

Abstract
Aging is a leading cause of human morbidity and mortality, but efforts to slow or reverse its effects are hampered by an incomplete understanding of its multi-faceted origins. Systems biology, the use of quantitative and computational methods to understand complex biological systems, offers a toolkit well suited to elucidating the root cause of aging. We describe the known components of the aging network and outline innovative techniques that open new avenues of investigation to the aging research community. We propose integration of the systems biology and aging fields, identifying areas of complementarity based on existing and impending technological capabilities.
Subject Areas: Gerontology, Biological Sciences, Systems Biology, Complex Systems
Graphical Abstract
Gerontology; Biological Sciences; Systems Biology; Complex Systems
Introduction
Aging, the time-dependent deterioration of function and physiology, is a major cause of human death and disability. Overcoming aging is a timeworn but elusive goal, dating back to the earliest of human writings. Our incomplete understanding of the aging process and its underlying complexities has restricted even modern efforts to broad-spectrum interventions with limited potential to slow the aging process. However, an ongoing renaissance in the field of aging research raises the possibility of intelligently designed targeted therapies that will slow or reverse the effects of aging.
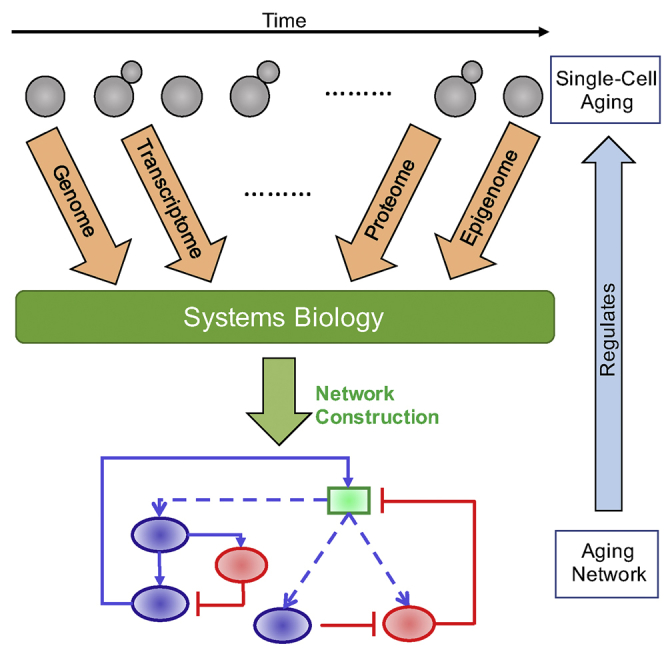
Although aging is commonly viewed at the organismal level (López-Otín et al., 2013), the outward appearance of aging results from the complex interplay of changes in multiple organ systems and their components. Indeed, the rate and characteristics of aging vary between tissue and organ systems, driven from both within cells and the extracellular environment. To fully understand the source of organismal aging, one must first grasp the contributions of its many ingredients. An approach in which each contributor is understood individually and subsequently synthesized into the whole simplifies this conceptual framework (Figure 1). We therefore focus this review on aging within single cells, but note the need for complementary reviews on the aging of the extracellular compartment, and the relative contributions of the intracellular and extracellular compartments in the aging of various organ systems.
Figure 1.

Layers of Organismal Aging
Aging is a complex phenotype that occurs in multiple organ systems through partially distinct mechanisms. Within a given organ system aging occurs both within the cell and in the extracellular space. Our review focuses on the causes of aging within the cell.
As organismal aging is a multi-faceted complex phenotype, so too is the aging of single cells. Its comprehensive understanding will require a similar approach in which its causative denominators are attacked individually or in small groups. A systems biology approach seeks insight into the fundamental underpinnings of a complex phenotype through computational modeling and quantitative analysis methods (Ideker et al., 2001, Kitano, 2002). First, experimental observations from many sources are computationally linked, creating an interconnected framework of key interactions. Components are treated not only as modular elements but also as cohesive parts of a whole that can create emergent properties when considered in the context of one another. Next, experimental validation is undertaken to validate the putative network, confirming its architecture in a directed, time-efficient manner. This approach has been highly successful where applied, for example, to guide our understanding of heart development and function (Sperling, 2011). The breadth and complexity of age-dependent physiological decline makes systems biology a natural companion to the aging field.
In this review, we discuss the emerging need for this interdisciplinary field at the cross section of cellular aging and systems biology, explaining key components to merge from each field and how integration could be accomplished. We summarize the existing literature on contributors to single-cell aging from two angles: a bottom-up approach to identify individual molecular and subsystem effectors of aging and a top-down approach to identify major pathways that contribute to aging. Next, we discuss how novel insights into the biology of single-cell aging can now be gathered using recently developed techniques that can quantify DNA alterations, transcript abundance, and epigenetic modifications at single-cell resolution, either by sampling cells from aging populations or by tracking individual cells while they age. Due to the scale and complexity of the resulting data, we discuss why a systems biology approach is essential to the comprehensive mapping of an aging network. The review ends with a summary of its main points together with a discussion of future research directions and promise of the new field.
Aging for Systems Biologists
Experimental enquiry can advance in either of two directions: top-down or bottom-up (Bruggeman and Westerhoff, 2007) (Figure 2). The top-down approach begins with a complex system and then iteratively breaks it down into component parts until its base elements are identified. Bottom-up experimentation begins from simple elements and their individual interactions and seeks emergent properties as these elements and interactions are combined into a more complex system. Aging research is conducted by distinct laboratory groups, each with its own special area of interest. As a result, the networks that have arisen are often disjointed and the connections between them unclear. We propose a systems approach to integrate the information from separate top-down and bottom-up inquiries, link these disparate networks, and direct the focus of the research community toward experimental validation of predicted intermediate elements.
Figure 2.

Directional Enquiry in Complex Systems
A complex system arises from interactions of its components, which can be grouped at various levels to comprise subsystems. Ultimately, successive breakdown of subsystems into their parts will yield inseparable base elements. System mapping can proceed either in a top-down fashion, advancing from more complex systems to understand their subsystems, or in a bottom-up fashion, relating base elements and subsystems into more complex networks.
In this section, we summarize the known pathways and effectors of single-cell aging. We synthesize information from yeast, worms, flies, mice, primates, and humans. Although not all aging effectors are universal, those discussed operate in most organisms, with particular examples of incomplete evolutionary conservation noted. The fragmentary nature of aging research precludes definitive categorization of aging factors and pathways, but the following section represents our best effort to define the limits of known aging networks. The complex and often contradictory interactions between components makes our categorization subjective and non-exhaustive; indeed, the central thesis of this review is that a computationally guided approach is essential to unify all components into a master network for single-cell aging.
Base Elements of Single-Cell Aging
Bottom-up investigations begin at the level of base elements and proceed upward. In the case of single-cell aging, bottom-up enquiry begins with a search for the specific molecular damages causative for aging (Figure 3). Numerous age-related changes have been described with varying degrees of evidence for their causative relationship with functional decline. The age-related changes discovered can be broadly classified as subsystem level or base element level. Base elements are the low-level components of a system; in this case, they are the fundamental units to which molecular damage occurs with age. However, if emergent properties affect aging, a combination of two or more base elements may be a functional effector. In this case, a subsystem, one node of a more complex aging pathway, is the causative effector.
Figure 3.

Base Elements of Aging
Multiple age-related alterations are likely to play a role in the aging process. This figure presents the most granular components that are likely to comprise the single-cell aging network.
Base element effectors of aging must constitute molecular damage to some specific cellular component. Most common cellular components can be classified as members of the genome, transcriptome, or proteome, and this review will be focused on these components.
Damage to the genome can take one of two forms: changes to the DNA molecules themselves or modifications of its associated chemical modifications and protein partners, the epigenome. Genetic mutations accumulate with age across phyla, arising from various sources of DNA damage (Moskalev et al., 2013). Increasing the rate of DNA damage through deletion of DNA repair genes or the addition of DNA-damaging agents manifests in symptoms of premature aging and a shortened lifespan (Chen et al., 2007, Shore, 1998), whereas overexpression of some DNA repair proteins (such as Rad51 in yeast) has been shown to increase lifespan (Pal et al., 2018). Moreover, some genes (such as lamin-associated genes) have been linked to both DNA damage and lifespan changes (Dechat et al., 2008). Therefore, although there is no definite mechanistic link between DNA damage and aging, their close correlation suggests that age-related changes to the nucleotide sequence may be responsible for impaired cellular function.
Telomeres are repetitive DNA elements that cap the ends of linear chromosomes and prevent misidentification of chromosome ends as DNA damage (De Lange, 2009). Standard DNA polymerases are incapable of replicating the ends of a linear chromosome, leading to a progressive loss of terminal nucleotides with each replication cycle. Telomeres solve this end-replication problem in conjunction with telomerase, a specialized RNA-dependent DNA polymerase that extends telomeres independent of mitosis (Greider and Blackburn, 1985), and telomere-protecting proteins like shelterins that prevent the cell from mistakenly recognizing telomeres as damaged DNA (Hewitt et al., 2012). Telomeres appear to be particularly susceptible to DNA damage themselves, however, at least in part because the same mechanism also prevents recognition of actual DNA damage to the telomeres (Fumagalli et al., 2012, Hewitt et al., 2012, Rochette and Brash, 2010). However, telomerase is not highly expressed in many cells, including most somatic tissue in mammals, leading to telomere shortening with age (Jiang et al., 2007). When telomeres become critically short, mammalian cells enact a senescence program, leading to irreversible arrest and a pro-inflammatory phenotype (Coppé et al., 2010). Interestingly, this phenotype is not universal in single-cell aging. S. cerevisiae telomeres do not shorten with age due to constitutive telomerase activity, and since C. elegans are entirely post-mitotic as an adult animal, they also exhibit no telomere shortening (Raices et al., 2005). Therefore, although telomere shortening may play a role in mammalian aging, it is not likely causative for aging in all organisms.
Age-related changes also exist in the epigenome, the molecular modifications and protein state that accompanies the genome (Pal and Tyler, 2016). Activating or repressing certain epigenetic processes increases lifespan, whereas others decrease lifespan. For example, loss of silencing at the silent mating type loci shortens the lifespan of haploid yeast (Kaeberlein et al., 1999), whereas H3K4 trimethylation has been shown to be inversely correlated with lifespan in C. elegans (Greer et al., 2010) and decreases in H3K27 trimethylation were shown to improve lifespan in Drosophila (Siebold et al., 2010). Some epigenetic signatures, like CpG methylation, are associated with age in some genomic loci, but youth in others (Maegawa et al., 2010). Therefore, although epigenetic changes drive aging, the precise epigenetic modifications that contribute to aging remain elusive. One means by which epigenetic modifications likely contribute to aging is through modifications of the transcriptome. The lifespan-affecting epigenetic alterations mentioned above, for instance, are all believed to operate via changes in transcriptional activity. Moreover, both protein coding (Harris et al., 2017, Janssens et al., 2015) and non-coding (Kim et al., 2016, Thum, 2014) RNA transcript levels change with age, and a meta-analysis has found aging-related changes in the transcription from genes associated with certain pathways (inflammation, mitochondria, and lysosome) across several species (de Magalhaes et al., 2009), although the extent to which these changes are causative for or responsive to aging is not clear. Although regulation of global transcriptional activity is a key mediator within longevity pathways (Filer et al., 2017), transcriptome modifications may be a second-order consequence of other effectors in the complex aging network. However, as potential contributors to emergent network properties, transcriptome modifications may indeed be important effectors of the aging phenotype.
Aged cells in many organisms exhibit an increase in misfolded proteins (Ben-Zvi et al., 2009), possibly due to impaired stress response (Calderwood et al., 2009). Interventions that eliminate (Kruegel et al., 2011) or repair misfolded proteins extend lifespan (Morley, 2003). However, it is unclear if aging is a function of damaged proteins per se, or if these interventions maintain a functional pool of some specific critical protein(s). Time-dependent functional decline in the performance of some long-lived proteins has been demonstrated. For example, in non-dividing cells nuclear pore proteins do not turn over and accumulate molecular damage and exhibit reduced functionality with time (D'Angelo et al., 2009). Even in dividing cells, asymmetric segregation of proteins may lead to accumulation of damage in the protein population (Thayer et al., 2014). Therefore, damage to some proteins can drive the aging phenotype, either through dysfunction of individual cell components or through deleterious effects from protein aggregates.
Subsystem effectors of aging are more difficult to semantically define than molecular effectors. However, several age-related changes likely converge on shared effectors to exert their harmful effects, and others may be damaging only in certain context. For example, mitochondrial dysfunction itself has been shown to drive cell aging phenotypes in proliferating human cells, and senescent cells with mitochondrial dysfunction have been observed in a progeroid mouse that rapidly accumulated mtDNA mutations (Wiley et al., 2016). On the other hand, some petite strains of yeast that lack fully functional mitochondria have been shown to have increased lifespan (Kirchman et al., 1999). Furthermore, mitochondrial dysfunction can arise from a range of molecular maladies including mitochondrial DNA damage and oxidative damage to protein components. Mapping the relationship between molecular effectors of aging, and identifying the emergent properties of their complex interactions, will be an important component to construct a comprehensive systems-level aging network.
Complex Systems in Single-Cell Aging
Top-down mapping of the single-cell aging network has identified three broad, interdependent pathways that collectively regulate organismal lifespan: the nutrient sensing pathway, the mitochondrial effector pathway, and the proteostasis pathway. Since the primary determinant(s) of cellular health remain elusive, it is unclear if these pathways converge on an effector preceding their shared phenotypic outcome, or if each modulates distinct age-related molecular changes. However, since each of these pathways is able to extend the lifespan of the unicellular eukaryote Saccharomyces cerevisiae, we can conclusively state that their effects are mediated at least in part through cell-autonomous mechanisms.
The nutrient sensing pathway is the best characterized pathway controlling the rate of cellular aging, with broad evolutionary conservation from yeast to humans (Fontana et al., 2010). A role for nutrient sensing in aging was first described in 1935, when rats fed a calorie-restricted diet were found to live longer than rats fed ad libitum (McCay et al., 1935). Anti-aging effects of calorie restriction have since been observed in multiple organisms (Blackwell et al., 1995, Chapman and Partridge, 1996, Colman et al., 2009, Hosono et al., 1989, Lin et al., 2000), and similar effects have been observed when other dietary components, such as the essential amino acid methionine, are restricted (Grandison et al., 2009). Genetic studies have expanded our knowledge of the network components underpinning dietary restriction, linking the pathway to nutrient-responsive target of rapamycin (TOR)/protein kinase A (PKA) signaling (Kaeberlein et al., 2005) and to the insulin/insulin growth factor (IGF)-like pathway (Narasimhan et al., 2009).
Mitochondrial perturbations extend lifespan (Dillin et al., 2002) via poorly understood mechanisms. Although a popular hypothesis has been that mitochondrial perturbations extend lifespan by activating the mitochondrial unfolded protein response (UPRmt) (Durieux et al., 2011, Virginija et al., 2014), recent work found no correlation between activation of UPRmt and lifetime extension in C. elegans (Bennett et al., 2014), calling this hypothesis into question. The mitochondrial effector pathway may be partially distinct from the nutrient sensing pathway. Both dietary restriction (López-Lluchh et al., 2006) and the insulin/IGF-like pathway (Lambert et al., 2004) affect mitochondrial metabolism, but dietary restriction extends lifespan in the absence of critical UPRmt effector genes (Cai et al., 2017, Jiang et al., 2000), and activation of certain mitochondrial effectors further extends lifespan in dietary restriction conditions (Jiang et al., 2000). Furthermore, sirtuins (Imai and Guarente, 2016), which appear partially independent from nutrient sensing pathways (Kaeberlein et al., 2004), act through the mitochondrial effector pathway to extend lifespan (Mouchiroud et al., 2013).
The third complex system involved in the single-cell aging process is proteostasis (Labbadia and Morimoto, 2014), or the maintenance of protein state within the cell through protein elimination and repair. Aged organisms exhibit an increase in misfolded proteins (Ben-Zvi et al., 2009). Folding state for a protein population is determined by the equilibrium between translation, folding, and elimination (Powers et al., 2009). Translation is a fundamental cellular property and is inhibited by at least some members of the nutrient sensing pathway (Wang and Proud, 2006). Protein folding is a function of protein-native thermodynamics and the contribution of molecular chaperones (Bukau et al., 2006, Horwich et al., 2007), overexpression of which is associated with insulin/IGF-like signaling and extends lifespan (Morley, 2003). Elimination is a complex process mediated by the proteasome (Amm et al., 2014) and the process of autophagy (Glick et al., 2010). In yeast, an age-related decline in vacuolar acidity, essential for autophagy (Li and Kane, 2009), has been linked to mitochondrial dysfunction and lifespan (Hughes and Gottschling, 2012). Calorie restriction increases vacuolar acidity through inhibition of TOR/PKA signaling and suppresses the onset of mitochondrial dysfunction (Hughes and Gottschling, 2012) and thus may exert its lifespan effect in part through this mechanism. Likewise, proteasome activation extends lifespan (Chondrogianni et al., 2015, Kruegel et al., 2011), although apparently independently of both the nutrient sensing pathway and at least some members of the mitochondrial effector pathway (Kruegel et al., 2011).
Systems Biology for Aging Experts
Systems biology is a multidisciplinary field specialized in the development and application of quantitative analysis methods and computational modeling approaches with the goal of gathering mechanistic insights from experimental data. It is a holistic approach to biology built around the idea that the whole is greater than the sum of its parts, and a full understanding of a biological system cannot be attained without studying it as a whole (Chuang et al., 2010, Ideker et al., 2001, Kitano, 2002). This puts it in sharp contrast with traditional reductionist approaches that study the individual parts of the system separately and thus can neglect complex behaviors emerging from the interactions of these individual parts. Given the extraordinary complexity of the aging phenotype and the wide variety of cellular systems it affects, it is all but certain that comprehensively studying aging will require the application of systems biology approaches.
A typical systems biology analysis integrates information from a wide variety of sources: quantitative measurements of the behavior of the components of interest; large-scale systematic measurements of key cellular building blocks like the genome, transcriptome, and proteome; and computational and mathematical modeling of the systems. A good exemplar is a recent work on dietary restriction in C. elegans (Hou et al., 2016a), in which the authors compared transcriptomic time courses of C. elegans fed ad libitum and under two forms of caloric restrictions to identify genes whose transcription are altered by caloric restriction, and then used the transcriptomic information of 73 genetically perturbed mutants to identify nine genes whose deletion caused changes in the transcriptome similar to those caused by caloric restriction, which were divided into three separate modules leveraging transcription factor (TF) binding information from modENCODE. A network model constructed for these modules is then used to make predictions about the behavior of the system in response to genetic perturbations in multiple modules, and the predictions were confirmed experimentally.
The potential impact for the systems biology of single-cell aging is extensive. Initially, it could direct limited resources toward more efficient, guided investigations, or reveal emergent properties of complex interactions that would go undiscovered with traditional reductionist investigation. As the field advances, we envision the development of a model comprising a unified aging network, enabling in silico prediction of longevity effects from genetic, environmental, or pharmacologic interventions. At its zenith, such a model might prove not only an academic resource, but a powerful tool to guide drug discovery in the many fields of age-related disease.
Methods to Study Single-Cell Aging
Many of the aging effectors and pathways were discovered through traditional reductionist investigation focused on one small part of aging at a time. Breakthroughs in sequencing and mass spectrometry technologies have since made large-scale genomic, transcriptomic, and proteomic data available. Advances in techniques to monitor, tag, and concentrate aged cells have enabled application of these techniques to the comparative study of young and old cells. Combinatorial application of these methods has delineated a broad framework of the aging network and formed a beachhead for further studies to elucidate its more complex architecture. In this section, we introduce the most salient techniques for the high-throughput study of the aging phenotype and provide selected examples of their application.
Omics Technologies
The advent of high-throughput sequencing technologies (Reuter et al., 2015) unlocked the ability to read and quantify an entire genome or transcriptome with relative ease. In recent years, single-cell sequencing technologies have extended the limits of this capability, permitting lower fidelity reading of both RNA and DNA within single cells (Mortazavi et al., 2008, Nawy, 2014). More recent technologies permit the simultaneous observation of both DNA and RNA within the same cell (Dey et al., 2015, Han et al., 2018, Macaulay et al., 2015, Macaulay et al., 2017), allowing correlations in genomic and transcriptomic variations to be directly explored on a single-cell level. Epigenomic studies, which map the chemical modifications that accompany DNA, are traditionally conducted in bulk samples containing many cells, but recent advances enable single-cell resolution for at least some epigenetic modifications as well (Clark et al., 2016, Guo et al., 2013, Rotem et al., 2015), including at least one study that also simultaneously measured genomic and transcriptomic information from the same cell (Hou et al., 2016b). Computational methods are already being combined with sequencing data from young and old cells to identify key differences and prioritize investigation into network actors that may modulate aging (Alfego et al., 2018, Boisvert et al., 2018). For example, cell-type-specific isolation and sequencing of mRNA (Sanz et al., 2009) from young and old astrocytes identified altered regulation of neuronal synapse control genes (Boisvert et al., 2018), suggesting a causative mechanism for age-related decline in neuronal synapse numbers (Pannese, 2011). Some studies have gone further, combining epigenomic and transcriptomic data to formulate causative hypotheses for age-related transcriptomic alterations (Slieker et al., 2016).
As high-throughput sequencing has permitted the global measurement of nucleotide components, advancements in shotgun proteomics enable quantification of most proteins within the cell (Nesvizhskii, 2010). This technique has been applied to compare the proteome of young and old cells in yeast (Janssens et al., 2015), worms (Walther et al., 2015), mice (Hwang et al., 2014), and humans (Waldera-Lupa et al., 2014). While providing important insights into general trends of proteomic expression, for example, that protein and transcript levels diverge in aged yeast (Janssens et al., 2015), this technology has not traditionally been capable of single-cell measurements. However, at least one pre-publication article has claimed technological advances that enable single-cell proteomics (Budnik et al., 2018), raising this possibility in the coming years. Likewise, single-cell multiomics, the simultaneous interrogation of the genome, transcriptome, epigenome, and proteome, is a rising capability (Macaulay et al., 2017) that may provide insight beyond current asynchronous studies.
Non-destructive Technologies
Existing omics techniques are destructive. They require killing of cells they are to measure and permit a snapshot of only a single point in time. This prevents many associative studies, for example, comparison of a cell's lifespan to its transcriptomic state. Many non-omics techniques capable of single-cell-level observation, such as single-cell qPCR to measure gene expression (Taniguchi et al., 2009) and flow-fluorescence in situ hybridization (which has been used to measure telomere length on a single-cell level [Baerlocher et al., 2006]), are similarly destructive. To study the dynamics of aging cells, multiple research groups have developed microfluidic technologies that permit microscopic monitoring of single cells throughout the lifespan (Chen et al., 2017). With these devices, one can observe cell morphology and expression dynamics for individual proteins over the entire course of a cell's life. Although this technology permits a single researcher to monitor hundreds of cells from tens of strain backgrounds or media conditions each week, a need for manual data analysis limits functional throughput. This technology is largely limited to yeast, because their natural unicellular state makes them amenable to single-cell observation and their short lifespan makes full-lifespan tracking of individual cells economically feasible.
Another common form of experimentation is directly evaluating the impact of a given intervention on organismal or cellular lifespan. Historically, direct measurement of lifespans has been an onerous and expensive process, generally requiring extensive manual labor in simple organisms (Steffen et al., 2009, Sutphin and Kaeberlein, 2009) or costly housing of more complex and longer-lived organisms. Recently, high-throughput lifespan measurement devices have been developed for organismal lifespan in worms (Stroustrup et al., 2013) and replicative lifespan in yeast (Sarnoski et al., 2017), which can reduce the effort and cost of lifespan experiments for these organisms by orders of magnitude. These technologies can transform genome-wide lifespan screens, at least for simpler organisms, from multi-year, eminently expensive projects (McCormick et al., 2015) to rapid and manageable experiments, not unlike the changes seen for sequencing in recent decades. Together, these advances in throughput and the achievable granularity of measurement techniques unlocked new doors in many fields of study.
Construction of an Aging Network from Single-Cell Data
Advances in high-throughput technologies have allowed researchers to gather copious amounts of genome-scale data of all kinds at the population level and many methods have been developed to analyze such data to identify genes and processes relevant to aging. The rise of single-cell data will further alter the landscape and permit analysis that were previously difficult or impossible.
Existing Network Construction Methods
Historically, a substantial number of aging regulators have been identified through traditional genetic approaches in model organisms examining the effect of genetic perturbations (knockout, knockdown, or overexpression) on specific genes (Johnson, 1990, Kaeberlein et al., 2005, Lin et al., 1997, Powers et al., 2006). The advent of high-throughput technologies allowed such experiments to be performed in a systematic, genome-wide manner. For example, genome-wide RNAi screens were used to identify some 160 aging-related genes in C. elegans (Curran and Ruvkun, 2007, Hamilton et al., 2005, Hansen et al., 2005), and more recently, McCormick et al. discovered some 238 genes whose deletion increases the replicative lifespan of S. cerevisiae, out of 4,698 single-gene deletion strains (McCormick et al., 2015). For humans, where application of genetic manipulation techniques is impossible for obvious reasons, observational techniques such as genome-wide association studies (GWAS), in which the genotype information of long-lived populations were compared with those of younger controls, have been used to identify SNPs that are associated with longevity (Anselmi et al., 2009, Flachsbart et al., 2009, Kojima et al., 2004, Li et al., 2009).
Another common avenue of attack is the transcriptome. Microarray analyses of age-related changes in gene expression have a comparatively long history—dating back to 1999 (Lee et al., 1999)—and have been carried out in numerous model organisms ranging from S. cerevisiae to rhesus monkeys (Bronikowski et al., 2003, Kayo et al., 2001, Lee et al., 1999, Lesur and Campbell, 2004, Lund et al., 2002, McCarroll et al., 2004, Melov and Hubbard, 2004, Pletcher et al., 2002) to humans (Lu et al., 2004, Rodwell et al., 2004, Welle et al., 2004). More recently, RNA sequencing (RNA-seq) has enabled the quantification of expression on the transcript level instead of whole genes, leading to the discovery of the so-called dark matter transcripts, which do not map to known exons (Wood et al., 2013). The expression level of some non-coding RNAs, such as microRNAs (miRNAs) has also been shown to vary with age (Li et al., 2011, Liu et al., 2012, Maes et al., 2008, Smith-Vikos et al., 2016), suggesting that they play some role in aging phenotypes. Although the exact function of many non-coding RNAs are not known, some miRNAs (such as miR-34 in Drosophila and a number of miRNAs in C. elegans) have been shown to modulate aging and lifespan (Boehm and Slack, 2005, de Lencastre et al., 2010, Liu et al., 2012, Smith-Vikos et al., 2016).
Methods have also been developed to integrate multiple sources of information. For instance, expression quantitative trait loci analysis, which integrates genomic and transcriptomic information by identifying SNPs that are associated with changes in gene expression level, has been applied to understand the genetic influence on the differential expression of genes in long-lived individuals (Häsler et al., 2017). Several methods have been developed to identify upstream regulatory factors based on transcriptional profiles and TF binding site information (Ernst et al., 2007, Segal et al., 2003). One example is a recent extension (Hou et al., 2016a, Li et al., 2013) that integrates transcriptomic data with TF binding, which was applied to identify three pathways in C. elegans whose simultaneous perturbation resulted in a phenotype closely resembling that resulting from dietary restriction (Hou et al., 2016a). Mukherjee et al. utilized a combination of GWAS and protein-protein interaction data to identify novel candidate genes that may contribute to late-onset Alzheimer disease, some of which were validated in C. elegans experiments (Mukherjee et al., 2017). Combining the topology of the protein-protein interaction network with knowledge of aging regulators gained from genetic studies can also help to identify potential additional regulators (Bell et al., 2009, Budovsky et al., 2007, Managbanag et al., 2008).
Extension to Single-Cell Aging Data
The ability to gather single-cell-level aging data could alter the landscape of experimental possibility in many ways. Elimination of the averaging effect allows new phenotypes like gene expression noise to be tracked on both intra- and intercellular basis, and their relationship with aging to be explored. The causal relationship among the various genotypical and phenotypical changes wrought by aging can be explored. For instance, a recent work based on single-cell RNA-seq data suggests that the increase in population-level noise and accumulation of somatic mutations during aging are likely to be independently acquired rather than causally linked (Enge et al., 2017). The ability to examine the contents of a specific cell is particularly valuable with an inherently stochastic phenotype like aging—it is well known that even an isogenic population of cells will have vastly different lifespans. Comparing the transcriptomic and proteomic content of isogenic cells of significantly different lifespans may identify genes whose differential expression contributes to premature death or exceptional longevity. Once such candidates have been identified, a bottom-up analysis using standard techniques such as fluorescent reporters or tags to monitor their expression levels in a single cell throughout the aging process can be performed to validate the association and potentially gather further information about their behavior. With advances in cellular barcoding technology (Pei et al., 2017, Perié et al., 2014), in which a heritable unique DNA sequence is introduced into a cell to identify its descendants via sequencing, daughter cells can be associated with their parents during single-cell aging experiments, and it may even be possible to use the daughters as a proxy to indirectly monitor the genomic, transcriptomic, and proteomic content of the same cell as it ages.
By extending the existing analysis techniques to single-cell-level data extracted from aging cells, we can identify genes whose participation in the aging process has been previously obscured by the averaging effects. For example, it seems plausible that the expression of a gene might shift during aging from a binary (bimodal) profile to a graded (unimodal) one, or vice versa, not unlike how isogenic cells grown in different conditions could respond to the same signal with either a graded or a binary response, but such changes would be difficult to detect in the absence of single-cell-level observations barring a marked shift in the average expression level (Biggar and Crabtree, 2001). By examining the phenotype resulting from perturbation of such genes, we can obtain information about its mechanism of action. One particularly interesting phenotype is the temporal scaling of lifespan distributions (Figure 4): Stroustrup and colleagues showed that the lifespan distributions of C. elegans under a wide array of interventions are differentiated only by an apparent stretching or shrinking of time (Stroustrup et al., 2016), and Liu and Acar found that the replicative lifespan distributions of a number of single-gene deletion strains of S. cerevisiae are similarly temporally scaled (Liu and Acar, 2018). Such lifespan distributions that differ only by temporal scaling have identical residual distributions under an accelerated failure time regression model (Swindell, 2009), which can be confirmed by statistical testing. Such temporal scaling suggests that the interventions at issue affect aging through a common effector, and if the lifespan distribution from a genetic perturbation lacks this scaling property—as was found to be the case for three interventions (eat-2(ad1116), nuo-6(qm200), and raising temperature from below to above 30°C) in C. elegans (Stroustrup et al., 2016)—then that would be strong evidence that the gene at issue is affecting lifespan via a different mechanism.
Figure 4.

Illustration of Temporal Scaling of Lifespan Distributions
Left: Four hypothetical viability curves corresponding to different interventions. Right: The same four curves are temporally rescaled based on the median lifespan value of each. The orange, yellow, and blue curves are identical after scaling, whereas the purple curve is different, indicating that the former three interventions act via the same underlying mechanism, whereas the last intervention operates via a different mechanism. To avoid exact overlap, the orange and yellow curves have been slightly shifted in the x direction.
Ultimately, by combining the top-down analysis of genome-scale information and bottom-up verifications of individual components identified, a master regulatory network—or perhaps several independent networks—governing aging can be constructed from the data gathered, assisted by known data such as TF-promoter binding (Contrino et al., 2012) and genetic (Costanzo et al., 2016, Mani et al., 2008) and protein-protein interactions (Chatr-aryamontri et al., 2017, Stark et al., 2006) among the genes identified as part of the network.
Validation of the Aging Network
These applications of computational modeling are best suited for hypothesis generation, creating putative networks for subsequent experimental validation. The inferred master network structure can be analyzed in a number of ways. In a top-down analysis, one might divide the component genes into individual pathways that affect aging based on functional and interaction information. Alternatively, a bottom-up analysis might examine the network for well-characterized motifs (Alon, 2007a, Alon, 2007b), which are recurring regulation patterns that occur within networks. Such regulation patterns include positive or negative feedback loops (in which a network component directly or indirectly regulates its own activity), feedforward loops (in which a network component regulates the activity of another network component via two distinct paths, usually one direct and one indirect), gene regulatory cascades, or special topologies that allow for network-dosage compensation where the output of a gene network is invariant to or independent of the number of copies of the network in the cell (Acar et al., 2010, Peng et al., 2016, Song et al., 2014) (Figure 5). As the effects of such motifs are well established, based on their presence or absence, it may be possible to predict the activity or phenotype of the network. For more complex networks with multiple interacting motifs, detailed computational simulations can also be performed for additional insight into the network's possible behavior (To and Maheshri, 2010).
Figure 5.

Identification of Network Motifs in a Complex Network
A hypothetical gene network is shown: rectangular boxes represent genes, whereas ovals represent proteins synthesized from such genes. Dotted arrows indicate transcription factor-promoter interaction, whereas dashed arrows indicate protein production after transcription and translation. Solid lines indicate protein-protein interaction. A positive feedback loop is highlighted in blue, and a negative feedback loop is highlighted in orange. Illustration of the potential effects of the positive feedback motif (bimodality) and the negative feedback motif (noise reduction) is shown next to the loop.
Experimental observations of the behavior of the predicted master regulatory network can be used to refine its regulatory structure. For instance, a positive feedback loop is usually required to produce a bimodal expression profile (i.e., where the expression level has two modes or peaks) with switch-like behavior (Mitrophanov and Groisman, 2008), whereas a negative feedback loop reduces the level of gene expression noise (Dublanche et al., 2006) and can lead to oscillations (Pigolotti et al., 2007). Thus, the presence of a bimodal expression profile may be an indicator that a core positive feedback loop module is present and exerting significant effects on the overall network, whereas a below-normal noise level or oscillation behavior might indicate the same thing for a negative feedback loop (Figure 5). Comparison of the experimental observations with computational simulation results may reveal that some edges in the network's regulatory graph are much more important than others, allowing for its simplification.
As previously mentioned, aging is an inherently stochastic process, and so one particularly interesting quantitative phenotype to track during aging is noise, or the variation in gene expression levels among different cells. Noise is usually divided into two components: extrinsic noise consists of variations due to cell-to-cell differences, such as ribosome copy number, whereas intrinsic noise is caused by the inherent randomness of the gene expression process (Elowitz et al., 2002, Fraser et al., 2004, Kærn et al., 2005). A number of studies (Bahar et al., 2006, Enge et al., 2017, Feser et al., 2010, Martinez-Jimenez et al., 2017, Raj and van Oudenaarden, 2008) that compared cells obtained from individual organisms of different age showed that the cell population obtained from older individuals generally have higher cell-to-cell variability. However, recent results from our laboratory demonstrated that, within a single cell, the level of variation in the expression of a protein steadily decreases in haploid S. cerevisiae until the final few generations of its replicative lifespan (Liu et al., 2017). We hypothesized that this was caused by increased rates of chromatin state transitions during aging, which is known to cause depletion of the histone supply (Feser et al., 2010, O'Sullivan et al., 2010) and formation of nucleosome-free patches (Ishimi et al., 1987). There are several possible ways to reconcile the apparently conflicting observations. The previous measurements are all on diploid organisms, where the increased ploidy and concomitant increase in the abundance of cellular machinery should cause a decrease in noise by buffering the impact of stochastic processes, which may well reduce or even eliminate the window of noise reduction available. Another possible explanation is that our measurement is on the level of variation within an individual cell, whereas the previous reports are based on a population-level measurement of total noise in distinct populations of cells (Figure 6). Thus, it may well be possible that the aging process causes an increase in cell-to-cell variations (extrinsic noise) but a decrease in intracellular variations (intrinsic noise) up until the final few generations of the cell's lifespan. Further experimentation to test this hypothesis—and to confirm the generality of the observations—is expected to be a fruitful research direction.
Figure 6.

Illustration of the Difference between Cell-to-Cell Variation and Intracellular Variation
Left: expression level trajectories of three hypothetical cells. Right top: level of cell-to-cell variation, expressed as coefficient of variation, increases over time. Right bottom: level of intracellular variation decreases over time. Dashed boxes illustrate the data points used to compute the coefficient of variation in each figure.
Conclusions and Perspectives
Why do cells grown in the same environment exhibit heterogeneous lifespans? Which gene networks are responsible for controlling the aging process? How can the healthy lifespan of a living system be maximized? Despite the fundamental nature of these questions, we have limited understanding on the mechanisms governing cellular aging, and such mechanistic understanding is even scarcer in the context of gene networks.
With recently developed techniques for performing genetic and phenotypic characterizations at single-cell resolution, comprehensive monitoring of time-dependent changes occurring in individual aging cells is now possible. For example, using single-cell RNA-seq, transcript levels associated with the expression of different proteasome subunits can now be probed in single cells to gather insights into how aging alters proteasome abundance. Also, locus-specific DNA alterations can be compared between young and old cells using single-cell DNA sequencing, which would elucidate how aging affects genome stability. The emergence of these powerful techniques is transforming the cellular aging field.
Systems biology is a multidisciplinary field (Ideker et al., 2001, Kitano, 2002) specialized in the development and application of quantitative analysis methods and computational modeling approaches with the goal of gathering mechanistic insights from experimental data. Since the beginning of the new millennium, systems biology approaches have been successfully used to gain insights into a myriad of cellular processes. However, aging remains one of the few research areas that has not seen extensive application of systems biology tools and methods. This review argues that the scale and complexity of age-related information collected through, for instance, high-throughput genetic screening or single-cell omics studies makes systems biology a natural and necessary companion to the cellular aging field. As an example, applying quantitative network construction methods based on the scalability of lifespan distributions would shed light on the modularity of the global gene network governing aging and organismal lifespan. The past several decades have witnessed the identification of many proteins affecting the lifespan of various model organisms. Despite recognizing that varying the expression levels of these proteins one-at-a-time impacts cellular lifespan, it is not fully understood how these proteins interact through, for example, feedback loops in a gene network setting. Systems biology approaches will be essential to map the structure of the aging networks.
As another example to future research directions at the cross section of systems biology and single-cell aging, one can track the changes occurring in global gene expression levels and protein interaction dynamics during the aging of single cells and build novel aging networks that would potentially dictate the rate of aging and lifespan. Here, any modular network structure would emerge as a result of the systems-level analysis of the global phenotypic information collected from aging cells. A multitude of existing quantitative analysis techniques on population-level genome-scale data can be extended and applied to single-cell-level data, aided by other previously gathered information concerning genetic and protein-protein interactions, to elucidate any underlying networks governing aging (Hou et al., 2012). The structure of such networks and interactions among the network components can be further validated through expected functional consequences by using insights and lessons accumulated through the use of bottom-up systems biology methods in the past two decades (Mitrophanov and Groisman, 2008). The knowledge about such structure-function relationships would confirm the interaction topology in any novel aging networks to be discovered through the use of top-down or global analysis methods.
The recent development of novel single-cell techniques to probe genetic and phenotypic changes occurring in aging cells and the application of systems biology methods on the resulting data are transforming the aging field into a new cross-disciplinary field that has great promise to provide insights into the long-standing unknowns of cellular aging and lifespan.
Acknowledgments
EAS acknowledges support through an NSF Graduate Research Fellowship. MA acknowledges funding from the National Institutes of Health (1DP2AG050461-01 and 1R01GM127870-01).
Author Contributions
R.S., E.A.S., and M.A. designed, prepared, read, and approved the manuscript.
References
- Acar M., Pando B.F., Arnold F.H., Elowitz M.B., van Oudenaarden A. A general mechanism for network-dosage compensation in gene circuits. Science. 2010;329:1656–1660. doi: 10.1126/science.1190544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfego D., Rodeck U., Kriete A. Global mapping of transcription factor motifs in human aging. PLoS One. 2018;13:e0190457. doi: 10.1371/journal.pone.0190457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon U. Chapman & Hall; 2007. An Introduction to Systems Biology: Design Principles of Biological Circuits. [Google Scholar]
- Alon U. Network motifs: theory and experimental approaches. Nat. Rev. Genet. 2007;8:450. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- Amm I., Sommer T., Wolf D.H. Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim. Biophys. Acta. 2014;1843:182–196. doi: 10.1016/j.bbamcr.2013.06.031. [DOI] [PubMed] [Google Scholar]
- Anselmi C.V., Malovini A., Roncarati R., Novelli V., Villa F., Condorelli G., Bellazzi R., Puca A.A. Association of the FOXO3A locus with extreme longevity in a southern Italian centenarian study. Rejuvenation Res. 2009;12:95–104. doi: 10.1089/rej.2008.0827. [DOI] [PubMed] [Google Scholar]
- Baerlocher G.M., Vulto I., de Jong G., Lansdorp P.M. Flow cytometry and FISH to measure the average length of telomeres (flow FISH) Nat. Protoc. 2006;1:2365–2376. doi: 10.1038/nprot.2006.263. [DOI] [PubMed] [Google Scholar]
- Bahar R., Hartmann C.H., Rodriguez K.A., Denny A.D., Busuttil R.A., Dolle M.E.T., Calder R.B., Chisholm G.B., Pollock B.H., Klein C.A. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature. 2006;441:1011–1014. doi: 10.1038/nature04844. [DOI] [PubMed] [Google Scholar]
- Bell R., Hubbard A., Chettier R., Chen D., Miller J.P., Kapahi P., Tarnopolsky M., Sahasrabuhde S., Melov S., Hughes R.E. A human protein interaction network shows conservation of aging processes between human and invertebrate species. PLoS Genet. 2009;5:e1000414. doi: 10.1371/journal.pgen.1000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Zvi A., Miller E.A., Morimoto R.I. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc. Natl. Acad. Sci. USA. 2009;106:14914–14919. doi: 10.1073/pnas.0902882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett C.F., Vander Wende H., Simko M., Klum S., Barfield S., Choi H., Pineda V.V., Kaeberlein M. Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nat. Commun. 2014;5:3483. doi: 10.1038/ncomms4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggar S.R., Crabtree G.R. Cell signaling can direct either binary or graded transcriptional responses. EMBO J. 2001;20:3167–3176. doi: 10.1093/emboj/20.12.3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell B.N., Bucci T.J., Hart R.W., Turturro A. Longevity, body weight, and neoplasia in ad libitum-fed and diet- restricted C57BL6 mice fed NIH-31 open formula diet. Toxicol. Pathol. 1995;23:570–582. doi: 10.1177/019262339502300503. [DOI] [PubMed] [Google Scholar]
- Boehm M., Slack F. A developmental timing microRNA and its target regulate life span in C. elegans. Science. 2005;310:1954. doi: 10.1126/science.1115596. [DOI] [PubMed] [Google Scholar]
- Boisvert M.M., Erikson G.A., Shokhirev M.N., Allen N.J. The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep. 2018;22:269–285. doi: 10.1016/j.celrep.2017.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronikowski A.M., Carter P.A., Morgan T.J., Garland T., Jr., Ung N., Pugh T.D., Weindruch R., Prolla T.A. Lifelong voluntary exercise in the mouse prevents age-related alterations in gene expression in the heart. Physiol. Genomics. 2003;12:129–138. doi: 10.1152/physiolgenomics.00082.2002. [DOI] [PubMed] [Google Scholar]
- Bruggeman F.J., Westerhoff H.V. The nature of systems biology. Trends Microbiol. 2007;15:45–50. doi: 10.1016/j.tim.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Budnik B., Levy E., Harmange G., Slavov N. Mass-spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. bioRxiv. 2018:102681. doi: 10.1186/s13059-018-1547-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budovsky A., Abramovich A., Cohen R., Chalifa-Caspi V., Fraifeld V. Longevity network: construction and implications. Mech. Ageing Dev. 2007;128:117–124. doi: 10.1016/j.mad.2006.11.018. [DOI] [PubMed] [Google Scholar]
- Bukau B., Weissman J., Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Cai H., Rasulova M., Vandemeulebroucke L., Meagher L., Vlaeminck C., Dhondt I., Braeckman B.P. Life-span extension by axenic dietary restriction is independent of the mitochondrial unfolded protein response and mitohormesis in Caenorhabditis elegans. J. Gerontol. A Biol. Sci. Med. Sci. 2017;72:1311–1318. doi: 10.1093/gerona/glx013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood S.K., Murshid A., Prince T. The shock of aging: molecular chaperones and the heat shock response in longevity and aging–a mini-review. Gerontology. 2009;55:550–558. doi: 10.1159/000225957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman T., Partridge L. Female fitness in Drosophila melanogaster: an interaction between the effect of nutrition and of encounter rate with males. Proc. Biol. Sci. 1996;263:755–759. doi: 10.1098/rspb.1996.0113. [DOI] [PubMed] [Google Scholar]
- Chatr-aryamontri A., Oughtred R., Boucher L., Rust J., Chang C., Kolas N.K., O’Donnell L., Oster S., Theesfeld C., Sellam A. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017;45:D369–D379. doi: 10.1093/nar/gkw1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.-H., Hales C.N., Ozanne S.E. DNA damage, cellular senescence and organismal ageing: causal or correlative? Nucleic Acids Res. 2007;35:7417–7428. doi: 10.1093/nar/gkm681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K.L., Crane M.M., Kaeberlein M. Elsevier; 2017. Microfluidic Technologies for Yeast Replicative Lifespan Studies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chondrogianni N., Georgila K., Kourtis N., Tavernarakis N., Gonos E.S. 20S proteasome activation promotes life span extension and resistance to proteotoxicity in Caenorhabditis elegans. FASEB J. 2015;29:611–622. doi: 10.1096/fj.14-252189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang H.-Y., Hofree M., Ideker T. A decade of systems biology. Annu. Rev. Cell Dev. Biol. 2010;26:721–744. doi: 10.1146/annurev-cellbio-100109-104122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark S.J., Lee H.J., Smallwood S.A., Kelsey G., Reik W. Single-cell epigenomics: powerful new methods for understanding gene regulation and cell identity. Genome Biol. 2016;17:72. doi: 10.1186/s13059-016-0944-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman R.J., Anderson R.M., Johnson S.C., Kastman E.K., Kosmatka K.J., Beasley T.M., Allison D.B., Cruzen C., Simmons H.A., Kemnitz J.W. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contrino S., Smith R.N., Butano D., Carr A., Hu F., Lyne R., Rutherford K., Kalderimis A., Sullivan J., Carbon S. modMine: flexible access to modENCODE data. Nucleic Acids Res. 2012;40:D1082. doi: 10.1093/nar/gkr921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé J.-P., Desprez P.-Y., Krtolica A., Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. Mech. Dis. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M., VanderSluis B., Koch E.N., Baryshnikova A., Pons C., Tan G., Wang W., Usaj M., Hanchard J., Lee S.D. A global genetic interaction network maps a wiring diagram of cellular function. Science. 2016;353:1381. doi: 10.1126/science.aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran S.P., Ruvkun G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 2007;3:e56. doi: 10.1371/journal.pgen.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Angelo M.A., Raices M., Panowski S.H., Hetzer M.W. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell. 2009;136:284–295. doi: 10.1016/j.cell.2008.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dechat T., Pfleghaar K., Sengupta K., Shimi T., Shumaker D.K., Solimando L., Goldman R.D. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008;22:832–853. doi: 10.1101/gad.1652708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey S.S., Kester L., Spanjaard B., Bienko M., van Oudenaarden A. Integrated genome and transcriptome sequencing of the same cell. Nat. Biotechnol. 2015;33:285–289. doi: 10.1038/nbt.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillin A., Hsu A.L., Arantes-Oliveira N., Lehrer-Graiwer J., Hsin H., Fraser A.G., Kamath R.S., Ahringer J., Kenyon C. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002;298:2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- Dublanche Y., Michalodimitrakis K., Kümmerer N., Foglierini M., Serrano L. Noise in transcription negative feedback loops: simulation and experimental analysis. Mol. Syst. Biol. 2006;2:41. doi: 10.1038/msb4100081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieux J., Wolff S., Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz M.B., Levine A.J., Siggia E.D., Swain P.S. Stochastic gene expression in a single cell. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- Enge M., Arda H.E., Mignardi M., Beausang J., Bottino R., Kim S.K., Quake S.R. Single-cell analysis of human pancreas reveals transcriptional signatures of aging and somatic mutation patterns. Cell. 2017;171:321–330.e14. doi: 10.1016/j.cell.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J., Vainas O., Harbison C.T., Simon I., Bar-Joseph Z. Reconstructing dynamic regulatory maps. Mol. Syst. Biol. 2007;3:74. doi: 10.1038/msb4100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feser J., Truong D., Das C., Carson J.J., Kieft J., Harkness T., Tyler J.K. Elevated histone expression promotes life span extension. Mol. Cell. 2010;39:724–735. doi: 10.1016/j.molcel.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filer D., Thompson M.A., Takhaveev V., Dobson A.J., Kotronaki I., Green J.W.M., Heinemann M., Tullet J.M.A., Alic N. RNA polymerase III limits longevity downstream of TORC1. Nature. 2017;552:263–267. doi: 10.1038/nature25007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flachsbart F., Caliebe A., Kleindorp R., Blanche H., von Eller-Eberstein H., Nikolaus S., Schreiber S., Nebel A. Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc. Natl. Acad. Sci. USA. 2009;106:2700–2705. doi: 10.1073/pnas.0809594106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L., Partridge L., Longo V.D. Dietary restriction, growth factors and aging: from yeast to humans. Science. 2010;328:321–326. doi: 10.1126/science.1172539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser H.B., Hirsh A.E., Giaever G., Kumm J., Eisen M.B. Noise minimization in eukaryotic gene expression. PLoS Biol. 2004;2:e137. doi: 10.1371/journal.pbio.0020137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M., Rossiello F., Clerici M., Barozzi S., Cittaro D., Kaplunov J.M., Bucci G., Dobreva M., Matti V., Beausejour C.M. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 2012;14:355–365. doi: 10.1038/ncb2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick D., Barth S., Macleod K.F. Autophagy: cellular and molecular mechanisms. J. Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandison R.C., Piper M.D.W., Partridge L. Amino-acid imbalance explains extension of lifespan by dietary restriction in Drosophila. Nature. 2009;462:1061–1064. doi: 10.1038/nature08619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer E.L., Maures T.J., Hauswirth A.G., Green E.M., Leeman D.S., Maro G.S., Han S., Banko M.R., Gozani O., Brunet A. Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature. 2010;466:383–387. doi: 10.1038/nature09195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greider C.W., Blackburn E.H. Identification of a specific telomere terminal transferase activity in tetrahymena extracts. Cell. 1985;43:405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- Guo H., Zhu P., Wu X., Li X., Wen L., Tang F. Single-Cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res. 2013;23:2126–2135. doi: 10.1101/gr.161679.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton B., Dong Y., Shindo M., Liu W., Odell I., Ruvkun G., Lee S.S. A systematic RNAi screen for longevity genes in C. elegans. Genes Dev. 2005;19:1544–1555. doi: 10.1101/gad.1308205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K.Y., Kim K.-T., Joung J.-G., Son D.-S., Kim Y.J., Jo A., Jeon H.-J., Moon H.-S., Yoo C.E., Chung W. SIDR: simultaneous isolation and parallel sequencing of genomic DNA and total RNA from single cells. Genome Res. 2018;28:75–87. doi: 10.1101/gr.223263.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M., Hsu A.L., Dillin A., Kenyon C. New genes tied to endocrine, metabolic, and dietary regulation of lifespan from a Caenorhabditis elegans genomic RNAi screen. PLoS Genet. 2005;1:119–128. doi: 10.1371/journal.pgen.0010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris S.E., Riggio V., Evenden L., Gilchrist T., McCafferty S., Murphy L., Wrobel N., Taylor A.M., Corley J., Pattie A. Age-related gene expression changes, and transcriptome wide association study of physical and cognitive aging traits, in the Lothian Birth Cohort 1936. Aging (Albany. NY) 2017;9:2489–2503. doi: 10.18632/aging.101333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häsler R., Venkatesh G., Tan Q., Flachsbart F., Sinha A., Rosenstiel P., Lieb W., Schreiber S., Christensen K., Christiansen L. Genetic interplay between human longevity and metabolic pathways — a large-scale eQTL study. Aging Cell. 2017;16:716–725. doi: 10.1111/acel.12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt G., Jurk D., Marques F.D.M., Correia-Melo C., Hardy T., Gackowska A., Anderson R., Taschuk M., Mann J., Passos J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012;3:708. doi: 10.1038/ncomms1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwich A.L., Fenton W.A., Chapman E., Farr G.W. Two Families of Chaperonin: physiology and mechanism. Annu. Rev. Cell Dev. Biol. 2007;23:115–145. doi: 10.1146/annurev.cellbio.23.090506.123555. [DOI] [PubMed] [Google Scholar]
- Hosono R., Nishimoto S., Kuno S. Alterations of life span in the nematode Caenorhabditis elegans under monoxenic culture conditions. Exp. Gerontol. 1989;24:251–264. doi: 10.1016/0531-5565(89)90016-8. [DOI] [PubMed] [Google Scholar]
- Hou L., Huang J., Green C.D., Boyd-Kirkup J., Zhang W., Yu X., Gong W., Zhou B., Han J.-D.J. Systems biology in aging: linking the old and the young. Curr. Genomics. 2012;13:558–565. doi: 10.2174/138920212803251418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L., Wang D., Chen D., Liu Y., Zhang Y., Cheng H., Xu C., Sun N., McDermott J., Mair W.B. A systems approach to reverse engineer lifespan extension by dietary restriction. Cell Metab. 2016;23:529. doi: 10.1016/j.cmet.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y., Guo H., Cao C., Li X., Hu B., Zhu P., Wu X., Wen L., Tang F., Huang Y. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016;26:304–319. doi: 10.1038/cr.2016.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes A.L., Gottschling D.E. An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature. 2012;492:261–265. doi: 10.1038/nature11654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang C.Y., Kim K., Choi J.Y., Bahn Y.J., Lee S.M., Kim Y.K., Lee C., Kwon K.S. Quantitative proteome analysis of age-related changes in mouse gastrocnemius muscle using mTRAQ. Proteomics. 2014;14:121–132. doi: 10.1002/pmic.201200497. [DOI] [PubMed] [Google Scholar]
- Ideker T., Galitski T., Hood L. A new approach to decoding life: systems biology. Annu. Rev. Genomics Hum. Genet. 2001;2:343–372. doi: 10.1146/annurev.genom.2.1.343. [DOI] [PubMed] [Google Scholar]
- Imai S., Guarente L. It takes two to tango: NAD+ and sirtuins in aging/longevity control. NPJ Aging Mech. Dis. 2016;2:16017. doi: 10.1038/npjamd.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimi Y., Kojima M., Takeuchi F., Miyamoto T., Yamada M.-A., Hanaoka F. Changes in chromatin structure during aging of human skin fibroblasts. Exp. Cell Res. 1987;169:458–467. doi: 10.1016/0014-4827(87)90206-0. [DOI] [PubMed] [Google Scholar]
- Janssens G.E., Meinema A.C., González J., Wolters J.C., Schmidt A., Guryev V., Bischoff R., Wit E.C., Veenhoff L.M., Heinemann M. Protein biogenesis machinery is a driver of replicative aging in yeast. Elife. 2015;4:e08527. doi: 10.7554/eLife.08527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H., Ju Z., Rudolph K.L. Telomere shortening and ageing. Z. Gerontol. Geriatr. 2007;40:314–324. doi: 10.1007/s00391-007-0480-0. [DOI] [PubMed] [Google Scholar]
- Jiang J.C., Jaruga E., Repnevskaya M.V., Jazwinski S.M. An intervention resembling caloric restriction prolongs life span and retards aging in yeast. FASEB J. 2000;14:2135–2137. doi: 10.1096/fj.00-0242fje. [DOI] [PubMed] [Google Scholar]
- Johnson T.E. Increased life-span of age-1 mutants in Caenorhabditis elegans and lower Gompertz rate of aging. Science. 1990;249:908–912. doi: 10.1126/science.2392681. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M., McVey M., Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M., Kirkland K.T., Fields S., Kennedy B.K. Sir2-independent life span extension by calorie restriction in yeast. PLoS Biol. 2004;2:E296. doi: 10.1371/journal.pbio.0020296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M., Powers R.W., Steffen K.K., Westman E.A., Hu D., Dang N., Kerr E.O., Kirkland K.T., Fields S., Kennedy B.K. Cell biology: regulation of yeast replicative life span by TOR and Sch9 response to nutrients. Science. 2005;310:1193–1196. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- Kærn M., Elston T.C., Blake W.J., Collins J.J. Stochasticity in gene expression: from theories to phenotypes. Nat. Rev. Genet. 2005;6:451–464. doi: 10.1038/nrg1615. [DOI] [PubMed] [Google Scholar]
- Kayo T., Allison D.B., Weindruch R., Prolla T.A. Influences of aging and caloric restriction on the transcriptional profile of skeletal muscle from rhesus monkeys. Proc. Natl. Acad. Sci. USA. 2001;98:5093–5098. doi: 10.1073/pnas.081061898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Kim K.M., Noh J.H., Yoon J.H., Abdelmohsen K., Gorospe M. Long noncoding RNAs in diseases of aging. Biochim. Biophys. Acta. 2016;1859:209–221. doi: 10.1016/j.bbagrm.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchman P.A., Kim S., Lai C.Y., Michal Jazwinski S. Interorganelle signaling is a determinant of longevity in Saccharomyces cerevisiae. Genetics. 1999;152:179–190. doi: 10.1093/genetics/152.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano H. Systems biology: a brief overview. Science. 2002;295:1662–1664. doi: 10.1126/science.1069492. [DOI] [PubMed] [Google Scholar]
- Kojima T., Kamei H., Aizu T., Arai Y., Takayama M., Nakazawa S., Ebihara Y., Inagaki H., Masui Y., Gondo Y. Association analysis between longevity in the Japanese population and polymorphic variants of genes involved in insulin and insulin-like growth factor 1 signaling pathways. Exp. Gerontol. 2004;39:1595–1598. doi: 10.1016/j.exger.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Kruegel U., Robison B., Dange T., Kahlert G., Delaney J.R., Kotireddy S., Tsuchiya M., Tsuchiyama S., Murakami C.J., Schleit J. Elevated proteasome capacity extends replicative lifespan in Saccharomyces cerevisiae. PLoS Genet. 2011;7:e1002253. doi: 10.1371/journal.pgen.1002253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J., Morimoto R.I. Proteostasis and longevity: when does aging really begin? F1000Prime Rep. 2014;6:7. doi: 10.12703/P6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert A.J., Wang B., Merry B.J. Exogenous insulin can reverse the effects of caloric restriction on mitochondria. Biochem. Biophys. Res. Commun. 2004;316:1196–1201. doi: 10.1016/j.bbrc.2004.03.005. [DOI] [PubMed] [Google Scholar]
- De Lange T. How telomeres solve the end-protection problem. Science. 2009;326:948–952. doi: 10.1126/science.1170633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.K., Klopp R.G., Weindruch R., Prolla T.A. Gene expression profile of aging and its retardation by caloric restriction. Science. 1999;285:1390–1393. doi: 10.1126/science.285.5432.1390. [DOI] [PubMed] [Google Scholar]
- de Lencastre A., Pincus Z., Zhou K., Kato M., Lee S.S., Slack F.J. MicroRNAs both promote and antagonize longevity in C. elegans. Curr. Biol. 2010;20:2159–2168. doi: 10.1016/j.cub.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesur I., Campbell J.L. The transcriptome of prematurely aging yeast cells is similar to that of telomerase-deficient cells. Mol. Biol. Cell. 2004;15:1297. doi: 10.1091/mbc.E03-10-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.C., Kane P.M. The yeast lysosome-like vacuole: endpoint and crossroads. Biochim. Biophys. Acta. 2009;1793:650–663. doi: 10.1016/j.bbamcr.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Liu Y., Liu M., Han J.D. Functional dissection of regulatory models using gene expression data of deletion mutants. PLoS Genet. 2013;9:e1003757. doi: 10.1371/journal.pgen.1003757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N., Bates D.J., An J., Terry D.A., Wang E. Up-regulation of key microRNAs, and inverse down-regulation of their predicted oxidative phosphorylation target genes, during aging in mouse brain. Neurobiol. Aging. 2011;32:944. doi: 10.1016/j.neurobiolaging.2009.04.020. [DOI] [PubMed] [Google Scholar]
- Li Y., Wang W.J., Cao H., Lu J., Wu C., Hu F.Y., Guo J., Zhao L., Yang F., Zhang Y.X. Genetic association of FOXO1A and FOXO3A with longevity trait in Han Chinese populations. Hum. Mol. Genet. 2009;18:4897–4904. doi: 10.1093/hmg/ddp459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K., Dorman J.B., Rodan A., Kenyon C. daf-16: an HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319–1322. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- Lin S.-J.J., Defossez P.-A.A., Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- Liu P., Acar M. The generational scalability of single-cell replicative aging. Sci. Adv. 2018;4:eaao4666. doi: 10.1126/sciadv.aao4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N., Landreh M., Cao K., Abe M., Hendriks G.J., Kennerdell J.R., Zhu Y., Wang L.S., Bonini N.M. The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila. Nature. 2012;482:519. doi: 10.1038/nature10810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P., Song R., Elison G.L., Peng W., Acar M. Noise reduction as an emergent property of single-cell aging. Nat. Commun. 2017;8:680. doi: 10.1038/s41467-017-00752-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Lluchh G., Hunt N., Jones B., Zhu M., Jamieson H., Hilmer S., Cascajo M.V., Allard J., Ingram D.K., Navas P. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc. Natl. Acad. Sci. USA. 2006;103:1768–1773. doi: 10.1073/pnas.0510452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Otín C., Blasco M.A., Partridge L., Serrano M., Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T., Pan Y., Kao S.Y., Li C., Kohane I., Chan J., Yankner B.A. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- Lund J., Tedesco P., Duke K., Wang J., Kim S.K., Johnson T.E. Transcriptional profile of aging in C. elegans. Curr. Biol. 2002;12:1566–1573. doi: 10.1016/s0960-9822(02)01146-6. [DOI] [PubMed] [Google Scholar]
- Macaulay I.C., Haerty W., Kumar P., Li Y.I., Hu T.X., Teng M.J., Goolam M., Saurat N., Coupland P., Shirley L.M. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat. Methods. 2015;12:519–522. doi: 10.1038/nmeth.3370. [DOI] [PubMed] [Google Scholar]
- Macaulay I.C., Ponting C.P., Voet T. Single-cell multiomics: multiple measurements from single cells. Trends Genet. 2017;33:155–168. doi: 10.1016/j.tig.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maegawa S., Hinkal G., Kim H.S., Shen L., Zhang L., Zhang J., Zhang N., Liang S., Donehower L.A., Issa J.P.J. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 2010;20:332–340. doi: 10.1101/gr.096826.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes O.C., An J., Sarojini H., Wang E. Murine microRNAs implicated in liver functions and aging process. Mech. Ageing Dev. 2008;129:534. doi: 10.1016/j.mad.2008.05.004. [DOI] [PubMed] [Google Scholar]
- de Magalhaes J.P., Curado J., Church G.M., de Magalhães J.P., Curado J., Church G.M. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics. 2009;25:875–881. doi: 10.1093/bioinformatics/btp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Managbanag J.R., Witten T.M., Bonchev D., Fox L.A., Tsuchiya M., Kennedy B.K., Kaeberlein M. Shortest-path network analysis is a useful approach toward identifying genetic determinants of longevity. PLoS One. 2008;3:e3802. doi: 10.1371/journal.pone.0003802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani R., St.Onge R.P., Hartman J.L., Giaever G., Roth F.P. Defining genetic interaction. Proc. Natl. Acad. Sci. USA. 2008;105:3461–3466. doi: 10.1073/pnas.0712255105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Jimenez C.P., Eling N., Chen H.-C., Vallejos C.A., Kolodziejczyk A.A., Connor F., Stojic L., Rayner T.F., Stubbington M.J.T., Teichmann S.A. Aging increases cell-to-cell transcriptional variability upon immune stimulation. Science. 2017;355:1433–1436. doi: 10.1126/science.aah4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll S.A., Murphy C.T., Zou S., Pletcher S.D., Chin C.S., Jan Y.N., Kenyon C., Bargmann C.I., Li H. Comparing genomic expression patterns across species identifies shared transcriptional profile in aging. Nat. Genet. 2004;36:197–204. doi: 10.1038/ng1291. [DOI] [PubMed] [Google Scholar]
- McCay C.M., Crowell M.F., Maynard L.A. The effect of retarded growth upon the length of life span and upon the ultimate body size. J. Nutr. 1935;10:63–79. [PubMed] [Google Scholar]
- McCormick M.A., Delaney J.R., Tsuchiya M., Tsuchiyama S., Shemorry A., Sim S., Chou A.C.Z., Ahmed U., Carr D., Murakami C.J. A Comprehensive analysis of replicative lifespan in 4,698 single-gene deletion strains uncovers conserved mechanisms of aging. Cell Metab. 2015;22:895–906. doi: 10.1016/j.cmet.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melov S., Hubbard A. Microarrays as a tool to investigate the biology of aging: a retrospective and a look to the future. Sci. Aging Knowledge Environ. 2004;2004:re7. doi: 10.1126/sageke.2004.42.re7. [DOI] [PubMed] [Google Scholar]
- Mitrophanov A.Y., Groisman E.A. Positive feedback in cellular control systems. Bioessays. 2008;30:542–555. doi: 10.1002/bies.20769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley J.F. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol. Biol. Cell. 2003;15:657–664. doi: 10.1091/mbc.E03-07-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Moskalev A.A., Shaposhnikov M.V., Plyusnina E.N., Zhavoronkov A., Budovsky A., Yanai H., Fraifeld V.E. The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res. Rev. 2013;12:661–684. doi: 10.1016/j.arr.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Mouchiroud L., Houtkooper R.H., Moullan N., Katsyuba E., Ryu D., Cantó C., Mottis A., Jo Y.S., Viswanathan M., Schoonjans K. XThe NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013;154:430–441. doi: 10.1016/j.cell.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S., Russell J.C., Carr D.T., Burgess J.D., Allen M., Serie D.J., Boehme K.L., Kauwe J.S., Naj A.C., Fardo D.W. Systems biology approach to late-onset Alzheimer’s disease genome-wide association study identifies novel candidate genes validated using brain expression data and Caenorhabditis elegans experiments. Alzheimers Dement. 2017;13:1133. doi: 10.1016/j.jalz.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan S.D., Yen K., Tissenbaum H.A. Converging pathways in lifespan regulation. Curr. Biol. 2009;19:R657–R666. doi: 10.1016/j.cub.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawy T. Single-cell sequencing. Nat. Methods. 2014;11:18. doi: 10.1038/nmeth.2771. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii A.I. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J. Proteomics. 2010;73:2092–2123. doi: 10.1016/j.jprot.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan R.J., Kubicek S., Schreiber S.L., Karlseder J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 2010;17:1218–1225. doi: 10.1038/nsmb.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S., Tyler J.K. Epigenetics and aging. Sci. Adv. 2016;2:e1600584. doi: 10.1126/sciadv.1600584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S., Postnikoff S.D., Chavez M., Tyler J.K. Impaired cohesion and homologous recombination during replicative aging in budding yeast. Sci. Adv. 2018;4:eaaq0236. doi: 10.1126/sciadv.aaq0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannese E. Morphological changes in nerve cells during normal aging. Brain Struct. Funct. 2011;216:85–89. doi: 10.1007/s00429-011-0308-y. [DOI] [PubMed] [Google Scholar]
- Pei W., Feyerabend T.B., Rössler J., Wang X., Postrach D., Busch K., Rode I., Klapproth K., Dietlein N., Quedenau C. Polylox barcoding reveals haematopoietic stem cell fates realized in vivo. Nature. 2017;548:456. doi: 10.1038/nature23653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W., Song R., Acar M. Noise reduction facilitated by dosage compensation in gene networks. Nat. Commun. 2016;7:12959. doi: 10.1038/ncomms12959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perié L., Hodgkin P.D., Naik S.H., Schumacher T.N., de Boer R.J., Duffy K.R. Determining lineage pathways from cellular barcoding experiments. Cell Rep. 2014;6:617–624. doi: 10.1016/j.celrep.2014.01.016. [DOI] [PubMed] [Google Scholar]
- Pigolotti S., Krishna S., Jensen M.H. Oscillation patterns in negative feedback loops. Proc. Natl. Acad. Sci. USA. 2007;104:6533–6537. doi: 10.1073/pnas.0610759104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletcher S.D., Macdonald S.J., Marguerie R., Certa U., Stearns S.C., Goldstein D.B., Partridge L. Genome-wide transcript profiles in aging and calorically restricted Drosophila melanogaster. Curr. Biol. 2002;12:712–723. doi: 10.1016/s0960-9822(02)00808-4. [DOI] [PubMed] [Google Scholar]
- Powers E.T., Morimoto R.I., Dillin A., Kelly J.W., Balch W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- Powers R.W., Kaeberlein M., Caldwell S.D., Kennedy B.K., Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006;20:174–184. doi: 10.1101/gad.1381406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raices M., Maruyama H., Dillin A., Kariseder J. Uncoupling of longevity and telomere length in C. elegans. PLoS Genet. 2005;1:295–301. doi: 10.1371/journal.pgen.0010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A., van Oudenaarden A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. 2008;135:216–226. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter J.A., Spacek D.V., Snyder M.P. High-throughput sequencing technologies. Mol. Cell. 2015;58:586–597. doi: 10.1016/j.molcel.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochette P.J., Brash D.E. Human telomeres are hypersensitive to UV-induced DNA damage and refractory to repair. PLoS Genet. 2010;6:e1000926. doi: 10.1371/journal.pgen.1000926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodwell G.E., Sonu R., Zahn J.M., Lund J., Wilhelmy J., Wang L., Xiao W., Mindrinos M., Crane E., Segal E. A transcriptional profile of aging in the human kidney. PLoS Biol. 2004;2:e427. doi: 10.1371/journal.pbio.0020427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotem A., Ram O., Shoresh N., Sperling R.A., Goren A., Weitz D.A., Bernstein B.E. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol. 2015;33:1165–1172. doi: 10.1038/nbt.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz E., Yang L., Su T., Morris D.R., McKnight G.S., Amieux P.S. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc. Natl. Acad. Sci. USA. 2009;106:13939–13944. doi: 10.1073/pnas.0907143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarnoski E.A., Liu P., Acar M. A high-throughput screen for yeast replicative lifespan identifies lifespan-extending compounds. Cell Rep. 2017;21:2639–2646. doi: 10.1016/j.celrep.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E., Shapira M., Regev A., Pe’er D., Botstein D., Koller D., Friedman N. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat. Genet. 2003;34:166–176. doi: 10.1038/ng1165. [DOI] [PubMed] [Google Scholar]
- Shore D. Cellular senescence: lessons from yeast for human aging? Curr. Biol. 1998;8:R192–R195. doi: 10.1016/s0960-9822(98)70122-8. [DOI] [PubMed] [Google Scholar]
- Siebold A.P., Banerjee R., Tie F., Kiss D.L., Moskowitz J., Harte P.J. Polycomb repressive complex 2 and trithorax modulate Drosophila longevity and stress resistance. Proc. Natl. Acad. Sci. USA. 2010;107:169–174. doi: 10.1073/pnas.0907739107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slieker R.C., van Iterson M., Luijk R., Beekman M., Zhernakova D.V., Moed M.H., Mei H., van Galen M., Deelen P., Bonder M.J. Age-related accrual of methylomic variability is linked to fundamental ageing mechanisms. Genome Biol. 2016;17:191. doi: 10.1186/s13059-016-1053-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Vikos T., Liu Z., Parsons C., Gorospe M., Ferrucci L., Gill T.M., Slack F.J. A serum miRNA profile of human longevity: findings from the Baltimore Longitudinal Study of Aging (BLSA) Aging (Albany NY) 2016;8:2971. doi: 10.18632/aging.101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song R., Liu P., Acar M. Network-dosage compensation topologies as recurrent network motifs in natural gene networks. BMC Syst. Biol. 2014;8:69. doi: 10.1186/1752-0509-8-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling S.R. Systems biology approaches to heart development and congenital heart disease. Cardiovasc. Res. 2011;91:269–278. doi: 10.1093/cvr/cvr126. [DOI] [PubMed] [Google Scholar]
- Stark C., Breitkreutz B.-J., Reguly T., Boucher L., Breitkreutz A., Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535–D539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen K.K., Kennedy B.K., Kaeberlein M. Measuring replicative life span in the budding yeast. J. Vis. Exp. 2009:e1209. doi: 10.3791/1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroustrup N., Ulmschneider B.E., Nash Z.M., López-Moyado I.F., Apfeld J., Fontana W. The Caenorhabditis elegans lifespan machine. Nat. Methods. 2013;10:665. doi: 10.1038/nmeth.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroustrup N., Anthony W.E., Nash Z.M., Gowda V., Gomez A., López-Moyado I.F., Apfeld J., Fontana W. The temporal scaling of Caenorhabditis elegans ageing. Nature. 2016;530:103–107. doi: 10.1038/nature16550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutphin G.L., Kaeberlein M. Measuring Caenorhabditis elegans life span on solid media. J. Vis. Exp. 2009:e1152. doi: 10.3791/1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swindell W.R. Accelerated failure time models provide a useful statistical framework for aging research. Exp. Gerontol. 2009;44:190–200. doi: 10.1016/j.exger.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi K., Kajiyama T., Kambara H. Quantitative analysis of gene expression in a single cell by qPCR. Nat. Methods. 2009;6:503–506. doi: 10.1038/nmeth.1338. [DOI] [PubMed] [Google Scholar]
- Thayer N.H., Leverich C.K., Fitzgibbon M.P., Nelson Z.W., Henderson K.A., Gafken P.R., Hsu J.J., Gottschling D.E. Identification of long-lived proteins retained in cells undergoing repeated asymmetric divisions. Proc. Natl. Acad. Sci. USA. 2014;111:14019–14026. doi: 10.1073/pnas.1416079111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thum T. Non-coding RNAs in ageing. Ageing Res. Rev. 2014;17:1–2. doi: 10.1016/j.arr.2014.08.001. [DOI] [PubMed] [Google Scholar]
- To T.-L., Maheshri N. Noise can induce bimodality in positive transcriptional feedback loops without bistability. Science. 2010;327:1142–1145. doi: 10.1126/science.1178962. [DOI] [PubMed] [Google Scholar]
- Virginija J., Laurent M., Johan A. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J. Exp. Biol. 2014;217:137–143. doi: 10.1242/jeb.090738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldera-Lupa D.M., Kalfalah F., Florea A.M., Sass S., Kruse F., Rieder V., Tigges J., Fritsche E., Krutmann J., Busch H. Proteome-wide analysis reveals an age-associated cellular phenotype of in situ aged human fibroblasts. Aging (Albany. NY) 2014;6:856–878. doi: 10.18632/aging.100698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther D.M., Kasturi P., Mann M., Hartl F.U., Walther D.M., Kasturi P., Zheng M., Pinkert S., Vecchi G., Ciryam P. Widespread proteome remodeling and aggregation in aging C. elegans. Cell. 2015;161:919–932. doi: 10.1016/j.cell.2015.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Proud C.G. The mTOR pathway in the control of protein synthesis. Physiology (Bethesda) 2006;21:362–369. doi: 10.1152/physiol.00024.2006. [DOI] [PubMed] [Google Scholar]
- Welle S., Brooks A.I., Delehanty J.M., Needler N., Bhatt K., Shah B., Thornton C.A. Skeletal muscle gene expression profiles in 20–29 year old and 65–71 year old women. Exp. Gerontol. 2004;39:369. doi: 10.1016/j.exger.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Wiley C.D., Velarde M.C., Lecot P., Liu S., Sarnoski E.A., Freund A., Shirakawa K., Lim H.W., Davis S.S., Ramanathan A. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016;23:303–314. doi: 10.1016/j.cmet.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood S.H., Craig T., Li Y., Merry B., de Magalhaes J.P. Whole transcriptome sequencing of the aging rat brain reveals dynamic RNA changes in the dark matter of the genome. Age. 2013;35:763. doi: 10.1007/s11357-012-9410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]