

Abstract
With an increase of resistance in bacteria there is an urgent need for alternative treatment methods that could complement conventional antibiotics. In the past two decades, focus has been drawn to Host Defense Peptides (HDPs) as potential antibiotic agents. Herein we reported our studies on the development of lipidated α/α-AA heterogeneous peptides as a new class of HDP mimetics. These compounds showed potent antimicrobial activity toward both Gram-positive and Gram-negative bacteria, and they also displayed excellent selectivity as they only exhibited limited hemolytic activity. The fluorescence microscopy suggested that the mechanism of action of these heterogeneous peptides is bacterial membrane disruption, which is believed to be the major reason why it is difficult for bacteria to develop resistance. The subsequent time kill studies suggested that these compounds could rapidly eradicate bacteria. Moreover, this class of compounds could also effectively clear biofilms formed by both Gram-positive and Gram-negative bacteria. These findings suggested that lipidated α/α-AA heterogeneous peptides, as a new class of peptidomimetics, are promising antibiotic agents combating antibiotic resistance.
Keywords: Peptidomimetics, Host Defense Peptides, Antimicrobial, Bacterial Resistance, Lipidation
Graphical Abstract
1. INTRODUCTION
Bacterial infections have been the cause of many diseases that could lead to severe conditions including death. For the past seventy years, antibiotics have been used to treat a wide range of bacterial infections. However, due to the prolonged use and frequent misuse of these conventional drugs, many microbes have become resistant to treatment. This problem has led to the loss of effectiveness in the antibiotics to treat infections, resulting in a global health concern.[1,2] Per the Center of Disease Control (CDC), over two million people become infected with antibiotic resistant bacteria every year and of these approximately twenty three thousand die.[3] The World Health Organization (WHO) also reports that microbes are developing new mechanisms of resistance globally and this has caused a threat in the ability to treat common diseases hence leading to prolonged illness, disability, and death.[4] There is an urgent need to search for the alternative approaches combating the emergent resistance in bacteria.
Host Defense Peptides (HDPs), such as cathelicidin and definsins,[5,6] are natural antimicrobial peptides which are produced in all classes of life. HDPs work by means of either antimicrobial or by immunomodulatory functions, or both.[7,8] Briefly, they could directly kill bacteria by compromising bacterial membranes.[9] Meanwhile, HDPs also utilize their immunomodulatory functions to regulate the release of immune cells that affect innate immunization against bacterial infections. For example, monocytes, lymphocytes and granulocytes all activate immune cells to control the rate of inflammation and improve the killing and removal of foreign bodies from the host after infection. [10–12] Although they are diverse in their three-dimensional structures, HDPs generally are short cationic sequences that make up of about no more than fifty amino acids and have both hydrophobic and hydrophilic faces in their structures. Their positive charge allows for selectivity toward bacteria which are all virtually negatively charged on their outer membrane leaflet, whereas the hydrophobic patches of HDPs are critical for penetration and disruption of the bacterial cell membranes.[13–15] Due to their low propensity to develop resistance as they lack specific targets on the bacterial membranes, their high selectivity for bacterial cells over mammalian cells, and their antimicrobial activity for a broad range of bacteria, significant efforts have been devoted to the development of HDPs and their mimics.[16,17]
Recently our group has designed a class of peptidomimetics termed α-AApeptides (Figure 1) whose backbone are derived from α chiral peptide nucleic acids (PNA).[18,19] The design has been shown to be advantageous as it exhibits enhanced proteolytic stability, bioavailability, and chemodiversity as the side chains can be diversified with a wide variety of unnatural functional groups to allow design adapted to different circumstances. Our previous studies show that α-AApeptides could be potential antimicrobial agents,[20] and their activity could have improved antimicrobial activity upon adding a lipid tail to the building block sequence.[21,22] We reasoned because adding a lipid tail to the sequence makes it more hydrophobic it increases its ability to interact with the lipid structures of the bacterial cell membranes.[21] In addition, we recently show that hybrid peptides could be potent and selective antimicrobial agents.[23] As such, it is envisioned that a new class of lipidated hybrid peptides consisting of both α-AApeptide building blocks and canonical amino acids could yield potent antibiotic agents with simple structures, and thereby more promise of future development for practical use in therapeutics.
Figure 1.
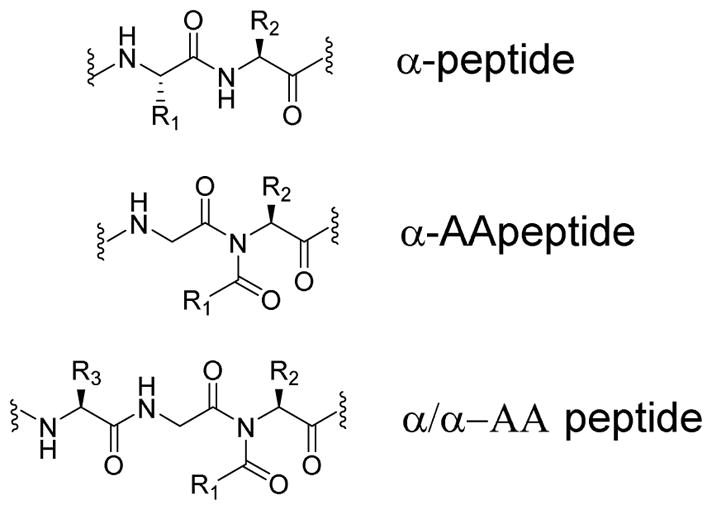
General structure of an α-peptide, an α-AApeptide, and an α/α-AA heterogeneous peptide.
2. RESULTS AND DISCUSSION
Building upon our previous findings[23], our design was aimed for molecular simplicity and strong membrane reactivity. To this end, lysine residue was included to give an overall positive charge, which is expected to attract the sequence to the surface of negatively charged bacterial membranes. A lipid tail was employed to endow hydrophobicity, facilitating sequence insertion into membranes. Furthermore, one or more amphiphilic α-AApeptide building blocks containing a positively charged side chain and a hydrophobic side chain were introduced to fine tune its membrane activity and selectivity (Scheme 1). All the sequences were synthesized on the solid phase using standard solid phase peptide synthesis (SPPS) protocol (Scheme 1. please see supporting information for details),[24] and their structures are shown in Figure 2.
Scheme 1.

Reaction scheme for solid phase synthesis of lipidated α/α-AA peptide hybrids.
Figure 2.

Structures of sequences synthesized in Scheme 1, with varying lengths of hydrocarbon tail.
These sequences were then tested for their antimicrobial activity against five reprehensive strains (Table 1): Gram-negative bacteria Escherichia coli, K. pneumonia and Gram-positive bacteria Methicillin resistant Staphylococcus aureus (MRSA), Methicillin Resistant S. epidermidis, and Vancomycin resistant E. faecalis.[25,26] We first examined the activity of sequences containing lysine residue, a lipid tail and one α-AA building block (n=1). Interestingly, sequences containing C6, C8, C10 and C12 lipid tails did not shown any activity under tested conditions. C14-K-BB1, bearing the C14 lipid tail, started to exhibit weak activity against MRSA and E. Coli. With the longer C16 lipid tail, C16-K-BB1 displayed broad spectrum activity against all the tested Gram-positive and Gram-negative strains. The results are indeed consistent to our previous findings that long lipid tails are needed to endow sequences with sufficient hydrophobicity to penetrate lipid bilayers in bacteria. However, to our surprise, C16-K-BB1 was highly hemolytic, probably due to its high hydrophobic content.
Table 1.
The antimicrobial activity and selectivity of α/α-AA peptide hybrid shown in Figure 2. “----” indicates the activity was not tested.
| MIC (μg/mL) | Hemolytic activity HC50 (μg/mL) | Selectivity (HC50/MIC of MRSA) | |||||
|---|---|---|---|---|---|---|---|
| Compound | Gram + | Gram − | |||||
| MRSA | MRSE | E. faecalis | E. coli | K. pneumoniae | |||
| C6-K-BB1 | >25 | >25 | >25 | >25 | >25 | ------- | ------- |
| C8-K-BB1 | >25 | >25 | >25 | >25 | >25 | ------- | ------- |
| C10-K-BB1 | >25 | >25 | >25 | >25 | >25 | ------- | ------- |
| C12-K-BB1 | >25 | >25 | >25 | >25 | >25 | ------- | ------- |
| C14-K-BB1 | 12–25 | >25 | >25 | 6–12 | >25 | ------- | ------- |
| C16-BB1 | 25 | >25 | >25 | >25 | >25 | ------- | ------- |
| C16-K-BB1 | 1.5–3 | 3–6 | 6–12 | 1.5–3 | 6–12 | 31.25 | 10.4 |
| C16-K-BB2 | <3 | >25 | >25 | 1.5–3 | >25 | 31.25 | 10.4 |
| C16-K-BB3 | 1.5–3 | 3–6 | 6–12 | 1.5–3 | >25 | 250 | 83 |
| C16-K-F-K | >25 | >25 | >25 | >25 | >25 | ------- | ------- |
To obtain more potent antimicrobial agents with enhanced selectivity, we introduced more α-AA building blocks (n=2, 3; m=15). Unfortunately, C16-K-BB2, only showed good activity against MRSA and E. Coli, but not other stains. Meanwhile, it was still highly hemolytic. However, it was intriguing that C16-K-BB3 showed excellent antimicrobial activity towards all tested strains except for K. pneumoniae. In addition, it exhibited high selectivity toward MRSA over mammalian cells. This demonstrated that proper ratio of hydrophobic groups and positively charged groups could lead to potent and selective antimicrobial α/α-AA hybrid. We next examined the impact of backbone on the antimicrobial activity. As seen in Scheme 1 and Table 1, C16-BB1, which does not contain the lysine residue, lost all the activity compared to C16-K-BB1. Whereas the other sequence C16-K-F-K, bearing purely canonic amino acid residue but with similar hydrophobicity and hydrophilicity, also did not show any antimicrobial activity. The results suggested that α/α-AA hybrid backbone are mandatory and unique for antimicrobial development.
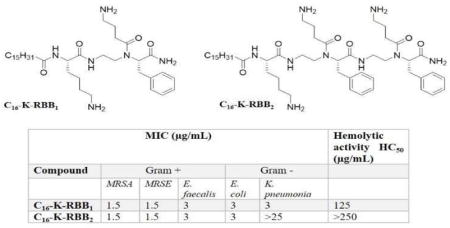
We then asked whether the position of the positive charge and the hydrophobic side chains would influence the antimicrobial activity of the compounds. Thus, we synthesized another three sequences bearing a C16 lipid tail, a lysine residue and one or more amphiphilic α-AA building blocks with reversed side chains (Scheme 2) compared to structures in Figure 2. Surprisingly, all three sequences exhibited broad-spectrum activity against both Gram-positive and Gram-negative bacteria. Among them, C16-K-RBB1 is the most potent one which showed excellent activity even toward K. pneumoniae whereas displayed limited cell toxicity. Although C16-K-RBB2 was not active toward K. pneumoniae, its high selectivity toward other bacterial strains including MRSA presents itself as a very promising antimicrobial agent. Our results suggest that not only the ratio of hydrophobicity and cationic charge is important for HDP mimetics, the three-dimensional distribution of hydrophobic and cationic groups is also highly critical for bacterial killing. Therefore, based on our above-mentioned findings, both C16-K-RBB1 and C16-K-RBB2 were chosen for the subsequent mechanistic studies.
Scheme 2.

Reaction Scheme for solid phase synthesis of the reverse antimicrobial sequences.
As C16-K-RBB2 having the best activity and selectivity toward E. coli and MRSA, we set out to explore its mechanism of action to kill bacterial cells using fluorescence microscopy (Figure 3). DAPI was used to stain both live and dead bacterial cells while PI was used to stain the dead cells only.[27,28] Upon treatment of E. coli and MRSA with C16-K-RBB2 for 2 h, the bacterial cells showed red fluorescence in the PI channel, indicating that the compound killed the bacterial cells by damaging their membranes.
Figure 3.

Fluorescence Microscopy of bacterial cells treated with C16-K-RBB2 for 2h (Scale bar 10μm). (1a) Control, DAPI stained, no treatment with compound. (1b) Control, PI stained, no treatment with compound. (2a) MRSA, DAPI stained, treated with compound. (2b) MRSA, PI stained, treated with compound. (3a) Control, DAPI stained, no treatment with compound. (3b) Control, PI stained, no treatment with compound. (4a) E. coli, DAPI stained, treated with compound. (4b) E. coli, PI stained, treated with compound.
The Time kill assay was subsequently conducted using the most potent compound C16-K-RBB1 to assess bacterial killing kinetics for this class of compounds. As shown in Figure 4A, it took 120 minutes for 12.5 μg/mL of C16-K-RBB1 to clear the bacteria E. Coli while it just took 60 minutes for 25 μg/mL and 50 μg/mL C16-K-RBB1 to eradicate the bacteria. In the case of MRSA (Figure 4B) the compound took 60 minutes for 12.5 μg/mL and 25 μg/mL whereas only 10 minutes for 50 μg/mL to completely get rid of the bacteria. The results demonstrated that the compounds could rapidly eradicate bacterial cells with bactericidal function.
Figure 4.

Time Kill Assay for Gram-negative E. coli (A) and MRSA (B).
One of the challenges faced when treating bacterial infections is their ability to develop biofilm. Bacterial biofilms are formed by the ability of bacteria to irreversibly bind to a surface and form a matrix that is made up of extra-polymer substances. They are complex as they use the environment that they are attached to develop channels that allow for the maintenance of the bacteria that are within the biofilm.[29,30] As such, they are harder to be eradicated compared to planktonic bacterial cells. Due to the ability of bacteria to bind surfaces such as medical devices and sites of infection, there is an urgent need for the identification of new antimicrobial agents that could inhibit the growth of bacterial biofilms. We thus evaluated the lead compound C16-K-RBB2 for its ability to decrease biofilm formation. Intriguingly, this compound was able to significantly reduce biofilm formation in both Gram-negative E. Coli (Figure 5A) and Gram-positive MRSA (Figure 5B) bacteria. The results further strength the potential of lipidated α/α-AA peptide hybrid as a new class of antimicrobial agents.
Figure 5.

Biofilm prevention assay for cells treated with C16-K-RBB2. Error bars based on standard deviation. E. coli (A) and MRSA (B).
3. EXPERIMENTAL PROCEDURE
3.1. General Information
Sequences were synthesized using solid phase synthesis. To do this Rink amide MBHA resin (100–200 mesh, 0.64 mmol/g) from Chem-Impex International, Inc was used. The α-AA peptides were synthesized by using a peptide reaction vessel clamped on a Burrell Wrist-Action Shaker and purified using High performance liquid chromatography (HPLC) equipped with a preparative C18 column (5μm, 9 × 250 mm). The molecular weight of the desired fraction was confirmed using MALDI-TOF (Bruker AutoFlex) mass spectrometer, and the product was dried on a labcono lyophilizer. All other chemicals were purchased for either Sigma Aldrich or Fischer unless otherwise stated.
3.2. Solid Phase Synthesis
[24] The solid phase peptide synthesis was conducted by using 100 mg of Rink amide MBHA resin in a peptide reaction vessel on a shaker. The resin was first treated with 20% piperidine in DMF for 15 min to remove Fmoc group. This step was repeated twice and after each time the beads were washed with 3 mL DMF and DCM alternatively. Subsequently, α-AA build block (2 equiv.), HOBT (6 euiv.) and DIC (6 equiv.) were dissolved in DMF and shaken for 10 min, and the mixture was added to the vessel and left to shake overnight. The beads were washed with DMF and DCM. After Fmoc group was removed, either one more α-AA building block, or the lysine amino acid, or a lipid tail, was coupled onto the beads, respectively depending on the sequences to be synthesized. The synthetic cycle was continued until the desired sequence was completed. The sequence was then cleaved from the beads using a 1:1 ratio of TFA: DCM for two hours. The solution was collected, dried over the air, and the residue was dissolved in water, which was subsequently purified by HPLC.
3.3. Minimum Inhibition Concentration (MIC) – Antimicrobial Assay: [22]
MIC is used to determine the minimum compound needed to inhibit both Gram-positive and Gram-negative bacteria. In the case of this experiment Escherichia coli (E. coli, JM109), and Klebsiella pneumoniae (K. pneumoniae, ATCC 13383) were used as the Gram-negative specimen, while Methicillin Resistant Staphylococcus aureus (MRSA, ATCC 33591), Methicillin Resistant Staphylococcus epidermidis (MRSE, RP62A), and Vancomycin Resistant Enterococcus faecalis (E. faecalis, ATCC 700802) were used as the Gram-positive specimen. To test the inhibition properties, one colony of bacteria was first taken from an agar plate and grown overnight in trypicase soy broth (TBS) media at 37 °C on a shaker. Subsequently, one colony was taken again and allowed to grow to mid-logarithm phase. Of this, 50 μL of 106 CFU/mL bacteria was added to each well of a 96 well plate along with 50μL of diluted lipidated α/α-AApeptides (100 to 0.5 μg/mL). After 18 hours of incubation at 37°C the MIC was determined by using a Biotek microplate reader with optical density of 600 nm. The lowest concentration of peptides to inhibit bacterial growth was considered as the MIC. All experiments were done three times, each time being duplicates. The control was the bacteria not treated with any peptides.
3.4. Hemolytic Studies: [26]
Freshly obtained human red blood cells were used for measurement of hemolytic activity of compounds. The cells were washed a few times with phosphate buffered saline (PBS) and centrifuged at 1000g for 10 minutes. The supernatant was then discarded and the red blood cells were resuspended in PBS to obtain a 5% volume by volume concentration. A 96 well plate was then used to contain a two-fold dilution of C16-K-RBB2 to make a total of 50μL in each well. To which 50μL of 5% volume by volume of red blood cells solution was added. The plate was then stored at for 1 hour at 37 °C and centrifuged for 10 minutes at 3500 rpm. The supernatant was collected and diluted with 100 μL of PBS to detect the hemoglobin using a Biotek microtiter plate reader (Synergy HT) at 540 nm. Controls were used to determine 0% hemolysis by incubation in PBS and 100% hemolysis by treatment with 0.1% Triton-X-100 in PBS. The percent hemolysis was determined by using the formula:
3.5. Fluorescence Microscopy: [25]
The double dyes staining method, using 4′,6 diamidino-2-phenylindole dihydrochloride (DAPI, Sigma, >98%) and propidium iodide (PI, Sigma), was employed to assess bacterial membrane integrity. DAPI is a cell permeable dye and has a great ability to bind to the A-T rich regions of double stranded DNA which allows for very strong fluorescence.[28] PI on the other hand is an ethidium derivative that are able to penetrate the cell walls of dead cells but not living cells. Therefore combination of both DAPI and PI were used to quantify the bacterial cells that became necrotic due to treatment with C16-K-RBB2.[27] These stains were used to determine the viability of both E. coli and MRSA. The bacterial cells were cultured to the mid-logarithmic phase, and then incubated for 4 h at 37 °C with C16-K-RBB2 (5 μg/mL). They were then centrifuged at 3000g for 15 minutes and the supernatant was removed. The pellets that were collected were washed with PBS then incubated with DAPI (10 μg/mL) for 15 minutes at 0°C in the dark. After 15 minutes the excess DAPI was washed a few times with 1 × PBS. The pellets were also treated with PI (5 μg/mL) and incubated at 0°C in the dark for 15 minutes, after which the excess PI was washed off with 1 × PBS. The controls were tested using the same method stated above, however, the cells were not incubated with C16-K-RBB2. The cells were then observed using a Zeiss Axio Imager Z1 optical microscope (100×).
3.6. Time Kill Studies: [26]
This assay was done in triplicates to determine the kinetics of the peptide C16-K-RBB1 as it relates to termination of bacteria. To determine this, the bacteria were incubated at 0, 10, 20, 30,60 and 120 minutes with various concentrations of the peptide. 100 μL of bacterial suspension was then taken from each time interval and further diluted. The diluted suspensions were incubated at 37°C on agar plates for 20 hours and the colonies counted.
3.7. Biofilm Assay:6
This assay was done in duplicates to determine the prevention of biofilm growth. MRSA and E. coli, were grown to 1X106 CFU/mL in a 96 well plate containing 50μL trypticase soy media, and 2:1 serial dilution of tested compound in 50μL of PBS. The plate was then incubated at 37°C for 24 hours and then 10 μL of crystal violet stain was added to each well at a concentration of 0.5% weight by volume. The plates were then washed with TBS to get rid of any cells that did not stick to the plate and 95% ethanol was added to the plates so as to dissolve the stain. A microtiter plate reader was then used to determine the formation of biofilm by taking readings at a wave length of 600 nm. The lesser the optical density at 600nm is equivalent to a decrease in biofilm formation.
4. CONCLUSION
In summary, we have developed a new class of lipidated α/α-AA heterogeneous peptides. They show excellent activity against both Gram-positive and Gram-negative bacteria, including clinically relevant bacterial strains. Our structure activity relationship studies suggest that lipidation, hydrophobicity, and positive charge, as well as their spatial arrangement, are of importance to selectively target bacterial cells over mammalian cells. Furthermore, our mechanistic studies show that the compounds could mimic HDPS and are capable of eradicating both Gram-positive and Gram-negative bacteria in a timely manner, while also eliminating the formation of biofilms. Therefore, lipidated α/α-AA heterogeneous peptides could be a new generation of antibiotic agents with potential therapeutic applications.
Supplementary Material
Table 2.
The antimicrobial activity and selectivity of α/α-AA peptide hybrid shown in Scheme 2.
| MIC (μg/mL) | Hemolytic activity HC50 (μg/mL) | Selectivity (HC50/MIC of MRSA) | |||||
|---|---|---|---|---|---|---|---|
| Compound | Gram + | Gram − | |||||
| MRSA | MRSE | E. faecalis | E. coli | K. pneumonia | |||
| C16-K-RBB1 | 1.5 | 1.5 | 3 | 3 | 3 | 125 | 83 |
| C16-K-RBB2 | 1.5 | 1.5 | 3 | 3 | >25 | >250 | >167 |
| C16-K-RBB3 | 3–6 | 3–6 | 6–12 | 6–12 | >25 | >250 | >42 |
HIGHLIGHTS.
Design and synthesis of 13 lipidated α/α-AA heterogenous peptides as mimics for Host Defense Peptides.
Compounds can efficiently kill both Gram-positive and Gram-negative bacteria and clear bacterial biofilm formation.
Compounds have the ability to select for bacterial cells over human red blood cells.
Acknowledgments
The work was supported by NSF 1708500 and NIH 1R01GM112652.
Footnotes
The supporting information contains information on purity of compounds as seen by High performance liquid chromatography (HPLC) and the molecular weight as seen by Matrix-assisted laser desorption ionization – Time of flight. (MALDI-TOF).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ventola CL. The Antibiotic Resistance. Crisis, Pharm Ther. 2015;40:277–283. [PMC free article] [PubMed] [Google Scholar]
- 2.Guilhelmelli F, Vilela N, Albuquerque P, da L, Derengowski S, Silva-Pereira I, Kyaw CM. Antibiotic development challenges: the various mechanisms of action of antimicrobial peptides and of bacterial resistance. Front Microbiol. 2013;4 doi: 10.3389/fmicb.2013.00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.She F, Nimmagadda A, Teng P, Su M, Zuo X, Cai J. Helical 1:1 α/Sulfono-γ-AA Heterogeneous Peptides with Antibacterial Activity. Biomacromolecules. 2016;17:1854–1859. doi: 10.1021/acs.biomac.6b00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bérdy J. Thoughts and facts about antibiotics: where we are now and where we are heading. J Antibiot (Tokyo) 2012;65:385–395. doi: 10.1038/ja.2012.27. [DOI] [PubMed] [Google Scholar]
- 5.Hancock REW, Haney EF, Gill EE. The immunology of host defence peptides: beyond antimicrobial activity. Nat Rev Immunol. 2016;16:321–334. doi: 10.1038/nri.2016.29. [DOI] [PubMed] [Google Scholar]
- 6.Nijnik A, Hancock R. Host defence peptides: antimicrobial and immunomodulatory activity and potential applications for tackling antibiotic-resistant infections. Emerg Health Threats J. 2009;2 doi: 10.3134/ehtj.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kasetty G, Papareddy P, Kalle M, Rydengård V, Mörgelin M, Albiger B, Malmsten M, Schmidtchen A. Structure-Activity Studies and Therapeutic Potential of Host Defense Peptides of Human Thrombin. Antimicrob Agents Chemother. 2011;55:2880–2890. doi: 10.1128/AAC.01515-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jenssen H, Hamill P, Hancock REW. Peptide Antimicrobial Agents. Clin Microbiol Rev. 2006;19:491–511. doi: 10.1128/CMR.00056-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee MO, Jang HJ, Rengaraj D, Yang SY, Han JY, Lamont SJ, Womack JE. Tissue expression and antibacterial activity of host defense peptides in chicken. BMC Vet Res. 2016;12 doi: 10.1186/s12917-016-0866-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mansour SC, Pena OM, Hancock REW. Host defense peptides: front-line immunomodulators. Trends Immunol. 2014;35:443–450. doi: 10.1016/j.it.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 11.Bals R, Wilson JM. Cathelicidins - a family of multifunctional antimicrobial peptides. Cell Mol Life Sci CMLS. 2003;60:711–720. doi: 10.1007/s00018-003-2186-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mattar EH, Almehdar HA, Yacoub HA, Uversky VN, Redwan EM. Antimicrobial potentials and structural disorder of human and animal defensins. Cytokine Growth Factor Rev. 2016;28:95–111. doi: 10.1016/j.cytogfr.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 13.Hilchie AL, Wuerth K, Hancock REW. Immune modulation by multifaceted cationic host defense (antimicrobial) peptides. Nat Chem Biol. 2013;9:761–768. doi: 10.1038/nchembio.1393. [DOI] [PubMed] [Google Scholar]
- 14.Huang W, Seo J, Willingham SB, Czyzewski AM, Gonzalgo ML, Weissman IL, Barron AE. Learning from Host-Defense Peptides: Cationic, Amphipathic Peptoids with Potent Anticancer Activity. PLoS ONE. 2014;9:e90397. doi: 10.1371/journal.pone.0090397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lo JCY, Lange D. Current and Potential Applications of Host-Defense Peptides and Proteins in Urology. BioMed Res Int. 2015;2015 doi: 10.1155/2015/189016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marr A, Gooderham W, Hancock R. Antibacterial peptides for therapeutic use: obstacles and realistic outlook. Curr Opin Pharmacol. 2006;6:468–472. doi: 10.1016/j.coph.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 17.Afacan NJ, Yeung ATY, Pena OM, Hancock REW. Therapeutic potential of host defense peptides in antibiotic-resistant infections. Curr Pharm Des. 2012;18:807–819. doi: 10.2174/138161212799277617. [DOI] [PubMed] [Google Scholar]
- 18.Niu Y, Wu H, Li Y, Hu Y, Padhee S, Li Q, Cao C, Cai J. AApeptides as a new class of antimicrobial agents. Org Biomol Chem. 2013;11:4283. doi: 10.1039/c3ob40444g. [DOI] [PubMed] [Google Scholar]
- 19.Nielsen PE. Peptide nucleic acids: on the road to new gene therapeutic drugs. Basic Clin Pharmacol Toxicol. 2000;86:3–7. doi: 10.1034/j.1600-0773.2000.pto860102.x. [DOI] [PubMed] [Google Scholar]
- 20.Hu Y, Li X, Sebti SM, Chen J, Cai J. Design and synthesis of AApeptides: A new class of peptide mimics. Bioorg Med Chem Lett. 2011;21:1469–1471. doi: 10.1016/j.bmcl.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 21.Hu Y, Amin MN, Padhee S, Wang RE, Qiao Q, Bai G, Li Y, Mathew A, Cao C, Cai J. Lipidated Peptidomimetics with Improved Antimicrobial Activity. ACS Med Chem Lett. 2012;3:683–686. doi: 10.1021/ml3001215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Smith C, Wu H, Padhee S, Manoj N, Cardiello J, Qiao Q, Cao C, Yin H, Cai J. Lipidated Cyclic γ-AApeptides Display Both Antimicrobial and Anti-inflammatory Activity. ACS Chem Biol. 2014;9:211–217. doi: 10.1021/cb4006613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Smith C, Wu H, Teng P, Shi Y, Padhee S, Jones T, Nguyen AM, Cao C, Yin H, Cai J. Short Antimicrobial Lipo-α/γ-AA Hybrid Peptides. ChemBioChem. 2014;15:2275–2280. doi: 10.1002/cbic.201402264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niu Y, Padhee S, Wu H, Bai G, Qiao Q, Hu Y, Harrington L, Burda WN, Shaw LN, Cao C, Cai J. Lipo-γ-AApeptides as a New Class of Potent and Broad-Spectrum Antimicrobial Agents. J Med Chem. 2012;55:4003–4009. doi: 10.1021/jm300274p. [DOI] [PubMed] [Google Scholar]
- 25.Padhee S, Smith C, Wu H, Li Y, Manoj N, Qiao Q, Khan Z, Cao C, Yin H, Cai J. The Development of Antimicrobial α-AApeptides that Suppress Proinflammatory Immune Responses. ChemBioChem. 2014;15:688–694. doi: 10.1002/cbic.201300709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Padhee S, Li Y, Cai J. Activity of lipo-cyclic γ-AApeptides against biofilms of Staphylococcus epidermidis and Pseudomonas aeruginosa. Bioorg Med Chem Lett. 2015;25:2565–2569. doi: 10.1016/j.bmcl.2015.04.039. [DOI] [PubMed] [Google Scholar]
- 27.Niu J, Li C, Wu H, Feng X, Su Q, Li S, Zhang L, Yew DTW, Cho EYP, Sha O. Propidium iodide (PI) stains Nissl bodies and may serve as a quick marker for total neuronal cell count. Acta Histochem. 2015;117:182–187. doi: 10.1016/j.acthis.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 28.Li M, Wu RS, Tsai JSC. DAPI derivative: A fluorescent DNA dye that Can be covalently attached to biomolecules. Bioorg Med Chem Lett. 2003;13:4351–4354. doi: 10.1016/j.bmcl.2003.09.038. [DOI] [PubMed] [Google Scholar]
- 29.Donlan RM. Biofilm Formation: A Clinically Relevant Microbiological Process. Clin Infect Dis. 2001;33:1387–1392. doi: 10.1086/322972. [DOI] [PubMed] [Google Scholar]
- 30.Chandki R, Banthia P, Banthia R. Biofilms: A microbial home. J Indian Soc Periodontol. 2011;15:111–114. doi: 10.4103/0972-124X.84377. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.