

Abstract
Repair of UV-induced DNA damage requires chromatin remodeling. How repair is initiated in chromatin remains largely unknown. We recently demonstrated that global genome–nucleotide excision repair (GG-NER) in chromatin is organized into domains in relation to open reading frames. Here, we define these domains, identifying the genomic locations from which repair is initiated. By examining DNA damage–induced changes in the linear structure of nucleosomes at these sites, we demonstrate how chromatin remodeling is initiated during GG-NER. In undamaged cells, we show that the GG-NER complex occupies chromatin, establishing the nucleosome structure at these genomic locations, which we refer to as GG-NER complex binding sites (GCBSs). We demonstrate that these sites are frequently located at genomic boundaries that delineate chromosomally interacting domains (CIDs). These boundaries define domains of higher-order nucleosome–nucleosome interaction. We demonstrate that initiation of GG-NER in chromatin is accompanied by the disruption of dynamic nucleosomes that flank GCBSs by the GG-NER complex.
The basic unit of primary chromatin structure is known as the nucleosome. It comprises a histone octamer, containing two copies each of the canonical four core histones, which are enveloped by the winding of 147 bp of DNA around the octamer. The chemical properties of histones can be altered following the post-translational modification of their N-terminal tails. In addition to the canonical histones H2A, H2B, H3, and H4, histone variants such as H2A.Z and H3.3 also exist (Buschbeck and Hake 2017; Talbert and Henikoff 2017). Decoration of histone tails with chemical moieties such as the ubiquitin, methyl, and acetyl groups, or indeed the exchange of histone variants within the octamer structure, alters the physicochemical properties of the nucleosome, imbuing it with biological information. It is known that the physical arrangement of the nucleosomes in the genome provides an important framework that supports the ordered modification of histone tails and variant turnover (Jiang and Pugh 2009; Zhang and Pugh 2011). This complex mechanism is known to be important in regulating many chromatin-related biological functions of the cell, including DNA replication, transcription, and DNA repair (Lai and Pugh 2017). Defects in such regulatory processes are also implicated in diseases associated with aging, including cancer (Luijsterburg and van Attikum 2011).
Several decades of research into the fundamental biochemical mechanisms of DNA repair have revealed the basic functions of the multiple pathways that evolved in cells to recognize, remove, and correct a bewildering variety of lesions that frequently occur in the genomes of cells (Friedberg et al. 1995). Such damage can be caused by influences of both the internal environment of cells, as well as the broader, external environment in which they exist (Lindahl 1993). One of the major DNA repair pathways is known as nucleotide excision repair (NER), and a great deal is known about its fundamental molecular mechanism (Friedberg 2003; Marteijn et al. 2014). Damaged DNA is excised from the genome as an oligonucleotide of ∼30 nucleotides (nt) in length by one of two subpathways: transcription-coupled NER (TC-NER) and global genome NER (GG-NER). The two pathways differ in the way in which DNA repair is initiated. In TC-NER, this is achieved following the recognition of damage-stalled RNA polymerase II (Pol II), as it encounters and is halted by DNA damage. Specific factors exist that mediate this mechanism, which manifests as a more rapid rate of removal of DNA damage from the transcribed strand of active genes. During GG-NER, which operates in all nontranscribing regions of the genome, repair is initiated by a different mechanism, involving a different set of DNA repair factors that function uniquely in GG-NER (Verhage et al. 1994; Reed et al. 1999; Marteijn et al. 2014).
Progress is being made with regard to discovering the histone modifications necessary to permit the efficient repair of DNA damage in chromatin, including those modifications that promote NER. A broad range of research has revealed a role for histone modification and variant exchange in a variety of DNA repair pathways (Adam et al. 2015; Polo 2015; Polo and Almouzni 2015). However, at this stage, the precise details of how these modifications enhance repair of damage in chromatin remain to be determined.
We previously purified a complex of proteins from yeast cells that is uniquely required for GG-NER in this simple eukaryote (Reed et al. 1999). The complex is composed of the SWI/SNF superfamily member, Rad16, the Rad7 protein, and the yeast general regulatory factor (GRF) Abf1. We later demonstrated that this complex interacts with the cullin, Cul3, and the elongin, Elc1, to form a UV-inducible E3 ubiquitin ligase that is required for efficient NER (Gillette et al. 2006). Importantly, it is established that Rad7 and Rad16 exist in a complex within the cell and that deletion strains of either component phenocopy (Verhage et al. 1994; Guzder et al. 1998; Reed et al. 1998). We demonstrated that the GG-NER complex regulates UV-induced histone H3 acetylation by controlling occupancy of the histone acetyl transferase, Gcn5, on chromatin (Teng et al. 2008). This UV-induced hyperacetylation of histones promotes an open chromatin conformation required for efficient repair of DNA damage (Yu et al. 2005; Teng et al. 2008).
We have developed genomic tools for the analysis of DNA damage and repair (Teng et al. 2011; Powell et al. 2015; Yu et al. 2016). We showed that enhanced DNA repair rates occur within open reading frames (ORFs), a result of the concerted action of both the TC-NER and GG-NER pathways in these regions. Inactivation of the GG-NER complex by deletion of the RAD7 or RAD16 genes resulted in a striking alteration to the genomic distribution of DNA repair rates (Yu et al. 2016), suggesting the existence of repair domains in relation to ORFs. In this study, we examine these domains in more detail, defining the genomic features that establish their identity. A paradigm is discussed by which the organization of GG-NER into higher-order chromatin domains reduces the genomic search space for DNA damage detection, thus promoting the efficient recognition and repair of DNA lesions throughout the genome.
Results
Identification of changes to the genome-wide linear arrangement of nucleosomes in response to UV damage
In order to determine how chromatin is remodeled in response to DNA damage, we first investigated the organization of nucleosome structure throughout the genome. The physical arrangement of nucleosomes can be thought of as a structured array of nucleosome units distributed throughout the genome. Within a population of cells, the precise translational setting of a nucleosome within its unit, in any given cell, may vary centering at a favored site, which is commonly referred to as its nucleosome position. A single nucleosome position, and its change in response to environmental conditions, can be characterized by a combination of three parameters that define it: first, the nucleosome position itself; second, its occupancy; and finally, its fuzziness, with the latter term meaning the degree of freedom that a nucleosome has to take up its unitary position within in a population of cells. This degree of freedom is high when a fuzzy nucleosome takes up a wider range of positions in a cell population, and vice versa for low fuzziness nucleosomes. In addition to describing the position and fuzziness score of a nucleosome unit, it is also possible to measure its occupancy, which is defined by its peak height as shown in Figure 1A. This refers to the frequency that the nucleosome unit is occupied by nucleosomes within the population of cells. Trans-acting factors can alter nucleosome structure by changing the position and/or fuzziness of a nucleosome, as well as affecting the nucleosome occupancy at any given position in response to environmental changes. Consequently, we used MNase-seq to map nucleosomes and measured alterations to their structure in wild-type cells before and after exposure to UV irradiation using a bioinformatics pipeline known as DANPOS. This software was specifically developed for determining genomic changes in nucleosome position, fuzziness, and occupancy in cells under different environmental conditions (Chen et al. 2013). In this pipeline, we consistently mapped more than 60,000 nucleosome positions with high accuracy (for full details, see Supplemental Methods). In response to UV irradiation, changes to nucleosome occupancy at various positions across the genome are readily observed when the mapped nucleosome traces are plotted in a linear fashion. A representative 8-kbp section of yeast Chromosome I is shown in Figure 1A (note that occupancies with or without UV exposure of cells are indicated in gray and black, respectively). The aggregate changes in nucleosome occupancy, fuzziness, and position for all of the more than 60,000 nucleosomes are summarized in Figure 1, B–D. It has been reported that certain types of DNA damage cause a significant loss of total nucleosomes from chromatin (Hauer et al. 2017). However, we observed no global change in nucleosome occupancy levels throughout the yeast genome in cells treated with UV radiation (Fig. 1B). We do detect a small increase in the frequency of low-occupancy nucleosomes (fewer than 350 normalized reads) immediately after UV irradiation (Fig. 1B, red line). Genome-wide nucleosome fuzziness, on the other hand, is altered to a greater extent in response to UV irradiation. For example, we observed an increase in the frequency of fuzzy nucleosomes detected, with a reciprocal decrease in the frequency of low-fuzziness nucleosomes both at 0 and 30 min after UV irradiation (Fig. 1C). Finally, we express the genomic position of nucleosomes as the internucleosomal distance, or nucleosome spacing, in base pairs. As expected, the average spacing for all nucleosomes, as defined by the length of linker and nucleosomal DNA, is enriched for distances of between 160 and 180 bp, as shown in Figure 1D. As a result of UV irradiation, a small loss of nucleosomes with this spacing is observed, with a reciprocal gain in more closely spaced nucleosomes also apparent (i.e., those with <140-bp spacing). Our observations thereby identify the UV-induced alteration of the linear nucleosome structure throughout the entire genome, revealing that chromatin is remodeled at only a subset of nucleosomes, via discrete local changes. Since UV-induced lesions are essentially distributed uniformly throughout the genome, our observations suggest that repair of damage may be initiated through nucleosome remodeling at specific sites in the linear genome in response to UV irradiation.
Figure 1.

UV-induced changes to the genome-wide nucleosome landscape. (A) Represented here are the nucleosome traces of wild-type cells before (black) and after UV irradiation (gray) in an 8-kbp region on Chromosome I (128,000–136,000). The genes and their systematic names are indicated by the black arrows underneath the traces. The y-axis on the left indicates the relative read-counts that define the nucleosome peaks in this region. (B) Genome-wide changes to wild-type nucleosome occupancy (peak height) in response to UV irradiation are quantified here. The distribution of relative occupancy (in reads) of all the more than 60,000 nucleosomes as a log-scale of percentage is shown here. (C) As B but now quantifying the degree of freedom a nucleosome has to occupy its unitary position, expressed as fuzziness of all nucleosomes in response to UV irradiation. (D) As B and C, but now quantifying the change in the distribution of nucleosome spacing, reflecting the position of nucleosomes in the linear genome, expressed in base pairs for all nucleosomes after UV irradiation.
Determining the chromatin context for the remodeling of nucleosomes after UV damage
Figure 1 reveals the genome-wide changes in nucleosome structure, as described by three parameters, following exposure of cells to UV. However, these changes are likely due to the collective effect of a variety of mechanisms in addition to that of DNA repair by GG-NER. For example, UV-induced changes to gene expression as part of the DNA damage response are also likely to cause alterations to nucleosome structure. We reasoned that UV-induced nucleosome remodeling might occur in relation to the genomic sites at which the GG-NER complex binds. We previously reported that GG-NER is organized into domains around ORF structure, thus promoting efficient repair (Yu et al. 2016). We showed that loss of GG-NER function severely affected repair rates around the promoter regions of genes containing Abf1 binding sites. Abf1 is a component of the GG-NER complex and is a known GRF in yeast. However, in this early study we relied upon Abf1 ChIP-chip data to identify the genomic locations of repair domains, reporting their effect on repair rates in relation to all approximately 4000 Abf1 binding sites that are located predominantly at gene promoters. These low-resolution data, however, preclude the accurate mapping of nucleosomes at these positions. Therefore, we performed Abf1 ChIP-seq experiments to map the genome-wide occupancy of Abf1 in chromatin at nucleotide resolution as a precursor to identifying the precise location of GG-NER complex binding. In agreement with our previously published Abf1 ChIP-chip data and other reports (Zentner et al. 2015; Yu et al. 2016), we found approximately 4000 Abf1 binding sites detected by MACS2 (Supplemental Methods; Supplemental Table S1; Zhang et al. 2008). It is well established that the GRF Abf1 exists in excess over the other GG-NER components (Rad7 and Rad16) in the cell and has a wide range of different functions outside of GG-NER (Yarragudi et al. 2007; Schlecht et al. 2008; Ganapathi et al. 2011; Zhang et al. 2012). Therefore, in order to examine nucleosome structure in relation to GG-NER function, we refined the list of approximately 4000 Abf1 binding sites to identify a novel set of GG-NER complex binding sites (Supplemental Table S1). To do this, we examined distinctive genomic features associated with Abf1 binding, including Abf1 consensus sequences (n = 1752) and genome-wide NFRs (n = 6589), with which Abf1 occupancy is frequently associated, as described in Figure 2A and Supplemental Figure S1 (Yarragudi et al. 2007; Hartley and Madhani 2009; Ganapathi et al. 2011; Ozonov and van Nimwegen 2013). This enabled us to categorize Abf1 binding sites according to these features. To subclassify the Abf1 sites associated with GG-NER, we used our previously published Rad16 ChIP-chip genome-wide occupancy data (Yu et al. 2016) to identify the set of Abf1 sites enriched for GG-NER complex binding. This yielded approximately 3600 Abf1 binding sites that are enriched for Rad16 (Supplemental Methods Fig. S1). The majority of these genomic positions (∼70%) are located in promoter regions upstream of genes, making it possible to examine the surrounding nucleosome structure (Fig. 2A; Supplemental Methods Fig. S2). We used this list to define a novel class of genomic features, which we now refer to as GG-NER complex binding sites (GCBSs; n = 2664). Full details of the analysis can be found in the accompanying Supplemental Methods. Annotating the data in this way now enables us to map nucleosomes directly at GCBSs or at GCBSs in relation to ORF structure. The strand information of the nearest annotated gene orientates the data in such a way that the ORFs are positioned downstream (i.e., to the right) from the GCBSs. By using this list, we generated composite plots of nucleosome positions directly at GCBSs (Fig. 2B) and at GCBS-adjacent ORFs (Fig. 2C). This reveals the position of an NFR at these locations, flanked by an array of positioned nucleosomes as others have reported previously (Lai and Pugh 2017). In Figure 2C, we noted that this class of GCBS-adjacent nucleosomes is located directly over the position of the TSS. Typically, the +1 nucleosome is positioned further into the ORF when nucleosomes are mapped to all TSSs (see Supplemental Fig. S2A). To determine whether this novel subset of TSS-positioned nucleosomes is unique to this class of GG-NER complex binding sites, we used k-means clustering of the individual nucleosome traces to identify a subclass of genomic positions that uniquely contain a +1 nucleosome at the TSS or whether this is a common feature among these genes. We find 13 clusters that all display a different distance between the TSS and the +1 nucleosome (Supplemental Fig. S3A). To represent the nucleosome structure independently of GCBS position, we used the coordinates of the NFR at these GCBSs and centered them. Plotting the MNase-seq data in this orientation uniformly aligns the nucleosome arrays, revealing the presence of only four clusters (Supplemental Fig. S3B). Therefore, we conclude that the +1 nucleosome position at the TSS as shown in Figure 2C is explained by the averaging of different nucleosome traces that exhibit a highly variable distance between the TSS and the +1-nucleosome position. This demonstrates that this is not a typical feature of these GCBS-associated regions (Supplemental Fig. S3) but simply reflects the variable distance between TSS and NFR in this class. Next, we investigated the effect of UV irradiation on nucleosome structure at these positions. In wild-type cells, loss of nucleosome occupancy at GCBS-adjacent +1 and −1 nucleosomes can be discerned immediately after UV irradiation (Fig. 2B,C, red line). Following 30 min of repair time, nucleosome occupancy is recovered to predamage levels (Fig. 2B, green line), with evidence of increased nucleosome occupancy at the +1 and +2 positions (Fig. 2C, green line). These experiments reveal the precise genomic location of UV-induced remodeled nucleosomes in relation to GCBSs.
Figure 2.
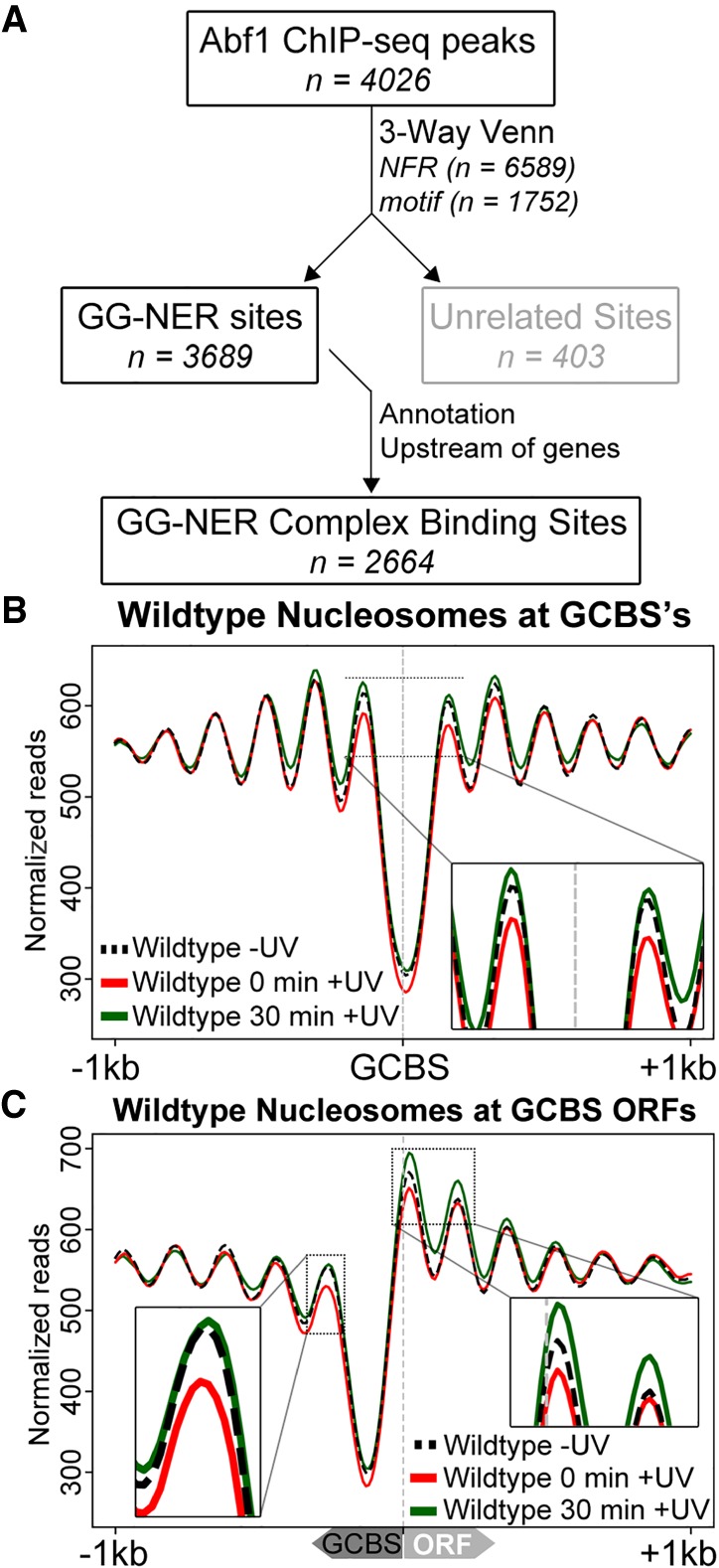
Identification of the genomic list of GCBSs and the nucleosome occupancy in relation to these sites. (A) Flow chart to illustrate the bioinformatics analysis performed to identify genome-wide GCBSs by refining and filtering the list of Abf1 ChIP-seq peaks using NFR positions, motif sites, and annotation information. (B) MNase-seq data of wild-type cells were used to plot cumulative nucleosome positions around GCBSs (n = 2664) in the absence of UV irradiation and at different intervals after UV irradiation, displaying regularly spaced nucleosome arrays at these genomic locations. The x-axis denotes the 2-kbp regions surrounding the GCBSs, while the y-axis indicates nucleosome occupancy as measured by normalized reads. (C) Nucleosome occupancy in wild-type cells before and after UV damage. MNase-seq data of untreated and UV-treated cells are shown as cumulative graphs around GCBSs in relation to ORF structure. The insets highlight the nucleosome remodeling at the −1 position (left) and the remodeling at positions +1 and +2 (right).
Damage-induced remodeling of nucleosome structure is not detected when events at all transcription start sites are examined
We considered whether the UV-induced changes in nucleosome structure described above can be observed when events are examined at all transcription start sites (TSSs) (Xu et al. 2009). We found that when all 5171 TSSs are examined, the nucleosome structure around these genomic features remains unaltered after UV irradiation (Supplemental Fig. S2A). This result demonstrates that no gross UV-induced changes to the nucleosome landscape occur in the context of this well-established genomic feature of nucleosome organization. Disruption of nucleosome structure at this feature has been previously described for mutants defective in certain essential SWI/SNF ATP-dependent chromatin remodelers, including CHD1, ISW1, or INO80 (van Bakel et al. 2013), which regulate gene expression by controlling nucleosome structure at these sites. Indeed, in yeast, nucleosome sliding, which shifts the translational setting of the nucleosome, altering its position, is a well-known mechanism to control gene expression (van Bakel et al. 2013). Part of the cellular response to DNA damage controls the gene expression of various DNA damage–responsive genes via this mechanism. Therefore, we investigated nucleosome sliding at a single UV-responsive gene by plotting nucleosomes at the DNA damage inducible locus, RAD51 (Shinohara et al. 1992), as shown in Supplemental Figure S2C. As expected, these data demonstrate that nucleosome sliding can be detected at this locus after DNA damage induction. The absence of linear nucleosome sliding when all TSSs are examined in aggregate, however, indicates that this mechanism of nucleosome remodeling does not occur globally throughout the genome in response to UV damage. Importantly, this class of remodeling occurs independently of GG-NER as shown by the presence of nucleosome sliding in cells deleted for RAD16, which are defective in the GG-NER process (Supplemental Fig. S2D). These results demonstrate that the UV-induced nucleosome remodeling reported in Figure 1 is the result of the collective activity of multiple mechanisms, as described in Figure 2, B and C, and Supplemental Figure S2, C and D.
GG-NER complex binding sites are located at the boundary regions of chromosomally interacting domains
We detected UV-induced nucleosome remodeling in only a small subset of GCBS-adjacent nucleosomes (Fig. 2B). Since GG-NER operates throughout the genome, we considered how such localized changes in nucleosome structure might contribute to chromatin remodeling in a wider context. We noted that standard MNase-seq only reveals changes in the linear arrangement of nucleosomes (Fig. 1A). Therefore, we investigated the genomic locations of GCBSs in relation to domains of higher-order chromatin structure. Recent advances in methods such as 3C and the related Hi-C, have led to the introduction of a chromatin capture method called Micro-C (Hsieh et al. 2015, 2016). This technique measures higher-order nucleosome–nucleosome interactions in chromatin. Micro-C follows the same principles as other 3C methods but uses MNase instead of restriction enzymes to digest cross-linked chromatin. This allows the detection of distal nucleosome–nucleosome interactions that have recently led to the discovery of chromosomally interacting domains (CIDs) at nucleosome resolution for the first time in yeast (Hsieh et al. 2015). These investigators reported that boundary sites that demarcate CIDs are often found upstream of highly expressed genes and are enriched for nucleosome pairs that flank NFRs in a similar fashion to the features we observe in relation to GCBSs. Therefore, we set out to determine the relationship between the genomic locations of the GCBSs identified above and the boundary sites of these newly described CIDs. To this end, we retrieved the genomic positions of the CID boundaries from published data (Hsieh et al. 2015) and calculated the overlap between these positions and the GCBSs. We found that GCBSs map predominantly to CID boundary positions, with ∼50% located precisely at these sites, as shown in Figure 3A. To examine the significance of this observation, we took a similar number of random genomic positions and calculated the overlap of boundaries and GCBSs with these sites. This revealed only two boundaries overlapping at these positions compared with over 1200 GCBSs found at CID boundaries (Fig. 3A). Figure 3B shows a representation of the newly discovered CID chromatin landscape in relation to the linear setting of nucleosomes (Fig. 3D) and also shows the position of two GCBSs at CID boundaries, as illustrated by the binding of Abf1 (Fig. 3C). The genomic location of the region is shown in Figure 3E. This confirms that GCBSs colocalize precisely at the boundary positions of a specific subset of CIDs and therefore occupy sites in the genome that demarcate regions of higher-order chromatin structure. Having established the key elements that define GCBSs in the three-way Venn diagram shown in Supplemental Figure S1, in Figure 3F, we now illustrate, using a five-way Venn diagram, the relationship of each individual element that contributes to the make-up of this novel class of genomic feature. The Venn depicts the combination of features that contribute to the class of GCBSs as highlighted in red. Importantly, outside of our GCBS set, it is apparent that significant numbers of sites in each of the separate classes exist (highlighted in white and blue). These genomic features are likely utilized in other cellular processes as defined by their original biological classification. This suggests that GG-NER may be organized and initiated from a subset of CID boundaries to which the GG-NER complex is bound in the absence of DNA damage.
Figure 3.

GCBSs are located at the boundaries of chromosomally interacting domains (CIDs). (A) Overlap calculations identified the number and identity of GCBSs (n = 2664) at CID boundaries (n = 3061) and at random sites (n = 3137). The percentage of GCBSs in each subcategory is indicated between brackets. (B) Micro-C data (Hsieh et al. 2016) were used to plot nucleosome–nucleosome interactions in a 11-kbp window on Chromosome I. The gray dashed lines indicate four boundary positions documented in the literature (Hsieh et al. 2015). The intensity of the heatmap is a measure for the normalized interactions indicated beneath the panel. (C) Abf1 ChIP-seq data are plotted here to highlight two GCBSs in this region of the genome labeled as GCBS 1 and 2. (D) The nucleosome landscape is presented here by plotting MNase-seq data at this genomic location. (E) Indicated in black bars are the genes located within this region of the genome. The labels on the x-axis highlight the genomic coordinates in kilobase pairs. The y-axis on each panel indicates peak height as normalized reads. (F) The combined positions of the five features that characterize GCBSs were used to generate a five-way Venn diagram to illustrate how each genomic feature contributes to the formation of a GCBS. Highlighted in red are the predominant classes that make up our list of GCBSs, with intensity signifying the amount of binding sites in each subclass. Conversely, in blue we highlight the Abf1 binding sites and other features that are not classified as GCBSs. Color intensity is used here to indicate the number of features in each subcategory. The number of each feature is listed between brackets.
The GG-NER complex remodels GCBS-adjacent nucleosomes in response to UV damage
Our results reveal the genomic location of UV-induced nucleosome remodeling in relation to GCBSs in wild-type cells (Fig. 2B,C). In order to determine whether this remodeling is dependent on the GG-NER complex, we investigated these events in GG-NER–defective, RAD16-deleted cells. To do this, we first mapped nucleosomes in untreated rad16 mutants and compared them to the wild-type pattern (Fig. 4A, gray line; Supplemental Fig. S4A). We observed reduced nucleosome occupancy at the positions immediately adjacent to the GCBSs in these GG-NER–defective cells. This demonstrates that the GG-NER complex is necessary for establishing the normal nucleosome structure adjacent to these locations in undamaged wild-type cells (cf. black line with gray line; Fig. 4A; Supplemental Fig. S4A). Next, we treated RAD16-deleted cells with UV irradiation and observed no loss of nucleosome occupancy at these positions (Fig. 4B; Supplemental Fig. S4B). This shows that the nucleosome remodeling observed at these sites in wild-type cells (Fig. 2B,C) is dependent on the GG-NER complex. To confirm that GG-NER dependent remodeling of nucleosomes is specific to GCBS sites, we analyzed nucleosomes at all TSSs in the RAD16-deleted strain and observe no UV-induced changes at these sites (Supplemental Fig. S2B). Nucleosomes accumulate at these sites 30 min after UV irradiation in GG-NER–defective cells (Fig. 4B, green line; Supplemental Fig. S4B, green line). We suggest that the nucleosome remodeling process observed at the GCBS-adjacent nucleosomes is a process that initiates the chromatin remodeling required in response to DNA damage (Weber et al. 2014).
Figure 4.
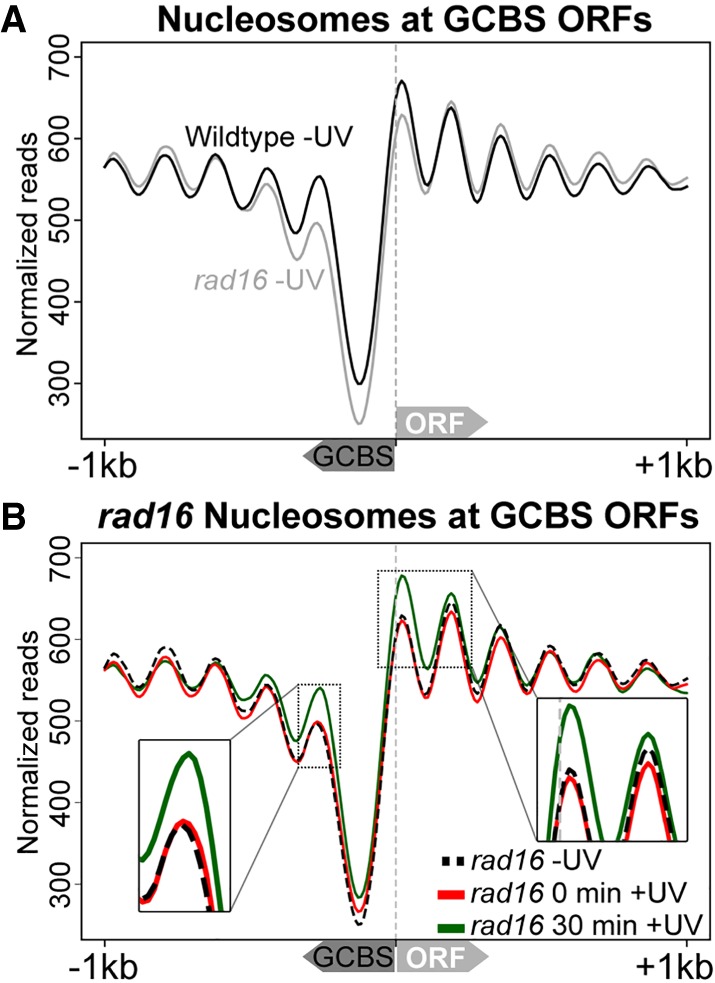
GG-NER complex adjacent nucleosomes are established and remodeled following UV irradiation in a Rad16-dependent fashion. (A) MNase-seq data of wild-type and rad16 mutant cells were used to plot cumulative nucleosome positions around GCBSs (n = 2664) in the absence of UV irradiation. The annotation of the nearest gene was used to infer strand information to align these genomic positions according to gene orientation as indicated by the arrows on the x-axis depicting the relative direction the GCBS and ORF. The x-axis denotes 2-kbp regions surrounding the GCBSs, while the y-axis indicates nucleosome occupancy as measured by normalized reads. (B) As described in A, but showing UV-induced changes to nucleosome positions around GCBSs and accompanying ORFs in rad16-mutated, GG-NER–defective cells.
GCBSs are flanked by histone H2A.Z-containing barrier nucleosomes that are remodeled by the GG-NER complex in response to UV damage
The UV-induced loss of nucleosome occupancy observed in wild-type cells is consistent with histone exchange events that occur at dynamic nucleosomes, as described by others in the context of gene transcription (van Bakel et al. 2013). Dynamic nucleosomes are often associated with the functional response of the cell to environmental change or stress (Lai and Pugh 2017). This physical organization of the chromatin controls the accessibility of binding proteins to the DNA in chromatin, such as transcription factors, thus regulating their activity in the cell. Such nucleosomes contain the histone variant H2A.Z and have been described previously as “barrier nucleosomes” that are highly dynamic in nature (Weber et al. 2014). As such, they represent nodes, inhibitory structures that must be altered to permit gene expression at such locations. A role for histone variants in DNA repair has been noted in both NER and other repair mechanisms (Adam et al. 2015). Indeed, in yeast, we have previously reported that histone H2A.Z is involved in NER (Yu et al. 2013). Therefore, we investigated the occupancy of histone H2A.Z at nucleosomes located immediately adjacent to GCBSs. To do this, we undertook ChIP-seq experiments using HA-tagged H2A.Z to map the positions of genome-wide H2A.Z-containing nucleosomes. We then measured the change in their occupancy in response to UV irradiation and detect approximately 16,000 H2A.Z-containing nucleosomes. Initial analysis of the genome-wide distribution of histone H2A.Z confirmed the presence of this histone variant predominantly at nucleosomes flanking NFRs upstream of genes. These display an asymmetric pattern of binding as described previously in the literature (Guillemette et al. 2005; Raisner et al. 2005; Albert et al. 2007; Weber et al. 2014). Next, we examined the H2A.Z occupancy in GCBS-adjacent nucleosomes and observed H2A.Z at both the +1 and −1 positions (Fig. 5A). Compared with the loss of nucleosomes observed earlier (Fig. 2C), we found that in the case of H2A.Z-containing histones, occupancy is lost uniquely from the +1 nucleosome position in response to UV irradiation (Fig. 5A, red line). In addition, after 60 min of repair time, H2A.Z occupancy returns to its predamage level (Fig. 5A, green line), which is consistent with the wild-type recovery of nucleosome occupancy shown in Figure 2C. These differences may reflect variations in the type and timing of the histone eviction/exchange events occurring in the +1 and −1 nucleosomes during repair. Collectively, our data demonstrate that nucleosome remodeling occurs at promoter NFRs adjacent to GG-NER complex binding sites. In response to UV irradiation, histone eviction or exchange occurs at these nucleosomes. In order to test the GG-NER complex dependence of H2A.Z loss at these sites, the H2A.Z ChIP-seq experiment was repeated in cells deleted for RAD16. We observed that in the absence of DNA damage, histone H2A.Z occupies GCBS-adjacent nucleosomes in rad16 mutant cells in a similar fashion to that observed in wild-type cells (Fig. 5, cf. panel A black dashed line with panel B black dashed line; see also Supplemental Fig. S5). However, in response to UV, no loss of histone H2A.Z occupancy from the +1 nucleosome can be detected in the GG-NER–defective RAD16-deleted cells (Fig. 5B, red line; Supplemental Fig. S5B, red line). Consistent with our findings for nucleosome occupancy observed in wild-type cells (Fig. 2B,C), 60 min after UV irradiation, H2A.Z occupancy also accumulates to levels higher than those observed prior to UV irradiation in wild-type cells (Fig. 5B, green line; Supplemental Fig. S5B, green line). The absence of H2A.Z loss from these sites in a GG-NER–defective mutant confirms a role for the GG-NER complex in this process. These data demonstrate that histone loss at H2A.Z-containing nucleosomes, adjacent to GCBSs, is driven by the GG-NER complex to alter chromatin structure during the initial stages of GG-NER in response to UV damage.
Figure 5.

UV-induced loss of H2A.Z occupancy requires GG-NER complex-dependent nucleosome remodeling around GCBSs. (A) The UV-induced change to H2A.Z occupancy in wild-type cells around GCBS-associated TSSs is shown here using H2A.Z ChIP-seq data, prior to UV irradiation and 0 or 60 min after UV damage. The light gray trace represents the nucleosome positioning in the absence of DNA damage retrieved from the data shown in Figure 4. The inset highlights the UV-induced changes to H2A.Z occupancy at the +1 position. (B) As described in A, but now representing the H2A.Z occupancy at GCBS-bound promoter regions in GG-NER–defective RAD16-deleted cells.
UV-induced changes in GG-NER complex chromatin occupancy at GCBSs
By using the list of GCBSs described above, we next investigated how the individual components of the GG-NER complex bind to chromatin in relation to the nucleosome structure that exists at these genomic features. Therefore, we used the Abf1 ChIP-seq and Rad16 ChIP-chip data to plot GG-NER complex chromatin occupancy at GCBS sites. As described previously, Abf1 occupancy occurs predominantly at promoter regions upstream of TSSs and is located precisely at the NFR (Fig. 6A). We observed a slight left-sided shoulder to the distribution of Abf1 occupancy upstream of the NFR, which reflects the variation in NFR sizes that exist upstream of the TSS (Supplemental Fig. S6). In the absence of UV irradiation, the Rad16 component of the GG-NER complex is enriched at these sites as previously reported (Yu et al. 2016). However, when the data are examined in this context, the precise location of Rad16 occupancy can now be discerned. Peaks of Rad16 occupancy are observed predominantly at the +1 and −1 nucleosomes, and Rad16 enrichment also extends to the −2 nucleosome (Fig. 6B). This is consistent with the upstream shoulder of Abf1 enrichment described earlier (Fig. 6A). Collectively, these data show that the Abf1 component of the GG-NER complex binds to chromatin at specific NFRs, which are flanked by nucleosomes where peaks of Rad16 binding are observed at the +1 and −1 positions. This arrangement suggests that Rad16 binds to the −1 nucleosome when the Abf1 component of the complex occupies broader NFRs that are located further upstream of the TSS. Indeed, this does account for the broader range of translational settings observed for Abf1, as shown in Supplemental Figure S6 (right-hand panel). In response to UV irradiation, the occupancy of the Rad16 component of the GG-NER complex is altered from its original position at the +1 and −1 nucleosomes within 30 min and is redistributed to sites more distally located at nucleosomes extending into the ORFs of genes, as well as the upstream regions of promoters (Fig. 6B, gray line). No change in Abf1 occupancy is observed as previously described (Yu et al. 2016). As expected, Rad7 chromatin occupancy before and after UV irradiation is similar to that observed for Rad16 (Supplemental Fig. S7). Our data demonstrate that GG-NER–dependent changes in GCBS-adjacent nucleosomes occur in conjunction with the redistribution of the Rad7 and Rad16 components of the GG-NER complex. A similar result is obtained when the data are analyzed in relation to GCBS-associated CID boundaries as shown in Supplemental Figure S8.
Figure 6.

GG-NER complex binding to chromatin around GCBS-associated promoter regions. (A) Genomic positions of GCBS-associated genes were used to plot the genome-wide Abf1 ChIP-seq data at TSSs to map the location of complex binding in relation to gene structure and nucleosome positions at these genes. The gray nucleosome trace represents nucleosome occupancy in the absence of UV irradiation at these loci. The y-axis represents the Z-score to compare data with different read-depths in the same plot. (B) GG-NER complex binding as assessed by Rad16 ChIP-chip data (Yu et al. 2016) plotted at GCBS-associated genes.
Rad16's ATPase function is required for the redistribution of the complex and nucleosome remodeling in response to UV damage
We considered whether the UV-induced redistribution of Rad16 away from its initial genomic occupancy, into the ORFs, is dependent on the primary biochemical function of Rad16 and its ability to remodel the GCBS-adjacent nucleosomes. To investigate this, we exploited the previously described strain of RAD16 that is specifically mutated in the ATPase domain and is defective in the DNA translocase function of the complex (Ramsey et al. 2004; Yu et al. 2011, 2016). This point mutation renders the strain UV sensitive and repair deficient, while maintaining the chromatin occupancy of this altered Rad16 protein (Yu et al. 2016). The use of this point mutant enabled us to measure the chromatin occupancy of the defective Rad16 protein and the nucleosome structure in this strain, both before and after exposure of these cells to UV irradiation. Figure 7A demonstrates similar chromatin occupancy of the Rad16 ATPase to that observed in wild-type untreated cells (Fig. 6B). Exposure of these cells to UV light does result in a loss of Rad16 occupancy but fails to redistribute the ATPase-defective Rad16 protein into the ORFs, as occurs in wild-type cells (cf. Fig. 7A and Fig. 6B). These observations are in agreement with similar findings reported previously (Yu et al. 2016). Figure 7B demonstrates that GCBS-adjacent nucleosomes are not remodeled in this strain in response to UV damage in the same way seen in wild-type cells. Furthermore, evidence of nucleosome accumulation at these positions is also observed in response to UV irradiation in a similar manner to that seen in RAD16-deleted cells (Fig. 4B). These observations demonstrate that the ATPase activity of Rad16 drives the redistribution of the GG-NER complex and the nucleosome remodeling observed in wild-type cells in response to UV irradiation. In addition, it also demonstrates that loss of occupancy of the complex in response to UV irradiation is not sufficient to induce nucleosome remodeling.
Figure 7.

GG-NER complex chromatin occupancy and nucleosome remodeling after UV irradiation requires the ATPase function of Rad16. (A) Chromatin occupancy of the Rad16 ATPase mutated protein at GCBSs is plotted here using ChIP-chip data (Yu et al. 2016). The pre-UV irradiation data are shown by the black trace, while the 15-min post-UV chromatin occupancy is presented by the gray trace. The nucleosome landscape in the ATPase mutant is shown by the gray shading at these of positions. (B) MNase-seq data from the RAD16 ATPase mutant cells are shown at GCBS-associated ORFs in a 2-kbp window. The arrows on the x-axis indicate the relative orientation of the GCBSs and ORFs. The insets highlight the UV-induced changes to the nucleosome occupancy at the positions immediately adjacent to the GG-NER complex–occupied promoter.
Chromatin remodeling during GG-NER is initiated from GCBSs that define origins of repair within the genome
Finally, we examined how the mechanism described above affects rates of DNA repair in relation to this novel class of genomic features. Therefore, we plotted relative repair rate data as determined by measuring CPD repair rates at the 2-h time point after UV damage (Yu et al. 2016) at GCBSs to establish how the GG-NER complex function regulates repair of UV-induced DNA damage. We are also now able to map relative rates of DNA repair in relation to nucleosome positions at GCBSs. In wild-type cells, relative repair rates are high across a 2-kbp window surrounding GCBSs (Fig. 8A; Supplemental Fig. S9C), with variation in the rates observed in relation to nucleosome positions as reported by others (Mao et al. 2016). However, in the absence of the GG-NER complex, relative CPD repair rates in the vicinity of GCBSs are severely reduced, as shown in RAD16-deleted cells (Fig. 8A, gray line; Supplemental Fig. S9C). Our data demonstrate that although relative rates of repair are most severely affected at GCBSs in GG-NER–defective mutants, the effect on repair extends well beyond the location of remodeled nucleosomes that are immediately adjacent to the GCBSs. This suggests that the role of the GG-NER complex in chromatin remodeling likely extends beyond the local alteration of GCBS-adjacent nucleosome structure in the linear genome. We speculate that this may involve the disruption of higher-order nucleosome interactions that comprise CIDs, the boundaries of which are frequently occupied by the GG-NER complex as described earlier (Fig. 3). We also used a set of genomic NFRs that are not associated with GG-NER complex binding as an in silico control, since no GG-NER–dependent nucleosome remodeling is expected to occur at these sites (n = 4415) (see Supplemental Fig. S1). Indeed, we observed no change in the nucleosome occupancy at these positions (Supplemental Fig. S9A,B). Relative rates of GG-NER at these sites in RAD16-deleted cells are not affected in the same way as those observed at GCBSs (Fig. 8B). The relative repair rate at these NFRs is reduced but similarly distributed in both RAD16-deleted cells and wild-type cells (Supplemental Fig. S9D). These observations confirm that the mechanism of repair organized and initiated from GCBSs is not a common feature of NFRs but is specifically dependent on the occupancy and function of the GG-NER complex at these sites. In conclusion, our studies show that GCBSs are novel genomic features that represent sites from which GG-NER is initiated following the remodeling of adjacent nucleosomes at these locations in response to UV damage.
Figure 8.

GG-NER complex binding at a subset of NFRs organizes repair in chromatin, but this is not a general feature of NFRs. (A) Relative CPD repair rates are plotted around GCBSs for both wild-type and rad16 GG-NER–defective mutant cells in relation to the nucleosome landscape, indicated as the gray shaded area. The x-axis indicates the orientation of both the GCBS and the ORF in relation to TSSs. (B) As described in A, but here plotting the relative repair rates at non-GCBS-associated NFRs (n = 4415) (see Supplemental Fig. S1), orienting the data in relation to the nearest gene aligning at the TSS with the NFR positioned upstream. The x-axis indicates regions 1 kbp upstream of and downstream from these positions. The gray shaded area represents the nucleosome data at these positions.
Discussion
In cells, maintaining the integrity of the genome is essential for life. Since DNA is constantly exposed to the deleterious effects of both the internal and external cellular environment, mechanisms have evolved to sense and repair the ensuing genetic damage. The ability to efficiently detect the presence of DNA damage that is packaged into chromatin is of paramount importance, and defects in the process are associated with a variety of diseases, including cancer.
The main findings of this report reveal that chromatin remodeling during repair of DNA damage by the NER is initiated from specific sites of GG-NER complex binding at the boundary sites of CIDs, which are genomic regions of higher-order nucleosome–nucleosome interaction. We show that in undamaged cells, the complex occupies these sites and is bounded by nucleosomes containing the histone variant H2A.Z. In response to DNA damage, we show that these boundary nucleosomes are remodeled in a GG-NER complex–dependent fashion, and this enables the Rad7 and Rad16 components of the complex to redistribute to more distal sites within the CID. Finally, we demonstrate the importance of this mechanism to the efficient removal of DNA damage by NER.
NER recognizes and repairs a broad range of lesions, including those induced by UV light and a variety of chemical carcinogens. Two subpathways of NER exist that differ in their mechanism of initiating damage recognition. During TC-NER, recognition is initiated by the stalling of RNA Pol II as it encounters the damaged DNA. This couples repair of DNA damage to the process of transcription and consequently to how this process is organized within the genome. This coupling results in an efficient mechanism for removing genetic damage and restoring gene expression to damaged transcribed DNA strands. Stalling of RNA Pol II subsequently recruits NER factors that function in later stages of the NER process. These factors are common to later-stage repair events in the GG-NER. However, less is known about how repair of DNA damage is initiated and organized in the GG-NER subpathway, which repairs all nontranscribed regions of the genome. In yeast, this pathway relies upon a protein complex that is unique for the function of GG-NER. By using genomic techniques, we recently showed that GG-NER is organized into domains related to the promoter regions of ORFs. We demonstrated that efficient DNA repair around these sites depends on the GG-NER complex regulating the histone acetylation status of nucleosomes in the vicinity, which alters the chromatin structure. However, until now, we have not established how chromatin remodeling is initiated during GG-NER by examining these events at the level of the nucleosome, the primary structural unit of chromatin.
To tackle this problem, we generated genome-wide nucleosome maps to analyze UV-induced changes to the nucleosome landscape. We analyzed the genomic distribution of changes to the three core nucleosome parameters that quantify occupancy, fuzziness, and position to identify a subset of nucleosomes that are altered in response to UV irradiation. These findings demonstrate that chromatin remodeling at this level occurs predominantly through dispersed local changes to nucleosome occupancy and fuzziness. However, nucleosome sliding in the context of gene expression can also be detected at known DNA-damage-responsive genes, in line with previously published data (Lai and Pugh 2017). Our data show that the remodeling of positioned nucleosomes adjacent to GCBSs at many hundreds of genomic features in aggregate does not occur via nucleosome sliding. These findings are consistent with our previous biochemical observations (Yu et al. 2004, 2009). The complex can translocate across DNA in vitro through the activity of the SWI/SNF and helicase domains of Rad16, but it cannot slide nucleosomes in vitro (Yu et al. 2009). This mechanism may also drive the nucleosome remodeling events we describe here. We demonstrated that the DNA translocase activity of the GG-NER complex contributes to the nucleosome remodeling demonstrated here. Furthermore, we show that the GG-NER complex binding sites identified in this study are not simply regions of repair initiation but also locations of UV-induced histone remodeling, involving nucleosomes containing the histone variant H2A.Z. We suggest that in undamaged cells, these H2A.Z-containing nucleosomes represent barriers that constrain and sequester the GG-NER complex at these genomic positions. DNA repair may be initiated by removal of these barriers, allowing the GG-NER complex to redistribute from its initial binding locations. The loss of H2A.Z-containing nucleosomes removes the barrier, permitting GG-NER complex redistribution. We speculate that this process might concurrently restrict RNA Pol II transcription that requires H2A.Z-containing nucleosomes for efficient gene transcription (Weber et al. 2014). Therefore, this mechanism may contribute to the inhibition of bulk transcription in response to DNA damage, while at the same time driving the search for DNA damage by the GG-NER complex. Shut-down and restoration of normal gene expression is an established hallmark in maintaining the stability of the genome in response to DNA damage (Ciccia and Elledge 2010).
The occupancy of the GG-NER complex upstream of genes in undamaged cells shows that it is an inherent component of chromatin as well as plays a role in repairing its structure in response to damage. We speculate that the E3 ligase function of the complex is important in this process. Our previous work showed that the E3 ligase activity of the GG-NER complex is essential for binding of the complex to the chromatin (Yu et al. 2016). By extension, we expect that this activity is pivotal in maintaining the chromatin structure around GCBSs in the absence of DNA damage. Future work will address this issue in order to understand how the E3-ligase activity maintains chromatin occupancy of the GG-NER complex and to determine which factors or components of the chromatin are its targets.
Higher-order chromatin structure in yeast has been identified following the introduction of methods that map distal nucleosome–nucleosome interactions, forming structural units that are classified as CIDs (Hsieh et al. 2015, 2016). These structures encompass one to five genes and range in size from a few kilobases up to 10 kbp. We find that ∼50% of our GCBSs can be found precisely at the boundaries between these genomic features. We speculate that the nucleosome–nucleosome interactions contained within CIDs may represent higher-order levels of structure that are remodeled during GG-NER. We plan to investigate this aspect in more detail in the future. We propose that the DNA translocase activity of the GG-NER complex could induce the remodeling of higher-order chromatin structure, similar to the loop-extrusion model suggested for CTCF-cohesin complexes in higher eukaryotes (Sanborn et al. 2015). In this model, two CTCF-cohesin complexes bind to the chromatin and extrude DNA through the cohesin ring structure until they encounter a CTCF binding site (Sanborn et al. 2015). The CTCF and cohesin factors reside at the base or boundary of these loop structures, which may be analogous to the boundary positions to which the GCBS complex binds in the yeast genome. Although the loop-extrusion model has not been demonstrated in yeast and although the lack of a yeast homolog for CTCF excludes the possibility of a direct parallel mechanism, we suggest that the redistribution of the GG-NER complex, by virtue of the DNA translocase activity of Rad16, could act as a wedge to disrupt the higher-order contacts that exist in the DNA loops that make up the CIDs. Future research aims to investigate the remodeling mechanism of higher-order chromatin structure using the micro-C methodology.
In summary, we conclude that our study demonstrates that in undamaged cells, DNA repair complexes are positioned at hundreds of boundary regions that define the presence of CIDs: genomic domains of higher-order nucleosome–nucleosome interactions. We suggest that this arrangement might represent origins of DNA repair initiation that promote the efficient repair of DNA damage in chromatin. We note that initiating chromatin remodeling from defined origins could effectively reduce the search space for DNA damage recognition by compartmentalizing the genome into functional modular chromatin structures that can be rapidly remodeled and efficiently repaired. Therefore, characteristic structural features of CIDs emerge when the genome is organized in this way—this ensures the rapid search and repair of genetic damage in chromatin.
Methods
Yeast strains used in this study
The yeast strains used in this study are listed, with their respective genotype, in Table 1.
Table 1.
Yeast strains and their respective genotypes used in this study

Preparation of MNase-seq and ChIP-seq samples
In order to prepare samples for MNase-seq and ChIP-seq, we UV irradiated yeast cells and generated DNA and chromatin extracts as described previously (Yu et al. 2011, 2016). MNase-seq samples were generated as described previously (Kent et al. 2011) and subsequently processed for NGS by following the library preparation protocol described below. The chromatin samples prepared for ChIP were used for pull-down as described previously (Teng et al. 2011; Yu et al. 2011, 2016). Immunoprecipitated DNA and related input DNA were used for library preparation for NGS as described below. For more information, see the Supplemental Methods.
Ion proton library preparation
The following protocol was adapted from that of Life Technologies (“Ion ChIP-Seq library preparation on the Ion Proton system,” publication no. 4473623, revision B), with minor modification to the blunt-ending, DNA purification, and amplification as described in the Supplemental Methods.
Data analysis
In short, NGS data were aligned to the reference genome (sacCer3) and processed for downstream analysis to detect peaks with MACS2 (Zhang et al. 2008) and map nucleosomes using DANPOS (Chen et al. 2013) or NFRs by applying HOMER (Heinz et al. 2010). Subsequent annotation data were assigned to relevant features using the ChIPpeakAnno package (Zhu et al. 2010) in R (R Core Team 2018), which was also used for the calculation of overlaps and drawing of Venn diagrams presented in this paper. Full details of all bioinformatics data analysis are included in the Supplemental Methods.
We obtained the Hi-C boundary positions from the supplemental data accompanying the work by Hsieh et al. (2015). A list of genome-wide NFRs was obtained from Yadon et al. (2010). The list of Abf1 consensus motifs can be obtained using the MEME fimo algorithm (Grant et al. 2011). The CPD repair rate and ChIP-chip data presented here that were published by our group previously (Yu et al. 2016) can be obtained from the ArrayExpress (http://www.ebi.ac.uk/arrayexpress/) database under accession number E-MTAB-4641.
Data access
The data from this study have been submitted to ArrayExpress (https://www.ebi.ac.uk/arrayexpress/) under accession number E-MTAB-6569.
Supplementary Material
Acknowledgments
We acknowledge our colleagues at the Wales Gene Park for their technical and bioinformatic support in generating the NGS data. We thank Stephen P. Jackson for critically reading the manuscript. The work was supported by a Medical Research Council (MRC) research grant MR/K000926/1 to S.H.R.
Author contributions: S.H.R. and P.V.E. conceived of the study and experiments. P.V.E. and S.P.N. performed the wet laboratory experiments. P.V.E. analyzed the data. S.H.R. and P.V.E. interpreted the data and wrote the manuscript. The other authors contributed ChIP-Chip or 3D-DIP-Chip experiments.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.237198.118.
Freely available online through the Genome Research Open Access option.
References
- Adam S, Dabin J, Polo SE. 2015. Chromatin plasticity in response to DNA damage: the shape of things to come. DNA Repair 32: 120–126. 10.1016/j.dnarep.2015.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, Schuster SC, Pugh BF. 2007. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature 446: 572–576. 10.1038/nature05632 [DOI] [PubMed] [Google Scholar]
- Buschbeck M, Hake SB. 2017. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat Rev Mol Cell Biol 18: 299–314. 10.1038/nrm.2016.166 [DOI] [PubMed] [Google Scholar]
- Chen K, Xi Y, Pan X, Li Z, Kaestner K, Tyler J, Dent S, He X, Li W. 2013. DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res 23: 341–351. 10.1101/gr.142067.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204. 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg EC. 2003. DNA damage and repair. Nature 421: 436–440. 10.1038/nature01408 [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W. 1995. DNA repair and mutagenesis. ASM Press, Washington, DC. [Google Scholar]
- Ganapathi M, Palumbo MJ, Ansari SA, He Q, Tsui K, Nislow C, Morse RH. 2011. Extensive role of the general regulatory factors, Abf1 and Rap1, in determining genome-wide chromatin structure in budding yeast. Nucleic Acids Res 39: 2032–2044. 10.1093/nar/gkq1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillette TG, Yu S, Zhou Z, Waters R, Johnston SA, Reed SH. 2006. Distinct functions of the ubiquitin–proteasome pathway influence nucleotide excision repair. EMBO J 25: 2529–2538. 10.1038/sj.emboj.7601120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, Noble WS. 2011. FIMO: scanning for occurrences of a given motif. Bioinformatics 27: 1017–1018. 10.1093/bioinformatics/btr064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemette B, Bataille AR, Gevry N, Adam M, Blanchette M, Robert F, Gaudreau L. 2005. Variant histone H2A.Z is globally localized to the promoters of inactive yeast genes and regulates nucleosome positioning. PLoS Biol 3: e384 10.1371/journal.pbio.0030384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzder SN, Sung P, Prakash L, Prakash S. 1998. The DNA-dependent ATPase activity of yeast nucleotide excision repair factor 4 and its role in DNA damage recognition. J Biol Chem 273: 6292–6296. 10.1074/jbc.273.11.6292 [DOI] [PubMed] [Google Scholar]
- Hartley PD, Madhani HD. 2009. Mechanisms that specify promoter nucleosome location and identity. Cell 137: 445–458. 10.1016/j.cell.2009.02.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauer MH, Seeber A, Singh V, Thierry R, Sack R, Amitai A, Kryzhanovska M, Eglinger J, Holcman D, Owen-Hughes T, et al. 2017. Histone degradation in response to DNA damage enhances chromatin dynamics and recombination rates. Nat Struct Mol Biol 24: 99–107. 10.1038/nsmb.3347 [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh TH, Weiner A, Lajoie B, Dekker J, Friedman N, Rando OJ. 2015. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell 162: 108–119. 10.1016/j.cell.2015.05.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh THS, Fudenberg G, Goloborodko A, Rando OJ. 2016. Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nat Methods 13: 1009–1011. 10.1038/nmeth.4025 [DOI] [PubMed] [Google Scholar]
- Jiang C, Pugh BF. 2009. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet 10: 161–172. 10.1038/nrg2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent NA, Adams S, Moorhouse A, Paszkiewicz K. 2011. Chromatin particle spectrum analysis: a method for comparative chromatin structure analysis using paired-end mode next-generation DNA sequencing. Nucleic Acids Res 39: e26 10.1093/nar/gkq1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai WKM, Pugh BF. 2017. Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat Rev Mol Cell Biol 18: 548–562. 10.1038/nrm.2017.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T. 1993. Instability and decay of the primary structure of DNA. Nature 362: 709–715. 10.1038/362709a0 [DOI] [PubMed] [Google Scholar]
- Luijsterburg MS, van Attikum H. 2011. Chromatin and the DNA damage response: the cancer connection. Mol Oncol 5: 349–367. 10.1016/j.molonc.2011.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P, Smerdon MJ, Roberts SA, Wyrick JJ. 2016. Chromosomal landscape of UV damage formation and repair at single-nucleotide resolution. Proc Natl Acad Sci 113: 9057–9062. 10.1073/pnas.1606667113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. 2014. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol 15: 465–481. 10.1038/nrm3822 [DOI] [PubMed] [Google Scholar]
- Ozonov EA, van Nimwegen E. 2013. Nucleosome free regions in yeast promoters result from competitive binding of transcription factors that interact with chromatin modifiers. PLoS Comput Biol 9: e1003181 10.1371/journal.pcbi.1003181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo SE. 2015. Reshaping chromatin after DNA damage: the choreography of histone proteins. J Mol Biol 427: 626–636. 10.1016/j.jmb.2014.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo SE, Almouzni G. 2015. Chromatin dynamics after DNA damage: the legacy of the access–repair–restore model. DNA Repair 36: 114–121. 10.1016/j.dnarep.2015.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JR, Bennett MR, Evans KE, Yu S, Webster RM, Waters R, Skinner N, Reed SH. 2015. 3D-DIP-Chip: a microarray-based method to measure genomic DNA damage. Sci Rep 5: 7975 10.1038/srep07975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . 2018. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: https://www.R-project.org/. [Google Scholar]
- Raisner RM, Hartley PD, Meneghini MD, Bao MZ, Liu CL, Schreiber SL, Rando OJ, Madhani HD. 2005. Histone variant H2A.Z marks the 5′ ends of both active and inactive genes in euchromatin. Cell 123: 233–248. 10.1016/j.cell.2005.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KL, Smith JJ, Dasgupta A, Maqani N, Grant P, Auble DT. 2004. The NEF4 complex regulates Rad4 levels and utilizes Snf2/Swi2-related ATPase activity for nucleotide excision repair. Mol Cell Biol 24: 6362–6378. 10.1128/MCB.24.14.6362-6378.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed SH, You Z, Friedberg EC. 1998. The yeast RAD7 and RAD16 genes are required for postincision events during nucleotide excision repair: in vitro and in vivo studies with rad7 and rad16 mutants and purification of a Rad7/Rad16-containing protein complex. J Biol Chem 273: 29481–29488. 10.1074/jbc.273.45.29481 [DOI] [PubMed] [Google Scholar]
- Reed SH, Akiyama M, Stillman B, Friedberg EC. 1999. Yeast autonomously replicating sequence binding factor is involved in nucleotide excision repair. Genes Dev 13: 3052–3058. 10.1101/gad.13.23.3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn AL, Rao SS, Huang SC, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, et al. 2015. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci 112: E6456–E6465. 10.1073/pnas.1518552112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlecht U, Erb I, Demougin P, Robine N, Borde V, van Nimwegen E, Nicolas A, Primig M. 2008. Genome-wide expression profiling, in vivo DNA binding analysis, and probabilistic motif prediction reveal novel Abf1 target genes during fermentation, respiration, and sporulation in yeast. Mol Biol Cell 19: 2193–2207. 10.1091/mbc.e07-12-1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara A, Ogawa H, Ogawa T. 1992. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell 69: 457–470. 10.1016/0092-8674(92)90447-K [DOI] [PubMed] [Google Scholar]
- Talbert PB, Henikoff S. 2017. Histone variants on the move: substrates for chromatin dynamics. Nat Rev Mol Cell Biol 18: 115–126. 10.1038/nrm.2016.148 [DOI] [PubMed] [Google Scholar]
- Teng Y, Liu H, Gill HW, Yu Y, Waters R, Reed SH. 2008. Saccharomyces cerevisiae Rad16 mediates ultraviolet-dependent histone H3 acetylation required for efficient global genome nucleotide-excision repair. EMBO Rep 9: 97–102. 10.1038/sj.embor.7401112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y, Bennett M, Evans KE, Zhuang-Jackson H, Higgs A, Reed SH, Waters R. 2011. A novel method for the genome-wide high resolution analysis of DNA damage. Nucleic Acids Res 39: e10 10.1093/nar/gkq1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bakel H, Tsui K, Gebbia M, Mnaimneh S, Hughes TR, Nislow C. 2013. A compendium of nucleosome and transcript profiles reveals determinants of chromatin architecture and transcription. PLoS Genet 9: e1003479 10.1371/journal.pgen.1003479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage R, Zeeman AM, Degroot N, Gleig F, Bang DD, Vandeputte P, Brouwer J. 1994. The RAD7 and RAD16 genes, which are essential for pyrimidine dimer removal from the silent mating type loci, are also required for repair of the nontranscribed strand of an active gene in Saccharomyces cerevisiae. Mol Cell Biol 14: 6135–6142. 10.1128/MCB.14.9.6135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber CM, Ramachandran S, Henikoff S. 2014. Nucleosomes are context-specific, H2A.Z-modulated barriers to RNA polymerase. Mol Cell 53: 819–830. 10.1016/j.molcel.2014.02.014 [DOI] [PubMed] [Google Scholar]
- Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Münster S, Camblong J, Guffanti E, Stutz F, Huber W, Steinmetz LM. 2009. Bidirectional promoters generate pervasive transcription in yeast. Nature 457: 1033–1037. 10.1038/nature07728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadon AN, Van de Mark D, Basom R, Delrow J, Whitehouse I, Tsukiyama T. 2010. Chromatin remodeling around nucleosome-free regions leads to repression of noncoding RNA transcription. Mol Cell Biol 30: 5110–5122. 10.1128/MCB.00602-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarragudi A, Parfrey LW, Morse RH. 2007. Genome-wide analysis of transcriptional dependence and probable target sites for Abf1 and Rap1 in Saccharomyces cerevisiae. Nucleic Acids Res 35: 193–202. 10.1093/nar/gkl1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Owen-Hughes T, Friedberg EC, Waters R, Reed SH. 2004. The yeast Rad7/Rad16/Abf1 complex generates superhelical torsion in DNA that is required for nucleotide excision repair. DNA Repair 3: 277–287. 10.1016/j.dnarep.2003.11.004 [DOI] [PubMed] [Google Scholar]
- Yu Y, Teng Y, Liu H, Reed SH, Waters R. 2005. UV irradiation stimulates histone acetylation and chromatin remodeling at a repressed yeast locus. Proc Natl Acad Sci 102: 8650–8655. 10.1073/pnas.0501458102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Smirnova JB, Friedberg EC, Stillman B, Akiyama M, Owen-Hughes T, Waters R, Reed SH. 2009. ABF1-binding sites promote efficient global genome nucleotide excision repair. J Biol Chem 284: 966–973. 10.1074/jbc.M806830200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Teng Y, Waters R, Reed SH. 2011. How chromatin is remodelled during DNA repair of UV-induced DNA damage in Saccharomyces cerevisiae. PLoS Genet 7: e1002124 10.1371/journal.pgen.1002124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Deng Y, Reed SH, Millar CB, Waters R. 2013. Histone variant Htz1 promotes histone H3 acetylation to enhance nucleotide excision repair in Htz1 nucleosomes. Nucleic Acids Res 41: 9006–9019. 10.1093/nar/gkt688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Evans K, van Eijk P, Bennett M, Webster RM, Leadbitter M, Teng Y, Waters R, Jackson SP, Reed SH. 2016. Global genome nucleotide excision repair is organized into domains that promote efficient DNA repair in chromatin. Genome Res 26: 1376–1387. 10.1101/gr.209106.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Kasinathan S, Xin B, Rohs R, Henikoff S. 2015. ChEC-seq kinetics discriminates transcription factor binding sites by DNA sequence and shape in vivo. Nat Commun 6: 8733 10.1038/ncomms9733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Pugh BF. 2011. High-resolution genome-wide mapping of the primary structure of chromatin. Cell 144: 175–186. 10.1016/j.cell.2011.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based Analysis of ChIP-Seq (MACS). Genome Biol 9: R137 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yu Q, Olsen L, Bi X. 2012. Functions of protosilencers in the formation and maintenance of heterochromatin in Saccharomyces cerevisiae. PLoS One 7: e37092 10.1371/journal.pone.0037092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LJ, Gazin C, Lawson ND, Pagès H, Lin SM, Lapointe DS, Green MR. 2010. ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics 11: 237 10.1186/1471-2105-11-237 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.