory Role. Biochem J. 1989;264:625–632. doi: 10.1042/bj2640625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547.Hansen RE, Roth D, Winther JR. Quantifying the Global Cellular Thiol-Disulfide Status. Proc Natl Acad Sci U S A. 2009;106:422–427. doi: 10.1073/pnas.0812149106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548.Nakabayashi T, Tsurugi J. Aralkyl Hydrodisulfides. II. The Thermal Decomposition of Benzhydryl Hydrodisulfide. J Org Chem. 1963;28:813–816. [Google Scholar]
- 549.Cuevasanta E, Möller MN, Alvarez B. Biological Chemistry of Hydrogen Sulfide and Persulfides. Arch Biochem Biophys. 2017;617:9–25. doi: 10.1016/j.abb.2016.09.018. [DOI] [PubMed] [Google Scholar]
- 550.Sawahata T, Neal RA. Use of 1-Fluoro-2,4-Dinitrobenzene as a Probe for the Presence of Hydrodisulfide Groups in Proteins. Anal Biochem. 1982;126:360–364. doi: 10.1016/0003-2697(82)90528-0. [DOI] [PubMed] [Google Scholar]
- 551.Karala A, Ruddock LW. Does S-Methyl Methanethiosulfonate Trap the Thiol-Disulfide State of Proteins? Antioxid Redox Signal. 2007;9:527–531. doi: 10.1089/ars.2006.1473. [DOI] [PubMed] [Google Scholar]
- 552.Sen N, Paul BD, Gadalla MM, Mustafa AK, Sen T, Xu R, Kim S, Snyder SH. Hydrogen Sulfide-Linked Sulfhydration of NF-κB Mediates Its Antiapoptotic Actions. Mol Cell. 2012;45:13–24. doi: 10.1016/j.molcel.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK, Barodka VM, Gazi FK, Barrow RK, Wang R, et al. Hydrogen Sulfide as Endothelium-Derived Hyperpolarizing Factor Sulfhydrates Potassium Channels. Circ Res. 2011;109:1259–1268. doi: 10.1161/CIRCRESAHA.111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554.Yang G, Zhao K, Ju Y, Mani S, Cao Q, Puukila S, Khaper N, Wu L, Wang R. Hydrogen Sulfide Protects against Cellular Senescence via S-Sulfhydration of Keap1 and Activation of Nrf2. Antioxid Redox Signal. 2013;18:1906–1919. doi: 10.1089/ars.2012.4645. [DOI] [PubMed] [Google Scholar]
- 555.Liu Y, Yang R, Liu X, Zhou Y, Qu C, Kikuiri T, Wang S, Zandi E, Du J, Ambudkar IS, et al. Hydrogen Sulfide Maintains Mesenchymal Stem Cell Function and Bone Homeostasis via Regulation of Ca(2+) Channel Sulfhydration. Cell Stem Cell. 2014;15:66–78. doi: 10.1016/j.stem.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556.Mir S, Sen T, Sen N. Cytokine-Induced GAPDH Sulfhydration Affects PSD95 Degradation and Memory. Mol Cell. 2014;56:786–795. doi: 10.1016/j.molcel.2014.10.019. [DOI] [PubMed] [Google Scholar]
- 557.Zhao K, Ju Y, Li S, Al Tanny Z, Wang R, Yang G. S-Sulfhydration of MEK1 Leads to PARP-1 Activation and DNA Damage Repair. EMBO Rep. 2014;15:792–800. doi: 10.1002/embr.201338213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558.Gao X-H, Krokowski D, Guan B-J, Bederman I, Majumder M, Parisien M, Diatchenko L, Kabil O, Willard B, Banerjee R, et al. Quantitative H2S-Mediated Protein Sulfhydration Reveals Metabolic Reprogramming during the Integrated Stress Response. eLife. 2015;4:e10067. doi: 10.7554/eLife.10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Longen S, Richter F, Köhler Y, Wittig I, Beck K-F, Pfeilschifter J. Quantitative Persulfide Site Identification (qPerS-SID) Reveals Protein Targets of H2S Releasing Donors in Mammalian Cells. Sci Rep. 2016;6:29808. doi: 10.1038/srep29808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Reisz JA, Bechtold E, King SB, Poole LB, Furdui CM. Thiol-Blocking Electrophiles Interfere with Labeling and Detection of Protein Sulfenic Acids. FEBS J. 2013;280:6150–6161. doi: 10.1111/febs.12535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.Krishnan N, Fu C, Pappin DJ, Tonks NK. H2S-Induced Sulfhydration of the Phosphatase PTP1B and Its Role in the Endoplasmic Reticulum Stress Response. Sci Signaling. 2011;4:ra86–ra86. doi: 10.1126/scisignal.2002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562.Parent A, Elduque X, Cornu D, Belot L, Le Caer J-P, Grandas A, Toledano MB, D’Autréaux B. Mammalian Frataxin Directly Enhances Sulfur Transfer of NFS1 Persulfide to Both ISCU and Free Thiols. Nat Commun. 2015;6:5686. doi: 10.1038/ncomms6686. [DOI] [PubMed] [Google Scholar]
- 563.Zhang D, Devarie-Baez NO, Li Q, Lancaster JR, Xian M. Methylsulfonyl Benzothiazole (MSBT): A Selective Protein Thiol Blocking Reagent. Org Lett. 2012;14:3396–3399. doi: 10.1021/ol301370s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564.Aroca A, Benito JM, Gotor C, Romero LC. Persulfidation Proteome Reveals the Regulation of Protein Function by Hydrogen Sulfide in Diverse Biological Processes in Arabidopsis. J Exp Bot. 2017;52:556–567. doi: 10.1093/jxb/erx294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Beinert H. Semi-Micro Methods for Analysis of Labile Sulfide and of Labile Sulfide plus Sulfane Sulfur in Unusually Stable Iron-Sulfur Proteins. Anal Biochem. 1983;131:373–378. doi: 10.1016/0003-2697(83)90186-0. [DOI] [PubMed] [Google Scholar]
- 566.Catsimpoolas N, Wood JL. The Reaction of Cyanide with Bovine Serum Albumin. J Biol Chem. 1964;239:4131–4137. [PubMed] [Google Scholar]
- 567.Fletcher JC, Robson A. The Occurrence of Bis-(2-Amino-2-Carboxyethyl) Trisulphide in Hydrolysates of Wool and Other Proteins. Biochem J. 1963;87:553–559. doi: 10.1042/bj0870553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Chen W, Liu C, Peng B, Zhao Y, Pacheco A, Xian M. New Fluorescent Probes for Sulfane Sulfurs and the Application in Bioimaging. Chem Sci. 2013;4:2892–2892. doi: 10.1039/C3SC50754H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569.Liu C, Chen W, Shi W, Peng B, Zhao Y, Ma H, Xian M. Rational Design and Bioimaging Applications of Highly Selective Fluorescence Probes for Hydrogen Polysulfides. J Am Chem Soc. 2014;136:7257–7260. doi: 10.1021/ja502968x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Chen W, Rosser EW, Matsunaga T, Pacheco A, Akaike T, Xian M. The Development of Fluorescent Probes for Visualizing Intracellular Hydrogen Polysulfides. Angew Chem, Int Ed. 2015;54:13961–13965. doi: 10.1002/anie.201506887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571.Chen W, Pacheco A, Takano Y, Day JJ, Hanaoka K, Xian M. A Single Fluorescent Probe to Visualize Hydrogen Sulfide and Hydrogen Polysulfides with Different Fluorescence Signals. Angew Chem, Int Ed. 2016;55:9993–9996. doi: 10.1002/anie.201604892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572.Han X, Yu F, Song X, Chen L. Quantification of Cysteine Hydropersulfide with a Ratiometric near-Infrared Fluorescent Probe Based on Selenium–sulfur Exchange Reaction. Chem Sci. 2016;7:5098–5107. doi: 10.1039/c6sc00838k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573.Kawagoe R, Takashima I, Uchinomiya S, Ojida A. Reversible Ratiometric Detection of Highly Reactive Hydropersulfides Using a FRET-Based Dual Emission Fluorescent Probe. Chem Sci. 2017;8:1134–1140. doi: 10.1039/c6sc03856e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574.Liu C, Zhang F, Munske G, Zhang H, Xian M. Isotope Dilution Mass Spectrometry for the Quantification of Sulfane Sulfurs. Free Radic Biol Med. 2014;76:200–207. doi: 10.1016/j.freeradbiomed.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Andrews JC. The Optical Activity of Cysteine. J Biol Chem. 1926;2:209–218. [Google Scholar]
- 576.Cavallini D, Federici G, Barboni E. Interaction of Proteins with Sulfide. Eur J Biochem. 1970;14:169–174. doi: 10.1111/j.1432-1033.1970.tb00275.x. [DOI] [PubMed] [Google Scholar]
- 577.Østergaard H, Tachibana C, Winther JR. Monitoring Disulfide Bond Formation in the Eukaryotic Cytosol. J Cell Biol. 2004;166:337–345. doi: 10.1083/jcb.200402120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578.Gutscher M, Pauleau A-L, Marty L, Brach T, Wabnitz GH, Samstag Y, Meyer AJ, Dick TP. Real-Time Imaging of the Intracellular Glutathione Redox Potential. Nat Methods. 2008;5:553–559. doi: 10.1038/nmeth.1212. [DOI] [PubMed] [Google Scholar]
- 579.Yang J, Gupta V, Carroll KS, Liebler DC. Site-Specific Mapping and Quantification of Protein S-Sulphenylation in Cells. Nat Commun. 2014;5:4776. doi: 10.1038/ncomms5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Paulsen CE, Carroll KS. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem Rev. 2013;113:4633–4679. doi: 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581.Devarie-Baez NO, Silva López EI, Furdui CM. Biological Chemistry and Functionality of Protein Sulfenic Acids and Related Thiol Modifications. Free Radical Res. 2016;50:172–194. doi: 10.3109/10715762.2015.1090571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Poole LB, Karplus PA, Claiborne A. Protein Sulfenic Acids in Redox Signaling. Annu Rev Pharmacol Toxicol. 2004;44:325–347. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 583.Malhotra JD, Kaufman RJ. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Cycle or a Double-Edged Sword? Antioxid Redox Signal. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 584.Poynton RA, Hampton MB. Peroxiredoxins as Biomarkers of Oxidative Stress. Biochim Biophys Acta, Gen Subj. 2014;1840:906–912. doi: 10.1016/j.bbagen.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 585.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin Evolution and the Regulation of Hydrogen Peroxide Signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 586.Biteau B, Labarre J, Toledano MB. ATP-Dependent Reduction of Cysteine–sulphinic Acid by S. Cerevisiae Sulphiredoxin. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 587.Woo HA, Chae HZ, Hwang SC, Yang K-S, Kang SW, Kim K, Rhee SG. Reversing the Inactivation of Peroxiredoxins Caused by Cysteine Sulfinic Acid Formation. Science. 2003;300:653–656. doi: 10.1126/science.1080273. [DOI] [PubMed] [Google Scholar]
- 588.Wong PS-Y, Hyun J, Fukuto JM, Shirota FN, DeMaster EG, Shoeman DW, Nagasawa HT. Reaction between S-Nitrosothiols and Thiols: Generation of Nitroxyl (HNO) and Subsequent Chemistry. Biochemistry. 1998;37:5362–5371. doi: 10.1021/bi973153g. [DOI] [PubMed] [Google Scholar]
- 589.Arnelle DR, Stamler JS. NO+, NO, and NO− Donation by S-Nitrosothiols: Implications for Regulation of Physiological Functions by S-Nitrosylation and Acceleration of Disulfide Formation. Arch Biochem Biophys. 1995;318:279–285. doi: 10.1006/abbi.1995.1231. [DOI] [PubMed] [Google Scholar]
- 590.Giustarini D, Milzani A, Aldini G, Carini M, Rossi R, Dalle-Donne I. S-Nitrosation versus S-Glutathionylation of Protein Sulfhydryl Groups by S-Nitrosoglutathione. Antioxid Redox Signal. 2005;7:930–939. doi: 10.1089/ars.2005.7.930. [DOI] [PubMed] [Google Scholar]
- 591.Talipov MR, Timerghazin QK. Protein Control of S-Nitrosothiol Reactivity: Interplay of Antagonistic Resonance Structures. J Phys Chem B. 2013;117:1827–1837. doi: 10.1021/jp310664z. [DOI] [PubMed] [Google Scholar]
- 592.Timerghazin QK, Talipov MR. Unprecedented External Electric Field Effects on S-Nitrosothiols: Possible Mechanism of Biological Regulation? J Phys Chem Lett. 2013;4:1034–1038. doi: 10.1021/jz400354m. [DOI] [PubMed] [Google Scholar]
- 593.Ivanova LV, Cibich D, Deye G, Talipov MR, Timerghazin QK. Modeling of S-Nitrosothiol-Thiol Reactions of Biological Significance: HNO Production by S-Thiolation Requires a Proton Shuttle and Stabilization of Polar Intermediates. ChemBioChem. 2017;18:726–738. doi: 10.1002/cbic.201600556. [DOI] [PubMed] [Google Scholar]
- 594.Vandiver MS, Paul BD, Xu R, Karuppagounder S, Rao F, Snowman AM, Seok Ko H, Il Lee Y, Dawson VL, Dawson TM, et al. Sulfhydration Mediates Neuroprotective Actions of Parkin. Nat Commun. 2013;4:1626. doi: 10.1038/ncomms2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595.Kimura Y, Mikami Y, Osumi K, Tsugane M, Oka J, Kimura H. Polysulfides Are Possible H2S-Derived Signaling Molecules in Rat Brain. FASEB J. 2013;27:2451–2457. doi: 10.1096/fj.12-226415. [DOI] [PubMed] [Google Scholar]
- 596.Greiner R, Dick TP. Real-Time Assays for Monitoring the Influence of Sulfide and Sulfane Sulfur Species on Protein Thiol Redox States. Methods Enzymol. 2015;555:57–77. doi: 10.1016/bs.mie.2014.11.020. [DOI] [PubMed] [Google Scholar]
- 597.Koike S, Ogasawara Y. Sulfur Atom in Its Bound State Is a Unique Element Involved in Physiological Functions in Mammals. Molecules. 2016;21:1753. doi: 10.3390/molecules21121753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598.Kimura H. Physiological Roles of Hydrogen Sulfide and Polysulfides. Handb Exp Pharmacol. 2015;230:61–81. doi: 10.1007/978-3-319-18144-8_3. [DOI] [PubMed] [Google Scholar]
- 599.Kimura Y, Toyofuku Y, Koike S, Shibuya N, Nagahara N, Lefer D, Ogasawara Y, Kimura H. Identification of H2S3 and H2S Produced by 3-Mercaptopyruvate Sulfurtransferase in the Brain. Sci Rep. 2015;5:14774. doi: 10.1038/srep14774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600.Steudel R. Inorganic Polysulfanes H2Sn with N > 1. In: Steudel R, editor. Elemental Sulfur und Sulfur-Rich Compounds II. Springer; Berlin: 2003. pp. 99–126. [Google Scholar]
- 601.Steudel R. Inorganic Polysulfides Sn (2–) and Radical Anions S N (•–) In: Steudel R, editor. Elemental Sulfur und Sulfur-Rich Compounds II. Springer; Berlin: 2003. pp. 127–152. [Google Scholar]
- 602.Teder A, et al. The Equilibrium Between Elementary Sulfur and Aqueous Polysulfide Solutions. Acta Chem Scand. 1971;25:1722–1728. [Google Scholar]
- 603.Steudel R, Holdt G, Göbel T. Ion-Pair Chromatographic Separation of Inorganic Sulphur Anions Including Polysulphide. J Chromatogr A. 1989;475:442–446. [Google Scholar]
- 604.Schwarzenbach G, Fischer A. Acidität Der Sulfane Und Zusammensetzung Der Wässerigen Alkalipolysulfidlösungen. Helv Chim Acta. 1960;43:1365–1390. [Google Scholar]
- 605.Fehér F, Baudler M. Beiträge Zur Chemie Des Schwefels, III. Mitteilung. Praparätive Darstellung Und Eigenschaften Des Wasser-stofftrisulfids. Z Anorg Chem. 1947;254:251–254. [Google Scholar]
- 606.Feher F, Berthold HJ. Beitraege Zur Chemie Des Schwefels, XIII Mitteillung, Die Titrimetriche Bestimmung Reiner Alkalipolysulfide. Fresenius’ Z Anal Chem. 1953;138:245–249. [Google Scholar]
- 607.Kamyshny A, Goifman A, Rizkov D, Lev O. Kinetics of Disproportionation of Inorganic Polysulfides in Undersaturated Aqueous Solutions at Environmentally Relevant Conditions. Aquat Geochem. 2003;9:291–304. [Google Scholar]
- 608.Cloke PL. The Geologic Role of polysulfides—Part I The Distribution of Ionic Species in Aqueous Sodium Polysulfide Solutions. Geochim Cosmochim Acta. 1963;27:1265–1298. [Google Scholar]
- 609.Giggenbach W. Optical Spectra and Equilibrium Distribution of Polysulfide Ions in Aqueous Solution at 20.deg. Inorg Chem. 1972;11:1201–1207. [Google Scholar]
- 610.Hahn J. Zusammensetzung von Rohsulfan, Nachweis Der Sulfane H2S9 Bis H2S35 Composition of Crude Sulfane Oil, Identification of the Sulfanes H2S9 to H2S35. Z Naturforsch, B: J Chem Sci. 1985;40:263–272. [Google Scholar]
- 611.Otto AH, Steudel R. The Gas Phase Acidities of the Sulfanes H2Sn (N = 1–4) Eur J Inorg Chem. 1999;1999:2057–2061. [Google Scholar]
- 612.Steudel R, Holdt G, Nagorka R. On the Autoxidation of Aqueous Sodium Polysulfide [1] Z Naturforsch, B: J Chem Sci. 1986;41:1519–1522. [Google Scholar]
- 613.Hanley AV, Czech FW. In: Analytical Chemistry of Sulfur and its Compounds, Part I. Karchmer JH, editor. Wiley; New York: 1970. p. 285. Chapter 5. [Google Scholar]
- 614.Steudel R. The Chemistry of Organic Polysulfanes R–S(n)–R (N > 2) Chem Rev. 2002;102:3905–3946. doi: 10.1021/cr010127m. [DOI] [PubMed] [Google Scholar]
- 615.Arndt S, Baeza-Garza CD, Logan A, Rosa T, Wedmann R, Prime TA, Martin JL, Saeb-Parsy K, Krieg T, Filipovic MR, et al. Assessment of H2S in Vivo Using the Newly Developed Mitochondria-Targeted Mass Spectrometry Probe MitoA. J Biol Chem. 2017;292:7761–7773. doi: 10.1074/jbc.M117.784678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616.Seel F, Guttler H-J, Simon G, Wieckowski A. Colored Sulfur Species in EPD-Solvents. Pure Appl Chem. 1977;49:45–54. [Google Scholar]
- 617.Iciek M, Kwiecien´ I, Wlodek L. Biological Properties of Garlic and Garlic-Derived Organosulfur Compounds. Environ Mol Mutagen. 2009;50:247–265. doi: 10.1002/em.20474. [DOI] [PubMed] [Google Scholar]
- 618.Benavides GA, Squadrito GL, Mills RW, Patel HD, Isbell TS, Patel RP, Darley-Usmar VM, Doeller JE, Kraus DW. Hydrogen Sulfide Mediates the Vasoactivity of Garlic. Proc Natl Acad Sci U S A. 2007;104:17977–17982. doi: 10.1073/pnas.0705710104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 619.Munday R, Munday JS, Munday CM. Comparative Effects of Mono-, Di-, Tri-, and Tetrasulfides Derived from Plants of the Allium Family: Redox Cycling in Vitro and Hemolytic Activity and Phase 2 Enzyme Induction in Vivo. Free Radic Biol Med. 2003;34:1200–1211. doi: 10.1016/s0891-5849(03)00144-8. [DOI] [PubMed] [Google Scholar]
- 620.Federici G, Duprè S, Matarese RM, Solinas SP, Cavallini D. Is the Alkaline Cleavage of Disulfide Bonds in Peptides an Alpha-Beta Elimination Reaction or a Hydrolysis? Int J Pept Protein Res. 1977;10:185–189. doi: 10.1111/j.1399-3011.1977.tb01732.x. [DOI] [PubMed] [Google Scholar]
- 621.Florence TM. Degradation of Protein Disulphide Bonds in Dilute Alkali. Biochem J. 1980;189:507–520. doi: 10.1042/bj1890507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622.Cavallini D, Mondovi B, De Marco C, Sciosciasantoro A. Inhibitory Effect of Mercaptoethanol and Hypotaurine on the Desulfhydration of Cysteine by Cystathionase. Arch Biochem Biophys. 1962;96:456–457. doi: 10.1016/0003-9861(62)90436-8. [DOI] [PubMed] [Google Scholar]
- 623.Flavin M. Microbial Transsulfuration: The Mechanism of an Enzymatic Disulfide Elimination Reaction. J Biol Chem. 1962;237:768–777. [PubMed] [Google Scholar]
- 624.Yamanishi T, Tuboi S. The Mechanism of the L-Cystine Cleavage Reaction Catalyzed by Rat Liver Gamma-Cystathionase. J Biochem. 1981;89:1913–1921. doi: 10.1093/oxfordjournals.jbchem.a133393. [DOI] [PubMed] [Google Scholar]
- 625.Zheng L, White RH, Cash VL, Jack RF, Dean DR. Cysteine Desulfurase Activity Indicates a Role for NIFS in Metallocluster Biosynthesis. Proc Natl Acad Sci U S A. 1993;90:2754–2758. doi: 10.1073/pnas.90.7.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 626.Frazzon J, Fick JR, Dean DR. Biosynthesis of Iron-Sulphur Clusters Is a Complex and Highly Conserved Process. Biochem Soc Trans. 2002;30:680–685. doi: 10.1042/bst0300680. [DOI] [PubMed] [Google Scholar]
- 627.Yuvaniyama P, Agar JN, Cash VL, Johnson MK, Dean DR. NifS-Directed Assembly of a Transient [2Fe-2S] Cluster within the NifU Protein. Proc Natl Acad Sci U S A. 2000;97:599–604. doi: 10.1073/pnas.97.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628.Zheng L, Dean DR. Catalytic Formation of a Nitrogenase Iron-Sulfur Cluster. J Biol Chem. 1994;269:18723–18726. [PubMed] [Google Scholar]
- 629.Flint DH. Escherichia Coli Contains a Protein That Is Homologous in Function and N-Terminal Sequence to the Protein Encoded by the nifS Gene of Azotobacter Vinelandii and That Can Participate in the Synthesis of the Fe-S Cluster of Dihydroxy-Acid Dehydratase. J Biol Chem. 1996;271:16068–16074. [PubMed] [Google Scholar]
- 630.Mihara H, Kurihara T, Yoshimura T, Soda K, Esaki N. Cysteine Sulfinate Desulfinase, a NIFS-like Protein of Escherichia Coli with Selenocysteine Lyase and Cysteine Desulfurase Activities. Gene Cloning, Purification, and Characterization of a Novel Pyridoxal Enzyme. J Biol Chem. 1997;272:22417–22424. doi: 10.1074/jbc.272.36.22417. [DOI] [PubMed] [Google Scholar]
- 631.Mihara H, Maeda M, Fujii T, Kurihara T, Hata Y, Esaki N. A nifS-like Gene, csdB, Encodes an Escherichia Coli Counterpart of Mammalian Selenocysteine Lyase. Gene Cloning, Purification, Characterization and Preliminary X-Ray Crystallographic Studies. J Biol Chem. 1999;274:14768–14772. doi: 10.1074/jbc.274.21.14768. [DOI] [PubMed] [Google Scholar]
- 632.Patzer SI, Hantke K. SufS Is a NifS-like Protein, and SufD Is Necessary for Stability of the [2Fe-2S] FhuF Protein in Escherichia Coli. J Bacteriol. 1999;181:3307–3309. doi: 10.1128/jb.181.10.3307-3309.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633.Lauhon CT, Kambampati R. The iscS Gene in Escherichia Coli Is Required for the Biosynthesis of 4-Thiouridine, Thiamin, and NAD. J Biol Chem. 2000;275:20096–20103. doi: 10.1074/jbc.M002680200. [DOI] [PubMed] [Google Scholar]
- 634.Mihara H, Kato S, Lacourciere GM, Stadtman TC, Kennedy RAJD, Kurihara T, Tokumoto U, Takahashi Y, Esaki N. The iscS Gene Is Essential for the Biosynthesis of 2-Selenouridine in tRNA and the Selenocysteine-Containing Formate Dehydrogenase H. Proc Natl Acad Sci U S A. 2002;99:6679–6683. doi: 10.1073/pnas.102176099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 635.Loiseau L, Ollagnier-de Choudens S, Lascoux D, Forest E, Fontecave M, Barras F. Analysis of the Heteromeric CsdA-CsdE Cysteine Desulfurase, Assisting Fe-S Cluster Biogenesis in Escherichia Coli. J Biol Chem. 2005;280:26760–26769. doi: 10.1074/jbc.M504067200. [DOI] [PubMed] [Google Scholar]
- 636.Mueller EG. Trafficking in Persulfides: Delivering Sulfur in Biosynthetic Pathways. Nat Chem Biol. 2006;2:185–194. doi: 10.1038/nchembio779. [DOI] [PubMed] [Google Scholar]
- 637.Schaedler TA, Thornton JD, Kruse I, Schwarzländer M, Meyer AJ, van Veen HW, Balk J. A Conserved Mitochondrial ATP-Binding Cassette Transporter Exports Glutathione Polysulfide for Cytosolic Metal Cofactor Assembly. J Biol Chem. 2014;289:23264–23274. doi: 10.1074/jbc.M114.553438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 638.Kessler D. Enzymatic Activation of Sulfur for Incorporation into Biomolecules in Prokaryotes. FEMS Microbiol Rev. 2006;30:825–840. doi: 10.1111/j.1574-6976.2006.00036.x. [DOI] [PubMed] [Google Scholar]
- 639.Kutney GW, Turnbull K. Compounds Containing the Sulfur-Sulfur Double Bond. Chem Rev. 1982;82:333–357. [Google Scholar]
- 640.Kutzelnigg W. Chemical Bonding in Higher Main Group Elements. Angew Chem, Int Ed Engl. 1984;23:272–295. [Google Scholar]
- 641.Steudel R, Drozdova Y, Miaskiewicz K, Hertwig RH, Koch W. How Unstable Are Thiosulfoxides? An Ab Initio MO Study of Various Disulfanes RSSR (R = H, Me, Pr, All), Their Branched Isomers R 2 SS, and the Related Transition States 1, 2. J Am Chem Soc. 1997;119:1990–1996. [Google Scholar]
- 642.Sörbo B. Enzymic Transfer of Sulfur from Mercaptopyruvate to Sulfite or Sulfinates. Biochim Biophys Acta. 1957;24:324–329. doi: 10.1016/0006-3002(57)90201-9. [DOI] [PubMed] [Google Scholar]
- 643.Valentine WN, Toohey JI, Paglia DE, Nakatani M, Brockway RA. Modification of Erythrocyte Enzyme Activities by Persulfides and Methanethiol: Possible Regulatory Role. Proc Natl Acad Sci U S A. 1987;84:1394–1398. doi: 10.1073/pnas.84.5.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 644.D’Imprima E, Mills DJ, Parey K, Brandt U, Kühlbrandt W, Zickermann V, Vonck J. Cryo-EM Structure of Respiratory Complex I Reveals a Link to Mitochondrial Sulfur Metabolism. Biochim Biophys Acta, Bioenerg. 2016;1857:1935–1942. doi: 10.1016/j.bbabio.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 645.Bonomi F, Pagani S, Cerletti P, Cannella C. Rhodanese-Mediated Sulfur Transfer to Succinate Dehydrogenase. Eur J Biochem. 1977;72:17–24. doi: 10.1111/j.1432-1033.1977.tb11219.x. [DOI] [PubMed] [Google Scholar]
- 646.Pagani S, Bonomi F, Cerletti P. Sulfide Insertion into Spinach Ferredoxin by Rhodanese. Biochim Biophys Acta, Protein Struct Mol Enzymol. 1982;700:154–164. [Google Scholar]
- 647.Bonomi F, Pagani S, Cerletti P. Insertion of Sulfide into Ferredoxins Catalyzed by Rhodanese. FEBS Lett. 1977;84:149–152. doi: 10.1016/0014-5793(77)81076-4. [DOI] [PubMed] [Google Scholar]
- 648.Tomati U, Matarese R, Federici G. Ferredoxin Activation by Rhodanese. Phytochemistry. 1974;13:1703–1706. [Google Scholar]
- 649.Cerletti P. Seeking a Better Job for an under-Employed Enzyme: Rhodanese. Trends Biochem Sci. 1986;11:369–372. [Google Scholar]
- 650.Agrò AF, Mavelli I, Cannella C, Federici G. Activation of Porcine Heart Mitochondrial Malate Dehydrogenase by Zero Valence Sulfur and Rhodanese. Biochem Biophys Res Commun. 1976;68:553–560. doi: 10.1016/0006-291x(76)91181-5. [DOI] [PubMed] [Google Scholar]
- 651.Pagani S, Galante YM. Interaction of Rhodanese with Mitochondrial NADH Dehydrogenase. Biochim Biophys Acta, Protein Struct Mol Enzymol. 1983;742:278–284. doi: 10.1016/0167-4838(83)90312-6. [DOI] [PubMed] [Google Scholar]
- 652.Krueger K, Koch K, Jühling A, Tepel M, Scholze A. Low Expression of Thiosulfate Sulfurtransferase (Rhodanese) Predicts Mortality in Hemodialysis Patients. Clin Biochem. 2010;43:95–101. doi: 10.1016/j.clinbiochem.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 653.Fauman EB, Cogswell JP, Lovejoy B, Rocque WJ, Holmes W, Montana VG, Piwnica-Worms H, Rink MJ, Saper MA. Crystal Structure of the Catalytic Domain of the Human Cell Cycle Control Phosphatase, Cdc25A. Cell. 1998;93:617–625. doi: 10.1016/s0092-8674(00)81190-3. [DOI] [PubMed] [Google Scholar]
- 654.Matthies A, Rajagopalan KV, Mendel RR, Leimkuhler S. Evidence for the Physiological Role of a Rhodanese-like Protein for the Biosynthesis of the Molybdenum Cofactor in Humans. Proc Natl Acad Sci U S A. 2004;101:5946–5951. doi: 10.1073/pnas.0308191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 655.Matthies A, Nimtz M, Leimkühler S. Molybdenum Cofactor Biosynthesis in Humans: Identification of a Persulfide Group in the Rhodanese-like Domain of MOCS3 by Mass Spectrometry. Biochemistry. 2005;44:7912–7920. doi: 10.1021/bi0503448. [DOI] [PubMed] [Google Scholar]
- 656.Cheng Z, Zhang J, Ballou DP, Williams CH. Reactivity of Thioredoxin as a Protein Thiol-Disulfide Oxidoreductase. Chem Rev. 2011;111:5768–5783. doi: 10.1021/cr100006x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 657.Lu J, Holmgren A. The Thioredoxin Antioxidant System. Free Radic Biol Med. 2014;66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 658.Buchanan BB, Holmgren A, Jacquot J-P, Scheibe R. Fifty Years in the Thioredoxin Field and a Bountiful Harvest. Biochim Biophys Acta, Gen Subj. 2012;1820:1822–1829. doi: 10.1016/j.bbagen.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 659.Lu J, Holmgren A. The Thioredoxin Superfamily in Oxidative Protein Folding. Antioxid Redox Signal. 2014;21:457–470. doi: 10.1089/ars.2014.5849. [DOI] [PubMed] [Google Scholar]
- 660.Li H, Hanson C, Fuchs JA, Woodward C, Thomas GJ. Determination of the pKa Values of Active-Center Cysteines, Cysteines-32 and –35, in Escherichia Coli Thioredoxin by Raman Spectroscopy. Biochemistry. 1993;32:5800–5808. doi: 10.1021/bi00073a012. [DOI] [PubMed] [Google Scholar]
- 661.Kallis GB, Holmgren A. Differential Reactivity of the Functional Sulfhydryl Groups of Cysteine-32 and Cysteine-35 Present in the Reduced Form of Thioredoxin from Escherichia Coli. J Biol Chem. 1980;255:10261–10265. [PubMed] [Google Scholar]
- 662.Jeng MF, Holmgren A, Dyson HJ. Proton Sharing between Cysteine Thiols in Escherichia Coli Thioredoxin: Implications for the Mechanism of Protein Disulfide Reduction. Biochemistry. 1995;34:10101–10105. doi: 10.1021/bi00032a001. [DOI] [PubMed] [Google Scholar]
- 663.Lennon BW, Williams CH. Enzyme-Monitored Turnover of Escherichia Coli Thioredoxin Reductase: Insights for Catalysis. Biochemistry. 1996;35:4704–4712. doi: 10.1021/bi952521i. [DOI] [PubMed] [Google Scholar]
- 664.Palde PB, Carroll KS. A Universal Entropy-Driven Mechanism for Thioredoxin–target Recognition. Proc Natl Acad Sci U S A. 2015;112:7960–7965. doi: 10.1073/pnas.1504376112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 665.Jeong W, Yoon HW, Lee S-R, Rhee SG. Identification and Characterization of TRP14, a Thioredoxin-Related Protein of 14 kDa. New Insights into the Specificity of Thioredoxin Function. J Biol Chem. 2004;279:3142–3150. doi: 10.1074/jbc.M307932200. [DOI] [PubMed] [Google Scholar]
- 666.Nakamura H, De Rosa S, Roederer M, Anderson MT, Dubs JG, Yodoi J, Holmgren A, Herzenberg LA, Herzenberg LA. Elevation of Plasma Thioredoxin Levels in HIV-Infected Individuals. Int Immunol. 1996;8:603–611. doi: 10.1093/intimm/8.4.603. [DOI] [PubMed] [Google Scholar]
- 667.Burke-Gaffney A, Callister MEJ, Nakamura H. Thioredoxin: Friend or Foe in Human Disease? Trends Pharmacol Sci. 2005;26:398–404. doi: 10.1016/j.tips.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 668.Nakamura H, De Rosa SC, Yodoi J, Holmgren A, Ghezzi P, Herzenberg LA, Herzenberg LA. Chronic Elevation of Plasma Thioredoxin: Inhibition of Chemotaxis and Curtailment of Life Expectancy in AIDS. Proc Natl Acad Sci U S A. 2001;98:2688–2693. doi: 10.1073/pnas.041624998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 669.Couto N, Wood J, Barber J. The Role of Glutathione Reductase and Related Enzymes on Cellular Redox Homoeostasis Network. Free Radic Biol Med. 2016;95:27–42. doi: 10.1016/j.freeradbiomed.2016.02.028. [DOI] [PubMed] [Google Scholar]
- 670.Toledano MB, Delaunay-Moisan A, Outten CE, Igbaria A. Functions and Cellular Compartmentation of the Thioredoxin and Glutathione Pathways in Yeast. Antioxid Redox Signal. 2013;18:1699–1711. doi: 10.1089/ars.2012.5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 671.Prast-Nielsen S, Huang H-H, Williams DL. Thioredoxin Glutathione Reductase: Its Role in Redox Biology and Potential as a Target for Drugs against Neglected Diseases. Biochim Biophys Acta, Gen Subj. 2011;1810:1262–1271. doi: 10.1016/j.bbagen.2011.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 672.Zhao W, Zhang J, Lu Y, Wang R. The Vasorelaxant Effect of H(2)S as a Novel Endogenous Gaseous K(ATP) Channel Opener. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 673.Wang R, Szabo C, Ichinose F, Ahmed A, Whiteman M, Papapetropoulos A. The Role of H2S Bioavailability in Endothelial Dysfunction. Trends Pharmacol Sci. 2015;36:568–578. doi: 10.1016/j.tips.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 674.Hybertson BM, Gao B, Bose SK, McCord JM. Oxidative Stress in Health and Disease: The Therapeutic Potential of Nrf2 Activation. Mol Aspects Med. 2011;32:234–246. doi: 10.1016/j.mam.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 675.Kaspar JW, Niture SK, Jaiswal AK. Nrf2:INrf2 (Keap1) Signaling in Oxidative Stress. Free Radic Biol Med. 2009;47:1304–1309. doi: 10.1016/j.freeradbiomed.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 676.Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang M-I, Kobayashi A, Yamamoto M, Kensler TW, Talalay P. Protection against Electrophile and Oxidant Stress by Induction of the Phase 2 Response: Fate of Cysteines of the Keap1 Sensor Modified by Inducers. Proc Natl Acad Sci U S A. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 677.Calvert JW, Elston M, Nicholson CK, Gundewar S, Jha S, Elrod JW, Ramachandran A, Lefer DJ. Genetic and Pharmacologic Hydrogen Sulfide Therapy Attenuates Ischemia-Induced Heart Failure in Mice. Circulation. 2010;122:11–19. doi: 10.1161/CIRCULATIONAHA.109.920991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 678.Calvert JW, Jha S, Gundewar S, Elrod JW, Ramachandran A, Pattillo CB, Kevil CG, Lefer DJ. Hydrogen Sulfide Mediates Cardioprotection through nrf2 Signaling. Circ Res. 2009;105:365–374. doi: 10.1161/CIRCRESAHA.109.199919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 679.Hourihan JM, Kenna JG, Hayes JD. The Gasotransmitter Hydrogen Sulfide Induces nrf2-Target Genes by Inactivating the keap1 Ubiquitin Ligase Substrate Adaptor through Formation of a Disulfide Bond between Cys-226 and Cys-613. Antioxid Redox Signal. 2013;19:465–481. doi: 10.1089/ars.2012.4944. [DOI] [PubMed] [Google Scholar]
- 680.Xie Z-Z, Shi M-M, Xie L, Wu Z-Y, Li G, Hua F, Bian J-S. Sulfhydration of p66Shc at Cysteine59 Mediates the Antioxidant Effect of Hydrogen Sulfide. Antioxid Redox Signal. 2014;21:2531–2542. doi: 10.1089/ars.2013.5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 681.Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, et al. Electron Transfer between Cytochrome c and p66Shc Generates Reactive Oxygen Species That Trigger Mitochondrial Apoptosis. Cell. 2005;122:221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 682.Zhou H, Ding L, Wu Z, Cao X, Zhang Q, Lin L, Bian J-S. Hydrogen Sulfide Reduces RAGE Toxicity through Inhibition of Its Dimer Formation. Free Radic Biol Med. 2017;104:262–271. doi: 10.1016/j.freeradbiomed.2017.01.026. [DOI] [PubMed] [Google Scholar]
- 683.Ramasamy R, Yan SF, Schmidt AM. Receptor for AGE (RAGE): Signaling Mechanisms in the Pathogenesis of Diabetes and Its Complications. Ann N Y Acad Sci. 2011;1243:88–102. doi: 10.1111/j.1749-6632.2011.06320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 684.Hetz C. The Unfolded Protein Response: Controlling Cell Fate Decisions under ER Stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 685.Feldhammer M, Uetani N, Miranda-Saavedra D, Tremblay ML. PTP1B: a simple enzyme for a complex world. Crit Rev Biochem Mol Biol. 2013;48:430–445. doi: 10.3109/10409238.2013.819830. [DOI] [PubMed] [Google Scholar]
- 686.Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS. Peroxide-Dependent Sulfenylation of the EGFR Catalytic Site Enhances Kinase Activity. Nat Chem Biol. 2011;8:57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 687.Peralta D, Bronowska AK, Morgan B, Dóka É, Van Laer K, Nagy P, Gräter F, Dick TP. A Proton Relay Enhances H2O2 Sensitivity of GAPDH to Facilitate Metabolic Adaptation. Nat Chem Biol. 2015;11:156–163. doi: 10.1038/nchembio.1720. [DOI] [PubMed] [Google Scholar]
- 688.Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, et al. S-Nitrosylated GAPDH Initiates Apoptotic Cell Death by Nuclear Translocation Following Siah1 Binding. Nat Cell Biol. 2005;7:665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 689.Jarosz AP, Wei W, Gauld JW, Auld J, Özcan F, Aslan M, Mutus B. Glyceraldehyde 3-Phosphate Dehydrogenase (GAPDH) Is Inactivated by S-Sulfuration in Vitro. Free Radic Biol Med. 2015;89:512–521. doi: 10.1016/j.freeradbiomed.2015.09.007. [DOI] [PubMed] [Google Scholar]
- 690.Napetschnig J, Wu H. Molecular Basis of NF-κB Signaling. Annu Rev Biophys. 2013;42:443–468. doi: 10.1146/annurev-biophys-083012-130338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 691.Aggarwal BB, Gupta SC, Kim JH. Historical Perspectives on Tumor Necrosis Factor and Its Superfamily: 25 Years Later, a Golden Journey. Blood. 2012;119:651–665. doi: 10.1182/blood-2011-04-325225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 692.Whiteman M, Winyard PG. Hydrogen Sulfide and Inflammation: The Good, the Bad, the Ugly and the Promising. Expert Rev Clin Pharmacol. 2011;4:13–32. doi: 10.1586/ecp.10.134. [DOI] [PubMed] [Google Scholar]
- 693.Du J, Huang Y, Yan H, Zhang Q, Zhao M, Zhu M, Liu J, Chen SX, Bu D, Tang C, et al. Hydrogen Sulfide Suppresses Oxidized Low-Density Lipoprotein (Ox-LDL)-Stimulated Monocyte Chemoattractant Protein 1 Generation from Macrophages via the Nuclear Factor B (NF-kB) Pathway. J Biol Chem. 2014;289:9741–9753. doi: 10.1074/jbc.M113.517995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 694.D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-Ribosyl)ation Reactions in the Regulation of Nuclear Functions. Biochem J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- 695.Cohen-Armon M, Visochek L, Rozensal D, Kalal A, Geistrikh I, Klein R, Bendetz-Nezer S, Yao Z, Seger R. DNA-Independent PARP-1 Activation by Phosphorylated ERK2 Increases Elk1 Activity: A Link to Histone Acetylation. Mol Cell. 2007;25:297–308. doi: 10.1016/j.molcel.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 696.Shulman JM, De Jager PL, Feany MB. Parkinson’s Disease: Genetics and Pathogenesis. Annu Rev Pathol: Mech Dis. 2011;6:193–222. doi: 10.1146/annurev-pathol-011110-130242. [DOI] [PubMed] [Google Scholar]
- 697.Moore DJ, West AB, Dawson VL, Dawson TM. Molecular Pathophysiology of Parkinson’s Disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- 698.Chung KKK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM. S-Nitrosylation of Parkin Regulates Ubiquitination and Compromises Parkin’s Protective Function. Science. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- 699.Scarffe LA, Stevens DA, Dawson VL, Dawson TM. Parkin and PINK1: Much More than Mitophagy. Trends Neurosci. 2014;37:315–324. doi: 10.1016/j.tins.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 700.Herrmann M, Widmann T, Colaianni G, Colucci S, Zallone A, Herrmann W. Increased Osteoclast Activity in the Presence of Increased Homocysteine Concentrations. Clin Chem. 2005;51:2348–2353. doi: 10.1373/clinchem.2005.053363. [DOI] [PubMed] [Google Scholar]
- 701.Xu Z-S, Wang X-Y, Xiao D-M, Hu L-F, Lu M, Wu Z-Y, Bian J-S. Hydrogen Sulfide Protects MC3T3-E1 Osteoblastic Cells against H2O2-Induced Oxidative Damage—implications for the Treatment of Osteoporosis. Free Radic Biol Med. 2011;50:1314–1323. doi: 10.1016/j.freeradbiomed.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 702.Gutteridge S, Tanner SJ, Bray RC. Comparison of the Molybdenum Centres of Native and Desulpho Xanthine Oxidase The Nature of the Cyanide-Labile Sulphur Atom and the Nature of the Proton-Accepting Group. Biochem J. 1978;175:887–897. doi: 10.1042/bj1750887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 703.Carrico RJ, Deutsch HF. Isolation of Human Hepatocuprein and Cerebrocuprein. Their Identity with Erythrocuprein. J Biol Chem. 1969;244:6087–6093. [PubMed] [Google Scholar]
- 704.de Beus MD, Chung J, Colón W. Modification of Cysteine 111 in Cu/Zn Superoxide Dismutase Results in Altered Spectroscopic and Biophysical Properties. Protein Sci. 2004;13:1347–1355. doi: 10.1110/ps.03576904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 705.Calabrese L, Federici G, Bannister WH, Bannister JV, Rotilio G, Finazzi-Agrò A. Labile Sulfur in Human Superoxide Dismutase. Eur J Biochem. 1975;56:305–309. doi: 10.1111/j.1432-1033.1975.tb02234.x. [DOI] [PubMed] [Google Scholar]
- 706.You Z, Cao X, Taylor AB, Hart PJ, Levine RL. Characterization of a Covalent Polysulfane Bridge in Copper–Zinc Superoxide Dismutase. Biochemistry. 2010;49:1191–1198. doi: 10.1021/bi901844d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 707.Kelly DP, Shergill JK, Lu WP, Wood AP. Oxidative Metabolism of Inorganic Sulfur Compounds by Bacteria. Antonie van Leeuwenhoek. 1997;71:95–107. doi: 10.1023/a:1000135707181. [DOI] [PubMed] [Google Scholar]
- 708.Kellogg WW, Cadle RD, Allen ER, Lazrus AL, Martell EA. The Sulfur Cycle. Science. 1972;175:587–596. doi: 10.1126/science.175.4022.587. [DOI] [PubMed] [Google Scholar]
- 709.Bang SW, Clark DS, Keasling JD. Engineering Hydrogen Sulfide Production and Cadmium Removal by Expression of the Thiosulfate Reductase Gene (phsABC) from Salmonella Enterica Serovar Typhimurium in Escherichia Coli. Appl Environ Microbiol. 2000;66:3939–3944. doi: 10.1128/aem.66.9.3939-3944.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 710.Peck HD. Enzymatic Basis for Assimilatory and Dissimilatory Sulfate Reduction. J Bacteriol. 1961;82:933–939. doi: 10.1128/jb.82.6.933-939.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 711.Carbonero F, Benefiel AC, Alizadeh-Ghamsari AH, Gaskins HR. Microbial Pathways in Colonic Sulfur Metabolism and Links with Health and Disease. Front Physiol. 2012;3:448. doi: 10.3389/fphys.2012.00448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 712.Singh S, Lin H. Hydrogen Sulfide in Physiology and Diseases of the Digestive Tract. Microorganisms. 2015;3:866–889. doi: 10.3390/microorganisms3040866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 713.Shatalin K, Shatalina E, Mironov A, Nudler E. H2S: A Universal Defense Against Antibiotics in Bacteria. Science. 2011;334:986–990. doi: 10.1126/science.1209855. [DOI] [PubMed] [Google Scholar]
- 714.Zhang H, Hu L-Y, Hu K-D, He Y-D, Wang S-H, Luo J-P. Hydrogen Sulfide Promotes Wheat Seed Germination and Alleviates Oxidative Damage against Copper Stress. J Integr Plant Biol. 2008;50:1518–1529. doi: 10.1111/j.1744-7909.2008.00769.x. [DOI] [PubMed] [Google Scholar]
- 715.Zhang H, Tang J, Liu X-P, Wang Y, Yu W, Peng W-Y, Fang F, Ma D-F, Wei Z-J, Hu L-Y. Hydrogen Sulfide Promotes Root Organogenesis in Ipomoea Batatas, Salix Matsudana and Glycine Max. J Integr Plant Biol. 2009;51:1086–1094. doi: 10.1111/j.1744-7909.2009.00885.x. [DOI] [PubMed] [Google Scholar]
- 716.Zhang H, Ye Y-K, Wang S-H, Luo J-P, Tang J, Ma D-F. Hydrogen Sulfide Counteracts Chlorophyll Loss in Sweetpotato Seedling Leaves and Alleviates Oxidative Damage against Osmotic Stress. Plant Growth Regul. 2009;58:243–250. [Google Scholar]
- 717.Zhang H, Dou W, Jiang C-X, Wei Z-J, Liu J, Jones RL. Hydrogen Sulfide Stimulates β-Amylase Activity during Early Stages of Wheat Grain Germination. Plant Signaling Behav. 2010;5:1031–1033. doi: 10.4161/psb.5.8.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 718.Zhang H, Hu S-L, Zhang Z-J, Hu L-Y, Jiang C-X, Wei Z-J, Liu J, Wang H-L, Jiang S-T. Hydrogen Sulfide Acts as a Regulator of Flower Senescence in Plants. Postharvest Biol Technol. 2011;60:251–257. [Google Scholar]
- 719.García-Mata C, Lamattina L. Hydrogen Sulphide, a Novel Gasotransmitter Involved in Guard Cell Signalling. New Phytol. 2010;188:977–984. doi: 10.1111/j.1469-8137.2010.03465.x. [DOI] [PubMed] [Google Scholar]
- 720.Jin Z, Xue S, Luo Y, Tian B, Fang H, Li H, Pei Y. Hydrogen Sulfide Interacting with Abscisic Acid in Stomatal Regulation Responses to Drought Stress in Arabidopsis. Plant Physiol Biochem. 2013;62:41–46. doi: 10.1016/j.plaphy.2012.10.017. [DOI] [PubMed] [Google Scholar]
- 721.Lisjak M, Srivastava N, Teklic T, Civale L, Lewandowski K, Wilson I, Wood ME, Whiteman M, Hancock JT. A Novel Hydrogen Sulfide Donor Causes Stomatal Opening and Reduces Nitric Oxide Accumulation. Plant Physiol Biochem. 2010;48:931–935. doi: 10.1016/j.plaphy.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 722.Lisjak M, Teklić T, Wilson ID, Wood M, Whiteman M, Hancock JT. Hydrogen Sulfide Effects on Stomatal Apertures. Plant Signaling Behav. 2011;6:1444–1446. doi: 10.4161/psb.6.10.17104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 723.Kimura H. Hydrogen Sulfide Induces Cyclic AMP and Modulates the NMDA Receptor. Biochem Biophys Res Commun. 2000;267:129–133. doi: 10.1006/bbrc.1999.1915. [DOI] [PubMed] [Google Scholar]
- 724.Nagai Y, Tsugane M, Oka J-I, Kimura H. Hydrogen Sulfide Induces Calcium Waves in Astrocytes. FASEB J. 2004;18:557–559. doi: 10.1096/fj.03-1052fje. [DOI] [PubMed] [Google Scholar]
- 725.Lee SW, Hu Y-S, Hu L-F, Lu Q, Dawe GS, Moore PK, Wong PT-H, Bian J-S. Hydrogen Sulphide Regulates Calcium Homeostasis in Microglial Cells. Glia. 2006;54:116–124. doi: 10.1002/glia.20362. [DOI] [PubMed] [Google Scholar]
- 726.Yong QC, Choo CH, Tan BH, Low C-M, Bian J-S. Effect of Hydrogen Sulfide on Intracellular Calcium Homeostasis in Neuronal Cells. Neurochem Int. 2010;56:508–515. doi: 10.1016/j.neuint.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 727.Lu M, Choo CH, Hu L-F, Tan BH, Hu G, Bian J-S. Hydrogen Sulfide Regulates Intracellular pH in Rat Primary Cultured Glia Cells. Neurosci Res. 2010;66:92–98. doi: 10.1016/j.neures.2009.09.1713. [DOI] [PubMed] [Google Scholar]
- 728.Matsunami M, Tarui T, Mitani K, Nagasawa K, Fukushima O, Okubo K, Yoshida S, Takemura M, Kawabata A. Luminal Hydrogen Sulfide Plays a Pronociceptive Role in Mouse Colon. Gut. 2009;58:751–761. doi: 10.1136/gut.2007.144543. [DOI] [PubMed] [Google Scholar]
- 729.Lee AT-H, Jitendrakumar Shah J, Li L, Cheng Y, Moore PK, Khanna S. A Nociceptive-Intensity-Dependent Role for Hydrogen Sulphide in the Formalin Model of Persistent Inflammatory Pain. Neuroscience. 2008;152:89–96. doi: 10.1016/j.neuroscience.2007.11.052. [DOI] [PubMed] [Google Scholar]
- 730.Nelson MT, Woo J, Kang H-W, Vitko I, Barrett PQ, Perez-Reyes E, Lee J-H, Shin H-S, Todorovic SM. Reducing Agents Sensitize C-Type Nociceptors by Relieving High-Affinity Zinc Inhibition of T-Type Calcium Channels. J Neurosci. 2007;27:8250–8260. doi: 10.1523/JNEUROSCI.1800-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 731.Kawabata A, Ishiki T, Nagasawa K, Yoshida S, Maeda Y, Takahashi T, Sekiguchi F, Wada T, Ichida S, Nishikawa H. Hydrogen Sulfide as a Novel Nociceptive Messenger. Pain. 2007;132:74–81. doi: 10.1016/j.pain.2007.01.026. [DOI] [PubMed] [Google Scholar]
- 732.Andersson DA, Gentry C, Bevan S. TRPA1 Has a Key Role in the Somatic Pro-Nociceptive Actions of Hydrogen Sulfide. PLoS One. 2012;7:e46917. doi: 10.1371/journal.pone.0046917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 733.Ogawa H, Takahashi K, Miura S, Imagawa T, Saito S, Tominaga M, Ohta T. H2S Functions as a Nociceptive Messenger through Transient Receptor Potential Ankyrin 1 (TRPA1) Activation. Neuroscience. 2012;218:335–343. doi: 10.1016/j.neuroscience.2012.05.044. [DOI] [PubMed] [Google Scholar]
- 734.Tsubota-Matsunami M, Noguchi Y, Okawa Y, Sekiguchi F, Kawabata A. Colonic Hydrogen Sulfide-Induced Visceral Pain and Referred Hyperalgesia Involve Activation of Both Ca(v)3.2 and TRPA1 Channels in Mice. J Pharmacol Sci. 2012;119:293–296. doi: 10.1254/jphs.12086sc. [DOI] [PubMed] [Google Scholar]
- 735.Cunha TM, Dal-Secco D, Verri WA, Guerrero AT, Souza GR, Vieira SM, Lotufo CM, Neto AF, Ferreira SH, Cunha FQ. Dual Role of Hydrogen Sulfide in Mechanical Inflammatory Hypernociception. Eur J Pharmacol. 2008;590:127–135. doi: 10.1016/j.ejphar.2008.05.048. [DOI] [PubMed] [Google Scholar]
- 736.Sachs D, Cunha FQ, Ferreira SH. Peripheral Analgesic Blockade of Hypernociception: Activation of arginine/NO/cGMP/protein Kinase G/ATP-Sensitive K+ Channel Pathway. Proc Natl Acad Sci U S A. 2004;101:3680–3685. doi: 10.1073/pnas.0308382101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 737.Distrutti E, Sediari L, Mencarelli A, Renga B, Orlandi S, Antonelli E, Roviezzo F, Morelli A, Cirino G, Wallace JL, et al. Evidence That Hydrogen Sulfide Exerts Antinociceptive Effects in the Gastrointestinal Tract by Activating KATP Channels. J Pharmacol Exp Ther. 2005;316:325–335. doi: 10.1124/jpet.105.091595. [DOI] [PubMed] [Google Scholar]
- 738.Whiteman M, Armstrong JS, Chu SH, Jia-Ling S, Wong B-S, Cheung NS, Halliwell B, Moore PK. The Novel Neuromodulator Hydrogen Sulfide: An Endogenous Peroxynitrite “Scavenger”? J Neurochem. 2004;90:765–768. doi: 10.1111/j.1471-4159.2004.02617.x. [DOI] [PubMed] [Google Scholar]
- 739.Lu M, Hu L-F, Hu G, Bian J-S. Hydrogen Sulfide Protects Astrocytes against H2O2-Induced Neural Injury via Enhancing Glutamate Uptake. Free Radic Biol Med. 2008;45:1705–1713. doi: 10.1016/j.freeradbiomed.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 740.Eto K, Asada T, Arima K, Makifuchi T, Kimura H. Brain Hydrogen Sulfide Is Severely Decreased in Alzheimer’s Disease. Biochem Biophys Res Commun. 2002;293:1485–1488. doi: 10.1016/S0006-291X(02)00422-9. [DOI] [PubMed] [Google Scholar]
- 741.Giuliani D, Ottani A, Zaffe D, Galantucci M, Strinati F, Lodi R, Guarini S. Hydrogen Sulfide Slows down Progression of Experimental Alzheimer’s Disease by Targeting Multiple Pathophysiological Mechanisms. Neurobiol Learn Mem. 2013;104:82–91. doi: 10.1016/j.nlm.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 742.Xuan A, Long D, Li J, Ji W, Zhang M, Hong L, Liu J. Hydrogen Sulfide Attenuates Spatial Memory Impairment and Hippocampal Neuroinflammation in β-Amyloid Rat Model of Alzheimer’s Disease. J Neuroinflammation. 2012;9:202. doi: 10.1186/1742-2094-9-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 743.Zhang H, Gao Y, Zhao F, Dai Z, Meng T, Tu S, Yan Y. Hydrogen Sulfide Reduces mRNA and Protein Levels of Beta-Site Amyloid Precursor Protein Cleaving Enzyme 1 in PC12 Cells. Neurochem Int. 2011;58:169–175. doi: 10.1016/j.neuint.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 744.Nagpure BV, Bian J-S. Hydrogen Sulfide Inhibits A2A Adenosine Receptor Agonist Induced β-Amyloid Production in SH-SY5Y Neuroblastoma Cells via a cAMP Dependent Pathway. PLoS One. 2014;9:e88508. doi: 10.1371/journal.pone.0088508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 745.Paul BD, Sbodio JI, Xu R, Vandiver MS, Cha JY, Snowman AM, Snyder SH. Cystathionine γ-Lyase Deficiency Mediates Neurodegeneration in Huntington’s Disease. Nature. 2014;509:96–100. doi: 10.1038/nature13136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 746.Sbodio JI, Snyder SH, Paul BD. Transcriptional Control of Amino Acid Homeostasis Is Disrupted in Huntington’s Disease. Proc Natl Acad Sci U S A. 2016;113:8843–8848. doi: 10.1073/pnas.1608264113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 747.Snijder PM, Baratashvili M, Grzeschik NA, Leuvenink HGD, Kuijpers L, Huitema S, Schaap O, Giepmans BNG, Kuipers J, Miljkovic JL, et al. Overexpression of Cystathionineγ-Lyase Suppresses Detrimental Effects of Spinocerebellar Ataxia Type 3. Mol Med. 2015;21:758. doi: 10.2119/molmed.2015.00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 748.Predmore BL, Lefer DJ. Development of Hydrogen Sulfide-Based Therapeutics for Cardiovascular Disease. J Cardiovasc Transl Res. 2010;3:487–498. doi: 10.1007/s12265-010-9201-y. [DOI] [PubMed] [Google Scholar]
- 749.Elsey DJ, Fowkes RC, Baxter GF. Regulation of Cardiovascular Cell Function by Hydrogen Sulfide (H2S) Cell Biochem Funct. 2010;28:95–106. doi: 10.1002/cbf.1618. [DOI] [PubMed] [Google Scholar]
- 750.Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, et al. Hydrogen Sulfide Attenuates Myocardial Ischemia-Reperfusion Injury by Preservation of Mitochondrial Function. Proc Natl Acad Sci U S A. 2007;104:15560–15565. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 751.Shen Y, Shen Z, Luo S, Guo W, Zhu YZ. The Cardioprotective Effects of Hydrogen Sulfide in Heart Diseases: From Molecular Mechanisms to Therapeutic Potential. Oxid Med Cell Longevity. 2015;2015:1–13. doi: 10.1155/2015/925167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 752.Szabó C, Papapetropoulos A. Hydrogen Sulphide and Angiogenesis: Mechanisms and Applications. Br J Pharmacol. 2011;164:853–865. doi: 10.1111/j.1476-5381.2010.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 753.Zhu YZ, Wang ZJ, Ho P, Loke YY, Zhu YC, Huang SH, Tan CS, Whiteman M, Lu J, Moore PK. Hydrogen Sulfide and Its Possible Roles in Myocardial Ischemia in Experimental Rats. J Appl Physiol. 2006;102:261–268. doi: 10.1152/japplphysiol.00096.2006. [DOI] [PubMed] [Google Scholar]
- 754.Zhao W, Wang R. H2S-Induced Vasorelaxation and Underlying Cellular and Molecular Mechanisms. Am J Physiol -Hear Circ Physiol. 2002;283:H474–H480. doi: 10.1152/ajpheart.00013.2002. [DOI] [PubMed] [Google Scholar]
- 755.Cheng Y, Ndisang JF, Tang G, Cao K, Wang R. Hydrogen Sulfide-Induced Relaxation of Resistance Mesenteric Artery Beds of Rats. Am J Physiol Hear Circ Physiol. 2004;287:H2316–H2323. doi: 10.1152/ajpheart.00331.2004. [DOI] [PubMed] [Google Scholar]
- 756.Jiang B, Tang G, Cao K, Wu L, Wang R. Molecular Mechanism for H2S-Induced Activation of K ATP Channels. Antioxid Redox Signal. 2010;12:1167–1178. doi: 10.1089/ars.2009.2894. [DOI] [PubMed] [Google Scholar]
- 757.Ishii I, Akahoshi N, Yamada H, Nakano S, Izumi T, Suematsu M. Cystathionine Gamma-Lyase-Deficient Mice Require Dietary Cysteine to Protect against Acute Lethal Myopathy and Oxidative Injury. J Biol Chem. 2010;285:26358–26368. doi: 10.1074/jbc.M110.147439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 758.Papapetropoulos A, Pyriochou A, Altaany Z, Yang G, Marazioti A, Zhou Z, Jeschke MG, Branski LK, Herndon DN, Wang R, et al. Hydrogen Sulfide Is an Endogenous Stimulator of Angiogenesis. Proc Natl Acad Sci U S A. 2009;106:21972–21977. doi: 10.1073/pnas.0908047106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 759.Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Zaid A, Giannogonas P, Cantalupo A, Dhayade S, Karalis KP, Wang R, et al. cGMP-Dependent Protein Kinase Contributes to Hydrogen Sulfide-Stimulated Vasorelaxation. PLoS One. 2012;7:e53319. doi: 10.1371/journal.pone.0053319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 760.Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, Murohara T, Predmore BL, Gojon G, Wang R, et al. H2S Protects against Pressure Overload-Induced Heart Failure via Upregulation of Endothelial Nitric Oxide Synthase. Circulation. 2013;127:1116–1127. doi: 10.1161/CIRCULATIONAHA.112.000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 761.Wang X, Wang Q, Guo W, Zhu YZ. Hydrogen Sulfide Attenuates Cardiac Dysfunction in a Rat Model of Heart Failure: A Mechanism through Cardiac Mitochondrial Protection. Biosci Rep. 2011;31:87–98. doi: 10.1042/BSR20100003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 762.Lin VS, Lippert AR, Chang CJ. Cell-Trappable Fluorescent Probes for Endogenous Hydrogen Sulfide Signaling and Imaging H2O2-Dependent H2S Production. Proc Natl Acad Sci U S A. 2013;110:7131–7135. doi: 10.1073/pnas.1302193110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 763.Tao B-B, Liu S-Y, Zhang C-C, Fu W, Cai W-J, Wang Y, Shen Q, Wang M-J, Chen Y, Zhang L-J, et al. VEGFR2 Functions As an H2S-Targeting Receptor Protein Kinase with Its Novel Cys1045–Cys1024 Disulfide Bond Serving As a Specific Molecular Switch for Hydrogen Sulfide Actions in Vascular Endothelial Cells. Antioxid Redox Signal. 2013;19:448–464. doi: 10.1089/ars.2012.4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 764.Wang Y, Zhao X, Jin H, Wei H, Li W, Bu D, Tang X, Ren Y, Tang C, Du J. Role of Hydrogen Sulfide in the Development of Atherosclerotic Lesions in Apolipoprotein E Knockout Mice. Arterioscler Thromb Vasc Biol. 2009;29:173–179. doi: 10.1161/ATVBAHA.108.179333. [DOI] [PubMed] [Google Scholar]
- 765.Mani S, Li H, Untereiner A, Wu L, Yang G, Austin RC, Dickhout JG, Lhotak S, Meng QH, Wang R. Decreased Endogenous Production of Hydrogen Sulfide Accelerates Atherosclerosis. Circulation. 2013;127:2523–2534. doi: 10.1161/CIRCULATIONAHA.113.002208. [DOI] [PubMed] [Google Scholar]
- 766.Bhatia M. H2S and Inflammation: An Overview. Handb Exp Pharmacol. 2015;230:165–180. doi: 10.1007/978-3-319-18144-8_8. [DOI] [PubMed] [Google Scholar]
- 767.Bhatia M, Wong FL, Fu D, Lau HY, Moochhala SM, Moore PK. Role of Hydrogen Sulfide in Acute Pancreatitis and Associated Lung Injury. FASEB J. 2005;19:623–625. doi: 10.1096/fj.04-3023fje. [DOI] [PubMed] [Google Scholar]
- 768.Ang AD, Rivers-Auty J, Hegde A, Ishii I, Bhatia M. The Effect of CSE Gene Deletion in Caerulein-Induced Acute Pancreatitis in the Mouse. Am J Physiol Gastrointest Liver Physiol. 2013;305:G712–G721. doi: 10.1152/ajpgi.00044.2013. [DOI] [PubMed] [Google Scholar]
- 769.Markó L, Szijártó IA, Filipovic MR, Kaßmann M, Balogh A, Park J-K, Przybyl L, N’diaye G, Krämer S, Anders J, et al. Role of Cystathionine Gamma-Lyase in Immediate Renal Impairment and Inflammatory Response in Acute Ischemic Kidney Injury. Sci Rep. 2016;6:27517. doi: 10.1038/srep27517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 770.Hui Y, Du J, Tang C, Bin G, Jiang H. Changes in Arterial Hydrogen Sulfide (H(2)S) Content during Septic Shock and Endotoxin Shock in Rats. J Infect. 2003;47:155–160. doi: 10.1016/s0163-4453(03)00043-4. [DOI] [PubMed] [Google Scholar]
- 771.Li L, Bhatia M, Zhu YZ, Zhu YC, Ramnath RD, Wang ZJ, Anuar FBM, Whiteman M, Salto-Tellez M, Moore PK. Hydrogen Sulfide Is a Novel Mediator of Lipopolysaccharide-Induced Inflammation in the Mouse. FASEB J. 2005;19:1196–1198. doi: 10.1096/fj.04-3583fje. [DOI] [PubMed] [Google Scholar]
- 772.Zhang H, Zhi L, Moore PK, Bhatia M. Role of Hydrogen Sulfide in Cecal Ligation and Puncture-Induced Sepsis in the Mouse. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1193–L1201. doi: 10.1152/ajplung.00489.2005. [DOI] [PubMed] [Google Scholar]
- 773.Liu Y, Kalogeris T, Wang M, Zuidema M, Wang Q, Dai H, Davis MJ, Hill MA, Korthuis RJ. Hydrogen Sulfide Preconditioning or Neutrophil Depletion Attenuates Ischemia-Reperfusion-Induced Mitochondrial Dysfunction in Rat Small Intestine. Am J Physiol Gastrointest Liver Physiol. 2012;302:G44–G54. doi: 10.1152/ajpgi.00413.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 774.Mard SA, Neisi N, Solgi G, Hassanpour M, Darbor M, Maleki M. Gastroprotective Effect of NaHS Against Mucosal Lesions Induced by Ischemia–Reperfusion Injury in Rat. Dig Dis Sci. 2012;57:1496–1503. doi: 10.1007/s10620-012-2051-5. [DOI] [PubMed] [Google Scholar]
- 775.Medeiros JVR, Bezerra VH, Gomes AS, Barbosa ALR, Lima-Junior RCP, Soares PMG, Brito GAC, Ribeiro RA, Cunha FQ, Souza MHLP. Hydrogen Sulfide Prevents Ethanol-Induced Gastric Damage in Mice: Role of ATP-Sensitive Potassium Channels and Capsaicin-Sensitive Primary Afferent Neurons. J Pharmacol Exp Ther. 2009;330:764–770. doi: 10.1124/jpet.109.152801. [DOI] [PubMed] [Google Scholar]
- 776.Fiorucci S, Antonelli E, Distrutti E, Rizzo G, Mencarelli A, Orlandi S, Zanardo R, Renga B, Di Sante M, Morelli A, et al. Inhibition of Hydrogen Sulfide Generation Contributes to Gastric Injury Caused by Anti-Inflammatory Nonsteroidal Drugs. Gastro-enterology. 2005;129:1210–1224. doi: 10.1053/j.gastro.2005.07.060. [DOI] [PubMed] [Google Scholar]
- 777.Zanardo RCO, Brancaleone V, Distrutti E, Fiorucci S, Cirino G, Wallace JL. Hydrogen Sulfide Is an Endogenous Modulator of Leukocyte-Mediated Inflammation. FASEB J. 2006;20:2118–2120. doi: 10.1096/fj.06-6270fje. [DOI] [PubMed] [Google Scholar]
- 778.Wallace JL, Dicay M, McKnight W, Martin GR. Hydrogen Sulfide Enhances Ulcer Healing in Rats. FASEB J. 2007;21:4070–4076. doi: 10.1096/fj.07-8669com. [DOI] [PubMed] [Google Scholar]
- 779.Flannigan KL, Ferraz JGP, Wang R, Wallace JL. Enhanced Synthesis and Diminished Degradation of Hydrogen Sulfide in Experimental Colitis: A Site-Specific, Pro-Resolution Mechanism. PLoS One. 2013;8:e71962. doi: 10.1371/journal.pone.0071962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 780.Whiteman M, Li L, Rose P, Tan C-H, Parkinson DB, Moore PK. The Effect of Hydrogen Sulfide Donors on Lipopolysaccharide-Induced Formation of Inflammatory Mediators in Macrophages. Antioxid Redox Signal. 2010;12:1147–1154. doi: 10.1089/ars.2009.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 781.Fang L, Li H, Tang C, Geng B, Qi Y, Liu X. Hydrogen Sulfide Attenuates the Pathogenesis of Pulmonary Fibrosis Induced by Bleomycin in Rats. Can J Physiol Pharmacol. 2009;87:531–538. doi: 10.1139/y09-039. [DOI] [PubMed] [Google Scholar]
- 782.Fang L-P, Lin Q, Tang C-S, Liu X-M. Hydrogen Sulfide Suppresses Migration, Proliferation and Myofibroblast Transdifferentiation of Human Lung Fibroblasts. Pulm Pharmacol Ther. 2009;22:554–561. doi: 10.1016/j.pupt.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 783.Perry MM, Hui CK, Whiteman M, Wood ME, Adcock I, Kirkham P, Michaeloudes C, Chung KF. Hydrogen Sulfide Inhibits Proliferation and Release of IL-8 from Human Airway Smooth Muscle Cells. Am J Respir Cell Mol Biol. 2011;45:746–752. doi: 10.1165/rcmb.2010-0304OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 784.Fang L-P, Lin Q, Tang C-S, Liu X-M. Hydrogen Sulfide Attenuates Epithelial–mesenchymal Transition of Human Alveolar Epithelial Cells. Pharmacol Res. 2010;61:298–305. doi: 10.1016/j.phrs.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 785.Castro-Piedras I, Perez-Zoghbi JF. Hydrogen Sulphide Inhibits Ca2+ Release through InsP3 Receptors and Relaxes Airway Smooth Muscle. J Physiol. 2013;591:5999–6015. doi: 10.1113/jphysiol.2013.257790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 786.Huang J, Luo Y, Hao Y, Zhang Y, Chen P, Xu J, Chen M, Luo Y, Zhong N, Xu J, et al. Cellular Mechanism Underlying Hydrogen Sulfide Induced Mouse Tracheal Smooth Muscle Relaxation: Role of BKCa. Eur J Pharmacol. 2014;741:55–63. doi: 10.1016/j.ejphar.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 787.Chen Y, Wang R. The Message in the Air: Hydrogen Sulfide Metabolism in Chronic Respiratory Diseases. Respir Physiol Neurobiol. 2012;184:130–138. doi: 10.1016/j.resp.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 788.Li H, Feng S-J, Zhang G-Z, Wang S-X. Correlation of Lower Concentrations of Hydrogen Sulfide with Atherosclerosis in Chronic Hemodialysis Patients with Diabetic Nephropathy. Blood Purif. 2015;38:188–194. doi: 10.1159/000368883. [DOI] [PubMed] [Google Scholar]
- 789.Yamamoto J, Sato W, Kosugi T, Yamamoto T, Kimura T, Taniguchi S, Kojima H, Maruyama S, Imai E, Matsuo S, et al. Distribution of Hydrogen Sulfide (H2S)-Producing Enzymes and the Roles of the H2S Donor Sodium Hydrosulfide in Diabetic Nephropathy. Clin Exp Nephrol. 2013;17:32–40. doi: 10.1007/s10157-012-0670-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 790.Kundu S, Pushpakumar SB, Tyagi A, Coley D, Sen U. Hydrogen Sulfide Deficiency and Diabetic Renal Remodeling: Role of Matrix Metalloproteinase-9. Am J Physiol Endocrinol Metab. 2013;304:E1365–E1378. doi: 10.1152/ajpendo.00604.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 791.Xia M, Chen L, Muh RW, Li P-L, Li N. Production and Actions of Hydrogen Sulfide, a Novel Gaseous Bioactive Substance, in the Kidneys. J Pharmacol Exp Ther. 2009;329:1056–1062. doi: 10.1124/jpet.108.149963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 792.Ge S-N, Zhao M-M, Wu D-D, Chen Y, Wang Y, Zhu J-H, Cai W-J, Zhu Y-Z, Zhu Y-C. Hydrogen Sulfide Targets EGFR Cys797/Cys798 Residues to Induce Na(+)/K(+)-ATPase Endocytosis and Inhibition in Renal Tubular Epithelial Cells and Increase Sodium Excretion in Chronic Salt-Loaded Rats. Antioxid Redox Signal. 2014;21:2061–2082. doi: 10.1089/ars.2013.5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 793.Lim JJ, Liu Y-H, Khin ESW, Bian J-S. Vasoconstrictive Effect of Hydrogen Sulfide Involves Downregulation of cAMP in Vascular Smooth Muscle Cells. Am J Physiol Cell Physiol. 2008;295:C1261–C1270. doi: 10.1152/ajpcell.00195.2008. [DOI] [PubMed] [Google Scholar]
- 794.Lu M, Liu Y-H, Ho CY, Tiong CX, Bian J-S. Hydrogen Sulfide Regulates cAMP Homeostasis and Renin Degranulation in As4.1 and Rat Renin-Rich Kidney Cells. Am J Physiol Cell Physiol. 2012;302:C59–C66. doi: 10.1152/ajpcell.00341.2010. [DOI] [PubMed] [Google Scholar]
- 795.Bos EM, Wang R, Snijder PM, Boersema M, Damman J, Fu M, Moser J, Hillebrands J-L, Ploeg RJ, Yang G, et al. Cystathionine γ-Lyase Protects against Renal Ischemia/Reperfusion by Modulating Oxidative Stress. J Am Soc Nephrol. 2013;24:759–770. doi: 10.1681/ASN.2012030268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 796.Luo Z-L, Tang L-J, Wang T, Dai R-W, Ren J-D, Cheng L, Xiang K, Tian F-Z. Effects of Treatment with Hydrogen Sulfide on Methionine-Choline Deficient Diet-Induced Non-Alcoholic Steatohepatitis in Rats. J Gastroenterol Hepatol. 2014;29:215–222. doi: 10.1111/jgh.12389. [DOI] [PubMed] [Google Scholar]
- 797.Shirozu K, Tokuda K, Marutani E, Lefer D, Wang R, Ichinose F. Cystathionine γ-Lyase Deficiency Protects Mice from Galactosamine/Lipopolysaccharide-Induced Acute Liver Failure. Antioxid Redox Signal. 2014;20:204–216. doi: 10.1089/ars.2013.5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 798.Peh MT, Anwar AB, Ng DSW, Atan MSBM, Kumar SD, Moore PK. Effect of Feeding a High Fat Diet on Hydrogen Sulfide (H2S) Metabolism in the Mouse. Nitric Oxide. 2014;41:138–145. doi: 10.1016/j.niox.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 799.Hwang S-Y, Sarna LK, Siow YL, O K. High-Fat Diet Stimulates Hepatic Cystathionine β-Synthase and Cystathionineγ-Lyase Expression. Can J Physiol Pharmacol. 2013;91:913–919. doi: 10.1139/cjpp-2013-0106. [DOI] [PubMed] [Google Scholar]
- 800.Fiorucci S, Antonelli E, Mencarelli A, Orlandi S, Renga B, Rizzo G, Distrutti E, Shah V, Morelli A. The Third Gas: H2S Regulates Perfusion Pressure in Both the Isolated and Perfused Normal Rat Liver and in Cirrhosis. Hepatology. 2005;42:539–548. doi: 10.1002/hep.20817. [DOI] [PubMed] [Google Scholar]
- 801.Zeng T, Zhang C-L, Zhu Z-P, Yu L-H, Zhao X-L, Xie K-Q. Diallyl Trisulfide (DATS) Effectively Attenuated Oxidative Stress-Mediated Liver Injury and Hepatic Mitochondrial Dysfunction in Acute Ethanol-Exposed Mice. Toxicology. 2008;252:86–91. doi: 10.1016/j.tox.2008.07.062. [DOI] [PubMed] [Google Scholar]
- 802.Gibson GR, Macfarlane GT, Cummings JH. Occurrence of Sulphate-Reducing Bacteria in Human Faeces and the Relationship of Dissimilatory Sulphate Reduction to Methanogenesis in the Large Gut. J Appl Bacteriol. 1988;65:103–111. doi: 10.1111/j.1365-2672.1988.tb01498.x. [DOI] [PubMed] [Google Scholar]
- 803.Suarez F, Furne J, Springfield J, Levitt M. Production and Elimination of Sulfur-Containing Gases in the Rat Colon. Am J Physiol. 1998;274:G727–G733. doi: 10.1152/ajpgi.1998.274.4.G727. [DOI] [PubMed] [Google Scholar]
- 804.Ohge H, Furne JK, Springfield J, Rothenberger DA, Madoff RD, Levitt MD. Association Between Fecal Hydrogen Sulfide Production and Pouchitis. Dis Colon Rectum. 2005;48:469–475. doi: 10.1007/s10350-004-0820-8. [DOI] [PubMed] [Google Scholar]
- 805.Flannigan KL, McCoy KD, Wallace JL. Eukaryotic and Prokaryotic Contributions to Colonic Hydrogen Sulfide Synthesis. Am J Physiol Gastrointest Liver Physiol. 2011;301:G188–G193. doi: 10.1152/ajpgi.00105.2011. [DOI] [PubMed] [Google Scholar]
- 806.Ohge H, Furne JK, Springfield J, Sueda T, Madoff RD, Levitt MD. The Effect of Antibiotics and Bismuth on Fecal Hydrogen Sulfide and Sulfate-Reducing Bacteria in the Rat. FEMS Microbiol Lett. 2003;228:137–142. doi: 10.1016/S0378-1097(03)00748-1. [DOI] [PubMed] [Google Scholar]
- 807.Loubinoux J, Bronowicki J-P, Pereira IAC, Mougenel J-L, Faou AE. Sulfate-Reducing Bacteria in Human Feces and Their Association with Inflammatory Bowel Diseases. FEMS Microbiol Ecol. 2002;40:107–112. doi: 10.1111/j.1574-6941.2002.tb00942.x. [DOI] [PubMed] [Google Scholar]
- 808.Langendijk PS, Hanssen JT, Van der Hoeven JS. Sulfate-Reducing Bacteria in Association with Human Periodontitis. J Clin Periodontol. 2000;27:943–950. doi: 10.1034/j.1600-051x.2000.027012943.x. [DOI] [PubMed] [Google Scholar]
- 809.Chassard C, Dapoigny M, Scott KP, Crouzet L, Del’Homme C, Marquet P, Martin JC, Pickering G, Ardid D, Eschalier A, et al. Functional Dysbiosis within the Gut Microbiota of Patients with Constipated-Irritable Bowel Syndrome. Aliment Pharmacol Ther. 2012;35:828–838. doi: 10.1111/j.1365-2036.2012.05007.x. [DOI] [PubMed] [Google Scholar]
- 810.Wang L, Zhang J, Guo Z, Kwok L, Ma C, Zhang W, Lv Q, Huang W, Zhang H. Effect of Oral Consumption of Probiotic Lactobacillus Planatarum P-8 on Fecal Microbiota, SIgA, SCFAs, and TBAs of Adults of Different Ages. Nutrition. 2014;30:776–783. doi: 10.1016/j.nut.2013.11.018. [DOI] [PubMed] [Google Scholar]
- 811.Sawin EA, De Wolfe TJ, Aktas B, Stroup BM, Murali SG, Steele JL, Ney DM. Glycomacropeptide Is a Prebiotic That Reduces Desulfovibrio Bacteria, Increases Cecal Short-Chain Fatty Acids, and Is Anti-Inflammatory in Mice. Am J Physiol Gastrointest Liver Physiol. 2015;309:G590–601. doi: 10.1152/ajpgi.00211.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 812.d’Emmanuele di Villa Bianca R, Sorrentino R, Maffia P, Mirone V, Imbimbo C, Fusco F, De Palma R, Ignarro LJ, Cirino G. Hydrogen Sulfide as a Mediator of Human Corpus Cavernosum Smooth-Muscle Relaxation. Proc Natl Acad Sci U S A. 2009;106:4513–4518. doi: 10.1073/pnas.0807974105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 813.Qiu X, Villalta J, Lin G, Lue TF. Role of Hydrogen Sulfide in the Physiology of Penile Erection. J Androl. 2012;33:529–535. doi: 10.2164/jandrol.111.014936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 814.Srilatha B, Adaikan PG, Moore PK. Possible Role for the Novel Gasotransmitter Hydrogen Sulphide in Erectile Dysfunction—a Pilot Study. Eur J Pharmacol. 2006;535:280–282. doi: 10.1016/j.ejphar.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 815.Srilatha B, Adaikan PG, Li L, Moore PK. Hydrogen Sulphide: A Novel Endogenous Gasotransmitter Facilitates Erectile Function. J Sex Med. 2007;4:1304–1311. doi: 10.1111/j.1743-6109.2007.00561.x. [DOI] [PubMed] [Google Scholar]
- 816.Sparatore A, Santus G, Giustarini D, Rossi R, Del Soldato P. Therapeutic Potential of New Hydrogen Sulfide-Releasing Hybrids. Expert Rev Clin Pharmacol. 2011;4:109–121. doi: 10.1586/ecp.10.122. [DOI] [PubMed] [Google Scholar]
- 817.Li G, Xie ZZ, Chua JMW, Wong PC, Bian J. Hydrogen Sulfide Protects Testicular Germ Cells against Heat-Induced Injury. Nitric Oxide. 2015;46:165–171. doi: 10.1016/j.niox.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 818.Liang R, Yu W, Du J, Yang L, Shang M, Guo J. Localization of Cystathionine Beta Synthase in Mice Ovaries and Its Expression Profile during Follicular Development. Chin Med J (Engl) 2006;119:1877–1883. [PubMed] [Google Scholar]
- 819.Patel P, Vatish M, Heptinstall J, Wang R, Carson RJ. The Endogenous Production of Hydrogen Sulphide in Intrauterine Tissues. Reprod Biol Endocrinol. 2009;7:10. doi: 10.1186/1477-7827-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 820.Srilatha B, Hu L, Adaikan GP, Moore PK. Initial Characterization of Hydrogen Sulfide Effects in Female Sexual Function. J Sex Med. 2009;6:1875–1884. doi: 10.1111/j.1743-6109.2009.01291.x. [DOI] [PubMed] [Google Scholar]
- 821.Wang K, Ahmad S, Cai M, Rennie J, Fujisawa T, Crispi F, Baily J, Miller MR, Cudmore M, Hadoke PWF, et al. Dysregulation of Hydrogen Sulfide Producing Enzyme Cystathionine γ-Lyase Contributes to Maternal Hypertension and Placental Abnormalities in Preeclampsia. Circulation. 2013;127:2514–2522. doi: 10.1161/CIRCULATIONAHA.113.001631. [DOI] [PubMed] [Google Scholar]
- 822.Guzmán MA, Navarro MA, Carnicer R, Sarría AJ, Acín S, Arnal C, Muniesa P, Surra JC, Arbonés-Mainar JM, Maeda N, et al. Cystathionine Beta-Synthase Is Essential for Female Reproductive Function. Hum Mol Genet. 2006;15:3168–3176. doi: 10.1093/hmg/ddl393. [DOI] [PubMed] [Google Scholar]
- 823.Ning N, Zhu J, Du Y, Gao X, Liu C, Li J, Wang R, Zhao W, Zhang J, Lu Y, et al. Dysregulation of Hydrogen Sulphide Metabolism Impairs Oviductal Transport of Embryos. Nat Commun. 2014;5:1792–1798. doi: 10.1038/ncomms5107. [DOI] [PubMed] [Google Scholar]
- 824.Hayden LJ, Franklin KJ, Roth SH, Moore GJ. Inhibition of Oxytocin-Induced but Not Angiotensin-Induced Rat Uterine Contractions Following Exposure to Sodium Sulfide. Life Sci. 1989;45:2557–2560. doi: 10.1016/0024-3205(89)90239-7. [DOI] [PubMed] [Google Scholar]
- 825.Sidhu R, Singh M, Samir G, Carson RJ. L-Cysteine and Sodium Hydrosulphide Inhibit Spontaneous Contractility in Isolated Pregnant Rat Uterine Strips in Vitro. Pharmacol Toxicol. 2001;88:198–203. doi: 10.1034/j.1600-0773.2001.d01-104.x. [DOI] [PubMed] [Google Scholar]
- 826.Robinson H, Wray S. A New Slow Releasing, H2S Generating Compound, GYY4137 Relaxes Spontaneous and Oxytocin-Stimulated Contractions of Human and Rat Pregnant Myometrium. PLoS One. 2012;7:e46278. doi: 10.1371/journal.pone.0046278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 827.Mijušković A, Oreščanin-Dušić Z, Nikolić-Kokić A, Slavić M, Spasić MB, Spasojević I, Blagojević D. Comparison of the Effects of Methanethiol and Sodium Sulphide on Uterine Contractile Activity. Pharmacol Rep. 2014;66:373–379. doi: 10.1016/j.pharep.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 828.Volpato GP, Searles R, Yu B, Scherrer-Crosbie M, Bloch KD, Ichinose F, Zapol WM. Inhaled Hydrogen Sulfide. Anesthesiology. 2008;108:659–668. doi: 10.1097/ALN.0b013e318167af0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 829.Peng Y-J, Nanduri J, Raghuraman G, Souvannakitti D, Gadalla MM, Kumar GK, Snyder SH, Prabhakar NR. H2S Mediates O2 Sensing in the Carotid Body. Proc Natl Acad Sci U S A. 2010;107:10719–10724. doi: 10.1073/pnas.1005866107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 830.Peng Y-J, Makarenko VV, Nanduri J, Vasavda C, Raghuraman G, Yuan G, Gadalla MM, Kumar GK, Snyder SH, Prabhakar NR. Inherent Variations in CO-H2S-Mediated Carotid Body O2 Sensing Mediate Hypertension and Pulmonary Edema. Proc Natl Acad Sci U S A. 2014;111:1174–1179. doi: 10.1073/pnas.1322172111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 831.Li Q, Sun B, Wang X, Jin Z, Zhou Y, Dong L, Jiang L-H, Rong W. A Crucial Role for Hydrogen Sulfide in Oxygen Sensing via Modulating Large Conductance Calcium-Activated Potassium Channels. Antioxid Redox Signal. 2010;12:1179–1189. doi: 10.1089/ars.2009.2926. [DOI] [PubMed] [Google Scholar]
- 832.Miller DL, Roth MB. Hydrogen Sulfide Increases Thermotolerance and Lifespan in Caenorhabditis Elegans. Proc Natl Acad Sci U S A. 2007;104:20618–20622. doi: 10.1073/pnas.0710191104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 833.Qabazard B, Li L, Gruber J, Peh MT, Ng LF, Kumar SD, Rose P, Tan C-H, Dymock BW, Wei F, et al. Hydrogen Sulfide Is an Endogenous Regulator of Aging in Caenorhabditis Elegans. Antioxid Redox Signal. 2014;20:2621–2630. doi: 10.1089/ars.2013.5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 834.Hellmich MR, Szabo C. Hydrogen Sulfide and Cancer. Handb Exp Pharmacol. 2015;230:233–241. doi: 10.1007/978-3-319-18144-8_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 835.Hellmich MR, Coletta C, Chao C, Szabo C. The Therapeutic Potential of Cystathionine β-Synthetase/Hydrogen Sulfide Inhibition in Cancer. Antioxid Redox Signal. 2015;22:424–448. doi: 10.1089/ars.2014.5933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 836.Szabo C, Ransy C, Módis K, Andriamihaja M, Murghes B, Coletta C, Olah G, Yanagi K, Bouillaud F. Regulation of Mitochondrial Bioenergetic Function by Hydrogen Sulfide. Part I. Biochemical and Physiological Mechanisms. Br J Pharmacol. 2014;171:2099–2122. doi: 10.1111/bph.12369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 837.Lee ZW, Zhou J, Chen C-S, Zhao Y, Tan C-H, Li L, Moore PK, Deng L-W. The Slow-Releasing Hydrogen Sulfide Donor, GYY4137, Exhibits Novel Anti-Cancer Effects In Vitro and In Vivo. PLoS One. 2011;6:e21077. doi: 10.1371/journal.pone.0021077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 838.Chattopadhyay M, Kodela R, Nath N, Barsegian A, Boring D, Kashfi K. Hydrogen Sulfide-Releasing Aspirin Suppresses NF-κB Signaling in Estrogen Receptor Negative Breast Cancer Cells in Vitro and in Vivo. Biochem Pharmacol. 2012;83:723–732. doi: 10.1016/j.bcp.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 839.Chattopadhyay M, Kodela R, Olson KR, Kashfi K. NOSH-Aspirin (NBS-1120), a Novel Nitric Oxide- and Hydrogen Sulfide-Releasing Hybrid Is a Potent Inhibitor of Colon Cancer Cell Growth in Vitro and in a Xenograft Mouse Model. Biochem Biophys Res Commun. 2012;419:523–528. doi: 10.1016/j.bbrc.2012.02.051. [DOI] [PubMed] [Google Scholar]
- 840.Sumi D, Ignarro LJ. Sp1 Transcription Factor Expression Is Regulated by Estrogen-Related Receptor alpha1. Biochem Biophys Res Commun. 2005;328:165–172. doi: 10.1016/j.bbrc.2004.12.165. [DOI] [PubMed] [Google Scholar]
- 841.Maor S, Mayer D, Yarden RI, Lee AV, Sarfstein R, Werner H, Papa MZ. Estrogen Receptor Regulates Insulin-like Growth Factor-I Receptor Gene Expression in Breast Tumor Cells: Involvement of Transcription Factor Sp1. J Endocrinol. 2006;191:605–612. doi: 10.1677/joe.1.07016. [DOI] [PubMed] [Google Scholar]
- 842.Wu D, Si W, Wang M, Lv S, Ji A, Li Y. Hydrogen Sulfide in Cancer: Friend or Foe? Nitric Oxide. 2015;50:38–45. doi: 10.1016/j.niox.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 843.Sen S, Kawahara B, Gupta D, Tsai R, Khachatryan M, Roy-Chowdhuri S, Bose S, Yoon A, Faull K, Farias-Eisner R, et al. Role of Cystathionine β-Synthase in Human Breast Cancer. Free Radic Biol Med. 2015;86:228–238. doi: 10.1016/j.freeradbiomed.2015.05.024. [DOI] [PubMed] [Google Scholar]
- 844.Thornburg JM, Nelson KK, Clem BF, Lane AN, Arumugam S, Simmons A, Eaton JW, Telang S, Chesney J. Targeting Aspartate Aminotransferase in Breast Cancer. Breast Cancer Res. 2008;10:R84. doi: 10.1186/bcr2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 845.Zhang W, Braun A, Bauman Z, Olteanu H, Madzelan P, Banerjee R. Expression Profiling of Homocysteine Junction Enzymes in the NCI60 Panel of Human Cancer Cell Lines. Cancer Res. 2005;65:1554–1560. doi: 10.1158/0008-5472.CAN-04-1554. [DOI] [PubMed] [Google Scholar]
- 846.Zhao H, Li Q, Wang J, Su X, Ng KM, Qiu T, Shan L, Ling Y, Wang L, Cai J, et al. Frequent Epigenetic Silencing of the Folate-Metabolising Gene Cystathionine-Beta-Synthase in Gastrointestinal Cancer. PLoS One. 2012;7:e49683. doi: 10.1371/journal.pone.0049683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 847.Takano N, Sarfraz Y, Gilkes DM, Chaturvedi P, Xiang L, Suematsu M, Zagzag D, Semenza GL. Decreased Expression of Cystathionine β-Synthase Promotes Glioma Tumorigenesis. Mol Cancer Res. 2014;12:1398–1406. doi: 10.1158/1541-7786.MCR-14-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 848.Kim J, Hong SJ, Park JH, Park SY, Kim SW, Cho EY, Do I-G, Joh J-W, Kim DS. Expression of Cystathionine Beta-Synthase Is Downregulated in Hepatocellular Carcinoma and Associated with Poor Prognosis. Oncol Rep. 2009;21:1449–1454. doi: 10.3892/or_00000373. [DOI] [PubMed] [Google Scholar]
- 849.Cao Q, Zhang L, Yang G, Xu C, Wang R. Butyrate-Stimulated H2S Production in Colon Cancer Cells. Antioxid Redox Signal. 2010;12:1101–1109. doi: 10.1089/ars.2009.2915. [DOI] [PubMed] [Google Scholar]
- 850.Fan K, Li N, Qi J, Yin P, Zhao C, Wang L, Li Z, Zha X. Wnt/β-Catenin Signaling Induces the Transcription of Cystathionine-γ-Lyase, a Stimulator of Tumor in Colon Cancer. Cell Signalling. 2014;26:2801–2808. doi: 10.1016/j.cellsig.2014.08.023. [DOI] [PubMed] [Google Scholar]
- 851.Pan Y, Ye S, Yuan D, Zhang J, Bai Y, Shao C. Hydrogen Sulfide (H2S)/cystathionine γ-Lyase (CSE) Pathway Contributes to the Proliferation of Hepatoma Cells. Mutat Res, Fundam Mol Mech Mutagen. 2014;763–764:10–18. doi: 10.1016/j.mrfmmm.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 852.Yin P, Zhao C, Li Z, Mei C, Yao W, Liu Y, Li N, Qi J, Wang L, Shi Y, et al. Sp1 Is Involved in Regulation of Cystathionine γ-Lyase Gene Expression and Biological Function by PI3K/Akt Pathway in Human Hepatocellular Carcinoma Cell Lines. Cell Signalling. 2012;24:1229–1240. doi: 10.1016/j.cellsig.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 853.Ma K, Liu Y, Zhu Q, Liu C, Duan J-L, Tan BK-H, Zhu YZ. H2S Donor, S-Propargyl-Cysteine, Increases CSE in SGC-7901 and Cancer-Induced Mice: Evidence for a Novel Anti-Cancer Effect of Endogenous H2S? PLoS One. 2011;6:e20525. doi: 10.1371/journal.pone.0020525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 854.Chattopadhyay M, Kodela R, Nath N, Dastagirzada YM, Velázquez-Martínez CA, Boring D, Kashfi K. Hydrogen Sulfide-Releasing NSAIDs Inhibit the Growth of Human Cancer Cells: A General Property and Evidence of a Tissue Type-Independent Effect. Biochem Pharmacol. 2012;83:715–722. doi: 10.1016/j.bcp.2011.12.018. [DOI] [PubMed] [Google Scholar]