

ABSTRACT
Evolving evidence links maternal stress exposure to changes in placental DNA methylation of specific genes regulating placental function that may have implications for the programming of a host of chronic disorders. Few studies have implemented an epigenome-wide approach. Using the Infinium HumanMethylation450 BeadChip (450K), we investigated epigenome-wide placental DNA methylation in relation to maternal experiences of traumatic and non-traumatic stressors over her lifetime assessed using the Life Stressor Checklist-Revised (LSC-R) survey (n = 207). We found differential DNA methylation at epigenome-wide statistical significance (FDR = 0.05) for 112 CpGs. Additionally, we observed three clusters that exhibited differential methylation in response to high maternal lifetime stress. Enrichment analyses, conducted at an FDR = 0.20, revealed lysine degradation to be the most significant pathway associated with maternal lifetimes stress exposure. Targeted enrichment analyses of the three largest clusters of probes, identified using the gap statistic, were enriched for genes associated with endocytosis (i.e., SMAP1, ANKFY1), tight junctions (i.e., EPB41L4B), and metabolic pathways (i.e., INPP5E, EEF1B2). These pathways, also identified in the top 10 KEGG pathways associated with maternal lifetime stress exposure, play important roles in multiple physiological functions necessary for proper fetal development. Further, two genes were identified to exhibit multiple probes associated with maternal lifetime stress (i.e., ANKFY1, TM6SF1). The methylation status of the probes belonging to each cluster and/or genes exhibiting multiple hits, may play a role in the pathogenesis of adverse health outcomes in children born to mothers with increased lifetime stress exposure.
KEYWORDS: DNA methylation, maternal stress, placenta, PRISM cohort, metabolism, endocytosis
Introduction
Maternal psychosocial stress exposure occurring preconceptionally and during gestation is associated with increased risk for a range of maladaptive outcomes in offspring, including lower birth weight, smaller head circumference, asthma, and altered stress-related hormone levels (e.g., cortisol) [1,2]. These links are likely mediated by stress-induced physiological changes that affect the in utero environment. Cumulative lifetime stress, particularly exposure to traumatic events, is especially likely to lead to persistent psychophysiological alterations in the mother [3,4] that, in turn, influence perinatal outcomes, offspring neurobehavioral development, and other complex disorders [5–7]. Notably, trauma exposures are more highly prevalent among low-income and racial/ethnic minority populations [8]. Thus, consideration of lifetime exposures to traumatic events and other stressors in these groups may be particularly informative of health disparities. However, the role of epigenetics in these associations remain poorly defined.
Epigenetics is properly defined as heritable changes in gene expression that occur without changes to the underlying DNA sequence. There are many types of epigenetic changes which include DNA methylation (the most widely studied epigenetic regulator), histone modifications, microRNA, and prions. By utilizing an epigenetic approach, we are able to incorporate the role of the environment as a potential regulator of gene expression that likely plays a key part in fetal programming [9,10]. Animal [9,11,12] and human [13,14] data show that epigenetic modifications of disease risk begin in utero and can undergo stable regulation that mediates persistent changes in biologic and behavioral phenotypes over the lifespan. The demonstration of inter-individual variation in DNA methylation profiles in newborns as a result of maternal lifetime stress is fundamental to establishing a role for epigenetic variation in response to maternal stress in the programming of human diseases.
Studies examining associations between prenatal maternal stress or stress correlates (e.g., maternal psychological functioning, socioeconomic adversity) and DNA methylation have largely examined epigenetic changes in cord blood at birth or child blood DNA and have focused on a handful of candidate genes [e.g., the promoter region of the glucocorticoid (GC) receptor, brain-derived neurotrophic factor (BDNF), imprinted genes such as insulin-like growth factor 2 (IGF2), nuclear Receptor Subfamily 3 Group C Member 1 (NRC31), and guanine nucleotide-binding protein, alpha stimulating extra-large (GNASXL)] [15–20]. Growing evidence suggests that the placenta may constitute a model organ to explore the role of epigenetics in linking maternal exposures to developmental programming of children’s health [21]. The placenta is a significant regulator of maternal-fetal signaling throughout pregnancy influencing a range of physiological functions via release of cytokines, neurosteroids, and neurotransmitters into the fetal circulation [22]. Moreover, environmentally induced perturbations in the maternal milieu can result in changes in placental function and signaling that impact fetal development [23]. While a growing number of animal and human studies have explored associations between prenatal environmental exposures and DNA methylation changes in the placenta, including particulate air pollution, endocrine disrupting chemicals, tobacco smoke, and nutrition [24–27], research considering prenatal maternal stress in this regard remains sparse. A study in rodents linked maternal stress during pregnancy with gene-specific (HSD11B2 gene promoter) changes in placental DNA methylation [28]. Further, two small human studies examined associations between prenatal maternal perceived stress (n = 61) [29] and chronic war-related stressors (n = 24) [30] and differences in placental DNA methylation in glucocorticoid pathway genes.
The goal of this study is to examine associations between maternal cumulative lifetime exposures to traumatic and non-traumatic stressors and epigenome-wide methylation in the placenta among women enrolled in a longitudinal ethnically diverse pregnancy cohort.
Results
Sample characteristics
Maternal and child characteristics for the full sample (n = 238) and the analytic sample (n = 207) are presented in Table 1. The analytic sample is composed of those participants with complete stress exposure data and placental tissue. In the full sample, the average maternal age at enrollment was 30.2 years; the majority of the sample were racial/ethnic minorities (Black/Haitian, 40%; Hispanic, 19%; multi-racial, 8%); 26% reported having less than or at most a high school degree; 54% of the children were male, and nearly all children (91%) were born full term. There were no significant differences between the full sample and the analytic sample on maternal age, race/ethnicity, or education or child sex or gestational age.
Table 1.
Sample characteristics.
| Full Sample (n = 238) |
Analytic Sample (n = 207) |
|
|---|---|---|
| Categorical Characteristics | No. (%) | No. (%) |
| Education | ||
| ≤High School degree | 62 (26) | 56 (27) |
| Missing | 7 (2) | 3 (1) |
| Race | ||
| White | 78 (33) | 70 (34) |
| Black | 94 (40) | 90 (43) |
| Hispanic | 46 (19) | 35 (17) |
| Other/Mixed | 20 (8) | 13 (6) |
| Child sex | ||
| Male | 129 (54) | 109 (52) |
| Child born full term | ||
| Yesa | 217 (91) | 189 (91) |
| Continuous Characteristics | mean ± SDb | mean ± SD |
| Age at enrollment in years | 30.2 ± 5.6 | 30.2 ± 5.7 |
| Gestational age at delivery in weeks | 39.0 ± 1.8 | 39.0 ± 1.8 |
| Life Stressor Checklist-Revised, weighted score (LSCRwt) | – | 12.5 ± 12.3 |
aChildren born from 37 to 42 weeks of gestation; bSD, Standard Deviation
Epigenome-wide analysis
Figure 1 outlines the probe filtering steps taken prior to analysis. We tested the association between methylation levels of 365,193 CpGs and maternal stress exposures using semi-continuous linear regression models. In these regression models, population structures were accounted for through incorporating four principle components of CpGs near cis-SNPs [31]; cell type heterogeneity was accounted for by including Reference-Free Adjustment for Cell Type Composition (ReFACTor) components [32]. The resulting P values and their distribution on the genome are illustrated in Figure 2. The genome inflation factor of the resulting P value distribution is 1.158 (Supplemental Figure S1 and Table S1), suggesting there is no or minimum inflation of type-I errors. Using FDR correction (Benjamini-Hochberg) for 365,193 tests, we observed epigenome-wide statistically significant (FDR = 0.05) associations between maternal lifetime traumatic and non-traumatic stressors and placental DNA methylation for 112 CpGs in covariate-adjusted analyses; 616 CpGs were differentially methylated at the FDR = 0.20 level (Supplement Table S2). The 616 CpGs were enriched for enhancers (P value <0.0001) and CpG islands (P value <0.0001) but not shores, shelves, or promoters; enrichment for CpGs located in enhancers, islands, shores, shelves, or promoters was not observed at the FDR = 0.05 level. Further stability selection and permutation analysis suggests the CpG probe, cg22065664, to be the most significantly associated with maternal lifetime stress (Supplement Table S2). This probe does not annotate to a gene but it is located in an enhancer near RNA, U7 small nuclear 24 pseudogene (U7). Results from all models [i.e., semi-continuous, simple linear regression and generalized additive model (GAM)] can be found in the online data supplement (Supplement Tables S1 and S2). The magnitude of methylation change was calculated as the absolute differences between averages of covariate adjusted methylation M-values from the subjects whose stress scores were in the upper 5th percentile and that from the subjects whose stress scores were in the lower 5th percentile (Supplement Table S2). For 111 out of the 112 probes significant at the FDR = 0.05 level, the magnitude of change was at least 0.05 folds of the standard deviation.
Figure 1.
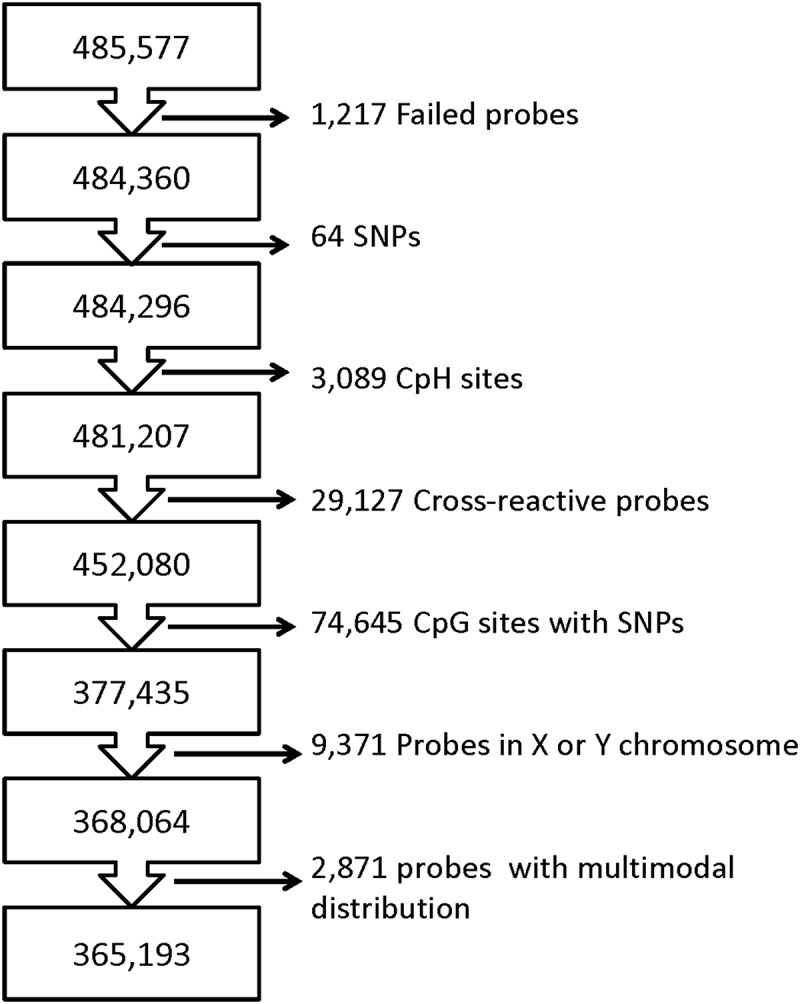
Flowchart of Probe Filtering. The flow chart outlines the filtering steps used prior to analysis.
Figure 2.
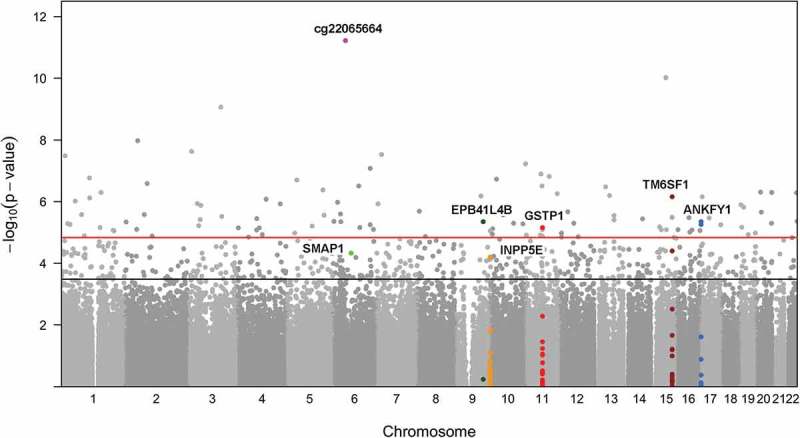
Epigenome-wide Association Results between Maternal Lifetime Stress and Methylation of 365,193 CpGs Measured in Placenta for the Semi-Continuous Model. Each point represents the genomic location (x-axis) and the – log10 P values based on the semi-continuous model for association test (y-axis) for a single probe. Horizontal lines depict the epigenome-wide significance level corresponding to FDR = 0.05 (red) and FDR = 0.20 (black). The set of probes/genes highlighted in the text are indicated by red triangles.
Epigenome-wide pathway enrichment analysis using an FDR level of 0.20
The enrichment analysis – based on the 616 probes that showed strong to moderate association with stress exposures (FDR = 0.20) and correspond to 459 genes – revealed 43 enriched pathways (Supplement Table S3). The top 10 significant KEGG biological pathways are listed in Table 2. The most significant KEGG pathway was Lysine Degradation (FDR = 3.04E-08), which includes 56 genes; 9 of those genes correspond to our set of significant probes. Other significant KEGG pathways encompass multiple KEGG classes including cellular processes, environmental information processing, human diseases, metabolism, and organismal systems and development.
Table 2.
Top 20 significant KEGG biological pathways affected by maternal lifetime stress, grouped by KEGG class.
| KEGG Class/Path ID | KEGG Pathway Name | n | DE | FDR |
|---|---|---|---|---|
| Cellular Processes | ||||
| hsa04530 | Tight junction | 162 | 9 | 2.11E-05 |
| hsa04144 | Endocytosis | 252 | 13 | 1.00E-06 |
| Environmental Information Processing | ||||
| hsa04151 | PI3K-Akt signaling pathway | 317 | 13 | 6.56E-06 |
| hsa04080 | Neuroactive ligand-receptor interaction | 257 | 11 | 5.67E-06 |
| Hsa04015 | Rap1 signaling pathway | 206 | 11 | 6.93E-06 |
| Human Diseases | ||||
| hsa05165 | Human papillomavirus infection | 302 | 11 | 0.0002 |
| hsa05200 | Pathways in cancer | 384 | 15 | 4.74E-06 |
| Metabolism | ||||
| hsa00310 | Lysine degradation | 56 | 9 | 3.04E-08 |
| hsa01100 | Metabolic pathways | 1190 | 20 | 6.41E-05 |
| Organismal Systems/Development | ||||
| hsa04914 | Progesterone-mediated oocyte maturation | 86 | 7 | 4.42E-05 |
Based on 616 significant probes (FDR = 0.20) using a background probe set based on the 365,193 probes used in association testing. Abbreviations: n, number of genes corresponding to the full set of probes used in our association test that belong to that pathway; DE, number of genes corresponding to our set of significant probes (FDR = 0.20) that belong to the KEGG pathway in question; FDR, false discovery rate using Benjamini-Hochberg correction.
Associations at FDR level equal to 0.20 with heatmap
For the 616 CpGs associated with maternal lifetime stress at the FDR level of 0.20 (Supplement Table S2), a heatmap was generated to visualize the placental DNA methylation pattern. The gap statistic [33] was used to identify the optimal number of clusters (Supplement Figure S2). Based on initial clustering into 10 groups (Supplement Figure S3), we selected the top 3 largest clusters for further analysis. We observed distinct patterns of demethylation among 2 clusters of probes and one cluster showed increased methylation in association with increased maternal lifetime stress (Figure 3). Pathway enrichment analyses conducted among each cluster of probes and their corresponding genes (top cluster = 112, middle cluster = 111, bottom cluster = 94) observed in the heatmap revealed further enrichment of 3 of our top 10 KEGG pathways: endocytosis (middle cluster), metabolic pathways (top cluster), and tight junction (bottom cluster) (Figure 3). The three most significant probes, mapping to genes associated with the three clusters, were cg13976799 [mapping to small ArfGAP 1 (SMAP1) as part of the endocytosis pathway], cg14024579 [mapping to inositol polyphosphate-5-phosphatase E (INPP5E) as part of the metabolic pathway], and cg13531977 [mapping to erythrocyte membrane protein band 4.1 like 4B (EPB41L4B) as part of the tight junction pathway]. All three probes were significant at the FDR = 0.10 level; cg13531977 (EPB41L4B) was significant at the FDR = 0.05 level. Generally speaking, CpG probes located in genes found in the endocytosis and metabolic pathways showed patterns of hypomethylation while probes associated with genes in the tight junction pathway were hypermethylated. Though there appeared to be additional regions of hyper- and hypomethylation among a small fraction of participants with low maternal lifetime stress, these regions were not associated with low maternal lifetime stress across all participants, nor were the patterns associated with covariates (i.e., maternal race/ethnicity, maternal age, child’s sex).
Figure 3.
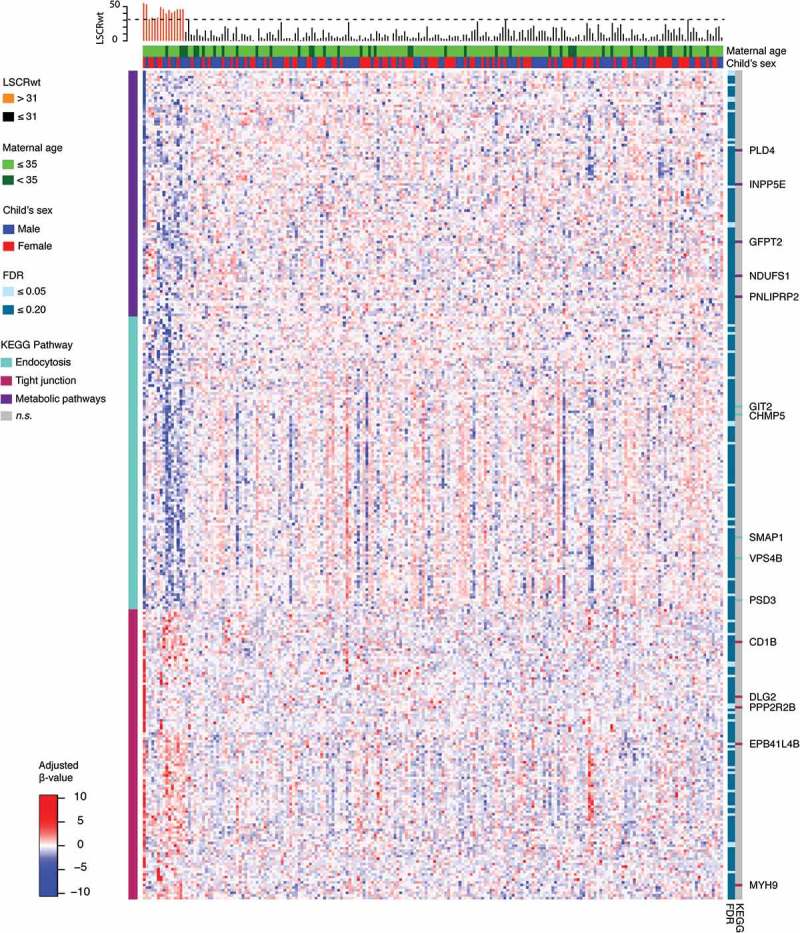
Heatmap of M-values for 130 Gene-annotated Probes Significantly Associated with Maternal Lifetime Stress at the FDR = 0.10 Level. Heatmap shows adjusted and normalized methylationβ-values with genetic background and cell heterogeneity factors removed, for the leading three clusters that consist of 317 probes of the 616 significant probes (FDR = 0.20). For each gene/probe (rows), the cluster membership is shown in the bar to the left and the significance level based on the semi-continuous regression model is shown to the immediate right. Pathway enrichment testing was conducted for the probes belonging to each cluster; genes belonging to enriched KEGG pathways (P <0.05) are highlighted in the bars to the far right. For each sample (column), the LSCRwt is plotted (top) and the child’s sex and maternal age is shown in the bars directly above the heatmap.
Identification of probes with ‘multiple’ hits
Of the 112 CpGs significant at the FDR = 0.05 level, 3 probes mapped to two genes where at least one other neighboring probe with an FDR value <0.05 was located. These include the following genes: transmembrane 6 superfamily member 1 (TM6SF1, probe cg03063639) and ankyrin repeat and FYVE domain containing 1 (ANKFY1, probes cg24084898 and 13,303,203). Locus-zoom-in plots of P value distributions along the genome were generated for TM6SF1 (Figure 4) and ANKFY1 (Figure 5). Within these genes, we observed spikes of multiple probes showing associations with higher maternal lifetime stress; the top most significant hits are highlighted in red (FDR = 0.10). Scatterplots showing the relationship between maternal lifetime stress and the methylation status of the significant probes listed above can be found in the supplement (Supplement Figure S4).
Figure 4.
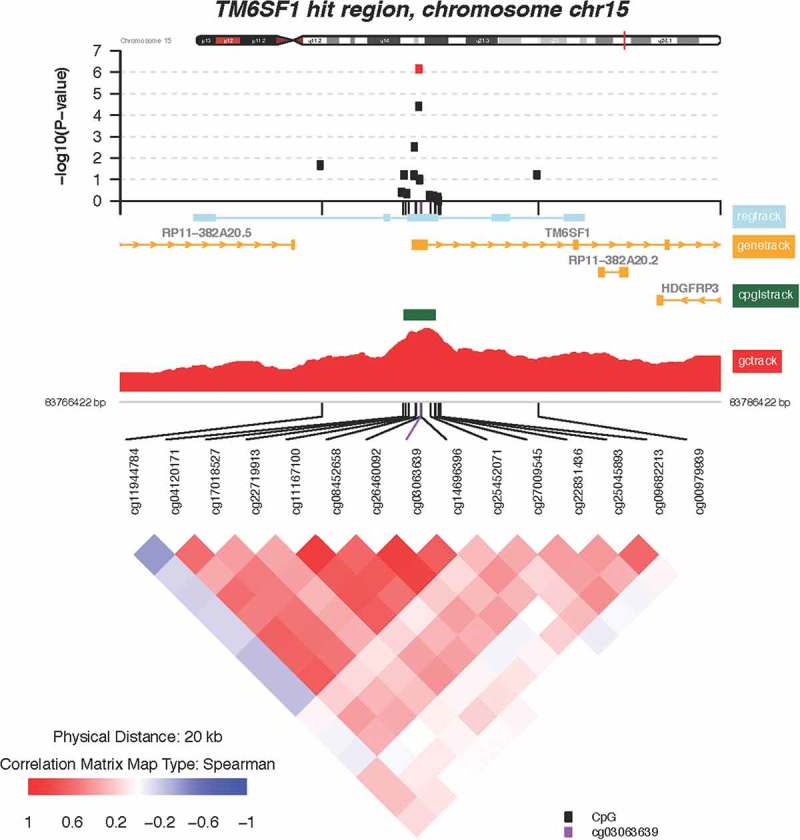
Zoomed-in plot featuring functional annotations for TM6SF1. Zoomed-in plot (top panel) shows the – log10 P values for the semi-continuous model within a 10 Kb neighbor of cg03063639/TM6SF1 on the genome. Functional annotations are derived from the ENCODE project and include regulatory, gene, CpG island, and GC content tracks. The lower panel shows pairwise correlations among the CpG sites selected. The plot was generated using the ‘CoMet’ packaged in R.
Figure 5.
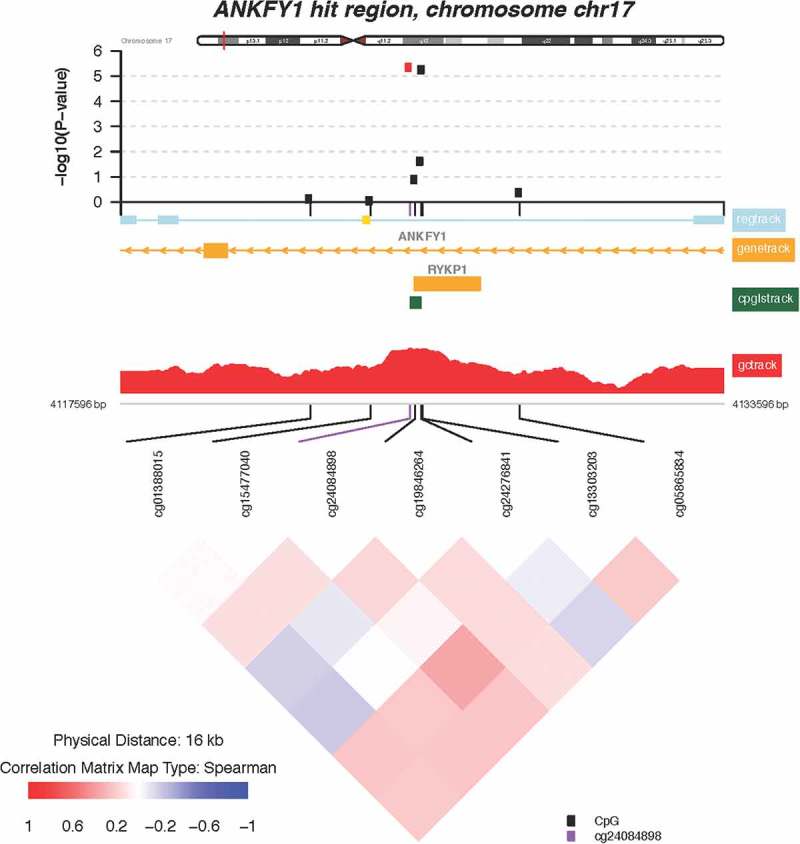
Zoomed-in plot featuring functional annotations for ANKFY1. Zoomed-in plot (top panel) shows the – log10 P values for the semi-continuous model within a 16 kb neighbor of cg13303203/ANKFY1 on the genome. Functional annotations are derived from the ENCODE project and include regulatory, gene, CpG island, and GC content tracks. The lower panel shows pairwise correlations among the CpG sites selected. The plot was generated using the ‘CoMet’ packaged in R.
Discussion
To our knowledge, this is the first human study to examine associations between cumulative lifetime maternal experiences of traumatic and non-traumatic stressors and epigenome-wide placental DNA methylation. We observed epigenome-wide statistically significant associations between increased maternal lifetime stress and placental methylation at 112 CpGs using a strict FDR of 0.05; a more liberal FDR of 0.20 resulted in an additional 504 significant CpGs, mapping to 459 genes.
Maternal lifetime exposure to stress was most significantly (FDR = 2.22E-6) associated with the placental methylation status of cg22065664. This finding was very robust, as it remained the top hit following our permutation analysis. While cg22065664 is not located within a gene, there are some characteristics that make it an interesting target for future studies. The probe is located in an intergenic enhancer and distal enhancers play a major role in the cell type-specific regulation of gene expression. While it is unclear whether the DNA methylation status of this upstream enhancer has an effect on gene expression, many intergenic enhancers have been associated with adverse birth outcomes (e.g., early-onset preeclampsia) and their methylation status has been shown to be retained in adult tissues [34,35]. Tissue-specific DNA methylation of intergenic enhancers are notably most variable in cancer cells [36] and global loss of DNA methylation in cancer cells has been shown to activate a large number of enhancer regions [37]. Whether maternal lifetime stress results in global hypomethylation, consequent activation of intergenic enhancers, and ultimately changes in gene expression in placenta requires further investigation. Further, the probe is located next to U7, a pseudogene. Although previously treated as ‘junk DNA’, it has been shown that some pseudogenes have important gene regulatory roles and are capable of being transcribed into RNA. How the methylation status of cg22065664 impacts the expression and function of U7 is unclear [38,39].
Enrichment analyses revealed the KEGG lysine degradation as the most significant pathway. In humans, lysine is an essential amino acid, and thus must come from food. Lysine is incorporated into collagen, one of the most important components of connective tissue; therefore, its supply is required during embryonic development and early childhood [40]. The main catabolic pathway for lysine is the Saccharopine (e-N-[L-Glutaryl-2]-L-lysine) pathway; this is a mitochondrial pathway leading to the formation of Acetyl-CoA (Acetyl-Coenzyme-A), which is oxidized and used for energy production [41]. Research suggests that decreases in lysine can lead to placental insufficiency, and reduced placental transport of lysine is observed in cases of intrauterine growth restriction [42,43]. Other significant KEGG pathways identified highlight the potential associations between maternal lifetime stress and cellular processes (tight junction and endocytosis), environmental information processing (PI3K-Akt signaling pathway, neuroactive ligand-receptor interaction, rap1 signaling pathway), human diseases (human papillomavirus infection, pathways in cancer), metabolism (metabolic pathways), and organismal systems and development (progesterone-mediated oocyte maturation). Interestingly, increased prenatal maternal perceived stress has been associated with multiple differentially methylated regions involved in the neuroactive ligand receptor interaction KEGG pathway in cord blood [44]. While the characterization of stress exposure and timing of exposure in the study by Trump and colleagues [44] differ from our study, these findings suggest some stressors may affect similar biological mechanisms (i.e., neuroactive ligand receptor interactions).
Targeted pathway enrichment analyses, conducted among the 3 largest clusters of probes observed in our heatmap, revealed further enrichment of three of our top 10 KEGG pathways: endocytosis, metabolic pathways, and tight junction. The most significant probe identified in the endocytosis cluster is located in the gene SMAP1. SMAP1 encodes for a type II membrane glycoprotein which plays an important role in the erythropoietic stimulatory activity of stromal cells whose deficiency can lead to embryonic lethality [45]. While there are no studies to support our finding that maternal lifetime stress has an impact on SMAP1 methylation and/or gene expression, other environmental exposures have been associated with SMAP1 gene expression, specifically metal mixture exposures [46]. The expression of INPP5E and EEF1B2, where two of the most significant probes identified in the metabolic cluster of our heatmap are mapped, has also been shown to be impacted by environmental exposures. INPP5E expression appears to be influenced by current smoking status [47], while EEF1B2 expression correlates with maternal tobacco smoke exposure during pregnancy [48]. Despite the fact that maternal lifetime stress is not a chemical exposure, both tobacco smoke and psychosocial stress have been shown to induce similar biological responses, such as oxidative stress [49–53]. Maternal lifetime stress was also associated with increased methylation of a probe located in the gene EPB41L4B. This gene is part of the tight junction cluster and plays important roles in cytoskeleton changes associated with steroid-induced cell differentiation and development [54,55]. This is likely due to its involvement in the glucocorticoid receptor (GR) pathway, a main target for the stress-related steroid hormone cortisol, where it serves as a GR stimulatory gene [56]. While glucocorticoids are vital to the development and survival of the fetus, exposure to maternal stress and excess glucocorticoids in utero can be harmful for fetal development and growth [57,58]. Given our results and the findings highlighted above, it is plausible that INPP5E, EEF1B2, and EPB41L4B play roles in environmentally-induced (via both physical and psychosocial exposures) placental changes that may lead to adverse perinatal outcomes.
The gene targets outlined below exhibited multiple probes whose methylation status is associated with increased maternal lifetime stress (Figures 4 and 5). TM6SF1 encodes a widely expressed lysosomal transmembrane protein which is vital for facilitating protein trafficking via organelle fusion [59]. Interestingly, lysosomal dysfunction has been associated with neonatal intestinal disorders preventing proper nutritional absorption [60]. Placental expression of TM6SF1 appears to be upregulated by environmental exposures such as prenatal alcohol consumption [61]. However, in some instances the regulation of TM6SF1, as a result of postnatal environmental insult (i.e., house dust mite exposure) does not appear to be associated with the methylation status of its promotor [62]. This raises the question of whether TM6SF1 expression is regulated by DNA methylation and thus warrants further investigation. The other gene to exhibit differential methylation of multiple cpg probes associated with maternal lifetime stress is ANKFY1, also known as Rabakyrin-5. ANKFY1 is widely expressed in adults and in fetal tissues during development and plays a major role in endocytosis [63]. Overexpression of ANKFY1 has been shown to increase macropinocytosis, the non-selective uptake of solute molecules and nutrients, while knockdown of ANKFY1 suppresses the process [63]. Macropinocytosis serves as a feeding mechanism and is particular relevant for nutrient-deprived environments [64] and nanoparticle uptake [65]. Nanoparticles can cross the placenta barrier [66] and cause fetotoxicity in offspring by disrupting placental function and structure [67]. Environmental exposure to nanoparticles can occur through many sources such as consumer spray products and cosmetics. [68,69] Thus, one could hypothesize that maternal lifetime stress exposure could result in increased uptake of nanoparticles at the maternal-fetal interface due to altered expression of ANKFY1 and its subsequent increase in macropinocytosis. Together, these findings underscore the potential importance of TM6SF1 and ANKFY1 in fetal and infant development consequent to maternal stress exposure.
This study has notable strengths. The study is comprised of a largely racial/ethnic minority population of pregnant women at increased risk for stress exposure and consequent adverse health outcomes among their offspring. Further, because PRISM is a longitudinal cohort, we will be able to examine the impact of these epigenetic changes on child developmental outcomes in future studies. Moreover, in the data analysis, we implemented a novel semi-continuous regression model to better detect associations with high stress exposures. When compared with traditional approaches, such as the ordinary linear regression and GAM (generalized additive model) on our data set, the semi-continuous regression model showed more favorable control of type-I errors and/or power (Supplementary Table S1).
There are also limitations worth noting. First, our study focused on maternal lifetime stress/trauma using a self-report survey of cumulative lifetime stress/trauma rather than examining specific timing of exposure. Despite many studies showing the negative impact chronic/lifetime stress has on health [3–7], using a measure of cumulative lifetime stress/trauma makes it challenging to identify the importance of timing, type, and duration of stress on placental DNA methylation. Second, our analyses were limited to placental samples. The effects of maternal lifetime stress/trauma on other easy-to-sample fetal tissues (e.g., umbilical cord blood) and maternal biological samples should be explored as differential effects across these various biosamples likely exist. [70,71] Third, the sample size prevented us from examining differences by sex and racial/ethnic groups; potential sex and racial/ethnic differences should be tested among larger diverse cohorts as they might provide insight into racial inequalities and sex differences in health. Fourth, it is important to replicate our EWAS and pathway analysis findings. To date, we have been unable to locate a cohort suitable for replication due to differences in tissue type and the operationalization (e.g., psychopathology, natural disaster) and timing (e.g., current vs. lifetime) of stressors. While we took precautions in avoiding erroneous associations in our pathway analysis by using missMethyl, follow-up studies would help confirm the importance of the identified pathways observed in this study. Fifth, the use of ReFACTor to adjust for cell-type heterogeneity in tissues other than whole blood needs further validation [72]. Lastly, it is important to explore the role of lifestyle and health related factors (e.g., diabetes, hypertension, obesity, smoking behaviors) in the associations between stress and placental DNA methylation. Cumulative lifetime trauma/stress can be related to maternal behavioral and health factors such as smoking and obesity, which can also influence methylation profiles. However, in this context these indicators may be considered as mediators (or in the pathway) so further consideration herein was beyond the scope of the intended analyses. It is also worth noting that in our sample of mothers, maternal smoking status (for which only 7% of participants reported smoking during pregnancy) was not associated with increased maternal exposure to traumatic and non-traumatic events (Fisher’s Exact P value = 0.09); this finding suggests smoking may not be a major confounder in our study. While we are unable to sufficiently explore these relationships given our sample size, larger studies could pursue these issues more definitively. Future studies should also explore the impact of the identified methylation marks on relevant birth and perinatal outcomes.
In summary, the placenta’s role in nutrient transfer and organ development is important for the proper development of the fetus. Epigenetic modification of the placental epigenome may provide a possible connection between maternal stress exposure and alteration in gene expression that might set in motion fetal programming signatures that shape early childhood conditions. Our study found that maternal lifetime stress/trauma is associated with changes in the epigenome-wide methylation status of multiple loci in genes that have key roles in cellular processes and metabolism. Both of these processes broadly speaking, and more specifically the genes for which the significant loci were mapped, appear to be important for early embryo development and metabolism. These findings may provide insight into possible biological mechanisms linking maternal stress exposure and adverse perinatal/child health outcomes.
Methods
Study population
Participants included women enrolled in the PRogramming of Intergenerational Stress Mechanisms (PRISM) study, a prospective pregnancy cohort of mother-child pairs originally designed to examine how perinatal stress influences child development as well as examining the role of the placenta in environmental programming. In brief, 238 women were recruited from prenatal clinics during the first or second trimester (27 ± 8 weeks of gestation) from the Beth Israel Deaconess Medical Center (BIDMC) from March 2011 to August 2012 and from the Icahn School of Medicine at Mount Sinai from November 2012 to August 2014. Eligibility criteria included: (i) English- or Spanish-speaking; (ii) age ≥18 years at enrollment; (iii) singleton pregnancy; and (iv) willingness to provide placenta samples at birth. Women who endorsed drinking more than seven alcoholic drinks per week prior to pregnancy recognition or any alcohol after pregnancy recognition and children born with congenital abnormalities that would impact neurodevelopment were excluded. Procedures were approved by the institutional review boards at the Brigham and Women’s Hospital (BWH) and the Icahn School of Medicine at Mount Sinai. BIDMC relied on BWH for review and oversight of the protocol. Written informed consent was obtained from all participants in their primary language.
Maternal lifetime stress
Maternal lifetime exposure to stress and potentially traumatic events was assessed in the second trimester using the validated 30-item Life Stressor Checklist-Revised (LSC-R) [73]. The LSC-R includes experiences particularly relevant to women (e.g., sexual assault, interpersonal violence) and questions reflecting the participant’s assessment of the severity of the negative impact [ranging from 1 (not at all) to 5 (extremely)] of each endorsed event during the past 12 months. The LSC-R has established test-retest reliability and validity in diverse populations [73,74]. A weighted score of all endorsed stressful life events (traumatic and non-traumatic) that considered the self-reported negative impact of each event was computed (weighted Life Stressor Checklist Revised, LSCRwt); scores could range from 0–150 (range in present sample = 0–96), with higher scores indicating greater exposure to and impact of exposure to stressful and traumatic events.
Placenta specimen collection and processing
Placentas were sampled per a published protocol [75] immediately after birth in 238 women. Each of four samples (~1 cm3) was taken on the fetal side ~4 cm from the cord insertion site and ~1–1.5 cm below the fetal membrane to avoid membrane contamination. The decidua and fetal membranes were removed, the sample was rinsed in a cold PBS bath, cut into smaller pieces (~0.1 cm3), and placed into 30 cc saline solution and placed at −80°C until DNA extraction.
DNA extraction and bisulfite treatment
DNA was isolated using the Gentra Puregene kit (Qiagen, Germantown, MD) and quantified using an Implen Nanophotometer Pearl (Westlake Village, CA). The origin of placental tissue from the fetal side of the organ was confirmed by the near-perfect agreement of placenta and cord blood samples in 64 genotyping probes used for identity verification. Across these 64 genotyping probes, the range of the Pearson correlation coefficients between cord blood and placenta within each participant was 0.99 to 1.00.
DNA (500 ng) was bisulfite-treated using the EZ DNA Methylation-Gold™ Kit (Zymo Research, Orange, CA) analyzed by the Infinium Methylation Assay. Samples were arranged on chips and plates with a stratified randomization followed by statistical checks for balance on birthweight z-score, gestational age, sex, and city of collection.
DNA methylation profiling
Illumina infinium humanmethylation450 array
HumanMethylation450 BeadChips (Illumina Inc., San Diego, CA, USA) were used to interrogate 485,577 DNA methylation sites and to generate a measure of the methylation proportion at each site. Specifically, single-CpG-site methylation values were quantified after bisulfite conversion using fluorescence measures at site-specific probes, which was computed as the methylated intensity divided by the sum of both the methylated and unmethylated intensities. Methylation values ranged from zero (for a fully unmethylated CpG site) to one (for a fully methylated CpG site). Extensive efforts were made to avoid confounding by batch effects by use of a stratified randomization of samples to chips and plates as well as a randomization of position within chip. Further, all samples were run consecutively at the Partners HealthCare Translational Genomics Core Labs.
Quality control, preprocessing and normalization
The presence of failed arrays or outliers was checked with detection P values (all samples passed with detection P values <0.05 in >99% of probes), through visualization of principal components analysis (PCA) and the clustering of PCs among the 10,000 highest varying probes, and evidence of sample swapping or contamination in 65 fingerprinting SNP probes (one sample was excluded due to maternal contamination). PCA plots and the analysis of five pairs of technical replicates that were arranged across chips and plates were further used to evaluate precision (of raw data and after pre-processing) and monitor for large batch effects. Sample identity was checked via imputed sex and agreement of genotype with paired tissues. Data were preprocessed using background correction [76], dye bias, and probe type adjustment [77]. BMIQ (Beta Mixture Quantile dilation) intra-sample normalization was applied to all probes to adjust the methylation values of Infinium II probes into a statistical distribution characteristic of Infinium I probes. The BMIQ function in the R package wateRmelon was used [78].
Probe filtering
Prediction models were not performed on all 485,577 methylation probes and the probe filtering steps used in PRISM has previously been reported [71]. In brief, probe filtering included the removal of 1217 probes with detection P value >0.05 in >1% of the samples, 64 SNPs, 3089 CpH sites, 29,127 cross-reactive probes, 74,645 CpG sites with variants within 10 base pairs common to Asian, American, African, and European populations (frequency >1%), and 9323 and 48 probes on chromosomes X and Y, respectively. An additional 2865 CpG sites with a multimodal distribution of methylation (Dip test’s P value <0.05) were excluded (Figure 1).
Statistical analysis
Placenta tissue, LSC-R data, and covariates were available for 207 participants.
ReFACTor analysis for cell heterogeneity adjustment
The cell heterogeneity adjustment was conducted using a Reference-Free Adjustment for Cell-Type composition (ReFACTor) analysis. ReFACTor is an unsupervised method for the correction of cell-type heterogeneity in epigenome-wide association studies (EWAS), which is based on a variant of principal component analysis (PCA) [32]. The ReFACTor algorithm requires a CpG sites-by-samples matrix of beta-normalized methylation levels, a number of assumed cell types (k) and a number of CpG sites to use for computing the ReFACTor components (t). As suggested by the authors of this method (https://github.com/cozygene/refactor), before running ReFACTor a subset of probes are defined that exclude problematic probes, as well as consistently methylated probes and consistently unmethylated probes. Specifically, filtered-out probes included 1,217 probes with a detection P value >0.05 in >1% of the samples, 64 genotyping SNPs, 3,089 CpH sites, 29,127 cross-reactive probes [2], 74,645 CpG sites with variants within 10 base pairs that are common to Asian, American, African and European populations with a frequency of >1% [3], 9,371 probes on chromosome X or Y, 99,878 and 141,322 CpG sites with mean methylation value >0.8 or <0.2, respectively. The resulting dataset for the ReFACTor analysis included 126,864 probes (i.e., reduced number of probes due to the additional filtering required by ReFACTor) and 238 unique samples. Guidelines for parameter selection guided us to set k and t equal to 3 and 500, respectively. Since the authors of this method observed that adjusting the methylation levels, before running ReFACTor, for genome-wide affecting factors, such as gender and global ancestry, improves the performance of ReFACTor, we decided to pass to ReFACTor also a samples-by-covariates matrix of covariates including gender and principal components for ancestry. The software output is a matrix with the first k ReFACTor components for each individual which can be included in downstream analyses as covariates. We noted from a scatterplot matrix that these three derived variables were uncorrelated with each other (as we would expect).
EPISTRUCTURE analysis for ancestry adjustment
The adjustment for population structure was conducted using principal components calculated on a subset of 4,905 CpG sites pre-screened to be highly correlated with local single nucleotide polymorphisms (cis-SNPs) [31]. Scatter plots showed the first four principal components of the between-probe relations of these CpGs capturing genetic information separated samples by the maternal self-reported race/ethnicity and thus were included in further association testing to avoid confounding due to population stratification. The four PCs included in association testing were not correlated (Pearson Correlation Coefficient P values ranged from 0.60 to 0.87)
Epigenome-wide association (EWAS) analysis
Association testing was performed using CpG methylation β-values based on data from 207 participants, excluding 1 individual with a very high stress score (LSCRwt >80) and 30 individuals with missing data on maternal lifetime stress. For each probe, outliers were evaluated on the methylation M-value scale. Methylation β-values were first transformed into M-values through logit transformation. Any M-value above or below the interquartile range (defined as having values outside 3 times the interquartile range below Q1 or above Q3) was truncated to its nearest range value prior to association testing. Evidence supports the notion that low to moderate levels of stress are normative (i.e., commonly encountered in the population) and thus, when stress exposure is within this range it is likely to have limited impact on epigenetic processes; however, when stress exposure and/or the perceived impact of the stressor is high or more severe, there is more likely to be biological consequences at the cell level [79,80]. This hypothesis is supported by the empirical distributions of LSCRwt scores and M-values observed in our study. As the examples in the supplement illustrate (Figure S4), the scatterplots of LSCRwt vs. the M-value of individual CpG probes across all subjects consistently shows a mass of data points at the low LSCRwt end (i.e., weaker effect is observed between LSCRwt and methylation). This observation motivated us to introduce two variables ( to better capture the information in the LSCRwt distribution above the threshold and at the same time to prevent the variability among small LSCRwt scores to dilute the signals in statistical tests. Ultimately, if there is a dosage effect in the high stress spectrum, the proposed semi-continuous model will better detect the effect than a model only using . Specifically, we define these variables as follows:
In the above definition, is a binary variable indicating whether the participant was exposed to high or low stress; while is a truncated continuous variable measuring the effect of extreme stress exposures among the high stress group.
We then used the following model to test for associations between methylation and stress, where represents the methylation M-value of individual i (1,…,207) for probe j (1,…,365,193):
Where the k = 1,…,8 covariates () included: child’s sex, 4 PCs to adjust for population stratification [31], and 3 PCs to adjust for cell heterogeneity [32].
Statistical testing was performed jointly on and using the ANOVA F-statistic, comparing the model stated above to a null model containing neither stress related term. We compared results from our semi-continuous model with those based on a simple linear model and a generalized additive model based on fitting a smoothed spline to the data as implemented in R the ‘gam’ package (Supplement Table S1). For the semi-continuous model we considered many values of α corresponding to percentile increments from the 80th to the 96st percentile of LSCRwt. For further investigations, we selected the semi-continuous model where α = 31 corresponding to the 92nd percentile of LSCRwt. This model shows low genomic inflation (λGC = 1.16, Supplement Figure S1), maintains an adequate number of samples above the threshold (n = 15), and demonstrates enhanced power to detect associations as compared to either the simple linear or the generalized additive model. The –log10 (ANOVA P values) from the semi-continuous linear regression for 365,193 CpGs across the genome were plotted (Figure 2). Information about the direction and strength of association for the binary and truncated stress terms was obtained using a multiple-partial F-test. To further substantiate our findings, we also conducted a stability selection [81] based permutation test to identify methylation probes significantly associated with LSCRwt scores (see Supplement Text). FDR adjusted P values from the ANOVA (FDRANOVA), as well as beta-coefficients for each stress term (βt and βb) and their associated P values (Pt and Pb), are included in Supplementary Table S2.
Heatmap visualization of methylation data
Methylation beta values for the 616 probes significant at FDR = 0.20 were first adjusted for genetic and cell background and then normalized before hierarchical clustering was performed (see Supplement Text). The number of clusters (K = 10) was selected based on gap statistic (Supplemental Figures S2 and S3) [33]. Visual inspection for patterns of methylation in association with maternal lifetime stress (LSCRwt) was conducted. Figure 3 presents a heatmap based on the top three largest clusters (K = 3).
Epigenome-wide enrichment analysis
We performed gene ontology (GO) and KEGG pathway analyses for the set of 616 probes significant at the FDR = 0.20 level (Supplementary Table S2) and for the each of the probe sets corresponding to the top 3 clusters shown in Figure 3. The epigenome-wide enrichment analyses for GO Terms and KEGG Pathways was conducted using the Bioconductor ‘missMethyl’ package for R [41]. Enrichment analyses were conducted based on a background probe set comprising the full set of 365,193 probes used in EWAS analysis. We utilized the modified hypergeometric test option of the ‘gometh’ function to account for selection bias due to the increased probability of finding an association for genes with a larger numbers of probes [41,82]. All significant KEGG biological pathways at an FDR = 0.05 can be found in the supplementary table S3. Figure 3 shows adjusted methylation values for the top 3 clusters, where cluster membership is shown on the left bar; the probes/genes belonging to each of the enriched pathways are highlighted in the bars to the right of the heatmap. We further performed pathway, CpG region, and promoter enrichment analysis using the two-sided doubling mid P value hypergeometric test [83] for probes selected for significance at the FDR = 0.05 (n = 112), FDR = 0.10 (n = 267), and FDR = 0.20 (n = 616) levels.
Funding Statement
This work was supported by the National Heart, Lung, and Blood Institute under grants R01HL095606 and R01HL114396; the National Institute of Environmental Health Sciences (NIEHS) under grants R00ES024116, R00ES023450, P30ES023515, and R01ES013744; the National Cancer Institute under grant U24CA160034; and the National Institute of General Medical Sciences under grants R01GM108711 and R01GM082802. Biobanking infrastructure was supported by the Mount Sinai Health System Clinical Translational Science Award from the National Center for Advancing Translational Sciences under grant UL1 TR001433.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- 1.Su Q, Zhang H, Zhang Y, et al. Maternal stress in gestation: birth outcomes and stress-related hormone response of the neonates. Pediatr Neonatol. 2015;56:376–381. [DOI] [PubMed] [Google Scholar]
- 2.Lee A, Mathilda Chiu YH, Rosa MJ, et al. Prenatal and postnatal stress and asthma in children: temporal- and sex-specific associations. J Allergy Clin Immunol. 2016;138(3):740–747 e743.PMC5011027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Christopher M. A broader view of trauma: A biopsychosocial-evolutionary view of the role of the traumatic stress response in the emergence of pathology and/or growth. Clin Psychol Rev. 2004;4(1):75–98. [DOI] [PubMed] [Google Scholar]
- 4.Tiedje LB. Psychosocial pathways to prematurity: changing our thinking toward a lifecourse and community approach. J Obstet Gynecol Neonatal Nurs. 2003;32(5):650–658. [DOI] [PubMed] [Google Scholar]
- 5.O’Donnell K, O’Connor TG, Glover V. Prenatal stress and neurodevelopment of the child: focus on the hpa axis and role of the placenta. Dev Neurosci. 2009;31(4):285–292. [DOI] [PubMed] [Google Scholar]
- 6.Henrichs J, Schenk JJ, Roza SJ, et al. Maternal psychological distress and fetal growth trajectories: the generation r study. Psychological medicine. 2010;40(4):633–643. [DOI] [PubMed] [Google Scholar]
- 7.Lane RH. Fetal programming, epigenetics, and adult onset disease. Clinics in perinatology. 2014;41(4):815–831. [DOI] [PubMed] [Google Scholar]
- 8.Hatch SL, Dohrenwend BP. Distribution of traumatic and other stressful life events by race/ethnicity, gender, ses and age: A review of the research. Am J Community Psychol. 2007;40(3–4):313–332. [DOI] [PubMed] [Google Scholar]
- 9.Santos F, Dean W. Epigenetic reprogramming during early development in mammals. Reproduction. 2004;127:643–651. [DOI] [PubMed] [Google Scholar]
- 10.Wadhwa PD, Buss C, Entringer S, et al. Developmental origins of health and disease: brief history of the approach and current focus on epigentic mechanisms. Semin Reprod Med. 2009;27:358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dolinoy DC, Weidman JR, Waterland RA, et al. Maternal geinstein alters coat color and protects avy mouse offspring from obesity by modifying the fetal epigenome. Environ Health Perspect. 2006;114:567–572.PMC1440782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meaney MJ. Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu Rev Neurosci. 2001;24:1161–1192. [DOI] [PubMed] [Google Scholar]
- 13.Oberlander TF, Weinberg J, Papsdorf M, et al. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (nr3c1) and infant cortisol stress responses. Epigenetics. 2008;3:97–106. [DOI] [PubMed] [Google Scholar]
- 14.Heijmans BT, Tobi EW, Stein AD, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Porceedings Natl Acad Sci. 2008;105:17046–17049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Appleton AA, Armstrong DA, Lesseaur C, et al. Patterning in placental 11-b hydroxysteroid dehydrogenase methylation according to prenatal socioeconomic adversity. PLoS One. 2013;8:e74691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vangeel EB, Izzi B, Hompes T, et al. DNA methylation in imprinted genes igf2 and gnasxl is associated with prenatal maternal stress. Psychoneuroendocrinology. 2015;61:16. [DOI] [PubMed] [Google Scholar]
- 17.Braithwaite EC, Kundakovic M, Ramchandani PG, et al. Maternal prenatal depressive symptoms predict infant nr3c1 1f and bdnf iv DNA methylation. Epigenetics. 2015;10(5):408–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palma-Gudiel H, Cordova-Palomera A, Eixarch E, et al. Maternal psychosocial stress during pregnancy alters the epigenetic signature of the glucocorticoid receptor gene promoter in their offspring: A meta-analysis. Epigenetics. 2015;10(10):893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kertes DA, Bhatt SS, Kamin HS, et al. Bndf methylation in mothers and newborns is associated with maternal exposure to war trauma. Clin Epigenetics. 2017;9(68):PMC5493129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mulligan CJ, D’Errico NC, Stees J, et al. Methylation changes at nr3c1 in newborns associate with maternal prenatal stress exposure and newborn birth weight. Epigenetics. 2012;7(8):853–857.PMC3427280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nugent BM, Bale TL. The omniscient placenta: metabolic and epigenetic regulation of fetal programming. Front Neuroendocrinol. 2015;39:28–37.PMC4681645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burton GJ, Fowden AL, Thornburg KL. Placental origins of chronic disease. Physiol Rev. 2016;96(4):1509–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Myllynen P, Pasanen M, Pelkonen O. Human placenta: A human organ for developmental toxicology research and biomonitoring. Placenta. 2005;26(5):361–371. [DOI] [PubMed] [Google Scholar]
- 24.Janssen BG, Godderis L, Pieters N, et al. Placental DNA hypomethylation in association with particulate air pollution in early life. Part Fibre Toxicol. 2013;10(22):PMC3686623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suter M, Ma J, Harris A, et al. Maternal tobacco use modestly alters correlated epigenome-wide placental DNA methylation and gene expression. Epigenetics. 2011;6(11):1284–1294.PMC3242811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallou-Kabani C, Gabory A, Tost J, et al. Sex- and diet-specific changes of imprinted gene expression and DNA methylation in mouse placenta under a high-fat diet. PLoS One. 2010;5(12):e14398.PMC3006175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hogg K, Price EM, Hanna CW, et al. Prenatal and perinatal environmental influences on the human fetal and placental epigenome. Clin Pharmacol Ther. 2012;92(6):716–726. [DOI] [PubMed] [Google Scholar]
- 28.Jensen Pena C, Monk C, Champagne FA. Epigenetic effects of prenatal stress on 11beta-hydroxysteroid dehydrogenase-2 in the placenta and fetal brain. PLoS One. 2012;7(6):e39791.PMC3383683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monk C, Feng T, Lee S, et al. Distress during pregnancy: epigenetic regulation of placenta glucocorticoid-related genes and fetal neurobehavior. Am J Psychiatry. 2016;173(7):705–713.PMC5026410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kertes DA, Kamin HS, Hughes DA, et al. Prenatal maternal stress predicts methylation of genes regulating the hypothalamic-pituitary-adrenocortical system in mothers and newborns in the democratic republic of congo. Child Dev. 2016;87(1):61–72.PMC4733886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rahmani E, Shenhav L, Schweiger R, et al. Genome-wide methylation data mirror ancestry information. Epigenetics Chromatin. 2017;10(1). PMC5267476 DOI: 10.1186/s13072-016-0108-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rahmani E, Zaitlen N, Baran Y, et al. Sparse pca corrects for cell type heterogeneity in epigenome-wide association studies. Nat Methods. 2016;13(5):443–445.PMC5548182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tibshirani R, Walther G, Hastie T. Estimating the number of clusters in a data set via the gap statistic. J Royal Stat Soc. 2001;63:411–423. [Google Scholar]
- 34.Blair JD, Yuen RK, Lim BK, et al. Widespread DNA hypomethylation at gene enhancer regions in placentas associated with early-onset pre-eclampsia. Mol Hum Reprod. 2013;19(10):697–708.PMC3779005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hon GC, Rajagopal N, Shen Y, et al. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat Genet. 2013;45(10):1198–1206.PMC4095776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hansen KD, Timp W, Bravo HC, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43(8):768–775.PMC3145050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blattler A, Yao L, Witt H, et al. Global loss of DNA methylation uncovers intronic enhancers in genes showing expression changes. Genome Biol. 2014;15(9):469.PMC4203885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tutar Y. Pseudogenes. Comp Funct Genomics. 2012;2012:424526.PMC3352212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soldati D, Schumperli D. Structures of four human pseudogenes for u7 small nuclear rna. Gene. 1990;95(2):305–306. [DOI] [PubMed] [Google Scholar]
- 40.Paik MJ, Lee HJ, Kim KR. Simultaneous retention index analysis of urinary amino acids and carboxylic acids for graphic recognition of abnormal state. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;821(1):94–104. [DOI] [PubMed] [Google Scholar]
- 41.Kanehisa M, Sato Y, Kawashima M, et al. Kegg as a reference resource for gene and protein annotation. Nucleic acids research. 2016;44(D1):D457-462;4702792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jansson T, Scholtbach V, Powell TL. Placental transport of leucine and lysine is reduced in intrauterine growth restriction. Pediatric research. 1998;44(4):532–537. [DOI] [PubMed] [Google Scholar]
- 43.Gabbe SG. Obstetrics: normal and problem pregnancies. 6th ed. Philadelphia: Elsevier/Saunders; 2012. [Google Scholar]
- 44.Trump S, Bieg M, Gu Z, et al. Prenatal maternal stress and wheeze in children: novel insights into epigenetic regulation. Scientific reports. 2016;6:28616; 4923849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sumiyoshi M, Masuda N, Tanuma N, et al. Mice doubly-deficient in the arf gaps smap1 and smap2 exhibit embryonic lethality. FEBS Lett. 2015;589(19Pt B):2754–2762. [DOI] [PubMed] [Google Scholar]
- 46.Martinez-Pacheco M, Hidalgo-Miranda A, Romero-Cordoba S, et al. Mrna and mirna expression patterns associated to pathways linked to metal mixture health effects. Gene. 2014;533(2):508–514. [DOI] [PubMed] [Google Scholar]
- 47.Ligthart S, Steenaard RV, Peters MJ, et al. Tobacco smoking is associated with DNA methylation of diabetes susceptibility genes. Diabetologia. 2016;59(5):998–1006.PMC4826423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Filis P, Nagrath N, Fraser M, et al. Maternal smoking dysregulates protein expression in second trimester human fetal livers in a sex-specific manner. J Clin Endocrinol Metab. 2015;100(6):E861–870.PMC4533306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Colaianna M, Schiavone S, Zotti M, et al. Neuroendocrine profile in a rat model of psychosocial stress: relation to oxidative stress. Antioxidants & redox signaling. 2013;18(12):1385–1399;3603501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jorgensen A. Oxidatively generated DNA/rna damage in psychological stress states. Danish medical journal. 2013;60(7):B4685. [PubMed] [Google Scholar]
- 51.Aschbacher K, O’Donovan A, Wolkowitz OM, et al. Good stress, bad stress and oxidative stress: insights from anticipatory cortisol reactivity. Psychoneuroendocrinology. 2013;38(9):1698–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gidron Y, Russ K, Tissarchondou H, et al. The relation between psychological factors and DNA-damage: A critical review. Biological psychology. 2006;72(3):291–304. [DOI] [PubMed] [Google Scholar]
- 53.Noakes PS, Thomas R, Lane C, et al. Association of maternal smoking with increased infant oxidative stress at 3 months of age. Thorax. 2007;62(8):714–717.PMC2117280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chauhan S, Pandey R, Way JF, et al. Androgen regulation of the human ferm domain encoding gene ehm2 in a cell model of steroid-induced differentiation. Biochem Biophys Res Commun. 2003;310(2):421–432. PMC2740477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eckert JJ, Fleming TP. Tight junction biogenesis during early development. Biochim Biophys Acta. 2008;1778(3):717–728. [DOI] [PubMed] [Google Scholar]
- 56.Wang JC, Derynck MK, Nonaka DF, et al. Chromatin immunoprecipitation (chip) scanning identifies primary glucocorticoid receptor target genes. Proc Natl Acad Sci U S A. 2004;101(44):15603–15608.PMC524211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Constantinof A, Moisiadis VG, Matthews SG. Programming of stress pathways: A transgenerational perspective. J Steroid Biochem Mol Biol. 2016;160:175–180. [DOI] [PubMed] [Google Scholar]
- 58.Murphy VE, Smith R, Giles WB, et al. Endocrine regulation of human fetal growth: the role of the mother, placenta, and fetus. Endocr Rev. 2006;27(2):141–169. [DOI] [PubMed] [Google Scholar]
- 59.Tam WY, Jiang L, Kwan KM. Transmembrane 6 superfamily 1 (tm6sf1) is a novel lysosomal transmembrane protein. Protoplasma. 2015;252(4):977–983. [DOI] [PubMed] [Google Scholar]
- 60.Remis NN, Wiwatpanit T, Castiglioni AJ, et al. Mucolipin co-deficiency causes accelerated endolysosomal vacuolation of enterocytes and failure-to-thrive from birth to weaning. PLoS Genet. 2014;10(12):e1004833.PMC4270466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rosenberg MJ, Wolff CR, El-Emawy A, et al. Effects of moderate drinking during pregnancy on placental gene expression. Alcohol. 2010;44(7–8):673–690.PMC3654802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shang Y, Das S, Rabold R, et al. Epigenetic alterations by DNA methylation in house dust mite-induced airway hyperresponsiveness. Am J Respir Cell Mol Biol. 2013;49(2):279–287.PMC3824034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schnatwinkel C, Christoforidis S, Lindsay MR, et al. The rab5 effector rabankyrin-5 regulates and coordinates different endocytic mechanisms. PLoS Biol. 2004;2(9):E261.PMC514490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Recouvreux MV, Commisso C. Macropinocytosis: A metabolic adaptation to nutrient stress in cancer. Frontiers in Endocrinology. 2017;8(261). DOI: 10.3389/fendo.2017.00261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ogden LE. Nanoparticles in the environment: tiny size, large consequences? BioScience. 2013;63(3):236. [Google Scholar]
- 66.Wick P, Malek A, Manser P, et al. Barrier capacity of human placenta for nanosized materials. Environ Health Perspect. 2010;118(3):432–436.PMC2854775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamashita K, Yoshioka Y, Higashisaka K, et al. Silica and titanium dioxide nanoparticles cause pregnancy complications in mice. Nat Nanotechnol. 2011;6(5):321–328. [DOI] [PubMed] [Google Scholar]
- 68.Riebeling C, Luch A, Gotz ME. Comparative modeling of exposure to airborne nanoparticles released by consumer spray products. Nanotoxicology. 2016;10(3):343–351. [DOI] [PubMed] [Google Scholar]
- 69.Bowman DM, van Calster G, Friedrichs S. Nanomaterials and regulation of cosmetics. Nat Nanotechnol. 2010;5(2):92. [DOI] [PubMed] [Google Scholar]
- 70.Rodney NC, Mulligan CJ. A biocultural study of the effects of maternal stress on mother and newborn health in the democratic republic of congo. Am J Phys Anthropol. 2014;155(2):200–209. [DOI] [PubMed] [Google Scholar]
- 71.De Carli MM, Baccarelli AA, Trevisi L, et al. Epigenome-wide cross-tissue predictive modeling and comparison of cord blood and placental methylation in a birth cohort. Epigenomics. 2017;9(3):231–240.PMC5331917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zheng SC, Beck S, Jaffe AE, et al. Correcting for cell-type heterogeneity in epigenome-wide association studies: revisiting previous analyses. Nat Methods. 2017;14(3):216–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wolfe J, Kimerling R. Gender issues in assessment of posttraumatic stress disorder. New York: Guilford; 1997. [Google Scholar]
- 74.McHugo GJ, Caspi Y, Kammerer N, et al. The assessment of trauma history in women with co-occurring substance abuse and mental disorders and a history of interpersonal violence. J Behav Health Serv Res. 2005;32(2):113–127. [DOI] [PubMed] [Google Scholar]
- 75.Janssen BG, Byun HM, Gyselaers W, et al. Placental mitochondrial methylation and exposure to airborne particulate matter in the early life environment: an environage birth cohort study. Epigenetics. 2015;10(6):536–544.PMC4623402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Triche TJ Jr., Weisenberger DJ, Van Den Berg D, et al. Low-level processing of illumina infinium DNA methylation beadarrays. Nucleic acids research. 2013;41(7):e90; 3627582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Teschendorff AE, Marabita F, Lechner M, et al. A beta-mixture quantile normalization method for correcting probe design bias in illumina infinium 450 k DNA methylation data. Bioinformatics. 2013;29(2):189–196;3546795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pidsley R, Cc Yw V, Lunnon M, et al. LC. A data-driven approach to preprocessing illumina 450k methylation array data. BMC Genomics. 2013;14:293.PMC3769145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kumsta R, Marzi SJ, Viana J, et al. Severe psychosocial deprivation in early childhood is associated with increased DNA methylation across a region spanning the transcription start site of cyp2e1. Transl Psychiatry. 2016;6(6):e830.PMC4931613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Romens SE, McDonald J, Svaren J, et al. Associations between early life stress and gene methylation in children. Child Dev. 2015;86(1):303–309.PMC4305348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Meinshausen N, Buhlmann P. Stability selection. J Royal Stat Soc. 2010;72:417–473. [Google Scholar]
- 82.Phipson B, Maksimovic J, Oshlack A. Missmethyl: an r package for analyzing data from illumina’s humanmethylation450 platform. Bioinformatics. 2016;32(2):286–288. [DOI] [PubMed] [Google Scholar]
- 83.Rivals I, Personnaz L, Taing L, et al. Enrichment or depletion of a go category within a class of genes: which test? Bioinformatics. 2007;23(4):401–407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.