

Abstract
A single oligonucleotide was covalently attached to a genetically engineered subunit of the heptameric protein pore, α-hemolysin, to allow DNA duplex formation inside the pore lumen. Single-channel current recording was used to study the properties of the modified pore. On addition of an oligonucleotide 8 bases in length and with a sequence complementary to the tethered DNA strand, current blockades with durations of hundreds of milliseconds occurred, representing hybridization events of individual oligonucleotides to the tethered DNA strand. Kinetic constants for DNA duplex formation at the single molecule level were derived and found to be consistent with established literature values for macroscopic duplex formation. The resultant equilibrium constant for duplex formation in the nanopore was found to be close to the experimentally derived constant for duplex formation in solution. A good agreement between the equilibrium constants for duplex formation in the nanopore and in solution was also found for two other oligonucleotide pairs. In addition, the nanopore recordings revealed details of the kinetics difficult to obtain by conventional methods, like surface plasmon resonance, which measure ensemble properties. By investigating the temperature dependence of DNA duplex formation at the single molecule level, the standard enthalpy and entropy of the interaction could be obtained.
Duplex formation by complementary RNA or DNA strands is a fundamental biochemical process. The physical chemistry of DNA duplex formation has been studied thoroughly with techniques that measure bulk properties (1–4). However, despite its importance, little is known about DNA duplex formation at the single molecule level. Individual polynucleotide strands can be studied by fluorescence-based detection (5–8) and force measurements (9, 10), and both techniques have been used to detect duplex formation within or between individual DNA strands (11–14). With the exception of unfolding of individual RNA molecules (14), detailed kinetic data about duplex formation at the single molecule level have not been obtained. Electrical recording is another attractive technique to study individual DNA strands (15–22). Electrical recordings measure the ionic current flowing through a single membrane-embedded nanopore, and the passage or binding of individual molecules, such as DNA, can be followed by the associated change in current.
In this work, single-channel current recording was used to detect and characterize the hybridization of individual DNA strands inside the lumen of the protein nanopore α-hemolysin. α-Hemolysin (αHL) is an exotoxin produced by Staphylococcus aureus. αHL monomers assemble to form a heptameric pore of known structure (23). The pore resembles a mushroom 10 nm in height and up to 10 nm in width. The cap of the mushroom contains an internal cavity 4.6 nm wide with entrances of 3 nm diameter at the cis opening and 1.4 nm diameter at the narrow inner constriction (Fig. 1A). Molecular graphics reveal that the internal cavity is big enough to accommodate a DNA duplex 10 bp in length. We covalently attached a single DNA oligonucleotide to a cysteine residue located at the cis opening of the pore and followed the hybridization of individual complementary DNA strands by single-channel current recording.
Figure 1.

Attachment of a single DNA oligonucleotide to the αHL pore. (A) A cross section of a model of a heteroheptameric αHL pore chemically modified with a single DNA oligonucleotide. The oligonucleotide is attached via a hexamethylene linker and a disulfide bond to cysteine residue 17 of a genetically engineered αHL subunit. ⊕ indicates the positive applied electrical potential that drives negatively charged molecules from the cis to the trans side of the bilayer. (B and C) Preparation of the αHL pore H6(17C-oligo-A)1. (B) Autoradiogram of an SDS–polyacrylamide gel after electrophoresis of a mixture of unmodified αHL monomers (H) and 17C-D4 monomers attached to oligo-A through a disulfide bond (17C-oligo-A-D4), in the absence (lane 1) and presence (lane 2) of the reducing agent DTT. (C) Autoradiogram of an SDS–polyacrylamide gel containing heteroheptamers formed by the assembly of a mixture of H and 17C-oligo-A-D4 monomers. Heptamers H7, H6(17C-oligo-A)1 and H5(17C-oligo-A)2 migrate in different gel bands because of an electrophoretic shift caused by the D4-tag in the 17C-oligo-A-D4 subunits. The positions of two molecular weight markers are indicated.
Materials and Methods
Preparation of αHL Pores Modified with a Single Oligonucleotide.
Oligonucleotides were first activated and then coupled to the single cysteine residue of αHL-17C-D4 or αHL-17C-D8-H6 by using a disulfide exchange reaction. 5′ thiol-modified DNA oligonucleotides with a hexamethylene linker were obtained from Research Genetics (Huntsville, AL). The thiol group was activated with 2,2′-dithiodipyridine to yield 5′-S-thiopyridyl oligonucleotides (24). The mutant αHL-17C-D4 was derived by site-directed mutagenesis from the engineered gene αHL-WT-RL-D4, which encodes the wild-type αHL protein and a C-terminal extension of four aspartate residues (gift from S. Cheley). This αHL gene contains silent mutations in the DNA sequence coding for the β-barrel. The mutant gene αHL-WT-RL-D8-H6 encodes a C-terminal extension of eight aspartates followed by six histidine residues to facilitate the purification of the mutant polypeptide (gift from S. Cheley). Coupled in vitro transcription/translation was used to generate 35S-labeled αHL polypeptides H (wild type), 17C-D4, and 17C-D8-H6 (25). To couple activated oligonucleotides to mutant 17C-D4, translation mixes of 17C-D4 (3 μl, ≈300 ng αHL protein) and of H (15 μl, ≈1.5 μg) were combined and separated from excess β-mercaptoethanol by using spin filter columns with a molecular weight cutoff of 10 kDa (no. 42407, Millipore). For this treatment, the combined mixes were diluted into 0.1 mM DTT (0.5 ml) and concentrated by centrifugation to a volume of 30 μl. The procedure was repeated twice. The retentate (30 μl) was then diluted 2-fold into ME buffer containing 10 mM Mops⋅NaOH, pH 7.4, 150 mM NaCl, and 0.5 mM EDTA, and reacted with 50 nmol 5′-S-thiopyridyl oligonucleotide for 10 min at 25°C. Before coupling activated oligonucleotides to mutant 17C-D8-H6, the translation mix of the mutant (4 μl) was diluted into 50 mM NaH2PO4/0.15 M NaCl, pH 8.0 (0.25 ml) containing Ni-NTA-agarose (10 μl of a 50% slurry) (Qiagen, Chatsworth, CA, no. 30210). The resin was washed and eluted twice with 50 mM NaH2PO4/0.15 M NaCl/0.25 M imidazole, pH 8.0 (10 μl), as described (26). The combined eluates were diluted 5-fold into ME buffer (100 μl), reacted with 10 nmol 5′-S-thiopyridyl oligonucleotide, and then mixed with polypeptide H (30 μl of translation mix), which had been separated from excess β-mercaptoethanol by using spin filter columns (see above). The monomeric subunits were then coassembled on rabbit erythrocyte membranes, and the resulting heptamers were purified by SDS/PAGE as described (27).
Single-Channel Current Recording.
Single-channel current recordings were performed by using a planar lipid bilayer apparatus. The apparatus was enclosed by a U-shaped heat-conducting element with circulating water, connected to a thermostat (Brinkmann, RE-106), and the recordings were carried out as described (28) at 20 ± 0.4°C, unless otherwise stated. The temperatures given are the values measured in the electrolyte by using a calibrated thermocouple. Briefly, a bilayer of 1,2-diphytanoyl-sn-glycerophosphocholine (Avanti Polar Lipids) was formed on an aperture (120 μm in diameter) in a Teflon septum (Goodfellow Corporation, Malvern, PA) separating the cis and trans chambers of the apparatus. Each compartment contained 1.3–1.5 ml of 2 M KCl/12 mM MgCl2/5 mM Tris titrated to pH 7.4 with HCl. To avoid evaporation of the buffer during recordings at temperatures >25°C, the chamber was covered with a Styrofoam block. Heptameric αHL protein (final concentration 0.01–0.1 ng/ml) was added to the cis compartment, and the electrolyte in the cis chamber was stirred until a single channel inserted into the bilayer. Transmembrane currents were recorded at a holding potential of +100 mV (with the cis side grounded) by using a patch–clamp amplifier (Axopatch 200B, Axon Instruments, Union City, CA). For analysis, currents were low-pass filtered with a built-in 4-pole Bessel filter at 10 kHz and sampled at 50 kHz by computer with a Digidata 1200 A/D converter (Axon Instruments), as described (29). Unless otherwise stated, unmodified DNA oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA) and used without further purification.
The purity of DNA oligonucleotides was determined by nondenaturing PAGE (30). Instead of TBE buffer (89 mM Tris/89 mM boric acid/2 mM EDTA, pH 8.3), buffer TAE (40 mM Tris acetate/2 mM EDTA, pH 8.5) was used to prepare the gel and the running buffer. After electrophoresis, the gels were stained for 2 min in a 0.2% aqueous solution of Stains-all (Sigma, E-9379) containing 50% formamide and destained in water for 20 min. To quantify the relative intensities of the DNA bands, destained gels were scanned and analyzed by using the software scion image (Scion, Frederick, MD). Commercial preparations of the 8-mers oligo-A, oligo-B, oligo-C, and oligo-D had purities of ≥98% and contained ≈1% 7-mer and ≤1% 6-mer.
Kinetic Analysis.
For the different oligonucleotide pairs, the association constants (kon) for duplex formation in the DNA nanopore were calculated from kon = 1/(c⋅τon), where τon is the inter-event interval and c the concentration of free oligonucleotide in the cis chamber. The strand dissociation constants (koff) were derived from the event lifetime (τoff): koff = 1/τoff (31).
For comparison with rate constants obtained from single-channel current recording, kinetic constants for duplex formation in homogenous solution were obtained for the same oligonucleotide pairs. The rate constants for duplex dissociation, k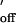 , in homogeneous solution were calculated by using the relation k
, in homogeneous solution were calculated by using the relation k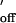 = k
= k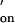 ⋅K
⋅K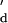 . The equilibrium dissociation constants K
. The equilibrium dissociation constants K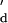 were experimentally derived by using a modified van't Hoff method on the basis of the dependence of the melting temperature, Tm, on the concentration of DNA strands (32) [see supporting information no. 3 on the PNAS web site (www.pnas.org)]. The association rate constant k
were experimentally derived by using a modified van't Hoff method on the basis of the dependence of the melting temperature, Tm, on the concentration of DNA strands (32) [see supporting information no. 3 on the PNAS web site (www.pnas.org)]. The association rate constant k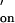 was assumed to be 107 M−1⋅s−1. Literature values for the association rate constant of complementary oligonucleotides range from 4 × 105 to 2 × 107 M−1⋅s−1 (33, 34) and are influenced by the ionic strength of the buffer, the base composition, and the length of the oligonucleotide. Short oligonucleotides (six to eight bases) with a GC content of 40–50% are reported to have k
was assumed to be 107 M−1⋅s−1. Literature values for the association rate constant of complementary oligonucleotides range from 4 × 105 to 2 × 107 M−1⋅s−1 (33, 34) and are influenced by the ionic strength of the buffer, the base composition, and the length of the oligonucleotide. Short oligonucleotides (six to eight bases) with a GC content of 40–50% are reported to have k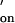 values of 106 M−1⋅s−1 (34, 35). The oligonucleotide pairs studied in this work have similar properties and an estimate of k
values of 106 M−1⋅s−1 (34, 35). The oligonucleotide pairs studied in this work have similar properties and an estimate of k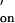 = 107 M−1⋅s−1 was made, which takes into account the high salt concentration (2 M KCl/12 mM MgCl2) in our system (1, 36).
= 107 M−1⋅s−1 was made, which takes into account the high salt concentration (2 M KCl/12 mM MgCl2) in our system (1, 36).
Results
Construction of a “DNA Nanopore.”
We generated an αHL pore carrying a single DNA oligonucleotide attached to a site located at the cis entrance of the lumen (Fig. 1A). The “DNA nanopore” was composed of six unmodified subunits and one subunit covalently modified with the oligonucleotide. Heptamers with this composition were obtained by assembly of unmodified αHL (H) and the cysteine mutant 17C-D4, which had been coupled through a disulfide linkage to oligo-A with the sequence 5′-CATTCACC-3′ (Fig. 1B). Heteroheptamer H6(17C-oligo-A)1 was separated from heptamers H7 and H5(17C-oligo-A)2, which also formed during the assembly process, by SDS/PAGE (Fig. 1C). The proteins in the electrophoretic bands were eluted, heated to dissociate the subunits, and analyzed by further electrophoresis to confirm the ratio of subunits in each heptamer (data not shown). The various heptamers migrated in separate bands by virtue of a gel shift caused by the C-terminal polypeptide extension of four aspartates (D4), present in 17-oligo-A-D4 but not in the H subunits. Interestingly, the modification of 17C-D4 with DNA caused the monomer to migrate more slowly (Fig. 1B, compare lanes 1 and 2) but did not alter the electrophoretic mobility of the heptamer.
We also generated a DNA nanopore containing a modified 17C-D8-H6 subunit instead of the 17C-D4 subunit. The cysteine mutant with the C-terminal His-tag was used to separate the mutant polypeptide from reducing agents and thereby facilitate the coupling to the oligonucleotide. The C-terminal extension did not influence the electrophysiological properties of the DNA nanopore (data not shown). Heptamers with the two different tags were used throughout the study and are, in the following, referred to as H6(17C-oligo-A)1.
Hybridization of Individual DNA Strands in the DNA Nanopore.
Planar lipid bilayer recordings were used to examine the single-channel properties of H6(17C-oligo-A)1 and its interaction with an oligonucleotide of complementary sequence added to the cis chamber. The pores were analyzed at an applied potential of +100 mV, which drives negatively charged molecules such as DNA from the cis to the trans side of the bilayer (Fig. 1A). In 2 M KCl/12 mM MgCl2/5 mM Tris⋅HCl, pH 7.4, the unitary conductance of the DNA nanopore was 1,580 ± 50 pS (n = 4). The single-channel currents were decorated with brief current fluctuations (mean lifetime, 0.13 ± 0.03 ms; amplitude, 100 ± 20 pS; frequency of occurrence, 8.1 ± 1.4 s−1, n = 4) (Fig. 2A). The conductance of H6(17C-oligo-A)1 is lower than the value for H7 channels (1,690 ± 30 pS, n = 4) or H6(17C-oligo-A)1 channels, which had been treated with DTT to cleave the disulfide bond between the oligonucleotide and αHL (1,670 ± 40 pS, n = 3). The reduced conductance of H6(17C-oligo-A)1 indicates that the tethered DNA oligonucleotide is likely to be inside the cavity (model in Fig. 2A); possible explanations for the reduced conductance include steric, electrostatic, or other factors. When 67 nM oligo-B, with a sequence fully complementary to the tethered oligo-A, was added to the cis side of the bilayer, two types of events occurred: negative current deflections (Fig. 2B, b), characterized by a duration of hundreds of milliseconds, a mean amplitude of 534 ± 18 pS, and a frequency of occurrence of 0.31 ± 0.03 s−1 (n = 4), and spike-like events (Fig. 2B, s), with a mean lifetime of 0.2 ± 0.03 ms, a mean amplitude of 870 ± 100 pS, and a frequency of occurrence of 0.080 ± 0.015 s−1 (n = 4). The current deflections (b) most likely represent single oligo-B molecules, which enter the DNA nanopore 5′-end first and form a duplex with the tethered complementary oligo-A. The spike at the end of each binding event (Fig. 2B) indicates that after dissociation oligo-B passes the inner constriction to exit on the trans side of the pore (model in Fig. 2B) (20). The individual spikes (s) most likely represent oligo-B strands that pass through the pore without binding to the tethered DNA strand. For example, binding to the tethered oligonucleotide might not occur, when oligo-B enters the pore with the 3′-end first. To prove that the current deflections (b) represent oligo-B binding to the tethered oligo-A, excess free oligo-A was added on the cis side. If the binding were specific, excess free oligo-A would compete for the binding of oligo-B to tethered oligo-A (Fig. 2C). Indeed, the frequency of occurrence of the proposed binding events was reduced 18-fold (to 0.017 s−1), whereas spikes, now presumably stemming from oligo-A transiting the lumen without binding (20, 37), appeared with a frequency of occurrence of 13.9 s−1 (Fig. 2C).
Figure 2.

An αHL pore modified with a single DNA oligonucleotide responds to individual binding events with oligonucleotides of complementary sequence. (A) Representative single-channel current trace of H6(17C-oligo-A)1 at a transmembrane potential of +100 mV relative to the cis side of the bilayer. (B) Representative trace of the same channel as in A in the presence of 67 nM oligo-B (green) in the cis chamber. Negative current deflections (b) represent individual binding events of oligo-B (green) to the tethered oligo-A (red). The short downward spikes (s) in the trace are translocation events of oligo-B that did not bind to the tethered oligonucleotide. (C) Trace of the same channel as in A and B with 67 nM oligo-B and 3.3 μM oligo-A in the cis chamber. Excess oligo-A hybridizes to oligo-B and thereby competes for the binding of oligo-B to the tethered oligonucleotide. The short downward spikes in the trace are translocation events of excess oligo-A molecules through the pore.
Kinetics of Duplex Formation on the Single Molecule Level.
Single-channel current recording was used to derive the kinetic constants for the association and the dissociation of individual DNA strands. Each binding event, oligo-B to H6(17C-oligo-A)1 (Fig. 2B), was characterized by its event amplitude IE and its event lifetime τoff (Fig. 3A). The two characteristic parameters for hundreds of individual binding events from one recording were plotted onto an event diagram, in which each point represents one event (Fig. 3B). Although the event amplitudes of the binding events were narrowly distributed (528 ± 23 pS), the event lifetimes were scattered between 10 and 4,000 ms with a mean value of 852 ms (Fig. 3B). Lifetime histogram analysis revealed that the event population was composed of two different event types with two different event lifetimes, τoff-1 and τoff-2 (Fig. 3C; see Inset for the event distribution with the shorter lifetime τoff-1). The mean τoff values of the two different event types obtained from four recordings with a total number of 7,000 events were τoff-1 = 84 ± 10 ms (11 ± 2% of the events) and τoff-2 = 821 ± 113 ms (89 ± 2%). The short τoff-1 does not stem from the short spikes (Fig. 2B, symbol s), as events with a lifetime shorter than 0.5 ms were not included in the event diagram and lifetime histogram. To account for the two τoff values, two simple kinetic models can be envisioned. In the first model, DNA duplex AB forms by the association of DNA strands A and B and is assumed to dissociate along two kinetically different routes (supporting information no. 1, www.pnas.org). The kinetic scheme and the observed lifetimes predict that the probabilities for duplex AB to dissociate along routes 1 and 2 are 0.91 and 0.09, respectively (see supporting information no. 1). However, these values are in clear contradiction to the experimentally found values of P1 = 0.11 and P2 = 0.89. Therefore, the observed kinetic parameters cannot be explained by the kinetic model I. More likely, hybridization follows kinetic model II, characterized by two completely distinct binding and dissociation events. Inserting the parameters τoff-1, τoff-2, P1, P2, and the interevent interval (τon), which showed a linear dependence on the concentration of oligo-B (Fig. 3D), into the second kinetic scheme yielded stability constants for the two classes of binding events (supporting information no. 1): Kd-1 = 7.4 × 10−6 M, Kd-2 = 9.2 × 10−8 M. Further investigation will be required to clarify the heterogeneity observed at the single molecule level.
Figure 3.

Statistical summary of the binding events of DNA oligonucleotide oligo-B to H6(17C-oligo-A)1. (A) Definition of event lifetime, τoff, and event amplitude, IE. (B) An event diagram shows the event lifetimes τoff and event amplitudes IE for a single-channel current recording of 3 min with 200 nM oligo-B in the cis chamber. Each point in the diagram represents an individual binding event of oligo-B to the tethered oligo-A in H6(17C-oligo-A)1. (C) Lifetime histogram (bin width, 200 ms) for the event diagram of the recording displayed in B showing the exponential fit for events with a longer lifetime τoff-2. (Inset) Histogram with a bin width of 30 ms showing the distribution of lifetimes for the shorter event, characterized by τoff-1. The exponential fits were obtained by using pstat of the pclamp software package and are weighted for the number of events. (D) The reciprocal of the interevent interval (τon) exhibits a linear dependence on the concentration of oligo-B up to 400 nM. (E) Effects of temperature on the kinetic constants kon and koff for the formation and dissociation of DNA duplexes in the lumen of the DNA nanopore H6(17C-oligo-A)1. The concentration of oligo-B in the cis chamber was 212 nM. The experiment was repeated and gave the same result.
It is clear that Kd-2 dominates the composite Kd obtained from the weighted lifetimes (supporting information no. 1, www.pnas.org), and kinetic constants for the dominant events with the longer lifetime are used from here on to simplify the analysis. Through the analysis of the inter-event intervals (τon) and the event lifetimes (τoff) of the pooled data from independent single-channel current recordings from four different H6(17C-oligo-A)1 heteroheptamers, we were able to obtain the kinetic constants for strand association (kon) and strand dissociation (koff). The value of kon was 1.3 × 107 M−1⋅s−1, and koff was 1.2 s−1. The value of kon is close to the value expected for duplex formation by a short oligonucleotide with 50% GC content at high ionic strength (see Materials and Methods and refs. 1, 34, and 35). koff is slightly higher than the value for dissociation in solution (0.4 s−1, calculated from an estimated k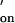 and the experimentally derived K
and the experimentally derived K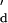 ; see supporting information no. 3, www.pnas.org).
; see supporting information no. 3, www.pnas.org).
Good agreement between the kinetic data for duplex formation in the nanopore and in solution was found for two other oligonucleotides with different affinities (Table 1). One oligonucleotide studied was oligo-D, an 8-mer-like oligo-B but with a different sequence (5′-TACGTGGA-3′). Oligo-D formed a duplex with the complementary oligonucleotide (5′-TCCACGTA-3′) tethered to the nanopore. As for oligo-B, two different τoff values were observed (37 ± 5 ms, 10 ± 3% of the events; and 290 ± 40 ms, 90 ± 3%). The rate constants for association and dissociation for the dominant events of longer lifetime were kon = 2.2 × 107 M−1⋅s−1 and koff = 3.4 s−1. These values are comparable to the rate constants for duplex formation of the same oligonucleotide pair in solution, k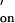 = 1.0 × 107 M−1⋅s−1 (estimated) and k
= 1.0 × 107 M−1⋅s−1 (estimated) and k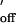 = 1.7 s−1 (calculated from k
= 1.7 s−1 (calculated from k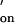 and the experimentally derived K
and the experimentally derived K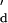 ). The equilibrium dissociation constants for the nanopore and in solution are similar, Kd = 1.5 × 10−7 M (from kon and koff) and K
). The equilibrium dissociation constants for the nanopore and in solution are similar, Kd = 1.5 × 10−7 M (from kon and koff) and K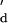 = 1.7 × 10−7 M. We also studied duplex formation with the 7-mer oligo-E, a truncated version of oligo-B with a lower binding affinity. Again, the rate constants and the equilibrium dissociation constants were comparable, e.g., Kd = 1.5 × 10−6 M and K
= 1.7 × 10−7 M. We also studied duplex formation with the 7-mer oligo-E, a truncated version of oligo-B with a lower binding affinity. Again, the rate constants and the equilibrium dissociation constants were comparable, e.g., Kd = 1.5 × 10−6 M and K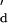 = 8.3 × 10−7 M (Table 1). Nanopore recordings did not yield two different τoff values for this oligonucleotide pair.
= 8.3 × 10−7 M (Table 1). Nanopore recordings did not yield two different τoff values for this oligonucleotide pair.
Table 1.
Comparison of the kinetics and thermodynamics of three oligonucleotide pairs studied by single channel current recordings with DNA-modified αHL pores and by ensemble melting curves
| Oligo | Sequence* | Values derived from nanopore recordings
|
Values derived from melting profiles in solution
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| kon [M−1⋅s−1]† | koff [s−1]† | Kd [M] | ΔG° [kcal mol−1]‡ | k′on [M−1⋅s−1]‡§ | k′off [s−1]‡¶ | K′d‡ | ΔG′° [kcal mol−1]‡ | ||
| Oligo-B | 5′-GGTGAATG-3′ | 1.3 × 107 | 1.2 | 9.2 × 10−8 | −9.2 | 107 | 0.4 | 3.6 × 10−8 | −9.8 |
| Oligo-D | 5′-TACGTGGA-3′ | 2.2 × 107 | 3.4 | 1.5 × 10−7 | −8.9 | 107 | 1.7 | 1.7 × 10−7 | −8.9 |
| Oligo-E | 5′-GGTGAAT-3′ | 1.1 × 107 | 16 | 1.5 × 10−6 | −7.7 | 107 | 8 | 8.3 × 10−7 | −8.0 |
The sequences of the DNA oligonucleotides added to the solution are given. The sequence of the corresponding tethered DNA strands were: for oligo-B, oligo-A (5′-CATTCACC-3′); for oligo-D, oligo-C (5′-TCCACGTA-3′); for oligo-E, oligo-A.
The values given are derived from the arithmetic mean of the exponential fits of at least three independent single channel current recordings performed at 20 ± 0.4°C. For oligo-E⋅oligo-A, the single exponential fits for the event lifetime, and for oligo-D⋅oligo-C, and oligo-B⋅oligo-A, the exponential fits for the dominant, longer lifetime were used. The single exponential fits for the inter-event interval were used in all cases, and the values were adjusted for the proportion factor P in the case of oligo-D⋅oligo-C and oligo-B⋅oligo-A. The association constant (kon) for duplex formation in the DNA-nanopore was calculated from kon = 1/(c⋅τon), where τon is the inter-event interval and c the concentration of free oligonucleotide in the cis chamber. The strand dissociation constant (koff) was derived from the event lifetime (τoff): koff = 1/τoff. The standard deviations of kon and koff were smaller than 15%.
The values are for 20°C. The values for the nanopore were derived experimentally at this temperature, and the values in solution were calculated for 20°C by using the experimentally derived thermodynamic parameters (Supporting information no. 3; www.pnas.org).
The association rate constants were assumed to be 107 M−1⋅s−1 (see Materials and Methods, Kinetic Analysis).
The rate constants for duplex dissociation in homogeneous solution were calculated by using the relation k′off = k′on⋅K′d. The equilibrium dissociation constant K′d was experimentally derived as described in Materials and Methods, Kinetic Analysis.
Hence, single-channel current experiments with a DNA nanopore give kinetic data in line with literature values and, in addition, offer the ability to detect properties (e.g., detailed kinetics) that are often difficult to investigate by conventional methods that measure ensemble properties. Our results indicate that the data obtained with DNA nanopores can approximate kinetic and thermodynamic data on DNA binding obtained from experiments in solution. However, it cannot be concluded that the two processes are identical. For instance, the kinetics might be affected by opposing but compensating factors, such as steric constraints or effects of the applied potential (38, 39).
Effects of Temperature on Hybridization in a Single Nanopore.
Next, we studied the temperature dependence of duplex formation of individual molecules by single-channel current recording. The interaction of the heteroheptameric pore H6(17C-oligo-A)1 with 212 nM oligo-B was examined for temperatures ranging from 10 to 40°C at 5° intervals. Through the analysis of the interevent intervals (τon) and the event lifetimes (τoff), the kinetic constants for strand association (kon) and dissociation (koff) were obtained and are plotted as a function of temperature (Fig. 3E). The plot for kon reveals a weak negative temperature dependence (linear fit: −0.007 M−1⋅s−1 deg−1). By contrast, koff increased exponentially with temperature (exponential constant: 0.19 deg−1). The weak temperature dependence of kon and the strong dependence of koff are in good agreement with the literature on duplex formation in solution (40).
From the temperature dependences of kon and koff, we derived the activation enthalpies ΔH‡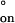 and ΔH‡
and ΔH‡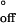 and the activation entropies ΔS‡
and the activation entropies ΔS‡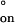 and ΔS‡
and ΔS‡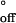 for the association and dissociation of oligo-A and oligo-B in the nanopore (supporting information no. 2, www.pnas.org). The values for duplex association were ΔH‡
for the association and dissociation of oligo-A and oligo-B in the nanopore (supporting information no. 2, www.pnas.org). The values for duplex association were ΔH‡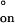 = −0.5 kcal mol−1 and ΔS‡
= −0.5 kcal mol−1 and ΔS‡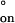 = −29 cal mol−1 deg−1. Small, negative values for ΔH‡
= −29 cal mol−1 deg−1. Small, negative values for ΔH‡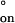 are often found for DNA duplex formation in solution (33, 41), and the negative sign of ΔH‡
are often found for DNA duplex formation in solution (33, 41), and the negative sign of ΔH‡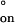 has been used to conclude that the rate-limiting step of duplex formation must involve at least two base pairs (33, 40). The observed value for ΔS‡
has been used to conclude that the rate-limiting step of duplex formation must involve at least two base pairs (33, 40). The observed value for ΔS‡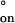 is close to the solution value for a DNA duplex of similar length (41). The values for the activation parameters for the duplex dissociation in the nanopore were ΔH‡
is close to the solution value for a DNA duplex of similar length (41). The values for the activation parameters for the duplex dissociation in the nanopore were ΔH‡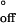 = 34 kcal mol−1 and ΔS‡
= 34 kcal mol−1 and ΔS‡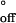 = 57 cal mol−1⋅deg−1. The value for the activation enthalpy is in line with literature values for duplex formation in solution (34, 40), and the value for the activation entropy is 40% lower than values for similar dissociation events in solution (41).
= 57 cal mol−1⋅deg−1. The value for the activation enthalpy is in line with literature values for duplex formation in solution (34, 40), and the value for the activation entropy is 40% lower than values for similar dissociation events in solution (41).
Similarly, we calculated the equilibrium constant for duplex formation from kinetic data and used the temperature dependence of the equilibrium constant to derive the standard enthalpy change ΔH° and the standard entropy change ΔS° for duplex formation in the nanopore (supporting information no. 2, www.pnas.org). This procedure yielded values of −34 kcal mol−1 for ΔH° and −84 cal mol−1⋅deg−1 for ΔS°, and a value of −9.4 kcal mol−1 for the standard Gibbs free energy change ΔG° at 20°C. By comparison, the thermodynamic parameters for duplex formation by oligo-A and oligo-B in solution were ΔH°′ = −42 kcal mol−1 and ΔS°′ = −110 cal mol−1⋅deg−1 as determined by analysis of the concentration dependence of the melting temperature (see supporting information no. 3, www.pnas.org). The resultant standard free energy ΔG°′ at 20°C was −9.8 kcal mol−1. The values for ΔG° and ΔG°′ are very close to each other, indicating that DNA duplex formation in a nanopore is not dissimilar, thermodynamically, to duplex formation in solution. The possibility that the similar free energy values for duplex formation in the nanopore and in solution result from the summations of contributions from various compensating factors cannot be ruled out.
Discussion
In this study, we describe the construction of DNA nanopores and their application in an examination of DNA duplex formation at the single molecule level. Nanopores have recently been used to identify single-base mismatches in individual DNA strands (20, 42). In the present work, electrical recordings of single DNA nanopores were used to study details of the kinetics of DNA duplex formation; this approach avoids problems associated with conventional techniques such as surface plasmon resonance (SPR). In SPR, the transport of the analyte to the sensor surface can be impeded by slow diffusion through the immobilization matrix (43). Indeed, kon values derived by SPR are reported to be around 104 M−1⋅s−1 (3), which is at least an order of magnitude lower than the values for duplex formation in solution at low salt concentrations (105 M−1⋅s−1 to 106 M−1⋅s−1) (1, 33, 34, 40). In comparison, our approach yields kinetic constants in good agreement with data for duplex formation in solution. Furthermore, the use of DNA nanopores provides kinetic details not readily obtained by conventional techniques, which measure ensemble properties. For example, we observed two different binding events characterized by their koff values.
The study of DNA duplex formation in a single nanopore also permits examination of the all-or-none model for duplex formation (33, 40) at the single molecule level. According to this kinetic model, the rate-limiting step of DNA recombination is the “nucleation” of a duplex with two or three base pairs followed by the rapid “zippering” of the two DNA strands to form the complete double helix. As a consequence, the model predicts that a population of complementary DNA strands is predominantly single-stranded and completely duplexed and contains few incomplete duplexes. Our single-channel current traces showing discrete DNA-binding events are in agreement with the predictions of this model. The start of each DNA-binding event is characterized by a sharp current drop indicating rapid formation of the duplex. The sharp current drop suggests that duplex zippering occurs at a rate faster than the resolution of the recording, which is 3 × 104 s−1 at the filtering frequency of 10 kHz (44). This result is in line with a reported zippering rate of 2 × 106 s−1 that was obtained for the hybridization of a different oligonucleotide pair studied by the temperature-jump method (45).
We also examined the effects of temperature on DNA duplex formation at the single molecule level and found that kon is almost independent of temperature, whereas koff is temperature dependent. These results are in excellent agreement with macroscopic kinetic data, demonstrating the potential application of DNA nanopores for kinetic studies. DNA duplex formation in a single nanopore also allows the exploration of the macroscopic concept of melting temperature for individual DNA molecules (see supporting information no. 4, www.pnas.org).
The transmembrane potential applied in a current recording might be expected to influence duplex formation by the charged DNA strands in the nanopore. Although we found this to be the case, our preliminary data indicate that the potential exerts only a minimal effect on the kinetics. For example, kon decreased by a factor of 1.7 when the applied potential was increased from +100 to +190 mV, whereas koff decreased linearly by a factor of 2.0 over the same voltage range (S.H. and H.B., unpublished work). Usually, the affinity of a charged blocker (in this case DNA) for a site within the lumen of a channel shows an exponential voltage dependence (46, 47). The observed weak voltage dependence of kon and koff might be explained by assuming that the transmembrane potential does not fall off across the internal cavity (Fig. 1A) but drops across the β-barrel of the αHL pore. In addition, the negative charge of the DNA backbone might be masked by Na+ and Mg2+ ions (48), minimizing the effect of the potential. Hence, the transmembrane voltage has a kinetically negligible effect on duplex formation and, therefore, does not limit the usefulness of DNA nanopores for kinetic studies.
Supplementary Material
Acknowledgments
We thank Stephen Cheley (Dept. of Medical Biochemistry and Genetics, The Texas A&M University System Health Science Center) for providing plasmids αHL-D4 and αHL-D8-H6, and Giovanni Gadda and Paul Nixon for help with melting profiles of oligonucleotides. This work was supported by the U.S. Department of Energy, the National Institutes of Health, the Office of Naval Research (Multidisciplinary University Research Initiative 1999), and the Texas Advanced Technology Program. S.H. currently holds a fellowship from the Max-Kade Foundation and was recipient of a postdoctoral scholarship from the Austrian Science Foundation (Fonds zur Förderung der wissenschaftlichen Forschung) during the earlier stages of the work.
Abbreviation
- αHL
α-hemolysin
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Braunlin W H, Bloomfield V A. Biochemistry. 1991;30:754–758. doi: 10.1021/bi00217a026. [DOI] [PubMed] [Google Scholar]
- 2.Parkhurst K M, Parkhurst L J. Biochemistry. 1995;34:285–292. doi: 10.1021/bi00001a035. [DOI] [PubMed] [Google Scholar]
- 3.Jensen K K, Orum H, Nielsen P E, Norden B. Biochemistry. 1997;36:5072–5077. doi: 10.1021/bi9627525. [DOI] [PubMed] [Google Scholar]
- 4.Wright Lucas S, Harding M M. Anal Biochem. 2000;282:70–79. doi: 10.1006/abio.2000.4568. [DOI] [PubMed] [Google Scholar]
- 5.Nie S, Zare R N. Annu Rev Biophys Biomol Struct. 1997;26:567–596. doi: 10.1146/annurev.biophys.26.1.567. [DOI] [PubMed] [Google Scholar]
- 6.Eigen M, Rigler R. Proc Natl Acad Sci USA. 1994;91:5740–5747. doi: 10.1073/pnas.91.13.5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ambrose W P, Goodwin P M, Jett J H, Van Orden A, Werner J H, Keller R A. Chem Rev. 1999;99:2929–2956. doi: 10.1021/cr980132z. [DOI] [PubMed] [Google Scholar]
- 8.Eggeling C, Fries J R, Brand L, Gunther R, Seidel C A. Proc Natl Acad Sci USA. 1998;95:1556–1561. doi: 10.1073/pnas.95.4.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith S B, Cui Y, Bustamante C. Science. 1996;271:795–799. doi: 10.1126/science.271.5250.795. [DOI] [PubMed] [Google Scholar]
- 10.Clausen-Schaumann H, Seitz M, Krautbauer R, Gaub H E. Curr Opin Chem Biol. 2000;4:524–530. doi: 10.1016/s1367-5931(00)00126-5. [DOI] [PubMed] [Google Scholar]
- 11.Strunz T, Oroszlan K, Schafer R, Guntherodt H J. Proc Natl Acad Sci USA. 1999;96:11277–11282. doi: 10.1073/pnas.96.20.11277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rief M, Clausen-Schaumann H, Gaub H E. Nat Struct Biol. 1999;6:346–349. doi: 10.1038/7582. [DOI] [PubMed] [Google Scholar]
- 13.Kinjo M, Rigler R. Nucleic Acids Res. 1995;23:1795–1799. doi: 10.1093/nar/23.10.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liphardt J, Onoa B, Smith S B, Tinoco I J, Bustamante C. Science. 2001;292:733–737. doi: 10.1126/science.1058498. [DOI] [PubMed] [Google Scholar]
- 15.Kasianowicz J J, Brandin E, Branton D, Deamer D W. Proc Natl Acad Sci USA. 1996;93:13770–13773. doi: 10.1073/pnas.93.24.13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akeson M, Branton D, Kasianowicz J J, Brandin E, Deamer D W. Biophys J. 1999;77:3227–3233. doi: 10.1016/S0006-3495(99)77153-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meller A, Nivon L, Brandin E, Golovchenko J, Branton D. Proc Natl Acad Sci USA. 2000;97:1079–1084. doi: 10.1073/pnas.97.3.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deamer D W, Akeson M. Trends Biotechnol. 2000;18:147–151. doi: 10.1016/s0167-7799(00)01426-8. [DOI] [PubMed] [Google Scholar]
- 19.Henrickson S E, Misakian M, Robertson B, Kasianowicz J J. Phys Rev Lett. 2000;85:3057–3060. doi: 10.1103/PhysRevLett.85.3057. [DOI] [PubMed] [Google Scholar]
- 20.Vercoutere W, Winters-Hilt S, Olsen H, Deamer D, Haussler D, Akeson M. Nat Biotechnol. 2001;19:248–252. doi: 10.1038/85696. [DOI] [PubMed] [Google Scholar]
- 21.Dumas F, Duckely M, Pelczar P, Van Gelder P, Hohn B. Proc Natl Acad Sci USA. 2001;98:485–490. doi: 10.1073/pnas.011477898. . (First Published January 9, 2001; 10.1073/pnas.011477898) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szabo I, Bathori G, Tombola F, Coppola A, Schmehl I, Brini M, Ghazi A, De Pinto V, Zoratti M. FASEB J. 1998;12:495–502. doi: 10.1096/fasebj.12.6.495. [DOI] [PubMed] [Google Scholar]
- 23.Song L, Hobaugh M R, Shustak C, Cheley S, Bayley H, Gouaux J E. Science. 1996;274:1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 24.Corey D R, Munoz-Medellin D, Huang A. Bioconjug Chem. 1995;6:93–100. doi: 10.1021/bc00031a011. [DOI] [PubMed] [Google Scholar]
- 25.Cheley S, Braha O, Lu X, Conlan S, Bayley H. Protein Sci. 1999;8:1257–1267. doi: 10.1110/ps.8.6.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Howorka S, Sára M, Wang Y, Kuen B, Sleytr U B, Lubitz W, Bayley H. J Biol Chem. 2000;48:37876–37886. doi: 10.1074/jbc.M003838200. [DOI] [PubMed] [Google Scholar]
- 27.Howorka S, Movileanu L, Lu X, Magnon M, Cheley S, Braha O, Bayley H. J Am Chem Soc. 2000;122:2411–2416. [Google Scholar]
- 28.Braha O, Walker B, Cheley S, Kasianowicz J J, Song L, Gouaux J E, Bayley H. Chem Biol. 1997;4:497–505. doi: 10.1016/s1074-5521(97)90321-5. [DOI] [PubMed] [Google Scholar]
- 29.Movileanu L, Howorka S, Braha O, Bayley H. Nat Biotechnol. 2000;18:1091–1095. doi: 10.1038/80295. [DOI] [PubMed] [Google Scholar]
- 30.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Current Protocols in Molecular Biology. Vol. 1. New York: Wiley; 1987. pp. 2.7.1–2.7.8. [Google Scholar]
- 31.Moczydlowski E. In: Ion Channel Reconstitution. Miller C, editor. New York: Plenum; 1986. pp. 75–113. [Google Scholar]
- 32.Martin F H, Uhlenbeck O C, Doty P. J Mol Biol. 1971;57:201–215. doi: 10.1016/0022-2836(71)90341-x. [DOI] [PubMed] [Google Scholar]
- 33.Cantor C R, Schimmel P R. Biophysical Chemistry Part III, The Behavior of Biological Macromolecules. New York: Freeman; 1980. [Google Scholar]
- 34.Riesner D, Romer R. In: Physico-Chemical Properties of Nucleic Acids. Duchesne J, editor. Vol. 2. New York: Academic; 1973. pp. 237–318. [Google Scholar]
- 35.Porschke D. Biopolymers. 1971;10:1989–1996. [Google Scholar]
- 36.Breslauer K J, Bina-Stein M. Biophys Chem. 1977;7:211–216. doi: 10.1016/0301-4622(77)87024-5. [DOI] [PubMed] [Google Scholar]
- 37.Meller A, Nivon L, Branton D. Phys Rev Lett. 2001;86:3435–3438. doi: 10.1103/PhysRevLett.86.3435. [DOI] [PubMed] [Google Scholar]
- 38.Gilles P N, Wu D J, Foster C B, Dillon P J, Chanock S J. Nat Biotechnol. 1999;17:365–370. doi: 10.1038/7921. [DOI] [PubMed] [Google Scholar]
- 39.Sosnowski R G, Tu E, Butler W F, O'Connell J P, Heller M J. Proc Natl Acad Sci USA. 1997;94:1119–1123. doi: 10.1073/pnas.94.4.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Porschke D, Eigen M. J Mol Biol. 1971;62:361–381. doi: 10.1016/0022-2836(71)90433-5. [DOI] [PubMed] [Google Scholar]
- 41.Nelson J W, Tinoco I., Jr Biochemistry. 1982;21:5289–5295. doi: 10.1021/bi00264a026. [DOI] [PubMed] [Google Scholar]
- 42.Howorka S, Cheley S, Bayley H. Nat Biotechnol. 2001;19:636–639. doi: 10.1038/90236. [DOI] [PubMed] [Google Scholar]
- 43.Schuck P. Annu Rev Biophys Biomol Struct. 1997;26:541–566. doi: 10.1146/annurev.biophys.26.1.541. [DOI] [PubMed] [Google Scholar]
- 44.Colquhoun D, Sigworth F J. In: Single-Channel Recording. Sakmann B, Neher E, editors. New York: Plenum; 1995. pp. 483–587. [Google Scholar]
- 45.Porschke D. Biophys Chem. 1974;2:97–101. doi: 10.1016/0301-4622(74)80029-3. [DOI] [PubMed] [Google Scholar]
- 46.Woodhull A M. J Gen Physiol. 1973;61:687–708. doi: 10.1085/jgp.61.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hille B. Ionic Channels of Excitable Membranes. Sunderland, MA: Sinauer; 1992. [Google Scholar]
- 48.Li A Z, Huang H, Re X, Qi L J, Marx K A. Biophys J. 1998;74:964–973. doi: 10.1016/S0006-3495(98)74019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.