

Abstract
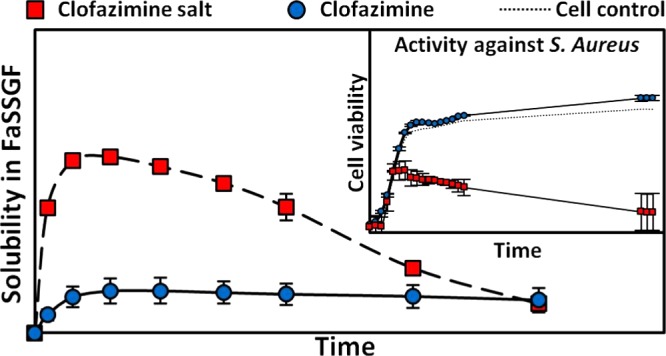
Clofazimine is an antimycobacterial agent that is routinely used for the treatment of leprosy. Clofazimine has also been shown to have high clinical potential for the treatment of many Gram-positive pathogens, including those that exhibit high levels of antibiotic resistance in the medical community. The use of clofazimine against these pathogens has largely been limited by the inherently poor water solubility of the drug substance. In this work, the possibility of repurposing and reformulating clofazimine to maximize its clinical potential is investigated. To achieve this, the potential of novel salt forms of clofazimine as supersaturating drug-delivery vehicles to enhance the aqueous solubility and gastrointestinal solubility of the drug substance was explored. The solution properties of seven novel salt forms, identified during an initial screening process, were examined in water and in a gastrointestinal-like media and were compared and contrasted with those of the free base, clofazimine, and the commercial formulation of the drug, Lamprene. The stability of the most promising solid forms was tested, and their bioactivity against Staphylococcus aureus was also compared with that of the clofazimine free base and Lamprene. Salts forms which showed superior stability as well as solubility and activity to the commercial drug formulation were fully characterized using a combination of spectroscopic techniques, including X-ray diffraction, solid-state NMR, and Fourier transform infrared spectroscopy.
Introduction
Antimicrobial resistance (AR) is a prominent global threat which has been accelerated by decades of constant selection pressures from the human use of antibiotic resources.1−3 It has been estimated that if action is not taken to slow the spread of AR, the financial cost to the global economy between now and 2050 will be around $100 trillion as well as result in the loss of countless human lives.4,5 AR has resulted in the emergence of multidrug resistant (MDR) strains of dangerous pathogens, such as Mycobacterium tuberculosis (MDR-TB) and Staphylococcus aureus,1−3 as well as an increase in drug resistance among bacteria that cause common healthcare-associated infections (HAIs).1−3 HAIs are largely the product of 19 pathogens that have developed worrying levels of resistance and are now capable of causing serious and difficult-to-treat infections.1 Six of these pathogens are known by the acronym “ESKAPE” and are the most common cause of life-threatening nosocomial infections amongst the critically ill and immunocomprised.5−9
There have been many suggestions on how best to tackle the looming AR problem, and the generation of novel antimicrobial agents with new and specific modes of actions is often suggested as the solution.10 Such an approach is likely to take decades for sufficient numbers of new antimicrobial drug substances to be identified and to get regulatory approval to treat the ever increasing number of pathogens that are showing dangerous levels of resistance.6 A more immediate strategy is to repurpose existing drug substances to fill the antibiotic discovery void until new treatments become available.6 This has proven successful in the past, for example, with the administration of β-lactamases inhibitors, such as clavulanic acid, along with β-lactamases antibiotics such as amoxicillin.6,11,12 Recently, researchers have begun to screen libraries of Food and Drug Administration (FDA) approved drugs against biological threats in vitro. Madrid et al. screened 1012 regulatory-approved drugs against various viral and bacterial agents, resulting in the identification of 333 unique hits,13 Chopra et al. identified five compounds that showed significant antibiotic activity against drug-resistant Acinetobacter baumannii,(14) and Kim et al. discovered 15 approved drugs that were able to kill Candida albicans in vitro, one of which showed a wide spectrum of antimicrobial activity.15
Clofazimine (CFZ) is an antimicrobial drug that is currently employed as part of the World Health Organization’s (WHO) triple drug regimen for the treatment of leprosy.16 CFZ is known to exhibit good in vitro activity against most Gram-positive bacteria species, including strains that are multidrug resistant, with minimum inhibitory concentrations (MICs) in the range 0.5–2 mg/L in most cases.17 Gram-negative bacteria, on the other hand, are uniformly resistant to CFZ.17,18 Gram-positive bacteria susceptible to CFZ include the aforementioned ESKAPE pathogens, Enterococcus faecium and S. aureus, as well as several pathogens which contribute to HAIs, namely Clostridium difficile, Enterococcus faecalis, Staphylococcus epidermis, and Streptococcus pneumoniae.(17−24) CFZ is also categorized as a group 5 medicine for the treatment of extensively drug-resistant TB and is included on the WHO list of essential medicines for both adults and children.17,25,26 The clinical use of CFZ in the treatment of many pathogens has been greatly limited due to its poor aqueous solubility (<0.01 mg/L).16,17,27,28 The limited water solubility and high lipophilicity (log P = 7.66) of CFZ result in the accumulation of this drug substance in fatty tissue.27,29−31 However, the antimicrobial potential of CFZ has resulted in a renewal of interest in the drug in recent times, and in 2016 CFZ was re-patented for the treatment of Clostridium difficile infections in humans.32,33 Despite the reports of good activity in vitro, CFZ has consistently performed poorly in vivo.17 For example, CFZ has never been successfully used in the treatment of MDR-TB despite being reportedly active against the pathogen in vitro and being recommended by the WHO to this end.17,28 To overcome the poor water solubility of CFZ, in vitro bioactivity assays are usually carried out using dimethyl sulfoxide (DMSO) as a solubilizing agent.17,19,23,28,34−37 Thus, the poor correlation between in vitro and in vivo activities is likely related to the poor water solubility of CFZ. If the aqueous solubility of CFZ could be improved, then the therapeutic potential of this drug substance might be reached in vivo.
Formulation strategies to improve solubility are broadly characterized as either solubilizing strategies (i.e., formulation with solubilizing agents) or supersaturating drug-delivery systems (SDDS).38,39 The latter involves the administration of high-energy solid forms, such as amorphous solids, crystalline salts or cocrystals and allows the generation of supersaturated solutions.38,39 The main disadvantage of these systems is their tendency to precipitate from solution and to revert to the equilibrium solubility of a more stable solid form.38,39 Several previous attempts to address the aqueous solubility of CFZ have been made with varying degrees of success. These included solubilizing strategies, such as the formulation of CFZ with bile salts,40 encapsulation of the drug into liposomes,41,42 formulation with water-soluble polymer carriers,43−45 conjugation onto modified cyclodextrin,46 and formulation as amorphous solid dispersions.47 The failure of these previous studies was likely due to the large hydrophobic structure of the CFZ molecules, and it associated inherently poor aqueous solubility. Of the various SDDS approaches, the use of crystalline salts is arguably the most well known. In the pharmaceutical industry, salt formation has become a commonly applied technique for increasing solubility and dissolution rates during the drug product development process.48 The resulting crystalline salts dissociate in solution to form ionized species that are more soluble in water compared to the unionized molecules.38,48 Even without crystallization inhibitors, pharmaceutical salts often generate supersaturation levels where precipitation out of solution is slow enough to improve drug bioavailability.49 CFZ itself is a weak base (pKa = 8.51129) and has previously been shown to readily form salts with a variety of acid coformers and with acidic polymers forming ionic solid dispersions.50,51 The salts were characterized, and their solution properties were studied in a dissolution medium of ethanol and water. This choice of media may have been due to the poor aqueous solubility of these CFZ salts, and the choice of counterion had no apparent rationality. In this study, the optimal solid form of CFZ is isolated based on its solubility and supersaturation in water and, more importantly, in a gastrointestinal-like environment. This should maximize the potential of the resulting solid form to increase absorption in the gastrointestinal tract and thus improve the bioavailability of the drug.
Results and Discussion
To improve the aqueous solubility of CFZ, a systematic screening process for new salt forms of the drug was developed based on supersaturation potential in water and in gastrointestinal-like media (Scheme 1). Acid coformers were selected such that ΔpKa, the difference between the pKa values of the acid coformers and that of CFZ (pKa = 8.51129), remained greater than 2 pKa units (Table 1). This is in keeping with the hypothesis that if ΔpKa is greater than 2, salt formation occurs.50,52 During these slurry experiments, the formation of a novel solid phase was confirmed by powder X-ray diffraction (PXRD) (Figure 1).
Scheme 1. Schematic Description of the Screening Procedure To Select Solid Forms Based on Solubility in Aqueous Media, in Fasted-State Simulated Gastric Fluid (FaSSGF) and in Fasted-State Simulated Intestinal Fluid (FaSSIF).

Step 1: screening CFZ for salt formation in various 1 M acidic solutions, isolation and monitoring of the solid phase present via vacuum filtration and PXRD. Step 2: determination of the supersaturation profile of the new solid form in deionized water, FaSSGF, and FaSSIF, followed by isolation and monitoring of the solid phase present via vacuum filtration and PXRD.
Table 1. List of Coformers Used in Solid Form Screening, Their pKa Values, and ΔpKa Relative to CFZa.
| coformers | pKa | ΔpKaa | abbreviation |
|---|---|---|---|
| hydrochloric acid | <−6 | 14.511 | HCl |
| sulfuric acid | –3 | 11.511 | Sul |
| nitric acid | –1.3 | 10.211 | Nit |
| oxalic acid | 1.27 | 7.241 | Oxa |
| phosphoric acid | 2.12 | 6.391 | Pho |
| citric acid | 3.1 | 5.411 | Cit |
| formic acid | 3.75 | 4.761 | For |
| acetic acid | 4.76 | 3.751 | Ace |
pKa CFZ taken as 8.511, as determined at 37 °C.29
Figure 1.

Comparison of PXRD diffractograms of the various CFZ polymorphs with the solid form present in solutions following slurry experiments.
Solution Properties of CFZ Solid Forms in Water
Following the identification of these new solid forms via PXRD, their melting points and solution properties in deionized water were studied (Figure 2, Table 2). From these supersaturation profiles values for maximum supersaturation (Smax), the time to reach Smax (tSmax) and initial dissolution rate (Dinitial) were obtained and are compared in Table 2. In deionized water, each new solid form of CFZ increased the aqueous concentration of CFZ, whereas the CFZ polymorphs or the commercial formulation (Lamprene) did not dissolve to any detectable extent (detection limit < 0.1 mg/L) (Figure 2). The order of supersaturation (relative to CFZ F III) for these salts observed in deionized water was seen to be phosphate > sulfate > acetate > formate > citrate > oxalate > nitrate > chloride ≫ CFZ (free base)/Lamprene. Following the dissolution of the various solid forms, de-supersaturation and precipitation occurred, removing CFZ from solution. The rate of precipitation from solution differed depending on the solid form administrated and the resulting supersaturation level in solution. For each new solid form administered to deionized water, the solid form precipitating out of solution was always found to be the triclinic polymorph of CFZ, F I (Figure 3). Crystallization of CFZ F I can only happen following the deprotonation of the CFZH+ solution species back to the unionized free base species, CFZ. Thus, the rate of precipitation likely depends on the rate at which CFZH+ converts back to CFZ. The unprotonated CFZ species is highly insoluble in water and therefore there is a strong driving force for it to crystallize. Interestingly, of the four CFZ polymorphs known, CFZ F I is the least thermodynamically stable and thus the first available crystal structure, according to the Oswald rule of stages.53
Figure 2.
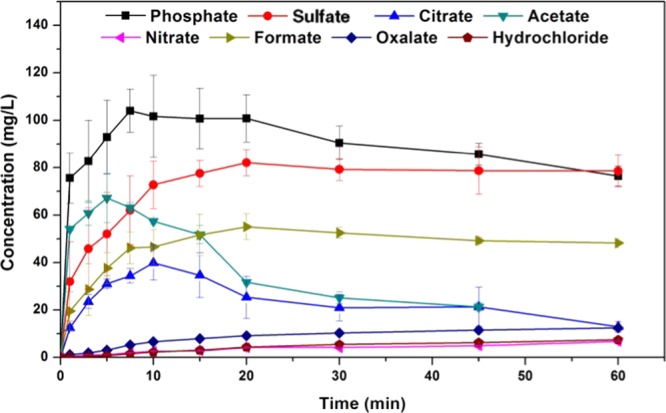
Comparison of the dissolution profiles of CFZ salt forms in deionized water at 37 °C and 150 rpm.
Table 2. Comparison of the Maximum Supersaturation (Smax), Time to Reach Smax (tSmax), Initial Dissolution Rate (Dinitial) over the First 30 s, and Melting Points (mp) of CFZ Solid Forms in Deionised Water, FaSSGF and FaSSIF Media.
| solid form | SH2Omax (mg/L) | tSmaxH2O (min) | DH2OInital (mg/L/min) | SFaSSGFmax (mg/L) | tSmaxFaSSGF (min) | DFaSSGFInitial (mg/L/min) | SFaSSIFmax (mg/L) | tSmaxFaSSIF (min) | DFaSSIFInitial (mg/L/min) | mp (°C) |
|---|---|---|---|---|---|---|---|---|---|---|
| CFZ F I | <0.01 | n/a | n/a | 6.942 ± 2.041 | 5 | 5.9516 | 2.236 ± 0.292 | 60 | 2.162 | 220.33 ± 0.58 |
| CFZ-HCl | 7.406 ± 0.683 | 60 | 0.301 | 261.67 ± 1.53 | ||||||
| CFZ-Sul | 82.052 ± 5.521 | 20 | 31.947 | 22.938 ± 2.436 | 1.5 | 27.371 | 8.916 ± 1.143 | 10 | 2.492 | 244 ± 0.82 |
| CFZ-Nit | 6.718 ± 0.230 | 60 | 0.316 | 232.33 ± 0.58 | ||||||
| CFZ-Oxa | 12.408 ± 0.035 | 60 | 1.189 | 194 ± 0.74 | ||||||
| CFZ-Pho | 104.02 ± 9.108 | 7.5 | 75.605 | 24.472 ± 4.259 | 1.5 | 47.305 | 6.056 ± 1.540 | 20 | 3.383 | 215.25 ± 0.96 |
| CFZ-Cit | 39.938 ± 7.515 | 20 | 12.389 | 21.689 ± 0.333 | 10 | 20.583 | 9.934 ± 0.630 | 10 | 6.155 | 203.50 ± 0.58 |
| CFZ-For | 55.649 ± 5.399 | 20 | 19.505 | 8.062 ± 1.738 | 3 | 6.823 | 183.33 ± 0.58 | |||
| CFZ-Ace | 67.215 ± 10.359 | 5 | 54.089 | 16.997 ± 1.339 | 15 | 6.5457 | n/a | |||
| Lamprene | 16.925 ± 2.342 | 1.5 | 28.933 | 3.812 ± 0.187 | 60 | 3.614 | n/a |
Figure 3.

PXRD comparison of solid form precipitating out of solutions of FaSSGF, FaSSIF, and deionized water after 30 min, following administration of the phosphate salt.
Solution Properties of CFZ Solid Form in FaSSGF
The solid forms with the best aqueous solubility, i.e., sulfate, phosphate, citrate, formate and acetate, were then screened for supersaturation potential in fasted-state simulated gastric fluid (FaSSGF), where they were once again compared with CFZ F I and the commercial formulation, Lamprene. From these supersaturation profiles, values for Smax, tSmax, and Dinitial were obtained for FaSSGF and are also presented in Table 2. The maximum supersaturation in FaSSGF followed the order, phosphate > sulfate > citrate > acetate > Lamprene > formate > CFZ F I, and these results are shown in Figure 4. In the FaSSGF media, there was also a stark reduction in the amount of CFZ that could be held in solution, or Smax, compared to that in deionized water (Table 2). The phosphate and sulfate salts, which reach concentrations as high as 104 and 82 mg/L respectively, in deionized water, struggled to surpass 20 mg/L in FaSSGF, and the stability of the resulting solutions was poor (Figure 4, Table 2). This apparent supersaturation limit of ∼20 mg/L was common across all novel salts of CFZ administered to FaSSGF media (Figure 4). Moreover, regardless of which solid form of CFZ was administered into the FaSSGF, the solid form which precipitated out of solution was found to be a hydrochloride salt of CFZ (Figure 3). The driving force for the observed conversion into the hydrochloric acid salt is likely the high chloride concentration of the FaSSGF. The fact that the CFZ F I also converts to the hydrochloric acid salt suggests that this salt form is a more thermodynamically stable crystalline form of the drug in the high chloride content of the FaSSGF media. De-supersaturation and crystallization from solution happened much faster for the salts in FaSSGF as compared to deionized water, and again this is likely due to the high chloride concentration of the FaSSGF. These findings are in agreement with a previous study in which this hydrochloride acid salt was first identified and was shown to be the form in which CFZ accumulates in animal tissues31 but contradicts dissolution studies carried out on amorphous CFZ loaded onto silica nanoparticles in a different gastrointestinal-like media.54 In FaSSGF media, the phosphate and sulfate salts reached the highest concentration but also precipitated out of solution faster than the citrate and acetate salts (Figure 4). Solution stability of all salts in the FaSSGF media appeared to be limited by supersaturation. If the concentration increased above ∼20 mg/L, rapid de-supersaturation occurred (Figure 4). The citrate and acetate salts maintain their supersaturation levels in FaSSGF better than the sulfate and phosphate, as they do not exceed this critical concentration threshold of ∼20 mg/L. The rapid onset of crystallization, which repeatedly occurs above a concentration of 20 mg/L, must be due to the supersaturation level entering the labile zone, where spontaneous nucleation occurs. Thus, there appears to be an inherent instability to solutions of CFZH+ in this FaSSGF medium at concentrations above 20 mg/L.
Figure 4.
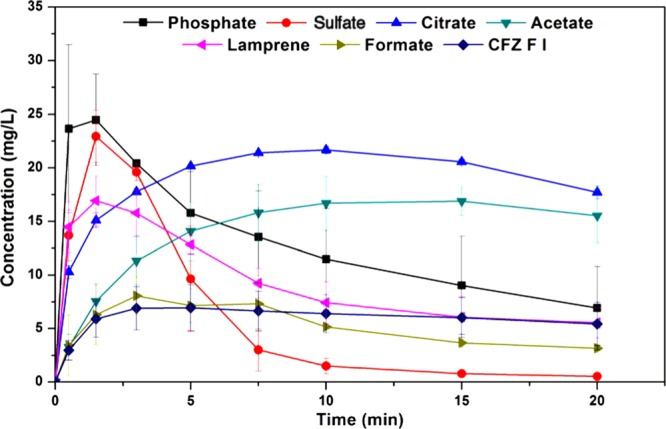
Comparison of the dissolution profiles in FaSSGF of the CFZ salts with CFZ F I and the commercial formulation of the drug (Lamprene).
Solution Properties of CFZ Solid Form in FaSSIF
One common failure of salt forms of basic drugs is their tendency to precipitate from solution in the more basic conditions of the intestinal tract. The intestine is the major location for the absorption of oral drugs, and thus dissolution and supersaturation properties in FaSSIF are arguably more relevant than in FaSSGF. Given the adequate stability and enhanced solution behavior of the phosphate, sulfate and citrate salts in FaSSGF, the solution properties of these three salts were examined in FaSSIF, where they were compared with CFZ F I and Lamprene (Figure 5). From their dissolution profiles, values for Smax, tSmax, and Dinitial were obtained as before and are presented in Table 2. In FaSSIF, the citrate, phosphate and sulfate salts all showed improved dissolution profiles compared to those of Lamprene or CFZ F I and supersaturation in this media followed the order, citrate > phosphate > sulfate > Lamprene > CFZ F I. Compared to the FaSSGF media, there was a significant reduction in the dissolution rates of the CFZ salts in FaSSIF and a significant reduction in the amount of CFZ which went into solution in the more basic FaSSIF (Figure 5 and Table 2). This was, however, unsurprising, as the fast dissolution rates associated with salt forms of basic drugs is a result of differences in the pH of the diffusion layer of the salt and the pH of the bulk media.55 If the latter is changed, then the dissolution rate of the salt in that media will consequently change. It was found to be CFZ F I, or the basic form of the drug, that precipitated from solution in the FaSSIF and not the chloride salt, as was the case in the lower pH FaSSGF media. This was unsurprising as significant work has been done to show how salts of basic drugs will convert to and precipitate from solution as their respective free bases above some theoretical pH value, termed pHmax.55 In the FaSSGF media, the high chloride concentration and fast dissolution rate of the salts result in a critical concentration of CFZH+ being reached around ∼20 mg/L, which results in rapid de-supersaturation. However, in the FaSSIF media, the maximum solution concentration reached was ∼10 mg/mL and thus de-supersaturation occurs slower even though the chloride concentration is higher. Chlorine concentration in the FaSSIF appears to have no effect on solution stability as it is CFZ F I and not the hydrochloride salt of CFZ which precipitates. Thus, the crystallization of CFZ F I from solution in FaSSIF is not driven by the common ion effect, as was the case in FaSSGF.
Figure 5.
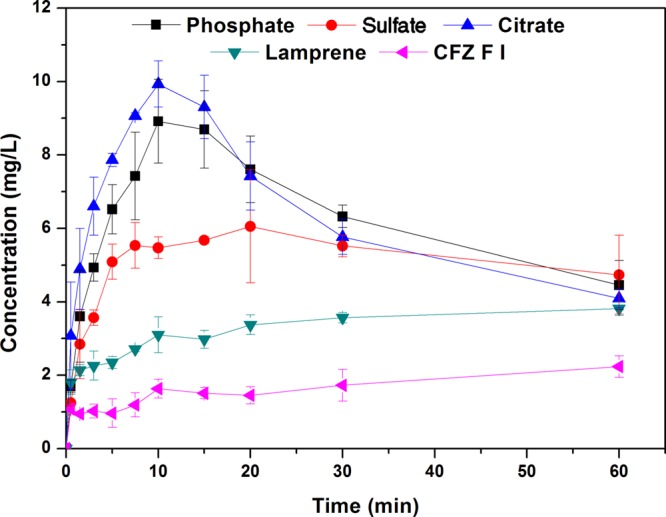
Comparison of the dissolution profiles in FaSSIF of CFZ salts with CFZ F I and the commercial formulation of the drug (Lamprene).
Antimicrobial Activity of Novel Salts
The antimicrobial activity of CFZ F I was compared to that of the salts that exhibited acceptable solution properties in FaSSGF and FaSSIF and long-term stability, i.e., the phosphate, sulfate, and citrate salts. These solid forms were tested for activity against S. aureus, and this data is presented in Figure 6. As mentioned previously, CFZ is routinely used in the treatment of leprosy; however, it has also been reported to display activity against S. aureus at concentrations of less than 5 μg/mL,23,56 albeit in the presence of DMSO, which acts as a solubilizing agent. During the microtiter activity studies of CFZ F I and the citrate, phosphate, and sulfate salts, varying degrees of inhibition of bacterial growth against S. aureus were observed. There was no inhibition of bacterial growth from bacteria incubated in the presence of CFZ F I at either 20 or 40 μg/mL (Figure 6). Bacterial growth initially was slower with the citrate salts for up to ∼8 h; however, final populations were similar in numbers to those of the control and therefore no MIC50 was determined (Figure 6). Similarly, initial growth of the bacteria in the presence of the sulfate salt at 20 μg/mL was much lower than that of the control; however, final populations after 24 h were similar (Figure 6). The sulfate salt caused a significant decrease in bacterial population when compared to that of the control at 24 h, giving an MIC50 of 40 μg/mL. In the case of the phosphate salts, an MIC50 was established at 20 μg/mL over the 24 h analysis when compared with the control (Figure 6). Although not all of the salts exhibited sufficient killing for the determination of a MIC50 value, each of them showed an improvement in activity against S. aureus when compared with CFZ F I. Indeed, without DMSO present in the assay to solubilize the CFZ, no activity was noted against S. aureus for the free base. There is a trend in the literature where antimicrobial activity assays for hydrophobic drugs such as CFZ are usually carried out by dissolving the drug substance in DMSO or ethanol, thereby overcoming the hydrophobic nature of the drug for the sake of the in vitro assay.17,19,23,28,34−37 The results of such tests has led to a library of low MIC values for CFZ against a range of different pathogens.17,19,23,28,34−37 Yet, in vivo activity data has remained largely inconclusive and these discrepancies have been alluded to in a recent review.17 The salt forms of CFZ isolated here overcome the hydrophobic nature of the pure drug temporarily, resulting in enhanced killing against S. aureus.
Figure 6.

Activity of CFZ F1 and citrate, phosphate and sulfate salts against S. aureus at 20 and 40 μg/mL. Cultures were incubated with the drugs for 24 h at 37 °C.
Solid-State Characterization
The solid forms generated in solutions with phosphoric acid, sulfuric acid, and citric acid coformers, having the best solution properties in the FaSSGF medium, were characterized using a combination of solid-state nuclear magnetic resonance (SSNMR) and Fourier transform infrared (FTIR) spectroscopy, X-ray diffraction (XRD) and stability testing. For a new solid form to make it from the lab to the market, it must be stable under storage conditions for prolonged periods of time. The salts with the most promising solubility in FaSSGF, i.e., the acetate, citrate, phosphate, and sulfate, were exposed to the FDA-recommended accelerated stability testing conditions.57 The results of these stability tests are summarized in Figure 7. The phosphate, citrate, and sulfate salts retained their respective structures under these conditions for the duration of the test (3 months). The acetate salt did not retain its structure during the testing and was seen to undergo a phase transformation into the monoclinic polymorph, CFZ F II, during the first week of the test. When a sample of the acetate salt was heated up to around 100 °C, it was found that acetate molecules evaporated to leave CFZ F II (Figure 8). Thus, the acetate salt would be unlikely to have a drug delivery application due to its poor stability and was excluded from further studies.
Figure 7.

Comparison of the PXRD diffractograms of the citrate, acetate, phosphate, and sulfate salts before storage at 40 °C/75%, with diffractograms collected after storage under these conditions for fixed times.
Figure 8.

Comparison of the PXRD diffractograms of the CFZ-acetate solid forms showing the effects of heat on the crystal structure.
Salts are defined by the transfer of a hydrogen atom from an acid to a base and thus the formation of an ionic bond.58,59 The challenge in characterizing a salt from a cocrystal stems from the difficulty in clearly defining hydrogen atom positions using XRD, although recently it has been suggested that in some cases, hydrogen atom positions can be accurately defined to within one standard deviation using the Hirshfeld atom refinement computational analysis.60 Nevertheless, it is also possible to characterize ionic bond formation indirectly using spectroscopic techniques, such as FTIR and NMR, as well as by monitoring changes in the crystal structures caused by protonation of molecules.
Crystallographic data obtained from single-crystal X-ray diffraction (SCXRD) experiments for the citrate, sulfate and phosphate is summarized in Table 3. For each of these salts, there was good correlation between the experimental PXRD diffractograms and those calculated from the single-crystal structures (Figure 9). The crystal structures of these salts and the previously reported CFZ polymorphs were analyzed using the Cambridge Structural Databank (CSD) software, Mercury. The bonding angle of the secondary ketimine groups (C=NC–C) in each crystal was measured. The bond angle C=NC–C, which undergoes protonation, was seen to increase from around 120° in each polymorph of CFZ to around 125° in each of the salt forms (Figure 10). However, the bond angle C–NB=C, which does not become protonated, remained unchanged in the range 118–119° in the various solid forms. Once protonated, the central nitrogen (NC), of the secondary ketimine (C=NC–C), develops a +1 charge and a new coordination number of 3, which explains the observed increase of the C=NC–C bond angle by about 5°. This increase in the bond angle was observed in the structure of each salt characterized here and is consistent with observations made during previous studies on the crystal structures of salts formed between pyridine derivatives and acidic coformers.61 When previously reported CFZ salts were analyzed in this way, a similar increase in the C=NC–C bonding angle was noted.50
Table 3. Summary of Crystallographic Data for CFZ Salts.
| solid form | CFZ-Cit | CFZ-Sul | CFZ-Pho |
| crystal system | triclinic | monoclinic | triclinic |
| space group | P1̅ | C2/c | P1̅ |
| a (Å) | 14.7405(6) | 18.9525(10) | 14.5795(12) |
| b (Å) | 15.8212(7) | 15.4712(10) | 15.6517(13) |
| c (Å) | 16.2505(9) | 20.0349(12) | 28.228(2) |
| α (deg) | 71.927(2) | 90 | 95.326(2) |
| β (deg) | 63.0413(13) | 100.360(2) | 92.101(2) |
| γ (deg) | 70.7443(15) | 90 | 112.174(2) |
| volume (Å3) | 3130.3 | 5778.82 | 5921.33 |
| Z | 4 | 8 | 8 |
| Dcald (g/cm) | 1.412 | 1.314 | 1.2818 |
| R-factor (%) | 6.53 | 12.21 | 13.61 |
| temp. (K) | 283–303 | 283–303 | 283–303 |
Figure 9.
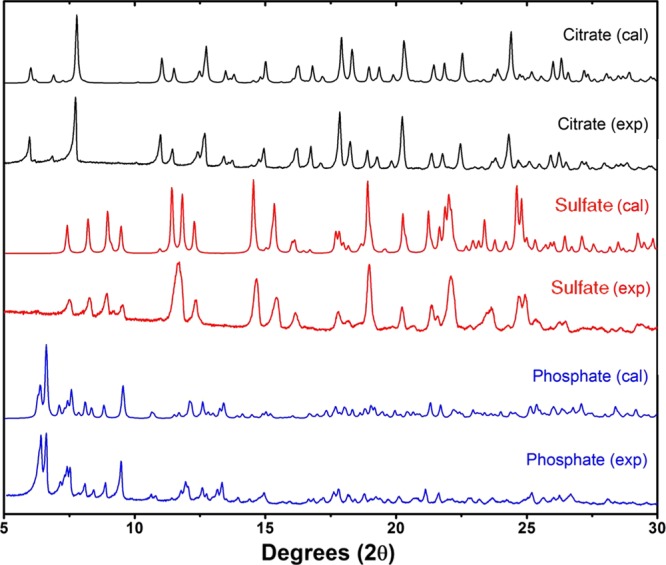
Comparison of the calculated and experimental PXRD diffractograms for the new salts: clofazimine citrate (black), clofazimine sulfate (red), and clofazimine phosphate (blue).
Figure 10.

Left: summary of secondary ketimine bonding angles in CFZ solid forms calculated using Mercury software from single-crystal cif files (average taken for solid Z′ numbers >1). Right: structure of CFZ(−H+) with nitrogen atoms labeled.
FTIR and SSNMR spectroscopy were used to further validate this salt formation hypothesis. Ionic bonds themselves do not resonate in the presence of infrared radiation, so FTIR spectroscopy was used to probe for a new NC+–H bond in the potential salt forms and to monitor the stretching of the C=NC double bond of the ketimine group. The FTIR spectra obtained for the different CFZ polymorphs were found to be indistinguishable (Figure S2), and thus only F III was compared with the potential salt forms. The presence of the new NC–H bond for each salt form was observable in the range 3250–3350 cm–1, and by comparison, no such peak was present in the free base (CFZ F III) (Figure 11). Protonation of NC caused a bathochromic shift in the peak representative of the C=NC double bond (1626.5 cm–1, in F III) to a lower wavenumber in each salt form of CFZ (Figure 11). This movement to a lower wavenumber indicates a weakening this C=NC double bond, giving further proof of protonation. These observations are consistent with accounts of reported ionic bond formation between CFZ and hypromellose phthalate as well as in other salts of CFZ with acidic coformers.50,51 A comparison of the full FTIR spectra for these solid forms can be found in the Supporting Information (Table S1, Figure S2).
Figure 11.

Overlay of the FTIR spectra of CFZ F III with sulfate, phosphate and citrate salts, showing evidence for (a) new NC–H bond formation in the range 3350–3250 cm–1 and (b) weakening of C=NC bond following protonation in the range 1650–1600 cm–1.
SSNMR was used to investigate changes in the chemical shifts of the carbon atoms of CFZ following salt formation. The peak positions of the carbon atoms in CFZ and its respective salt forms were assigned through a combination of molecular modeling and solution NMR. These peak assignments were in agreement with previous studies.27,51,6213C cross-polarization magic-angle spinning (CPMAS) solid-state NMR spectra were collected for the phosphate, sulfate and citrate salts as well as CFZ F III. During these 13C CPMAS experiments, the chemical shift of the ketimine carbons (NB=C and NC=C) were monitored in the range 140–155 ppm, whereas the chemical shift of the isopropyl carbon adjacent to NC was monitored in the range 40–65 ppm. The resulting peak position from the 13C CPMAS spectra is shown in Figure 12. A full comparison of the 13C CPMAS spectra for these salts with CFZ F III can be found in the Supporting Information as well as a table of the chemical shifts of the carbon atoms bound to the nitrogen atoms in the secondary ketimine functional groups of CFZ, i.e., C=NC–C and C–NB=C, and their respective bond lengths from the crystal structures of solid form (Table S2, Figure S3). These new salts of CFZ had several drug molecules in their unit cells, some of these being chemically inequivalent, leading to broad or several peaks for individual carbons in the 13C CPMAS spectra (Figure S3). In general, following salt formation, the chemical shift of the isopropyl carbon (NC–C) shifted upfield (Figure 12), indicating shielding of this carbon nucleus from the magnetic field of the spectrometer. This shielding is likely due to electron density being donated from the adjacent methyl groups in response to the protonation of NC. A similar upfield shift was seen for the ketimine carbon bonded to NC (C=NC) (Figure 12). This shift is expected due to electron density migrating from the phenazine nucleus toward the protonated nitrogen (NC+–H), causing local shielding of the ketimine carbon (C=NC). By comparison, the carbon atom in the conjugated ketimine (C–NB=C) did not shift upfield following protonation and was seen to be resonant at ∼151 ppm in the four CFZ salts (Figure 12). These observations are in agreement with previous studies where protonation of NC in CFZ occurring from HPMCP resulted in similar upfield shifts in the chemical shift of these ketimine carbons.51
Figure 12.

Comparison of the 13C CPMAS spectra obtained for the citrate, phosphate, and sulfate salts with CFZ F III (top) in the range 140–155 ppm to monitor the chemical shift of the ketimine carbons highlighted and (bottom) in the range 40–65 ppm, to monitor the chemical shift of the isopropyl carbon highlighted.
Thus, the increase in C=NC–C bonding angle following interaction with acidic coformers, the subsequent weakening of the C=NC bond, and presence of a new NC–H bond in the FTIR spectra of these solid forms, as well as the shielding of both the ketimine carbon and the isopropyl carbon adjacent to the protonation site (C=NC–C) indicate that salt formation occurs between phosphoric acid, sulfuric acid, and citric acid with CFZ, as was suspected from the ΔpKa of CFZ with these acid coformers.50,52
Conclusions
Novel solid forms of drugs are of value provided that, compared with the existing solid forms, their physicochemical properties are more favorable. For many drugs, solubility and dissolution rates are properties that will determine the drug bioavailability in vivo, and thus being able to manipulate these properties is of great importance. Eight salts of CFZ were identified using an in situ salt screening approach during this work, seven of which were novel salts, and three of these were then selected for further characterization based on their solubility and dissolution rates in simulated gastrointestinal fluids. The CFZ salts exhibited fast dissolution rate and enhanced solution properties in the low pH FaSSGF media, but there was an intrinsic instability to supersaturated solutions of CFZH+ above ∼20 mg/L in this medium. In the FaSSIF media, each salt tested showed a much reduced dissolution rate and a lower maximum solution concentration compared to those in FaSSGF media. This was due to the higher pH of the FaSSIF. However, in every case, the CFZ salts exhibited increased dissolution rates and increased solution concentrations compared with the commercial formulation or the basic drug. The improved solubility profiles of the sulfate, phosphate, and citrate CFZ salts also enhanced killing against S. aureus compared with the free base (CFZ F I) in vitro. Thus, supersaturating delivery systems can improve the solubility and antimicrobial activity of CFZ in vitro but whether this is sufficient to enable the antimicrobial potential of CFZ in vivo remains to be seen.
Materials and Methods
Materials
Clofazimine (CAS registry number 2030-63-9) was purchased from Beijing Mesochem Technology Co., Ltd. Coformers, including hydrochloric acid (37%, Sigma-Aldrich), sulfuric acid (95–97%, Sigma-Aldrich), nitric acid (70%, Sigma-Aldrich), oxalic acid anhydrous (>99%, Sigma-Aldrich), ortho-phosphoric acid (85%, Sigma-Aldrich), citric acid monohydrate (>99%, Sigma-Aldrich), formic acid (acetic acid (>95%, Sigma-Aldrich)), and acetic acid (99.8%, Sigma-Aldrich) were used as received. Methanol (>99%) being of HPLC grade and used without further purification was also purchased from Sigma-Aldrich. Ingredients for the dissolution media; pepsin (extracted from porcine mucosa, Amresco), sodium taurocholate hydrate (NaTc, >97%, Sigma-Aldrich), L-α-phosphatidylcholine (lecithin, ∼ 99% purity, from bovine brain, Sigma-Aldrich), sodium chloride (Fisher Scientific), maleic acid (>99%, Sigma-Aldrich), and hydrochloric acid (37%, Sigma-Aldrich) were used as received from suppliers. Brain Heart Infusion Broth (Sigma-Aldrich) was used as a medium for bioactivity assays.
Screening for Salt Formation
Ten milliliters of 1 M acid solution was placed into 20 mL vials along with a magnetic stir bar and 100 mg of CFZ (F I). The resulting suspensions were stirred at 400 rpm for 24 h, at which point the solid form was monitored. Twenty milligrams of the solid was removed, dried under vacuum, and then immediately analyzed using reflection PXRD.
Preparation of Salts
One gram of CFZ (F I) and 1.2 equiv of coformer were added to a 100 mL Duran flask containing 50 mL of methanol. The solution was slurried for 24 h and then filtered under vacuum. The resulting crystalline powder was placed in an oven at 40 °C overnight to dry.
Growth of Single Crystals
Hundred milligrams of CFZ F I and coformer (1:1 molar ratio) were added to 100 mL of methanol and heated to 50 °C and stirred at 600 rpm for 1 h. The solution was then filtered (using preheated syringe and filter) into a second 100 mL Duran flask, which had been preheated to 50 °C, sealed with parafilm, and pierced with a single hole. The solution was left to cool and evaporated until ∼25 mL of solvent remained, which point the crystals were harvested under vacuum.
Preparation of Simulated Body Fluid (FaSSGF and FaSSIF)
Fasted-state simulated gastric fluid (FaSSGF) was prepared at pH 1.6 using a previously reported recipe,63 with a small modification to control the final chloride-ion concentration. The final composition of the FaSSGF consisted of pepsin (0.1 mg/mL), sodium taurocholate (80 μM), lecithin (20 μM), sodium chloride (7.2 mM), and hydrochloric acid (25 mM). Fasted-state simulated intestinal fluid (FaSSIF) was prepared at pH 6.5, in accordance with a recipe previously reported by Marques et al.64 The final composition of the FaSSIF consisted of sodium taurocholate (3 mM), lecithin (0.2 mM), maleic acid (19.12 mM), sodium hydroxide (34.8 mM), and sodium chloride (68.62 mM).
Supersaturation Studies on CFZ Solid Forms
Dissolution experiments were carried out in deionized water, FaSSGF, and FaSSIF. These experiments were conducted in 100 mL Duran flasks containing poly(tetrafluoroethylene) (PTFE) stir bars and 100 mL of dissolution medium. Solutions were stirred at 150 rpm on a submersible stir plate in a water bath at 37 °C. All samples were ground and passed through particle size sieves (Fisher scientific) to obtain a particle size distribution of 63–90 μm. Following grinding and sieving of the solids, reflection PXRD confirmed no change in their solid form. For the dissolution experiments, samples containing 20 mg of CFZ were added to the dissolution medium. The flask was inverted several times due to the poor wettability of CFZ. Samples were withdrawn at predetermined time intervals using preheated (40 °C) 5 mL syringes and hypodermic needles and then filtered using preheated (40 °C) PTFE syringe filters (0.2 μm, 25 mm diameter, Fisher Scientific). The CFZ concentration was then immediately determined using a double beam UV–vis spectrophotometer (Shimadzu, UV-1800) at a detection wavelength of 488 nm. For calibration of the instrument, standard solutions were prepared in the concentration range 0.1–20 mg/L and good linearity was observed (R2 > 0.999).
Slurry Experiments to Monitor Solid Form
Slurry experiments were carried out in the various dissolution media to determine the solid phase present during and after the dissolution experiments. Fifty milligrams of the solid sample was added to 20 mL of the dissolution medium to mimic supersaturation conditions of the dissolution experiments. This was inverted several times and then stirred on a submersible stir plate at 150 rpm and 37 °C. Solid samples were removed via filtration under vacuum at predetermined time points and characterized by reflection PXRD.
Stability Testing of Optimal Salt Forms
Accelerated stability testing was carried out for selected CFZ salts at 40 °C/75% relative humidity in a sealed humidity chamber. Glass vials, without caps, containing 20–30 mg of a salt were kept in the humidity chamber and then removed at intervals of 1, 2, 4, 8, and 12 weeks. Upon removal, each solid sample was characterized via PXRD, where full retention of the PXRD pattern indicated compound stability.
Antibacterial Activity Assays
The minimum inhibitory concentration (MIC) against S. aureus (DSMZ 20231) was calculated using a microtiter assay. More specifically, the minimum concentration required to kill 50% of the bacterial population, the MIC50, in this case after 24 h, was calculated. An overnight culture was grown up in Brain Heart Infusion Broth at 37 °C, shaking at 250 rpm. First, the CFZ F I and CFZ salts were dissolved/suspended in water and shaken at room temperature for 10 min. Following this, the drug solution/suspension was added to a 96-well plate to give a final concentration of 20 or 40 μg/mL of CFZ in each well. Sterile water was added to each well to bring the volume to 20 μL. Then, 180 μL of the diluted bacterial culture (OD = 0.1) was added, so the total volume in each well was 200 μL. Blanks were set up with media only and media plus different test concentrations of each salt. Control wells were also set up with bacterial culture only. The 96-well plates were incubated in a Biotek ELx808 Ultra microplate reader (Mason Technologies, Dublin, Ireland) at 37 °C for 24 h, and wavelength was read at 590 nm. The plate was shaken mildly before each reading, and readings were taken every 30 min. Three wells were set up for each test concentration, control and blank.
Solid-State Characterization
Powder X-ray diffraction (PXRD) was performed using an Empyrean diffractometor (PANalytical, Phillips) in reflection mode, with Cu Kα radiation (γ = 1.5406 Å) operating at 40 kV and 40 mA and at room temperature. Samples were scanned from 4 to 35° (2θ) at 4 rpm and scan conditions of a scan step size of 0.0131° (2θ) and 48.195 s per step.
Single-crystal X-ray diffraction (SCXRD) measurements were collected at room temperature (299.86 K), on a Bruker Quest D8 Mo Sealed Tube (λ = 0.71073 Å), equipped with a CMOS Photon Detector. Data were corrected for absorption using empirical methods (SADABS) based upon symmetry-equivalent reflections combined with measurements at different azimuthal angles. Crystal structures were solved and refined against all F2 values using the SHELX interfaced with the X-SEED program. Nonhydrogen atoms were refined anisotropically, and hydrogen atoms were placed in calculated positions refined using idealized geometries (riding model) and assigned fixed isotropic displacement parameters.
Melting points of solid forms were determined using a Stuart melting point apparatus (SMP10). Solid samples in the size range 63–90 μm were added into capillary tubes and heated until melting was observed.
Fourier transform infrared (FTIR) spectra of the solid samples were collected on a PerkinElmer Spectrum 100 FTIR spectrometer equipped with a Universal ATR sampling accessory (single reflection and diamond/zinc selenide material). Spectra for each solid form were collected at room temperature using an average of 32 scans and a spectral resolution of 4 cm–1 in the spectral region 4000–650 cm–1.
Carbon-13 solid-state nuclear magnetic resonance (SSNMR) spectra were acquired on a Bruker Avance III HD NMR spectrometer operating at B0 = 9.4 T, with corresponding 1H and 13C resonance frequencies of ν0(1H) = 400.1 MHz and ν0(13C) = 100.6 MHz. CFZ solid forms were packed in 4 mm o.d. zirconia rotors with Kel-F caps under ambient atmosphere, and experimental 13C NMR spectra were acquired at natural abundance using a 4 mm triple channel (H/X/Y) Bruker MAS probe operating in double resonance mode. The magic angle was optimized using a rotor packed with KBr and spun at 5 kHz. NMR spectra were referenced to TMS at δiso = 0 ppm by setting the high frequency 13C resonance in adamantane to 38.48 ppm.65 The 13C CPMAS NMR spectra were acquired in a single spectral window using the cross-polarization pulse sequence, with a magic-angle spinning (MAS) rotor frequency of 10 kHz, a 1H 90° pulse width of 2.5 μs, and 50 kHz 1H decoupling during acquisition. Proton decoupling was carried out with the SPINAL6466 decoupling sequence at 100%. For each sample, the 1H T1 relaxation time(s) were checked using the saturation recovery pulse sequence to ensure that the recycle delay allowed for adequate relaxation between the collection of subsequent transients. 13C CPMAS spectra were collected using optimized contact times (2.5 μs) and relaxation delays (at least 1.4 × T1 values) for each sample. The optimized parameters for CFZ F III were a contact time of 2 ms, relaxation delay of 6 s, and 128 scans; for CFZ phosphate, contact time was 3 ms, relaxation delay 2 s, and 500 scans; for CFZ citrate, contact time was 4 ms, relaxation delay 7 s, and 500 scans; for CFZ sulfate, contact time was 1 ms, relaxation delay 3 s, and 500 scans.
Acknowledgments
This project was funded directly by the Science Foundation Ireland through 13/CDA/2122, and we also acknowledge the Program for Research in Third-Level Institutions (PRTLI) Cycle 5 for its role in funding certain elements of this project.
Glossary
Abbreviations
- AR
antimicrobial resistance
- MDR
multidrug resistance
- HAI
healthcare-associated infections
- FDA
food and drug administration
- CFZ
clofazimine
- WHO
World Health Organization
- MIC
minimum inhibitor concentration
- DMSO
dimethyl sulfoxide
- SDDS
supersaturating drug-delivery systems
- XRD
X-ray diffraction
- PXRD
powder X-ray diffraction
- SCXRD
single-crystal X-ray diffraction
- FaSSGF
fasted-state simulated gastric fluid
- FaSSIF
fasted-state simulated intestinal fluid
- FTIR
Fourier transform infrared
- (SS)NMR
(solid-state) nuclear magnetic resonance
- CPMAS
cross-polarization/magic-angle spinning
- CSD
Cambridge structure database
- Smax
maximum supersaturation
- tSmax
time to reach Smax
- Dinitial
initial dissolution rate
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b01454.
Summary of peak portions for CFZ solid forms from FTIR spectra, summary of bond lengths for single-crystal structures and chemical shifts from CPMAS 13C spectra for CFZ solid forms, comparison of the FTIR spectra of CFZ polymorphs, comparison of the FTIR spectra of CFZ salts, and comparison of the CPMAS 13C spectra of CFZ salts (PDF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Davies J.; Davies D. Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. 10.1128/MMBR.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecchini M.; Langer J.; Slawomirski L.. Organisation for Economic Co-operation and Development (OECD); Online Report. Resistance in G7 Countries and Beyond: Economic Issues, Policies, 2015.
- World Health Organization. Antimicrobial Resistance: A Global Report on Surveillance, 2014.
- O ’Neill J.Online Report Commissioned by UK Government and the Welcome Trust. Tackling Drug-Resistant Infections Globally: An Overview of Our Work the Review on Antimicrobial Resistance, 2016.
- Santajit S.; Indrawattana N. Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. BioMed Res. Int. 2016, 2016, 1–8. 10.1155/2016/2475067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D. Antibiotic Resistance Breakers: Can Repurposed Drugs Fill the Antibiotic Discovery Void?. Nat. Rev. Drug Discovery 2015, 14, 821–832. 10.1038/nrd4675. [DOI] [PubMed] [Google Scholar]
- Rice L. B. Federal Funding for the Study of Antimicrobial Resistance in Nosocomial Pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- Bush K.; Jacoby G. A. Updated Functional Classification of Beta-Lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. 10.1128/AAC.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice L. B. Progress and Challenges in Implementing the Research on ESKAPE Pathogens. Infect. Control Hosp. Epidemiol. 2010, 31, S7–S10. 10.1086/655995. [DOI] [PubMed] [Google Scholar]
- Chellat M. F.; Raguž L.; Riedl R. Targeting Antibiotic Resistance. Angew. Chem., Int. Ed. 2016, 55, 6600–6626. 10.1002/anie.201506818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White A. R.; Kaye C.; Poupard J.; Pypstra R.; Woodnutt G.; Wynne B. Augmentin (Amoxicillin/Clavulanate) in the Treatment of Community Aquired Respiratory Tract Infection. J. Antimicrob. Chemother. 2004, 53, i3–i20. 10.1093/jac/dkh050. [DOI] [PubMed] [Google Scholar]
- Prabhudesai P. P.; Jain S.; Keshvani A.; Kulkarni K. P. The Efficacy and Safety of Amoxicillin-Clavulanic Acid 1000/125 mg Twice Daily Extended Release (XR) Tablet for the Treatment of Bacterial Community-Acquired Pneumonia in Adults. J. Indian Med. Assoc. 2011, 109, 124–127. [PubMed] [Google Scholar]
- Madrid P. B.; Chopra S.; Manger I. D.; Gilfillan L.; Keepers T. R.; Shurtleff A. C.; Green C. E.; Iyer L. V.; Dilks H. H.; Davey R. A.; Kolokoltsov A. A.; Carrion R.; Patterson J. L.; Bavari S.; Panchal R. G.; Warren T. K.; Wells J. B.; Moos W. H.; Burke R. L. L.; Tanga M. J. A Systematic Screen of FDA-Approved Drugs for Inhibitors of Biological Threat Agents. PLoS One 2013, 8, e60579 10.1371/journal.pone.0060579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra S.; Torres-Ortiz M.; Hokama L.; Madrid P.; Tanga M.; Mortelmans K.; Kodukula K.; Galande A. K. Repurposing FDA-Approved Drugs to Combat Drug-Resistant Acinetobacter Baumannii. J. Antimicrob. Chemother. 2010, 65, 2598–2601. 10.1093/jac/dkq353. [DOI] [PubMed] [Google Scholar]
- Kim K.; Zilbermintz L.; Martchenko M. Repurposing FDA Approved Drugs against the Human Fungal Pathogen, Candida albicans. Ann. Clin. Microbiol. Antimicrob. 2015, 14, 32. 10.1186/s12941-015-0090-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clofazimine - A Review. Tuberculosis 2008, 88, 96–99. 10.1016/S1472-9792(08)70006-4. [DOI] [PubMed] [Google Scholar]
- Cholo M. C.; Steel H. C.; Fourie P. B.; Germishuizen W. A.; Anderson R. Clofazimine: Current Status and Future Prospects. J. Antimicrob. Chemother. 2012, 67, 290–298. 10.1093/jac/dkr444. [DOI] [PubMed] [Google Scholar]
- van Ingen J.; van der Laan T.; Dekhuijzen R.; Boeree M.; van Soolingen D. In Vitro Drug Susceptibility of 2275 Clinical Non-Tuberculous Mycobacterium Isolates of 49 Species in The Netherlands. Int. J. Antimicrob. Agents 2010, 35, 169–173. 10.1016/j.ijantimicag.2009.09.023. [DOI] [PubMed] [Google Scholar]
- Shen G.-H.; Wu B.-D.; Hu S.-T.; Lin C.-F.; Wu K.-M.; Chen J.-H. High Efficacy of Clofazimine and Its Synergistic Effect with Amikacin against Rapidly Growing Mycobacteria. Int. J. Antimicrob. Agents 2010, 35, 400–404. 10.1016/j.ijantimicag.2009.12.008. [DOI] [PubMed] [Google Scholar]
- Van Rensburg C. E.; Joone G. K.; O’Sullivan J. F.; Anderson R. Antimicrobial Activities of Clofazimine and B669 Are Mediated by Lysophospholipids. Antimicrob. Agents Chemother. 1992, 36, 2729–2735. 10.1128/AAC.36.12.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rensburg C. E.; van Straten A. M. An in Vitro Investigation of the Susceptibility of Enterococcus faecalis to Clofazimine and B669. J. Antimicrob. Chemother. 1994, 33, 356–358. 10.1093/jac/33.2.356-a. [DOI] [PubMed] [Google Scholar]
- Huygens F.; O’Sullivan J. F.; van Rensburg C. E. J. Antimicrobial Activities of Seven Novel Tetramethylpiperidine-Substituted Phenazines against Multiple-Drug-Resistant Gram-Positive Bacteria. Chemotherapy 2005, 51, 263–267. 10.1159/000087454. [DOI] [PubMed] [Google Scholar]
- Oliva B.; O’Neill A. J.; Miller K.; Stubbings W.; Chopra I. Anti-Staphylococcal Activity and Mode of Action of Clofazimine. J. Antimicrob. Chemother. 2004, 53, 435–440. 10.1093/jac/dkh114. [DOI] [PubMed] [Google Scholar]
- O’Neill A. J.; Miller K.; Oliva B.; Chopra I. Comparison of Assays for Detection of Agents Causing Membrane Damage in Staphylococcus aureus. J. Antimicrob. Chemother. 2004, 54, 1127–1129. 10.1093/jac/dkh476. [DOI] [PubMed] [Google Scholar]
- World Health Organization . WHO Model List of Essential Medicines - 19th List (April 2015), 2015.
- World Health Organization, WHO . WHO Model List of Essential Medicines for Children (4th List), 2013.
- Bannigan P.; Zeglinski J.; Lusi M.; O’Brien J.; Hudson S. P. Investigation into the Solid and Solution Properties of Known and Novel Polymorphs of the Antimicrobial Molecule Clofazimine. Cryst. Growth Des. 2016, 16, 7240–7250. 10.1021/acs.cgd.6b01411. [DOI] [Google Scholar]
- Acharya B. K.; Robson J. M.; Sullivan F. M. Antituberculous Activity of a Phenazine Derivitive (B663). Am. Rev. Respir. Dis. 1959, 80, 871–875. [Google Scholar]
- Quigley J. M.; Fahelelbom K. M. S.; Timoney R. F.; Corrigan O. I. Temperature Dependence and Thermodynamics of Partitioning of Clofazimine Analogues in the N-Octanol/water System. Int. J. Pharm. 1990, 58, 107–113. 10.1016/0378-5173(90)90247-2. [DOI] [Google Scholar]
- Baik J.; Rosania G. R. Macrophages Sequester Clofazimine in an Intracellular Liquid Crystal-Like Supramolecular Organization. PLoS One 2012, 7, e47494 10.1371/journal.pone.0047494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keswani R. K.; Baik J.; Yeomans L.; Hitzman C.; Johnson A. M.; Pawate A. S.; Kenis P. J. A.; Rodriguez-Hornedo N.; Stringer K. A.; Rosania G. R. Chemical Analysis of Drug Biocrystals: A Role for Counterion Transport Pathways in Intracellular Drug Disposition. Mol. Pharmaceutics 2015, 12, 2528–2536. 10.1021/acs.molpharmaceut.5b00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy V. M.; Prensky W.; Vedbrat S.. Methods and Compositions for Treating Clostridium difficile Associated Disease. 2016081825, 2016.
- Wu X.; Cherian P. T.; Lee R. E.; Hurdle J. G. The Membrane as a Target for Controlling Hypervirulent Clostridium Difficile Infections. J. Antimicrob. Chemother. 2013, 68, 806–815. 10.1093/jac/dks493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Li T.; Qu G.; Pang Y.; Zhao Y. In Vitro Synergistic Activity of Clofazimine and Other Antituberculous Drugs against Multidrug-Resistant Mycobacterium tuberculosis Isolates. Int. J. Antimicrob. Agents 2015, 45, 71–75. 10.1016/j.ijantimicag.2014.09.012. [DOI] [PubMed] [Google Scholar]
- Reddy V. M.; O’Sullivan J. F.; Gangadharam P. R. J. Antimycobacterial Activities of Riminophenazines. J. Antimicrob. Chemother. 1999, 43, 615–623. 10.1093/jac/43.5.615. [DOI] [PubMed] [Google Scholar]
- Barry V. C.; Belton J. G.; Conalty M. L.; Denneny J. M.; Edward D. W.; O’Sullivan J. F.; Twomey D.; Winder F. A New Series of Phenazines (Rimino-Compounds) With High Antituberculosis Activity. Nature 1957, 179, 1013–1015. 10.1038/1791013a0. [DOI] [PubMed] [Google Scholar]
- Barry V. C.; Conalty M. L.; Gaffney E. E. Antituberculosis Activity in the Phenazine Series. Isomeric Pigments Obtained By Oxidation of O-Phenylenediamine Derivatives. J. Pharm. Pharmacol. 1956, 8, 1089–1096. 10.1111/j.2042-7158.1956.tb12238.x. [DOI] [PubMed] [Google Scholar]
- Almeida e Sousa L.; Reutzel-Edens S. M.; Stephenson G. A.; Taylor L. S. Supersaturation Potential of Salt, Co-Crystal, and Amorphous Forms of a Model Weak Base. Cryst. Growth Des. 2016, 16, 737–748. 10.1021/acs.cgd.5b01341. [DOI] [Google Scholar]
- Xu S.; Dai W.-G. Drug Precipitation Inhibitors in Supersaturable Formulations. Int. J. Pharm. 2013, 453, 36–43. 10.1016/j.ijpharm.2013.05.013. [DOI] [PubMed] [Google Scholar]
- O’Reilly J. R.; Corrigan O. I.; O’Driscoll C. M. The Effect of Simple Micellar Systems on the Solubility and Intestinal Absorption of Clofazimine (B663) in the Anaesthetised Rat. Int. J. Pharm. 1994, 105, 137–146. 10.1016/0378-5173(94)90459-6. [DOI] [Google Scholar]
- Patel V. B.; Misra A. N. Encapsulation and Stability of Clofazimine Liposomes. J. Microencapsulation 1999, 16, 357–367. 10.1080/026520499289077. [DOI] [PubMed] [Google Scholar]
- Mehta R. T. Liposome Encapsulation of Clofazimine Reduces Toxicity in Vitro and in Vivo and Improves Therapeutic Efficacy in the Beige Mouse Model of Disseminated Mycobacterium avium-M. intracellulare Complex Infection. Antimicrob. Agents Chemother. 1996, 40, 1893–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott M. A.; Domurado M.; Leclercq L.; Barbaud C.; Domurado D. Solubilization of Water-Insoluble Drugs due to Random Amphiphilic and Degradable Poly(dimethylmalic Acid) Derivatives. Biomacromolecules 2013, 14, 1936–1944. 10.1021/bm400323c. [DOI] [PubMed] [Google Scholar]
- Bakkour Y.; Darcos V.; Coumes F.; Li S.; Coudane J. Brush-like Amphiphilic Copolymers Based on Polylactide and Poly(ethylene Glycol): Synthesis, Self-Assembly and Evaluation as Drug Carrier. Polymer 2013, 54, 1746–1754. 10.1016/j.polymer.2013.01.042. [DOI] [Google Scholar]
- Darcos V.; El Habnouni S.; Nottelet B.; El Ghzaoui A.; Coudane J. Well-Defined PCL-Graft-PDMAEMA Prepared by Ring-Opening Polymerisation and Click Chemistry. Polym. Chem. 2010, 1, 280. 10.1039/c0py00004c. [DOI] [Google Scholar]
- Salem I. I.; Steffan G.; Düzgünes N. Efficacy of Clofazimine-Modified Cyclodextrin against Mycobacterium avium Complex in Human Macrophages. Int. J. Pharm. 2003, 260, 105–114. 10.1016/S0378-5173(03)00236-9. [DOI] [PubMed] [Google Scholar]
- Narang A. S.; Srivastava A. K. Evaluation of Solid Dispersions of Clofazimine. Drug Dev. Ind. Pharm. 2002, 28, 1001–1013. 10.1081/DDC-120006431. [DOI] [PubMed] [Google Scholar]
- Serajuddin A. T. M. Salt Formation to Improve Drug Solubility. Adv. Drug Delivery Rev. 2007, 59, 603–616. 10.1016/j.addr.2007.05.010. [DOI] [PubMed] [Google Scholar]
- Mannava M. K. C.; Suresh K.; Nangia A. Enhanced Bioavailability in the Oxalate Salt of the Anti-Tuberculosis Drug Ethionamide. Cryst. Growth Des. 2016, 16, 1591–1598. 10.1021/acs.cgd.5b01700. [DOI] [Google Scholar]
- Bolla G.; Nangia A. Clofazimine Mesylate: A High Solubility Stable Salt. Cryst. Growth Des. 2012, 12, 6250–6259. 10.1021/cg301463z. [DOI] [Google Scholar]
- Nie H.; Su Y.; Zhang M.; Song Y.; Leone A.; Taylor L. S.; Marsac P. J.; Li T.; Byrn S. R. Solid-State Spectroscopic Investigation of Molecular Interactions between Clofazimine and Hypromellose Phthalate in Amorphous Solid Dispersions. Mol. Pharmaceutics 2016, 13, 3964–3975. 10.1021/acs.molpharmaceut.6b00740. [DOI] [PubMed] [Google Scholar]
- Duggirala N. K.; Perry M. L.; Almarsson Ö; Zaworotko M. J. Pharmaceutical Cocrystals: Along the Path to Improved Medicines. Chem. Commun. 2016, 52, 640–655. 10.1039/C5CC08216A. [DOI] [PubMed] [Google Scholar]
- Feenstra T.; De Bruyn P. The Ostwald Rule of Stages in Precipitation from Highly Supersaturated Solutions: A Model and Its Application to the Formation of the Nonstoichiometric Amorphous Calcium Phosphate Precursor Phase. J. Colloid Interface Sci. 1981, 84, 66–72. 10.1016/0021-9797(81)90260-5. [DOI] [Google Scholar]
- Valetti S.; Xia X.; Costa-Gouveia J.; Brodin P.; Bernet-Camard M.-F.; Andersson M.; Feiler A. Clofazimine Encapsulation in Nanoporous Silica Particles for the Oral Treatment of Antibiotic-Resistant Mycobacterium tuberculosis Infections. Nanomedicine 2017, 12, 831–844. 10.2217/nnm-2016-0364. [DOI] [PubMed] [Google Scholar]
- Serajuddin A. T. M. Salt Formation to Improve Drug Solubility. Adv. Drug Delivery Rev. 2007, 59, 603–616. 10.1016/j.addr.2007.05.010. [DOI] [PubMed] [Google Scholar]
- Oliva B.; Comanducci A.; Chopra I. Antibacterial Spectra of Drugs Used for Chemotherapy of Mycobacterial Infections. Tubercle Lung Dis. 1998, 79, 107–109. 10.1054/tuld.1998.0012. [DOI] [PubMed] [Google Scholar]
- Food and Drug Administration . Guidance for Industry of New Drug Substances and Products Guidance for Industry Q1A ( R2 ) Stability Testing of New Drug Substances and Products, 2003.
- Braga D. Crystal Engineering. A Textbook. By Gautam R. Desiraju, Jagadese J. Vittal and Arunachalam Ramanan. Angew. Chem., Int. Ed. 2012, 51, 3516–3516. 10.1002/anie.201200277. [DOI] [Google Scholar]
- Elder D. P.; Holm R.; de Diego H. L. Use of Pharmaceutical Salts and Cocrystals to Address the Issue of Poor Solubility. Int. J. Pharm. 2013, 453, 88–100. 10.1016/j.ijpharm.2012.11.028. [DOI] [PubMed] [Google Scholar]
- Woinska M.; Grabowsky S.; Dominiak P. M.; Wozniak K.; Jayatilaka D. Hydrogen Atoms Can Be Located Accurately and Precisely by X-Ray Crystallography. Sci. Adv. 2016, 2, e1600192 10.1126/sciadv.1600192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bis J. A.; Zaworotko M. J. The 2-Aminopyridinium-Carboxylate Supramolecular Heterosynthon: A Robust Motif for Generation of Multiple-Component Crystals. Cryst. Growth Des. 2005, 5, 1169–1179. 10.1021/cg049622c. [DOI] [Google Scholar]
- Nie H.; Mo H.; Zhang M.; Song Y.; Fang K.; Taylor L. S.; Li T.; Byrn S. R. Investigating the Interaction Pattern and Structural Elements of a Drug-Polymer Complex at the Molecular Level. Mol. Pharmaceutics 2015, 12, 2459–2468. 10.1021/acs.molpharmaceut.5b00162. [DOI] [PubMed] [Google Scholar]
- Vertzoni M.; Dressman J.; Butler J.; Hempenstall J.; Reppas C. Simulation of Fasting Gastric Conditions and Its Importance for the in Vivo Dissolution of Lipophilic Compounds. Eur. J. Pharm. Biopharm. 2005, 60, 413–417. 10.1016/j.ejpb.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Marques M. R. C.; Loebenberg R.; Almukainzi M. Simulated Biological Fluids with Possible Application in Dissolution Testing. Dissolution Technol. 2011, 18, 15–28. 10.14227/DT180311P15. [DOI] [Google Scholar]
- Morcombe C. R.; Zilm K. W. Chemical Shift Referencing in MAS Solid State NMR. J. Magn. Reson. 2003, 162, 479–486. 10.1016/S1090-7807(03)00082-X. [DOI] [PubMed] [Google Scholar]
- Fung B. M.; Khitrin A. K.; Ermolaev K. An Improved Broadband Decoupling Sequence for Liquid Crystals and Solids. J. Magn. Reson. 2000, 142, 97–101. 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.