

Abstract
New approaches to the neurobiology of posttraumatic stress disorder (PTSD) are needed to address the reported crisis in PTSD drug development. These new approaches may require the field to move beyond a narrow fear-based perspective, as fear-based medications have not yet demonstrated compelling efficacy. Antidepressants, particularly recent rapid-acting antidepressants, exert complex effects on brain function and structure that build on novel aspects of the biology of PTSD, including a role for stress-related synaptic dysconnectivity in the neurobiology and treatment of PTSD. Here, we integrate this perspective within a broader framework, i.e., a dual pathology model of (1) stress-related synaptic loss arising from amino acid-based pathology, and (2) stress-related synaptic gain related to monoamine-based pathology. Then, we summarize the standard and experimental (e.g., ketamine) pharmacotherapeutic options for PTSD, and discuss their putative mechanism of action and clinical efficacy.
1. Introduction
Posttraumatic stress disorder (PTSD) is a debilitating, and often chronic, psychiatric disorder that develops following exposure to severe trauma. PTSD is associated with intrusive memories, distressing dreams, dissociative reactions, avoidance of trauma-related stimuli, negative cognition and mood, increased arousal and irritability, and clinically significant distress and impairment in functioning. It is estimated that 70% of the world population have been exposed to trauma and that approximately 6% of trauma-exposed individuals develop PTSD (1). The prevalence is even higher in select populations with high trauma exposure. For example, the prevalence of PTSD is close to 25% in combat-exposed Veterans (2). Yet, unfortunately, to date the neurobiology of the disorder is not fully understood and available treatment options are limited, with only two FDA approved medications – both of which are slow-acting antidepressants (SAAD) (3).
Perhaps the greatest advancement in understanding the neurobiology of PTSD has been in the field of fear regulation. PTSD was found to be associated with deficits in fear extinction, increased generalization of fear, and a negative bias of viewing threat from neutral stimuli and feeling danger in a safe environment. These fear conditioning disturbances are believed to underlie many of the symptoms of PTSD and to correlate with some of the biological abnormalities identified in patients with PTSD (4–6). However, the mechanisms through which trauma induces fear dysregulation and extinction deficits are not entirely clear. In addition, to date, the drug development of fear-based pharmacotherapeutic approaches (e.g. d-cycloserine or propranolol) has been challenging (7–9), raising concerns whether narrowly targeting fear memory is the optimal path for drug development. Moreover, the only PTSD drugs with reproducible efficacy are antidepressants. These drugs are believed to target the reversal of trauma- and stress-induced synaptic dysconnectivity and have shown efficacy across a number of stress-related disorders with no known prominent fear dysregulation (10–13).
Convergent evidence implicates stress in the pathophysiology of trauma-related impairment in fear extinction (4; 14). Moreover, accumulating literature increasingly implicates glutamate dysregulation and synaptic loss in the pathophysiology and treatment of PTSD (11; 12). Together, these findings have led to the proposition of a synaptic connectivity model of PTSD based on impairment in stress response, resulting in sustained threat paradigm and chronic stress pathology (CSP) (11–13). The synaptic dysconnectivity model provides a framework for investigating and understanding the biological predispositions, pathophysiology, and treatment of PTSD (Fig. 1). In the current review, we present a synaptic and a network-based models of PTSD, describe a “vicious cycle” of chronic stress pathology, and propose a dual pathology model associating (1) the stress-related synaptic loss in the prefrontal cortex and hippocampus with amino acid-based pathology, and (2) the stress-related synaptic gain in the nucleus accumbens, and presumably the basolateral amygdala, with monoamine-based pathology. Then, we summarize the standard and experimental (e.g., ketamine) pharmacotherapeutic options for PTSD, and discuss their putative mechanism of action and clinical efficacy.
Figure 1. The “vicious cycle” of chronic stress pathology: A synaptic model of posttraumatic stress disorder (PTSD).
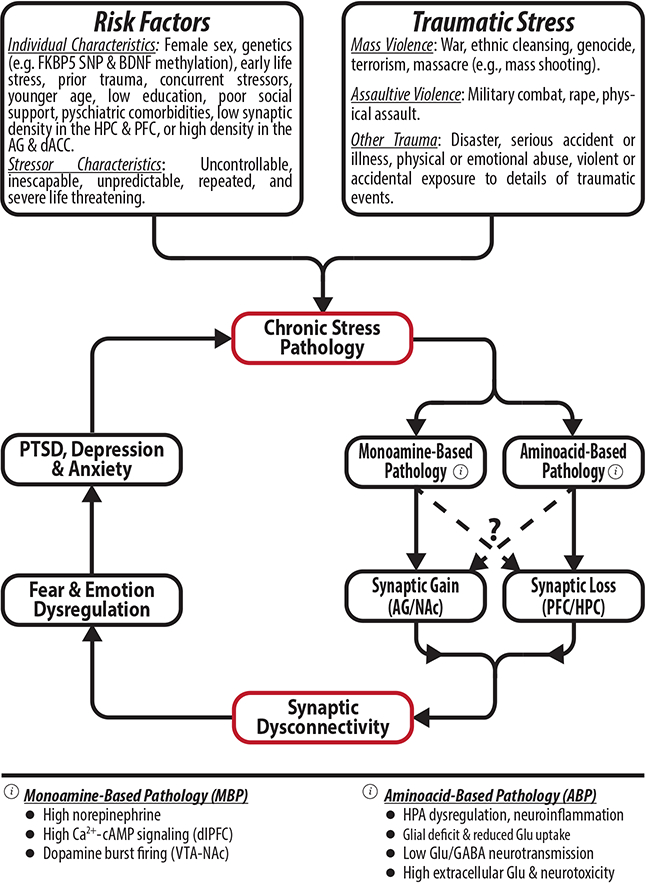
It is believed that traumatic stress interacts with predisposing factors to precipitate chronic stress pathology, consistent with localized synaptic loss and/or gain, leading to behavioral disruptions, which further exacerbate the chronic stress pathology. Abbreviations: FKBP5 = FK506-binding protein 5; SNP = single nucleotide polymorphism; BDNF = brain derived neurotrophic factor; HPC = hippocampus; PFC = prefrontal cortex; AG = amygdala; dACC = dorsal anterior cingulate; NAc = nucleus accumbens; dlPFC = dorsolateral PFC; VTA = ventral tegmental area; HPA = hypothalamic-pituitary axis; Glu = glutamate; The figure was adapted with permission from the Emerge Research Program (emerge.care).
2. Neurobiology of PTSD
2.a. Stress Response
Life threatening traumatic events, associated with PTSD psychopathology, typically induce a prolonged stress response, whether the stressor was acute (e.g., mass shooting) or chronic (e.g., combat). Thus, while transient (minutes to hours) stress responses may enhance plasticity, improve cognition, and promote resilience (15–17), traumatic stressors are often associated with chronic (days to weeks) stress responses that are detrimental to the brain and are often accompanied with behavioral disturbances (18; 19). In animals, stress-related synaptic loss and the associated behavioral changes are evident within days to weeks of traumatic stress, and are reversible typically within 2–4 weeks after the stress (18; 20). In humans, most trauma-exposed individuals transiently experience symptoms associated with PTSD. However, individuals still symptomatic 4 weeks after the trauma may have lasting illness (1; 21).
The propensity to exhibit lasting stress responses is complex and reflects genetics, history of environmental exposures, stage of life, and features of the traumatic stress and post-traumatic social context (21). Psychopathology risk increases in proportion to the severity of the stressor. Inescapable, uncontrollable, unpredictable, repeated, and severe stressors pose an increased probability to initiate a sustained threat paradigm leading to chronic stress responses with subsequent biological and behavioral abnormalities (17; 18; 20; 22). Similarly, biopsychosocial vulnerabilities (e.g., female sex, history of early life stress, or predisposing neuronal disturbances) represent an increased risk for fear-related disturbances leading to sustained threat paradigm and prolonged stress responses following severe traumatic events (21; 23; 24).
This complexity contributes to variability in the reported rates of PTSD. On average, fewer than 10% of individuals exposed to extreme stress develop PTSD. But the PTSD rate is 20% following assaultive violence and up to 50% in rape victims (25). Among refugees, PTSD rates range from 4 to 86%, with higher rates associated with the severity of the trauma exposure (26). Among tattooed Auschwitz Holocaust survivors, 80% suffered intrusive recollections, 90% experienced recurrent nightmares, and 100% endured sleep disturbances (27). Together, the data suggest that, despite the level of resiliency of an individual, increasing the magnitude of the traumatic load would eventually precipitate psychopathology. Thus, the variability in posttraumatic outcomes could be due to an individualized threshold at which a person develops a prolonged stress response and subsequent psychopathology. In this context, resiliency could be conceptualized as not only whether a trauma victim would suffer from PTSD symptoms, but rather expressed as reduced PTSD severity, reduced duration of PTSD symptoms, or reduced impact of PTSD symptoms on overall quality of life (i.e., less disability, less disruption of social relationships, and less development of comorbid diagnoses such as addiction).
2.b. Synaptic Model of Chronic Stress Pathology (CSP)
Extreme stressors, particularly when repeated, induce neuronal remodeling associated with regional reductions and increases in synaptic density. In animal studies, the CSP reduction in synaptic connectivity has been mostly demonstrated in the prefrontal cortex (PFC) and the hippocampus, while the increases in synaptic connectivity were most evident in the nucleus accumbens (NAc) and the basolateral amygdala (18; 28).
In the PFC and hippocampus, prolonged stress responses have been associated with disruption in glucocorticoid signaling, increased neuroinflammation, reduced brain derived neurotrophic factor (BDNF), and astrocytic deficits along with reduced uptake of synaptically-released glutamate, leading to increased extracellular glutamate and excitotoxicity (10; 29–31). Particularly, prolonged stress response maintains a paradoxical increase in extracellular glutamate despite a considerable reduction in glutamate neurotransmission, N-methyl-D-aspartate receptors (NMDARs), and α-amino-3-hydroxy-5-methyl-4-isoxazloepropionic acid receptors (AMPARs) (19; 32; 33). These molecular changes precipitate neuronal atrophy consistent with reduced dendritic length and arborization, and reduction in synaptic density and neurotransmission strength. In preclinical studies, this synaptic loss and hypoconnectivity is directly associated with behavioral abnormalities, including mood and anxiety dysregulation (18; 34). Moreover, these CSP behavioral disturbances are normalized following the reversal of the synaptic deficit, by both SAAD and rapid-acting antidepressants (RAAD; e.g. ketamine) (29; 35).
In the amygdala, CSP is associated with functional and structural changes consistent with reduced synaptic connectivity in the medial amygdala, but increased BDNF and synaptic connectivity in the basolateral amygdala (36–38). Notably, a single stressor is sufficient to upregulate BDNF in the basolateral amygdala, which is evident 1 day post-stress and lasts for at least 10 days (38). The single stressor also gradually increases basolateral amygdala synaptogenesis over the 10-day period, which is paralleled by a gradual increase in anxiety-like behavior (39). Similarly, a single large dose of corticosterone induces sustained anxiety-like behavior and increased basolateral amygdala hypertrophy, lasting more than 10 days (40). Moreover, after withholding the stressor, hippocampal/PFC synaptic loss, downregulation of BDNF, and related behavioral disturbances recover within 2–4 weeks, while the basolateral amygdala synaptic hyperconnectivity, upregulation of BDNF, and related anxiety-like behavior are not reversible within the same time period (38; 41–43). In the NAc, CSP is also associated with neuronal hypertrophy, including increased BDNF, dendritic length and branching, and synaptic density and strength (44–52). Similarly, the NAc hypertrophy is associated with behavioral impairment and is reversed by both SAAD and RAAD (28; 53; 54).
2.c. Dual Pathology Model: Amino Acid-Based vs. Monoamine-Based Pathology
A major challenge in PTSD, depression, and anxiety research is apparent “contradictory” biological findings across human studies. This is best exemplified in the reports of reduced hippocampal volume in some, but not all PTSD cohorts (55; 56). Similarly, increased amygdala volume was found in some, but not all PTSD patients (57; 58). Together, these seemingly contradictory findings raise the question whether trauma and stress psychopathology is associated with two distinct underlying pathophysiological processes, one related to synaptic loss and another related to synaptic gain.
Indeed, as described earlier, the synaptic model of CSP provides abundant evidence associating localized synaptic loss with Amino Acid-Based Pathology (ABP), consistent with glutamate dysregulation and excitotoxicity. Conversely, synaptic gain – particularly in the NAc – is associated with Monoamine-Based Pathology (MBP), consistent with disruption in catecholamine (i.e., adrenaline, noradrenaline, and dopamine) signaling (Fig. 1). Integrating these preclinical data along with accumulating clinical evidence, we recently described a dual pathology model proposing trauma and stress pathology is independently associated with both ABP and MBP (59). However, the characteristic of the stressor and the individual biopsychosocial predispositions may differentially lead to prominent amino acid disruption and synaptic loss (i.e., ABP) or monoamine dysregulation and synaptic gain (i.e., MBP) (59).
In the dual pathology model, ABP would be more closely associated with prominent glutamate dysregulation and synaptic loss, as evident by treatment resistance to monoaminergic drugs, alterations in glutamate and GABA markers and signaling, and gross structural correlates of synaptic loss on magnetic resonance imaging (MRI) (59–61). In contrast, MBP would be more closely associated with monoamine dysregulation and synaptic gain, as evident by enhanced response to monoaminergic drugs, signs of autonomic dysregulation, and gray matter hypertrophy [reviewed in (29; 59)}]. Consistent with this model, animal studies have shown that the type and magnitude of a stressor determine the extent and nature of the dopaminergic response in the NAc (62; 63), and its related behavioral pathology (50; 64). Moreover, it has been also shown that only a subgroup of animals develops MBP NAc hypertrophy and related behavioral disturbances (65). Therefore, the stressor characteristics, along with individual predisposition, may dictate the pattern of biological injury (i.e., MBP vs. ABP) and related behavioral abnormalities (e.g., PTSD or depression). For example, certain stressors, such as single prolonged stress, are more likely to recapitulate key symptoms of PTSD (66; 67). Comparably, differing brain regions may have variable response to a unique stress, for example, brief uncontrollable stress was found to induce synaptic loss in the infralimbic (medial PFC, involved in extinction), but not prelimbic area (dorsal PFC, involved in fear acquisition) (68).
The dual pathology model was proposed initially for major depressive disorder (MDD) (29; 59; 61). However, various lines of evidence also support the applicability of this model for PTSD. First, traumatic stress may trigger either PTSD and/or depression, as well as increase the risk of several other stress-related disorders (69). Second, the preclinical CSP literature summarized earlier clearly implicates both ABP and MBP in PTSD. Third, in addition to the extensive human evidence relating synaptic loss and ABP to PTSD (70–75), there is strong evidence of catecholamine dysregulation and MBP in patients with PTSD (76–78) – where increases in catecholamine are believed to simultaneously weaken dorsolateral PFC (dlPFC) synaptic connectivity and strengthen neuronal activity in the amygdala and striatum (which includes the NAc) (50–52; 79). Notably, the “viscous cycle” of chronic stress pathology (Fig. 1) suggests that initial MBP behavioral disturbances further exacerbate the stress magnitude, which could eventually lead to ABP and synaptic loss. Thus, the discrepancy in the amygdala findings in PTSD (i.e., both hypertrophy and hypotrophy) may also reflect the time course of the disorder, with chronic suffering from severe PTSD leading to more prominent ABP and synaptic loss. This hypothesized time course effect of an MBP to ABP switch is supported by the fact that compiled data of multiple cohorts or meta-analyses often succeed in demonstrating the ABP-related biomarkers (e.g., gray matter deficits), but fails to show statistically significant structural evidence of synaptic gain (80–83).
Finally, considering the noticeable role of chronic stress pathology across many psychiatric disorders, the dual pathology model presented is unlikely to be limited to depression and PTSD, but rather common to several psychiatric disorders, including generalized anxiety disorder (GAD), obsessive compulsive disorder (OCD), panic disorder, and bipolar depression (60; 84–92). Together, the presented models hypothesize that CSP, and related synaptic dysconnectivity (Fig. 1), are common across many psychiatric disorders. Most importantly, targeting synaptic connectivity is a convergent pathway across antidepressants – a class of medications that has shown efficacy in alleviating symptoms of major depression, PTSD, GAD, OCD, panic disorder, bipolar depression, and other disorders with a considerable chronic stress component. Therefore, while the synaptic pathology described by ABP and MBP is not specific to PTSD, it is increasingly evident that targeting these synaptic disturbances is essential for successful pharmacological treatment (e.g., both traditional and rapid acting antidepressants – drugs that show efficacy in PTSD Treatment – are believed to exert their effects by affecting neuroplasticity and synaptic restructuring (18; 29; 30; 93)). Moreover, there is well replicated evidence that markers of MBP (e.g., lack gray matter deficits) predicts better response to monoaminergic antidepressants, with some studies associating ABP biomarkers (e.g., smaller hippocampal volume) with enhanced response to amino acid-based antidepressants (e.g., ketamine or riluzole) (60; 94).
2.d. Network-Based Model of PTSD
Trauma-related structural and functional synaptic dysconnectivity has been demonstrated in animals and in humans within brain regions critical to anxiety, mood, and fear regulation (95). Thus, the question is raised how the pattern of CSP synaptic dysconnectivity may dictate the prominent clinical presentation, being it PTSD, depression, or another stress-related psychopathology.
In the PTSD literature, early models presented an elegant circuit-based hypothesis supported by a wealth of neuroimaging studies (95). It is believed that reduced hippocampal, but increased amygdala, synaptic connectivity might be predisposing factors that are exacerbated by the index traumatic event to induce disruption in fear memory function (96; 97). This pattern of synaptic dysconnectivity would favor striatal-dependent “habitual” over hippocampal-dependent “cognitive” memory (98–100). In patients with PTSD, it is thought that the heightened fear and arousal are correlates of underlying hyperactivity in the amygdala and dorsal anterior cingulate, putatively due to loss of top-down control from a hypoactive PFC, as well as due to fear extinction deficits driven by dysfunction in the medial PFC and hippocampus (95; 101).
Recently, a more comprehensive model – known as the triple network model – has been proposed (102; 103). Similar to the synaptic model of CSP, the triple network model is not specific to PTSD, but rather to a broad range of psychopathological presentations (102). This network-based model extends the circuit-based hypothesis in PTSD by incorporating the wide disruption in three large-scale intrinsic connectivity networks, that are the default mode network, the central executive network, and the salience network (Fig. 2) (104). The default mode network spatially spans important regions in the posterior cingulate cortex, medial PFC, and medial temporal lobe including the hippocampus (105). It is known to engage in self-referential, introspective processes, and autobiographical memory. Consistent with its function, it is most active at rest, and hypoactive during goal-oriented tasks. In individuals with PTSD, the default mode is known to be hypoactive and weakly interconnected (106; 107), which is thought to parallel symptoms of dissociation, avoidance, and intrusiveness (102; 103).
Figure 2. A network-based model of posttraumatic stress disorder (PTSD).
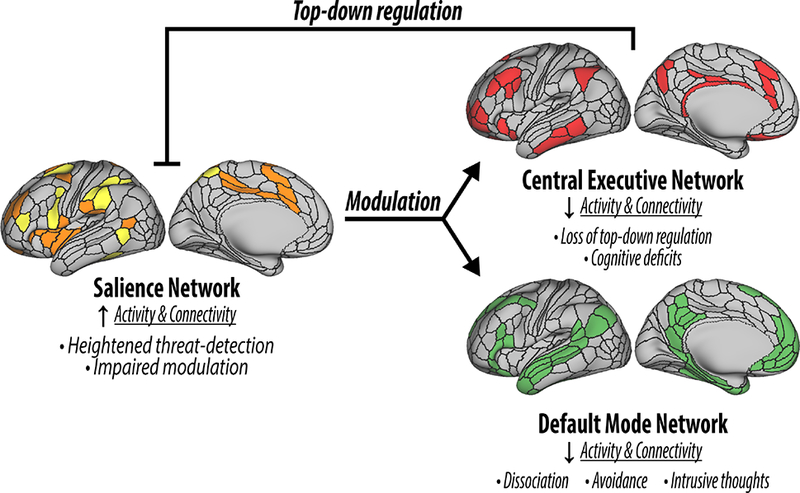
The figure depicts the cortical representations of the salience network (SN; orange and yellow), central executive network (CEN; red), and default mode network (DMN; green). PTSD has been associated with hyperactive SN leading to heightened threat-detection and impaired modulation of the CEN and DMN. In turn, CEN and DMN deficits are associated with disruption in top-control, as well as several PTSD related symptomatology. The figure was adapted with permission from the Emerge Research Program (emerge.care).
The central executive network, which is anchored primarily in the dlPFC, is known to engage in goal- directed behavior and top-down regulation of emotions. Here, evidence of dysconnectivity in PTSD is thought to mirror a loss of modulation over fear/threat-detection circuits, and deficits in cognition and executive function. In line with this hypothesis, enhanced connectivity between the central executive network and default mode network was found in individuals who respond to exposure therapy, suggesting a compensatory role (108; 109). The salience network, has important nodes in the insula, dorsal anterior cingulate cortex, and possibly the amygdala. The salience network is implicated to the response to subjective salience and arbitrates between central executive network (task-positive) and default mode network (task-negative) accordingly (103; 110). Dysconnectivity in the salience network is thought to impair this arbitration function, resulting in a low threshold for saliency and a hypervigilant state (107; 111). In summary, by extending the fronto-limbic fear circuitry model, the triple network model may better account for the varying endophenotypes in PTSD, as well as the symptomology of common comorbidities (102; 103). In addition, it provides a construct for better understanding of the interplay between the pattern of microstructural synaptic disturbances and the complexity of clinical psychopathological presentations.
3. Pharmacotherapy of PTSD
3.a. Complexity of PTSD symptomatology
PTSD is distinctive among psychiatric disorders because by definition it depends on a type of environmental exposure. Moreover, in adults, the PTSD diagnosis has higher test-retest reliability compared to most psychiatric disorders (112). While these characteristics foretell reproducible neurobiological and pharmacotherapeutic studies, the complexity of PTSD symptomatology and high comorbidity considerably hindered the consistency of most biological findings in PTSD, including the clinical efficacy of standard and investigational medications. In fact, the constellation of PTSD symptoms appears to recapitulate core symptomatology of most psychiatric disorders including anxiety, depression, addictive, impulsive, obsessive-compulsive (e.g., intrusive thoughts), and dissociative/psychotic disorders. Furthermore, the presence of “pure” PTSD in the absence of psychiatric comorbidities is often the exception rather than the rule (112). Together, this complexity has resulted in a pattern of PTSD drugs showing promise in early clinical trials, then later fail to show consistent efficacy in replication and/or larger trials (113; 114). Moreover, the efficacy of many of the most commonly prescribed medications for PTSD has never been tested adequately (e.g., trazodone, quetiapine, mirtazapine, gabapentin, etc.). This state of affairs has been described as a crisis requiring a national commitment to PTSD pharmacotherapy research (3).
Currently, PTSD focused psychotherapies are considered first line of treatment, with pharmacotherapy as first or second line in some guidelines (115). Here, we present a list of standard and investigational drugs that were studied in PTSD. Of these, only paroxetine and sertraline – both are serotonin reuptake inhibitors – are FDA approved for PTSD treatment. In addition, to date, meta-analyses have overall only supported the clinical efficacy of few drugs – all of which are antidepressants, which underscores the prospect of developing drugs that target trauma- and stress-related pathology (e.g., ketamine targeting synaptic dysconnectivity) compared to PTSD specific abnormalities (e.g., D-cycloserine or propranolol targeting fear circuitry).
3.b. Traditional Drugs: Slow-Acting Antidepressants
In the 1950s, it was serendipitously discovered that iproniazid and imipramine have antidepressant effects, drugs that were being developed to treat tuberculosis and schizophrenia, respectively. Subsequently, other antidepressants that possess a common mechanism of increasing synaptic serotonin have been developed. Among these drugs are the serotonin reuptake inhibitors, the tricyclic antidepressants (e.g., imipramine), and monoamine oxidase inhibitors (e.g., iproniazid). These traditional drugs are SAAD requiring weeks to months to achieve full therapeutic benefit. SAAD have shown efficacy in several depressive, anxiety, and trauma-related disorders. The mechanisms through which SAAD exert their therapeutic effects are not fully known. The drug’s in vitro pharmacodynamics of increasing synaptic serotonin, combined with laboratory studies associating serotonin depletion with depressive symptoms, have resulted in the rise of the “serotonin deficiency” hypothesis of depression (116). However, several lines of evidence have questioned this model, including the discrepancy between the acute increase in serotonin and the delayed therapeutic response following treatment with SAAD (117). More recently, accumulating evidence in animal models have shown that repeated, but not acute, treatment with SAAD reverses the depressive-like behavior by increasing BDNF in brain structures known to have stress-related synaptic loss (30; 118). Consistent with the synaptic model of CSP, the neurotrophic hypothesis of SAAD provides putative explanation for the delayed onset of treatment response, as well as for the efficacy of SAAD in several psychiatric disorders with a prominent stress-related pathology.
In PTSD, the highest evidence of clinical efficacy is mostly for trials investigating paroxetine, sertraline, and venlafaxine (119). Other SAAD have shown efficacy in some meta-analyses, but these SAAD were investigated mostly in small samples clinical trials (120). Although some SAAD are more effective than placebo in reducing PTSD symptoms, the effect size of these drugs is often small, with high treatment resistance in certain population – e.g., in Veterans (120–122).
3.c. Novel Drugs: Rapid-Acting Antidepressants
In the late 1990s, a study exploring the role of NMDARs in the pathology of depression unexpectedly found RAAD effects following infusion of a single subanesthetic dose of ketamine (123). This finding has since been replicated in several studies and other RAAD are being developed. Ketamine is typically administered intravenously over 40 minutes, with the antidepressant effects evident within few hours and lasting for approximately two weeks following single infusion. These effects are maintained by repeated 2–3 times per week administration in short-term studies (124; 125). Ketamine is believed to exert its RAAD effects by inducing a transient surge in post-synaptic glutamate activation, leading to upregulation of BDNF and increased synaptic formation (126). In animals, it was shown that ketamine reverses the stress-related synaptic loss/gain within 24h of administration (54; 127). Supportive evidence, paralleling the preclinical findings, have been reported in humans suggesting a rapid structural and functional reversal of synaptic dysconnectivity within 24h of treatment of depressed patients (59; 128; 129). Consistent with the synaptic model of CSP, ketamine’s therapeutic effects extend beyond depression and have been suggested in several stress-related disorders (130–133), particularly in tre2ating suicidal ideations (134; 135).
In PTSD, a pilot randomized controlled trial showed moderate to large effect size in reducing PTSD symptoms following administration of a single infusion of ketamine compared to an active placebo control (136). Other retrospective or case report studies have supported the potential utility of subanesthetic doses of ketamine in treating PTSD (124; 137). However, it remains to be seen whether these early reports would be replicated in future confirmatory randomized controlled trials (clinicaltrials.gov/ct2/show/NCT02655692). In addition, ketamine induces transient perceptual disturbances during infusion and is used as a recreational drug. Thus, the safety and efficacy of repeated ketamine administration would need to be confirmed prior to adopting it as standard treatment.
3.d. Other Drugs
In addition to SAAD and RAAD, several drugs have been tested in PTSD. Unfortunately, many of these drugs have failed to show reproducible therapeutic benefit. Despite the failure in confirmatory clinical trials and/or in meta-analyses, several of these drugs continue to be frequently used as augmentation in treating PTSD, particularly second-generation antipsychotics (e.g., risperidone), prazosin, and benzodiazepines. Risperidone is the most studied antipsychotic in PTSD, with early promising result, yet failed in a large confirmatory augmentation randomized controlled trial (113). Prazosin, an α1-adrenoreceptor antagonist, showed early promise in treating PTSD, particularly sleep-related symptoms. However, several studies have failed to reproduce these effects and a recent large randomized controlled trial has shown no differences between prazosin and placebo in treating overall PTSD or sleep-related symptoms (114). Benzodiazepine failed to show therapeutic effects in PTSD, with evidence that they may worsen symptoms or increase risk of developing PTSD (138). Other drugs that have been tested with limited to no evidence to support their use, include: topiramate, buproprion, divaloproex, NK1R antagonists, and ganaxolone (120; 121).
3.e. PTSD Prevention
Hydrocortisone and β-adrenergic receptor blockers (e.g., propranolol) are the main pharmacological approaches investigated to prevent the development of PTSD following trauma. Based on evidence of low cortisol in patient with PTSD (139), it was suggested that administering hydrocortisone shortly after the traumatic event may reduce the risk of developing PTSD. Similarly, preclinical evidence has shown that propranolol can block consolidation of traumatic memory, which raised the possibility that administering propranolol shortly after the trauma may reduce the risk of PTSD. To date, randomized controlled trials found no preventive effects following propranolol administration post trauma exposure, although these were mostly small trials with varied administration regimen and timing (8; 9). Conversely, there is preliminary evidence to suggest that hydrocortisone may have beneficial effects in reducing PTSD risk (8; 9). Other pharmacological preventive approaches that were reported, but which currently have no strong evidence to support their use, include serotonin reuptake inhibitors, temazepam, gabapentin, albuterol, morphine, and oxytocin (9; 140; 141).
3.f. Psychotherapy Augmentation
Preclinical studies have shown that the partial NMDAR agonist D-cycloserine facilitates the excitation of fear (142). Considering the hypothesized extinction failure in PTSD, several studies have investigated whether administration of D-cycloserine would augment the therapeutic effect of fear exposure in psychotherapy for PTSD. To date, the results have been mixed, with some studies showing beneficial effects, while others showing worsening of symptoms (7; 11). It has been suggested that pretreatment with D-cycloserine might indiscriminately enhance the consolidation of memory, leading to improvement if the patient successfully achieved extinction during the fear exposure session or to inadvertently enhance reconsolidation of trauma memory and worsening of symptoms if the exposure session was unsuccessful (143). A recent small randomized controlled trial suggested beneficial effects combining propranolol with trauma reactivation, a pilot finding that awaits replication (144). Another approach for PTSD psychotherapy augmentation is the administration of 3,4-methylenedioxy-methamphetamine (MDMA), a recreational drug known as “ecstasy”, which induces transient serotonin and norepinephrine release (145; 146). MDMA is currently in phase 3 trials and has received an FDA breakthrough designation, which facilitates the approval of the drug if the randomized controlled trial results were confirmatory. Considering the addiction liability of the drug, strong evidence of safety and therapeutic benefit would be required.
4. Conclusions
Psychiatric illnesses are often conceptualized as entities with complex interaction between biological, psychological, and social components. Notably, chronic stress – as predisposing, triggering, or perpetuating factor – is a major component of most psychiatric disorders. Moreover, biomarkers suggestive of stress-related synaptic dysconnectivity (e.g., gray matter deficits) were reported across most severe psychiatric disorders. Furthermore, antidepressants, that are believed to exert their clinical benefit through the reversal of stress-related synaptic dysconnectivity, have shown clinical efficacy in several psychiatric disorders. Together, this converging evidence strongly suggests CSP is a common pathophysiological process across multiple disorders. In recent years, clinical neuroscience research has begun to unravel the unique dysconnectivity patterns through which CSP contribute to distinct clinical presentations, including PTSD – highlighting a triple network model of psychopathology. Finally, although pharmacotherapeutic options to alleviate PTSD are available, these drugs have minimal (e.g., serotonin reuptake inhibitors) or inconsistent efficacy (e.g., prazosin or risperidone). While SAAD have minimal effects on synaptic remodeling and PTSD symptoms, RAAD exert strong synaptic remodeling and pilot evidence suggest high efficacy in alleviating PTSD. It remains to be seen whether the initial promise of these novel drugs would eventually translate into robust and effective pharmacological treatment for millions of individuals suffering from PTSD.
Table 1. Drugs commonly used to treat PTSD.
List of commonly used pharmacological agents in the treatment of PTSD, their putative mechanism of action, and their clinical therapeutic aims. * indicates FDA approval for PTSD.
| Medication class | Commonly used agents | Putative mechanism | Therapeutic aim |
|---|---|---|---|
| Antidepressants (slow-acting) |
Paroxetine*; Sertraline*; fluoxetine |
Serotonin reuptake inhibition | Overall symptoms (115; 120; 121) |
| Venlafaxine | Serotonin-norepinephrine reuptake inhibition | Overall symptoms (115; 120; 121); possibly less effective for hyperarousal symptoms (147) |
|
| Mirtazapine | Serotonin 5-HT2 and 5- HT3 and adrenoreceptor α2 antagonism | Overall symptoms (115; 120) | |
| Desipramine | Norepinephrine reuptake inhibition | Overall symptoms (115; 120) | |
| Phenelzine | Monoamine oxidase inhibition | Overall symptoms (115; 120) | |
| Antipsychotics (second-generation) |
Risperidone; quetiapine |
Dopamine D2 and serotonin 5-HT2 antagonism | Primarily used for sleep symptoms with questionable evidence of efficacy (113); may possibly improve other symptoms (121; 148) |
| Anxiolytics or sedative-hypnotics | Prazosin | α1-adrenoreceptor antagonism |
Primarily used for sleep symptoms with inconsistent evidence of efficacy (114; 149); may possibly improve other symptoms (150) |
| Alprazolam; clonazepam |
GABAA receptor agonism | No evidence of efficacy; possible worsening of symptoms (138) |
Footnotes
Conflict of Interest statement
CGA has served as a consultant and/or on advisory boards for Genentech and Janssen, and editor of Chronic Stress for Sage Publications, Inc.; JHK is a consultant for AbbVie, Inc., Amgen, Astellas Pharma Global Development, Inc., AstraZeneca Pharmaceuticals, Biomedisyn Corporation, Bristol-Myers Squibb, Eli Lilly and Company, Euthymics Bioscience, Inc., Neurovance, Inc., FORUM Pharmaceuticals, Janssen Research & Development, Lundbeck Research USA, Novartis Pharma AG, Otsuka America Pharmaceutical, Inc., Sage Therapeutics, Inc., Sunovion Pharmaceuticals, Inc., and Takeda Industries; is on the Scientific Advisory Board for Lohocla Research Corporation, Mnemosyne Pharmaceuticals, Inc., Naurex, Inc., and Pfizer; is a stockholder in Biohaven Pharmaceuticals; holds stock options in Mnemosyne Pharmaceuticals, Inc.; holds patents for Dopamine and Noradrenergic Reuptake Inhibitors in Treatment of Schizophrenia, U.S. Patent No. 5,447,948 (issued Sep 5, 1995), and Glutamate Modulating Agents in the Treatment of Mental Disorders, U.S. Patent No. 8,778,979 (issued Jul 15, 2014); and filed a patent for Intranasal Administration of Ketamine to Treat Depression. U.S. Application No. 14/197,767 (filed on Mar 5, 2014); U.S. application or Patent Cooperation Treaty international application No. 14/306,382 (filed on Jun 17, 2014); All other authors disclose no conflict of interest.
References
- 1.Koenen KC, Ratanatharathorn A, Ng L, McLaughlin KA, Bromet EJ, et al. 2017. Posttraumatic stress disorder in the World Mental Health Surveys. Psychol Med 47:2260–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fulton JJ, Calhoun PS, Wagner HR, Schry AR, Hair LP, et al. 2015. The prevalence of posttraumatic stress disorder in Operation Enduring Freedom/Operation Iraqi Freedom (OEF/OIF) Veterans: a meta-analysis. J Anxiety Disord 31:98–107 [DOI] [PubMed] [Google Scholar]
- 3.Krystal JH, Davis LL, Neylan TC, M AR, Schnurr PP, et al. 2017. It Is Time to Address the Crisis in the Pharmacotherapy of Posttraumatic Stress Disorder: A Consensus Statement of the PTSD Psychopharmacology Working Group. Biol Psychiatry 82:e51–e9 [DOI] [PubMed] [Google Scholar]
- 4.Maeng LY, Milad MR. 2017. Post-Traumatic Stress Disorder: The Relationship Between the Fear Response and Chronic Stress. Chronic Stress 1:2470547017713297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.VanElzakker MB, Dahlgren MK, Davis FC, Dubois S, Shin LM. 2014. From Pavlov to PTSD: the extinction of conditioned fear in rodents, humans, and anxiety disorders. Neurobiol Learn Mem 113:3–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maren S, Phan KL, Liberzon I. 2013. The contextual brain: implications for fear conditioning, extinction and psychopathology. Nat Rev Neurosci 14:417–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker JF, Cates ME, Luthin DR. 2017. D-cycloserine in the treatment of posttraumatic stress disorder. Mental Health Clinician 7:88–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sijbrandij M, Kleiboer A, Bisson JI, Barbui C, Cuijpers P. 2015. Pharmacological prevention of post-traumatic stress disorder and acute stress disorder: a systematic review and meta-analysis. Lancet Psychiatry 2:413–21 [DOI] [PubMed] [Google Scholar]
- 9.Amos T, Stein DJ, Ipser JC. 2014. Pharmacological interventions for preventing post-traumatic stress disorder (PTSD). Cochrane Database Syst Rev:CD006239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abdallah CG, Sanacora G, Duman RS, Krystal JH. 2015. Ketamine and rapid-acting antidepressants: a window into a new neurobiology for mood disorder therapeutics. Annu Rev Med 66:509–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Averill LA, Purohit P, Averill CL, Boesl MA, Krystal JH, Abdallah CG. 2017. Glutamate dysregulation and glutamatergic therapeutics for PTSD: Evidence from human studies. Neurosci Lett 649:147–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abdallah CG, Southwick SM, Krystal JH. 2017. Neurobiology of posttraumatic stress disorder (PTSD): A path from novel pathophysiology to innovative therapeutics. Neurosci Lett 649:130–2 [DOI] [PubMed] [Google Scholar]
- 13.Krystal JH, Abdallah CG, Averill LA, Kelmendi B, Harpaz-Rotem I, et al. 2017. Synaptic Loss and the Pathophysiology of PTSD: Implications for Ketamine as a Prototype Novel Therapeutic. Curr Psychiatry Rep 19:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maren S, Holmes A. 2016. Stress and Fear Extinction. Neuropsychopharmacology 41:58–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuen EY, Liu W, Karatsoreos IN, Feng J, McEwen BS, Yan Z. 2009. Acute stress enhances glutamatergic transmission in prefrontal cortex and facilitates working memory. Proc Natl Acad Sci U S A 106:14075–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuen EY, Liu W, Karatsoreos IN, Ren Y, Feng J, et al. 2011. Mechanisms for acute stress-induced enhancement of glutamatergic transmission and working memory. Molecular Psychiatry 16:156–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Averill LA, Averill CL, Kelmendi B, Abdallah CG, Southwick S. 2018. Stress Response Modulation Underlying the Psychobiology of Resilience. Curr Psychiatry Rep:In Press [DOI] [PubMed] [Google Scholar]
- 18.McEwen BS. 2017. Neurobiological and Systemic Effects of Chronic Stress. Chronic Stress 1:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuen EY, Wei J, Liu W, Zhong P, Li X, Yan Z. 2012. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron 73:962–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Popoli M, Yan Z, McEwen BS, Sanacora G. 2012. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci 13:22–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ozer EJ, Best SR, Lipsey TL, Weiss DS. 2003. Predictors of posttraumatic stress disorder and symptoms in adults: a meta-analysis. Psychol Bull 129:52–73 [DOI] [PubMed] [Google Scholar]
- 22.Sutanto W, de Kloet ER. 1994. The use of various animal models in the study of stress and stress-related phenomena. Lab Anim 28:293–306 [DOI] [PubMed] [Google Scholar]
- 23.Zoladz PR, Diamond DM. 2013. Current status on behavioral and biological markers of PTSD: a search for clarity in a conflicting literature. Neurosci Biobehav Rev 37:860–95 [DOI] [PubMed] [Google Scholar]
- 24.Admon R, Milad MR, Hendler T. 2013. A causal model of post-traumatic stress disorder: disentangling predisposed from acquired neural abnormalities. Trends Cogn Sci 17:337–47 [DOI] [PubMed] [Google Scholar]
- 25.Breslau N 2012. Epidemiology of posttraumatic stress disorder in adults The Oxford Handbook of Traumatic Stress Disorders. Oxford University Press: New York:84–97 [Google Scholar]
- 26.Bogic M, Njoku A, Priebe S. 2015. Long-term mental health of war-refugees: a systematic literature review. BMC Int Health Hum Rights 15:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuch K, Cox BJ. 1992. Symptoms of PTSD in 124 survivors of the Holocaust. Am J Psychiatry 149:337–40 [DOI] [PubMed] [Google Scholar]
- 28.Russo SJ, Nestler EJ. 2013. The brain reward circuitry in mood disorders. Nat Rev Neurosci 14:609–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abdallah CG, Sanacora G, Duman RS, Krystal JH. 2018. The neurobiology of depression, ketamine and rapid-acting antidepressants: Is it glutamate inhibition or activation? Pharmacol Ther [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duman RS, Aghajanian GK. 2012. Synaptic dysfunction in depression: potential therapeutic targets. Science 338:68–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanacora G, Banasr M. 2013. From pathophysiology to novel antidepressant drugs: glial contributions to the pathology and treatment of mood disorders. Biol Psychiatry 73:1172–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li SX, Han Y, Xu LZ, Yuan K, Zhang RX, et al. 2018. Uncoupling DAPK1 from NMDA receptor GluN2B subunit exerts rapid antidepressant-like effects. Mol Psychiatry 23:597–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Banasr M, Chowdhury GM, Terwilliger R, Newton SS, Duman RS, et al. 2010. Glial pathology in an animal model of depression: reversal of stress-induced cellular, metabolic and behavioral deficits by the glutamate-modulating drug riluzole. Molecular Psychiatry 15:501–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duman RS, Aghajanian GK, Sanacora G, Krystal JH. 2016. Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat Med 22:238–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hare B, Ghosal S, Duman R. 2017. Rapid acting antidepressants in chronic stress models: molecular and cellular mechanisms. Chronic Stress 1:1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S. 2002. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci 22:6810–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bennur S, Shankaranarayana Rao BS, Pawlak R, Strickland S, McEwen BS, Chattarji S. 2007. Stress-induced spine loss in the medial amygdala is mediated by tissue-plasminogen activator. Neuroscience 144:8–16 [DOI] [PubMed] [Google Scholar]
- 38.Lakshminarasimhan H, Chattarji S. 2012. Stress leads to contrasting effects on the levels of brain derived neurotrophic factor in the hippocampus and amygdala. PLoS One 7:e30481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mitra R, Jadhav S, McEwen BS, Vyas A, Chattarji S. 2005. Stress duration modulates the spatiotemporal patterns of spine formation in the basolateral amygdala. Proc Natl Acad Sci U S A 102:9371–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mitra R, Sapolsky RM. 2008. Acute corticosterone treatment is sufficient to induce anxiety and amygdaloid dendritic hypertrophy. Proc Natl Acad Sci U S A 105:5573–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conrad CD, LeDoux JE, Magarinos AM, McEwen BS. 1999. Repeated restraint stress facilitates fear conditioning independently of causing hippocampal CA3 dendritic atrophy. Behav Neurosci 113:902–13 [DOI] [PubMed] [Google Scholar]
- 42.Radley JJ, Rocher AB, Janssen WG, Hof PR, McEwen BS, Morrison JH. 2005. Reversibility of apical dendritic retraction in the rat medial prefrontal cortex following repeated stress. Exp Neurol 196:199–203 [DOI] [PubMed] [Google Scholar]
- 43.Vyas A, Pillai AG, Chattarji S. 2004. Recovery after chronic stress fails to reverse amygdaloid neuronal hypertrophy and enhanced anxiety-like behavior. Neuroscience 128:667–73 [DOI] [PubMed] [Google Scholar]
- 44.Christoffel DJ, Golden SA, Heshmati M, Graham A, Birnbaum S, et al. 2012. Effects of inhibitor of kappaB kinase activity in the nucleus accumbens on emotional behavior. Neuropsychopharmacology 37:2615–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muhammad A, Carroll C, Kolb B. 2012. Stress during development alters dendritic morphology in the nucleus accumbens and prefrontal cortex. Neuroscience 216:103–9 [DOI] [PubMed] [Google Scholar]
- 46.Warren BL, Sial OK, Alcantara LF, Greenwood MA, Brewer JS, et al. 2014. Altered gene expression and spine density in nucleus accumbens of adolescent and adult male mice exposed to emotional and physical stress. Dev Neurosci 36:250–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Christoffel DJ, Golden SA, Dumitriu D, Robison AJ, Janssen WG, et al. 2011. IkappaB kinase regulates social defeat stress-induced synaptic and behavioral plasticity. J Neurosci 31:314–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Campioni MR, Xu M, McGehee DS. 2009. Stress-induced changes in nucleus accumbens glutamate synaptic plasticity. J Neurophysiol 101:3192–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coplan JD, Lu D, El Sehamy AM, Tang C, Jackowski AP, et al. 2018. Early Life Stress Associated with Increased Striatal N-acetylaspartate (NAA): Cerebrospinal Fluid (CSF) Corticotropin-Releasing Factor (CRF) Concentrations, Hippocampal Volume, Body Mass and Behavioral Correlates. Chronic Stress 2:In Press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chaudhury D, Walsh JJ, Friedman AK, Juarez B, Ku SM, et al. 2013. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature 493:532–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walsh JJ, Friedman AK, Sun H, Heller EA, Ku SM, et al. 2014. Stress and CRF gate neural activation of BDNF in the mesolimbic reward pathway. Nat Neurosci 17:27–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wook Koo J, Labonte B, Engmann O, Calipari ES, Juarez B, et al. 2016. Essential Role of Mesolimbic Brain-Derived Neurotrophic Factor in Chronic Social Stress-Induced Depressive Behaviors. Biol Psychiatry 80:469–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krishnan V, Nestler EJ. 2008. The molecular neurobiology of depression. Nature 455:894–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Melo A, Kokras N, Dalla C, Ferreira C, Ventura-Silva AP, et al. 2015. The positive effect on ketamine as a priming adjuvant in antidepressant treatment. Translational psychiatry 5:e573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bremner JD, Randall P, Scott TM, Bronen RA, Seibyl JP, et al. 1995. MRI-based measurement of hippocampal volume in patients with combat-related posttraumatic stress disorder. Am J Psychiatry 152:973–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yehuda R, Golier JA, Tischler L, Harvey PD, Newmark R, et al. 2007. Hippocampal volume in aging combat veterans with and without post-traumatic stress disorder: relation to risk and resilience factors. J Psychiatr Res 41:435–45 [DOI] [PubMed] [Google Scholar]
- 57.Kuo JR, Kaloupek DG, Woodward SH. 2012. Amygdala volume in combat-exposed veterans with and without posttraumatic stress disorder: a cross-sectional study. Arch Gen Psychiatry 69:1080–6 [DOI] [PubMed] [Google Scholar]
- 58.Morey RA, Gold AL, LaBar KS, Beall SK, Brown VM, et al. 2012. Amygdala volume changes in posttraumatic stress disorder in a large case-controlled veterans group. Arch Gen Psychiatry 69:1169–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abdallah CG, Jackowski A, Salas R, Gupta S, Sato JR, et al. 2017. The Nucleus Accumbens and Ketamine Treatment in Major Depressive Disorder. Neuropsychopharmacology 42:1739–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abdallah CG, Coplan JD, Jackowski A, Sato JR, Mao X, et al. 2013. A pilot study of hippocampal volume and N-acetylaspartate (NAA) as response biomarkers in riluzole-treated patients with GAD. Eur Neuropsychopharmacol 23:276–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abdallah CG, Jackowski A, Sato JR, Mao X, Kang G, et al. 2015. Prefrontal cortical GABA abnormalities are associated with reduced hippocampal volume in major depressive disorder. Eur Neuropsychopharmacol 25:1082–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Valenti O, Gill KM, Grace AA. 2012. Different stressors produce excitation or inhibition of mesolimbic dopamine neuron activity: response alteration by stress pre-exposure. Eur J Neurosci 35:1312–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Holly EN, Miczek KA. 2016. Ventral tegmental area dopamine revisited: effects of acute and repeated stress. Psychopharmacology (Berl) 233:163–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tye KM, Mirzabekov JJ, Warden MR, Ferenczi EA, Tsai HC, et al. 2013. Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature 493:537–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krishnan V, Han MH, Graham DL, Berton O, Renthal W, et al. 2007. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 131:391–404 [DOI] [PubMed] [Google Scholar]
- 66.Flandreau EI, Toth M. 2017. Animal Models of PTSD: A Critical Review. Current topics in behavioral neurosciences [DOI] [PubMed] [Google Scholar]
- 67.Goswami S, Rodriguez-Sierra O, Cascardi M, Pare D. 2013. Animal models of post-traumatic stress disorder: face validity. Front Neurosci 7:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Izquierdo A, Wellman CL, Holmes A. 2006. Brief uncontrollable stress causes dendritic retraction in infralimbic cortex and resistance to fear extinction in mice. J Neurosci 26:5733–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shalev AY, Freedman S, Peri T, Brandes D, Sahar T, et al. 1998. Prospective study of posttraumatic stress disorder and depression following trauma. Am J Psychiatry 155:630–7 [DOI] [PubMed] [Google Scholar]
- 70.Abdallah CG, Wrocklage KM, Averill CL, Akiki T, Schweinsburg B, et al. 2017. Anterior hippocampal dysconnectivity in posttraumatic stress disorder: a dimensional and multimodal approach. Translational psychiatry 7:e1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Akiki TJ, Averill CL, Wrocklage KM, Schweinsburg B, Scott JC, et al. 2017. The Association of PTSD Symptom Severity with Localized Hippocampus and Amygdala Abnormalities. Chronic Stress 1:2470547017724069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Averill CL, Satodiya RM, Scott JC, Wrocklage KM, Schweinsburg B, et al. 2017. Posttraumatic Stress Disorder and Depression Symptom Severities Are Differentially Associated With Hippocampal Subfield Volume Loss in Combat Veterans. Chronic Stress 1:2470547017744538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Averill LA, Abdallah CG, Pietrzak RH, Averill CL, Southwick SM, et al. 2017. Combat Exposure Severity is Associated with Reduced Cortical Thickness in Combat Veterans: A Preliminary Report. Chronic Stress 1:2470547017724714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pietrzak RH, Averill LA, Abdallah CG, Neumeister A, Krystal JH, et al. 2015. Amygdala-hippocampal volume and the phenotypic heterogeneity of posttraumatic stress disorder: a cross-sectional study. JAMA psychiatry 72:396–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wrocklage KM, Averill LA, Cobb Scott J, Averill CL, Schweinsburg B, et al. 2017. Cortical thickness reduction in combat exposed U.S. veterans with and without PTSD. Eur Neuropsychopharmacol 27:515–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Southwick SM, Krystal JH, Bremner JD, Morgan CA 3rd, Nicolaou AL, et al. 1997. Noradrenergic and serotonergic function in posttraumatic stress disorder. Arch Gen Psychiatry 54:749–58 [DOI] [PubMed] [Google Scholar]
- 77.Southwick SM, Krystal JH, Morgan CA, Johnson D, Nagy LM, et al. 1993. Abnormal noradrenergic function in posttraumatic stress disorder. Arch Gen Psychiatry 50:266–74 [DOI] [PubMed] [Google Scholar]
- 78.Abdallah CG, Averill LA, Krystal JH, Southwick SM, Arnsten AF. 2016. Glutamate and norepinephrine interaction: Relevance to higher cognitive operations and psychopathology. Behav Brain Sci 39:e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Arnsten AF. 2015. Stress weakens prefrontal networks: molecular insults to higher cognition. Nat Neurosci 18:1376–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Logue MW, van Rooij SJH, Dennis EL, Davis SL, Hayes JP, et al. 2018. Smaller Hippocampal Volume in Posttraumatic Stress Disorder: A Multisite ENIGMA-PGC Study: Subcortical Volumetry Results From Posttraumatic Stress Disorder Consortia. Biol Psychiatry 83:244–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schmaal L, Veltman DJ, van Erp TG, Samann PG, Frodl T, et al. 2016. Subcortical brain alterations in major depressive disorder: findings from the ENIGMA Major Depressive Disorder working group. Mol Psychiatry 21:806–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.O’Doherty DC, Chitty KM, Saddiqui S, Bennett MR, Lagopoulos J. 2015. A systematic review and meta-analysis of magnetic resonance imaging measurement of structural volumes in posttraumatic stress disorder. Psychiatry Res 232:1–33 [DOI] [PubMed] [Google Scholar]
- 83.Kempton MJ, Salvador Z, Munafo MR, Geddes JR, Simmons A, et al. 2011. Structural neuroimaging studies in major depressive disorder. Meta-analysis and comparison with bipolar disorder. Archives of General Psychiatry 68:675–90 [DOI] [PubMed] [Google Scholar]
- 84.Kwon JS, Shin YW, Kim CW, Kim YI, Youn T, et al. 2003. Similarity and disparity of obsessive-compulsive disorder and schizophrenia in MR volumetric abnormalities of the hippocampus-amygdala complex. J Neurol Neurosurg Psychiatry 74:962–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Anticevic A, Brumbaugh MS, Winkler AM, Lombardo LE, Barrett J, et al. 2013. Global prefrontal and fronto-amygdala dysconnectivity in bipolar I disorder with psychosis history. Biol Psychiatry 73:565–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Anticevic A, Hu S, Zhang S, Savic A, Billingslea E, et al. 2014. Global resting-state functional magnetic resonance imaging analysis identifies frontal cortex, striatal, and cerebellar dysconnectivity in obsessive-compulsive disorder. Biol Psychiatry 75:595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Haukvik UK, Westlye LT, Morch-Johnsen L, Jorgensen KN, Lange EH, et al. 2015. In vivo hippocampal subfield volumes in schizophrenia and bipolar disorder. Biol Psychiatry 77:581–8 [DOI] [PubMed] [Google Scholar]
- 88.Syed SA, Nemeroff CB. 2017. Early Life Stress, Mood, and Anxiety Disorders. Chronic Stress 1:2470547017694461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Matosin N, Cruceanu C, Binder EB. 2017. Preclinical and Clinical Evidence of DNA Methylation Changes in Response to Trauma and Chronic Stress. Chronic Stress 1:2470547017710764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sheth C, McGlade E, Yurgelun-Todd D. 2017. Chronic Stress in Adolescents and Its Neurobiological and Psychopathological Consequences: An RDoC Perspective. Chronic Stress 1:2470547017715645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Adams TG, Kelmendi B, Brake CA, Gruner P, Badour CL, Pittenger C. 2018. The Role of Stress in the Pathogenesis and Maintenance of Obsessive-Compulsive Disorder. Chronic Stress 2:2470547018758043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Goddard AW. 2017. The Neurobiology of Panic: A Chronic Stress Disorder. Chronic Stress 1:2470547017736038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zanos P, Thompson SM, Duman RS, Zarate CA Jr., Gould TD. 2018. Convergent Mechanisms Underlying Rapid Antidepressant Action. CNS Drugs 32:197–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Abdallah CG, Salas R, Jackowski A, Baldwin P, Sato JR, Mathew SJ. 2015. Hippocampal volume and the rapid antidepressant effect of ketamine. J Psychopharmacol 29:591–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pitman RK, Rasmusson AM, Koenen KC, Shin LM, Orr SP, et al. 2012. Biological studies of post-traumatic stress disorder. Nat Rev Neurosci 13:769–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Admon R, Lubin G, Stern O, Rosenberg K, Sela L, et al. 2009. Human vulnerability to stress depends on amygdala’s predisposition and hippocampal plasticity. Proc Natl Acad Sci U S A 106:14120–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gilbertson MW, Shenton ME, Ciszewski A, Kasai K, Lasko NB, et al. 2002. Smaller hippocampal volume predicts pathologic vulnerability to psychological trauma. Nat Neurosci 5:1242–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schwabe L, Dalm S, Schachinger H, Oitzl MS. 2008. Chronic stress modulates the use of spatial and stimulus-response learning strategies in mice and man. Neurobiol Learn Mem 90:495–503 [DOI] [PubMed] [Google Scholar]
- 99.Schwabe L, Joels M, Roozendaal B, Wolf OT, Oitzl MS. 2012. Stress effects on memory: an update and integration. Neurosci Biobehav Rev 36:1740–9 [DOI] [PubMed] [Google Scholar]
- 100.de Quervain D, Schwabe L, Roozendaal B. 2017. Stress, glucocorticoids and memory: implications for treating fear-related disorders. Nat Rev Neurosci 18:7–19 [DOI] [PubMed] [Google Scholar]
- 101.Koch SB, van Zuiden M, Nawijn L, Frijling JL, Veltman DJ, Olff M. 2016. Aberrant Resting-State Brain Activity in Posttraumatic Stress Disorder: A Meta-Analysis and Systematic Review. Depress Anxiety 33:592–605 [DOI] [PubMed] [Google Scholar]
- 102.Menon V 2011. Large-scale brain networks and psychopathology: a unifying triple network model. Trends Cogn Sci 15:483–506 [DOI] [PubMed] [Google Scholar]
- 103.Akiki TJ, Averill CL, Abdallah CG. 2017. A Network-Based Neurobiological Model of PTSD: Evidence From Structural and Functional Neuroimaging Studies. Curr Psychiatry Rep 19:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Akiki TJ, Abdallah CG. 2018. Determining the Hierarchical Architecture of the Human Brain Using Subject-Level Clustering of Functional Networks. bioRxiv [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Buckner RL, Andrews-Hanna JR, Schacter DL. 2008. The brain’s default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci 1124:1–38 [DOI] [PubMed] [Google Scholar]
- 106.Akiki TJ, Averill CL, Wrocklage KM, Scott JC, Averill LA, et al. 2018. Default mode network abnormalities in posttraumatic stress disorder: A novel network-restricted topology approach. Neuroimage 176:489–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sripada RK, King AP, Welsh RC, Garfinkel SN, Wang X, et al. 2012. Neural dysregulation in posttraumatic stress disorder: evidence for disrupted equilibrium between salience and default mode brain networks. Psychosom Med 74:904–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.King AP, Block SR, Sripada RK, Rauch S, Giardino N, et al. 2016. Altered Default Mode Network (Dmn) Resting State Functional Connectivity Following a Mindfulness-Based Exposure Therapy for Posttraumatic Stress Disorder (Ptsd) in Combat Veterans of Afghanistan and Iraq. Depress Anxiety 33:289–99 [DOI] [PubMed] [Google Scholar]
- 109.Cisler JM, Scott Steele J, Smitherman S, Lenow JK, Kilts CD. 2013. Neural processing correlates of assaultive violence exposure and PTSD symptoms during implicit threat processing: a network-level analysis among adolescent girls. Psychiatry Res 214:238–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Goulden N, Khusnulina A, Davis NJ, Bracewell RM, Bokde AL, et al. 2014. The salience network is responsible for switching between the default mode network and the central executive network: replication from DCM. Neuroimage 99:180–90 [DOI] [PubMed] [Google Scholar]
- 111.Brown VM, LaBar KS, Haswell CC, Gold AL, Mid-Atlantic MW, et al. 2014. Altered resting-state functional connectivity of basolateral and centromedial amygdala complexes in posttraumatic stress disorder. Neuropsychopharmacology 39:351–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Regier DA, Narrow WE, Clarke DE, Kraemer HC, Kuramoto SJ, et al. 2013. DSM-5 field trials in the United States and Canada, Part II: test-retest reliability of selected categorical diagnoses. Am J Psychiatry 170:59–70 [DOI] [PubMed] [Google Scholar]
- 113.Krystal JH, Rosenheck RA, Cramer JA, Vessicchio JC, Jones KM, et al. 2011. Adjunctive risperidone treatment for antidepressant-resistant symptoms of chronic military service-related PTSD: a randomized trial. JAMA 306:493–502 [DOI] [PubMed] [Google Scholar]
- 114.Raskind MA, Peskind ER, Chow B, Harris C, Davis-Karim A, et al. 2018. Trial of Prazosin for Post-Traumatic Stress Disorder in Military Veterans. N Engl J Med 378:507–17 [DOI] [PubMed] [Google Scholar]
- 115.Forbes D, Creamer M, Bisson JI, Cohen JA, Crow BE, et al. 2010. A guide to guidelines for the treatment of PTSD and related conditions. J Trauma Stress 23:537–52 [DOI] [PubMed] [Google Scholar]
- 116.Delgado PL, Miller HL, Salomon RM, Licinio J, Krystal JH, et al. 1999. Tryptophan-depletion challenge in depressed patients treated with desipramine or fluoxetine: implications for the role of serotonin in the mechanism of antidepressant action. Biol Psychiatry 46:212–20 [DOI] [PubMed] [Google Scholar]
- 117.Coplan JD, Gopinath S, Abdallah CG, Berry BR. 2014. A neurobiological hypothesis of treatment-resistant depression - mechanisms for selective serotonin reuptake inhibitor non-efficacy. Frontiers in behavioral neuroscience 8:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Duman RS, Heninger GR, Nestler EJ. 1997. A molecular and cellular theory of depression. Arch Gen Psychiatry 54:597–606 [DOI] [PubMed] [Google Scholar]
- 119.Friedman MJ, Bernardy NC. 2016. Considering future pharmacotherapy for PTSD. Neurosci Lett [DOI] [PubMed] [Google Scholar]
- 120.Cipriani A, Williams T, Nikolakopoulou A, Salanti G, Chaimani A, et al. 2017. Comparative efficacy and acceptability of pharmacological treatments for post-traumatic stress disorder in adults: a network meta-analysis. Psychol Med:1–10 [DOI] [PubMed] [Google Scholar]
- 121.Watts BV, Schnurr PP, Mayo L, Young-Xu Y, Weeks WB, Friedman MJ. 2013. Meta-analysis of the efficacy of treatments for posttraumatic stress disorder. J Clin Psychiatry 74:e541–50 [DOI] [PubMed] [Google Scholar]
- 122.Bernardy NC, Friedman MJ. 2017. Pharmacological management of posttraumatic stress disorder. Curr Opin Psychol 14:116–21 [DOI] [PubMed] [Google Scholar]
- 123.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, et al. 2000. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 47:351–4 [DOI] [PubMed] [Google Scholar]
- 124.Abdallah CG, Averill LA, Krystal JH. 2015. Ketamine as a promising prototype for a new generation of rapid-acting antidepressants. Ann N Y Acad Sci 1344:66–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.McGirr A, Berlim MT, Bond DJ, Fleck MP, Yatham LN, Lam RW. 2015. A systematic review and meta-analysis of randomized, double-blind, placebo-controlled trials of ketamine in the rapid treatment of major depressive episodes. Psychol Med 45:693–704 [DOI] [PubMed] [Google Scholar]
- 126.Abdallah CG, De Feyter HM, Averill LA, Jiang L, Averill CL, et al. 2018. The effects of ketamine on prefrontal glutamate neurotransmission in healthy and depressed subjects. Neuropsychopharmacology [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, et al. 2011. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry 69:754–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Abdallah CG, Averill CL, Salas R, Averill LA, Baldwin PR, et al. 2017. Prefrontal Connectivity and Glutamate Transmission: Relevance to Depression Pathophysiology and Ketamine Treatment. Biol Psychiatry Cogn Neurosci Neuroimaging 2:566–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Abdallah CG, Averill LA, Collins KA, Geha P, Schwartz J, et al. 2017. Ketamine Treatment and Global Brain Connectivity in Major Depression. Neuropsychopharmacology 42:1210–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Glue P, Medlicott NJ, Harland S, Neehoff S, Anderson-Fahey B, et al. 2017. Ketamine’s dose-related effects on anxiety symptoms in patients with treatment refractory anxiety disorders. J Psychopharmacol 31:1302–5 [DOI] [PubMed] [Google Scholar]
- 131.Ivan Ezquerra-Romano I, Lawn W, Krupitsky E, Morgan CJA. 2018. Ketamine for the treatment of addiction: Evidence and potential mechanisms. Neuropharmacology [DOI] [PubMed] [Google Scholar]
- 132.Rodriguez CI, Kegeles LS, Levinson A, Feng T, Marcus SM, et al. 2013. Randomized controlled crossover trial of ketamine in obsessive-compulsive disorder: proof-of-concept. Neuropsychopharmacology 38:2475–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Taylor JH, Landeros-Weisenberger A, Coughlin C, Mulqueen J, Johnson JA, et al. 2018. Ketamine for Social Anxiety Disorder: A Randomized, Placebo-Controlled Crossover Trial. Neuropsychopharmacology 43:325–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wilkinson ST, Ballard ED, Bloch MH, Mathew SJ, Murrough JW, et al. 2018. The Effect of a Single Dose of Intravenous Ketamine on Suicidal Ideation: A Systematic Review and Individual Participant Data Meta-Analysis. Am J Psychiatry 175:150–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Grunebaum MF, Galfalvy HC, Choo TH, Keilp JG, Moitra VK, et al. 2018. Ketamine for Rapid Reduction of Suicidal Thoughts in Major Depression: A Midazolam-Controlled Randomized Clinical Trial. Am J Psychiatry 175:327–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Feder A, Parides MK, Murrough JW, Perez AM, Morgan JE, et al. 2014. Efficacy of intravenous ketamine for treatment of chronic posttraumatic stress disorder: a randomized clinical trial. JAMA psychiatry 71:681–8 [DOI] [PubMed] [Google Scholar]
- 137.Hartberg J, Garrett-Walcott S, De Gioannis A. 2018. Impact of oral ketamine augmentation on hospital admissions in treatment-resistant depression and PTSD: a retrospective study. Psychopharmacology (Berl) 235:393–8 [DOI] [PubMed] [Google Scholar]
- 138.Guina J, Rossetter SR, De RB, Nahhas RW, Welton RS. 2015. Benzodiazepines for PTSD: A Systematic Review and Meta-Analysis. J Psychiatr Pract 21:281–303 [DOI] [PubMed] [Google Scholar]
- 139.Yehuda R, Southwick SM, Nussbaum G, Wahby V, Giller EL Jr., Mason JW 1990. Low urinary cortisol excretion in patients with posttraumatic stress disorder. J Nerv Ment Dis 178:366–9 [DOI] [PubMed] [Google Scholar]
- 140.Howlett JR, Stein MB. 2016. Prevention of Trauma and Stressor-Related Disorders: A Review. Neuropsychopharmacology 41:357–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sippel LM, Allington CE, Pietrzak RH, Harpaz-Rotem I, Mayes LC, Olff M. 2017. Oxytocin and Stress-related Disorders: Neurobiological Mechanisms and Treatment Opportunities. Chronic Stress 1:2470547016687996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Walker DL, Ressler KJ, Lu KT, Davis M. 2002. Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of D-cycloserine as assessed with fear-potentiated startle in rats. J Neurosci 22:2343–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Litz BT, Salters-Pedneault K, Steenkamp MM, Hermos JA, Bryant RA, et al. 2012. A randomized placebo-controlled trial of D-cycloserine and exposure therapy for posttraumatic stress disorder. J Psychiatr Res 46:1184–90 [DOI] [PubMed] [Google Scholar]
- 144.Brunet A, Saumier D, Liu A, Streiner DL, Tremblay J, Pitman RK. 2018. Reduction of PTSD Symptoms With Pre-Reactivation Propranolol Therapy: A Randomized Controlled Trial. Am J Psychiatry:appiajp201717050481 [DOI] [PubMed] [Google Scholar]
- 145.Kelmendi B, Adams TG, Yarnell S, Southwick S, Abdallah CG, Krystal JH. 2016. PTSD: from neurobiology to pharmacological treatments. Eur J Psychotraumatol 7:31858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Amoroso T, Workman M. 2016. Treating posttraumatic stress disorder with MDMA-assisted psychotherapy: A preliminary meta-analysis and comparison to prolonged exposure therapy. J Psychopharmacol 30:595–600 [DOI] [PubMed] [Google Scholar]
- 147.Davidson J, Baldwin D, Stein DJ, Kuper E, Benattia I, et al. 2006. Treatment of posttraumatic stress disorder with venlafaxine extended release: a 6-month randomized controlled trial. Arch Gen Psychiatry 63:1158–65 [DOI] [PubMed] [Google Scholar]
- 148.Villarreal G, Hamner MB, Canive JM, Robert S, Calais LA, et al. 2016. Efficacy of Quetiapine Monotherapy in Posttraumatic Stress Disorder: A Randomized, Placebo-Controlled Trial. Am J Psychiatry 173:1205–12 [DOI] [PubMed] [Google Scholar]
- 149.Khachatryan D, Groll D, Booij L, Sepehry AA, Schutz CG. 2016. Prazosin for treating sleep disturbances in adults with posttraumatic stress disorder: a systematic review and meta-analysis of randomized controlled trials. Gen Hosp Psychiatry 39:46–52 [DOI] [PubMed] [Google Scholar]
- 150.Raskind MA, Peterson K, Williams T, Hoff DJ, Hart K, et al. 2013. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry 170:1003–10 [DOI] [PubMed] [Google Scholar]