

Abstract
Purkinje neurons, the sole output of the cerebellar cortex, deliver GABA-mediated inhibition to the deep cerebellar nuclei. To subserve this critical function, Purkinje neurons fire repetitively, and at high frequencies, features that have been linked to the unique properties of the voltage-gated sodium (Nav) channels expressed. In addition to the rapidly activating and inactivating, or transient, component of the Nav current (INaT) present in many types of central and peripheral neurons, Purkinje neurons, also expresses persistent (INaP) and resurgent (INaR) Nav currents. Considerable progress has been made in detailing the biophysical properties and identifying the molecular determinants of these discrete Nav current components, as well as defining their roles in the regulation of Purkinje neuron excitability. Here, we review this important work and highlight the remaining questions about the molecular mechanisms controlling the expression and the functioning of Nav currents in Purkinje neurons. We also discuss the impact of the dynamic regulation of Nav currents on the functioning of individual Purkinje neurons and cerebellar circuits.
Keywords: Conductance, Accessory subunit, Cerebellum, Intrinsic excitability
Introduction
In most mammalian central neurons, voltage-gated sodium (Nav) currents underlie the generation and propagation of action potentials and regulate repetitive firing rates [1, 2]. The detailed time- and voltage-dependent properties of the Nav currents, in concert with the many other voltage- and non-voltage-gated currents expressed, underlie the generation of cell-type-specific differences in the waveforms of individual action potentials and the rates and patterns of repetitive firing [1, 2]. In cerebellar Purkinje neurons, which fire repetitively at high frequency in vivo [3] and in vitro [4], independent of synaptic inputs [4, 5], for example, three Nav current components have been distinguished: (1) a rapidly activating and inactivating, i.e., transient, component, INaT; (2) a non-inactivating, persistent component, INaP [6]; and (3) a resurgent component, INaR [5]. These Nav current components all contribute to shape the waveforms of action potentials and control the repetitive firing rates of Purkinje neurons [5, 7, 8]. Deletion or mutation of Scn1a or Scn8a, which encode the Nav channel pore-forming (α) subunits Nav1.1 or Nav1.6, respectively, attenuates Nav currents and repetitive firing rates in Purkinje neurons, the sole output neurons of the cerebellar cortex, and impairs motor performance [9–12]. Interestingly, modulation of the voltage-dependent properties of Nav currents in Purkinje neurons also disrupts high-frequency firing and affects motor coordination and balance [13].
Here, we review the time- and voltage-dependent properties of the Nav currents expressed in cerebellar Purkinje neurons and explore the roles of the three Nav current components, INaT, INaP, and INaR, in shaping the firing properties and the functioning of these cells. We also discuss recent studies focused on identifying the molecular determinants of native Nav currents in Purkinje neurons and the mechanisms contributing to INaT, INaP, and INaR gating. Finally, we discuss upstream mechanisms that may contribute to Nav current modulation and important, unanswered questions about the regulation of Nav channel expression, properties, and functioning in cerebellar Purkinje neurons.
Purkinje neurons are the sole output of the cerebellar cortex
In pioneering work conducted during the 1960s, Eccles, Llinás, and Sasaki directly linked the cerebellum to motor function and mapped the physiological underpinnings of the cerebellar circuit [14–21]. Purkinje neurons function as the final destination of sensory information processing in the cerebellar cortex (Fig. 1) and the relay of information from the cerebellar cortex to downstream targets [22]. Sensory information enters the cerebellum via mossy fibers (Fig. 1), originating from over two-dozen brain and spinal cord nuclei [22], that synapse on and excite granule neurons [14]. The granule neurons, in turn, project parallel fiber axon bundles into the molecular layer of the cerebellar cortex (Fig. 1), where they make excitatory synapses on the large dendritic trees of Purkinje neurons [15]. The axons of Purkinje neurons exit the cerebellar cortex (Fig. 1) and provide GABA-mediated inhibition to neurons in the deep cerebellar nuclei [23–25]. In addition to granule cell inputs, Purkinje neurons also receive excitatory synaptic climbing fiber inputs from neurons in the inferior olivary nucleus [16] and local GABAergic inhibitory inputs from cerebellar basket [26] and stellate [17] cells (Fig. 1).
Fig. 1.
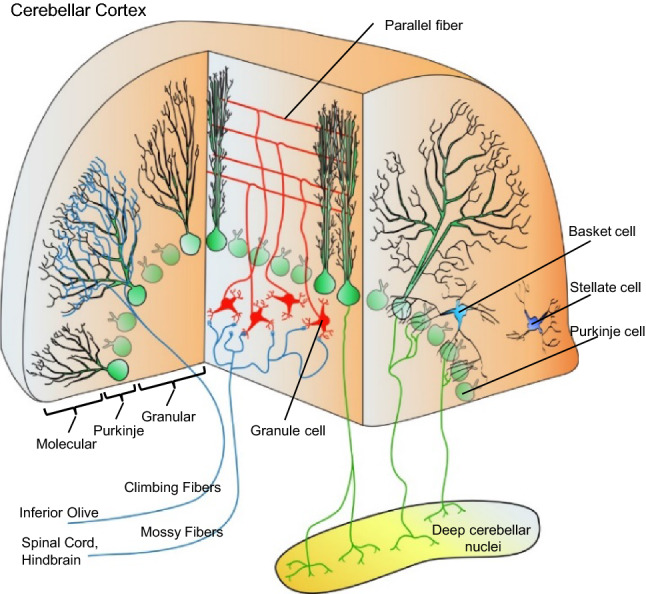
Schematic of the cerebellar circuit shown in sagittal and coronal planes. Climbing fibers, originating in the inferior olive, form excitatory synapses on the dendrites of Purkinje neurons (green). Mossy fibers, which originate in multiple hindbrain and spinal cord nuclei, form excitatory synapses on granule neurons (red) in the cerebellar cortex. The granule cells, in turn, project parallel fiber axons into the molecular layer and form excitatory synapses on the dendrites of Purkinje neurons. Stellate and basket cells (blue), in contrast, form inhibitory synapses on Purkinje neurons, the sole output of the cerebellar cortex, which project inhibitory axons to the deep cerebellar nuclei
The interplay of inhibitory and excitatory synaptic inputs modulates the repetitive firing rates and can cause brief pauses in the firing of individual Purkinje neurons [19, 27], as well as synchronize the repetitive firing of adjacent Purkinje neurons [28, 29]. The synchronized firing of Purkinje neurons is important in relaying spike timing information to neurons in the deep cerebellar nuclei [30], and is evident during cerebellar-related motor behaviors [31, 32]. In the absence of synaptic inputs, however, Purkinje cells continue to fire action potentials spontaneously and repetitively at high frequencies [4, 5]. This sustained, high-frequency repetitive firing, which depends on the unique intrinsic membrane properties of Purkinje neurons, is vital to the functioning of the cerebellar cortex. Indeed, alterations in the rates and patterns of repetitive firing of Purkinje neurons impact balance and motor performance [9–13].
Nav currents underlie high-frequency repetitive firing in cerebellar Purkinje neurons
In 1980, Llinas and Sugimori demonstrated that the high-frequency repetitive firing of action potentials observed in Purkinje neurons in acute cerebellar slices requires subthreshold Na+-dependent, tetrodotoxin (TTX)-sensitive membrane depolarizations [6, 33]. Combining voltage-clamp and action potential-clamp recordings on isolated Purkinje neurons, Raman and Bean detailed the time- and voltage-dependent properties of the critical subthreshold currents underlying spontaneous action potential firing, providing new insights into the roles of these currents in shaping the waveforms of individual action potentials and in controlling the rates and patterns of repetitive firing [5, 7, 34]. Importantly, these experiments revealed that the intrinsic, high-frequency repetitive firing of action potentials in cerebellar Purkinje neurons is not dependent on calcium currents (ICa) or hyperpolarization-activated cation currents (Ih) [7], conductance pathways often associated with pacemaker activity [35–37]. Instead, TTX-sensitive Nav conductances were identified as the primary source of subthreshold membrane depolarizations, both during the action potential and the inter-spike interval, and to be critical in controlling the repetitive firing of Purkinje neurons [5, 7]. Indeed, spontaneous, high-frequency repetitive firing persists in isolated Purkinje neurons with pharmacological inhibition of ICa and Ih, but not in the presence of TTX [7].
Action potential-clamp recordings also revealed that the TTX-sensitive inward Nav currents are activated in Purkinje neurons at subthreshold membrane potentials during the upstroke of individual action potentials, as well as during the inter-spike interval [7]. Combining action potential-clamp and voltage-clamp recordings, Carter and Bean [38] went on to examine INa availability during action potentials and during inter-spike intervals in Purkinje neurons firing at different rates. To quantify INa as a function of time and voltage, they applied steady-state depolarizing voltage steps at different times during action potentials [38]. These experiments revealed that Purkinje neurons continue to fire at > 100 Hz even with ~ 75% of the Nav channels inactivated. In Purkinje neurons firing at 300 Hz, INa availability fell to 15% of its (original) maximal value [38].
Multiple Nav current components drive and fine tune repetitive firing in Purkinje neurons
Steady-state voltage-clamp recordings revealed that the critical Nav conductance in cerebellar Purkinje neurons included a rapidly activating and inactivating, i.e., a transient Nav current component, INaT, and a non-inactivating, i.e., persistent Nav current component, INaP [5, 7, 9, 39], as illustrated in Fig. 2a. In mouse Purkinje neurons, INaP, which is observed at voltages as negative as − 80 mV, was shown to contribute to the regulation of action potential thresholds and the amplification of dendritic excitatory and inhibitory synaptic potentials [40]. Voltage-clamp experiments also revealed that the decay of INaT in cerebellar Purkinje neurons follows a bi-exponential time course [9]. At room temperature, the fast component of INaT decay inactivates with a time constant of ~ 1 ms, and the slow component of INaT decay inactivates with a time constant of ~ 8 ms [9]. Recovery of the fast inactivating component of INaT (from inactivation) is best described by a single exponential; the time constant of recovery at − 40 mV (and at − 80 mV) at room temperature, for example, is 3.8 ms [5, 41]. This rate is considerably faster than the rate of INaT recovery observed in other mammalian central neurons. In hippocampal CA1 pyramidal cells, for example, the time constant of INaT recovery from inactivation at − 70 mV at room temperature was reported to be 9 ms [42].
Fig. 2.

Three Nav current components, INaT, INaP, and INaR, resolved in steady-state voltage-clamp experiments. In the example voltage-clamp paradigm illustrated, INaT and INaP, are revealed during the depolarizing voltage step to + 10 mV (a), and INaR is observed on membrane repolarization (from + 10 mV) to − 40 mV (b). In both the left and right panels, the waveform of the evoked Nav currents is shown below the voltage-clamp paradigm, and schematics of Nav channel gating states are presented; the intrinsic channel inactivation gate is shown in red and the open-channel blocking particle is depicted in blue
Voltage-clamp analyses also led to the discovery of a distinct “resurgent” Nav current component, INaR, in Purkinje neurons, revealed on membrane repolarizations following depolarizing voltage steps [5, 43], as illustrated in Fig. 2b. INaR, therefore, would be expected to provide depolarizing, inward (Na+) current during the falling phase of action potentials and inter-spike intervals in Purkinje neurons [8, 41]. Action potential-clamp recordings from Purkinje neurons indeed revealed an increase in TTX-sensitive inward Nav current during the action potential downstroke [7, 38, 44, 45].
To explore the functional role(s) of the individual Nav current components in the regulation of high-frequency repetitive firing in Purkinje neurons, Khaliq et al. [8] constructed a conductance-based Purkinje neuron model based on the experimentally determined properties of the three Nav current components, INaT, INaP, and INaR, together with the three voltage-gated potassium (Kv) currents, two voltage-gated calcium (Cav) currents, and the calcium-activated potassium current, identified in these cells [7–9, 34]. The Nav currents were simulated using a Markov model [34] that included an ‘open blocked’ state to simulate INaR. This conductance-based model allowed manipulation of INaR and INaT densities and revealed that decreasing INaR reduced repetitive firing rates in model cells [8]. Using this model [34] to simulate INaR, Ransdell et al. [41] demonstrated that (positively) scaling the magnitude of INaR in dynamic-clamp recordings [46, 47] increased instantaneous and evoked firing rates in Purkinje neurons in acute cerebellar slices.
Nav channel gating in cerebellar Purkinje neurons
Each Nav channel pore-forming (α) subunit contains four homologous domains (domains I–IV), each with six transmembrane segments (S1–S6) that are linked by cytosolic peptides (Fig. 3a) [48]. The S5 and S6 transmembrane domains contribute to the formation of the Na+-selective pore (Fig. 3b). The voltage-sensing, S4, segment in each domain has multiple positively charged residues, repeated at three-residue intervals, followed by two hydrophobic amino acids [48, 49]. The positively charged amino acid residues in the S4 domain respond to changes in membrane voltage and move (outward) on membrane depolarization [50]. This charge movement opens the Nav channel pore, allowing Na+ influx. Following opening, Nav channels inactivate, mediated by the cytosolic DIII–DIV linker peptide, specifically by three residues in the DIII–DIV linker that form the hydrophobic (IFM) motif [51], terminating Na+ influx. The IFM motif binds to a site in the channel pore that is only made available after the outward movement (opening) of the DIV S4 segment [52–54]. Importantly, this means that Nav channel inactivation, although tightly linked to voltage-dependent Nav channel activation [55], is a voltage-independent process [52, 56].
Fig. 3.

Schematic of the membrane topology of Nav α and β subunits. a Nav α subunits have four homologous (DI–DIV) domains, each of which has six transmembrane segments; the voltage-sensing S4 segment in each domain is red and the two (S5 and S6) transmembrane domains contributing to the pore are brown. The inactivation gate (grey) in the DIII–DIV linker and the Nav channel transmembrane accessory (β) subunits, Navβ1–Navβ4, are also shown. Navβ2 and Navβ4 (green) form covalent disulfide bonds with Nav α subunits, whereas Navβ1 and Navβ3 (blue) interact non-covalently with Nav α subunits. Binding sites for additional Nav channel interacting proteins, including AnkyrinG (purple) in the DII–DIII cytosolic linker and iFGF14 (blue) in the C-terminus, are also indicated. b The 24 transmembrane spanning segments of a single Nav α subunit assemble to form an Na+ selective pore (brown)
Interestingly, the S4 voltage sensors in each of the four domains of an Nav channel α subunit have different voltage sensitivities [53]. Because Nav channel inactivation requires the movement or activation of the domain IV voltage sensor [54], and Nav channels conduct Na+ after activation of domains I, II, and III [57, 58], INaP can arise at membrane potentials that activate the domain I, II, and III voltage sensors if domain IV remains deactivated. Importantly, these gating mechanisms indicate that differential regulation of the voltage sensitivities of the domain I–IV voltage sensors may regulate the proportion/density and voltage dependence of INaP and INaR.
The “resurgent” component of the Nav current in Purkinje neurons, INaR, is evident on membrane repolarization following brief depolarizations to positive test potentials (Fig. 2b) [5, 43]. In Purkinje neurons, INaR, peaks at − 35 to − 45 mV, activates with a time constant of 5–6 ms and inactivates with a time constant of ~ 20 ms [5, 41, 43]. Importantly, the fact that INaR is not revealed instantaneously on membrane repolarization, but rather takes time to activate, reveals that it (INaR) does not reflect INaP tail currents. Rather, INaR must be mediated by newly opened or recovered Nav channels. In Purkinje neurons, prolonged (40 ms), moderate depolarizations (e.g., to − 30 mV) result in INaR (at − 40 mV) that is much smaller than INaR (also measured at − 40 mV) after a short (5 ms) depolarizing step to more positive (e.g., + 30 mV) potentials [34]. Using (both) voltage-clamp protocols mimicking these paradigms on the same cell, Raman and Bean [34] showed that INaT recovery paralleled INaR, i.e., INaT recovery was greater when measured after a hyperpolarizing voltage step producing a large INaR, than following steps yielding little INaR. This result has several important implications. First, because the magnitude of INaR was dependent on the duration and potential of the pre-step, with shorter, more depolarized potentials resulting in larger INaR, Raman and Bean reasoned that there were two competing Nav channel inactivation mechanisms [34]. Specifically, they suggested that Nav channels undergo conventional inactivation (mediated by the cytosolic DIII–DIV linker) at more hyperpolarized potentials and a novel form of Nav channel inactivation at more depolarized potentials.
This novel mechanism of Nav channel inactivation was referred to as “open-channel block”, i.e., activated Nav channels opened at depolarized membrane potentials were blocked by a particle distinct from the DIII–DIV linker. Grieco et al. [59] specifically suggested that a blocking particle, not part of the Nav α subunit, competes with conventional inactivation, occludes the Nav channel pore during strong depolarizations, and is expelled from the pore on membrane repolarization allowing ‘resurgent’ Na+ influx (before the Nav channels enter a conventional inactivated state). In the open-channel block mechanism, Nav channels, or at least the subset of Nav channels that undergo open-channel block, forego conventional inactivation allowing faster recovery of INaT during hyperpolarization [34].
The proposed mechanism for INaR generation requires the presence/functioning of a particle that competes with conventional inactivation and occludes the Nav channel pore at depolarized potentials. The Nav channel β4 subunit (Navβ4), which binds covalently to Nav α subunits [60, 61] and has a short (20 residues) intracellular C-terminus, which includes several positively charged amino acids, in addition to a phenylalanine residue, was suggested by Lewis and Raman to function as an endogenous “blocking” particle [62]. In a landmark study, Grieco et al. [63] demonstrated that INaR, measured in membrane patches excised from Purkinje neurons, was abolished when the intracellular face of the membrane was exposed to proteases specific for cleavage at positively charged (trypsin) or aromatic (chymotrypsin) residues. In addition, following protease treatment, INaR was rescued by intracellular application of a synthetic peptide, referred to as β4peptide, homologous to the Navβ4 C-terminus [63]. Interestingly, although co-expression of Nav α subunits with Navβ4 in heterologous cells results in Nav currents that lack INaR [64–66], intracellular applications of the β4peptide to HEK-293 cells expressing Nav1.1, 1.4, 1.5, or 1.7 produce robust INaR [65–68]. It has also been reported that shRNA-mediated ‘knockdown’ of Navβ4 reduced or eliminated INaR in cerebellar granule neurons [69], as well as in dorsal root ganglion neurons [70]. More recently, however, it was shown that targeted deletion of Navβ4 (Scn4b−/−) reduces, but does not abolish INaR in striatal medium spiny neurons [71] or in cerebellar Purkinje neurons [41]. In fact, INaR amplitudes/densities in Scn4b−/− Purkinje neurons were ~ 50% of wild-type INaR levels, and in addition, the time- and voltage-dependent properties of the residual INaR were indistinguishable from wild-type INaR [41]. These results clearly indicate that Navβ4-independent mechanisms contribute to the generation of native INaR.
An alternative, or perhaps additional, mechanism for INaR generation is that there is a range of membrane voltages and/or a distinct subset of Nav channels in which the rate of Nav channel deactivation is slower than the rate of channel recovery from inactivation [41, 43]. If Nav channel closing (deactivation), for example, is slower than recovery from inactivation at membrane potentials between − 50 and − 30 mV, Na+ influx through recovered Nav channels will occur. This mechanism was first suggested by the results of experiments demonstrating that β-toxins from Centruroides scorpions [72] slow the kinetics of deactivation of heterologously expressed Nav1.6 channels in HEK-293 cells [73, 74]. The change in deactivation kinetics resulted in the generation of resurgent Nav currents in cells exposed to the toxin [73, 74]. It has also been reported that exposure of rat cerebellar Purkinje neurons to Cn2 toxin, a β-toxin from Centruroides noxius, produced an additional Na+-mediated resurgent current with voltage-dependent activation properties distinct from native INaR [73]. It remains to be determined if there are intrinsic mechanisms in Purkinje neurons, or other neuronal cell types, which similarly modulate Nav current deactivation kinetics to produce resurgent Nav currents.
A fascinating aspect of INaR in Purkinje neurons that has not been thoroughly explored to date is that, at negative potentials, the magnitude and the rate of decay of INaR parallel the magnitude and the rate of decay of the slow component of INaT inactivation. Representative recordings from an isolated Purkinje neuron demonstrate that the magnitude and decay phase of INaR at − 40 mV (from a depolarized membrane potential) are indistinguishable from the slow component of INaT inactivation evoked directly on membrane depolarization to − 40 mV from a hyperpolarized membrane potential (Fig. 4). Noting this property, Raman and Bean proposed that, rather than competing with conventional inactivation, Nav channels responsible for INaR may undergo open-channel block at all voltages that activate INaT [34].
Fig. 4.
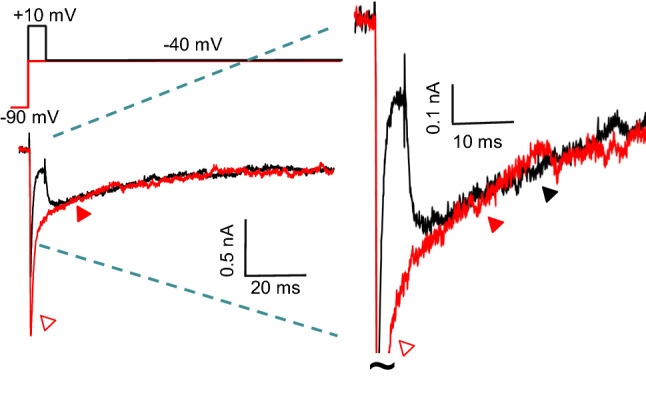
Decay phases of INaT and INaR. In the panel on the left, Nav current waveforms recorded from an isolated neonatal Purkinje neuron during two different voltage-clamp paradigms are shown; the voltage-clamp paradigms are illustrated above the current records. In the first case, membrane depolarization to − 40 mV from a holding potential (HP) of − 90 mV (red) evoked INaT (red); note the fast (red, open arrow) and slow (red, filled arrow) components of inactivation of INaT. Membrane repolarization to − 40 mV following a brief (5 ms) strong (+ 10 mV) depolarization from the same HP revealed INaR (black). Note that the time course of INaR decay (black, filled arrow) and the slow component of INaT inactivation (red, filled arrow) are indistinguishable. The currents are shown on expanded amplitude and time scales in the records shown in the panel on the right
Nav channel alpha and accessory subunits in the functioning of Purkinje neurons
Transcripts encoding the Nav1.6 (SCN8A/Scn8a) and Nav1.1 (SCN1A/Scn1a) pore-forming α subunits are robustly expressed in cerebellar Purkinje neurons [75]. In situ hybridization studies have provided conflicting results [76–78] regarding the expression of Scn2a, which encodes the Nav1.2 α subunit, in cerebellar Purkinje neurons. In addition, although readily detected in cerebellar granule neurons, the Nav1.2 protein was not detected on Purkinje neuron somata, dendrites and axons [12]. In contrast, robust anti-Nav1.6 and anti-Nav1.1 protein labeling has been reported in Purkinje neurons [79, 80], and the Nav1.6 and Nav1.1 proteins are differentially localized. Anti-Nav1.1 labeling, for example, was observed on the cell bodies and dendrites of Purkinje neurons [79] and, in addition, was detected in the segment of the axon proximal to the soma [81]. In contrast, anti-Nav1.6 staining was reported to be localized to the axon initial segment (AIS) [80, 81]. Focal application of TTX (to block Nav currents) or β-pompilidotoxin (to increase INaR) [82] revealed that action potentials in Purkinje neurons are initiated in the distal portion of the AIS, although when AIS Nav channels were blocked, somatic Nav channels in Purkinje neurons were sufficient to generate repetitive firing [83]. Simultaneous extracellular recordings from Purkinje neuron somata and different sites along the axon [84], in combination with the results of antibody labeling [80, 81], suggest that Nav1.6 plays a critical role in the initiation of action potentials in the AIS and the regulation of repetitive firing in Purkinje neurons.
In studies designed to explore directly the contributions of individual Nav α subunits to the regulation of action potential generation and repetitive firing in Purkinje neurons, the functional consequences of the targeted disruption of the genes encoding individual Nav α subunits were examined. The first of these studies used the naturally occurring Scn8amed mutant mouse [85], which results in the functional ‘knockout’ of Scn8a and loss of the Nav1.6 protein [9, 85, 86]. As might have been expected, loss of Nav1.6 resulted in attenuation of repetitive firing in Purkinje neurons. Recordings from cells isolated from 14- to 19-day-old mice revealed spontaneous firing rates reduced to 9 Hz in Scn8amed, compared with 35 Hz in wild type, Purkinje neurons [8]. In addition, peak INaT densities in Scn8Amed Purkinje neurons were ~ 65% of wild-type levels [9]. Interestingly, INaP and INaR densities were reduced to 30 and 10–20%, respectively, of wild-type levels in Scn8Amed Purkinje neurons [9]. Targeted disruption of Scn1a and loss of Nav1.1 also reportedly reduced spontaneous, repetitive firing rates in acutely isolated postnatal day 13–14 Purkinje neurons to 45 Hz, compared with 69 Hz in wild-type cells [12]. Loss of Nav1.1 resulted in INaT, INaP, and INaR densities that were 42, 41, and 31% of those measured in wild-type Purkinje neurons, respectively.
Taken together, these observations suggest that both Nav1.1 and Nav1.6 contribute to the high rates of repetitive firing in Purkinje neurons and animals lacking either of these subunits have severe deficits in balance and motor coordination [10, 12]. In addition, both the Nav1.1 and Nav1.6 α subunits contribute to INaT, INaP, and INaR, although loss of Nav1.6 disproportionately affects INaR and INaP. Using the Cre–lox system to eliminate Scn8a expression selectively in Purkinje or in cerebellar granule neurons, Levin and colleagues demonstrated that loss of Nav1.6 in granule neurons resulted in only minor behavioral defects, whereas mice lacking Nav1.6 in Purkinje neurons exhibited ataxia, tremors, and impaired coordination [11].
It is important to note that the biophysical properties of native INaP and INaR in Purkinje neurons are distinct from those of heterologously expressed Nav1.1- or Nav1.6-encoded Nav currents. Heterologous expression of Nav1.1, for example, revealed persistent Nav current amplitudes corresponding to 2–13% of INaT [87]. It has been estimated that Nav1.1-mediated INaP in cerebellar Purkinje neurons is ~ 1.4% of Nav1.1-mediated INaT [12]. Even more striking is the fact that neither Nav1.6 nor Nav1.1 produces resurgent Nav currents in heterologous expression systems [64, 65].
Accumulated evidence suggests that native neuronal Nav channels, like many other types of ion channels, function in macromolecular protein complexes comprising the pore-forming α subunits and a complement of accessory and regulatory proteins [88–90]. Various Nav channel accessory proteins have been linked to the regulation of Nav channel trafficking, localization, and gating [65, 91–93], increasing the complexities by which Nav channel regulation can influence intrinsic neuronal excitability, action potential waveforms, and repetitive firing. Clear roles for the intracellular fibroblast growth factors (iFGF11–14), also referred to as fibroblast homologous factors (FHFs) [13, 81, 94, 95], and the Nav channel β subunits (Navβ1–4) [41, 43, 63] in the regulation of Purkinje neuron Nav channels, action potential waveforms, and repetitive firing patterns, for example, have been provided. FGF14 is the locus of mutations in spinal cerebellar ataxia type 27 (SCA27) [96], an autosomal dominant disorder characterized by progressive ataxia and cognitive decline [96]. iFGFs are unique from the canonical FGFs in that they lack a signal sequence for secretion and they do not activate any known FGF receptors [97, 98]. Rather, the iFGFs function intracellularly, binding to the C-terminus of Nav α subunits/channels [99–101].
iFGF14 is robustly expressed in Purkinje neurons and localizes with Nav1.6 at the AIS [81, 102]. iFGF14 null (Fgf14−/−) mice display severe deficits in balance and motor coordination [103] and the vast majority of Fgf14−/− Purkinje neurons are quiescent and lack the ability to fire repetitively, even in response to depolarizing current injections [95]. Interestingly, shRNA-mediated ‘knockdown’ of iFGF14 in adult Purkinje neurons also resulted in deficits in balance and motor coordination and markedly attenuated firing in Purkinje neurons [13]. Additional experiments revealed that attenuated firing in Fgf14−/− Purkinje neurons could be attributed to a hyperpolarizing shift in the voltage-dependence of steady-state inactivation of INaT and that firing could be rescued by membrane hyperpolarizations to membrane potentials that allow inactivated Nav channels to recover. It has also been reported that shRNA-mediated ‘knockdown’ of iFGF14 in neonatal Purkinje neurons in culture results in reduced INaR [94]. Loss of iFGF14, however, does not measurably affect INaR in acutely dissociated Purkinje neurons (unpublished). Additional iFGFs may also serve important roles in the regulation of Nav currents and the excitability of Purkinje neurons. Goldfarb and colleagues [102] demonstrated, for example, that animals lacking both iFGF12 and iFGF14 (Fgf12−/−, Fgf14−/−) display significantly worse motor defects than Fgf14−/− animals and that granule neuron excitability in Fgf12−/−, Fgf14−/− mice was markedly attenuated. The role of iFGF12 in Purkinje neurons, however, has not been explored to date.
Transcripts (Scn1b–Scn4b) encoding Navβ1–4 are expressed in developing cerebellar Purkinje neurons [41, 61, 93, 104, 105], although Scn3b (Navβ3) is undetectable in mature Purkinje neurons [105]. As discussed above, Navβ4 contributes to INaR and repetitive firing rates are attenuated in adult Scn4b−/− Purkinje neurons. In marked contrast, repetitive firing rates and properties in neonatal (P14–P15) Scn4b−/− and wild-type Purkinje neurons are indistinguishable [41], suggesting that the functional effects of Navβ4 on the repetitive firing rates of Purkinje neurons are developmentally regulated. Adult Scn4b−/− animals also have deficits in balance and motor coordination [41], although not as severe as those observed in Fgf14−/− animals [13, 103].
In contrast with expectations, voltage-clamp experiments revealed that INaR is readily detected in Scn4b−/− Purkinje neurons and that the mean amplitude of the current is ~ 50% of the magnitude of INaR in wild-type cells [41]. These observations demonstrate that additional, perhaps compensatory, mechanisms contribute to the generation of INaR in Purkinje neurons. Although the critical, alternate mechanism(s) remain to be identified, there are several possibilities. As mentioned above, it has been reported that shRNA-mediated ‘knockdown’ of iFGFf14 in neonatal Purkinje neurons in dissociated cell culture resulted in reduced INaR [94]. In addition, INaR has been reported to be attenuated (compared with wild-type levels) in neonatal Scn1b−/− granule neurons in culture [93]. Importantly, no changes in INaP or INaT densities or properties were observed in Scn1b−/− granule neurons [93], indicating specificity for INaR. In contrast, Navβ2 (Scn2b) more closely resembles Navβ4 than Navβ1 and includes multiple positively charged residues in the C-terminus [62]. However, relative INaR (to INaT) amplitudes are similar in Scn2b−/− and wild-type Purkinje neurons [63]. The effects of the additional loss of Navβ1 or Navβ2 with Navβ4 on Nav currents in Purkinje neurons have not been investigated to date.
Teasing out the additional molecular determinants of INaR and determining if there are physiologically relevant mechanisms distinct from open-channel block of Nav channels that contribute to INaR will be important in efforts to provide a detailed understanding of the diverse roles of Nav currents in controlling the physiology and functioning of cerebellar Purkinje neurons.
Summary, open questions, and future directions
The discovery and detailed characterization of the biophysical properties of INaP [6] and INaR [5] have provided important insights into the contributions of these Nav current components, together with INaT, to regulate spontaneous, high-frequency, repetitive firing in Purkinje neurons [8, 9, 38, 41, 44]. Cerebellar Purkinje neurons are also being utilized to uncover the important contributions of distinct Nav channel accessory proteins on the properties of the Nav currents and on the regulation of action potential waveforms and repetitive firing properties [13, 63, 94, 106]. However, this work is still in its infancy. At present, for example, the only Nav channel accessory proteins shown to contribute to the regulation of the excitability and the functioning of cerebellar Purkinje neurons are iFGF14 and Navβ4. In addition to the contribution of the many other Nav channel accessory proteins that are expressed in Purkinje neurons (including Navβ1, Navβ2, iFGF11, iFGF12, and iFGF13), we should also consider the possibility that multiple proteins are involved and that this could result in non-linear contributions to membrane excitability. Binding of calmodulin on the C-terminus of Nav1.4, which is expressed primarily in skeletal smooth muscle, for example, has been reported to affect channel permeability [107]. Over-expression of calmodulin has also been shown to attenuate Nav1.6-mediated INaP in Purkinje neurons in vitro [106]. It is certainly possible that calmodulin and iFGF14 (or other iFGFs), both of which bind to the C-terminus of Nav α subunits (Fig. 3), act competitively or synergistically to modulate Nav channel gating properties. Interestingly, mice with global deletion of Fgf12 do not exhibit changes in motor coordination or granule neuron excitability, whereas loss of Fgf14 caused deficits in both animal coordination and granule neuron firing [102]. The combined deletion of Fgf14 and Fgf12, however, resulted in a more severe deficit in both these phenotypes than Fgf14 deletion alone [102]. These observations suggest that the expression/functioning of these two Nav channel accessory proteins may be linked in ways that are physiologically relevant. An important area for future research will be to explore the functional relationships among the various Nav channel accessory subunits (Fig. 3) and to define their roles in the dynamic regulation of Purkinje neuronal excitability.
Beyond the combined and/or non-linear impact of Nav channel accessory subunits, there are also compelling recent studies that have identified potential upstream regulators that promote the binding of iFGF14 to Nav α subunits. These include glycogen synthase kinase 3 (GSKIII) [108, 109] and, more recently, casein kinase II (CKII) [110], which is a priming kinase for GSKIII. Inhibition of either of these kinases in hippocampal neurons disrupts iFGF14:Nav α subunit complexes, specifically Nav1.6- and Nav1.2-channel complexes [108, 110], and alters Nav channel localization and membrane excitability [110]. While GSKIII is required during neuronal development and actively regulates neuronal proliferation and migration [111–113], it also functions in the regulation of synaptic transmission [114]. GSKIII is detected in both pre-synaptic and post-synaptic neuronal compartments [115], and interestingly, GSKII has been found to participate in both long-term synaptic depression [116] and potentiation [117]. The finding that GSKIII also functions in the regulation of iFGF14:Nav α subunit complexes may signify a mechanism by which network-level activity regulates or modulates the intrinsic excitability of individual neurons. Although this hypothesis has not been tested at the network level, Purkinje neurons and the cerebellar circuit are an ideal model system to examine these processes.
Acknowledgements
The authors thank Richard Wilson for technical assistance in creating figures. Financial support provided by the National Institutes of Health (R01NS065761 to J.M.N., F32NS090765 to J.L.R.) is also gratefully acknowledged.
References
- 1.Llinas RR. The intrinsic electrophysiological properties of mammalian neurons: insights into central nervous system function. Science. 1988;242(4886):1654–1664. doi: 10.1126/science.3059497. [DOI] [PubMed] [Google Scholar]
- 2.Bean BP. The action potential in mammalian central neurons. Nat Rev Neurosci. 2007;8(6):451–465. doi: 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- 3.Thach WT. Discharge of Purkinje and cerebellar nuclear neurons during rapidly alternating arm movements in the monkey. J Neurophysiol. 1968;31(5):785–797. doi: 10.1152/jn.1968.31.5.785. [DOI] [PubMed] [Google Scholar]
- 4.Hausser M, Clark BA. Tonic synaptic inhibition modulates neuronal output pattern and spatiotemporal synaptic integration. Neuron. 1997;19(3):665–678. doi: 10.1016/S0896-6273(00)80379-7. [DOI] [PubMed] [Google Scholar]
- 5.Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17(12):4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Llinas R, Sugimori M. Electrophysiological properties of in vitro Purkinje cell dendrites in mammalian cerebellar slices. J Physiol. 1980;305:197–213. doi: 10.1113/jphysiol.1980.sp013358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raman IM, Bean BP. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci. 1999;19(5):1663–1674. doi: 10.1523/JNEUROSCI.19-05-01663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khaliq ZM, Gouwens NW, Raman IM. The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: an experimental and modeling study. J Neurosci. 2003;23(12):4899–4912. doi: 10.1523/JNEUROSCI.23-12-04899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raman IM, Sprunger LK, Meisler MH, Bean BP. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron. 1997;19(4):881–891. doi: 10.1016/S0896-6273(00)80969-1. [DOI] [PubMed] [Google Scholar]
- 10.Meisler MH, Plummer NW, Burgess DL, Buchner DA, Sprunger LK. Allelic mutations of the sodium channel SCN8A reveal multiple cellular and physiological functions. Genetica. 2004;122(1):37–45. doi: 10.1007/s10709-004-1441-9. [DOI] [PubMed] [Google Scholar]
- 11.Levin SI, Khaliq ZM, Aman TK, Grieco TM, Kearney JA, Raman IM, Meisler MH. Impaired motor function in mice with cell-specific knockout of sodium channel Scn8a (NaV1.6) in cerebellar Purkinje neurons and granule cells. J Neurophysiol. 2006;96(2):785–793. doi: 10.1152/jn.01193.2005. [DOI] [PubMed] [Google Scholar]
- 12.Kalume F, Yu FH, Westenbroek RE, Scheuer T, Catterall WA. Reduced sodium current in Purkinje neurons from Nav1.1 mutant mice: implications for ataxia in severe myoclonic epilepsy in infancy. J Neurosci. 2007;27(41):11065–11074. doi: 10.1523/JNEUROSCI.2162-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bosch MK, Carrasquillo Y, Ransdell JL, Kanakamedala A, Ornitz DM, Nerbonne JM. Intracellular FGF14 (iFGF14) is required for spontaneous and evoked firing in cerebellar Purkinje neurons and for motor coordination and balance. J Neurosci. 2015;35(17):6752–6769. doi: 10.1523/JNEUROSCI.2663-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eccles JC, Llinas R, Sasaki K. The mossy fibre-granule cell relay of the cerebellum and its inhibitory control by Golgi cells. Exp Brain Res. 1966;1(1):82–101. doi: 10.1007/BF00235211. [DOI] [PubMed] [Google Scholar]
- 15.Eccles JC, Llinas R, Sasaki K. Parallel fibre stimulation and the responses induced thereby in the Purkinje cells of the cerebellum. Exp Brain Res. 1966;1(1):17–39. doi: 10.1007/BF00235207. [DOI] [PubMed] [Google Scholar]
- 16.Eccles J, Llinas R, Sasaki K. Excitation of cerebellar Purkinje cells by the climbing fibres. Nature. 1964;203:245–246. doi: 10.1038/203245a0. [DOI] [PubMed] [Google Scholar]
- 17.Eccles JC, Llinas R, Sasaki K. The inhibitory interneurones within the cerebellar cortex. Exp Brain Res. 1966;1(1):1–16. doi: 10.1016/0006-8993(66)90102-8. [DOI] [PubMed] [Google Scholar]
- 18.Eccles J, Llinas R, Sasaki K. Golgi cell inhibition in the cerebellar cortex. Nature. 1964;204:1265–1266. doi: 10.1038/2041265a0. [DOI] [PubMed] [Google Scholar]
- 19.Eccles JC, Llinas R, Sasaki K. The excitatory synaptic action of climbing fibres on the Purkinje cells of the cerebellum. J Physiol. 1966;182(2):268–296. doi: 10.1113/jphysiol.1966.sp007824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eccles JC, Llinas R, Sasaki K. Intracellularly recorded responses of the cerebellar Purkinje cells. Exp Brain Res. 1966;1(2):161–183. doi: 10.1007/BF00236869. [DOI] [PubMed] [Google Scholar]
- 21.Eccles JC, Llinas R, Sasaki K. The action of antidromic impulses on the cerebellar Purkinje cells. J Physiol. 1966;182(2):316–345. doi: 10.1113/jphysiol.1966.sp007826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Itō M. The cerebellum and neural control. 1. New York: Raven Press; 1984. [Google Scholar]
- 23.Ito M, Yoshida M, Obata K. Monosynaptic inhibition of the intracerebellar nuclei induced rom the cerebellar cortex. Experientia. 1964;20(10):575–576. doi: 10.1007/BF02150304. [DOI] [PubMed] [Google Scholar]
- 24.Obata K, Ito M, Ochi R, Sato N. Pharmacological properties of the postsynaptic inhibition by Purkinje cell axons and the action of gamma-aminobutyric acid on deiters neurones. Exp Brain Res. 1967;4(1):43–57. doi: 10.1007/BF00235216. [DOI] [PubMed] [Google Scholar]
- 25.Obata K, Takeda K, Shinozaki H. Further study on pharmacological properties of the cerebellar-induced inhibition of deiters neurones. Exp Brain Res. 1970;11(4):327–342. doi: 10.1007/BF00237907. [DOI] [PubMed] [Google Scholar]
- 26.Andersen P, Eccles JC, Voorhoeve PE. Postsynaptic inhibition of cerebellar Purkinje cells. J Neurophysiol. 1964;27:1138–1153. doi: 10.1152/jn.1964.27.6.1138. [DOI] [PubMed] [Google Scholar]
- 27.McKay BE, Engbers JD, Mehaffey WH, Gordon GR, Molineux ML, Bains JS, Turner RW. Climbing fiber discharge regulates cerebellar functions by controlling the intrinsic characteristics of Purkinje cell output. J Neurophysiol. 2007;97(4):2590–2604. doi: 10.1152/jn.00627.2006. [DOI] [PubMed] [Google Scholar]
- 28.Bell CC, Grimm RJ. Discharge properties of Purkinje cells recorded on single and double microelectrodes. J Neurophysiol. 1969;32(6):1044–1055. doi: 10.1152/jn.1969.32.6.1044. [DOI] [PubMed] [Google Scholar]
- 29.Heck DH, Thach WT, Keating JG. On-beam synchrony in the cerebellum as the mechanism for the timing and coordination of movement. Proc Natl Acad Sci USA. 2007;104(18):7658–7663. doi: 10.1073/pnas.0609966104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Person AL, Raman IM. Purkinje neuron synchrony elicits time-locked spiking in the cerebellar nuclei. Nature. 2012;481(7382):502–505. doi: 10.1038/nature10732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Welsh JP, Lang EJ, Suglhara I, Llinas R. Dynamic organization of motor control within the olivocerebellar system. Nature. 1995;374(6521):453–457. doi: 10.1038/374453a0. [DOI] [PubMed] [Google Scholar]
- 32.Lang EJ, Apps R, Bengtsson F, Cerminara NL, De Zeeuw CI, Ebner TJ, Heck DH, Jaeger D, Jorntell H, Kawato M, Otis TS, Ozyildirim O, Popa LS, Reeves AM, Schweighofer N, Sugihara I, Xiao J. The roles of the olivocerebellar pathway in motor learning and motor control. A consensus paper. Cerebellum. 2017;16(1):230–252. doi: 10.1007/s12311-016-0787-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Llinas R, Sugimori M. Electrophysiological properties of in vitro Purkinje cell somata in mammalian cerebellar slices. J Physiol. 1980;305:171–195. doi: 10.1113/jphysiol.1980.sp013357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raman IM, Bean BP. Inactivation and recovery of sodium currents in cerebellar Purkinje neurons: evidence for two mechanisms. Biophys J. 2001;80(2):729–737. doi: 10.1016/S0006-3495(01)76052-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jahnsen H, Llinas R. Ionic basis for the electro-responsiveness and oscillatory properties of guinea-pig thalamic neurones in vitro. J Physiol. 1984;349:227–247. doi: 10.1113/jphysiol.1984.sp015154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huguenard JR. Low-threshold calcium currents in central nervous system neurons. Annu Rev Physiol. 1996;58:329–348. doi: 10.1146/annurev.ph.58.030196.001553. [DOI] [PubMed] [Google Scholar]
- 37.Pape HC. Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu Rev Physiol. 1996;58:299–327. doi: 10.1146/annurev.ph.58.030196.001503. [DOI] [PubMed] [Google Scholar]
- 38.Carter BC, Bean BP. Incomplete inactivation and rapid recovery of voltage-dependent sodium channels during high-frequency firing in cerebellar Purkinje neurons. J Neurophysiol. 2011;105(2):860–871. doi: 10.1152/jn.01056.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gahwiler BH, Llano I. Sodium and potassium conductances in somatic membranes of rat Purkinje cells from organotypic cerebellar cultures. J Physiol. 1989;417:105–122. doi: 10.1113/jphysiol.1989.sp017793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carter BC, Giessel AJ, Sabatini BL, Bean BP. Transient sodium current at subthreshold voltages: activation by EPSP waveforms. Neuron. 2012;75(6):1081–1093. doi: 10.1016/j.neuron.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ransdell JL, Dranoff E, Lau B, Lo WL, Donermeyer DL, Allen PM, Nerbonne JM. Loss of Navbeta4-mediated regulation of sodium currents in adult Purkinje neurons disrupts firing and impairs motor coordination and balance. Cell Rep. 2017;19(3):532–544. doi: 10.1016/j.celrep.2017.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuo CC, Bean BP. Na+ channels must deactivate to recover from inactivation. Neuron. 1994;12(4):819–829. doi: 10.1016/0896-6273(94)90335-2. [DOI] [PubMed] [Google Scholar]
- 43.Lewis AH, Raman IM. Resurgent current of voltage-gated Na(+) channels. J Physiol. 2014;592(22):4825–4838. doi: 10.1113/jphysiol.2014.277582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raman IM, Bean BP. Properties of sodium currents and action potential firing in isolated cerebellar Purkinje neurons. Ann N Y Acad Sci. 1999;868:93–96. doi: 10.1111/j.1749-6632.1999.tb11279.x. [DOI] [PubMed] [Google Scholar]
- 45.Carter BC, Bean BP. Sodium entry during action potentials of mammalian neurons: incomplete inactivation and reduced metabolic efficiency in fast-spiking neurons. Neuron. 2009;64(6):898–909. doi: 10.1016/j.neuron.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prinz AA, Abbott LF, Marder E. The dynamic clamp comes of age. Trends Neurosci. 2004;27(4):218–224. doi: 10.1016/j.tins.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 47.Sharp AA, O’Neil MB, Abbott LF, Marder E. The dynamic clamp: artificial conductances in biological neurons. Trends Neurosci. 1993;16(10):389–394. doi: 10.1016/0166-2236(93)90004-6. [DOI] [PubMed] [Google Scholar]
- 48.Noda M, Shimizu S, Tanabe T, Takai T, Kayano T, Ikeda T, Takahashi H, Nakayama H, Kanaoka Y, Minamino N, et al. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature. 1984;312(5990):121–127. doi: 10.1038/312121a0. [DOI] [PubMed] [Google Scholar]
- 49.Catterall WA. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron. 2010;67(6):915–928. doi: 10.1016/j.neuron.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bezanilla F. The voltage sensor in voltage-dependent ion channels. Physiol Rev. 2000;80(2):555–592. doi: 10.1152/physrev.2000.80.2.555. [DOI] [PubMed] [Google Scholar]
- 51.West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proc Natl Acad Sci USA. 1992;89(22):10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Armstrong CM, Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. J Gen Physiol. 1977;70(5):567–590. doi: 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bosmans F, Martin-Eauclaire MF, Swartz KJ. Deconstructing voltage sensor function and pharmacology in sodium channels. Nature. 2008;456(7219):202–208. doi: 10.1038/nature07473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Capes DL, Goldschen-Ohm MP, Arcisio-Miranda M, Bezanilla F, Chanda B. Domain IV voltage-sensor movement is both sufficient and rate limiting for fast inactivation in sodium channels. J Gen Physiol. 2013;142(2):101–112. doi: 10.1085/jgp.201310998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu FH, Catterall WA. Overview of the voltage-gated sodium channel family. Genome Biol. 2003;4(3):207. doi: 10.1186/gb-2003-4-3-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bezanilla F, Armstrong CM. Inactivation of the sodium channel. I. Sodium current experiments. J Gen Physiol. 1977;70(5):549–566. doi: 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chanda B, Bezanilla F. Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J Gen Physiol. 2002;120(5):629–645. doi: 10.1085/jgp.20028679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Horn R, Ding S, Gruber HJ. Immobilizing the moving parts of voltage-gated ion channels. J Gen Physiol. 2000;116(3):461–476. doi: 10.1085/jgp.116.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grieco TM, Afshari FS, Raman IM. A role for phosphorylation in the maintenance of resurgent sodium current in cerebellar Purkinje neurons. J Neurosci. 2002;22(8):3100–3107. doi: 10.1523/JNEUROSCI.22-08-03100.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen C, Calhoun JD, Zhang Y, Lopez-Santiago L, Zhou N, Davis TH, Salzer JL, Isom LL. Identification of the cysteine residue responsible for disulfide linkage of Na+ channel alpha and beta2 subunits. J Biol Chem. 2012;287(46):39061–39069. doi: 10.1074/jbc.M112.397646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu FH, Westenbroek RE, Silos-Santiago I, McCormick KA, Lawson D, Ge P, Ferriera H, Lilly J, DiStefano PS, Catterall WA, Scheuer T, Curtis R. Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2. J Neurosci. 2003;23(20):7577–7585. doi: 10.1523/JNEUROSCI.23-20-07577.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lewis AH, Raman IM. Cross-species conservation of open-channel block by Na channel beta4 peptides reveals structural features required for resurgent Na current. J Neurosci. 2011;31(32):11527–11536. doi: 10.1523/JNEUROSCI.1428-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grieco TM, Malhotra JD, Chen C, Isom LL, Raman IM. Open-channel block by the cytoplasmic tail of sodium channel beta4 as a mechanism for resurgent sodium current. Neuron. 2005;45(2):233–244. doi: 10.1016/j.neuron.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 64.Chen Y, Yu FH, Sharp EM, Beacham D, Scheuer T, Catterall WA. Functional properties and differential neuromodulation of Na(v)1.6 channels. Mol Cell Neurosci. 2008;38(4):607–615. doi: 10.1016/j.mcn.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aman TK, Grieco-Calub TM, Chen C, Rusconi R, Slat EA, Isom LL, Raman IM. Regulation of persistent Na current by interactions between beta subunits of voltage-gated Na channels. J Neurosci. 2009;29(7):2027–2042. doi: 10.1523/JNEUROSCI.4531-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Theile JW, Jarecki BW, Piekarz AD, Cummins TR. Nav1.7 mutations associated with paroxysmal extreme pain disorder, but not erythromelalgia, enhance Navbeta4 peptide-mediated resurgent sodium currents. J Physiol. 2011;589(Pt 3):597–608. doi: 10.1113/jphysiol.2010.200915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang GK, Edrich T, Wang SY. Time-dependent block and resurgent tail currents induced by mouse beta4(154–167) peptide in cardiac Na+ channels. J Gen Physiol. 2006;127(3):277–289. doi: 10.1085/jgp.200509399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lewis AH, Raman IM. Interactions among DIV voltage-sensor movement, fast inactivation, and resurgent Na current induced by the NaVbeta4 open-channel blocking peptide. J Gen Physiol. 2013;142(3):191–206. doi: 10.1085/jgp.201310984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bant JS, Raman IM. Control of transient, resurgent, and persistent current by open-channel block by Na channel beta4 in cultured cerebellar granule neurons. Proc Natl Acad Sci USA. 2010;107(27):12357–12362. doi: 10.1073/pnas.1005633107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barbosa C, Tan ZY, Wang R, Xie W, Strong JA, Patel RR, Vasko MR, Zhang JM, Cummins TR. Navbeta4 regulates fast resurgent sodium currents and excitability in sensory neurons. Mol Pain. 2015;11:60. doi: 10.1186/s12990-015-0063-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miyazaki H, Oyama F, Inoue R, Aosaki T, Abe T, Kiyonari H, Kino Y, Kurosawa M, Shimizu J, Ogiwara I, Yamakawa K, Koshimizu Y, Fujiyama F, Kaneko T, Shimizu H, Nagatomo K, Yamada K, Shimogori T, Hattori N, Miura M, Nukina N. Singular localization of sodium channel beta4 subunit in unmyelinated fibres and its role in the striatum. Nat Commun. 2014;5:5525. doi: 10.1038/ncomms6525. [DOI] [PubMed] [Google Scholar]
- 72.Cahalan MD. Modification of sodium channel gating in frog myelinated nerve fibres by Centruroides sculpturatus scorpion venom. J Physiol. 1975;244(2):511–534. doi: 10.1113/jphysiol.1975.sp010810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schiavon E, Sacco T, Cassulini RR, Gurrola G, Tempia F, Possani LD, Wanke E. Resurgent current and voltage sensor trapping enhanced activation by a beta-scorpion toxin solely in Nav1.6 channel. Significance in mice Purkinje neurons. J Biol Chem. 2006;281(29):20326–20337. doi: 10.1074/jbc.M600565200. [DOI] [PubMed] [Google Scholar]
- 74.Schiavon E, Pedraza-Escalona M, Gurrola GB, Olamendi-Portugal T, Corzo G, Wanke E, Possani LD. Negative-shift activation, current reduction and resurgent currents induced by beta-toxins from Centruroides scorpions in sodium channels. Toxicon. 2012;59(2):283–293. doi: 10.1016/j.toxicon.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 75.Vega-Saenz de Miera EC, Rudy B, Sugimori M, Llinas R. Molecular characterization of the sodium channel subunits expressed in mammalian cerebellar Purkinje cells. Proc Natl Acad Sci USA. 1997;94(13):7059–7064. doi: 10.1073/pnas.94.13.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schaller KL, Caldwell JH. Expression and distribution of voltage-gated sodium channels in the cerebellum. Cerebellum. 2003;2(1):2–9. doi: 10.1080/14734220309424. [DOI] [PubMed] [Google Scholar]
- 77.Jarnot M, Corbett AM. Immunolocalization of NaV1.2 channel subtypes in rat and cat brain and spinal cord with high affinity antibodies. Brain Res. 2006;1107(1):1–12. doi: 10.1016/j.brainres.2006.05.090. [DOI] [PubMed] [Google Scholar]
- 78.Felts PA, Yokoyama S, Dib-Hajj S, Black JA, Waxman SG. Sodium channel alpha-subunit mRNAs I, II, III, NaG, Na6 and hNE (PN1): different expression patterns in developing rat nervous system. Brain Res Mol Brain Res. 1997;45(1):71–82. doi: 10.1016/S0169-328X(96)00241-0. [DOI] [PubMed] [Google Scholar]
- 79.Vacher H, Mohapatra DP, Trimmer JS. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol Rev. 2008;88(4):1407–1447. doi: 10.1152/physrev.00002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lorincz A, Nusser Z. Cell-type-dependent molecular composition of the axon initial segment. J Neurosci. 2008;28(53):14329–14340. doi: 10.1523/JNEUROSCI.4833-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xiao M, Bosch MK, Nerbonne JM, Ornitz DM. FGF14 localization and organization of the axon initial segment. Mol Cell Neurosci. 2013;56:393–403. doi: 10.1016/j.mcn.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Grieco TM, Raman IM. Production of resurgent current in NaV1.6-null Purkinje neurons by slowing sodium channel inactivation with beta-pompilidotoxin. J Neurosci. 2004;24(1):35–42. doi: 10.1523/JNEUROSCI.3807-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Khaliq ZM, Raman IM. Relative contributions of axonal and somatic Na channels to action potential initiation in cerebellar Purkinje neurons. J Neurosci. 2006;26(7):1935–1944. doi: 10.1523/JNEUROSCI.4664-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Palmer LM, Clark BA, Grundemann J, Roth A, Stuart GJ, Hausser M. Initiation of simple and complex spikes in cerebellar Purkinje cells. J Physiol. 2010;588(Pt 10):1709–1717. doi: 10.1113/jphysiol.2010.188300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Burgess DL, Kohrman DC, Galt J, Plummer NW, Jones JM, Spear B, Meisler MH. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant ‘motor endplate disease’. Nat Genet. 1995;10(4):461–465. doi: 10.1038/ng0895-461. [DOI] [PubMed] [Google Scholar]
- 86.Kohrman DC, Plummer NW, Schuster T, Jones JM, Jang W, Burgess DL, Galt J, Spear BT, Meisler MH. Insertional mutation of the motor endplate disease (med) locus on mouse chromosome 15. Genomics. 1995;26(2):171–177. doi: 10.1016/0888-7543(95)80198-U. [DOI] [PubMed] [Google Scholar]
- 87.Mantegazza M, Yu FH, Powell AJ, Clare JJ, Catterall WA, Scheuer T. Molecular determinants for modulation of persistent sodium current by G-protein betagamma subunits. J Neurosci. 2005;25(13):3341–3349. doi: 10.1523/JNEUROSCI.0104-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Meadows LS, Isom LL. Sodium channels as macromolecular complexes: implications for inherited arrhythmia syndromes. Cardiovasc Res. 2005;67(3):448–458. doi: 10.1016/j.cardiores.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 89.Brackenbury WJ. Voltage-gated sodium channels and metastatic disease. Channels (Austin) 2012;6(5):352–361. doi: 10.4161/chan.21910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Abriel H, Rougier JS, Jalife J. Ion channel macromolecular complexes in cardiomyocytes: roles in sudden cardiac death. Circ Res. 2015;116(12):1971–1988. doi: 10.1161/CIRCRESAHA.116.305017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Goldfarb M. Voltage-gated sodium channel-associated proteins and alternative mechanisms of inactivation and block. Cell Mol Life Sci. 2012;69(7):1067–1076. doi: 10.1007/s00018-011-0832-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Patino GA, Isom LL. Electrophysiology and beyond: multiple roles of Na+ channel beta subunits in development and disease. Neurosci Lett. 2010;486(2):53–59. doi: 10.1016/j.neulet.2010.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brackenbury WJ, Calhoun JD, Chen C, Miyazaki H, Nukina N, Oyama F, Ranscht B, Isom LL. Functional reciprocity between Na+ channel Nav1.6 and beta1 subunits in the coordinated regulation of excitability and neurite outgrowth. Proc Natl Acad Sci USA. 2010;107(5):2283–2288. doi: 10.1073/pnas.0909434107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yan H, Pablo JL, Wang C, Pitt GS. FGF14 modulates resurgent sodium current in mouse cerebellar Purkinje neurons. Elife. 2014;3:e04193. doi: 10.7554/eLife.04193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shakkottai VG, Xiao M, Xu L, Wong M, Nerbonne JM, Ornitz DM, Yamada KA. FGF14 regulates the intrinsic excitability of cerebellar Purkinje neurons. Neurobiol Dis. 2009;33(1):81–88. doi: 10.1016/j.nbd.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.van Swieten JC, Brusse E, de Graaf BM, Krieger E, van de Graaf R, de Koning I, Maat-Kievit A, Leegwater P, Dooijes D, Oostra BA, Heutink P. A mutation in the fibroblast growth factor 14 gene is associated with autosomal dominant cerebellar ataxia [corrected] Am J Hum Genet. 2003;72(1):191–199. doi: 10.1086/345488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Goldfarb M. Fibroblast growth factor homologous factors: evolution, structure, and function. Cytokine Growth Factor Rev. 2005;16(2):215–220. doi: 10.1016/j.cytogfr.2005.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Olsen SK, Garbi M, Zampieri N, Eliseenkova AV, Ornitz DM, Goldfarb M, Mohammadi M. Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J Biol Chem. 2003;278(36):34226–34236. doi: 10.1074/jbc.M303183200. [DOI] [PubMed] [Google Scholar]
- 99.Liu C, Dib-Hajj SD, Waxman SG. Fibroblast growth factor homologous factor 1B binds to the C terminus of the tetrodotoxin-resistant sodium channel rNav1.9a (NaN) J Biol Chem. 2001;276(22):18925–18933. doi: 10.1074/jbc.M101606200. [DOI] [PubMed] [Google Scholar]
- 100.Liu CJ, Dib-Hajj SD, Renganathan M, Cummins TR, Waxman SG. Modulation of the cardiac sodium channel Nav1.5 by fibroblast growth factor homologous factor 1B. J Biol Chem. 2003;278(2):1029–1036. doi: 10.1074/jbc.M207074200. [DOI] [PubMed] [Google Scholar]
- 101.Wittmack EK, Rush AM, Craner MJ, Goldfarb M, Waxman SG, Dib-Hajj SD. Fibroblast growth factor homologous factor 2B: association with Nav1.6 and selective colocalization at nodes of Ranvier of dorsal root axons. J Neurosci. 2004;24(30):6765–6775. doi: 10.1523/JNEUROSCI.1628-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Goldfarb M, Schoorlemmer J, Williams A, Diwakar S, Wang C, Huan X, Giza J, Tchetchik D, Kelley K, Vega A, Matthews G, Rossi P, Ornitz DM, D’Angelo E. Fibroblast growth factor homologous factors control neuronal excitability through modulation of voltage-gated sodium channels. Neuron. 2007;55(3):449–463. doi: 10.1016/j.neuron.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang Q, Bardgett ME, Wong M, Wozniak DF, Lou J, McNeil BD, Chen C, Nardi A, Reid DC, Yamada K, Ornitz DM. Ataxia and paroxysmal dyskinesia in mice lacking axonally transported FGF14. Neuron. 2002;35(1):25–38. doi: 10.1016/S0896-6273(02)00744-4. [DOI] [PubMed] [Google Scholar]
- 104.Kazen-Gillespie KA, Ragsdale DS, D’Andrea MR, Mattei LN, Rogers KE, Isom LL. Cloning, localization, and functional expression of sodium channel beta1A subunits. J Biol Chem. 2000;275(2):1079–1088. doi: 10.1074/jbc.275.2.1079. [DOI] [PubMed] [Google Scholar]
- 105.Shah BS, Stevens EB, Pinnock RD, Dixon AK, Lee K. Developmental expression of the novel voltage-gated sodium channel auxiliary subunit beta3, in rat CNS. J Physiol. 2001;534(Pt 3):763–776. doi: 10.1111/j.1469-7793.2001.t01-1-00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yan H, Wang C, Marx SO, Pitt GS. Calmodulin limits pathogenic Na+ channel persistent current. J Gen Physiol. 2017;149(2):277–293. doi: 10.1085/jgp.201611721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ben-Johny M, Yang PS, Niu J, Yang W, Joshi-Mukherjee R, Yue DT. Conservation of Ca2+/calmodulin regulation across Na and Ca2+ channels. Cell. 2014;157(7):1657–1670. doi: 10.1016/j.cell.2014.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shavkunov AS, Wildburger NC, Nenov MN, James TF, Buzhdygan TP, Panova-Elektronova NI, Green TA, Veselenak RL, Bourne N, Laezza F. The fibroblast growth factor 14.voltage-gated sodium channel complex is a new target of glycogen synthase kinase 3 (GSK3) J Biol Chem. 2013;288(27):19370–19385. doi: 10.1074/jbc.M112.445924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shavkunov A, Panova N, Prasai A, Veselenak R, Bourne N, Stoilova-McPhie S, Laezza F. Bioluminescence methodology for the detection of protein–protein interactions within the voltage-gated sodium channel macromolecular complex. Assay Drug Dev Technol. 2012;10(2):148–160. doi: 10.1089/adt.2011.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hsu WC, Scala F, Nenov MN, Wildburger NC, Elferink H, Singh AK, Chesson CB, Buzhdygan T, Sohail M, Shavkunov AS, Panova NI, Nilsson CL, Rudra JS, Lichti CF, Laezza F. CK2 activity is required for the interaction of FGF14 with voltage-gated sodium channels and neuronal excitability. FASEB J. 2016;30(6):2171–2186. doi: 10.1096/fj.201500161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Eom TY, Jope RS. Blocked inhibitory serine-phosphorylation of glycogen synthase kinase-3alpha/beta impairs in vivo neural precursor cell proliferation. Biol Psychiatry. 2009;66(5):494–502. doi: 10.1016/j.biopsych.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Doble BW, Patel S, Wood GA, Kockeritz LK, Woodgett JR. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell. 2007;12(6):957–971. doi: 10.1016/j.devcel.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, Tassa C, Berry EM, Soda T, Singh KK, Biechele T, Petryshen TL, Moon RT, Haggarty SJ, Tsai LH. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell. 2009;136(6):1017–1031. doi: 10.1016/j.cell.2008.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cuesto G, Jordan-Alvarez S, Enriquez-Barreto L, Ferrus A, Morales M, Acebes A. GSK3beta inhibition promotes synaptogenesis in Drosophila and mammalian neurons. PLoS One. 2015;10(3):e0118475. doi: 10.1371/journal.pone.0118475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Smillie KJ, Cousin MA. The role of GSK3 in presynaptic function. Int J Alzheimers Dis. 2011;2011:263673. doi: 10.4061/2011/263673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T, Matthews P, Isaac JT, Bortolotto ZA, Wang YT, Collingridge GL. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron. 2007;53(5):703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- 117.Hooper C, Markevich V, Plattner F, Killick R, Schofield E, Engel T, Hernandez F, Anderton B, Rosenblum K, Bliss T, Cooke SF, Avila J, Lucas JJ, Giese KP, Stephenson J, Lovestone S. Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur J Neurosci. 2007;25(1):81–86. doi: 10.1111/j.1460-9568.2006.05245.x. [DOI] [PubMed] [Google Scholar]