

Abstract
Loss of cell cycle control is a hallmark of cancer, which can be targeted with agents, including Cyclin Dependent Kinase-4/6 (CDK4/6) kinase inhibitors that impinge upon the G1-S cell cycle checkpoint via maintaining activity of the retinoblastoma tumor suppressor (RB). This class of drugs is under clinical investigation for various solid tumor types, and has recently been FDA-approved for treatment of breast cancer. However, development of therapeutic resistance is not uncommon. In this study, palbociclib (a CDK4/6 inhibitor) resistance was established in models of early stage, RB-positive cancer. This study demonstrates that acquired palbociclib resistance renders cancer cells broadly resistant to CDK4/6 inhibitors. Acquired resistance was associated with aggressive in vitro and in vivo phenotypes, including proliferation, migration, and invasion. Integration of RNA sequencing analysis and phospho-proteomics profiling revealed rewiring of the kinome, with a strong enrichment for enhanced MAPK signaling across all resistance models, which resulted in aggressive in vitro and in vivo phenotypes and pro-metastatic signaling. However, CDK4/6 inhibitor resistant models were sensitized to MEK inhibitors, revealing reliance on active MAPK signaling to promote tumor cell growth and invasion. In sum, these studies identify MAPK reliance in acquired CDK4/6 inhibitor resistance that promotes aggressive disease, while nominating MEK inhibition as putative novel therapeutic strategy to treat or prevent CDK4/6 inhibitor resistance in cancer.
Keywords: CDK4, Cyclin Dependent Kinase, cell cycle, MAPK, ERK, kinase inhibitor, drug resistance, prostate cancer, RB, palbociclib, ribociclib
INTRODUCTION
Dysregulation of the cell cycle is a hallmark of cancer. While initial attempts to target the cell cycle with non-specific Cyclin Dependent Kinase (CDK) inhibitors were clinically unsuccessful, a new generation of selective CDK4/6 inhibitors has emerged that has shown clinical promise across multiple cancer types, including breast, melanoma and colorectal cancer(1,2). Currently, three CDK4/6 inhibitors have entered clinical trials: palbociclib (Ibrance), ribociclib (Kisqali) and abemaciclib(1,3). Notably, these three inhibitors have recently received accelerated FDA-approval for treatment of hormone-receptor positive (HR+), Her2-negative breast cancer in combination with endocrine therapy based on the PALOMA-2 (palbociclib), the MONALEESA-2 (ribociclib), and the MONARCH-2 (abemaciclib) Phase III clinical trials(4).
Despite evidence of clinical response, development of resistance is common, and the underlying mechanisms that lead to resistance, are poorly understood. Consistent with the known functions of CDK4/6 in promoting cell cycle progression through phosphorylation and inactivation of the retinoblastoma tumor suppressor protein (RB), CDK4/6 inhibitors require an active RB pathway to elicit anti-tumor effects. As such, resistance to CDK4/6 inhibitors can occur through disruption of the RB pathway, as mediated by loss of the retinoblastoma tumor suppressor (RB), Cyclin D1 and CDK6 amplification, and Cyclin E-CDK2 activation(3,5,6). Other mechanisms of resistance have also been reported that are independent of RB pathway alteration, including alterations or activation of the PI3K/AKT/mTOR pathway(7,8). Given the promise of CDK4/6 inhibitors in the clinical setting, there is an increasing need to discern mechanisms of bypass, and identify mechanisms to anticipate and prevent therapeutic resistance.
Therapeutic opportunities for implementation of CDK4/6 inhibitors in the clinical setting are widest in tumors for which RB pathway disruption is infrequent or occurs late in tumor progression. As such, studies herein were conducted using prostate adenocarcinoma (PCa) as a tumor paradigm, in which a functional RB pathway is largely intact in early stage disease. Prior to androgen deprivation therapy, which is the first line of therapeutic intervention for disseminated disease, almost all tumors retain RB function, with 5% RB loss observed in primary tumors(9). By contrast, RB loss is enriched in metastatic disease, occurring in 37% of metastatic cases in a retrospective cohort, and in 21% of metastatic castration-resistant prostate cancer (CRPC)(10), and is found to be causative for the transition to CRPC(11), the lethal stage of disease. Thus, CDK4/6 inhibitors are under clinical testing in both hormone-naïve metastatic PCa (NCT02059213) and metastatic CRPC (NCT02555189) as adjuvant therapy for first- and second-line hormone therapy, respectively.
As shown herein, acquired palbociclib resistance not only rendered cancer cells broadly resistant to CDK4/6 inhibitors, but also promoted aggressive phenotypes, including accelerated growth in vitro and in vivo, as well as enhanced invasion and clonogenic capacity. Unbiased global gene expression and phosphoproteomic profiling were utilized to interrogate the molecular alterations of acquired resistance to CDK4/6 inhibition in multiple models of palbociclib resistance. These integrated approaches revealed a reduction of RB function resulting in CDK4/6 inhibitor resistance. Additionally, acquired CDK4/6 resistance was associated with activation of the MAPK signaling pathway, which conferred sensitization to MEK inhibitors. In sum, these studies demonstrate partially retained, hyperphosphorylated Rb in acquired CDK4/6 inhibitor resistance, and nominate MEK inhibitors as a new treatment strategy for advanced cancers upon developing CDK4/6 inhibitor resistance.
MATERIALS AND METHODS
Tissue culture and establishing resistance lines
LNCaP and LAPC4-derived cells were cultured in improved minimum essential media (IMEM; Corning, Manassis, VA) or Iscove’s modified Dulbecco media (IMDM; Corning, Manassis, VA), respectively, supplemented with 5% heat-inactivated fetal bovine serum (FBS; HyClone, USA), 1% L-glutamine and 1% penicillin/streptomycin(11). Unless otherwise described, cells were plated overnight, and treated with 0.5μM PD0233991 (PD, palbociclib, SelleckChem), LEE011 (LEE, ribociclib, Novartis) or U0126 (Promega). Palbociclib resistant cell models were generated from LNCaP and LAPC4 cells by sustained treatment with 0.5μM PD (Schematic in Figure 1A), and maintained under selection when resistance, measured via BrdU incorporation, was achieved after ~3 months. Cells were authenticated by ATCC and checked for mycoplasma upon thawing and at termination of maintenance after <20 passages (ATCC 30-1012K).
Figure 1. Acquired resistance to palbociclib results in broad CDK4/6 inhibitor resistance.
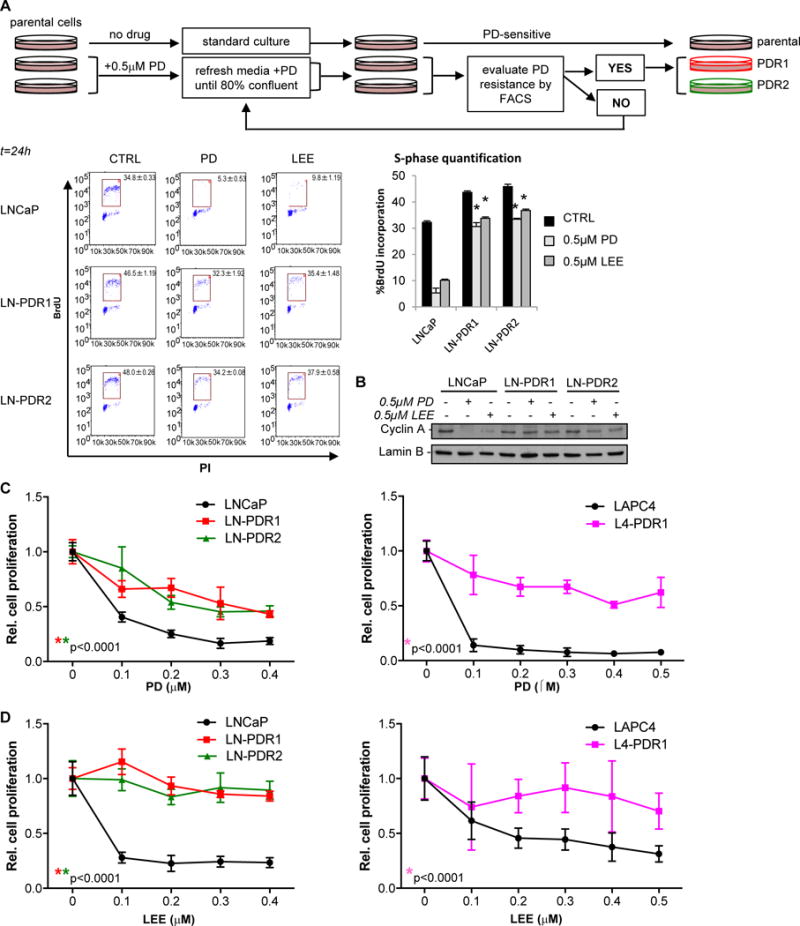
A. Palbociclib (PD) resistant (PDR) prostate cancer cells were generated via continuous selection with 0.5μM PD for 2-3 months and evaluated regularly via flow cytometry (top). PD resistance was determined by treating biological triplicate parental or PDR cells with 0.5μM PD, LEE (ribociclib), or CTRL (no drug) for 24h and measuring BrdU incorporation with a flow cytometer after a 2 hour pulse labeling, fixation in EtOH and staining with a secondary FITC-mouse-anti-BrdU antibody. FACS analysis was performed by gating for the BrdU+ S-phase population (representative flow traces for biological triplicates are shown on the left), quantified in a bar graph on the right as an indication of cell proliferation. FACS analysis showed that PD and ribociclib (LEE) fail to induce cell cycle arrest in the G1-phase in LNCaP PDR lines. B. Cells were treated for 24h with CTRL, 0.5μM PD, or LEE and immunoblotted for Cyclin A, demonstrating a reduction in Cyclin A protein upon exposure to PD or LEE only in the parental cells, indicating biochemical resistance in LNCaP PDR cells. C. Cell counting via Trypan blue exclusion of quadruplet samples with dose escalation treatment with PD of t=6 days (LNCaP) or t=13 days (LAPC4) shows a significantly reduced response to PD in the PDR models, compared to the parental cells. D. Acquired resistance to PD results in broad CDK4/6 inhibitor resistance as shown by a LEE dose escalation. *Significance for dose response curves was determined by a Two-way ANOVA analysis performing a multivariate comparison of mean per dose for PDR vs parental data.
Trypan Blue Exclusion
Cells were treated with a dose range of indicated doses of PD, LEE or U0126 for up to 6 days, with drugs refreshed every other day. Cells were trypsinized, counted twice on a hemacytometer using the Trypan blue exclusion method, normalized to a drug-free control. Experiments were performed at least twice in quadruplicate.
Flow Cytometry
Cells were treated with 0.5μM PD or LEE for 24h, and after a 2-hour pulse labeling of bromodeoxyuridine (BrdU; Amersham, RPN201), adherent cells were harvested and fixed with 100% ethanol. Proliferation was measured by bivariate flow cytometry using BrdU and propidium-iodide (PI) staining. A Millipore Guava EasyCyte flow cytometer captured 10,000 BrdU/PI events and the Millipore InCyte software was used to gate for the percentage of BrdU incorporation. Experiments were performed at least twice in biological triplicate.
RT-PCR
Cells were treated with 0.5μM PD or CTRL for 24h, and processed to assess RB1 expression, as described previously(12), performed at least twice in biological triplicate.
Immunoblotting
Protein harvesting and immunoblotting were performed as previously described(12), quantified in ImageJ. Antibodies used are mouse-α-RB (BD-Biosciences, 554136), rabbit-α-GAPDH (Santa Cruz, SC-25778), rabbit-α-CyclinA (Santa Cruz, SC-596), rabbit-α-ERK1 (Santa Cruz, SC-94), rabbit-α-phospho-p44/p42(T202/Y204) (Cell Signaling, 4370S), goat-α-LaminB (Santa Cruz, SC-6217). Phospho-blots were blocked and immunoblotted in 2.5% BSA, all other blots with 5% milk in PBS-Tween.
Migration and Invasion Assays
Corning FluoroBlok 24-Multiwell and Corning BioCoat Tumor Invasion System plates were utilized for migration and invasion assays, according to protocols provided by the company, performed at least twice in quadruplicate. Bottom wells were filled with IMEM/20%FBS. 50,000 cells (passed 2x, 40um cell strainers) were seeded in the top compartment of each well in serum-free IMEM and incubated for 48h (migration) or 72h (invasion). Cells were stained with Corning Calcein AM Fluorescent Dye and measured on a BioTek SynergyHT plate reader.
Clonogenic Assays and FIJI
The bottom layer of 1% agar/PBS was mixed 1:1 with culture media supplemented with 20% FBS and poured into 6-well plates, after which a second layer with 0.6% agar/FBS mixed with a cell-media/20%FBS suspension was poured on top. Where mentioned, drugs were mixed into both agar layers. 200ul media +/− drugs was added on top and refreshed twice a week. 5000 cells were seeded per well in triplicate. Plates were incubated for 3 weeks, after which each well was photographed. Images were analyzed with FIJI (ImageJ) as follows: select each well transform to gray scale, adjust the threshold to remove background, and detect colonies, Analyze Particles to count total colonies and pixel sizes of each colony. Experiments were performed at least twice in triplicate.
In vivo studies
Xenograft studies were performed in accordance with NIH Guidelines and animal protocols were approved by IACUC at Thomas Jefferson University. Cells (3×106 per injection) suspended in PBS were combined 1:1 with Matrigel (BD Biosciences, 354234) and injected subcutaneously into the flanks of 5-6-week-old, intact-male athymic nude mice (Charles River Laboratories). Tumor development was monitored over time by palpation. Where indicated, mice received AIN-76A diet laced with 6.7mg/kg trametinib or control (kindly provided by the laboratory of Dr Andrew Aplin, Thomas Jefferson University).
RNA sequencing (RNAseq) and GSEA analyses
RNA was extracted with the RNeasy kit (Qiagen) from PDR and parental LNCaP or LAPC4 cells pre-treated 24h with 0.5μM PD or vehicle (CTRL). 100-200 ng of total RNA was used to prepare RNAseq libraries using the TruSeq RNA Sample Prep Kit V2 (Illumina, San Diego, CA), following the protocol described by the manufacturer. High throughput sequencing (HTS) was performed using an Illumina HiSeq2500 with each sample sequenced to a minimum depth of ~50 million reads. A paired end 2×125 cycle sequencing strategy was employed. Data were subjected to Illumina quality control (QC) procedures (>80% of the data yielded a Phred score of 30). Secondary analysis was carried out on an OnRamp Bioinformatics Genomics Research Platform (OnRamp Bioinformatics, San Diego, CA)(13). OnRamp’s advanced Genomics Analysis Engine utilized an automated RNAseq workflow to process the data(13,14), including (1) data validation and quality control, (2) read alignment to the human genome (hg19) using TopHat2(15), which revealed >90% mapping of the paired end reads, (3) generation of gene-level count data with HTSeq, and (4) differential expression analysis with DEseq2(15), which enabled the inference of differential signals with robust statistical power. (Genomics Research Platform with RNAseq workflow v1.0.1, including FastQValidator v0.1.1a, Fastqc v0.11.3, Bowtie2 v2.1.0, TopHat2 v2.0.9, HTSeq v0.6.0, DEseq v1.8.0).
The resulting BAM files were sorted and inputted into the Python package HTSeq to generate count data for gene-level differential expression analyses. To infer differential signal within the data sets with robust statistical power, DEseq2 was utilized(15). Transcript count data from DESeq2 analysis of the samples were sorted according to their adjusted p-value or q-value, the smallest false discovery rate (FDR) at which a transcript is called significant (q<0.1). FDR is the expected fraction of false positive tests among significant tests and was calculated using the Benjamini-Hochberg multiple testing adjustment procedure. LNCaP (LN) and LAPC4 (L4) sequencing data are deposited NCBI’s Gene Expression Omnibus(16), accessible through GEO Series accession number GSE99675.
Analysis of Phosphotyrosine, Phosphoserine and Phosphothreonine Peptides by Quantitative Mass Spectrometry
PDR and parental LNCaP or LAPC4 cells were treated 24h with 0.5μM PD or CTRL, scraped, pelleted, and snap frozen. Protein digestion and phosphopeptide enrichment were performed as previously described(17–19) with minor modifications. Briefly, cells were lysed in 6M guanidinium hydrochloride buffer (6M Guanidinium chloride, 100mM Tris pH8.5, 10mM Tris (2-carboxyethyl) phosphine, 40mM 2-chloroacetamide, 2mM Vanadate, 2.5mM NaPyrophosphate, 1mM Beta-glycerophosphate, 10mg/ml N-octyl-glycoside). Lysates were sonicated, cleared, and protein was measured. 5 mg of protein was digested with trypsin and the resulting phosphopeptides were subjected to phosphotyrosine antibody-based enrichment via immunoprecipitation. The immunoprecipitate was washed and phospho-Tyrosine (pY) peptides were eluted. The supernatant from the pY immunoprecipitations was kept for phospho-Serine/Threonine (pST) peptide enrichment. 2.5 mg of pST peptides were de-salted using C18 columns and then separated using strong cation exchange chromatography. In separate reactions the pY and pST peptides were then further enriched using titanium dioxide columns to remove existing non-phosphorylated peptides. The pY and pST peptides were then de-salted using C18 tips prior to submission on the mass spectrometer. Samples were analyzed by LC-MS/MS using a dual pump nanoRSLC system (Dionex, Sunnyvale CA) interfaced with a Q Exactive HF (ThermoFisher, San Jose, CA). Samples were run in technical duplicates, and data were searched using MaxQuant Andromeda version 1.5.3.30(20) against the Uniprot human reference proteome database with canonical and isoform sequences (downloaded September 2016 from http://uniprot.org). MaxQuant Andromeda parameters were set as previously described(21). Data are deposited in the ProteomeXchange Consortion via the PRIDE partner repository, accessible through dataset identifier PXD006561(22).
MS data analysis was performed as previously described(23). For clustering, pY data were filtered using an FDR-corrected ANOVA p-value of 0.2, pS/pT data were filtered using an FDR-corrected ANOVA p-value of 0.05. Hierarchical clustering was performed using the Cluster version 3.0 with the Pearson correlation and pairwise complete linkage analysis(24). Java TreeView version 1.1.6r4 was used to visualize clustering results(25).
Kinase Substrate Enrichment Analysis (KSEA)
KSEA was performed as previously described(18). Briefly, phosphopeptides were rank-ordered by average fold change between PDR and parental cells. The enrichment score was calculated using the Kolmogorov-Smirnov statistic. Statistical significance was calculated via permutation analysis. The normalized enrichment score (NES) was calculated by taking the enrichment score and dividing by the mean of the absolute values of all enrichment scores from the permutation analysis. The Benjamini-Hochberg procedure was utilized to calculate false discovery rate for each kinase. For pY analyses, cutoffs of FDR<0.05, hits>4, and NES>1.3 were used. For pST analyses, cutoffs of FDR<0.02, hits>5, and NES>2 were used.
RESULTS
While CDK4/6 inhibitors have shown promise in clinical trials for cancer treatment, acquired resistance is common. Thus, the present study interrogated the underpinnings of therapeutic resistance, to identify markers for therapeutic outcome and develop new strategies when resistance develops.
Acquired resistance to palbociclib results in broad CDK4/6 inhibitor resistance
Palbociclib-resistant (PDR) PCa cell models generated from hormone-therapy sensitive PCa cells (LNCaP and LAPC4) demonstrated retained BrdU incorporation after PD treatment (LNCaP-parental: 5.3%; LN-PDR1: 32.3%; LN-PDR2: 34.2%), indicative of a G1/S cell cycle checkpoint bypass (Figure 1A, Supplementary Figure 1A). Retained Cyclin A (an RB/E2F target gene) protein levels after 24 hours treatment with 0.5 μM PD confirmed acquired resistance (Figure 1B, Supplementary Figure 1B). Ribociclib (LEE011, LEE) is similar to PD in chemical structure and likewise targets the ATP binding pocket, and thus it was not surprising that the PDR models were resistant to LEE, as observed via retained BrdU incorporation (LNCaP-parental: 9.8%; LN-PDR1: 35.4%, LN-PDR2: 37.9%) and Cyclin A (Figure 1A/B, Supplementary Figure 1A/B). Dose response curves confirmed that PDR models continued to proliferate, while the parental cells were arrested at sub-micromolar doses of PD or LEE (Figure 1C/D). These findings indicate that acquired resistance to palbociclib confers broad resistance to this class of agents, suggestive of common mechanisms of bypass.
Acquired CDK4/6 inhibitor resistance is associated with rewired transcriptional programs
To identify transcriptional alterations underlying CDK4/6 inhibitor resistance, PDR and parental cells were treated for 24 hours with 0.5μM PD or control (CTRL), and RNA sequencing was performed. The MA plots in Supplementary Figures 2A (right) and 3A represent the log ratio (M) of the PD versus CTRL values over the average log intensity (A) of each transcript, visualizing a global reduction of differentially expressed genes in PDR models compared to the parental cells after PD treatment. Strikingly, the large cluster of downregulated genes in the parental model (indicated by the blue arrow) was absent in the resistant cells, confirming that PD is unable to significantly inhibit this gene cluster. When comparing PDR1 and PDR2 profiles to parental cells, in both PD-treated and CTRL conditions, vast transcriptomic changes were observed in both the total number of differentially expressed transcripts and log ratio amplitudes (Figure 2A, left, Supplementary Figure 3B). While PD treatment showed limited impact on the transcriptome of the PDR lines compared to parental cells, these models demonstrated extensive alteration in the transcriptome to adapt to extended PD exposure. The majority of transcripts (>1.5-fold, q-value<0.1) were common between the two independently generated LNCaP PDR models in both CTRL and PD conditions (Figure 2A, right). Although the PDR models showed a few distinctions, deregulated signaling pathways common between PDR models are most likely to contribute to resistance.
Figure 2. Acquired CDK4/6 inhibitor resistance is associated with rewired transcriptional programs including RB function.
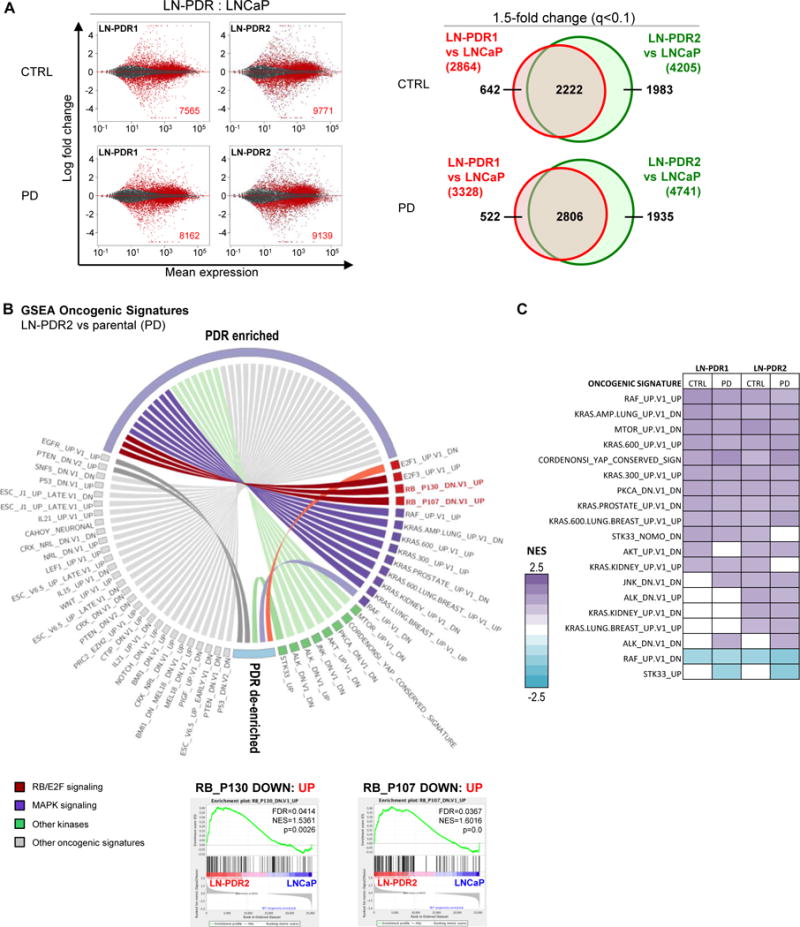
A. RNAseq was performed on PDR1/2 and parental LNCaP cells treated 24h with 0.5μM PD or vehicle (CTRL). The MA plots (right) represent the log ratio (M) of PDR versus parental values over the average log intensity (A) of each transcript, which visualizes vast differences between the PDR vs parental data (red dots and inset numbers indicate significant hits, q-value<0.1). Venn diagrams show overlap between PDR1 and PDR2 of genes >1.5x differentially expressed compared to the parental cells (q-value<0.1; right). B. The complete RNAseq profiles for each PDR vs parental model comparison were subjected to unbiased Gene Set Enrichment Analysis (GSEA, MSigDB) to determine enrichment of predefined Oncogenic Signatures in both PDR1 and PDR2 compared to parental cells under at least one condition (CTRL/PD) with a False Discovery Rate or FDR<0.25 (Complete list in Supplementary Figure 2B). The Oncogenic Signatures enriched in the PDR models included two signatures defined by RB knockdown, suggesting the PDR models have upregulated genes that are induced by RB knockdown. Representative GSEA plots of the RB knockdown signatures are shown for PDR2 vs WT after PD treatment. C. GSEA Oncogenic Signature altered kinase signatures in the LNCaP PDR models for all conditions.
To identify common pathways enriched in the PDR models, complete transcriptional profiles were utilized in Gene Set Enrichment Analyses (GSEA: www.broadinstitute.org/GSEA) interrogating the predefined Oncogenic Signatures and Hallmarks from the Molecular Signature Database (MSigDB) (Figure 2B; Supplementary Figures 2B/3D). Gene sets enriched in both PDR models under at least one condition were selected to highlight pathways that are most likely to contribute to acquired CDK4/6 inhibitor resistance. Enriched Hallmark gene sets included G2M Checkpoint, supporting the contention that resistant cells bypass the G1/S checkpoint and progress through cell cycle. Enrichment of E2F Targets in PDR lines suggests increased E2F activity, potentially mediated through a bypass of RB. Genomic RB loss has been observed in ~30% of advanced PCa across different patient cohorts, and as RB is the main target of CDK4/6-mediated phosphorylation, loss of this gene has been attributed to resistance to CDK4/6 inhibitors(5,26). Concordantly, RB knockdown Oncogenic Signatures were also enriched, further highlighting that RB function may be reduced (Figure 2B). RT-PCR and immunoblotting demonstrated that total RB mRNA and protein were retained in all PDR lines, albeit reduced to 29-56% (Supplementary Figures 2C/3C top). Retained RB protein in the PDR models remained hyperphosphorylated (Supplementary Figures 2C/3C bottom, upper band total RB (tRB)) in presence of a CDK4/6 inhibitor, which attenuates RB function, confirming aberrant inactivation of RB in presence of CDK4/6 inhibitors that likely causes bypass of the G1/S checkpoint and activation of E2F transcription factors. As the kinome forms an intricate network of interactions(27), it was postulated that bypass of CDK4/6 inhibition likely causes kinome rewiring. Notably, RNA sequencing uncovered enrichment of numerous kinase related signatures (Figure 2B/C, Supplementary Figure 4A). These findings served as the impetus for further exploration to identify specific kinase pathway(s) that could present novel targets for therapeutic intervention.
MAPK activation is a hallmark for CDK4/6 inhibitor resistance
Phosphoproteomic mass spectrometry profiling provided an unbiased approach to investigate the PDR-associated kinome. Cells were treated for 24h with either 0.5μM PD or control, lysed and digested (Figure 3A). Phospho-Tyrosine (pY) peptides were immunoprecipitated with an anti-pY antibody, while phospho-Serine/Threonine peptides (pST) remained in the supernatant, allowing analysis of both peptide populations via Liquid Chromatography with tandem Mass Spectrometry (LC-MS/MS)(23). Hierarchical clustering revealed altered phosphorylation levels of peptides across models and treatments, while duplicate samples cluster together (Figure 3A, Supplementary Figure 4B). Phospho-peptide profiles were subjected to kinase substrate enrichment analyses (KSEA). The pY peptides unveiled multiple motifs targeted by Src and its SH2-domain (Figure 3B, Supplementary Figure 4C), which is linked to cancer progression(28). The pST peptides were strongly enriched for motifs targeted by ERK1 and ERK2 (aka MAPK3 and MAPK1; Figure 3C, Supplementary Figure 4D). To prioritize changes in the phosphoproteome most likely to link to CDK4/6 inhibitor resistance, results were compared with the gene expression data. Intriguingly, KRAS and RAF kinases, both of which were highly represented in the Oncogenic Signatures from the RNAseq data, regulate ERK1/2 via Mitogen-Activated Protein Kinase Kinase, or MEK(29). These data suggest that kinase-signaling cascades play an important role in acquired CDK4/6 inhibitor resistance in these models. Moreover, the MAPK pathway is targetable with clinically tested pharmacologic agents, such as MEK inhibitors that are already FDA-approved for some cancers (e.g. melanoma)(29), nominating this pathway for further study. Together, these data indicate that acquired resistance to CDK4/6 inhibitors was associated with induced MAPK pathway activity.
Figure 3. Integrative transcriptome and kinome profiling identifies differential MAPK stimulus as a hallmark of CDK4/6 inhibitor resistance.
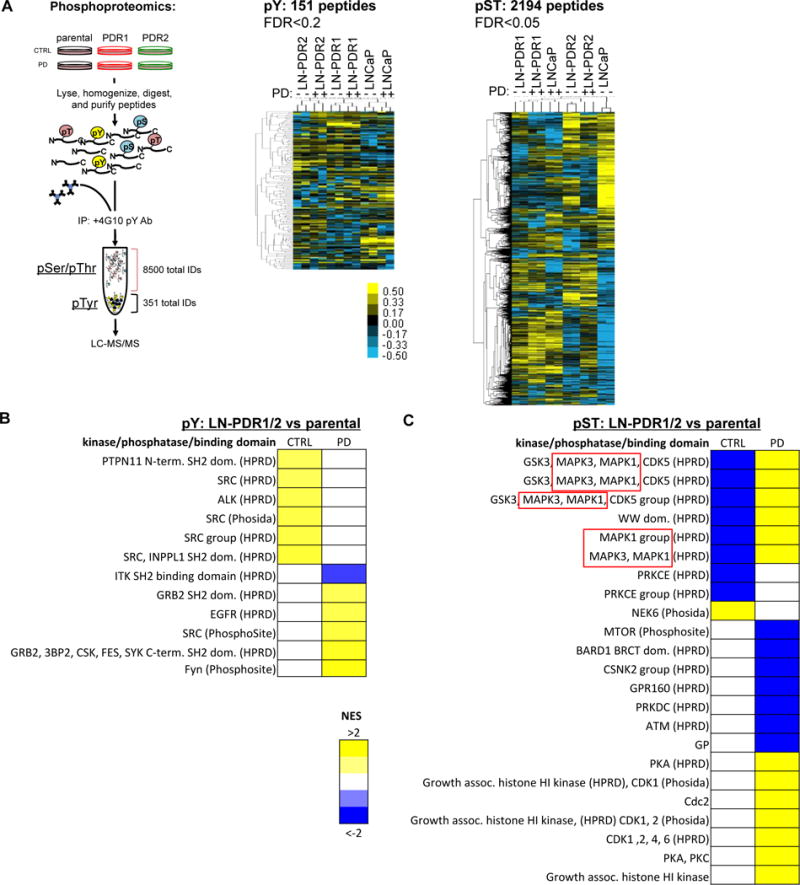
A. LNCaP PDR and parental cells were treated for 24h with 0.5uM PD or control, snap frozen and lysed. After peptide digestion, phospho-Tyrosine peptides (pY) were immunoprecipitated, while phospho-Serine/Threonine peptides (pST) remained in the upper fraction (see schematic, more details in Materials and Methods). Duplicates of both peptide fractions were utilized in an unbiased phosphoproteomics approach to identify altered phosphorylation of Tyrosine and Serine/Threonine peptides across PDR models compared to the parental cells, displayed by hierarchical clustering on the right. B. Kinase/Substrate Enrichment Analysis (KSEA) defined enriched peptide motifs for phosphorylated Tyrosine (pY) hits and mapped them to kinases that are most likely to target these motifs. This revealed enrichment for Src and Src Homology (SH2) domain target motifs in PDR1/2 compared to parental cells (p=0.2). C. KSEA analysis for pST hits showed enrichment for altered phosphorylation of MAPK3 and MAPK1 (ERK1/2) target motifs (p=0.05), indicative of differential MAPK signaling.
Activated ERK is associated with aggressive phenotypes
As described above, Oncogenic Signatures representative of altered KRAS and RAF signaling were highly enriched as a function of acquired CDK4/6 inhibitor resistance. KRAS acts upstream of RAF family kinases, regulating their activity, and in turn the RAF family kinases directly activate ERK1/2 kinases through phosphorylation(29). Indeed, while overall ERK1/2 levels were unchanged, ERK1/2 kinases were hyper-phosphorylated in the resistance models (Figure 4A, Supplementary Figure 5A), which would explain the observation that ERK1/2 motifs are differentially phosphorylated in the phosphoproteomics analysis. While canonical MAPK activating growth factor receptors (EGFR and FGFR families(30)) displayed no consensus in differential expression, EGF transcripts were consistently upregulated in the PDR models, which was associated with altered EGFR activity in the phosphoproteomic data (Figure 3B and Suppl. Figure 4C). Elevated EGF protein levels were confirmed via western blotting, which likely causes MAPK activation in these models (Figure 4B, Supplementary Figure 5B). Combined, these data strongly suggest that acquired CDK4/6 inhibitor resistance results in increased EGF production, leading to hyperactivation of the MAPK pathway.
Figure 4. Acquired CDK4/6 inhibitor resistance promotes aggressive phenotypes.
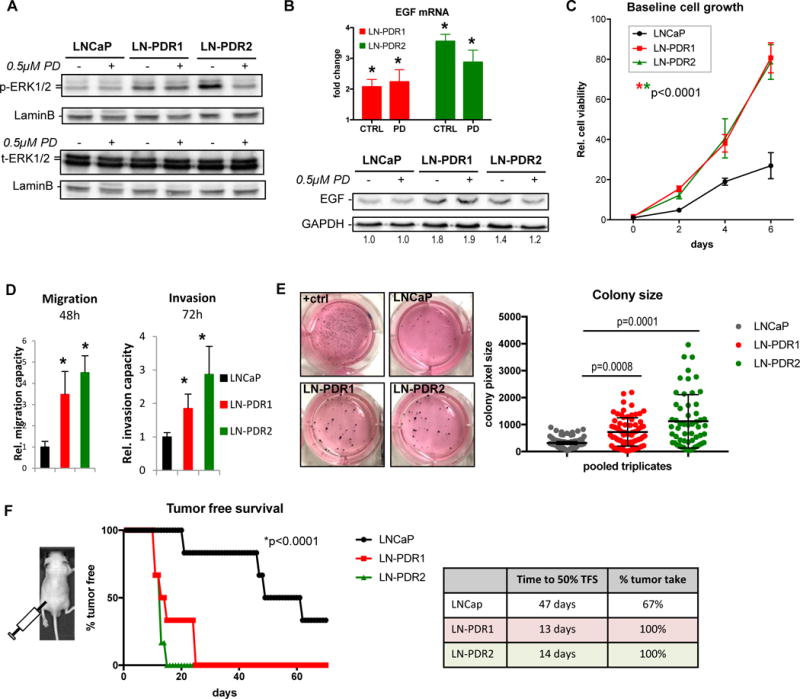
A. LNCaP PDR and parental cells were treated for 24h with 0.5μM PD or CTRL, lysed and immunoblotted for phospho-ERK1/2 (P-ERK1/2) and total ERK1/2 (t-ERK1/2). Results show hyperphosphorylation of ERK1/2 in the PDR lines compared to parental cells, while total protein levels are unchanged across the different models and conditions. These data indicate that the MAPK pathway is activated in the PDR models. B. EGF transcript level fold changes of LNCaP PDR1/2 relative to parental from RNAseq data (top, *q-value<0.1; error bars: standard error) show elevated EGF mRNA, resulting in increased EGF protein expression via Western blotting (bottom, numbers represent EGF quantification normalized to GAPDH, relative to parental CTRL. C. A time course experiment to assess cell proliferation via Trypan blue exclusion revealed that the PDR cells off PD selection are hyperproliferative compared to parental cells (*Significance was determined by a Two-way ANOVA analysis performing a multivariate comparison of mean per time point for PDR vs parental; p<0.0001). D. Cells were seeded in 6 replicates in serum-free media in a Fluoroblok transwell migration (without matrigel) or invasion (with matrigel) plates and allowed to migrate for 48h or invade for 72h to serum-rich (20%) media in the bottom of the well. PDR cells display enhanced migratory capacity and invasion through matrigel (*Student’s t-test: p<0.05 vs parental). E. Clonogenic capacity of the PDR and parental lines was assessed by seeding cells at low density (5,000/well in a 6-wells plate) in media supplemented with 0.6% agar and left to grow 3 weeks. The PDR lines displayed the ability to grow larger 3D colonies in agar. Images (left) show representative wells of triplicates. “+ctrl” are MAF cells with known clonogenicity. Colony sizes were determined via image pixel counts in FIJI (see Materials and Methods), triplicates were pooled and plotted (right) as single dots according to size (Y-axis). Line and whiskers show median and 95% Confidence Interval (CI, *Statistical significance determined by a One-way ANOVA compared to parental). F. Sub-cutaneous xenograft tumor growth of LNCaP PDR and parental cells injected at 3×106 cells in the flanks of athymic nude mice (n=6 per group) reveal accelerated tumor formation in vivo, graphed as % tumor free survival (TFS) over time (left). *Statistical significance determined via One-way ANOVA compared to parental. Table (right) shows reduced time to 50% tumor free survival (TFS) and enhanced overall tumor take.
As activated MAPK signaling is known to induce proliferation via induction of D-type Cyclins and prevent apoptosis, and thus can drive cancer progression(29,31–33), the PDR models were further characterized biologically. Baseline growth (off PD selection) was compared to the parental cells, demonstrating an almost three-fold higher growth rate for the LNCaP-derived models (Figure 4C; no growth difference was observed for LAPC4 cells, data not shown). Trans-well chemotaxis assays from 0% to 20% serum conditions revealed an increased migratory capacity and enhanced invasion in the PDR models (Figure 4D, Supplementary Figure 5C). Consonantly, transcriptome data revealed enrichment of the GSEA Hallmark for Epithelial Mesenchymal Transition (EMT) in the PDR lines (Supplementary Figure 2D/3E), which could underlie the aggressive, migratory and invasive characteristics these models have obtained upon acquiring PD resistance. To assess clonogenic growth capacity, cells were suspended at low density in agar-containing culture media and incubated for three weeks to allow colony formation. HRAS-transformed Mouse Adult Fibroblasts (MAF cells) served as a positive control due to previously described capacity to grow 3D colonies(34). Representative images of culture wells for each cell line are shown in Figure 4E (left). To utilize an unbiased approach for colony formation analyses, a macro was developed for FIJI (ImageJ), which counts total colonies per well and measures the pixel size of each colony. Although there was no significant difference in total colonies formed by the LN-PDR or parental LNCaP cells (Supplementary Figure 5D; note that LAPC4 and derivatives did not form colonies after >1 month), median colony sizes in the LN-PDR1 and LN-PDR2 models were significantly larger than the parental cells (p=0.0008 and p=0.0001 respectively, One-way ANOVA; Figure 4E, right). Combined, these findings reveal that acquired resistance to CDK4/6 inhibitors is associated with phenotypes linked to aggressive tumor behavior.
To challenge these concepts in vivo, cells were injected subcutaneously on the flanks of athymic nude mice (n=6 per group) and monitored over time for tumor take. Strikingly, the PDR models reached a 50% tumor take after only 13-14 days, while the first parental tumor formed after 20 days, and a 50% tumor take was not reached until 47 days post-injection. Moreover, 100% of the mice injected with PDR cells formed palpable tumors, compared to only 67% in the parental model (Figure 4F). Combined, acquired CDK4/6 inhibitor resistance is associated with hyperproliferation, enhanced migration and invasion, enlarged colonies, and accelerated, more efficient in vivo tumor take, thus promoting aggressive tumor phenotypes.
Acquired CDK4/6i resistance leads to dependence on MAPK signaling
With clinical trials testing a MEK inhibitor already underway for PCa (trametinib: NCT01990196, NCT02881242), and the observation that the direct downstream targets of MEK (i.e. ERK1/2) are hyperactivated upon CDK4/6 inhibitor resistance, the PDR models were assessed for sensitization to MEK inhibition. Cells treated overnight with a pre-clinical MEK inhibitor (U0126) showed reduction in phosphorylation of ERK1/2, the primary target proteins for MEK kinases (Figure 5A, Supplementary Figure 6A). To assess sensitivity to MEK inhibition, cells were treated for 6 days with U0126, and quantified via Trypan blue exclusion, demonstrating that the CDK4/6 inhibitor resistant cells are sensitized to MEK inhibition (Figure 5B, Supplementary Figure 6B). Additionally, MEK inhibition reduced the invasion capacity of the PDR lines, whereas the parental cells were unresponsive (Figure 5C). The MEK inhibitor hampered the clonogenic capacity in the PDR models, reducing not only the size of the colonies, but also total numbers of colonies formed, while the parental cells were not affected (Figure 5D). Combining MEK and CDK4/6 inhibitors resulted in a cooperative effect (Figure 5E). Sensitization to MEK inhibition was validated in vivo (Figure 5F), wherein mice with palpable xenograft tumors received a diet laced with trametinib (6.7mg/kg AIN-76A diet) or control diet (AIN-76A). These data demonstrated that tumor growth of the CDK4/6 inhibitor resistant models was reduced, while parental tumors were unresponsive. Together, these findings suggests that activation of the MAPK pathway not only promotes cell growth in the PDR models, but also the more aggressive, invasive phenotypes observed. Additionally, the CDK4/6i resistant cells appear to have become more reliant on MAPK signaling and as such are sensitized to MEK inhibition.
Figure 5. CDK4/6i resistant models become reliant on activated MAPK pathway and sensitized to MEK inhibition.
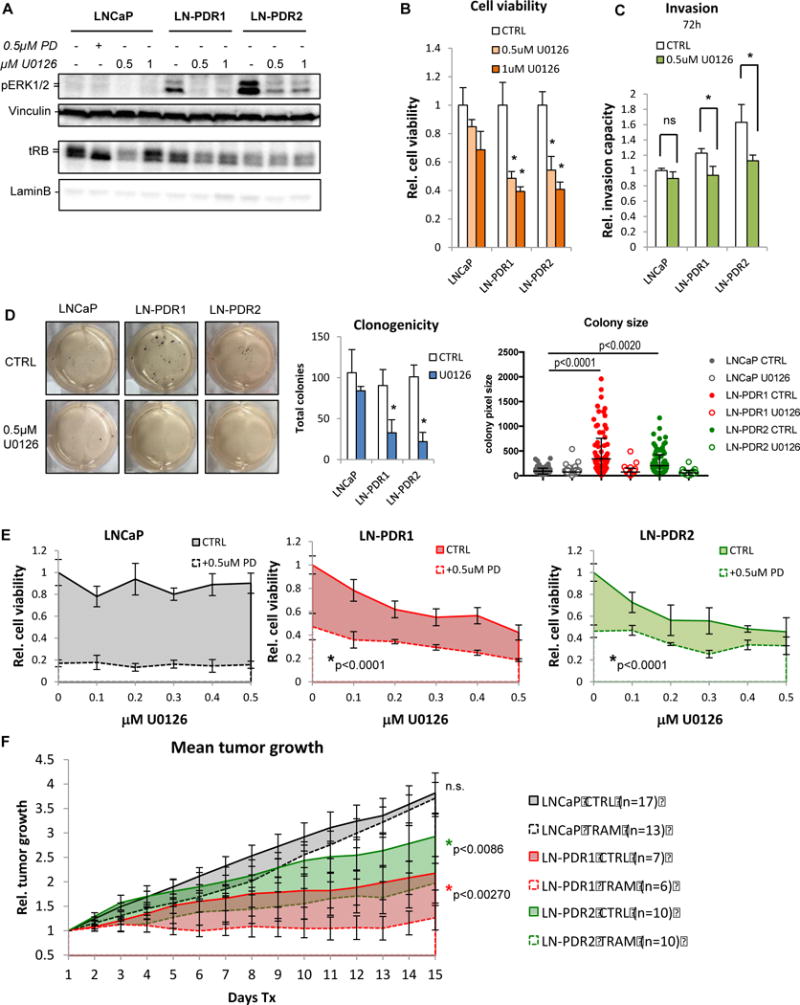
A. LNCaP cells were treated 24h with 0.5 or 1μM MEK inhibitor (U0126) or CTRL and immunoblotted for p-ERK1/2, demonstrating loss of the hyperphosphorylation of ERK1/2 observed in the LNCaP PDR models when MEK is inhibited, confirming that the MAPK pathway is activated in the PDR models. MEK inhibition does not affect RB hyperphosphorylation (upper band tRB) or CDK4 phosphorylation, indicating a bypass of the RB cell cycle checkpoint. B. Cell counting via Trypan blue exclusion after 6 days of 0.5 or 1μM U0126 treatment reveals that the PDR models are sensitized to MEK inhibition (*significant difference compared to corresponding treatment in parental cells). C. Invasion of PDR cells, but not parental cells, through matrigel in a Fluoroblok transwell system decreases with a MEK inhibitor (*p<0.05 vs parental). D. Clonogenic assay shows reduction in both size (center) and total numbers of colonies (right) formed by the PDR models, whereas the parental cells are unresponsive to MEK inhibition (0.5μM U0128). E. Cell counting with escalating doses of U0126 (0-0.5uM, t=6d) shows a cooperative effect with 0.5uM PD in PDR cells. F. LNCaP parental and PDR cells were injected sub-Q into the flanks of athymic nude mice. When tumors reached ~150mm3, mice were treated with chow laced with trametinib (TRAM). Caliper measurements revealed that, while the parental tumors were unresponsive to the MEK inhibitor,, the PDR models are sensitized to trametinib in vivo (error bars represent SEM).
Although KRAS activating mutations are not commonly observed in PCa, the MAPK pathway can be activated via other mechanisms, such as RAF fusions and overexpression(35). To investigate the clinical relevance of potential MAPK activation in prostate cancer, human primary PCa (TCGA(36)) and metastatic CRPC (SU2C/PCF(10)) datasets were interrogated for alterations of the following components of the MAPK signaling pathway (Figure 6AB): RAS family (KRAS, HRAS, NRAS), RAF family (c-RAF/RAF1, ARAF, BRAF), MEK family (MAP2K1, MAP2K2) and ERK1/2 (or MAPK3 and MAPK1). The primary PCa dataset displayed alterations (mutations, CNA, mRNA z-score:2.0) in any of these kinases in 38% of all patients, the majority of which were mRNA upregulation (Figure 6A/B, Supplementary Figure 7). The frequency of MAPK related upregulation was confirmed in the metastatic CRPC cohort, in which 47% presented with alterations, including RAF amplifications and gene fusions that may lead to MAPK activation. Interestingly, RAF alterations are more common than RAS perturbations in either cohort (Figure 6B). Taken together, data herein suggest that acquired CDK4/6 inhibitor resistance is associated with reduced RB expression and loss of function, and this coincides with MAPK activation that may bypass CDK4/6-RB signaling to induce phenotypes of aggressive features and metastasis; however, this dependence confers sensitivity to MEK inhibitors that may be exploited therapeutically.
Figure 6. cBioportal analysis of clinical datasets reveals MAPK subfamily alterations in primary and metastatic tumors.
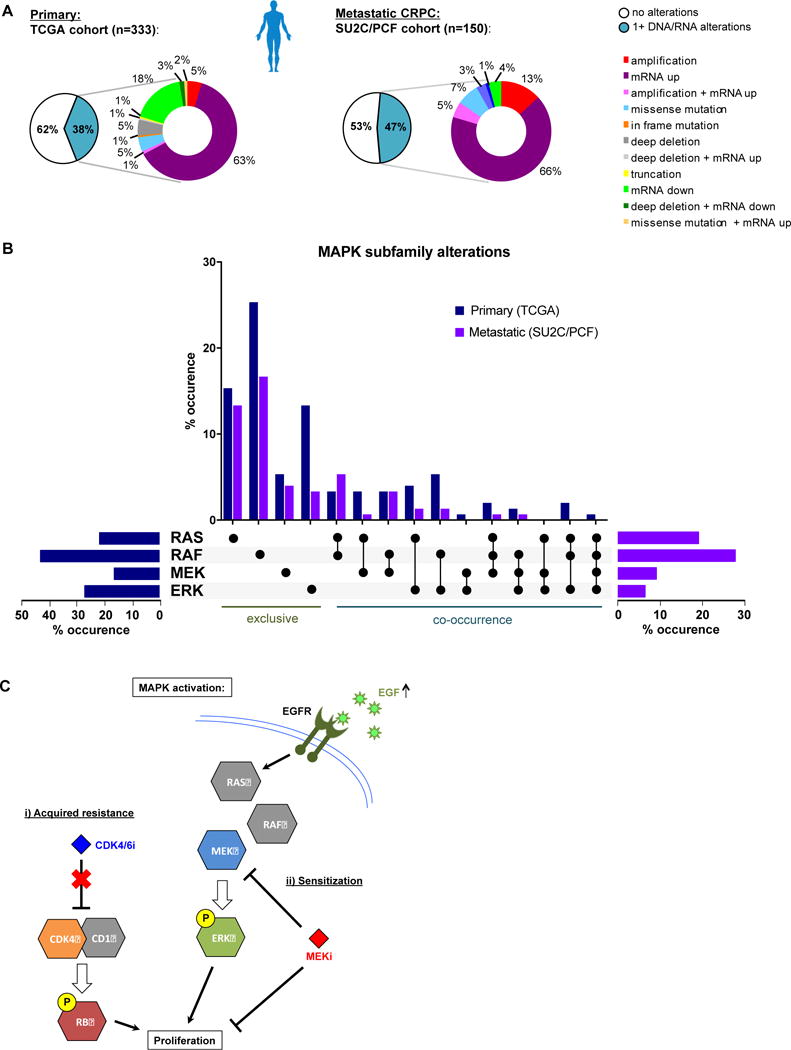
A. cBioportal analyses of the TCGA clinical cohort shows alterations in DNA and/or RNA for MAPK pathway genes in 128/333 primary prostate cancer patients (38%). Similarly, SU2C/PCF cohort reveals alterations in 71/150 patients with advanced prostate cancer (~47%). The majority of alterations in these patient datasets are gene amplification and/or transcriptional upregulation (~68% and ~85%, respectively). B. MAPK subfamily kinase analysis of alterations observed in primary (blue) and metastatic (purple) showing total percentages per subfamily (horizontal bar graphs), and exclusive versus co-occuring subfamily alterations (vertical bar graph, black dots indicate alteration, details per patient in Supplementary Figure 7). C. Schematic of acquired CDK4/6 inhibitor resistance. The PDR models presented here have acquired resistance to CDK4/6 inhibition, resulting in proliferation and aggressive phenotypes. This acquired resistance is associated with MAPK pathway activation, creating a delicate reliance on this pathway that is independent of the RB cell cycle checkpoint. This MAPK activation resulted in sensitization to MEK inhibition. EGF upregulation was observed in the PDR models, which activates the EGF Receptor (EGFR), and likely activates MAPK signaling downstream. While CDK4/6 inhibitors fail to block tumor growth in these models of acquired resistance, the sensitization to MEK inhibition provides new rationale for treating cancers that have acquired resistance to CDK4/6 inhibitors.
Discussion
CDK4/6 inhibitors have shown clinical benefit in multiple tumor types, including breast cancer; however, these agents are unlikely to provide a durable cure, and development of resistance is anticipated. Studies herein developed pre-clinical models of CDK4/6 inhibitor resistance to identify major mechanisms and consequences of CDK4/6 bypass, wherein: i) Acquired resistance to palbociclib resulted in broad CDK4/6 inhibitor resistance; ii) Resistance was associated with MAPK activation, which iii) conferred aggressive in vitro and in vivo phenotypes; and iv) MAPK-activated, CDK4/6 inhibitor resistant cancer cells are sensitized to MEK inhibitors. In summary, these studies identify MAPK induction in acquired CDK4/6 inhibitor resistance, and nominate MEK inhibitors as a means to either to prevent or treat CDK4/6 inhibitor resistant cancer.
Several previous reports have approached CDK4/6 inhibitor resistance in various cancers from different perspectives, including clinical correlations with genetic alterations(6), correlation of RB loss of function gene signature(37), and an siRNA screen identifying PDK1 by targeting the whole kinome(7), which was not observed in the current study. To date, few studies have reported on acquired resistance modeling(8,26,31,38), which largely focused on genomic alterations, gene amplifications or deletions that could lead to CDK4/6 inhibitor resistance (including RB1 loss and other aberrations in the CDK4/6-RB pathway). Although the observed genetic alterations are poised to affect CDK4/6 inhibitor efficacy, these may not reflect the molecular underpinnings of disease progression in patients that initially respond. Data herein suggest that cancer cells acquired resistance through aberrant inactivation of retained (albeit reduced) RB protein, likely via rewiring of the kinome to bypass CDK4/6 inhibition. Induction of the MAPK pathway was observed in CDK4/6 inhibitor resistance, determined by comparing phosphoproteomic and transcriptomic data and selecting alterations observed across PDR models. While genomic and functional RB loss are associated with poor cancer outcomes(39) and progression to castration resistance in the context of PCa(11,12,40), the role for MAPK signaling in regulation of RB remains somewhat elusive. MAPK controls expression and function of D-type Cyclins, and CDK4/6 requires direct binding to D-type Cyclins to exert kinase function and phosphorylate RB, and therefore it could be speculated that the observed elevated MAPK signaling bypasses CDK4/6 inhibitors via Cyclin D (Figure 6C). Cyclin D1 elevation has been linked to early adaptation to palbociclib in breast cancer models, where it induced a non-canonical Cyclin D1/CDK2 complex that restored RB phosphorylation under CDK4/6 inhibition(8). However, the CDK4/6 inhibitor resistant models presented herein displayed no change in Cyclin D1 levels (data not shown), and MEK inhibition did not affect hyperphosphorylation of RB (Figure 5A, Supplementary Figure 6A), demonstrating that MAPK activation does not mediate CDK4/6 inhbitor resistance directly, but likely presents a kinome bypass to promote tumor progression. Some studies have demonstrated intrinsic insensitivity to CDK4/6 inhibitors in cancer models with MAPK-activating mutations(41). Whole Exome Sequencing revealed no reproducible alterations (data not shown), and none had relevance to MAPK signaling. Taken together, non-genomic MAPK activation, e.g. through upregulation of EGF observed in these models, likely mediates acquired resistance.
MAPK was identified as a major factor in kinome rewiring upon acquired CDK4/6 inhibitor resistance, and associated with aggressive tumor phenotypes in vitro and in vivo. Activated MAPK signaling has been previously shown to promote prometastatic signaling and EMT(42). Invasion was attributed to ATF-2-mediated MMP-2 activation across various cancer models, including prostate(43). Moreover, MAPK activation has been associated with advanced stages of solid tumors (e.g. prostate, breast, lung)(43), and therefore it is not surprising that the PDR models displayed more aggressive phenotypes than the parental cells, and thus it is crucial to identify vulnerabilities of disease that progresses on CDK4/6 inhibitors, and develop alternative treatment regimens. Strikingly, activation of the MAPK pathway sensitized the CDK4/6 inhibitor resistant cells to MEK inhibition, providing new rationale for testing MEK inhibitors in advanced prostate and other cancers that do not necessarily activate MAPK via classical KRAS mutations or RAF fusions. Moreover, phospho-ERK1/2, indicative of MAPK activity, can be determined via immuno-labeling in clinical specimens from clinical trials (NCT02881242). It would be of interest to explore phospho-ERK1/2 as a clinical biomarker for resistance to CDK4/6 inhibitors. Alternatively, CDKs and MAPKs are known to form complex kinase networks that interact and regulate cellular processes involved in cell growth and death(27), and thus targeting these two nodes of the kinome would provide a novel opportunity for therapeutic intervention that could extend CDK4/6 inhibitor response or potentially prevent resistance altogether. These drug combinations are demonstrated to synergize in models of colorectal cancer(33), neuroblastoma(41), and even in MEK-inhibitor-resistant melanoma models(32,44). BRAF-inhibitor-resistant melanoma may still respond to CDK4/6 inhibitors combined with mTOR inhibitors(45). One activated-RAS melanoma study shows MEK and CDK4/6 inhibitors target complementary downstream networks inducing apoptosis and cell cycle arrest, causing in vivo tumor regression(46). KRAS-mutant non-small-cell lung cancer patients showed improved progression-free survival on palbociclib and a MEK inhibitor (PD-0325901)(47). Considering the cooperative effect of a CDK4/6 and MEK inhibitor observed in the PDR models, these combinations merit prioritization for further preclinical cancer studies with the potential for future clinical trial development, either upfront to delay onset of CDK4/6 inhibitor resistance or when cancer progresses. As trametinib has already entered clinical trials in PCa (NCT02881242), and has been FDA-approved for melanoma(44), its clinical application could be considered in the context of CDK4/6 inhibitor resistance.
Clinically, MEK inhibitors are likely to be combined with AR signaling targeted therapeutics (e.g. enzalutamide). Interestingly, the PDR models herein display a de-enrichment of the Androgen Response Hallmark signature, suggesting a reduced reliance on AR, which could result in castration resistance. Therefore, it is paramount to assess whether acquired CDK4/6 inhibitor resistance can lead to enzalutamide resistance, and whether this can be circumvented with the addition of a MEK inhibitor, to define optimal strategies for clinical implementation.
While induction of MAPK signaling, associated with aggressive phenotypes, was commonly observed across the PDR models, this does not rule out that other kinases identified in either of the RNAseq and phosphoproteomics datasets may contribute to the development of resistance and cancer progression. Notably, Src motifs were highly enriched in the phosphoproteomic data alone, yet Src kinase is known to impinge on CyclinD/CDK4, and is an actionable target for which clinical agents (i.e. dasatinib) have already been developed(28). Although dasatinib in combination with abiraterone did not improve progression free survival in metastatic CRPC(48), it merits further preclinical investigation in the context of CDK4/6 inhibitor resistance with the potential for additional therapeutic application.
In summary, the findings presented demonstrate that acquired CDK4/6 inhibitor resistance resulted in a kinome rewiring that not only promotes therapeutic resistance, but conferred aggressive phenotypes associated with tumor cell proliferation and invasion. Mechanistic investigation identified a reliance on MAPK activation, and therefore nominates the MAPK signaling pathway as a potential therapeutic target for tumors bypassing CDK4/6 inhibition. These collective observations not only provide insight into the molecular underpinnings of acquired resistance to cell cycle targeted therapies, but provide the basis for the next line of pre-clinical investigation, and a rationale to develop novel combinatorial or sequential therapeutic strategies in the clinic.
Supplementary Material
TRANSLATIONAL RELEVANCE.
In light of recent successes with the clinical application of Cyclin Dependent Kinase-4/6 (CDK4/6) inhibitors in breast cancer and with clinical trials underway in multiple tumor types, it is anticipated that this class of drugs will become standard of care for a variety of malignancies. Unfortunately, development of therapeutic resistance is common, and therefore it is imperative to understand mechanisms allowing cancer progression. This study demonstrates in preclinical models that acquired CDK4/6 inhibitor resistance is associated with a rewired kinome, which includes activation of the MAPK signaling pathway as a common occurrence across models, which conferred aggressive in vitro phenotypes, pro-metastatic signaling, and enhanced tumor take in vivo. However, this MAPK signaling dependence resulted in sensitization to MEK inhibitors, nominating MEK inhibition as a potential therapeutic approach to treat CDK4/6 inhibitor resistant cancers.
Acknowledgments
This study was funded by the NIH R01CA176401 (KE Knudsen), R01CA217329 (KE Knudsen, WK Kelly), Prostate Cancer Foundation Young Investigator Awards (R de Leeuw: Marjorie Katz Foundation 2016, JM Drake: 2015), Prostate Cancer Foundation Challenge Award (WK Kelly, KE Knudsen), Novartis (JA Diehl, KE Knudsen), R01CA093237 (JA Diehl), National Institute of General Medical Sciences of the National Institutes of Health T32 GM008339 (LC Cheng), Department of Defense Prostate Cancer Research Program W81XWH-15-1-0236 (JM Drake), New Jersey Health Foundation grant (JM Drake).
Footnotes
COI: The authors declare no potential conflicts of interest.
References
- 1.Barroso-Sousa R, Shapiro GI, Tolaney SM. Clinical Development of the CDK4/6 Inhibitors Ribociclib and Abemaciclib in Breast Cancer. Breast Care (Basel) 2016;11:167–173. doi: 10.1159/000447284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tripathy D, Bardia A, Sellers WR. Ribociclib (LEE011): mechanism of action and clinical impact of this selective cyclin-dependent kinase 4/6 inhibitor in various solid tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017 doi: 10.1158/1078-0432.CCR-16-3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garrido-Castro AC, Shom Goel S. CDK4/6 Inhibition in Breast Cancer: Mechanisms of Response and Treatment Failure. Curr Breast Cancer Rep. 2017:26–33. doi: 10.1007/s12609-017-0232-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vijayaraghavan S, Moulder S, Keyomarsi K, Layman RM. Inhibiting CDK in Cancer Therapy: Current Evidence and Future Directions. Target Oncol. 2017 doi: 10.1007/s11523-017-0541-2. [DOI] [PubMed] [Google Scholar]
- 5.Comstock CE, Augello MA, Goodwin JF, de Leeuw R, Schiewer MJ, Ostrander WF, Jr, et al. Targeting cell cycle and hormone receptor pathways in cancer. Oncogene. 2013;32:5481–5491. doi: 10.1038/onc.2013.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gu G, Dustin D, Fuqua SA. Targeted therapy for breast cancer and molecular mechanisms of resistance to treatment. Curr Opin Pharmacol. 2016;31:97–103. doi: 10.1016/j.coph.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 7.Jansen VM, Bhola NE, Bauer JA, Formisano L, Lee KM, Hutchinson KE, et al. Kinome-Wide RNA Interference Screen Reveals a Role for PDK1 in Acquired Resistance to CDK4/6 Inhibition in ER-Positive Breast Cancer. Cancer research. 2017;77:2488–2499. doi: 10.1158/0008-5472.CAN-16-2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer research. 2016;76:2301–2313. doi: 10.1158/0008-5472.CAN-15-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma A, Yeow WS, Ertel A, Coleman I, Clegg N, Thangavel C, et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. The Journal of clinical investigation. 2010;120:4478–4492. doi: 10.1172/JCI44239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Leeuw R, Berman-Booty LD, Schiewer MJ, Ciment SJ, Den RB, Dicker AP, et al. Novel actions of next-generation taxanes benefit advanced stages of prostate cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21:795–807. doi: 10.1158/1078-0432.CCR-14-1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis-Turak J, Courtney SM, Hazard ES, Glen WB, Jr, da Silveira WA, Wesselman T, et al. Genomics pipelines and data integration: challenges and opportunities in the research setting. Expert Rev Mol Diagn. 2017;17:225–237. doi: 10.1080/14737159.2017.1282822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardiman G, Savage SJ, Hazard ES, Wilson RC, Courtney SM, Smith MT, et al. Systems analysis of the prostate transcriptome in African-American men compared with European-American men. Pharmacogenomics. 2016;17:1129–1143. doi: 10.2217/pgs-2016-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Humphrey SJ, Azimifar SB, Mann M. High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nat Biotechnol. 2015;33:990–995. doi: 10.1038/nbt.3327. [DOI] [PubMed] [Google Scholar]
- 18.Drake JM, Graham NA, Stoyanova T, Sedghi A, Goldstein AS, Cai H, et al. Oncogene-specific activation of tyrosine kinase networks during prostate cancer progression. Proc Natl Acad Sci U S A. 2012;109:1643–1648. doi: 10.1073/pnas.1120985109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zimman A, Chen SS, Komisopoulou E, Titz B, Martinez-Pinna R, Kafi A, et al. Activation of aortic endothelial cells by oxidized phospholipids: a phosphoproteomic analysis. J Proteome Res. 2010;9:2812–2824. doi: 10.1021/pr901194x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scheltema RA, Hauschild JP, Lange O, Hornburg D, Denisov E, Damoc E, et al. The Q Exactive HF, a Benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field Orbitrap analyzer. Mol Cell Proteomics. 2014;13:3698–3708. doi: 10.1074/mcp.M114.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 22.Vizcaino JA, Csordas A, Del-Toro N, Dianes JA, Griss J, Lavidas I, et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44:11033. doi: 10.1093/nar/gkw880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drake JM, Paull EO, Graham NA, Lee JK, Smith BA, Titz B, et al. Phosphoproteome Integration Reveals Patient-Specific Networks in Prostate Cancer. Cell. 2016;166:1041–1054. doi: 10.1016/j.cell.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saldanha AJ. Java Treeview–extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- 26.Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene. 2010;29:4018–4032. doi: 10.1038/onc.2010.154. [DOI] [PubMed] [Google Scholar]
- 27.Varjosalo M, Keskitalo S, Van Drogen A, Nurkkala H, Vichalkovski A, Aebersold R, et al. The protein interaction landscape of the human CMGC kinase group. Cell Rep. 2013;3:1306–1320. doi: 10.1016/j.celrep.2013.03.027. [DOI] [PubMed] [Google Scholar]
- 28.Patel A, Sabbineni H, Clarke A, Somanath PR. Novel roles of Src in cancer cell epithelial-to-mesenchymal transition, vascular permeability, microinvasion and metastasis. Life Sci. 2016;157:52–61. doi: 10.1016/j.lfs.2016.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tran KA, Cheng MY, Mitra A, Ogawa H, Shi VY, Olney LP, et al. MEK inhibitors and their potential in the treatment of advanced melanoma: the advantages of combination therapy. Drug Des Devel Ther. 2016;10:43–52. doi: 10.2147/DDDT.S93545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butti R, Das S, Gunasekaran VP, Yadav AS, Kumar D, Kundu GC. Receptor tyrosine kinases (RTKs) in breast cancer: signaling, therapeutic implications and challenges. Mol Cancer. 2018;17:34. doi: 10.1186/s12943-018-0797-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taylor-Harding B, Aspuria PJ, Agadjanian H, Cheon DJ, Mizuno T, Greenberg D, et al. Cyclin E1 and RTK/RAS signaling drive CDK inhibitor resistance via activation of E2F and ETS. Oncotarget. 2015;6:696–714. doi: 10.18632/oncotarget.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yadav V, Burke TF, Huber L, Van Horn RD, Zhang Y, Buchanan SG, et al. The CDK4/6 inhibitor LY2835219 overcomes vemurafenib resistance resulting from MAPK reactivation and cyclin D1 upregulation. Mol Cancer Ther. 2014;13:2253–2263. doi: 10.1158/1535-7163.MCT-14-0257. [DOI] [PubMed] [Google Scholar]
- 33.Lee MS, Helms TL, Feng N, Gay J, Chang QE, Tian F, et al. Efficacy of the combination of MEK and CDK4/6 inhibitors in vitro and in vivo in KRAS mutant colorectal cancer models. Oncotarget. 2016;7:39595–39608. doi: 10.18632/oncotarget.9153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Augello MA, Berman-Booty LD, Carr R, 3rd, Yoshida A, Dean JL, Schiewer MJ, et al. Consequence of the tumor-associated conversion to cyclin D1b. EMBO Mol Med. 2015;7:628–647. doi: 10.15252/emmm.201404242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palanisamy N, Ateeq B, Kalyana-Sundaram S, Pflueger D, Ramnarayanan K, Shankar S, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med. 2010;16:793–798. doi: 10.1038/nm.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cancer Genome Atlas Research N. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malorni L, Piazza S, Ciani Y, Guarducci C, Bonechi M, Biagioni C, et al. A gene expression signature of retinoblastoma loss-of-function is a predictive biomarker of resistance to palbociclib in breast cancer cell lines and is prognostic in patients with ER positive early breast cancer. Oncotarget. 2016;7:68012–68022. doi: 10.18632/oncotarget.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang C, Li Z, Bhatt T, Dickler M, Giri D, Scaltriti M, et al. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene. 2017;36:2255–2264. doi: 10.1038/onc.2016.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McNair C, Xu K, Mandigo AC, Benelli M, Leiby B, Rodrigues D, et al. Differential impact of RB status on E2F1 reprogramming in human cancer. The Journal of clinical investigation. 2018;128:341–358. doi: 10.1172/JCI93566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bosco EE, Knudsen ES. RB in breast cancer: at the crossroads of tumorigenesis and treatment. Cell Cycle. 2007;6:667–671. doi: 10.4161/cc.6.6.3988. [DOI] [PubMed] [Google Scholar]
- 41.Hart LS, Rader J, Raman P, Batra V, Russell MR, Tsang M, et al. Preclinical Therapeutic Synergy of MEK1/2 and CDK4/6 Inhibition in Neuroblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23:1785–1796. doi: 10.1158/1078-0432.CCR-16-1131. [DOI] [PubMed] [Google Scholar]
- 42.Mulholland DJ, Kobayashi N, Ruscetti M, Zhi A, Tran LM, Huang J, et al. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer research. 2012;72:1878–1889. doi: 10.1158/0008-5472.CAN-11-3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koul HK, Pal M, Koul S. Role of p38 MAP Kinase Signal Transduction in Solid Tumors. Genes Cancer. 2013;4:342–359. doi: 10.1177/1947601913507951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Teh JL, Purwin TJ, Greenawalt EJ, Chervoneva I, Goldberg A, Davies MA, et al. An In Vivo Reporter to Quantitatively and Temporally Analyze the Effects of CDK4/6 Inhibitor-Based Therapies in Melanoma. Cancer research. 2016;76:5455–5466. doi: 10.1158/0008-5472.CAN-15-3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoshida A, Lee EK, Diehl JA. Induction of Therapeutic Senescence in Vemurafenib-Resistant Melanoma by Extended Inhibition of CDK4/6. Cancer research. 2016;76:2990–3002. doi: 10.1158/0008-5472.CAN-15-2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kwong LN, Costello JC, Liu H, Jiang S, Helms TL, Langsdorf AE, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med. 2012;18:1503–1510. doi: 10.1038/nm.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shapiro GI, Hilton J, Gandi L, Chau N, Cleary J, Wolansky A, et al. paper presented at the AACR Annual Meeting 2017. Washington, DC: 2017. [Google Scholar]
- 48.Dorff TB, Quinn DI, Pinski JK, Goldkorn A, Sadeghi S, Tsao-Wei D, et al. Randomized phase II trial of abiraterone +/− dasatinib for patients with metastatic castration-resistant prostate cancer (mCRPC) ASCO Genitourinary Cancers Symposium. 2017 doi: 10.1016/j.clgc.2019.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.